
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain and Structure information: based on WT protein | Annotated databases | Deep learning-based pathogenicity predictions | Folding stability-based pathogenicity predictions | Sequence/structure-based pathogenicity predictions | Phase Separation | Evolutionary/physical properties | Molecular Dynamics-based analysis | DOI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | IUPred2 | ANCHOR2 | AlphaFold | MobiDB | PhosphoSitePlus | ClinVar | gnomAD | ESM1b | AlphaMissense | FoldX | Rosetta | Foldetta | PremPS | REVEL | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PSMutPred | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | |||||||||||||||||||||||||||||||||||||||||||||
| Score | Prediction | Score | Prediction | pLDDT | disorder | disorder | LTP | HTP | KL | PTM | Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | IP RF | SP RF | Prediction | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.3064C>A | L1022I 2D  AIThe SynGAP1 missense variant L1022I is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions strongly supports a benign classification, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.859585 | Disordered | 0.986981 | Binding | 0.339 | 0.752 | 0.500 | -4.624 | Likely Benign | 0.166 | Likely Benign | Likely Benign | 0.025 | Likely Benign | -0.91 | Neutral | 0.114 | Benign | 0.072 | Benign | 2.58 | Benign | 0.11 | Tolerated | 0.1081 | 0.4395 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3064C>G | L1022V 2D  AIThe SynGAP1 missense variant L1022V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, creating a single discordant prediction. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.859585 | Disordered | 0.986981 | Binding | 0.339 | 0.752 | 0.500 | -3.957 | Likely Benign | 0.174 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -1.22 | Neutral | 0.664 | Possibly Damaging | 0.260 | Benign | 2.66 | Benign | 0.07 | Tolerated | 0.1702 | 0.4045 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3064C>T | L1022F 2D  AIThe SynGAP1 missense variant L1022F is reported in gnomAD (variant ID 6‑33443616‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion is not contradicted by any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.859585 | Disordered | 0.986981 | Binding | 0.339 | 0.752 | 0.500 | 6-33443616-C-T | 2 | 1.24e-6 | -5.035 | Likely Benign | 0.351 | Ambiguous | Likely Benign | 0.072 | Likely Benign | -1.84 | Neutral | 0.971 | Probably Damaging | 0.801 | Possibly Damaging | 2.51 | Benign | 0.07 | Tolerated | 3.77 | 5 | 0.0730 | 0.4231 | 0 | 2 | -1.0 | 34.02 | ||||||||||||||||||||||||||||||||||
| c.3065T>A | L1022H 2D  AIThe SynGAP1 missense variant L1022H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie (2 pathogenic, 2 benign) and is therefore inconclusive. Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the majority of conventional predictors lean toward pathogenicity, whereas the single high‑accuracy tool predicts benign and the consensus is unresolved. Thus, the variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.859585 | Disordered | 0.986981 | Binding | 0.339 | 0.752 | 0.500 | -2.473 | Likely Benign | 0.589 | Likely Pathogenic | Likely Benign | 0.140 | Likely Benign | -2.21 | Neutral | 0.999 | Probably Damaging | 0.944 | Probably Damaging | 2.49 | Pathogenic | 0.00 | Affected | 0.1165 | 0.1313 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3065T>C | L1022P 2D  AIThe SynGAP1 missense variant L1022P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic, two benign), and Foldetta data are unavailable. Overall, the balance of evidence favors a pathogenic interpretation, and this assessment does not conflict with ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.859585 | Disordered | 0.986981 | Binding | 0.339 | 0.752 | 0.500 | -2.532 | Likely Benign | 0.643 | Likely Pathogenic | Likely Benign | 0.177 | Likely Benign | -2.22 | Neutral | 0.995 | Probably Damaging | 0.925 | Probably Damaging | 2.49 | Pathogenic | 0.01 | Affected | 0.3332 | 0.1589 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3065T>G | L1022R 2D  AIThe SynGAP1 missense variant L1022R is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict the ClinVar status, which currently contains no classification for L1022R. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.859585 | Disordered | 0.986981 | Binding | 0.339 | 0.752 | 0.500 | -2.875 | Likely Benign | 0.659 | Likely Pathogenic | Likely Benign | 0.183 | Likely Benign | -1.96 | Neutral | 0.986 | Probably Damaging | 0.894 | Possibly Damaging | 2.51 | Benign | 0.01 | Affected | 0.1279 | 0.1387 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3067T>A | S1023T 2D  AIThe SynGAP1 missense variant S1023T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, and Foldetta results are unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no conflict with ClinVar status. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.831250 | Disordered | 0.990262 | Binding | 0.322 | 0.750 | 0.500 | -5.573 | Likely Benign | 0.360 | Ambiguous | Likely Benign | 0.092 | Likely Benign | -1.62 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 2.49 | Pathogenic | 0.04 | Affected | 0.1299 | 0.5370 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3067T>C | S1023P 2D  AIThe SynGAP1 missense variant S1023P is reported in gnomAD (ID 6‑33443619‑T‑C) but has no ClinVar entry (ClinVar status: not reported). Functional prediction tools are split: benign calls come from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls come from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, SGM Consensus remains inconclusive, and Foldetta (combining FoldX‑MD and Rosetta) has no available result. Overall, the majority of standard predictors lean toward pathogenicity, whereas the few high‑accuracy tools do not support a pathogenic verdict. Thus, the variant is most likely pathogenic based on the aggregate predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.831250 | Disordered | 0.990262 | Binding | 0.322 | 0.750 | 0.500 | 6-33443619-T-C | 2 | 1.24e-6 | -5.634 | Likely Benign | 0.679 | Likely Pathogenic | Likely Benign | 0.146 | Likely Benign | -2.11 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.43 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.1818 | 0.4616 | -1 | 1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||
| c.3067T>G | S1023A 2D  AIThe SynGAP1 missense variant S1023A is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign, while the majority of pathogenic predictors—polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT—suggest a damaging impact. The AlphaMissense‑Default score is uncertain, and Foldetta stability analysis is unavailable. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy tools therefore lean toward a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus indicates likely benign, and no Foldetta data is available. Overall, the computational evidence supports a benign classification, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.990262 | Binding | 0.322 | 0.750 | 0.500 | -6.031 | Likely Benign | 0.356 | Ambiguous | Likely Benign | 0.098 | Likely Benign | -1.59 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 2.53 | Benign | 0.04 | Affected | 0.4427 | 0.4386 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3068C>T | S1023L 2D  AIThe SynGAP1 missense variant S1023L is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, while the SGM‑Consensus remains Likely Pathogenic; Foldetta stability analysis is unavailable. Overall, the balance of evidence from the majority of tools and the SGM‑Consensus indicates a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.831250 | Disordered | 0.990262 | Binding | 0.322 | 0.750 | 0.500 | -5.735 | Likely Benign | 0.705 | Likely Pathogenic | Likely Benign | 0.204 | Likely Benign | -3.47 | Deleterious | 0.991 | Probably Damaging | 0.991 | Probably Damaging | 2.47 | Pathogenic | 0.01 | Affected | 0.1080 | 0.4923 | -3 | -2 | 4.6 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.306G>C | L102F 2D AIThe SynGAP1 missense variant L102F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.696014 | Binding | 0.357 | 0.885 | 0.625 | -4.712 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.144 | Likely Benign | -0.80 | Neutral | 0.984 | Probably Damaging | 0.969 | Probably Damaging | 4.13 | Benign | 0.00 | Affected | 0.0863 | 0.3205 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.306G>T | L102F 2D AIThe SynGAP1 missense variant L102F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.696014 | Binding | 0.357 | 0.885 | 0.625 | -4.712 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.153 | Likely Benign | -0.80 | Neutral | 0.984 | Probably Damaging | 0.969 | Probably Damaging | 4.13 | Benign | 0.00 | Affected | 0.0863 | 0.3205 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3070C>A | L1024I 2D AIThe SynGAP1 missense variant L1024I is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -4.794 | Likely Benign | 0.260 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -0.99 | Neutral | 0.959 | Probably Damaging | 0.642 | Possibly Damaging | 2.45 | Pathogenic | 0.11 | Tolerated | 0.0992 | 0.3735 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3070C>G | L1024V 2D AIThe SynGAP1 missense variant L1024V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for the variant. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -3.758 | Likely Benign | 0.244 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.33 | Neutral | 0.907 | Possibly Damaging | 0.642 | Possibly Damaging | 2.47 | Pathogenic | 0.08 | Tolerated | 0.1459 | 0.3185 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3070C>T | L1024F 2D AIThe SynGAP1 missense variant L1024F is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it contains both benign and pathogenic calls. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence favors a benign interpretation, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -5.133 | Likely Benign | 0.544 | Ambiguous | Likely Benign | 0.059 | Likely Benign | -2.08 | Neutral | 0.994 | Probably Damaging | 0.924 | Probably Damaging | 2.40 | Pathogenic | 0.07 | Tolerated | 0.0706 | 0.3708 | 2 | 0 | -1.0 | 34.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3071T>A | L1024H 2D AIThe SynGAP1 missense variant L1024H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized returns an Uncertain result, SGM‑Consensus indicates Likely Pathogenic, and Foldetta data are unavailable. Overall, the majority of evidence points toward a deleterious effect, suggesting the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -3.271 | Likely Benign | 0.868 | Likely Pathogenic | Ambiguous | 0.123 | Likely Benign | -3.09 | Deleterious | 1.000 | Probably Damaging | 0.981 | Probably Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.1198 | 0.1483 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3071T>C | L1024P 2D AIThe SynGAP1 missense variant L1024P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of standard prediction tools lean toward pathogenicity, while high‑accuracy methods are inconclusive. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -4.385 | Likely Benign | 0.730 | Likely Pathogenic | Likely Benign | 0.149 | Likely Benign | -2.42 | Neutral | 0.999 | Probably Damaging | 0.974 | Probably Damaging | 2.43 | Pathogenic | 0.03 | Affected | 0.3187 | 0.2033 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3071T>G | L1024R 2D AIThe SynGAP1 missense variant L1024R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (5 pathogenic vs. 3 benign) lean toward a pathogenic interpretation. This assessment does not contradict ClinVar status, as the variant is not yet catalogued there. Thus, based on current computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -3.434 | Likely Benign | 0.841 | Likely Pathogenic | Ambiguous | 0.148 | Likely Benign | -2.41 | Neutral | 0.997 | Probably Damaging | 0.962 | Probably Damaging | 2.40 | Pathogenic | 0.02 | Affected | 0.1335 | 0.1557 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3073C>A | Q1025K 2D  AIThe SynGAP1 missense variant Q1025K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: benign calls come from REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas the only pathogenic call is from polyPhen‑2 HumDiv. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which has no pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -4.510 | Likely Benign | 0.529 | Ambiguous | Likely Benign | 0.041 | Likely Benign | -1.09 | Neutral | 0.649 | Possibly Damaging | 0.353 | Benign | 2.78 | Benign | 0.22 | Tolerated | 0.1690 | 0.4438 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3073C>G | Q1025E 2D  AIThe SynGAP1 missense variant Q1025E is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only polyPhen‑2 HumDiv predicts it as pathogenic. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -3.010 | Likely Benign | 0.254 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.60 | Neutral | 0.649 | Possibly Damaging | 0.353 | Benign | 2.79 | Benign | 1.00 | Tolerated | 0.1391 | 0.2269 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3074A>C | Q1025P 2D  AIThe SynGAP1 missense variant Q1025P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -2.151 | Likely Benign | 0.211 | Likely Benign | Likely Benign | 0.153 | Likely Benign | -1.22 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.72 | Benign | 0.11 | Tolerated | 0.2211 | 0.5196 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3074A>G | Q1025R 2D  AIThe SynGAP1 missense variant Q1025R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (both HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -4.435 | Likely Benign | 0.480 | Ambiguous | Likely Benign | 0.101 | Likely Benign | -1.28 | Neutral | 0.818 | Possibly Damaging | 0.453 | Possibly Damaging | 2.72 | Benign | 0.10 | Tolerated | 0.1342 | 0.2656 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3074A>T | Q1025L 2D  AIThe SynGAP1 missense variant Q1025L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -6.460 | Likely Benign | 0.463 | Ambiguous | Likely Benign | 0.117 | Likely Benign | -2.48 | Neutral | 0.901 | Possibly Damaging | 0.534 | Possibly Damaging | 2.70 | Benign | 0.05 | Affected | 0.0798 | 0.5497 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3075G>C | Q1025H 2D  AIThe SynGAP1 missense variant Q1025H is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence supports a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -4.976 | Likely Benign | 0.405 | Ambiguous | Likely Benign | 0.051 | Likely Benign | -1.43 | Neutral | 0.014 | Benign | 0.012 | Benign | 2.68 | Benign | 0.05 | Affected | 0.1332 | 0.3852 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3075G>T | Q1025H 2D AIThe SynGAP1 missense variant Q1025H is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence supports a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -4.976 | Likely Benign | 0.405 | Ambiguous | Likely Benign | 0.051 | Likely Benign | -1.43 | Neutral | 0.014 | Benign | 0.012 | Benign | 2.68 | Benign | 0.05 | Affected | 0.1332 | 0.3852 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3076G>A | D1026N 2D  AIThe SynGAP1 D1026N variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and therefore reports a likely benign outcome. AlphaMissense‑Optimized also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -4.166 | Likely Benign | 0.608 | Likely Pathogenic | Likely Benign | 0.117 | Likely Benign | -2.07 | Neutral | 0.411 | Benign | 0.239 | Benign | 2.55 | Benign | 0.01 | Affected | 0.1240 | 0.5060 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3076G>C | D1026H 2D  AIThe SynGAP1 missense variant D1026H is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443628‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | 6-33443628-G-C | 1 | 6.20e-7 | -4.412 | Likely Benign | 0.900 | Likely Pathogenic | Ambiguous | 0.105 | Likely Benign | -2.03 | Neutral | 0.832 | Possibly Damaging | 0.600 | Possibly Damaging | 2.48 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1470 | 0.5345 | -1 | 1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||
| c.3076G>T | D1026Y 2D  AIThe SynGAP1 missense variant D1026Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default all classify the variant as damaging. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -6.999 | Likely Benign | 0.892 | Likely Pathogenic | Ambiguous | 0.200 | Likely Benign | -3.08 | Deleterious | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 2.47 | Pathogenic | 0.00 | Affected | 0.0676 | 0.4936 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.3077A>C | D1026A 2D  AIThe SynGAP1 D1026A variant is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM, while those that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a tie, and Foldetta results are not available. Overall, the majority of standard tools favor a benign interpretation, and no ClinVar entry contradicts this assessment. Thus, the variant is most likely benign based on current predictions, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -4.211 | Likely Benign | 0.849 | Likely Pathogenic | Ambiguous | 0.070 | Likely Benign | -2.69 | Deleterious | 0.112 | Benign | 0.061 | Benign | 2.53 | Benign | 0.02 | Affected | 0.3392 | 0.5279 | 0 | -2 | 5.3 | -44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3077A>G | D1026G 2D  AIThe SynGAP1 missense variant D1026G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign, two pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -4.125 | Likely Benign | 0.691 | Likely Pathogenic | Likely Benign | 0.098 | Likely Benign | -2.84 | Deleterious | 0.001 | Benign | 0.005 | Benign | 2.67 | Benign | 0.01 | Affected | 0.3377 | 0.5042 | 1 | -1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3077A>T | D1026V 2D  AIThe SynGAP1 missense variant D1026V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Tools that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and therefore also indicates a likely pathogenic outcome. AlphaMissense‑Optimized is uncertain, and no Foldetta stability result is available, so it does not contribute evidence. Overall, the majority of reliable predictors classify the variant as pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -5.871 | Likely Benign | 0.900 | Likely Pathogenic | Ambiguous | 0.144 | Likely Benign | -3.13 | Deleterious | 0.004 | Benign | 0.004 | Benign | 2.48 | Pathogenic | 0.00 | Affected | 0.0957 | 0.5236 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3078C>A | D1026E 2D  AIThe SynGAP1 missense variant D1026E is listed in ClinVar (ID 4746138) as benign and is not reported in gnomAD. Across the spectrum of in‑silico predictors, every tool examined—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classifies the substitution as benign. No tool predicts pathogenicity. Grouping by agreement, all predictors fall into the benign category, with no pathogenic calls. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status remains unavailable. Overall, the computational evidence strongly supports a benign classification, fully consistent with the ClinVar designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | Benign | 1 | -3.431 | Likely Benign | 0.190 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.37 | Neutral | 0.001 | Benign | 0.005 | Benign | 2.73 | Benign | 0.56 | Tolerated | 0.1449 | 0.4783 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.3078C>G | D1026E 2D AIThe SynGAP1 missense variant D1026E is not reported in ClinVar and is absent from gnomAD. All available in silico predictors classify the change as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly supports a benign classification, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -3.431 | Likely Benign | 0.190 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.37 | Neutral | 0.001 | Benign | 0.005 | Benign | 2.73 | Benign | 0.56 | Tolerated | 0.1449 | 0.4783 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3079A>C | N1027H 2D  AIThe SynGAP1 missense variant N1027H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions, including the high‑accuracy tools, indicate that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -4.185 | Likely Benign | 0.193 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -1.44 | Neutral | 0.970 | Probably Damaging | 0.799 | Possibly Damaging | 2.74 | Benign | 0.07 | Tolerated | 0.1345 | 0.7176 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||||||||||
| c.3079A>G | N1027D 2D  AIThe SynGAP1 missense variant N1027D is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all score benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also classifies it as likely benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, and AlphaMissense‑Default remains uncertain. High‑accuracy tools reinforce the benign assessment: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence supports a benign classification, and this is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -2.891 | Likely Benign | 0.458 | Ambiguous | Likely Benign | 0.073 | Likely Benign | -1.27 | Neutral | 0.649 | Possibly Damaging | 0.353 | Benign | 2.74 | Benign | 0.27 | Tolerated | 0.1854 | 0.4004 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3079A>T | N1027Y 2D  AIThe SynGAP1 missense variant N1027Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -5.799 | Likely Benign | 0.626 | Likely Pathogenic | Likely Benign | 0.074 | Likely Benign | -2.15 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.70 | Benign | 0.03 | Affected | 0.0611 | 0.6133 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.307G>A | G103S 2D  AIThe SynGAP1 missense variant G103S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.177 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -0.03 | Neutral | 0.565 | Possibly Damaging | 0.207 | Benign | 4.32 | Benign | 0.00 | Affected | 0.2816 | 0.4867 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.307G>C | G103R 2D  AIThe SynGAP1 missense variant G103R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.384 | Likely Benign | 0.710 | Likely Pathogenic | Likely Benign | 0.100 | Likely Benign | 0.00 | Neutral | 0.949 | Possibly Damaging | 0.708 | Possibly Damaging | 4.27 | Benign | 0.00 | Affected | 0.1198 | 0.4362 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.307G>T | G103C 2D  AIThe SynGAP1 missense variant G103C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect for G103C, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -5.681 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -1.12 | Neutral | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 4.17 | Benign | 0.00 | Affected | 0.1494 | 0.3767 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3080A>C | N1027T 2D  AIThe SynGAP1 missense variant N1027T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also reports Likely Benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is consistent with the absence of any ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -3.604 | Likely Benign | 0.199 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.71 | Neutral | 0.481 | Possibly Damaging | 0.220 | Benign | 2.75 | Benign | 0.13 | Tolerated | 0.1239 | 0.7188 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||||||||||
| c.3080A>G | N1027S 2D  AIThe SynGAP1 missense variant N1027S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -2.443 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.35 | Neutral | 0.068 | Benign | 0.039 | Benign | 2.77 | Benign | 0.50 | Tolerated | 0.3540 | 0.6874 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3080A>T | N1027I 2D  AIThe SynGAP1 missense variant N1027I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -5.847 | Likely Benign | 0.751 | Likely Pathogenic | Likely Benign | 0.065 | Likely Benign | -2.36 | Neutral | 0.970 | Probably Damaging | 0.726 | Possibly Damaging | 2.71 | Benign | 0.02 | Affected | 0.0677 | 0.5724 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3081C>A | N1027K 2D  AIThe SynGAP1 missense variant N1027K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus result is benign; Foldetta predictions are unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -3.177 | Likely Benign | 0.841 | Likely Pathogenic | Ambiguous | 0.063 | Likely Benign | -0.64 | Neutral | 0.481 | Possibly Damaging | 0.220 | Benign | 2.81 | Benign | 0.65 | Tolerated | 0.1808 | 0.6079 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3081C>G | N1027K 2D AIThe SynGAP1 missense variant N1027K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and AlphaMissense‑Optimized is uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -3.177 | Likely Benign | 0.841 | Likely Pathogenic | Ambiguous | 0.063 | Likely Benign | -0.64 | Neutral | 0.481 | Possibly Damaging | 0.220 | Benign | 2.81 | Benign | 0.65 | Tolerated | 0.1808 | 0.6079 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3082C>A | L1028M 2D  AIThe SynGAP1 missense variant L1028M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for the variant, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -4.591 | Likely Benign | 0.229 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.07 | Neutral | 0.986 | Probably Damaging | 0.825 | Possibly Damaging | 2.70 | Benign | 0.18 | Tolerated | 0.0776 | 0.3124 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3082C>G | L1028V 2D AIThe SynGAP1 missense variant L1028V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of computational evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -3.992 | Likely Benign | 0.217 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -0.68 | Neutral | 0.737 | Possibly Damaging | 0.376 | Benign | 2.74 | Benign | 0.26 | Tolerated | 0.1328 | 0.2715 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3083T>A | L1028Q 2D  AIThe SynGAP1 missense variant L1028Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -2.718 | Likely Benign | 0.612 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -0.19 | Neutral | 0.986 | Probably Damaging | 0.825 | Possibly Damaging | 2.80 | Benign | 0.38 | Tolerated | 0.1110 | 0.1403 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3083T>C | L1028P 2D AIThe SynGAP1 missense variant L1028P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Taken together, the majority of evidence points to a benign impact for this variant, and there is no ClinVar annotation to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -3.080 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 0.215 | Likely Benign | -0.42 | Neutral | 0.004 | Benign | 0.010 | Benign | 2.70 | Benign | 0.25 | Tolerated | 0.2846 | 0.1763 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3083T>G | L1028R 2D AIThe SynGAP1 missense variant L1028R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -2.204 | Likely Benign | 0.793 | Likely Pathogenic | Ambiguous | 0.167 | Likely Benign | -0.24 | Neutral | 0.960 | Probably Damaging | 0.761 | Possibly Damaging | 2.75 | Benign | 0.84 | Tolerated | 0.1328 | 0.1603 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3085C>A | Q1029K 2D AIThe SynGAP1 missense variant Q1029K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. AlphaMissense‑Default remains uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while Foldetta data is missing. Overall, the majority of evidence points to a benign impact for Q1029K, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.698 | Likely Benign | 0.516 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -1.18 | Neutral | 0.771 | Possibly Damaging | 0.482 | Possibly Damaging | 2.79 | Benign | 1.00 | Tolerated | 0.1656 | 0.4196 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3085C>G | Q1029E 2D AIThe SynGAP1 missense variant Q1029E is reported in gnomAD (ID 6‑33443637‑C‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, creating a single discordant call. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | 6-33443637-C-G | 17 | 1.05e-5 | -3.660 | Likely Benign | 0.281 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.92 | Neutral | 0.625 | Possibly Damaging | 0.258 | Benign | 2.83 | Benign | 0.26 | Tolerated | 3.77 | 5 | 0.1327 | 0.2433 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||||||
| c.3086A>C | Q1029P 2D AIThe SynGAP1 missense variant Q1029P is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available for this variant. Overall, the consensus of all available predictions strongly supports a benign impact, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -2.940 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.105 | Likely Benign | -1.23 | Neutral | 0.005 | Benign | 0.015 | Benign | 2.68 | Benign | 0.15 | Tolerated | 0.1966 | 0.4986 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3086A>G | Q1029R 2D AIThe SynGAP1 missense variant Q1029R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (both HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign; Foldetta results are unavailable. Based on the preponderance of evidence from both general and high‑accuracy predictors, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.437 | Likely Benign | 0.420 | Ambiguous | Likely Benign | 0.073 | Likely Benign | -0.72 | Neutral | 0.961 | Probably Damaging | 0.677 | Possibly Damaging | 2.73 | Benign | 0.88 | Tolerated | 0.1377 | 0.2420 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3086A>T | Q1029L 2D AIThe SynGAP1 missense variant Q1029L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a benign outcome (2 benign vs. 1 pathogenic, with one uncertain). AlphaMissense‑Default remains uncertain, and Foldetta results are unavailable. High‑accuracy predictions therefore point to a benign impact: AlphaMissense‑Optimized is benign, SGM Consensus is benign, and no Foldetta data are available. Overall, the computational evidence indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.984 | Likely Benign | 0.364 | Ambiguous | Likely Benign | 0.067 | Likely Benign | -2.65 | Deleterious | 0.891 | Possibly Damaging | 0.587 | Possibly Damaging | 2.70 | Benign | 0.16 | Tolerated | 0.0685 | 0.5866 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3087G>C | Q1029H 2D AIThe SynGAP1 missense variant Q1029H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar classification, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.984 | Likely Benign | 0.308 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.83 | Neutral | 0.989 | Probably Damaging | 0.879 | Possibly Damaging | 2.74 | Benign | 0.15 | Tolerated | 0.1268 | 0.4184 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3087G>T | Q1029H 2D AIThe SynGAP1 missense variant Q1029H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of the available predictions indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.984 | Likely Benign | 0.308 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.83 | Neutral | 0.989 | Probably Damaging | 0.879 | Possibly Damaging | 2.74 | Benign | 0.15 | Tolerated | 0.1268 | 0.4184 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3088C>A | H1030N 2D  AIThe SynGAP1 missense variant H1030N is not reported in ClinVar and is absent from gnomAD. Consensus prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign classification, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -3.454 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.033 | Likely Benign | -0.88 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.78 | Benign | 0.04 | Affected | 0.1553 | 0.3195 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.3088C>G | H1030D 2D  AIThe SynGAP1 missense variant H1030D is reported in gnomAD (variant ID 6‑33443640‑C‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign. Only SIFT predicts a pathogenic effect, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | 6-33443640-C-G | 1 | 6.19e-7 | -3.500 | Likely Benign | 0.424 | Ambiguous | Likely Benign | 0.189 | Likely Benign | -0.85 | Neutral | 0.126 | Benign | 0.066 | Benign | 2.78 | Benign | 0.05 | Affected | 3.77 | 5 | 0.2273 | 0.2422 | -1 | 1 | -0.3 | -22.05 | ||||||||||||||||||||||||||||||||||
| c.3088C>T | H1030Y 2D  AIThe SynGAP1 missense variant H1030Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for H1030Y, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -5.365 | Likely Benign | 0.268 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -1.73 | Neutral | 0.812 | Possibly Damaging | 0.298 | Benign | 2.73 | Benign | 0.01 | Affected | 0.0874 | 0.4236 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.3089A>C | H1030P 2D  AIThe SynGAP1 missense variant H1030P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of available predictions indicates that H1030P is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -2.185 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.154 | Likely Benign | -0.78 | Neutral | 0.812 | Possibly Damaging | 0.298 | Benign | 2.77 | Benign | 0.02 | Affected | 0.1872 | 0.4225 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3089A>G | H1030R 2D  AIThe SynGAP1 missense variant H1030R is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -2.312 | Likely Benign | 0.340 | Likely Benign | Likely Benign | 0.031 | Likely Benign | -1.08 | Neutral | 0.224 | Benign | 0.066 | Benign | 2.85 | Benign | 0.06 | Tolerated | 0.1872 | 0.2955 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||||||||||||
| c.3089A>T | H1030L 2D  AIThe SynGAP1 missense variant H1030L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that H1030L is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -3.513 | Likely Benign | 0.270 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -2.35 | Neutral | 0.224 | Benign | 0.120 | Benign | 2.76 | Benign | 0.02 | Affected | 0.0968 | 0.5710 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.308G>A | G103D 2D  AIThe SynGAP1 missense variant G103D is not reported in ClinVar and has no entry in gnomAD. Consensus from multiple in‑silico predictors shows a predominance of benign calls: REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT each predict a pathogenic impact. The AlphaMissense‑Default tool remains uncertain, and no Foldetta stability assessment is available. High‑accuracy tools specifically highlight benign predictions: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta data are missing. Overall, the balance of evidence points to a benign effect for G103D, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.523 | Likely Benign | 0.477 | Ambiguous | Likely Benign | 0.110 | Likely Benign | -0.10 | Neutral | 0.949 | Possibly Damaging | 0.617 | Possibly Damaging | 4.26 | Benign | 0.00 | Affected | 0.2237 | 0.2896 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.308G>C | G103A 2D  AIThe SynGAP1 missense variant G103A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.549 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -0.08 | Neutral | 0.008 | Benign | 0.008 | Benign | 4.31 | Benign | 0.00 | Affected | 0.3941 | 0.3784 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.308G>T | G103V 2D  AIThe SynGAP1 missense variant G103V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.584 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.126 | Likely Benign | -0.96 | Neutral | 0.820 | Possibly Damaging | 0.376 | Benign | 4.22 | Benign | 0.00 | Affected | 0.1420 | 0.3404 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3090C>A | H1030Q 2D 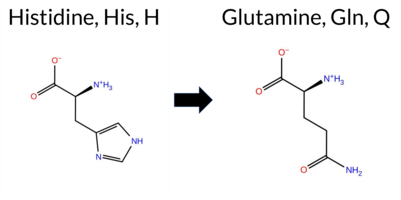 AIThe SynGAP1 missense variant H1030Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -2.548 | Likely Benign | 0.185 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.01 | Neutral | 0.004 | Benign | 0.004 | Benign | 2.88 | Benign | 0.53 | Tolerated | 0.1538 | 0.4001 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3090C>G | H1030Q 2D AIThe SynGAP1 missense variant H1030Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -2.548 | Likely Benign | 0.185 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.01 | Neutral | 0.004 | Benign | 0.004 | Benign | 2.88 | Benign | 0.53 | Tolerated | 0.1538 | 0.4001 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3091A>C | M1031L 2D 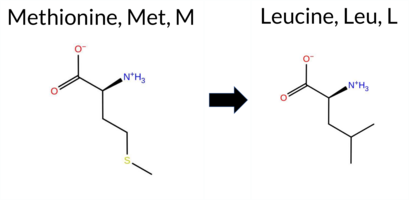 AIThe SynGAP1 missense variant M1031L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -2.213 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.82 | Neutral | 0.044 | Benign | 0.018 | Benign | 2.84 | Benign | 0.33 | Tolerated | 0.1220 | 0.4050 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3091A>G | M1031V 2D 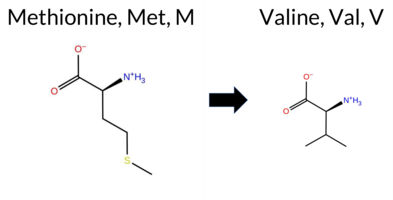 AIThe SynGAP1 missense variant M1031V is catalogued in gnomAD (ID 6‑33443643‑A‑G) but has no ClinVar entry. Across the spectrum of in‑silico predictors, every tool listed—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently scores the variant as benign. No pathogenic predictions are reported. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the uniform benign predictions and the lack of any ClinVar pathogenic classification, the variant is most likely benign and does not contradict existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | 6-33443643-A-G | 7 | 4.34e-6 | -2.815 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -0.81 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.72 | Benign | 0.44 | Tolerated | 3.77 | 5 | 0.2305 | 0.3388 | 1 | 2 | 2.3 | -32.06 | ||||||||||||||||||||||||||||||||||
| c.3091A>T | M1031L 2D AIThe SynGAP1 missense variant M1031L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -2.213 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.82 | Neutral | 0.044 | Benign | 0.018 | Benign | 2.84 | Benign | 0.33 | Tolerated | 0.1220 | 0.4050 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3092T>A | M1031K 2D 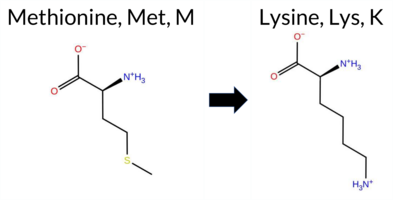 AIThe SynGAP1 missense variant M1031K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -1.940 | Likely Benign | 0.709 | Likely Pathogenic | Likely Benign | 0.114 | Likely Benign | -0.80 | Neutral | 0.325 | Benign | 0.098 | Benign | 2.66 | Benign | 0.18 | Tolerated | 0.1224 | 0.1412 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.3092T>C | M1031T 2D  AIThe SynGAP1 missense variant M1031T is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443644‑T‑C). In silico prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts a pathogenic outcome; the only inconclusive result is AlphaMissense‑Default, which is treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus is “Likely Benign,” and Foldetta data are not available. **Thus, the variant is most likely benign, and this conclusion does not contradict the ClinVar “Uncertain” status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | Uncertain | 1 | 6-33443644-T-C | 2 | 1.24e-6 | -1.863 | Likely Benign | 0.540 | Ambiguous | Likely Benign | 0.085 | Likely Benign | -0.24 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.67 | Benign | 1.00 | Tolerated | 3.77 | 5 | 0.1587 | 0.2264 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||
| c.3092T>G | M1031R 2D  AIThe SynGAP1 missense variant M1031R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Taken together, the majority of evidence points to a benign impact for M1031R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -1.365 | Likely Benign | 0.673 | Likely Pathogenic | Likely Benign | 0.117 | Likely Benign | -0.85 | Neutral | 0.325 | Benign | 0.129 | Benign | 2.64 | Benign | 0.12 | Tolerated | 0.1368 | 0.1212 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||||||||||||
| c.3093G>A | M1031I 2D  AIThe SynGAP1 missense variant M1031I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a benign impact for M1031I. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -3.312 | Likely Benign | 0.739 | Likely Pathogenic | Likely Benign | 0.026 | Likely Benign | -0.99 | Neutral | 0.095 | Benign | 0.027 | Benign | 2.71 | Benign | 0.25 | Tolerated | 0.1128 | 0.3324 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3093G>C | M1031I 2D AIThe SynGAP1 missense variant M1031I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a benign impact for M1031I. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -3.312 | Likely Benign | 0.739 | Likely Pathogenic | Likely Benign | 0.025 | Likely Benign | -0.99 | Neutral | 0.095 | Benign | 0.027 | Benign | 2.71 | Benign | 0.25 | Tolerated | 0.1128 | 0.3324 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3093G>T | M1031I 2D AIThe SynGAP1 missense variant M1031I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a benign impact for M1031I. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -3.312 | Likely Benign | 0.739 | Likely Pathogenic | Likely Benign | 0.026 | Likely Benign | -0.99 | Neutral | 0.095 | Benign | 0.027 | Benign | 2.71 | Benign | 0.25 | Tolerated | 0.1128 | 0.3324 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3094C>A | L1032M 2D AIThe SynGAP1 missense variant L1032M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of any ClinVar pathogenic annotation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -4.353 | Likely Benign | 0.219 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -0.09 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 2.66 | Benign | 0.10 | Tolerated | 0.0935 | 0.4719 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3094C>G | L1032V 2D AIThe SynGAP1 missense variant L1032V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and polyPhen‑2 HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for the variant, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -4.123 | Likely Benign | 0.176 | Likely Benign | Likely Benign | 0.075 | Likely Benign | 0.16 | Neutral | 0.889 | Possibly Damaging | 0.514 | Possibly Damaging | 2.78 | Benign | 0.78 | Tolerated | 0.1637 | 0.4353 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3095T>A | L1032Q 2D AIThe SynGAP1 missense variant L1032Q is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar) and SIFT. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the aggregate evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -2.992 | Likely Benign | 0.467 | Ambiguous | Likely Benign | 0.134 | Likely Benign | -0.78 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 2.66 | Benign | 0.03 | Affected | 0.1217 | 0.1677 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3095T>C | L1032P 2D AIThe SynGAP1 missense variant L1032P has no ClinVar entry and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and the SGM‑Consensus score all classify the change as benign or likely benign. AlphaMissense‑Optimized also predicts a benign outcome, whereas SIFT uniquely flags the variant as pathogenic. The AlphaMissense‑Default assessment is uncertain, and no Foldetta stability data are available. High‑accuracy analyses reinforce the benign consensus: AlphaMissense‑Optimized reports a benign effect, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta results are missing, so they do not influence the interpretation. Overall, the majority of computational evidence supports a benign classification, which is consistent with the absence of a ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this conclusion does not contradict ClinVar status, as no ClinVar pathogenic claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -2.560 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.142 | Likely Benign | -0.71 | Neutral | 0.012 | Benign | 0.017 | Benign | 2.64 | Benign | 0.05 | Affected | 0.3024 | 0.2330 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3095T>G | L1032R 2D AIThe SynGAP1 missense variant L1032R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -2.658 | Likely Benign | 0.707 | Likely Pathogenic | Likely Benign | 0.099 | Likely Benign | -1.03 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 2.66 | Benign | 0.02 | Affected | 0.1350 | 0.1919 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3097T>A | S1033T 2D AIThe SynGAP1 missense variant S1033T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -3.702 | Likely Benign | 0.140 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -0.05 | Neutral | 0.568 | Possibly Damaging | 0.171 | Benign | 2.73 | Benign | 0.58 | Tolerated | 0.1379 | 0.6089 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3097T>C | S1033P 2D AIThe SynGAP1 missense variant S1033P is not reported in ClinVar and is absent from gnomAD, indicating no documented population frequency. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are not available, so they do not influence the overall assessment. Based on the consensus of all available predictions, the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -2.046 | Likely Benign | 0.268 | Likely Benign | Likely Benign | 0.068 | Likely Benign | 0.05 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.68 | Benign | 0.27 | Tolerated | 0.1889 | 0.5486 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3097T>G | S1033A 2D AIThe SynGAP1 missense variant S1033A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions indicates that the variant is most likely benign, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -3.107 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.06 | Neutral | 0.220 | Benign | 0.085 | Benign | 2.81 | Benign | 1.00 | Tolerated | 0.4387 | 0.5282 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3098C>A | S1033Y 2D  AIThe SynGAP1 missense variant S1033Y is reported in gnomAD (ID 6‑33443650‑C‑A) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | 6-33443650-C-A | 1 | 6.19e-7 | -4.857 | Likely Benign | 0.564 | Ambiguous | Likely Benign | 0.034 | Likely Benign | -1.01 | Neutral | 0.021 | Benign | 0.008 | Benign | 2.69 | Benign | 0.04 | Affected | 3.77 | 5 | 0.0708 | 0.5262 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||
| c.3098C>G | S1033C 2D  AIThe SynGAP1 missense variant S1033C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for S1033C, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -6.263 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.39 | Neutral | 0.992 | Probably Damaging | 0.750 | Possibly Damaging | 2.68 | Benign | 0.12 | Tolerated | 0.1031 | 0.5704 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.3098C>T | S1033F 2D  AIThe SynGAP1 missense variant S1033F is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus is Likely Benign, and Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -5.013 | Likely Benign | 0.555 | Ambiguous | Likely Benign | 0.022 | Likely Benign | -1.02 | Neutral | 0.440 | Benign | 0.185 | Benign | 2.70 | Benign | 0.05 | Affected | 0.0699 | 0.5546 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.3100C>A | P1034T 2D  AIThe SynGAP1 missense variant P1034T is not reported in ClinVar and has no entry in gnomAD, indicating it is not catalogued in these databases. Prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only two tools predict a pathogenic outcome: SIFT and FATHMM. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions support a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -4.367 | Likely Benign | 0.285 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -2.00 | Neutral | 0.126 | Benign | 0.096 | Benign | 2.44 | Pathogenic | 0.03 | Affected | 0.1518 | 0.6620 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3100C>G | P1034A 2D  AIThe SynGAP1 missense variant P1034A is listed in ClinVar as Benign (ClinVar ID 1901716.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome, representing the sole discordant signal. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates a benign impact. This prediction aligns with the ClinVar status, with no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | Benign | 1 | -4.174 | Likely Benign | 0.178 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -2.44 | Neutral | 0.059 | Benign | 0.061 | Benign | 2.47 | Pathogenic | 0.06 | Tolerated | 3.77 | 5 | 0.3028 | 0.5622 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||
| c.3100C>T | P1034S 2D  AIThe SynGAP1 missense variant P1034S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for P1034S, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -3.730 | Likely Benign | 0.262 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -2.28 | Neutral | 0.011 | Benign | 0.015 | Benign | 2.44 | Pathogenic | 0.05 | Affected | 0.3044 | 0.5853 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3101C>A | P1034H 2D  AIThe SynGAP1 missense variant P1034H is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. three benign) lean toward pathogenicity, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -4.634 | Likely Benign | 0.540 | Ambiguous | Likely Benign | 0.083 | Likely Benign | -3.17 | Deleterious | 0.938 | Possibly Damaging | 0.750 | Possibly Damaging | 2.38 | Pathogenic | 0.02 | Affected | 0.1776 | 0.5451 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3101C>G | P1034R 2D  AIThe SynGAP1 P1034R variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic; Foldetta stability analysis is unavailable. Overall, the predictions are mixed, with a slight tilt toward pathogenicity due to the SGM Consensus result and the number of pathogenic calls. The variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -3.666 | Likely Benign | 0.676 | Likely Pathogenic | Likely Benign | 0.073 | Likely Benign | -3.04 | Deleterious | 0.002 | Benign | 0.005 | Benign | 2.40 | Pathogenic | 0.02 | Affected | 0.1366 | 0.4182 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3101C>T | P1034L 2D  AIThe SynGAP1 missense variant P1034L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default is uncertain. The high‑accuracy AlphaMissense‑Optimized model classifies the variant as benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that P1034L is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -4.204 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.067 | Likely Benign | -3.24 | Deleterious | 0.001 | Benign | 0.005 | Benign | 2.53 | Benign | 0.01 | Affected | 0.2267 | 0.6937 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3103C>A | P1035T 2D AIThe SynGAP1 missense variant P1035T is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default remains uncertain, and no Foldetta stability assessment is available. High‑accuracy evidence from AlphaMissense‑Optimized and the SGM‑Consensus both support a benign classification, while the absence of a Foldetta result does not alter this view. Overall, the majority of predictions indicate a benign effect, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | Uncertain | 1 | -4.447 | Likely Benign | 0.426 | Ambiguous | Likely Benign | 0.087 | Likely Benign | -0.96 | Neutral | 0.901 | Possibly Damaging | 0.537 | Possibly Damaging | 2.72 | Benign | 0.23 | Tolerated | 3.77 | 5 | 0.1628 | 0.7220 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||
| c.3103C>G | P1035A 2D AIThe SynGAP1 missense variant P1035A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, creating a single discordant signal. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -4.293 | Likely Benign | 0.241 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.02 | Neutral | 0.481 | Possibly Damaging | 0.222 | Benign | 2.74 | Benign | 0.41 | Tolerated | 0.3188 | 0.6022 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3103C>T | P1035S 2D AIThe SynGAP1 missense variant P1035S is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Only polyPhen‑2 HumDiv flags it as pathogenic, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign; Foldetta results are unavailable. Taken together, the preponderance of evidence points to a benign impact for P1035S, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -3.678 | Likely Benign | 0.341 | Ambiguous | Likely Benign | 0.059 | Likely Benign | -0.97 | Neutral | 0.818 | Possibly Damaging | 0.355 | Benign | 2.79 | Benign | 0.28 | Tolerated | 0.3257 | 0.6253 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3104C>A | P1035H 2D AIThe SynGAP1 missense variant P1035H is not reported in ClinVar and has no entries in gnomAD. Consensus predictions from multiple in‑silico tools are mixed: benign calls come from SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -5.333 | Likely Benign | 0.608 | Likely Pathogenic | Likely Benign | 0.058 | Likely Benign | -0.76 | Neutral | 0.997 | Probably Damaging | 0.889 | Possibly Damaging | 2.68 | Benign | 0.15 | Tolerated | 0.1893 | 0.5669 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3104C>G | P1035R 2D AIThe SynGAP1 missense variant P1035R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -4.534 | Likely Benign | 0.604 | Likely Pathogenic | Likely Benign | 0.119 | Likely Benign | 1.07 | Neutral | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 2.89 | Benign | 0.93 | Tolerated | 0.1424 | 0.4201 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3104C>T | P1035L 2D AIThe SynGAP1 missense variant P1035L is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -5.694 | Likely Benign | 0.675 | Likely Pathogenic | Likely Benign | 0.071 | Likely Benign | -2.11 | Neutral | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 2.70 | Benign | 0.21 | Tolerated | 0.2444 | 0.7354 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3106C>A | Q1036K 2D AIThe SynGAP1 missense variant Q1036K is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -3.757 | Likely Benign | 0.646 | Likely Pathogenic | Likely Benign | 0.079 | Likely Benign | -1.72 | Neutral | 0.011 | Benign | 0.005 | Benign | 2.58 | Benign | 0.05 | Affected | 0.2096 | 0.5220 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3106C>G | Q1036E 2D AIThe SynGAP1 missense variant Q1036E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome. When predictions are grouped by consensus, the benign group contains nine tools, while the pathogenic group contains one (SIFT). High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable, so they do not influence the assessment. Overall, the evidence strongly suggests the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -3.797 | Likely Benign | 0.313 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -1.22 | Neutral | 0.264 | Benign | 0.062 | Benign | 2.59 | Benign | 0.03 | Affected | 0.1629 | 0.3484 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3107A>C | Q1036P 2D AIThe SynGAP1 missense variant Q1036P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a damaging or pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple prediction algorithms and the consensus score indicates that the variant is most likely benign, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -2.491 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -2.00 | Neutral | 0.802 | Possibly Damaging | 0.238 | Benign | 2.57 | Benign | 0.01 | Affected | 0.2018 | 0.5564 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3107A>G | Q1036R 2D AIThe SynGAP1 missense variant Q1036R is listed in ClinVar (ID 4746139) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to a likely benign classification (3 benign vs. 1 pathogenic). High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | Uncertain | 1 | -3.816 | Likely Benign | 0.596 | Likely Pathogenic | Likely Benign | 0.083 | Likely Benign | -1.32 | Neutral | 0.292 | Benign | 0.071 | Benign | 2.54 | Benign | 0.04 | Affected | 0.1710 | 0.3071 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||
| c.3107A>T | Q1036L 2D AIThe SynGAP1 missense variant Q1036L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors a benign outcome, with two benign votes versus one pathogenic and one uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -4.389 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.092 | Likely Benign | -2.92 | Deleterious | 0.152 | Benign | 0.045 | Benign | 2.52 | Benign | 0.01 | Affected | 0.1069 | 0.5996 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3108G>C | Q1036H 2D AIThe SynGAP1 missense variant Q1036H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -4.189 | Likely Benign | 0.536 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.18 | Neutral | 0.977 | Probably Damaging | 0.615 | Possibly Damaging | 2.50 | Benign | 0.29 | Tolerated | 3.77 | 5 | 0.1641 | 0.4835 | 0 | 3 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||
| c.3108G>T | Q1036H 2D AIThe SynGAP1 missense variant Q1036H is listed in gnomAD (ID 6‑33443660‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and Foldetta (FoldX‑MD/Rosetta stability assessment) has no available result. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta data are missing. Overall, the majority of reliable predictors indicate a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | 6-33443660-G-T | 1 | 6.20e-7 | -4.189 | Likely Benign | 0.536 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.18 | Neutral | 0.977 | Probably Damaging | 0.615 | Possibly Damaging | 2.50 | Benign | 0.29 | Tolerated | 3.77 | 5 | 0.1641 | 0.4835 | 0 | 3 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||
| c.3109A>C | I1037L 2D  AIThe SynGAP1 missense variant I1037L is not reported in ClinVar and is absent from gnomAD, indicating no known clinical or population evidence. Functional prediction tools uniformly favor a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity, and the only uncertain call comes from AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Benign.” High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that I1037L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -2.563 | Likely Benign | 0.448 | Ambiguous | Likely Benign | 0.074 | Likely Benign | -0.46 | Neutral | 0.421 | Benign | 0.128 | Benign | 2.82 | Benign | 0.81 | Tolerated | 0.0939 | 0.4302 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3109A>G | I1037V 2D  AIThe SynGAP1 missense variant I1037V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that I1037V is most likely benign, and this conclusion is not contradicted by ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -2.326 | Likely Benign | 0.461 | Ambiguous | Likely Benign | 0.085 | Likely Benign | -0.09 | Neutral | 0.421 | Benign | 0.128 | Benign | 2.76 | Benign | 0.93 | Tolerated | 0.1229 | 0.3985 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3109A>T | I1037F 2D  AIThe SynGAP1 missense variant I1037F is not reported in ClinVar and has no entries in gnomAD. Consensus from multiple in‑silico predictors shows a split: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy tools further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the majority of evidence points to a benign effect, and this assessment is consistent with the absence of a ClinVar pathogenic claim. Thus, the variant is most likely benign, and this conclusion does not contradict ClinVar, which has no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -3.517 | Likely Benign | 0.653 | Likely Pathogenic | Likely Benign | 0.115 | Likely Benign | -0.97 | Neutral | 0.977 | Probably Damaging | 0.632 | Possibly Damaging | 2.71 | Benign | 0.16 | Tolerated | 0.0653 | 0.3722 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.310C>A | R104S 2D  AIThe SynGAP1 missense variant R104S has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | -2.790 | Likely Benign | 0.771 | Likely Pathogenic | Likely Benign | 0.143 | Likely Benign | -0.23 | Neutral | 0.625 | Possibly Damaging | 0.118 | Benign | 4.13 | Benign | 0.00 | Affected | 0.2490 | 0.3806 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.310C>G | R104G 2D 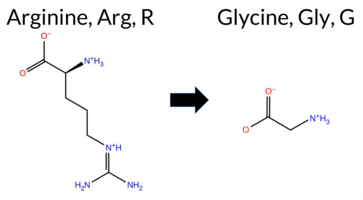 AIThe SynGAP1 missense variant R104G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R104G, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | -2.373 | Likely Benign | 0.452 | Ambiguous | Likely Benign | 0.109 | Likely Benign | -0.90 | Neutral | 0.835 | Possibly Damaging | 0.165 | Benign | 4.03 | Benign | 0.00 | Affected | 0.3111 | 0.3625 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.310C>T | R104C 2D 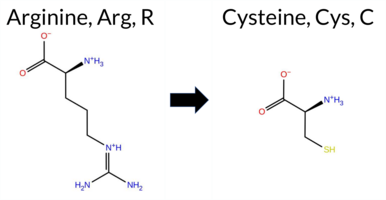 AIThe SynGAP1 missense variant R104C has no ClinVar entry and is present in gnomAD (ID 6‑33432175‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | 6-33432175-C-T | 2 | 1.24e-6 | -5.716 | Likely Benign | 0.475 | Ambiguous | Likely Benign | 0.109 | Likely Benign | -1.41 | Neutral | 0.993 | Probably Damaging | 0.446 | Benign | 3.99 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2954 | 0.3292 | -3 | -4 | 7.0 | -53.05 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||||
| c.3110T>A | I1037N 2D 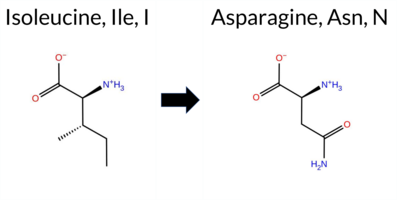 AIThe SynGAP1 missense variant I1037N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a Likely Benign classification, while AlphaMissense‑Optimized remains uncertain. Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -3.955 | Likely Benign | 0.832 | Likely Pathogenic | Ambiguous | 0.131 | Likely Benign | 1.56 | Neutral | 0.666 | Possibly Damaging | 0.211 | Benign | 2.81 | Benign | 0.34 | Tolerated | 0.1022 | 0.1140 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3110T>C | I1037T 2D 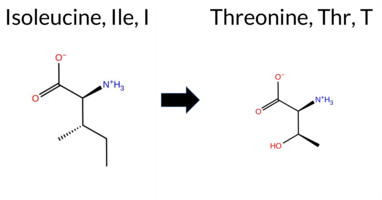 AIThe SynGAP1 missense variant I1037T is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33443662‑T‑C). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are AlphaMissense‑Default and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains Likely Benign; Foldetta results are unavailable. Overall, the majority of predictions (seven benign vs. two pathogenic) indicate that the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | 6-33443662-T-C | 1 | 6.21e-7 | -2.565 | Likely Benign | 0.973 | Likely Pathogenic | Likely Pathogenic | 0.066 | Likely Benign | 0.40 | Neutral | 0.292 | Benign | 0.110 | Benign | 2.79 | Benign | 0.34 | Tolerated | 3.77 | 5 | 0.1071 | 0.2175 | -1 | 0 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||||
| c.3110T>G | I1037S 2D  AIThe SynGAP1 missense variant I1037S is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which classifies the variant as Likely Benign. Only AlphaMissense‑Default predicts a pathogenic outcome, while AlphaMissense‑Optimized is uncertain and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Based on the overall consensus of the majority of tools, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -2.247 | Likely Benign | 0.935 | Likely Pathogenic | Ambiguous | 0.120 | Likely Benign | 0.43 | Neutral | 0.032 | Benign | 0.017 | Benign | 2.83 | Benign | 0.27 | Tolerated | 0.2634 | 0.1110 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3111C>G | I1037M 2D  AIThe SynGAP1 missense variant I1037M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Only two tools, polyPhen‑2 HumDiv and polyPhen‑2 HumVar, predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence supports a benign classification, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -3.849 | Likely Benign | 0.333 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -0.40 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 2.71 | Benign | 0.18 | Tolerated | 0.0775 | 0.3885 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3112A>C | T1038P 2D  AIThe SynGAP1 missense variant T1038P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (both HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -2.196 | Likely Benign | 0.390 | Ambiguous | Likely Benign | 0.116 | Likely Benign | -1.10 | Neutral | 0.965 | Probably Damaging | 0.611 | Possibly Damaging | 2.66 | Benign | 0.26 | Tolerated | 0.1856 | 0.4680 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3112A>G | T1038A 2D  AIThe SynGAP1 missense variant T1038A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, whereas only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign impact, and this is consistent with the lack of ClinVar evidence, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -3.544 | Likely Benign | 0.265 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.79 | Neutral | 0.649 | Possibly Damaging | 0.209 | Benign | 2.81 | Benign | 0.15 | Tolerated | 0.3429 | 0.3983 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3112A>T | T1038S 2D  AIThe SynGAP1 missense variant T1038S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -2.693 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -0.17 | Neutral | 0.649 | Possibly Damaging | 0.209 | Benign | 2.98 | Benign | 0.74 | Tolerated | 0.2879 | 0.4035 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3113C>A | T1038N 2D  AIThe SynGAP1 missense variant T1038N is not reported in ClinVar and has no entries in gnomAD, indicating it has not been catalogued in these databases. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the substitution as benign, while the majority‑vote consensus from SGM (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, and AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign. No Foldetta stability analysis is available, so folding‑stability evidence is lacking. Overall, the preponderance of computational evidence supports a benign classification for T1038N, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this is not contradicted by ClinVar, which has no pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -3.837 | Likely Benign | 0.390 | Ambiguous | Likely Benign | 0.054 | Likely Benign | -1.36 | Neutral | 0.818 | Possibly Damaging | 0.355 | Benign | 2.68 | Benign | 0.10 | Tolerated | 0.1184 | 0.4490 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||||||||||||
| c.3113C>G | T1038S 2D AIThe SynGAP1 missense variant T1038S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -2.693 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.102 | Likely Benign | -0.17 | Neutral | 0.649 | Possibly Damaging | 0.209 | Benign | 2.98 | Benign | 0.74 | Tolerated | 0.2879 | 0.4035 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3113C>T | T1038I 2D  AIThe SynGAP1 T1038I missense variant has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which classifies the variant as Likely Benign. Tools that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is Uncertain, and the Foldetta protein‑folding stability assessment is unavailable. Overall, the balance of evidence leans toward a benign interpretation, with one high‑accuracy tool inconclusive and no conflicting ClinVar annotation. Thus, the variant is most likely benign, and there is no ClinVar status that contradicts this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -4.552 | Likely Benign | 0.877 | Likely Pathogenic | Ambiguous | 0.093 | Likely Benign | -2.02 | Neutral | 0.990 | Probably Damaging | 0.637 | Possibly Damaging | 2.69 | Benign | 0.04 | Affected | 0.1012 | 0.5036 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||||||||||||
| c.3115A>C | I1039L 2D AIThe SynGAP1 missense variant I1039L is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the consensus of all available predictions strongly supports a benign classification, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -2.035 | Likely Benign | 0.269 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -0.14 | Neutral | 0.264 | Benign | 0.048 | Benign | 2.82 | Benign | 0.98 | Tolerated | 0.1057 | 0.4605 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3115A>G | I1039V 2D AIThe SynGAP1 missense variant I1039V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -2.455 | Likely Benign | 0.164 | Likely Benign | Likely Benign | 0.060 | Likely Benign | 0.21 | Neutral | 0.264 | Benign | 0.048 | Benign | 2.76 | Benign | 0.41 | Tolerated | 0.1405 | 0.4112 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3115A>T | I1039F 2D AIThe SynGAP1 missense variant I1039F is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign interpretation: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign, while the majority of other predictors (polyPhen‑2 HumDiv and HumVar) indicate pathogenic. When predictions are grouped by agreement, the benign‑oriented tools outnumber the pathogenic ones, and the single uncertain call from AlphaMissense‑Default does not alter this balance. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Benign. Foldetta data are unavailable. Overall, the computational evidence overwhelmingly supports a benign effect, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -2.872 | Likely Benign | 0.545 | Ambiguous | Likely Benign | 0.124 | Likely Benign | -1.16 | Neutral | 0.925 | Possibly Damaging | 0.510 | Possibly Damaging | 2.68 | Benign | 0.13 | Tolerated | 0.0710 | 0.4368 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3116T>A | I1039N 2D AIThe SynGAP1 missense variant I1039N has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -3.469 | Likely Benign | 0.951 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.24 | Neutral | 0.011 | Benign | 0.010 | Benign | 2.67 | Benign | 0.02 | Affected | 0.1151 | 0.1311 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3116T>C | I1039T 2D AIThe SynGAP1 missense variant I1039T is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443668‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a likely benign outcome. Only AlphaMissense‑Default predicts a pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | Uncertain | 2 | 6-33443668-T-C | 12 | 7.43e-6 | -2.465 | Likely Benign | 0.645 | Likely Pathogenic | Likely Benign | 0.193 | Likely Benign | 0.45 | Neutral | 0.004 | Benign | 0.008 | Benign | 2.75 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1248 | 0.2293 | -1 | 0 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||
| c.3116T>G | I1039S 2D AIThe SynGAP1 missense variant I1039S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM all classify the substitution as benign. In contrast, SIFT and AlphaMissense‑Default predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. No Foldetta stability assessment is available. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -1.688 | Likely Benign | 0.829 | Likely Pathogenic | Ambiguous | 0.171 | Likely Benign | -0.20 | Neutral | 0.032 | Benign | 0.008 | Benign | 2.76 | Benign | 0.03 | Affected | 0.2917 | 0.1294 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3117T>G | I1039M 2D AIThe SynGAP1 missense variant I1039M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for I1039M, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -3.444 | Likely Benign | 0.292 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.38 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 2.68 | Benign | 0.16 | Tolerated | 0.0926 | 0.4392 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3118G>A | G1040S 2D AIThe SynGAP1 missense variant G1040S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence (six benign versus four pathogenic predictions) points to a benign impact for G1040S. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -2.179 | Likely Benign | 0.307 | Likely Benign | Likely Benign | 0.653 | Likely Pathogenic | -1.81 | Neutral | 0.827 | Possibly Damaging | 0.375 | Benign | -0.74 | Pathogenic | 0.02 | Affected | 0.2332 | 0.5298 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3118G>C | G1040R 2D AIThe SynGAP1 missense variant G1040R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Benign predictions are limited to polyPhen‑2 HumVar and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus remains pathogenic. Foldetta, a protein‑folding stability predictor, has no available result for this residue. Taken together, the majority of evidence supports a pathogenic classification, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -2.901 | Likely Benign | 0.949 | Likely Pathogenic | Ambiguous | 0.704 | Likely Pathogenic | -3.00 | Deleterious | 0.463 | Possibly Damaging | 0.194 | Benign | -0.74 | Pathogenic | 0.00 | Affected | 0.0924 | 0.4415 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3118G>T | G1040C 2D AIThe SynGAP1 missense variant G1040C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all predict pathogenicity, while ESM1b and AlphaMissense‑Optimized predict a benign outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized reports a benign prediction, whereas the SGM‑Consensus remains Likely Pathogenic; the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence from multiple in silico tools indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -6.272 | Likely Benign | 0.620 | Likely Pathogenic | Likely Benign | 0.744 | Likely Pathogenic | -3.04 | Deleterious | 0.999 | Probably Damaging | 0.917 | Probably Damaging | -0.74 | Pathogenic | 0.00 | Affected | 0.1155 | 0.4556 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3119G>A | G1040D 2D AIThe SynGAP1 missense variant G1040D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only ESM1b, whereas the remaining tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic impact. The high‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus (a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it likely pathogenic, and the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence indicates that G1040D is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -4.154 | Likely Benign | 0.956 | Likely Pathogenic | Likely Pathogenic | 0.761 | Likely Pathogenic | -2.82 | Deleterious | 0.986 | Probably Damaging | 0.787 | Possibly Damaging | -0.74 | Pathogenic | 0.00 | Affected | 0.1677 | 0.2042 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.3119G>C | G1040A 2D AIThe SynGAP1 missense variant G1040A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL and FATHMM, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign prediction (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation, as the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -2.625 | Likely Benign | 0.409 | Ambiguous | Likely Benign | 0.606 | Likely Pathogenic | -1.65 | Neutral | 0.114 | Benign | 0.030 | Benign | -0.74 | Pathogenic | 0.30 | Tolerated | 0.3246 | 0.5331 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3119G>T | G1040V 2D AIThe SynGAP1 missense variant G1040V is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443671‑G‑T). Prediction tools that agree on a benign effect are ESM1b and AlphaMissense‑Optimized; those that predict a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta results are unavailable. Overall, the majority of predictions indicate a pathogenic impact, and this is not in conflict with the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | Uncertain | 1 | 6-33443671-G-T | 4 | 2.48e-6 | -3.453 | Likely Benign | 0.645 | Likely Pathogenic | Likely Benign | 0.774 | Likely Pathogenic | -2.89 | Deleterious | 0.827 | Possibly Damaging | 0.456 | Possibly Damaging | -0.74 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1239 | 0.4213 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||
| c.311G>A | R104H 2D  AIThe SynGAP1 missense variant R104H is reported in gnomAD (variant ID 6‑33432176‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is also benign. Foldetta results are not available for this variant. Overall, the preponderance of evidence indicates that R104H is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | 6-33432176-G-A | 2 | 1.24e-6 | -4.126 | Likely Benign | 0.268 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -1.42 | Neutral | 0.066 | Benign | 0.004 | Benign | 4.02 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2431 | 0.2102 | 0 | 2 | 1.3 | -19.05 | ||||||||||||||||||||||||||||||||||
| c.311G>C | R104P 2D  AIThe SynGAP1 missense variant R104P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | -3.184 | Likely Benign | 0.510 | Ambiguous | Likely Benign | 0.200 | Likely Benign | -0.88 | Neutral | 0.947 | Possibly Damaging | 0.410 | Benign | 4.01 | Benign | 0.00 | Affected | 0.1759 | 0.4498 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.311G>T | R104L 2D  AIThe SynGAP1 missense variant R104L is listed in ClinVar (ID 2746314.0) as Benign and is present in gnomAD (6‑33432176‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is benign, and the SGM‑Consensus (majority vote) is also benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the ClinVar benign classification and does not contradict the existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | Benign | 1 | 6-33432176-G-T | 1 | 6.20e-7 | -3.563 | Likely Benign | 0.578 | Likely Pathogenic | Likely Benign | 0.170 | Likely Benign | -1.38 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.05 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1681 | 0.4894 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||
| c.3121C>A | P1041T 2D AIThe SynGAP1 missense variant P1041T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -5.009 | Likely Benign | 0.187 | Likely Benign | Likely Benign | 0.361 | Likely Benign | -2.45 | Neutral | 0.051 | Benign | 0.025 | Benign | 5.50 | Benign | 0.10 | Tolerated | 0.1632 | 0.6699 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3121C>G | P1041A 2D AIThe SynGAP1 missense variant P1041A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -4.597 | Likely Benign | 0.157 | Likely Benign | Likely Benign | 0.331 | Likely Benign | -2.73 | Deleterious | 0.798 | Possibly Damaging | 0.283 | Benign | 5.53 | Benign | 0.24 | Tolerated | 0.3154 | 0.5939 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3121C>T | P1041S 2D AIThe SynGAP1 missense variant P1041S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443673‑C‑T). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | Conflicting | 2 | 6-33443673-C-T | 1 | 6.20e-7 | -4.246 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.344 | Likely Benign | -2.72 | Deleterious | 0.664 | Possibly Damaging | 0.283 | Benign | 5.48 | Benign | 0.11 | Tolerated | 3.77 | 5 | 0.3199 | 0.6120 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||
| c.3122C>A | P1041H 2D AIThe SynGAP1 missense variant P1041H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, and Foldetta’s protein‑folding stability result is unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no conflict with ClinVar status because the variant has not been reported there. Thus, based on current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -5.312 | Likely Benign | 0.377 | Ambiguous | Likely Benign | 0.476 | Likely Benign | -3.32 | Deleterious | 0.999 | Probably Damaging | 0.917 | Probably Damaging | 5.45 | Benign | 0.03 | Affected | 0.1883 | 0.5434 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3122C>G | P1041R 2D AIThe SynGAP1 missense variant P1041R is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The AlphaMissense‑Default result is uncertain, and no Foldetta stability data are available, so these are treated as unavailable. Overall, six tools support a benign classification while three support pathogenicity, and the high‑accuracy AlphaMissense‑Optimized and SGM Consensus both predict benign. Therefore, the variant is most likely benign based on the current predictions, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -4.808 | Likely Benign | 0.545 | Ambiguous | Likely Benign | 0.443 | Likely Benign | -3.33 | Deleterious | 0.986 | Probably Damaging | 0.787 | Possibly Damaging | 5.46 | Benign | 0.07 | Tolerated | 0.1401 | 0.4292 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||||
| c.3122C>T | P1041L 2D AIThe SynGAP1 missense variant P1041L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also resolves to benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact. This conclusion does not contradict ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -4.901 | Likely Benign | 0.399 | Ambiguous | Likely Benign | 0.403 | Likely Benign | -3.14 | Deleterious | 0.905 | Possibly Damaging | 0.375 | Benign | 5.46 | Benign | 1.00 | Tolerated | 0.2357 | 0.6664 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3124C>A | Q1042K 2D AIThe SynGAP1 missense variant Q1042K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -4.331 | Likely Benign | 0.498 | Ambiguous | Likely Benign | 0.304 | Likely Benign | -1.52 | Neutral | 0.224 | Benign | 0.091 | Benign | 5.44 | Benign | 0.13 | Tolerated | 0.2338 | 0.5202 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3124C>G | Q1042E 2D AIThe SynGAP1 missense variant Q1042E is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -4.231 | Likely Benign | 0.262 | Likely Benign | Likely Benign | 0.267 | Likely Benign | -1.12 | Neutral | 0.224 | Benign | 0.077 | Benign | 5.44 | Benign | 0.15 | Tolerated | 0.2042 | 0.3276 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3125A>C | Q1042P 2D AIThe SynGAP1 missense variant Q1042P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -2.085 | Likely Benign | 0.060 | Likely Benign | Likely Benign | 0.461 | Likely Benign | -0.73 | Neutral | 0.586 | Possibly Damaging | 0.223 | Benign | 5.42 | Benign | 0.31 | Tolerated | 0.2336 | 0.5762 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3125A>G | Q1042R 2D AIThe SynGAP1 missense variant Q1042R is listed in ClinVar (ID 2662705.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33443677‑A‑G). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Overall, the majority of evidence supports a benign impact for Q1042R, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | Uncertain | 2 | 6-33443677-A-G | 2 | 1.24e-6 | -2.928 | Likely Benign | 0.413 | Ambiguous | Likely Benign | 0.300 | Likely Benign | -1.39 | Neutral | 0.586 | Possibly Damaging | 0.120 | Benign | 5.48 | Benign | 0.12 | Tolerated | 3.77 | 5 | 0.1929 | 0.3887 | 1 | 1 | -1.0 | 28.06 | ||||||||||||||||||||||||||||||||
| c.3125A>T | Q1042L 2D AIThe SynGAP1 missense variant Q1042L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -3.796 | Likely Benign | 0.203 | Likely Benign | Likely Benign | 0.338 | Likely Benign | -2.47 | Neutral | 0.369 | Benign | 0.120 | Benign | 5.47 | Benign | 0.05 | Affected | 0.1469 | 0.6276 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3126G>C | Q1042H 2D AIThe SynGAP1 missense variant Q1042H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -4.258 | Likely Benign | 0.335 | Likely Benign | Likely Benign | 0.302 | Likely Benign | -1.54 | Neutral | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 5.42 | Benign | 0.03 | Affected | 0.2207 | 0.4671 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3126G>T | Q1042H 2D AIThe SynGAP1 missense variant Q1042H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -4.258 | Likely Benign | 0.335 | Likely Benign | Likely Benign | 0.302 | Likely Benign | -1.54 | Neutral | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 5.42 | Benign | 0.03 | Affected | 0.2207 | 0.4671 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3127A>G | R1043G 2D AIThe SynGAP1 missense variant R1043G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -2.168 | Likely Benign | 0.201 | Likely Benign | Likely Benign | 0.383 | Likely Benign | -3.17 | Deleterious | 0.130 | Benign | 0.049 | Benign | 5.94 | Benign | 0.00 | Affected | 0.3179 | 0.3697 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3127A>T | R1043W 2D  AIThe SynGAP1 missense variant R1043W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction; Foldetta results are unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -6.382 | Likely Benign | 0.523 | Ambiguous | Likely Benign | 0.444 | Likely Benign | -3.43 | Deleterious | 0.971 | Probably Damaging | 0.729 | Possibly Damaging | 5.38 | Benign | 0.00 | Affected | 0.1403 | 0.3696 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3128G>A | R1043K 2D  AIThe SynGAP1 missense variant R1043K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar or gnomAD entries—there is no contradiction with existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -4.038 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.376 | Likely Benign | -1.41 | Neutral | 0.069 | Benign | 0.033 | Benign | 5.39 | Benign | 0.00 | Affected | 0.5141 | 0.4070 | Weaken | 3 | 2 | 0.6 | -28.01 | ||||||||||||||||||||||||||||||||||||||
| c.3128G>C | R1043T 2D  AIThe SynGAP1 missense variant R1043T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -3.928 | Likely Benign | 0.298 | Likely Benign | Likely Benign | 0.463 | Likely Benign | -1.77 | Neutral | 0.001 | Benign | 0.003 | Benign | 5.39 | Benign | 0.00 | Affected | 0.1975 | 0.5513 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||||||||||||
| c.3128G>T | R1043M 2D  AIThe SynGAP1 missense variant R1043M is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign, while polyPhen‑2 HumDiv and SIFT predict pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus also indicates Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for R1043M, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -4.800 | Likely Benign | 0.510 | Ambiguous | Likely Benign | 0.471 | Likely Benign | -1.98 | Neutral | 0.744 | Possibly Damaging | 0.229 | Benign | 5.38 | Benign | 0.00 | Affected | 0.1982 | 0.4468 | 0 | -1 | 6.4 | -24.99 | |||||||||||||||||||||||||||||||||||||||
| c.3129G>C | R1043S 2D AIThe SynGAP1 missense variant R1043S is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools cluster into two groups: benign predictions include PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from REVEL and SIFT. AlphaMissense‑Default is uncertain, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores benign, SGM‑Consensus is likely benign, and Foldetta results are unavailable. Taken together, the majority of evidence points to a benign impact for R1043S. This conclusion is consistent with the absence of a ClinVar assertion, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -3.223 | Likely Benign | 0.457 | Ambiguous | Likely Benign | 0.509 | Likely Pathogenic | -2.10 | Neutral | 0.036 | Benign | 0.018 | Benign | 5.42 | Benign | 0.00 | Affected | 3.77 | 5 | 0.2727 | 0.4628 | -1 | 0 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||
| c.3129G>T | R1043S 2D AIThe SynGAP1 missense variant R1043S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443681‑G‑T). Prediction tools that agree on a benign effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are REVEL and SIFT. AlphaMissense‑Default remains uncertain, and no Foldetta stability result is available. High‑accuracy assessments: AlphaMissense‑Optimized classifies the variant as benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome; Foldetta data are missing, so it does not contribute to the evaluation. Based on the collective predictions, the variant is most likely benign, which is consistent with its ClinVar “Uncertain” classification and does not contradict the available evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | Uncertain | 1 | 6-33443681-G-T | 2 | 1.24e-6 | -3.223 | Likely Benign | 0.457 | Ambiguous | Likely Benign | 0.509 | Likely Pathogenic | -2.10 | Neutral | 0.036 | Benign | 0.018 | Benign | 5.42 | Benign | 0.00 | Affected | 3.77 | 5 | 0.2727 | 0.4628 | -1 | 0 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||
| c.3130C>A | P1044T 2D AIThe SynGAP1 missense variant P1044T is catalogued in gnomAD (ID 6‑33443682‑C‑A) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool reports a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the set predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.952126 | Binding | 0.331 | 0.855 | 0.750 | 6-33443682-C-A | 1 | 6.20e-7 | -4.605 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.372 | Likely Benign | -1.02 | Neutral | 0.126 | Benign | 0.096 | Benign | 5.53 | Benign | 0.17 | Tolerated | 3.77 | 5 | 0.1681 | 0.6860 | -1 | 0 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||
| c.3130C>G | P1044A 2D AIThe SynGAP1 missense variant P1044A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.952126 | Binding | 0.331 | 0.855 | 0.750 | -3.957 | Likely Benign | 0.059 | Likely Benign | Likely Benign | 0.359 | Likely Benign | -1.07 | Neutral | 0.059 | Benign | 0.061 | Benign | 5.50 | Benign | 0.18 | Tolerated | 0.3194 | 0.5850 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3130C>T | P1044S 2D AIThe SynGAP1 missense variant P1044S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.952126 | Binding | 0.331 | 0.855 | 0.750 | -4.114 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.311 | Likely Benign | -0.79 | Neutral | 0.011 | Benign | 0.015 | Benign | 5.51 | Benign | 0.14 | Tolerated | 0.3194 | 0.6081 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3131C>A | P1044Q 2D  AIThe SynGAP1 missense variant P1044Q is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.952126 | Binding | 0.331 | 0.855 | 0.750 | -4.439 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.338 | Likely Benign | 0.51 | Neutral | 0.004 | Benign | 0.015 | Benign | 5.43 | Benign | 0.45 | Tolerated | 0.1544 | 0.5591 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3131C>G | P1044R 2D AIThe SynGAP1 missense variant P1044R is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.952126 | Binding | 0.331 | 0.855 | 0.750 | -3.969 | Likely Benign | 0.242 | Likely Benign | Likely Benign | 0.368 | Likely Benign | 0.79 | Neutral | 0.259 | Benign | 0.140 | Benign | 5.45 | Benign | 1.00 | Tolerated | 0.1451 | 0.4270 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3131C>T | P1044L 2D AIThe SynGAP1 missense variant P1044L is not represented in ClinVar (no ClinVar ID) and is absent from gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic or likely pathogenic outcome. High‑accuracy assessments reinforce this benign prediction: AlphaMissense‑Optimized indicates benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.952126 | Binding | 0.331 | 0.855 | 0.750 | -4.327 | Likely Benign | 0.144 | Likely Benign | Likely Benign | 0.418 | Likely Benign | -1.64 | Neutral | 0.411 | Benign | 0.187 | Benign | 5.43 | Benign | 0.15 | Tolerated | 0.2264 | 0.6586 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3133G>A | A1045T 2D  AIThe SynGAP1 missense variant A1045T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975609 | Disordered | 0.948874 | Binding | 0.352 | 0.882 | 0.750 | -4.531 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.059 | Likely Benign | 0.03 | Neutral | 0.004 | Benign | 0.010 | Benign | 2.68 | Benign | 0.52 | Tolerated | 0.1857 | 0.6682 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3133G>C | A1045P 2D  AIThe SynGAP1 missense variant A1045P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975609 | Disordered | 0.948874 | Binding | 0.352 | 0.882 | 0.750 | -2.260 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.099 | Likely Benign | 1.09 | Neutral | 0.586 | Possibly Damaging | 0.223 | Benign | 2.64 | Benign | 0.24 | Tolerated | 0.2130 | 0.5332 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3133G>T | A1045S 2D  AIThe SynGAP1 missense variant A1045S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975609 | Disordered | 0.948874 | Binding | 0.352 | 0.882 | 0.750 | -3.724 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.11 | Neutral | 0.011 | Benign | 0.010 | Benign | 2.66 | Benign | 0.46 | Tolerated | 0.2648 | 0.5493 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3134C>A | A1045D 2D  AIThe SynGAP1 missense variant A1045D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach consensus all indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all score the variant as benign. AlphaMissense‑Optimized also predicts a benign outcome, whereas AlphaMissense‑Default remains uncertain. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” No tools predict pathogenicity. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective predictions strongly suggest that A1045D is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975609 | Disordered | 0.948874 | Binding | 0.352 | 0.882 | 0.750 | -5.734 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.066 | Likely Benign | -1.00 | Neutral | 0.411 | Benign | 0.172 | Benign | 2.64 | Benign | 0.16 | Tolerated | 0.1954 | 0.2102 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.3134C>G | A1045G 2D  AIThe SynGAP1 missense variant A1045G is reported in ClinVar as Benign (ClinVar ID 416778.0) and is present in the gnomAD database (gnomAD ID 6‑33443686‑C‑G). Prediction tools that assess pathogenicity all converge on a benign outcome: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. No tool in the dataset predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this prediction aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975609 | Disordered | 0.948874 | Binding | 0.352 | 0.882 | 0.750 | Benign/Likely benign | 7 | 6-33443686-C-G | 1407 | 8.72e-4 | -3.246 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.024 | Likely Benign | -1.21 | Neutral | 0.224 | Benign | 0.066 | Benign | 2.64 | Benign | 0.33 | Tolerated | 3.77 | 5 | 0.2246 | 0.4798 | 1 | 0 | -2.2 | -14.03 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||
| c.3134C>T | A1045V 2D  AIThe SynGAP1 missense variant A1045V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975609 | Disordered | 0.948874 | Binding | 0.352 | 0.882 | 0.750 | -4.229 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -0.35 | Neutral | 0.011 | Benign | 0.017 | Benign | 3.06 | Benign | 1.00 | Tolerated | 0.1487 | 0.5474 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.3136C>A | P1046T 2D AIThe SynGAP1 missense variant P1046T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | -5.249 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.052 | Likely Benign | -1.18 | Neutral | 0.411 | Benign | 0.131 | Benign | 2.37 | Pathogenic | 0.21 | Tolerated | 0.1430 | 0.5934 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3136C>G | P1046A 2D AIThe SynGAP1 missense variant P1046A is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443688‑C‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | Uncertain | 1 | 6-33443688-C-G | 1 | 6.20e-7 | -3.246 | Likely Benign | 0.048 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.67 | Neutral | 0.001 | Benign | 0.008 | Benign | 2.39 | Pathogenic | 0.29 | Tolerated | 3.77 | 5 | 0.2976 | 0.5358 | -1 | 1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||
| c.3136C>T | P1046S 2D AIThe SynGAP1 missense variant P1046S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence indicates a benign effect, and this consensus does not contradict any ClinVar status (none is available). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | -3.909 | Likely Benign | 0.059 | Likely Benign | Likely Benign | 0.096 | Likely Benign | -0.72 | Neutral | 0.126 | Benign | 0.096 | Benign | 2.39 | Pathogenic | 0.70 | Tolerated | 0.2929 | 0.5744 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3137C>A | P1046H 2D AIThe SynGAP1 missense variant P1046H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for P1046H. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | -5.715 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -1.64 | Neutral | 0.832 | Possibly Damaging | 0.670 | Possibly Damaging | 2.33 | Pathogenic | 0.07 | Tolerated | 0.1585 | 0.5250 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3137C>G | P1046R 2D AIThe SynGAP1 missense variant P1046R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all classify the change as benign or likely benign. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM‑Consensus also indicates a likely benign status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | -4.929 | Likely Benign | 0.222 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -1.79 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.38 | Pathogenic | 0.07 | Tolerated | 0.1267 | 0.3841 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3137C>T | P1046L 2D AIThe SynGAP1 missense variant P1046L is reported in gnomAD (ID 6‑33443689‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, SIFT and FATHMM predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors and the consensus analysis points to a benign classification. This conclusion is consistent with the absence of a ClinVar pathogenic report, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | 6-33443689-C-T | 1 | 6.20e-7 | -5.022 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -2.11 | Neutral | 0.001 | Benign | 0.005 | Benign | 2.35 | Pathogenic | 0.05 | Affected | 3.77 | 5 | 0.2036 | 0.6442 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.3139T>A | S1047T 2D AIThe SynGAP1 missense variant S1047T is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.963420 | Disordered | 0.933764 | Binding | 0.409 | 0.909 | 0.750 | -4.222 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.029 | Likely Benign | 0.23 | Neutral | 0.069 | Benign | 0.049 | Benign | 2.63 | Benign | 0.63 | Tolerated | 0.1796 | 0.6265 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3139T>C | S1047P 2D AIThe SynGAP1 missense variant S1047P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as likely benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions points to a benign impact, and this is consistent with the lack of any ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.963420 | Disordered | 0.933764 | Binding | 0.409 | 0.909 | 0.750 | -2.415 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.064 | Likely Benign | 0.52 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.62 | Benign | 0.03 | Affected | 0.2373 | 0.5656 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3139T>G | S1047A 2D AIThe SynGAP1 missense variant S1047A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.963420 | Disordered | 0.933764 | Binding | 0.409 | 0.909 | 0.750 | -3.503 | Likely Benign | 0.060 | Likely Benign | Likely Benign | 0.040 | Likely Benign | -0.50 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.65 | Benign | 0.07 | Tolerated | 0.4460 | 0.5548 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.313T>A | S105T 2D AIThe SynGAP1 missense variant S105T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a benign effect. AlphaMissense‑Optimized independently scores the variant as benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.669201 | Binding | 0.364 | 0.870 | 0.625 | -4.166 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.54 | Neutral | 0.012 | Benign | 0.007 | Benign | 4.06 | Benign | 0.00 | Affected | 0.1583 | 0.5212 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.313T>C | S105P 2D AIThe SynGAP1 missense variant S105P is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only two tools—polyPhen‑2 HumDiv and SIFT—predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.669201 | Binding | 0.364 | 0.870 | 0.625 | Uncertain | 1 | -3.631 | Likely Benign | 0.166 | Likely Benign | Likely Benign | 0.204 | Likely Benign | 0.03 | Neutral | 0.808 | Possibly Damaging | 0.212 | Benign | 4.00 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2236 | 0.4584 | -1 | 1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||
| c.313T>G | S105A 2D AIThe SynGAP1 missense variant S105A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect. The predictions do not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.669201 | Binding | 0.364 | 0.870 | 0.625 | -3.779 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.67 | Neutral | 0.012 | Benign | 0.002 | Benign | 4.11 | Benign | 0.00 | Affected | 0.5299 | 0.4214 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.3140C>T | S1047L 2D AIThe SynGAP1 missense variant S1047L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443692‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta stability analysis is not available for this variant. Overall, the preponderance of computational evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.963420 | Disordered | 0.933764 | Binding | 0.409 | 0.909 | 0.750 | Uncertain | 1 | 6-33443692-C-T | 1 | 6.20e-7 | -4.062 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.032 | Likely Benign | -0.63 | Neutral | 0.144 | Benign | 0.058 | Benign | 2.60 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1575 | 0.5555 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||
| c.3142G>A | G1048R 2D AIThe SynGAP1 missense variant G1048R is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split: pathogenic calls come from REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar, whereas benign calls are made by PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a benign majority, and AlphaMissense‑Optimized itself predicts benign. The SGM‑Consensus, which aggregates these four predictors, reports a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar assertion; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | -4.305 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.503 | Likely Pathogenic | -0.54 | Neutral | 0.919 | Possibly Damaging | 0.728 | Possibly Damaging | 2.54 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0956 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3142G>C | G1048R 2D AIThe SynGAP1 missense variant G1048R is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM‑Consensus also indicates a likely benign outcome; the Foldetta protein‑folding stability assessment is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | Uncertain | 1 | -4.305 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.503 | Likely Pathogenic | -0.54 | Neutral | 0.919 | Possibly Damaging | 0.728 | Possibly Damaging | 2.54 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0956 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.3142G>T | G1048W 2D  AIThe SynGAP1 missense variant G1048W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | -8.803 | Likely Pathogenic | 0.340 | Likely Benign | Likely Benign | 0.498 | Likely Benign | -1.52 | Neutral | 0.996 | Probably Damaging | 0.961 | Probably Damaging | 2.54 | Benign | 0.02 | Affected | 0.0852 | 0.4046 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.3143G>A | G1048E 2D  AIThe SynGAP1 missense variant G1048E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only REVEL predicts a pathogenic outcome, while ESM1b is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | -7.028 | In-Between | 0.331 | Likely Benign | Likely Benign | 0.529 | Likely Pathogenic | -0.62 | Neutral | 0.018 | Benign | 0.030 | Benign | 2.54 | Benign | 0.10 | Tolerated | 0.1444 | 0.4063 | 0 | -2 | -3.1 | 72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3143G>C | G1048A 2D AISynGAP1 missense variant G1048A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only polyPhen‑2 HumDiv predicts it as pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” status. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that G1048A is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | -4.821 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.456 | Likely Benign | -0.14 | Neutral | 0.573 | Possibly Damaging | 0.358 | Benign | 2.57 | Benign | 0.64 | Tolerated | 0.3301 | 0.5138 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3143G>T | G1048V 2D AIThe SynGAP1 missense variant G1048V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | -6.108 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.520 | Likely Pathogenic | -0.59 | Neutral | 0.958 | Probably Damaging | 0.787 | Possibly Damaging | 2.54 | Benign | 0.11 | Tolerated | 0.1312 | 0.3688 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3145C>A | P1049T 2D AIThe SynGAP1 missense variant P1049T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta results are unavailable, so no additional stability evidence is present. Overall, the consensus of available predictions indicates that P1049T is most likely benign, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.917915 | Binding | 0.428 | 0.920 | 0.750 | -5.100 | Likely Benign | 0.056 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -1.18 | Neutral | 0.519 | Possibly Damaging | 0.222 | Benign | 2.74 | Benign | 0.02 | Affected | 0.1791 | 0.5514 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3145C>G | P1049A 2D AIThe SynGAP1 missense variant P1049A is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.917915 | Binding | 0.428 | 0.920 | 0.750 | -3.870 | Likely Benign | 0.049 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -1.43 | Neutral | 0.180 | Benign | 0.171 | Benign | 2.81 | Benign | 0.06 | Tolerated | 0.3306 | 0.4897 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3145C>T | P1049S 2D AIThe SynGAP1 missense variant P1049S is reported in gnomAD (variant ID 6‑33443697‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the majority of predictions, including the high‑accuracy tools, indicate that P1049S is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.917915 | Binding | 0.428 | 0.920 | 0.750 | 6-33443697-C-T | 2 | 1.24e-6 | -2.351 | Likely Benign | 0.053 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -1.53 | Neutral | 0.519 | Possibly Damaging | 0.303 | Benign | 2.76 | Benign | 0.04 | Affected | 3.77 | 5 | 0.3144 | 0.5083 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.3146C>A | P1049H 2D AIThe SynGAP1 missense variant P1049H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.917915 | Binding | 0.428 | 0.920 | 0.750 | -5.427 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -2.06 | Neutral | 0.978 | Probably Damaging | 0.750 | Possibly Damaging | 2.76 | Benign | 0.01 | Affected | 0.1800 | 0.5070 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3146C>G | P1049R 2D AIThe SynGAP1 missense variant P1049R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.917915 | Binding | 0.428 | 0.920 | 0.750 | -5.144 | Likely Benign | 0.145 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -1.90 | Neutral | 0.791 | Possibly Damaging | 0.500 | Possibly Damaging | 2.74 | Benign | 0.03 | Affected | 0.1376 | 0.3769 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3146C>T | P1049L 2D AIThe SynGAP1 missense variant P1049L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.917915 | Binding | 0.428 | 0.920 | 0.750 | -4.819 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -2.37 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.71 | Benign | 0.02 | Affected | 0.2267 | 0.5838 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3148G>A | G1050R 2D AIThe SynGAP1 missense variant G1050R is catalogued in gnomAD (ID 6‑33443700‑G‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign verdict, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | 6-33443700-G-A | 3 | 1.86e-6 | -6.637 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.045 | Likely Benign | -0.68 | Neutral | 0.009 | Benign | 0.008 | Benign | 2.48 | Pathogenic | 0.06 | Tolerated | 3.77 | 5 | 0.0939 | 0.4532 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.3148G>C | G1050R 2D AIThe SynGAP1 missense variant G1050R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority vote. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact; this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | -6.637 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.045 | Likely Benign | -0.68 | Neutral | 0.009 | Benign | 0.008 | Benign | 2.48 | Pathogenic | 0.06 | Tolerated | 3.77 | 5 | 0.0939 | 0.4532 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3149G>A | G1050E 2D AIThe SynGAP1 missense variant G1050E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect. There is no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | -8.175 | Likely Pathogenic | 0.266 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -0.28 | Neutral | 0.411 | Benign | 0.171 | Benign | 2.50 | Benign | 0.08 | Tolerated | 0.1418 | 0.4258 | 0 | -2 | -3.1 | 72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3149G>C | G1050A 2D AIThe SynGAP1 missense variant G1050A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | -5.514 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.047 | Likely Benign | 0.18 | Neutral | 0.000 | Benign | 0.004 | Benign | 2.68 | Benign | 1.00 | Tolerated | 0.3349 | 0.5133 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3149G>T | G1050V 2D AIThe SynGAP1 missense variant G1050V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no classification for G1050V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | -6.450 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.83 | Neutral | 0.126 | Benign | 0.096 | Benign | 2.49 | Pathogenic | 0.13 | Tolerated | 0.1260 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.314C>G | S105W 2D  AIThe SynGAP1 missense variant S105W is catalogued in gnomAD (ID 6‑33432179‑C‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” status, reflecting the majority of benign calls. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also leans benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.669201 | Binding | 0.364 | 0.870 | 0.625 | 6-33432179-C-G | 2 | 1.24e-6 | -5.600 | Likely Benign | 0.606 | Likely Pathogenic | Likely Benign | 0.177 | Likely Benign | -2.28 | Neutral | 0.998 | Probably Damaging | 0.844 | Possibly Damaging | 3.97 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0705 | 0.4984 | -3 | -2 | -0.1 | 99.14 | ||||||||||||||||||||||||||||||||||
| c.314C>T | S105L 2D AIThe SynGAP1 missense variant S105L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33432179‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy methods both support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.669201 | Binding | 0.364 | 0.870 | 0.625 | Uncertain | 2 | 6-33432179-C-T | 4 | 2.48e-6 | -3.710 | Likely Benign | 0.233 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -1.52 | Neutral | 0.828 | Possibly Damaging | 0.048 | Benign | 4.06 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1161 | 0.5023 | -3 | -2 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||
| c.3151G>A | G1051S 2D AIThe SynGAP1 missense variant G1051S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only FATHMM predicts a pathogenic outcome. When the predictions are grouped, the benign consensus includes eight tools, whereas the pathogenic consensus contains a single tool. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign effect, and the SGM‑Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates a likely benign outcome. Foldetta data are unavailable, so no stability inference can be drawn. Overall, the computational evidence overwhelmingly supports a benign classification for G1051S, and this conclusion is consistent with the absence of any ClinVar annotation. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.900141 | Binding | 0.358 | 0.936 | 0.875 | -4.742 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.445 | Likely Benign | 0.10 | Neutral | 0.245 | Benign | 0.096 | Benign | -0.74 | Pathogenic | 0.61 | Tolerated | 0.2446 | 0.4911 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3151G>C | G1051R 2D AIThe SynGAP1 missense variant G1051R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. Two tools (ESM1b and AlphaMissense‑Default) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it contains one pathogenic, one benign, and two uncertain calls, and Foldetta results are unavailable. Overall, the balance of evidence favors a benign classification. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.900141 | Binding | 0.358 | 0.936 | 0.875 | -7.907 | In-Between | 0.346 | Ambiguous | Likely Benign | 0.438 | Likely Benign | 0.20 | Neutral | 0.761 | Possibly Damaging | 0.305 | Benign | -0.74 | Pathogenic | 0.20 | Tolerated | 0.0956 | 0.4342 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3151G>T | G1051C 2D AIThe SynGAP1 missense variant G1051C is listed in ClinVar as Pathogenic and is not reported in gnomAD. Functional prediction tools show a split assessment: benign calls come from REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls come from polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. High‑accuracy methods give a benign result from AlphaMissense‑Optimized; the SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is tied (2 benign vs. 2 pathogenic) and therefore inconclusive, and Foldetta’s stability prediction is unavailable. Overall, the majority of predictions lean toward a benign effect, which contradicts the ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.900141 | Binding | 0.358 | 0.936 | 0.875 | Likely Pathogenic | 1 | -9.050 | Likely Pathogenic | 0.122 | Likely Benign | Likely Benign | 0.497 | Likely Benign | -0.90 | Neutral | 0.971 | Probably Damaging | 0.750 | Possibly Damaging | -0.74 | Pathogenic | 0.10 | Tolerated | 3.77 | 5 | 0.1322 | 0.4612 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||
| c.3152G>A | G1051D 2D AISynGAP1 missense variant G1051D is listed in ClinVar as Benign and is present in gnomAD (variant ID 6‑33443704‑G‑A). Prediction tools that classify the variant as benign include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are polyPhen‑2 HumDiv, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign versus two pathogenic votes), and Foldetta stability analysis is unavailable. Overall, the balance of evidence favors a benign effect, consistent with the ClinVar annotation and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.900141 | Binding | 0.358 | 0.936 | 0.875 | Benign | 1 | 6-33443704-G-A | 2 | 1.24e-6 | -9.379 | Likely Pathogenic | 0.311 | Likely Benign | Likely Benign | 0.445 | Likely Benign | -0.31 | Neutral | 0.761 | Possibly Damaging | 0.239 | Benign | -0.74 | Pathogenic | 0.39 | Tolerated | 3.77 | 5 | 0.1872 | 0.2235 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||
| c.3152G>C | G1051A 2D AIThe SynGAP1 missense variant G1051A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence indicates a benign effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.900141 | Binding | 0.358 | 0.936 | 0.875 | -6.406 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.406 | Likely Benign | -0.14 | Neutral | 0.009 | Benign | 0.004 | Benign | -0.74 | Pathogenic | 1.00 | Tolerated | 0.3353 | 0.4944 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3152G>T | G1051V 2D AIThe SynGAP1 missense variant G1051V is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443704‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction (2 benign vs. 1 pathogenic, 1 uncertain). Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for G1051V, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.900141 | Binding | 0.358 | 0.936 | 0.875 | 6-33443704-G-T | 1 | 6.20e-7 | -7.098 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.460 | Likely Benign | -0.62 | Neutral | 0.245 | Benign | 0.096 | Benign | -0.74 | Pathogenic | 0.17 | Tolerated | 3.77 | 5 | 0.1274 | 0.3680 | -3 | -1 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||
| c.3154G>A | G1052R 2D AISynGAP1 missense variant G1052R is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores the variant as benign, and the SGM consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar uncertain status but provides additional support toward a likely benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | Uncertain | 1 | -9.050 | Likely Pathogenic | 0.383 | Ambiguous | Likely Benign | 0.497 | Likely Benign | -0.41 | Neutral | 0.990 | Probably Damaging | 0.798 | Possibly Damaging | 3.90 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0976 | 0.4142 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||
| c.3154G>C | G1052R 2D AIThe SynGAP1 missense variant G1052R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and ESM1b predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | -9.050 | Likely Pathogenic | 0.383 | Ambiguous | Likely Benign | 0.497 | Likely Benign | -0.41 | Neutral | 0.990 | Probably Damaging | 0.798 | Possibly Damaging | 3.90 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0976 | 0.4142 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3154G>T | G1052W 2D AIThe SynGAP1 missense variant G1052W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | -11.322 | Likely Pathogenic | 0.309 | Likely Benign | Likely Benign | 0.457 | Likely Benign | -0.90 | Neutral | 0.997 | Probably Damaging | 0.946 | Probably Damaging | 3.90 | Benign | 0.02 | Affected | 0.0872 | 0.4046 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.3155G>A | G1052E 2D AIThe SynGAP1 missense variant G1052E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a neutral impact. In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b predict a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign effect. Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign classification, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | -9.869 | Likely Pathogenic | 0.287 | Likely Benign | Likely Benign | 0.448 | Likely Benign | -0.64 | Neutral | 0.901 | Possibly Damaging | 0.537 | Possibly Damaging | 3.90 | Benign | 0.12 | Tolerated | 0.1405 | 0.3873 | 0 | -2 | -3.1 | 72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3155G>C | G1052A 2D AIThe SynGAP1 missense variant G1052A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, creating a single discordant signal. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence supports a benign classification, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | -6.945 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.382 | Likely Benign | -0.14 | Neutral | 0.649 | Possibly Damaging | 0.287 | Benign | 3.93 | Benign | 1.00 | Tolerated | 0.3259 | 0.4949 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3155G>T | G1052V 2D AIThe SynGAP1 missense variant G1052V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign, while the high‑accuracy AlphaMissense‑Optimized score is benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign. In contrast, polyPhen‑2 HumDiv and HumVar both predict pathogenic, and ESM1b remains uncertain. No Foldetta stability assessment is available, so it does not influence the overall interpretation. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | -7.717 | In-Between | 0.094 | Likely Benign | Likely Benign | 0.452 | Likely Benign | -0.12 | Neutral | 0.901 | Possibly Damaging | 0.619 | Possibly Damaging | 3.90 | Benign | 0.19 | Tolerated | 0.1329 | 0.3499 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3157A>C | S1053R 2D  AIThe SynGAP1 missense variant S1053R is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign, while the majority‑vote SGM‑Consensus also reports a likely benign outcome. In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic impact. The AlphaMissense‑Default score is uncertain, and no Foldetta stability assessment is available. High‑accuracy analyses reinforce the benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely benign, with no contradictory Foldetta data. Overall, the preponderance of evidence points to a benign effect for S1053R, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | -6.421 | Likely Benign | 0.359 | Ambiguous | Likely Benign | 0.225 | Likely Benign | 0.43 | Neutral | 0.969 | Probably Damaging | 0.581 | Possibly Damaging | 5.33 | Benign | 0.59 | Tolerated | 3.77 | 5 | 0.1321 | 0.3820 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.3157A>G | S1053G 2D  AIThe SynGAP1 missense variant S1053G is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions strongly suggests that S1053G is most likely benign, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | -1.016 | Likely Benign | 0.050 | Likely Benign | Likely Benign | 0.220 | Likely Benign | -0.57 | Neutral | 0.001 | Benign | 0.002 | Benign | 5.32 | Benign | 0.59 | Tolerated | 0.2724 | 0.5046 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3157A>T | S1053C 2D AIThe SynGAP1 missense variant S1053C is catalogued in gnomAD (ID 6‑33443709‑A‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The ESM1b score is uncertain, providing no clear direction. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant. Overall, the majority of reliable predictors classify S1053C as benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | 6-33443709-A-T | -7.574 | In-Between | 0.095 | Likely Benign | Likely Benign | 0.220 | Likely Benign | -0.61 | Neutral | 0.977 | Probably Damaging | 0.777 | Possibly Damaging | 5.30 | Benign | 0.11 | Tolerated | 3.77 | 5 | 0.1675 | 0.5895 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.3158G>A | S1053N 2D  AIThe SynGAP1 missense variant S1053N is reported in gnomAD (variant ID 6‑33443710‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, while the consensus score from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus also indicates a likely benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | 6-33443710-G-A | 1 | 6.21e-7 | -6.282 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.208 | Likely Benign | -0.54 | Neutral | 0.625 | Possibly Damaging | 0.193 | Benign | 5.30 | Benign | 0.34 | Tolerated | 3.77 | 5 | 0.1953 | 0.4605 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||||
| c.3158G>C | S1053T 2D AIThe SynGAP1 missense variant S1053T is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the preponderance of evidence points to a benign impact for S1053T, and this conclusion is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | -6.209 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.223 | Likely Benign | -0.25 | Neutral | 0.625 | Possibly Damaging | 0.249 | Benign | 5.32 | Benign | 0.70 | Tolerated | 0.2055 | 0.5976 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3158G>T | S1053I 2D  AIThe SynGAP1 missense variant S1053I is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that S1053I is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | -6.572 | Likely Benign | 0.177 | Likely Benign | Likely Benign | 0.250 | Likely Benign | -0.46 | Neutral | 0.925 | Possibly Damaging | 0.413 | Benign | 5.32 | Benign | 0.10 | Tolerated | 0.1536 | 0.4780 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3159C>A | S1053R 2D AIThe SynGAP1 missense variant S1053R is reported in gnomAD (variant ID 6‑33443711‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. Overall, the majority of high‑accuracy predictors and consensus analyses indicate a benign impact. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | 6-33443711-C-A | 1 | 1.10e-6 | -6.421 | Likely Benign | 0.359 | Ambiguous | Likely Benign | 0.303 | Likely Benign | 0.43 | Neutral | 0.969 | Probably Damaging | 0.581 | Possibly Damaging | 5.33 | Benign | 0.59 | Tolerated | 3.77 | 5 | 0.1321 | 0.3820 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||
| c.3159C>G | S1053R 2D AIThe SynGAP1 missense variant S1053R is reported in gnomAD (ID 6‑33443711‑C‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and the high‑accuracy consensus methods give a benign verdict: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.981594 | Disordered | 0.885608 | Binding | 0.399 | 0.944 | 0.875 | 6-33443711-C-G | -6.421 | Likely Benign | 0.359 | Ambiguous | Likely Benign | 0.304 | Likely Benign | 0.43 | Neutral | 0.969 | Probably Damaging | 0.581 | Possibly Damaging | 5.33 | Benign | 0.59 | Tolerated | 3.77 | 5 | 0.1321 | 0.3820 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||
| c.3160G>A | G1054S 2D AIThe SynGAP1 missense variant G1054S is listed in ClinVar (ID 699126.0) as Benign and is present in gnomAD (variant ID 6‑33443712‑G‑A). All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic or likely pathogenic outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts Benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the computational evidence strongly supports a benign effect, consistent with the ClinVar designation and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | Benign | 1 | 6-33443712-G-A | 32 | 1.99e-5 | -5.294 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.160 | Likely Benign | 0.21 | Neutral | 0.121 | Benign | 0.013 | Benign | 4.04 | Benign | 0.63 | Tolerated | 3.77 | 5 | 0.2506 | 0.5311 | 1 | 0 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||||
| c.3160G>C | G1054R 2D AIThe SynGAP1 missense variant G1054R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for G1054R, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | -8.863 | Likely Pathogenic | 0.326 | Likely Benign | Likely Benign | 0.234 | Likely Benign | 0.29 | Neutral | 0.988 | Probably Damaging | 0.589 | Possibly Damaging | 4.05 | Benign | 0.42 | Tolerated | 0.1164 | 0.4342 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3160G>T | G1054C 2D AIThe SynGAP1 missense variant G1054C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | -9.548 | Likely Pathogenic | 0.118 | Likely Benign | Likely Benign | 0.297 | Likely Benign | -0.59 | Neutral | 0.999 | Probably Damaging | 0.907 | Possibly Damaging | 4.00 | Benign | 0.10 | Tolerated | 0.1413 | 0.4427 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3161G>A | G1054D 2D AISynGAP1 missense variant G1054D is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely benign, which does not contradict the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | Uncertain | 1 | -10.385 | Likely Pathogenic | 0.351 | Ambiguous | Likely Benign | 0.279 | Likely Benign | -0.26 | Neutral | 0.818 | Possibly Damaging | 0.266 | Benign | 4.07 | Benign | 0.37 | Tolerated | 3.77 | 5 | 0.1824 | 0.2035 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||
| c.3161G>C | G1054A 2D AIThe SynGAP1 missense variant G1054A is catalogued in gnomAD (ID 6‑33443713‑G‑C) but has no ClinVar entry. Across a broad panel of in‑silico predictors, every tool reports a benign outcome: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity, so the pathogenic‑prediction group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant, so its status is unavailable. Based on the unanimous benign predictions and the lack of any ClinVar pathogenic classification, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | 6-33443713-G-C | -6.786 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.243 | Likely Benign | -0.05 | Neutral | 0.288 | Benign | 0.071 | Benign | 4.03 | Benign | 1.00 | Tolerated | 3.77 | 5 | 0.3305 | 0.5144 | 0 | 1 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||||||
| c.3161G>T | G1054V 2D AIThe SynGAP1 missense variant G1054V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also reports likely benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign impact, and this is consistent with the lack of ClinVar evidence; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | -6.994 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.171 | Likely Benign | -0.22 | Neutral | 0.818 | Possibly Damaging | 0.221 | Benign | 4.01 | Benign | 0.18 | Tolerated | 0.1578 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3163G>A | G1055R 2D AIThe SynGAP1 missense variant G1055R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.982235 | Disordered | 0.872113 | Binding | 0.379 | 0.935 | 0.875 | -8.778 | Likely Pathogenic | 0.375 | Ambiguous | Likely Benign | 0.275 | Likely Benign | -0.09 | Neutral | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 3.31 | Benign | 0.08 | Tolerated | 0.1013 | 0.4733 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3163G>C | G1055R 2D AIThe SynGAP1 missense variant G1055R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. AlphaMissense‑Default is uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign outcome (2 benign vs. 1 pathogenic, 1 uncertain). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.982235 | Disordered | 0.872113 | Binding | 0.379 | 0.935 | 0.875 | -8.778 | Likely Pathogenic | 0.375 | Ambiguous | Likely Benign | 0.275 | Likely Benign | -0.09 | Neutral | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 3.31 | Benign | 0.08 | Tolerated | 0.1013 | 0.4733 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3163G>T | G1055W 2D AIThe SynGAP1 missense variant G1055W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.982235 | Disordered | 0.872113 | Binding | 0.379 | 0.935 | 0.875 | -11.436 | Likely Pathogenic | 0.317 | Likely Benign | Likely Benign | 0.367 | Likely Benign | -0.77 | Neutral | 0.997 | Probably Damaging | 0.946 | Probably Damaging | 3.28 | Benign | 0.01 | Affected | 0.0899 | 0.4246 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.3164G>A | G1055E 2D AIThe SynGAP1 missense variant G1055E is catalogued in gnomAD (variant ID 6‑33443716‑G‑A) but has no ClinVar entry. In silico prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a benign effect. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.982235 | Disordered | 0.872113 | Binding | 0.379 | 0.935 | 0.875 | 6-33443716-G-A | 1 | 6.21e-7 | -10.951 | Likely Pathogenic | 0.290 | Likely Benign | Likely Benign | 0.320 | Likely Benign | -0.03 | Neutral | 0.901 | Possibly Damaging | 0.456 | Possibly Damaging | 3.30 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1525 | 0.4459 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.3164G>C | G1055A 2D AIThe SynGAP1 missense variant G1055A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also reports Likely Benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.982235 | Disordered | 0.872113 | Binding | 0.379 | 0.935 | 0.875 | -6.835 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.244 | Likely Benign | 0.15 | Neutral | 0.649 | Possibly Damaging | 0.148 | Benign | 3.30 | Benign | 1.00 | Tolerated | 0.3350 | 0.5144 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3164G>T | G1055V 2D AIThe SynGAP1 missense variant G1055V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while ESM1b remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.982235 | Disordered | 0.872113 | Binding | 0.379 | 0.935 | 0.875 | -7.434 | In-Between | 0.114 | Likely Benign | Likely Benign | 0.399 | Likely Benign | 0.26 | Neutral | 0.818 | Possibly Damaging | 0.222 | Benign | 3.28 | Benign | 0.17 | Tolerated | 0.1399 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3166G>A | G1056S 2D AIThe SynGAP1 missense variant G1056S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -5.252 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.339 | Likely Benign | -0.28 | Neutral | 0.451 | Benign | 0.149 | Benign | 1.87 | Pathogenic | 0.55 | Tolerated | 0.2497 | 0.5702 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3166G>C | G1056R 2D AIThe SynGAP1 missense variant G1056R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and FATHMM, while AlphaMissense‑Default remains uncertain. High‑accuracy assessment shows AlphaMissense‑Optimized as benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors a pathogenic outcome, and Foldetta data are unavailable. Overall, the majority of conventional predictors indicate a benign impact, whereas the SGM Consensus suggests pathogenicity. Given the preponderance of benign predictions and the lack of ClinVar evidence, the variant is most likely benign, and this assessment does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -9.358 | Likely Pathogenic | 0.390 | Ambiguous | Likely Benign | 0.410 | Likely Benign | 0.12 | Neutral | 0.011 | Benign | 0.010 | Benign | 1.83 | Pathogenic | 0.13 | Tolerated | 0.1127 | 0.4533 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3166G>T | G1056C 2D AIThe SynGAP1 missense variant G1056C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign impact. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -9.974 | Likely Pathogenic | 0.122 | Likely Benign | Likely Benign | 0.432 | Likely Benign | -0.70 | Neutral | 0.994 | Probably Damaging | 0.777 | Possibly Damaging | 1.83 | Pathogenic | 0.06 | Tolerated | 0.1414 | 0.4439 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.3167G>A | G1056D 2D AIThe SynGAP1 missense variant G1056D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs. 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs. three pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -10.352 | Likely Pathogenic | 0.328 | Likely Benign | Likely Benign | 0.380 | Likely Benign | 0.09 | Neutral | 0.666 | Possibly Damaging | 0.193 | Benign | 1.83 | Pathogenic | 0.92 | Tolerated | 0.1912 | 0.2626 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3167G>C | G1056A 2D AIThe SynGAP1 missense variant G1056A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while ESM1b remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign consensus (2 benign vs. 1 pathogenic, with one uncertain). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -7.458 | In-Between | 0.086 | Likely Benign | Likely Benign | 0.325 | Likely Benign | -0.25 | Neutral | 0.264 | Benign | 0.097 | Benign | 1.85 | Pathogenic | 0.49 | Tolerated | 0.3325 | 0.4957 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3167G>T | G1056V 2D AIThe SynGAP1 missense variant G1056V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence (7 benign vs 2 pathogenic) supports a benign classification. This consensus does not contradict ClinVar status, which has no entry for this variant. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -8.130 | Likely Pathogenic | 0.097 | Likely Benign | Likely Benign | 0.448 | Likely Benign | -0.24 | Neutral | 0.292 | Benign | 0.110 | Benign | 1.83 | Pathogenic | 0.08 | Tolerated | 0.1488 | 0.3507 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3169A>C | S1057R 2D AIThe SynGAP1 missense variant S1057R is catalogued in gnomAD (ID 6‑33443721‑A‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | 6-33443721-A-C | -6.648 | Likely Benign | 0.379 | Ambiguous | Likely Benign | 0.221 | Likely Benign | -0.24 | Neutral | 0.677 | Possibly Damaging | 0.168 | Benign | 5.30 | Benign | 0.21 | Tolerated | 3.77 | 5 | 0.1584 | 0.3620 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||
| c.3169A>G | S1057G 2D AIThe SynGAP1 missense variant S1057G is reported in gnomAD (ID 6‑33443721‑A‑G) but has no ClinVar entry. All evaluated in‑silico predictors uniformly classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | 6-33443721-A-G | 1 | 7.20e-7 | -1.005 | Likely Benign | 0.049 | Likely Benign | Likely Benign | 0.211 | Likely Benign | -0.47 | Neutral | 0.421 | Benign | 0.111 | Benign | 5.76 | Benign | 0.52 | Tolerated | 3.77 | 5 | 0.2654 | 0.4846 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||
| c.3169A>T | S1057C 2D AIThe SynGAP1 missense variant S1057C is reported in gnomAD (ID 6‑33443721‑A‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Two tools, polyPhen‑2 HumDiv and polyPhen‑2 HumVar, predict a pathogenic impact, while ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | 6-33443721-A-T | -7.529 | In-Between | 0.100 | Likely Benign | Likely Benign | 0.258 | Likely Benign | -0.64 | Neutral | 0.977 | Probably Damaging | 0.683 | Possibly Damaging | 5.23 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1856 | 0.6106 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.316A>G | R106G 2D AIThe SynGAP1 missense variant R106G is listed in gnomAD (ID 6‑33432181‑A‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Those that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar classification to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | 6-33432181-A-G | 1 | 6.20e-7 | -2.617 | Likely Benign | 0.835 | Likely Pathogenic | Ambiguous | 0.161 | Likely Benign | -2.21 | Neutral | 0.421 | Benign | 0.050 | Benign | 3.65 | Benign | 0.00 | Affected | 4.05 | 3 | 0.3680 | 0.3980 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||
| c.316A>T | R106W 2D AIThe SynGAP1 missense variant R106W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, more tools predict pathogenicity (5) than benign (3), and no ClinVar evidence contradicts this assessment. Thus, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | -5.350 | Likely Benign | 0.875 | Likely Pathogenic | Ambiguous | 0.240 | Likely Benign | -3.31 | Deleterious | 0.983 | Probably Damaging | 0.624 | Possibly Damaging | 3.62 | Benign | 0.00 | Affected | 0.1369 | 0.3995 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3170G>A | S1057N 2D AIThe SynGAP1 missense variant S1057N is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All available in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No tool predicts pathogenicity. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | Uncertain | 1 | -6.386 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.218 | Likely Benign | -0.41 | Neutral | 0.451 | Benign | 0.129 | Benign | 5.25 | Benign | 0.28 | Tolerated | 0.2232 | 0.4605 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||
| c.3170G>C | S1057T 2D AIThe SynGAP1 missense variant S1057T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that the variant is most likely benign, and this assessment is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | -6.375 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.241 | Likely Benign | -0.18 | Neutral | 0.625 | Possibly Damaging | 0.170 | Benign | 5.26 | Benign | 0.60 | Tolerated | 0.2289 | 0.5976 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3170G>T | S1057I 2D AIThe SynGAP1 missense variant S1057I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to the variant being most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | -6.887 | Likely Benign | 0.186 | Likely Benign | Likely Benign | 0.259 | Likely Benign | -0.87 | Neutral | 0.925 | Possibly Damaging | 0.238 | Benign | 5.24 | Benign | 0.07 | Tolerated | 0.1802 | 0.4980 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3171C>A | S1057R 2D AIThe SynGAP1 missense variant S1057R is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Only polyPhen‑2 HumDiv flags it as pathogenic, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | -6.648 | Likely Benign | 0.379 | Ambiguous | Likely Benign | 0.272 | Likely Benign | -0.24 | Neutral | 0.677 | Possibly Damaging | 0.168 | Benign | 5.30 | Benign | 0.21 | Tolerated | 3.77 | 5 | 0.1584 | 0.3620 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.3171C>G | S1057R 2D AIThe SynGAP1 missense variant S1057R is catalogued in gnomAD (ID 6‑33443723‑C‑G) but has no ClinVar submission. Functional prediction tools largely converge on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all report benign or tolerated. Only polyPhen‑2 HumDiv flags it as pathogenic, while AlphaMissense‑Default remains uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus confirms a benign likelihood; Foldetta data are unavailable, so no stability evidence is provided. Taken together, the preponderance of evidence indicates the variant is most likely benign, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.869507 | Binding | 0.413 | 0.927 | 0.875 | 6-33443723-C-G | -6.648 | Likely Benign | 0.379 | Ambiguous | Likely Benign | 0.272 | Likely Benign | -0.24 | Neutral | 0.677 | Possibly Damaging | 0.168 | Benign | 5.30 | Benign | 0.21 | Tolerated | 3.77 | 5 | 0.1584 | 0.3620 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||
| c.3172G>A | G1058S 2D AIThe SynGAP1 missense variant G1058S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6-33443724-G-A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. Foldetta results are unavailable. Overall, the majority of computational evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.980739 | Disordered | 0.885724 | Binding | 0.407 | 0.929 | 0.875 | Conflicting | 3 | 6-33443724-G-A | 114 | 7.08e-5 | -5.178 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.108 | Likely Benign | 0.26 | Neutral | 0.001 | Benign | 0.001 | Benign | 5.38 | Benign | 0.04 | Affected | 3.77 | 5 | 0.2488 | 0.5511 | 1 | 0 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||||
| c.3172G>C | G1058R 2D AIThe SynGAP1 missense variant G1058R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only two predictors—SIFT and ESM1b—suggest a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.980739 | Disordered | 0.885724 | Binding | 0.407 | 0.929 | 0.875 | -8.967 | Likely Pathogenic | 0.339 | Likely Benign | Likely Benign | 0.138 | Likely Benign | 0.34 | Neutral | 0.174 | Benign | 0.140 | Benign | 5.29 | Benign | 0.00 | Affected | 0.1145 | 0.4342 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3172G>T | G1058C 2D AIThe SynGAP1 missense variant G1058C is reported in gnomAD (ID 6‑33443724‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and ESM1b. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of computational evidence points to a benign effect, and this is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.980739 | Disordered | 0.885724 | Binding | 0.407 | 0.929 | 0.875 | 6-33443724-G-T | 1 | 6.21e-7 | -9.384 | Likely Pathogenic | 0.132 | Likely Benign | Likely Benign | 0.264 | Likely Benign | -0.19 | Neutral | 0.600 | Possibly Damaging | 0.433 | Benign | 5.19 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1456 | 0.4627 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.3173G>A | G1058D 2D AIThe SynGAP1 missense variant G1058D is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443725‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and ESM1b, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.980739 | Disordered | 0.885724 | Binding | 0.407 | 0.929 | 0.875 | 6-33443725-G-A | 1 | 6.21e-7 | -10.344 | Likely Pathogenic | 0.391 | Ambiguous | Likely Benign | 0.177 | Likely Benign | -0.33 | Neutral | 0.077 | Benign | 0.042 | Benign | 5.20 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1889 | 0.2435 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||
| c.3173G>C | G1058A 2D AIThe SynGAP1 missense variant G1058A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.980739 | Disordered | 0.885724 | Binding | 0.407 | 0.929 | 0.875 | -6.823 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.159 | Likely Benign | 0.21 | Neutral | 0.000 | Benign | 0.002 | Benign | 5.29 | Benign | 0.55 | Tolerated | 0.3288 | 0.5144 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3173G>T | G1058V 2D AIThe SynGAP1 missense variant G1058V is reported in gnomAD (variant ID 6‑33443725‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.980739 | Disordered | 0.885724 | Binding | 0.407 | 0.929 | 0.875 | 6-33443725-G-T | 1 | 6.21e-7 | -6.877 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.152 | Likely Benign | -0.12 | Neutral | 0.000 | Benign | 0.001 | Benign | 5.22 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1561 | 0.3694 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||
| c.3175G>A | G1059R 2D AIThe SynGAP1 missense variant G1059R is listed in ClinVar with an uncertain significance and is present in the gnomAD database (ID 6‑33443727‑G‑A). Consensus among most in silico predictors leans toward a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized all report benign. In contrast, SIFT and ESM1b predict pathogenicity, while AlphaMissense‑Default remains uncertain. High‑accuracy assessment consolidates this view: AlphaMissense‑Optimized is benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign classification. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its influence is unavailable. Overall, the preponderance of computational evidence supports a benign interpretation, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.983019 | Disordered | 0.898939 | Binding | 0.399 | 0.926 | 0.875 | Conflicting | 2 | 6-33443727-G-A | 68 | 4.23e-5 | -8.452 | Likely Pathogenic | 0.376 | Ambiguous | Likely Benign | 0.333 | Likely Benign | -0.55 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.53 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1052 | 0.4342 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||
| c.3175G>C | G1059R 2D AIThe SynGAP1 missense variant G1059R is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors shows a predominance of benign calls: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized all predict a benign effect, whereas SIFT and ESM1b predict pathogenicity; AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized is benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also returns a benign prediction. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence points to a benign impact for G1059R, and this conclusion is consistent with the absence of any ClinVar annotation or gnomAD observation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.983019 | Disordered | 0.898939 | Binding | 0.399 | 0.926 | 0.875 | -8.452 | Likely Pathogenic | 0.376 | Ambiguous | Likely Benign | 0.333 | Likely Benign | -0.55 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.53 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1052 | 0.4342 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3175G>T | G1059W 2D AIThe SynGAP1 missense variant G1059W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.898939 | Binding | 0.399 | 0.926 | 0.875 | -11.549 | Likely Pathogenic | 0.312 | Likely Benign | Likely Benign | 0.446 | Likely Benign | -1.18 | Neutral | 0.983 | Probably Damaging | 0.813 | Possibly Damaging | 2.53 | Benign | 0.00 | Affected | 0.0925 | 0.4046 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.3176G>A | G1059E 2D AIThe SynGAP1 missense variant G1059E is reported in gnomAD (variant ID 6‑33443728‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only two tools—SIFT and ESM1b—predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.898939 | Binding | 0.399 | 0.926 | 0.875 | 6-33443728-G-A | 1 | 6.22e-7 | -10.459 | Likely Pathogenic | 0.297 | Likely Benign | Likely Benign | 0.390 | Likely Benign | -0.71 | Neutral | 0.126 | Benign | 0.066 | Benign | 2.53 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1464 | 0.4069 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.3176G>C | G1059A 2D AIThe SynGAP1 missense variant G1059A is listed in ClinVar (ID 1420036.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33443728‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of computational evidence supports a benign impact for G1059A, which does not contradict the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.898939 | Binding | 0.399 | 0.926 | 0.875 | Uncertain | 1 | 6-33443728-G-C | 4 | 2.49e-6 | -6.754 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.329 | Likely Benign | -0.17 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.56 | Benign | 0.00 | Affected | 4.32 | 2 | 0.3295 | 0.4944 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||
| c.3176G>T | G1059V 2D AIThe SynGAP1 missense variant G1059V is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score benign, while the majority‑vote SGM‑Consensus also classifies it as likely benign. Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy tools corroborate the benign prediction: AlphaMissense‑Optimized is benign and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely benign; Foldetta results are not available. Overall, the preponderance of evidence supports a benign classification for G1059V, and this assessment does not conflict with ClinVar, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.898939 | Binding | 0.399 | 0.926 | 0.875 | -7.242 | In-Between | 0.106 | Likely Benign | Likely Benign | 0.478 | Likely Benign | -0.82 | Neutral | 0.259 | Benign | 0.066 | Benign | 2.54 | Benign | 0.00 | Affected | 0.1462 | 0.3494 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3178G>A | G1060S 2D AIThe SynGAP1 missense variant G1060S is listed in ClinVar with an uncertain significance (ClinVar ID 1512003.0) and is present in gnomAD (variant ID 6‑33443730‑G‑A). All evaluated in‑silico predictors classify the change as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this benign view: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | Uncertain | 1 | 6-33443730-G-A | -4.759 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.376 | Likely Benign | -0.08 | Neutral | 0.271 | Benign | 0.054 | Benign | 2.69 | Benign | 0.49 | Tolerated | 4.32 | 2 | 0.2468 | 0.5311 | 1 | 0 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||||||
| c.3178G>C | G1060R 2D AIThe SynGAP1 missense variant G1060R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for G1060R, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | -8.225 | Likely Pathogenic | 0.323 | Likely Benign | Likely Benign | 0.362 | Likely Benign | -0.29 | Neutral | 0.971 | Probably Damaging | 0.580 | Possibly Damaging | 2.63 | Benign | 0.17 | Tolerated | 0.1038 | 0.4342 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3178G>T | G1060C 2D AIThe SynGAP1 missense variant G1060C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for G1060C, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | -9.630 | Likely Pathogenic | 0.116 | Likely Benign | Likely Benign | 0.363 | Likely Benign | -0.60 | Neutral | 0.999 | Probably Damaging | 0.917 | Probably Damaging | 2.63 | Benign | 0.12 | Tolerated | 0.1340 | 0.4227 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3179G>A | G1060D 2D AIThe SynGAP1 missense variant G1060D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also leans benign (2 benign vs 1 pathogenic). Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | -9.824 | Likely Pathogenic | 0.342 | Ambiguous | Likely Benign | 0.391 | Likely Benign | -0.58 | Neutral | 0.905 | Possibly Damaging | 0.538 | Possibly Damaging | 2.63 | Benign | 0.20 | Tolerated | 0.1703 | 0.2035 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3179G>C | G1060A 2D AIThe SynGAP1 missense variant G1060A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, creating a single discordant signal. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | -6.539 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.341 | Likely Benign | 0.30 | Neutral | 0.664 | Possibly Damaging | 0.283 | Benign | 2.69 | Benign | 0.98 | Tolerated | 0.3335 | 0.4944 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3179G>T | G1060V 2D AIThe SynGAP1 missense variant G1060V is listed in ClinVar as benign (ClinVar ID 1345112.0) and is observed in gnomAD (6‑33443731‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic effect. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as likely benign, and AlphaMissense‑Optimized also reports a benign outcome. No Foldetta stability assessment is available for this variant. Overall, the majority of evidence points to a benign impact, aligning with the ClinVar designation and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | Benign | 1 | 6-33443731-G-T | 1 | 6.22e-7 | -6.966 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.369 | Likely Benign | -0.73 | Neutral | 0.986 | Probably Damaging | 0.728 | Possibly Damaging | 2.63 | Benign | 0.33 | Tolerated | 4.32 | 2 | 0.1453 | 0.3494 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||
| c.317G>A | R106K 2D AIThe SynGAP1 missense variant R106K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus indicates Likely Benign, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for R106K, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | -4.312 | Likely Benign | 0.513 | Ambiguous | Likely Benign | 0.150 | Likely Benign | -1.25 | Neutral | 0.004 | Benign | 0.001 | Benign | 3.82 | Benign | 0.00 | Affected | 0.5602 | 0.4129 | Weaken | 3 | 2 | 0.6 | -28.01 | ||||||||||||||||||||||||||||||||||||||
| c.317G>C | R106T 2D AIThe SynGAP1 missense variant R106T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. Separately, the high‑accuracy consensus (SGM‑Consensus) classifies the variant as Likely Benign, while AlphaMissense‑Optimized remains uncertain and Foldetta data are missing. Based on the overall pattern of predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | -4.197 | Likely Benign | 0.950 | Likely Pathogenic | Ambiguous | 0.229 | Likely Benign | -2.30 | Neutral | 0.004 | Benign | 0.002 | Benign | 3.67 | Benign | 0.00 | Affected | 0.1941 | 0.4742 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||||||||||||
| c.317G>T | R106M 2D AIThe SynGAP1 missense variant R106M is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2), and Foldetta data are unavailable. Because the majority of tools (five of nine) predict pathogenicity and the most accurate predictor (AlphaMissense‑Optimized) also indicates pathogenicity, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | -4.804 | Likely Benign | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.184 | Likely Benign | -2.65 | Deleterious | 0.940 | Possibly Damaging | 0.360 | Benign | 3.64 | Benign | 0.00 | Affected | 0.1971 | 0.4146 | 0 | -1 | 6.4 | -24.99 | ||||||||||||||||||||||||||||||||||||||||
| c.3181G>A | G1061S 2D AIThe SynGAP1 missense variant G1061S is listed in ClinVar (ID 3571724.0) with an uncertain significance designation and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only SIFT indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta stability analysis is unavailable. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods supports a benign classification for G1061S, which is consistent with its ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | Uncertain | 1 | -4.891 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.283 | Likely Benign | -0.68 | Neutral | 0.004 | Benign | 0.004 | Benign | 4.00 | Benign | 0.00 | Affected | 0.2404 | 0.5300 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||
| c.3181G>C | G1061R 2D AIThe SynGAP1 missense variant G1061R is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it contains two benign and two uncertain calls, and Foldetta results are unavailable. Overall, the balance of evidence favors a benign classification. This conclusion does not contradict ClinVar status, as the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | -7.721 | In-Between | 0.343 | Ambiguous | Likely Benign | 0.315 | Likely Benign | -0.17 | Neutral | 0.411 | Benign | 0.132 | Benign | 3.99 | Benign | 0.00 | Affected | 0.1037 | 0.4332 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3181G>T | G1061C 2D AIThe SynGAP1 missense variant G1061C is listed in ClinVar (ID 536997.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33443733‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence (six benign vs. four pathogenic predictions) and the two high‑accuracy tools support a benign classification. This conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | Conflicting | 2 | 6-33443733-G-T | 6 | 3.73e-6 | -9.511 | Likely Pathogenic | 0.119 | Likely Benign | Likely Benign | 0.409 | Likely Benign | -1.46 | Neutral | 0.938 | Possibly Damaging | 0.665 | Possibly Damaging | 3.97 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1283 | 0.4227 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||
| c.3182G>A | G1061D 2D AIThe SynGAP1 missense variant G1061D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and ESM1b, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign prediction (2 benign vs. 1 pathogenic, 1 uncertain). Foldetta results are unavailable. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | -9.481 | Likely Pathogenic | 0.346 | Ambiguous | Likely Benign | 0.375 | Likely Benign | -1.32 | Neutral | 0.224 | Benign | 0.120 | Benign | 4.01 | Benign | 0.00 | Affected | 0.1671 | 0.2024 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3182G>C | G1061A 2D AIThe SynGAP1 missense variant G1061A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | -6.328 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.244 | Likely Benign | -0.34 | Neutral | 0.004 | Benign | 0.002 | Benign | 4.01 | Benign | 0.00 | Affected | 0.3289 | 0.5133 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3182G>T | G1061V 2D AIThe SynGAP1 missense variant G1061V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence indicates that G1061V is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | -6.709 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.307 | Likely Benign | -1.41 | Neutral | 0.224 | Benign | 0.066 | Benign | 3.98 | Benign | 0.00 | Affected | 0.1431 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3184G>A | G1062R 2D AIThe SynGAP1 missense variant G1062R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443736‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, polyPhen‑2 HumVar, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.936972 | Binding | 0.368 | 0.917 | 0.875 | Conflicting | 2 | 6-33443736-G-A | 7 | 4.35e-6 | -6.933 | Likely Benign | 0.353 | Ambiguous | Likely Benign | 0.403 | Likely Benign | -0.34 | Neutral | 0.797 | Possibly Damaging | 0.139 | Benign | 4.10 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1013 | 0.4342 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||
| c.3184G>C | G1062R 2D AIThe SynGAP1 missense variant G1062R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicating a likely benign outcome, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.936972 | Binding | 0.368 | 0.917 | 0.875 | -6.933 | Likely Benign | 0.353 | Ambiguous | Likely Benign | 0.413 | Likely Benign | -0.34 | Neutral | 0.797 | Possibly Damaging | 0.139 | Benign | 4.10 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1013 | 0.4342 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3184G>T | G1062W 2D AIThe SynGAP1 missense variant G1062W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which labels the variant as “Likely Benign.” Pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b, all of which classify the change as damaging. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.936972 | Binding | 0.368 | 0.917 | 0.875 | -9.667 | Likely Pathogenic | 0.315 | Likely Benign | Likely Benign | 0.401 | Likely Benign | -1.38 | Neutral | 0.993 | Probably Damaging | 0.890 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 0.0908 | 0.4246 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.3185G>A | G1062E 2D AIThe SynGAP1 missense variant G1062E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.936972 | Binding | 0.368 | 0.917 | 0.875 | -8.185 | Likely Pathogenic | 0.272 | Likely Benign | Likely Benign | 0.383 | Likely Benign | -1.02 | Neutral | 0.126 | Benign | 0.041 | Benign | 4.10 | Benign | 0.01 | Affected | 0.1490 | 0.4069 | 0 | -2 | -3.1 | 72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3185G>C | G1062A 2D AIThe SynGAP1 missense variant G1062A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.936972 | Binding | 0.368 | 0.917 | 0.875 | -6.124 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.350 | Likely Benign | 0.20 | Neutral | 0.059 | Benign | 0.028 | Benign | 4.20 | Benign | 0.51 | Tolerated | 0.3305 | 0.5144 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3185G>T | G1062V 2D AIThe SynGAP1 missense variant G1062V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.936972 | Binding | 0.368 | 0.917 | 0.875 | -6.598 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.377 | Likely Benign | -0.78 | Neutral | 0.259 | Benign | 0.066 | Benign | 4.12 | Benign | 0.01 | Affected | 0.1441 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3187G>A | G1063S 2D AIThe SynGAP1 missense variant G1063S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign. Foldetta results are unavailable, so they do not influence the overall assessment. Consequently, the variant is most likely benign based on the collective predictions, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975134 | Disordered | 0.945784 | Binding | 0.394 | 0.913 | 0.875 | -4.707 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.052 | Likely Benign | 0.20 | Neutral | 0.004 | Benign | 0.003 | Benign | 4.33 | Benign | 0.09 | Tolerated | 0.2491 | 0.5702 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3187G>C | G1063R 2D AIThe SynGAP1 missense variant G1063R is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective evidence strongly supports a benign classification, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975134 | Disordered | 0.945784 | Binding | 0.394 | 0.913 | 0.875 | -5.711 | Likely Benign | 0.391 | Ambiguous | Likely Benign | 0.078 | Likely Benign | 0.55 | Neutral | 0.411 | Benign | 0.114 | Benign | 4.28 | Benign | 0.09 | Tolerated | 0.1037 | 0.5133 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3187G>T | G1063C 2D AIThe SynGAP1 missense variant G1063C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which itself is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975134 | Disordered | 0.945784 | Binding | 0.394 | 0.913 | 0.875 | -8.315 | Likely Pathogenic | 0.106 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -1.07 | Neutral | 0.938 | Possibly Damaging | 0.477 | Possibly Damaging | 4.19 | Benign | 0.01 | Affected | 0.1440 | 0.4639 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3188G>A | G1063D 2D AIThe SynGAP1 missense variant G1063D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the computational evidence overwhelmingly supports a benign impact for G1063D, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975134 | Disordered | 0.945784 | Binding | 0.394 | 0.913 | 0.875 | -6.950 | Likely Benign | 0.367 | Ambiguous | Likely Benign | 0.057 | Likely Benign | -0.30 | Neutral | 0.411 | Benign | 0.058 | Benign | 4.24 | Benign | 0.08 | Tolerated | 0.1878 | 0.3026 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.3188G>C | G1063A 2D AIThe SynGAP1 missense variant G1063A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975134 | Disordered | 0.945784 | Binding | 0.394 | 0.913 | 0.875 | -5.373 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.045 | Likely Benign | 0.33 | Neutral | 0.000 | Benign | 0.002 | Benign | 4.30 | Benign | 0.12 | Tolerated | 0.3348 | 0.4957 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3188G>T | G1063V 2D AIThe SynGAP1 missense variant G1063V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority of the high‑accuracy tools) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools indicates that G1063V is most likely benign, with no ClinVar status to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975134 | Disordered | 0.945784 | Binding | 0.394 | 0.913 | 0.875 | -6.228 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.82 | Neutral | 0.004 | Benign | 0.002 | Benign | 4.29 | Benign | 0.03 | Affected | 0.1434 | 0.3707 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.318G>C | R106S 2D AIThe SynGAP1 missense variant R106S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as benign, and the Foldetta stability analysis is unavailable. Overall, the majority of predictions lean toward a benign impact, but the high‑accuracy tools provide conflicting evidence. Thus, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | -2.651 | Likely Benign | 0.961 | Likely Pathogenic | Likely Pathogenic | 0.093 | Likely Benign | -1.87 | Neutral | 0.131 | Benign | 0.026 | Benign | 3.68 | Benign | 0.00 | Affected | 0.3050 | 0.4129 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.318G>T | R106S 2D AIThe SynGAP1 missense variant R106S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as benign, and the Foldetta stability analysis is unavailable. Overall, the majority of predictions lean toward a benign impact, but the high‑accuracy tools provide conflicting evidence. Thus, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | -2.651 | Likely Benign | 0.961 | Likely Pathogenic | Likely Pathogenic | 0.093 | Likely Benign | -1.87 | Neutral | 0.131 | Benign | 0.026 | Benign | 3.68 | Benign | 0.00 | Affected | 0.3050 | 0.4129 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3190C>A | Q1064K 2D AIThe SynGAP1 missense variant Q1064K is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective predictions strongly support a benign classification, and this conclusion is consistent with the lack of any ClinVar pathogenic annotation, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.953106 | Binding | 0.378 | 0.914 | 0.875 | -3.592 | Likely Benign | 0.234 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.32 | Neutral | 0.224 | Benign | 0.120 | Benign | 4.23 | Benign | 0.24 | Tolerated | 0.2758 | 0.4192 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3190C>G | Q1064E 2D AIThe SynGAP1 missense variant Q1064E is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are not available. Based on the unanimous benign predictions and lack of ClinVar evidence, the variant is most likely benign and does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.953106 | Binding | 0.378 | 0.914 | 0.875 | -4.277 | Likely Benign | 0.184 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.48 | Neutral | 0.203 | Benign | 0.077 | Benign | 4.22 | Benign | 0.32 | Tolerated | 0.2259 | 0.2843 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3191A>C | Q1064P 2D AIThe SynGAP1 missense variant Q1064P is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.953106 | Binding | 0.378 | 0.914 | 0.875 | -2.032 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.270 | Likely Benign | 0.93 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.23 | Benign | 0.20 | Tolerated | 0.2819 | 0.4766 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3191A>G | Q1064R 2D AIThe SynGAP1 missense variant Q1064R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and there is no conflict with ClinVar status because the variant is not yet classified there. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.953106 | Binding | 0.378 | 0.914 | 0.875 | -2.981 | Likely Benign | 0.202 | Likely Benign | Likely Benign | 0.143 | Likely Benign | -0.28 | Neutral | 0.586 | Possibly Damaging | 0.159 | Benign | 4.19 | Benign | 0.16 | Tolerated | 0.2257 | 0.3654 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3191A>T | Q1064L 2D AIThe SynGAP1 missense variant Q1064L is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the consensus of all available predictions strongly supports a benign classification, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.953106 | Binding | 0.378 | 0.914 | 0.875 | -3.492 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.133 | Likely Benign | -1.16 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.20 | Benign | 0.13 | Tolerated | 0.1817 | 0.5485 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3192G>C | Q1064H 2D AIThe SynGAP1 missense variant Q1064H is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence—including high‑accuracy tools—points to a benign effect, and this conclusion does not contradict the current ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.953106 | Binding | 0.378 | 0.914 | 0.875 | Uncertain | 1 | -4.576 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.66 | Neutral | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 4.15 | Benign | 0.05 | Affected | 0.2467 | 0.4243 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||
| c.3192G>T | Q1064H 2D AIThe SynGAP1 missense variant Q1064H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q1064H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.953106 | Binding | 0.378 | 0.914 | 0.875 | -4.576 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.66 | Neutral | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 4.15 | Benign | 0.05 | Affected | 0.2467 | 0.4243 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3193C>A | P1065T 2D AIThe SynGAP1 missense variant P1065T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign classification. There is no ClinVar status to contradict this assessment, so the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -5.392 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -1.23 | Neutral | 0.770 | Possibly Damaging | 0.481 | Possibly Damaging | 2.04 | Pathogenic | 0.00 | Affected | 0.1577 | 0.6788 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3193C>G | P1065A 2D AIThe SynGAP1 missense variant P1065A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for P1065A, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -4.043 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.050 | Likely Benign | -1.85 | Neutral | 0.580 | Possibly Damaging | 0.184 | Benign | 2.19 | Pathogenic | 0.00 | Affected | 0.3140 | 0.5790 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3193C>T | P1065S 2D AIThe SynGAP1 missense variant P1065S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -5.512 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -2.07 | Neutral | 0.770 | Possibly Damaging | 0.255 | Benign | 2.06 | Pathogenic | 0.00 | Affected | 0.3172 | 0.6021 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3194C>A | P1065Q 2D AIThe SynGAP1 missense variant P1065Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -3.928 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -2.44 | Neutral | 0.102 | Benign | 0.057 | Benign | 2.00 | Pathogenic | 0.00 | Affected | 0.1487 | 0.5478 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3194C>G | P1065R 2D AIThe SynGAP1 missense variant P1065R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -3.237 | Likely Benign | 0.228 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -2.46 | Neutral | 0.102 | Benign | 0.052 | Benign | 2.00 | Pathogenic | 0.00 | Affected | 0.1439 | 0.4369 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3194C>T | P1065L 2D AIThe SynGAP1 missense variant P1065L is listed in ClinVar as Benign and is present in gnomAD (ID 6‑33443746‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no reported result for this variant. Overall, the balance of evidence (5 benign vs. 4 pathogenic predictions) and the high‑accuracy benign call support a benign classification, aligning with the ClinVar status and indicating no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | Likely Benign | 1 | 6-33443746-C-T | 14 | 8.71e-6 | -5.085 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -2.94 | Deleterious | 0.950 | Possibly Damaging | 0.419 | Benign | 2.01 | Pathogenic | 0.00 | Affected | 4.32 | 2 | 0.2286 | 0.6922 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||
| c.3196C>A | P1066T 2D AIThe SynGAP1 missense variant P1066T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.967676 | Disordered | 0.968838 | Binding | 0.403 | 0.913 | 0.875 | -5.973 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.167 | Likely Benign | -2.61 | Deleterious | 0.996 | Probably Damaging | 0.928 | Probably Damaging | 2.66 | Benign | 0.00 | Affected | 0.1652 | 0.7240 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3196C>G | P1066A 2D AIThe SynGAP1 missense variant P1066A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.967676 | Disordered | 0.968838 | Binding | 0.403 | 0.913 | 0.875 | -4.856 | Likely Benign | 0.050 | Likely Benign | Likely Benign | 0.149 | Likely Benign | -2.35 | Neutral | 0.972 | Probably Damaging | 0.802 | Possibly Damaging | 2.78 | Benign | 0.00 | Affected | 0.3228 | 0.6042 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3196C>T | P1066S 2D AIThe SynGAP1 missense variant P1066S is listed in ClinVar as Pathogenic (ClinVar ID 1343237.0) and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, which contradicts the ClinVar pathogenic classification. Thus, based on current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.967676 | Disordered | 0.968838 | Binding | 0.403 | 0.913 | 0.875 | Likely Pathogenic | 1 | -4.746 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -2.47 | Neutral | 0.972 | Probably Damaging | 0.850 | Possibly Damaging | 2.74 | Benign | 0.00 | Affected | 4.32 | 2 | 0.3304 | 0.6353 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||
| c.3197C>A | P1066H 2D AIThe SynGAP1 missense variant P1066H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.967676 | Disordered | 0.968838 | Binding | 0.403 | 0.913 | 0.875 | -6.034 | Likely Benign | 0.185 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -2.90 | Deleterious | 1.000 | Probably Damaging | 0.975 | Probably Damaging | 2.61 | Benign | 0.00 | Affected | 0.1984 | 0.5861 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3197C>G | P1066R 2D AIThe SynGAP1 missense variant P1066R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, tools that predict a pathogenic outcome are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.967676 | Disordered | 0.968838 | Binding | 0.403 | 0.913 | 0.875 | -5.154 | Likely Benign | 0.292 | Likely Benign | Likely Benign | 0.176 | Likely Benign | -2.89 | Deleterious | 0.992 | Probably Damaging | 0.873 | Possibly Damaging | 2.63 | Benign | 0.00 | Affected | 0.1523 | 0.4742 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3197C>T | P1066L 2D AIThe SynGAP1 missense variant P1066L is listed in ClinVar as a benign variant (ClinVar ID 951518.0) and is present in gnomAD (ID 6‑33443749‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, which is consistent with the ClinVar classification and does not contradict the reported status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.967676 | Disordered | 0.968838 | Binding | 0.403 | 0.913 | 0.875 | Likely Benign | 1 | 6-33443749-C-T | 14 | 8.71e-6 | -5.478 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -3.68 | Deleterious | 0.996 | Probably Damaging | 0.903 | Possibly Damaging | 2.72 | Benign | 0.00 | Affected | 4.32 | 2 | 0.2269 | 0.6780 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||
| c.3199C>A | P1067T 2D AIThe SynGAP1 missense variant P1067T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 HumDiv and SIFT predict pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.966441 | Disordered | 0.975099 | Binding | 0.459 | 0.907 | 0.875 | -4.898 | Likely Benign | 0.084 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -1.94 | Neutral | 0.827 | Possibly Damaging | 0.375 | Benign | 2.79 | Benign | 0.04 | Affected | 0.1449 | 0.6132 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3199C>G | P1067A 2D AIThe SynGAP1 missense variant P1067A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, representing the sole discordant prediction. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence supports a benign classification for P1067A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.966441 | Disordered | 0.975099 | Binding | 0.459 | 0.907 | 0.875 | -4.639 | Likely Benign | 0.052 | Likely Benign | Likely Benign | 0.073 | Likely Benign | -2.05 | Neutral | 0.664 | Possibly Damaging | 0.283 | Benign | 2.87 | Benign | 0.07 | Tolerated | 0.3097 | 0.5322 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3199C>T | P1067S 2D AIThe SynGAP1 missense variant P1067S is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.966441 | Disordered | 0.975099 | Binding | 0.459 | 0.907 | 0.875 | -4.673 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -1.43 | Neutral | 0.271 | Benign | 0.054 | Benign | 2.91 | Benign | 0.48 | Tolerated | 0.3086 | 0.5753 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.319A>G | R107G 2D AIThe SynGAP1 missense variant R107G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM, while those that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a tie and thus unavailable, and Foldetta stability analysis is missing. Overall, the majority of tools (five benign vs. three pathogenic) suggest a benign impact, but the lack of consensus from the most accurate predictors means the variant’s effect remains uncertain. This assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.663448 | Binding | 0.331 | 0.863 | 0.875 | -3.486 | Likely Benign | 0.948 | Likely Pathogenic | Ambiguous | 0.180 | Likely Benign | -3.15 | Deleterious | 0.421 | Benign | 0.050 | Benign | 2.98 | Benign | 0.00 | Affected | 0.3280 | 0.4000 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.319A>T | R107W 2D AIThe SynGAP1 missense variant R107W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two pathogenic vs two benign), and Foldetta results are unavailable. Overall, the majority of evidence (six pathogenic vs three benign) points to a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.663448 | Binding | 0.331 | 0.863 | 0.875 | -4.963 | Likely Benign | 0.965 | Likely Pathogenic | Likely Pathogenic | 0.328 | Likely Benign | -2.99 | Deleterious | 0.983 | Probably Damaging | 0.624 | Possibly Damaging | 2.95 | Benign | 0.00 | Affected | 0.1146 | 0.4228 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.31G>A | G11R 2D AIThe SynGAP1 missense variant G11R is catalogued in gnomAD (ID 6‑33420295‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | 6-33420295-G-A | -3.418 | Likely Benign | 0.428 | Ambiguous | Likely Benign | 0.102 | Likely Benign | -0.47 | Neutral | 0.498 | Possibly Damaging | 0.026 | Benign | 3.92 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1022 | 0.4596 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||
| c.31G>C | G11R 2D AIThe SynGAP1 missense variant G11R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | -3.418 | Likely Benign | 0.428 | Ambiguous | Likely Benign | 0.109 | Likely Benign | -0.47 | Neutral | 0.498 | Possibly Damaging | 0.026 | Benign | 3.92 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1022 | 0.4596 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.31G>T | G11W 2D AIThe SynGAP1 missense variant G11W is catalogued in gnomAD (ID 6‑33420295‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | 6-33420295-G-T | -5.819 | Likely Benign | 0.403 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -0.67 | Neutral | 0.959 | Probably Damaging | 0.318 | Benign | 3.87 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0747 | 0.4731 | -2 | -7 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||||||||
| c.3200C>A | P1067Q 2D AIThe SynGAP1 missense variant P1067Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for P1067Q, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.966441 | Disordered | 0.975099 | Binding | 0.459 | 0.907 | 0.875 | -4.767 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.136 | Likely Benign | -2.39 | Neutral | 0.463 | Possibly Damaging | 0.087 | Benign | 2.84 | Benign | 0.01 | Affected | 0.1369 | 0.5415 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3200C>G | P1067R 2D AIThe SynGAP1 missense variant P1067R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign, while Foldetta’s stability analysis is unavailable. Taken together, the majority of reliable predictors and the high‑accuracy tools indicate a benign impact. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.966441 | Disordered | 0.975099 | Binding | 0.459 | 0.907 | 0.875 | -4.878 | Likely Benign | 0.376 | Ambiguous | Likely Benign | 0.167 | Likely Benign | -2.74 | Deleterious | 0.971 | Probably Damaging | 0.580 | Possibly Damaging | 2.78 | Benign | 0.01 | Affected | 0.1300 | 0.3651 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||||
| c.3200C>T | P1067L 2D AIThe SynGAP1 missense variant P1067L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of predictions and the consensus analysis indicate a benign impact. This conclusion is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.966441 | Disordered | 0.975099 | Binding | 0.459 | 0.907 | 0.875 | -4.461 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.157 | Likely Benign | -3.01 | Deleterious | 0.951 | Possibly Damaging | 0.619 | Possibly Damaging | 2.76 | Benign | 0.01 | Affected | 0.2047 | 0.6198 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3202T>A | L1068M 2D AIThe SynGAP1 missense variant L1068M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.947281 | Disordered | 0.981041 | Binding | 0.362 | 0.907 | 0.875 | -5.739 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -0.22 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 2.49 | Pathogenic | 0.00 | Affected | 0.0930 | 0.4582 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3202T>G | L1068V 2D AIThe SynGAP1 missense variant L1068V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.947281 | Disordered | 0.981041 | Binding | 0.362 | 0.907 | 0.875 | -5.325 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -0.58 | Neutral | 0.451 | Benign | 0.110 | Benign | 2.54 | Benign | 0.00 | Affected | 0.1619 | 0.4404 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3203T>C | L1068S 2D  AIThe SynGAP1 missense variant L1068S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign, while only SIFT predicts it as pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.947281 | Disordered | 0.981041 | Binding | 0.362 | 0.907 | 0.875 | -6.297 | Likely Benign | 0.211 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -0.57 | Neutral | 0.032 | Benign | 0.017 | Benign | 2.59 | Benign | 0.00 | Affected | 0.2810 | 0.1853 | -3 | -2 | -4.6 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3203T>G | L1068W 2D  AIThe SynGAP1 missense variant L1068W is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized remains benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) resolves as pathogenic, and Foldetta’s protein‑folding stability analysis is unavailable. Overall, the majority of evidence points toward a pathogenic impact for L1068W. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.947281 | Disordered | 0.981041 | Binding | 0.362 | 0.907 | 0.875 | -8.574 | Likely Pathogenic | 0.342 | Ambiguous | Likely Benign | 0.151 | Likely Benign | -1.96 | Neutral | 0.994 | Probably Damaging | 0.884 | Possibly Damaging | 2.46 | Pathogenic | 0.00 | Affected | 0.0742 | 0.3763 | -2 | -2 | -4.7 | 73.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3204G>C | L1068F 2D AIThe SynGAP1 missense variant L1068F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) favor a benign classification. Thus, the variant is most likely benign based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.947281 | Disordered | 0.981041 | Binding | 0.362 | 0.907 | 0.875 | -6.338 | Likely Benign | 0.182 | Likely Benign | Likely Benign | 0.107 | Likely Benign | -1.38 | Neutral | 0.934 | Possibly Damaging | 0.537 | Possibly Damaging | 2.49 | Pathogenic | 0.00 | Affected | 0.0754 | 0.4202 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3204G>T | L1068F 2D AIThe SynGAP1 missense variant L1068F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) favor a benign classification. Thus, the variant is most likely benign based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.947281 | Disordered | 0.981041 | Binding | 0.362 | 0.907 | 0.875 | -6.338 | Likely Benign | 0.182 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -1.38 | Neutral | 0.934 | Possibly Damaging | 0.537 | Possibly Damaging | 2.49 | Pathogenic | 0.00 | Affected | 0.0754 | 0.4202 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3205C>A | Q1069K 2D AIThe SynGAP1 missense variant Q1069K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of computational evidence points to a benign effect, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.943310 | Disordered | 0.981477 | Binding | 0.333 | 0.906 | 0.875 | -5.080 | Likely Benign | 0.542 | Ambiguous | Likely Benign | 0.099 | Likely Benign | -0.88 | Neutral | 0.625 | Possibly Damaging | 0.266 | Benign | 2.77 | Benign | 0.28 | Tolerated | 0.1904 | 0.5071 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3205C>G | Q1069E 2D AIThe SynGAP1 missense variant Q1069E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.943310 | Disordered | 0.981477 | Binding | 0.333 | 0.906 | 0.875 | -5.165 | Likely Benign | 0.322 | Likely Benign | Likely Benign | 0.107 | Likely Benign | -0.68 | Neutral | 0.451 | Benign | 0.266 | Benign | 2.76 | Benign | 0.33 | Tolerated | 0.1460 | 0.3127 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3206A>C | Q1069P 2D AIThe SynGAP1 missense variant Q1069P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.943310 | Disordered | 0.981477 | Binding | 0.333 | 0.906 | 0.875 | -3.458 | Likely Benign | 0.056 | Likely Benign | Likely Benign | 0.211 | Likely Benign | 0.75 | Neutral | 0.977 | Probably Damaging | 0.722 | Possibly Damaging | 2.73 | Benign | 1.00 | Tolerated | 0.2179 | 0.5944 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3206A>G | Q1069R 2D AIThe SynGAP1 missense variant Q1069R is not reported in ClinVar (ClinVar ID = None) and has no entries in gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable. Overall, the preponderance of evidence points to the variant being most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.943310 | Disordered | 0.981477 | Binding | 0.333 | 0.906 | 0.875 | -3.257 | Likely Benign | 0.467 | Ambiguous | Likely Benign | 0.094 | Likely Benign | -1.17 | Neutral | 0.666 | Possibly Damaging | 0.355 | Benign | 2.73 | Benign | 0.21 | Tolerated | 0.1557 | 0.3114 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3206A>T | Q1069L 2D AIThe SynGAP1 missense variant Q1069L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.943310 | Disordered | 0.981477 | Binding | 0.333 | 0.906 | 0.875 | -4.278 | Likely Benign | 0.141 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -0.96 | Neutral | 0.003 | Benign | 0.008 | Benign | 2.83 | Benign | 0.10 | Tolerated | 0.0936 | 0.6624 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3207G>C | Q1069H 2D AIThe SynGAP1 missense variant Q1069H is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.943310 | Disordered | 0.981477 | Binding | 0.333 | 0.906 | 0.875 | -4.723 | Likely Benign | 0.306 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -1.31 | Neutral | 0.006 | Benign | 0.008 | Benign | 2.69 | Benign | 0.06 | Tolerated | 0.1560 | 0.4678 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3207G>T | Q1069H 2D AIThe SynGAP1 missense variant Q1069H is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.943310 | Disordered | 0.981477 | Binding | 0.333 | 0.906 | 0.875 | -4.723 | Likely Benign | 0.306 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -1.31 | Neutral | 0.006 | Benign | 0.008 | Benign | 2.69 | Benign | 0.06 | Tolerated | 0.1560 | 0.4678 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3208A>G | R1070G 2D AIThe SynGAP1 missense variant R1070G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Consensus from standard in silico predictors shows five tools (REVEL, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized) classifying it as benign, while four (PROVEAN, polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default) predict pathogenicity. High‑accuracy assessments give a benign result from AlphaMissense‑Optimized; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 vs 2), and Foldetta data are unavailable. Overall, the balance of evidence leans toward a benign effect. This conclusion does not conflict with ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.930790 | Disordered | 0.982693 | Binding | 0.297 | 0.906 | 0.875 | -4.731 | Likely Benign | 0.568 | Likely Pathogenic | Likely Benign | 0.149 | Likely Benign | -2.88 | Deleterious | 0.789 | Possibly Damaging | 0.258 | Benign | 3.75 | Benign | 0.01 | Affected | 0.3150 | 0.4070 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3208A>T | R1070W 2D AIThe SynGAP1 missense variant R1070W is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant as benign, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates it is likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this residue. Overall, the majority of tools and the SGM‑Consensus support a pathogenic effect, so the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.930790 | Disordered | 0.982693 | Binding | 0.297 | 0.906 | 0.875 | -8.063 | Likely Pathogenic | 0.743 | Likely Pathogenic | Likely Benign | 0.139 | Likely Benign | -3.42 | Deleterious | 0.999 | Probably Damaging | 0.956 | Probably Damaging | 3.72 | Benign | 0.00 | Affected | 0.1195 | 0.4293 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3209G>A | R1070K 2D AIThe SynGAP1 missense variant R1070K is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All evaluated in‑silico predictors agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” High‑accuracy tools reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta results are unavailable. Based on the unanimous benign predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.982693 | Binding | 0.297 | 0.906 | 0.875 | Conflicting | 2 | -5.093 | Likely Benign | 0.326 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -1.42 | Neutral | 0.049 | Benign | 0.048 | Benign | 3.86 | Benign | 0.09 | Tolerated | 3.77 | 5 | 0.4997 | 0.4867 | 3 | 2 | 0.6 | -28.01 | |||||||||||||||||||||||||||||||||||
| c.3209G>C | R1070T 2D AIThe SynGAP1 missense variant R1070T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are not available. Overall, the balance of evidence leans toward a benign impact, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.982693 | Binding | 0.297 | 0.906 | 0.875 | -5.093 | Likely Benign | 0.860 | Likely Pathogenic | Ambiguous | 0.144 | Likely Benign | -2.35 | Neutral | 0.948 | Possibly Damaging | 0.507 | Possibly Damaging | 3.78 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1634 | 0.4727 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||||||||||
| c.3209G>T | R1070M 2D AIThe SynGAP1 missense variant R1070M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence, including the consensus benign call and the absence of pathogenic predictions from the most reliable tools, suggests the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.982693 | Binding | 0.297 | 0.906 | 0.875 | -6.455 | Likely Benign | 0.917 | Likely Pathogenic | Ambiguous | 0.183 | Likely Benign | -2.27 | Neutral | 0.995 | Probably Damaging | 0.907 | Possibly Damaging | 3.74 | Benign | 0.00 | Affected | 0.1661 | 0.4324 | 0 | -1 | 6.4 | -24.99 | |||||||||||||||||||||||||||||||||||||||
| c.3209_3210delinsCA | R1070T 2D AIThe SynGAP1 missense variant R1070T is listed in ClinVar (ID 2759838.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Prediction tools that agree on a benign effect include PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (which aggregates these three benign calls with the pathogenic AlphaMissense‑Default to yield a Likely Benign verdict). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized returns an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and no Foldetta stability data is available. Overall, the balance of evidence leans toward a benign impact, which is consistent with the ClinVar “Uncertain” status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.982693 | Binding | 0.297 | 0.906 | 0.875 | Uncertain | 1 | -5.093 | Likely Benign | 0.860 | Likely Pathogenic | Ambiguous | -2.35 | Neutral | 0.948 | Possibly Damaging | 0.507 | Possibly Damaging | 3.78 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1634 | 0.4727 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||||||||||
| c.320G>A | R107K 2D AIThe SynGAP1 missense variant R107K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R107K, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.666105 | Disordered | 0.663448 | Binding | 0.331 | 0.863 | 0.875 | -3.941 | Likely Benign | 0.758 | Likely Pathogenic | Likely Benign | 0.153 | Likely Benign | -1.33 | Neutral | 0.004 | Benign | 0.001 | Benign | 3.07 | Benign | 0.00 | Affected | 0.5618 | 0.4185 | Weaken | 3 | 2 | 0.6 | -28.01 | ||||||||||||||||||||||||||||||||||||||
| c.320G>C | R107T 2D AIThe SynGAP1 missense variant R107T is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as pathogenic, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 pathogenic vs. 2 benign) and Foldetta results are unavailable. Overall, the majority of predictions (five benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.663448 | Binding | 0.331 | 0.863 | 0.875 | -2.902 | Likely Benign | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.191 | Likely Benign | -2.63 | Deleterious | 0.421 | Benign | 0.050 | Benign | 2.98 | Benign | 0.00 | Affected | 0.1549 | 0.4761 | -1 | -1 | 3.8 | -55.08 | ||||||||||||||||||||||||||||||||||||||||
| c.320G>T | R107M 2D AIThe SynGAP1 missense variant R107M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta results are unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not conflict with the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.663448 | Binding | 0.331 | 0.863 | 0.875 | -4.873 | Likely Benign | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.233 | Likely Benign | -2.65 | Deleterious | 0.028 | Benign | 0.011 | Benign | 2.96 | Benign | 0.00 | Affected | 0.1605 | 0.4415 | 0 | -1 | 6.4 | -24.99 | ||||||||||||||||||||||||||||||||||||||||
| c.3210G>C | R1070S 2D AIThe SynGAP1 missense variant R1070S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.982693 | Binding | 0.297 | 0.906 | 0.875 | -4.311 | Likely Benign | 0.913 | Likely Pathogenic | Ambiguous | 0.091 | Likely Benign | -2.07 | Neutral | 0.789 | Possibly Damaging | 0.258 | Benign | 3.85 | Benign | 0.01 | Affected | 0.2640 | 0.4042 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3210G>T | R1070S 2D AIThe SynGAP1 missense variant R1070S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.982693 | Binding | 0.297 | 0.906 | 0.875 | -4.311 | Likely Benign | 0.913 | Likely Pathogenic | Ambiguous | 0.091 | Likely Benign | -2.07 | Neutral | 0.789 | Possibly Damaging | 0.258 | Benign | 3.85 | Benign | 0.01 | Affected | 0.2640 | 0.4042 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3211G>A | G1071S 2D AIThe SynGAP1 missense variant G1071S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign impact for G1071S, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983740 | Binding | 0.313 | 0.905 | 0.875 | -1.139 | Likely Benign | 0.168 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -1.06 | Neutral | 0.692 | Possibly Damaging | 0.222 | Benign | 4.10 | Benign | 0.28 | Tolerated | 0.2468 | 0.5451 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3211G>C | G1071R 2D AIThe SynGAP1 missense variant G1071R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie and thus unavailable; Foldetta results are not provided and are therefore unavailable. Overall, more tools predict pathogenicity (5) than benignity (3), and no ClinVar entry contradicts this assessment. **The variant is most likely pathogenic based on the available predictions.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.919029 | Disordered | 0.983740 | Binding | 0.313 | 0.905 | 0.875 | -3.052 | Likely Benign | 0.886 | Likely Pathogenic | Ambiguous | 0.135 | Likely Benign | -2.61 | Deleterious | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 4.06 | Benign | 0.00 | Affected | 0.0980 | 0.4482 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3211G>T | G1071C 2D AIThe SynGAP1 missense variant G1071C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence—including the consensus and high‑accuracy predictions—supports a benign classification, and this is consistent with the lack of ClinVar annotation. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983740 | Binding | 0.313 | 0.905 | 0.875 | -5.364 | Likely Benign | 0.305 | Likely Benign | Likely Benign | 0.182 | Likely Benign | -2.16 | Neutral | 0.997 | Probably Damaging | 0.889 | Possibly Damaging | 4.01 | Benign | 0.00 | Affected | 0.1323 | 0.4227 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3212G>A | G1071D 2D AIThe SynGAP1 missense variant G1071D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the majority of evidence points toward a benign impact, with no conflict with ClinVar status (which has no entry). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983740 | Binding | 0.313 | 0.905 | 0.875 | -4.704 | Likely Benign | 0.866 | Likely Pathogenic | Ambiguous | 0.101 | Likely Benign | -1.92 | Neutral | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 4.05 | Benign | 0.01 | Affected | 0.1760 | 0.2175 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.3212G>C | G1071A 2D AIThe SynGAP1 missense variant G1071A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983740 | Binding | 0.313 | 0.905 | 0.875 | -1.825 | Likely Benign | 0.229 | Likely Benign | Likely Benign | 0.070 | Likely Benign | -1.34 | Neutral | 0.025 | Benign | 0.022 | Benign | 4.12 | Benign | 0.05 | Affected | 0.3376 | 0.5084 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3212G>T | G1071V 2D AIThe SynGAP1 missense variant G1071V is catalogued in gnomAD (ID 6‑33443764‑G‑T) but has no ClinVar entry. In silico prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or likely benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, supports a benign interpretation. This prediction does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983740 | Binding | 0.313 | 0.905 | 0.875 | 6-33443764-G-T | 3 | 1.87e-6 | -2.901 | Likely Benign | 0.300 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -2.42 | Neutral | 0.057 | Benign | 0.022 | Benign | 4.14 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1408 | 0.3634 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||
| c.3214A>C | K1072Q 2D  AIThe SynGAP1 missense variant K1072Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.984675 | Binding | 0.307 | 0.907 | 0.750 | -1.631 | Likely Benign | 0.589 | Likely Pathogenic | Likely Benign | 0.081 | Likely Benign | -0.63 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 3.95 | Benign | 0.12 | Tolerated | 0.4465 | 0.1804 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3214A>G | K1072E 2D  AIThe SynGAP1 missense variant K1072E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions are returned by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments indicate that AlphaMissense‑Optimized classifies the variant as pathogenic, whereas the SGM‑Consensus (derived from the same set of predictors) labels it as likely benign. No Foldetta stability analysis is available for this residue. Overall, the majority of tools lean toward a benign effect, but the presence of pathogenic calls from several high‑confidence predictors suggests uncertainty. The variant is most likely benign based on the preponderance of evidence, and this conclusion does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.984675 | Binding | 0.307 | 0.907 | 0.750 | -3.889 | Likely Benign | 0.961 | Likely Pathogenic | Likely Pathogenic | 0.105 | Likely Benign | -0.90 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 3.96 | Benign | 0.08 | Tolerated | 0.3636 | 0.1874 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3215A>C | K1072T 2D  AIThe SynGAP1 missense variant K1072T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus result is benign; Foldetta predictions are unavailable. Overall, the balance of evidence leans toward a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.984675 | Binding | 0.307 | 0.907 | 0.750 | -2.557 | Likely Benign | 0.834 | Likely Pathogenic | Ambiguous | 0.082 | Likely Benign | -1.31 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 3.92 | Benign | 0.06 | Tolerated | 0.1967 | 0.4307 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||||||||||||
| c.3215A>G | K1072R 2D  AIThe SynGAP1 missense variant K1072R is reported in gnomAD (ID 6‑33443767‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. When predictions are grouped by consensus, the benign group contains seven tools, whereas the pathogenic group contains two. High‑accuracy assessments reinforce the benign view: AlphaMissense‑Optimized reports a benign outcome, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. No Foldetta stability data are available, so folding‑stability evidence is unavailable. Overall, the majority of evidence points to a benign impact, and this is not in conflict with ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.984675 | Binding | 0.307 | 0.907 | 0.750 | 6-33443767-A-G | 1 | 6.23e-7 | -2.458 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.15 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 4.16 | Benign | 0.88 | Tolerated | 3.77 | 5 | 0.4655 | 0.2061 | 2 | 3 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||
| c.3215A>T | K1072M 2D  AIThe SynGAP1 K1072M missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points toward a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current predictive data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.984675 | Binding | 0.307 | 0.907 | 0.750 | -2.821 | Likely Benign | 0.928 | Likely Pathogenic | Ambiguous | 0.144 | Likely Benign | -1.37 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.88 | Benign | 0.02 | Affected | 0.1266 | 0.4877 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||||||||||||
| c.3216G>C | K1072N 2D  AIThe SynGAP1 missense variant K1072N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are not available. Overall, the balance of evidence leans toward a benign impact, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.984675 | Binding | 0.307 | 0.907 | 0.750 | -2.215 | Likely Benign | 0.953 | Likely Pathogenic | Ambiguous | 0.078 | Likely Benign | -1.18 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 3.92 | Benign | 0.05 | Affected | 0.3449 | 0.2391 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3216G>T | K1072N 2D AIThe SynGAP1 missense variant K1072N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are not available. Overall, the balance of evidence leans toward a benign impact, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.984675 | Binding | 0.307 | 0.907 | 0.750 | -2.215 | Likely Benign | 0.953 | Likely Pathogenic | Ambiguous | 0.078 | Likely Benign | -1.18 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 3.92 | Benign | 0.05 | Affected | 0.3449 | 0.2391 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3217T>A | S1073T 2D AIThe SynGAP1 missense variant S1073T is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.985818 | Binding | 0.313 | 0.905 | 0.750 | -5.203 | Likely Benign | 0.169 | Likely Benign | Likely Benign | 0.161 | Likely Benign | -0.26 | Neutral | 0.025 | Benign | 0.026 | Benign | 4.06 | Benign | 0.55 | Tolerated | 0.1696 | 0.6509 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3217T>C | S1073P 2D AIThe SynGAP1 missense variant S1073P is reported in gnomAD (variant ID 6‑33443769‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign impact for S1073P, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.985818 | Binding | 0.313 | 0.905 | 0.750 | 6-33443769-T-C | 1 | 6.23e-7 | -4.520 | Likely Benign | 0.338 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.76 | Neutral | 0.006 | Benign | 0.008 | Benign | 3.85 | Benign | 0.01 | Affected | 3.77 | 5 | 0.2243 | 0.5900 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||
| c.3217T>G | S1073A 2D AIThe SynGAP1 missense variant S1073A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign, while only SIFT predicts it as pathogenic. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) confirms a benign status. Foldetta’s protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools suggests that the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.985818 | Binding | 0.313 | 0.905 | 0.750 | -5.333 | Likely Benign | 0.220 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -0.96 | Neutral | 0.447 | Benign | 0.103 | Benign | 3.95 | Benign | 0.02 | Affected | 0.4571 | 0.5883 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3218C>A | S1073Y 2D AIThe SynGAP1 missense change S1073Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.985818 | Binding | 0.313 | 0.905 | 0.750 | -6.768 | Likely Benign | 0.752 | Likely Pathogenic | Likely Benign | 0.165 | Likely Benign | -2.43 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 3.81 | Benign | 0.00 | Affected | 0.0977 | 0.5684 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||||
| c.3218C>G | S1073C 2D AISynGAP1 missense variant S1073C is recorded in gnomAD (ID 6‑33443770‑C‑G) but has no ClinVar submission. Functional prediction tools show mixed results: benign calls come from REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The AlphaMissense‑Default score is uncertain. A consensus from the SGM framework (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because the votes are split. High‑accuracy methods give a benign prediction from AlphaMissense‑Optimized; the SGM Consensus remains ambiguous, and Foldetta stability analysis is unavailable. Consequently, the evidence does not strongly favor either outcome. The variant is most likely of uncertain significance, with no contradiction to ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.916840 | Disordered | 0.985818 | Binding | 0.313 | 0.905 | 0.750 | 6-33443770-C-G | 1 | 6.23e-7 | -8.862 | Likely Pathogenic | 0.461 | Ambiguous | Likely Benign | 0.137 | Likely Benign | -1.52 | Neutral | 0.997 | Probably Damaging | 0.840 | Possibly Damaging | 3.81 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1343 | 0.6088 | -1 | 0 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||
| c.3218C>T | S1073F 2D AIThe SynGAP1 missense variant S1073F is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.916840 | Disordered | 0.985818 | Binding | 0.313 | 0.905 | 0.750 | -6.783 | Likely Benign | 0.836 | Likely Pathogenic | Ambiguous | 0.161 | Likely Benign | -2.58 | Deleterious | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 3.81 | Benign | 0.00 | Affected | 0.0952 | 0.5957 | -3 | -2 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||||||
| c.321G>C | R107S 2D AIThe SynGAP1 missense variant R107S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as pathogenic, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome; Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, and this is not contradicted by any ClinVar annotation. Thus, the variant is most likely benign, although high‑accuracy tools provide conflicting evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.666105 | Disordered | 0.663448 | Binding | 0.331 | 0.863 | 0.875 | -1.162 | Likely Benign | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.147 | Likely Benign | -2.37 | Neutral | 0.231 | Benign | 0.037 | Benign | 2.99 | Benign | 0.00 | Affected | 0.2771 | 0.4148 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.321G>T | R107S 2D AIThe SynGAP1 missense variant R107S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome; Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, but the high‑accuracy tools provide conflicting evidence. Thus, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.666105 | Disordered | 0.663448 | Binding | 0.331 | 0.863 | 0.875 | -1.162 | Likely Benign | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.147 | Likely Benign | -2.37 | Neutral | 0.231 | Benign | 0.037 | Benign | 2.99 | Benign | 0.00 | Affected | 0.2771 | 0.4148 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3220C>A | Q1074K 2D AIThe SynGAP1 missense variant Q1074K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion is consistent with the lack of any ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.987006 | Binding | 0.339 | 0.897 | 0.750 | -6.162 | Likely Benign | 0.712 | Likely Pathogenic | Likely Benign | 0.110 | Likely Benign | -0.88 | Neutral | 0.011 | Benign | 0.006 | Benign | 2.75 | Benign | 0.36 | Tolerated | 0.1865 | 0.4600 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3220C>G | Q1074E 2D AIThe SynGAP1 missense variant Q1074E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions strongly suggests that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.987006 | Binding | 0.339 | 0.897 | 0.750 | -2.815 | Likely Benign | 0.419 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -0.79 | Neutral | 0.264 | Benign | 0.103 | Benign | 2.79 | Benign | 0.24 | Tolerated | 0.1386 | 0.2856 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3221A>C | Q1074P 2D AIThe SynGAP1 missense variant Q1074P is listed in gnomAD (ID 6‑33443773‑A‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or tolerated. Only polyPhen‑2 HumDiv flags it as pathogenic, creating a single outlier. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.987006 | Binding | 0.339 | 0.897 | 0.750 | 6-33443773-A-C | 1 | 6.23e-7 | -4.259 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.188 | Likely Benign | 0.52 | Neutral | 0.925 | Possibly Damaging | 0.432 | Benign | 2.66 | Benign | 0.13 | Tolerated | 3.77 | 5 | 0.2046 | 0.5784 | -1 | 0 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||
| c.3221A>G | Q1074R 2D AIThe SynGAP1 missense variant Q1074R is listed in gnomAD (ID 6‑33443773‑A‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar status (none reported). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.987006 | Binding | 0.339 | 0.897 | 0.750 | 6-33443773-A-G | -5.710 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 0.124 | Likely Benign | -0.54 | Neutral | 0.292 | Benign | 0.157 | Benign | 2.70 | Benign | 0.28 | Tolerated | 3.77 | 5 | 0.1523 | 0.2643 | 1 | 1 | -1.0 | 28.06 | ||||||||||||||||||||||||||||||||||||
| c.3221A>T | Q1074L 2D AIThe SynGAP1 missense variant Q1074L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.987006 | Binding | 0.339 | 0.897 | 0.750 | -3.561 | Likely Benign | 0.259 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -1.29 | Neutral | 0.625 | Possibly Damaging | 0.266 | Benign | 2.68 | Benign | 1.00 | Tolerated | 0.0840 | 0.6293 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3222G>C | Q1074H 2D AIThe SynGAP1 missense variant Q1074H is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools uniformly favor a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity, and the only uncertain call comes from AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Benign.” High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.987006 | Binding | 0.339 | 0.897 | 0.750 | -5.051 | Likely Benign | 0.381 | Ambiguous | Likely Benign | 0.053 | Likely Benign | -0.12 | Neutral | 0.006 | Benign | 0.008 | Benign | 2.67 | Benign | 0.07 | Tolerated | 0.1436 | 0.4407 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3222G>T | Q1074H 2D AIThe SynGAP1 missense variant Q1074H is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools uniformly favor a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity, and the only uncertain call comes from AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Benign.” High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.987006 | Binding | 0.339 | 0.897 | 0.750 | -5.051 | Likely Benign | 0.381 | Ambiguous | Likely Benign | 0.053 | Likely Benign | -0.12 | Neutral | 0.006 | Benign | 0.008 | Benign | 2.67 | Benign | 0.07 | Tolerated | 0.1436 | 0.4407 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3223C>A | Q1075K 2D AIThe SynGAP1 missense variant Q1075K (ClinVar ID 2762879.0) is listed as “Uncertain” in ClinVar and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign” because three of the four contributing tools predict benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) as benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.988305 | Binding | 0.354 | 0.894 | 0.750 | Uncertain | 1 | -5.135 | Likely Benign | 0.728 | Likely Pathogenic | Likely Benign | 0.134 | Likely Benign | -0.67 | Neutral | 0.963 | Probably Damaging | 0.959 | Probably Damaging | 2.75 | Benign | 1.00 | Tolerated | 3.77 | 5 | 0.1898 | 0.4411 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||
| c.3223C>G | Q1075E 2D AIThe SynGAP1 missense variant Q1075E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. AlphaMissense‑Default remains uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. High‑accuracy assessments therefore indicate a benign outcome: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is Likely Benign, and Foldetta data is missing. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.988305 | Binding | 0.354 | 0.894 | 0.750 | -2.275 | Likely Benign | 0.401 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -0.70 | Neutral | 0.963 | Probably Damaging | 0.959 | Probably Damaging | 2.75 | Benign | 0.25 | Tolerated | 0.1459 | 0.2463 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3224A>C | Q1075P 2D AIThe SynGAP1 missense variant Q1075P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.988305 | Binding | 0.354 | 0.894 | 0.750 | -1.854 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.175 | Likely Benign | 0.78 | Neutral | 0.996 | Probably Damaging | 0.988 | Probably Damaging | 2.68 | Benign | 0.28 | Tolerated | 0.2288 | 0.5741 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3224A>G | Q1075R 2D AIThe SynGAP1 missense variant Q1075R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar status, as none is assigned to the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.988305 | Binding | 0.354 | 0.894 | 0.750 | -5.039 | Likely Benign | 0.697 | Likely Pathogenic | Likely Benign | 0.142 | Likely Benign | -0.37 | Neutral | 0.985 | Probably Damaging | 0.973 | Probably Damaging | 2.72 | Benign | 0.93 | Tolerated | 0.1504 | 0.2650 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3224A>T | Q1075L 2D AIThe SynGAP1 missense variant Q1075L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy methods confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence supports a benign impact for Q1075L, and this conclusion is consistent with the absence of a ClinVar assertion. The variant is most likely benign based on predictions, and there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.988305 | Binding | 0.354 | 0.894 | 0.750 | -3.976 | Likely Benign | 0.209 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -2.10 | Neutral | 0.985 | Probably Damaging | 0.973 | Probably Damaging | 2.72 | Benign | 0.15 | Tolerated | 0.0888 | 0.6454 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3225G>C | Q1075H 2D AIThe SynGAP1 missense variant Q1075H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM‑Consensus also indicates likely benign, while Foldetta data is missing. Overall, the majority of reliable predictors classify the variant as benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.988305 | Binding | 0.354 | 0.894 | 0.750 | -4.616 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.072 | Likely Benign | -1.06 | Neutral | 0.996 | Probably Damaging | 0.992 | Probably Damaging | 2.68 | Benign | 0.13 | Tolerated | 0.1457 | 0.4214 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3225G>T | Q1075H 2D AIThe SynGAP1 missense variant Q1075H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM‑Consensus also indicates likely benign, while Foldetta data is missing. Overall, the majority of reliable predictors classify the variant as benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.988305 | Binding | 0.354 | 0.894 | 0.750 | -4.616 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.072 | Likely Benign | -1.06 | Neutral | 0.996 | Probably Damaging | 0.992 | Probably Damaging | 2.68 | Benign | 0.13 | Tolerated | 0.1457 | 0.4214 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3226T>A | L1076M 2D AIThe SynGAP1 missense variant L1076M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.989617 | Binding | 0.301 | 0.892 | 0.750 | -5.047 | Likely Benign | 0.320 | Likely Benign | Likely Benign | 0.087 | Likely Benign | 0.02 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.42 | Pathogenic | 0.04 | Affected | 0.0886 | 0.4147 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3226T>G | L1076V 2D AIThe SynGAP1 missense variant L1076V is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.989617 | Binding | 0.301 | 0.892 | 0.750 | -3.615 | Likely Benign | 0.344 | Ambiguous | Likely Benign | 0.097 | Likely Benign | -0.56 | Neutral | 0.995 | Probably Damaging | 0.982 | Probably Damaging | 2.53 | Benign | 0.11 | Tolerated | 0.1632 | 0.3781 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3227T>C | L1076S 2D AIThe SynGAP1 missense variant L1076S is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b, whereas polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default all predict a pathogenic outcome. High‑accuracy assessments are less decisive: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic votes); and Foldetta results are unavailable. Consequently, the evidence is evenly split between benign and pathogenic predictions. The variant is therefore most likely of uncertain significance, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.989617 | Binding | 0.301 | 0.892 | 0.750 | -2.975 | Likely Benign | 0.913 | Likely Pathogenic | Ambiguous | 0.225 | Likely Benign | 0.55 | Neutral | 0.999 | Probably Damaging | 0.983 | Probably Damaging | 2.43 | Pathogenic | 0.75 | Tolerated | 0.3045 | 0.1178 | -3 | -2 | -4.6 | -26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3227T>G | L1076W 2D AIThe SynGAP1 missense variant L1076W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated predictors (five pathogenic vs. three benign) lean toward a pathogenic interpretation. This prediction is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.989617 | Binding | 0.301 | 0.892 | 0.750 | -6.377 | Likely Benign | 0.822 | Likely Pathogenic | Ambiguous | 0.183 | Likely Benign | -1.40 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.38 | Pathogenic | 0.02 | Affected | 0.0749 | 0.3740 | -2 | -2 | -4.7 | 73.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3228G>C | L1076F 2D AIThe SynGAP1 missense variant L1076F is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that classify the variant as benign include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions (5 pathogenic vs. 4 benign) lean toward pathogenicity, and this is consistent with the lack of ClinVar annotation. Thus, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.989617 | Binding | 0.301 | 0.892 | 0.750 | -4.606 | Likely Benign | 0.675 | Likely Pathogenic | Likely Benign | 0.092 | Likely Benign | -0.92 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.42 | Pathogenic | 0.03 | Affected | 0.0759 | 0.3767 | 2 | 0 | -1.0 | 34.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3228G>T | L1076F 2D AIThe SynGAP1 missense variant L1076F is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that classify the variant as benign include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions (5 pathogenic vs. 4 benign) lean toward pathogenicity, and this is consistent with the lack of ClinVar annotation. Thus, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.989617 | Binding | 0.301 | 0.892 | 0.750 | -4.606 | Likely Benign | 0.675 | Likely Pathogenic | Likely Benign | 0.092 | Likely Benign | -0.92 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.42 | Pathogenic | 0.03 | Affected | 0.0759 | 0.3767 | 2 | 0 | -1.0 | 34.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3229A>C | T1077P 2D AIThe SynGAP1 missense variant T1077P is reported in ClinVar as “Not submitted” and is not present in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and no Foldetta stability assessment is available. High‑accuracy evidence shows AlphaMissense‑Optimized classifying the variant as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicating a likely benign outcome, and Foldetta results are unavailable. Overall, the majority of predictions and the high‑accuracy consensus point to a benign impact. This conclusion is not contradicted by ClinVar status, which has no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.988141 | Binding | 0.329 | 0.892 | 0.750 | -2.436 | Likely Benign | 0.345 | Ambiguous | Likely Benign | 0.164 | Likely Benign | -0.91 | Neutral | 0.970 | Probably Damaging | 0.787 | Possibly Damaging | 4.16 | Benign | 0.04 | Affected | 0.1876 | 0.4788 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3229A>G | T1077A 2D AIThe SynGAP1 missense variant T1077A is catalogued in gnomAD (ID 6‑33443781‑A‑G) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool reports a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the set predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.988141 | Binding | 0.329 | 0.892 | 0.750 | 6-33443781-A-G | -3.303 | Likely Benign | 0.280 | Likely Benign | Likely Benign | 0.153 | Likely Benign | -0.60 | Neutral | 0.288 | Benign | 0.194 | Benign | 4.25 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.3373 | 0.4393 | 0 | 1 | 2.5 | -30.03 | ||||||||||||||||||||||||||||||||||||
| c.3229A>T | T1077S 2D AIThe SynGAP1 missense variant T1077S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.988141 | Binding | 0.329 | 0.892 | 0.750 | -2.929 | Likely Benign | 0.201 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -0.45 | Neutral | 0.068 | Benign | 0.025 | Benign | 4.32 | Benign | 0.06 | Tolerated | 0.2788 | 0.4635 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.322A>C | K108Q 2D AIThe SynGAP1 missense variant K108Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.626927 | Disordered | 0.673331 | Binding | 0.338 | 0.858 | 0.875 | -3.676 | Likely Benign | 0.639 | Likely Pathogenic | Likely Benign | 0.168 | Likely Benign | -0.73 | Neutral | 0.998 | Probably Damaging | 0.981 | Probably Damaging | 4.09 | Benign | 0.06 | Tolerated | 0.4843 | 0.1322 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||||
| c.322A>G | K108E 2D AIThe SynGAP1 K108E missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus indicates a likely benign outcome; Foldetta results are unavailable. Overall, the predictions are split evenly between benign and pathogenic, with no clear majority. Consequently, the variant’s impact remains uncertain, and there is no contradiction with ClinVar status, which currently lists no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.626927 | Disordered | 0.673331 | Binding | 0.338 | 0.858 | 0.875 | -3.679 | Likely Benign | 0.960 | Likely Pathogenic | Likely Pathogenic | 0.166 | Likely Benign | -1.24 | Neutral | 0.993 | Probably Damaging | 0.956 | Probably Damaging | 4.12 | Benign | 0.04 | Affected | 0.4145 | 0.1166 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3230C>A | T1077K 2D  AIThe SynGAP1 missense variant T1077K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are not available. Overall, the majority of evidence—including the SGM‑Consensus—suggests a benign impact, and this conclusion does not contradict the absence of a ClinVar entry. Thus, based on current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.988141 | Binding | 0.329 | 0.892 | 0.750 | -4.196 | Likely Benign | 0.928 | Likely Pathogenic | Ambiguous | 0.110 | Likely Benign | -1.44 | Neutral | 0.818 | Possibly Damaging | 0.460 | Possibly Damaging | 4.21 | Benign | 0.03 | Affected | 0.1176 | 0.3968 | 0 | -1 | -3.2 | 27.07 | |||||||||||||||||||||||||||||||||||||||
| c.3230C>G | T1077R 2D  AIThe SynGAP1 missense variant T1077R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), which collectively classify the variant as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and no Foldetta stability assessment is available. Overall, the balance of evidence leans toward a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.988141 | Binding | 0.329 | 0.892 | 0.750 | -4.109 | Likely Benign | 0.890 | Likely Pathogenic | Ambiguous | 0.121 | Likely Benign | -1.01 | Neutral | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 4.18 | Benign | 0.03 | Affected | 0.1028 | 0.3491 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||||||||||||
| c.3230C>T | T1077I 2D AIThe SynGAP1 missense variant T1077I is listed in gnomAD (ID 6‑33443782‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions (REVEL, PROVEAN, SIFT, ESM1b, FATHMM) and pathogenic predictions (PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, AlphaMissense‑Default). The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to a likely benign verdict (3 benign vs. 1 pathogenic). High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized returns an uncertain result, while Foldetta (combining FoldX‑MD and Rosetta outputs) is not available for this residue. Taken together, the majority of evidence points toward a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification, and there is no contradiction with existing database annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.988141 | Binding | 0.329 | 0.892 | 0.750 | 6-33443782-C-T | 1 | 6.25e-7 | -4.710 | Likely Benign | 0.919 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.11 | Neutral | 0.970 | Probably Damaging | 0.787 | Possibly Damaging | 4.19 | Benign | 0.33 | Tolerated | 3.77 | 5 | 0.1135 | 0.5870 | -1 | 0 | 5.2 | 12.05 | ||||||||||||||||||||||||||||||||||
| c.3232G>A | V1078I 2D  AIThe SynGAP1 missense variant V1078I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as likely benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and consensus methods suggests that V1078I is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.986989 | Binding | 0.294 | 0.898 | 0.750 | -3.652 | Likely Benign | 0.200 | Likely Benign | Likely Benign | 0.120 | Likely Benign | -0.16 | Neutral | 0.625 | Possibly Damaging | 0.266 | Benign | 3.98 | Benign | 0.07 | Tolerated | 0.0771 | 0.4693 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3232G>C | V1078L 2D  AIThe SynGAP1 missense variant V1078L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign classification. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective evidence strongly suggests that V1078L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.986989 | Binding | 0.294 | 0.898 | 0.750 | -2.547 | Likely Benign | 0.523 | Ambiguous | Likely Benign | 0.091 | Likely Benign | -0.16 | Neutral | 0.451 | Benign | 0.209 | Benign | 4.13 | Benign | 0.56 | Tolerated | 0.0889 | 0.5349 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3232G>T | V1078F 2D  AIThe SynGAP1 missense variant V1078F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect for V1078F, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.986989 | Binding | 0.294 | 0.898 | 0.750 | -3.768 | Likely Benign | 0.570 | Likely Pathogenic | Likely Benign | 0.142 | Likely Benign | -0.97 | Neutral | 0.977 | Probably Damaging | 0.722 | Possibly Damaging | 3.87 | Benign | 0.02 | Affected | 0.0677 | 0.4561 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.3233T>A | V1078D 2D  AIThe SynGAP1 missense variant V1078D is listed in ClinVar (ID 2993122.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Tools that predict a pathogenic effect are AlphaMissense‑Default, AlphaMissense‑Optimized, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the majority of predictions lean toward a benign impact, and this is consistent with the ClinVar “Uncertain” designation; there is no contradiction with the existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.986989 | Binding | 0.294 | 0.898 | 0.750 | Uncertain | 1 | -5.155 | Likely Benign | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.158 | Likely Benign | -1.45 | Neutral | 0.003 | Benign | 0.008 | Benign | 3.84 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1570 | 0.1173 | -3 | -2 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||
| c.3233T>C | V1078A 2D  AIThe SynGAP1 missense variant V1078A is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign. Only two tools predict pathogenicity: SIFT and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of predictions indicate that V1078A is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.986989 | Binding | 0.294 | 0.898 | 0.750 | -2.794 | Likely Benign | 0.690 | Likely Pathogenic | Likely Benign | 0.089 | Likely Benign | -0.27 | Neutral | 0.011 | Benign | 0.006 | Benign | 3.94 | Benign | 0.02 | Affected | 0.2679 | 0.2546 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.3233T>G | V1078G 2D  AIThe SynGAP1 missense variant V1078G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign (3 benign vs 1 pathogenic). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence indicates a benign impact. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.986989 | Binding | 0.294 | 0.898 | 0.750 | -3.270 | Likely Benign | 0.699 | Likely Pathogenic | Likely Benign | 0.168 | Likely Benign | -0.54 | Neutral | 0.157 | Benign | 0.292 | Benign | 3.85 | Benign | 0.00 | Affected | 0.2041 | 0.2693 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3235A>C | S1079R 2D AIThe SynGAP1 missense variant S1079R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized returns an uncertain result. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983887 | Binding | 0.307 | 0.900 | 0.750 | -4.579 | Likely Benign | 0.955 | Likely Pathogenic | Ambiguous | 0.163 | Likely Benign | -1.81 | Neutral | 0.177 | Benign | 0.075 | Benign | 3.86 | Benign | 0.00 | Affected | 3.77 | 5 | 0.0811 | 0.4028 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.3235A>G | S1079G 2D AIThe SynGAP1 missense variant S1079G is reported in gnomAD (variant ID 6‑33443787‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign status. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983887 | Binding | 0.307 | 0.900 | 0.750 | 6-33443787-A-G | 6 | 3.76e-6 | -3.552 | Likely Benign | 0.177 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -1.50 | Neutral | 0.036 | Benign | 0.018 | Benign | 3.87 | Benign | 0.00 | Affected | 3.77 | 5 | 0.2151 | 0.4623 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||
| c.3235A>T | S1079C 2D AIThe SynGAP1 missense variant S1079C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. Two tools—ESM1b and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive, and Foldetta data are unavailable. Overall, the balance of evidence slightly favors a pathogenic interpretation, with four tools supporting pathogenicity versus three supporting benignity. This assessment does not contradict ClinVar status, as the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.919029 | Disordered | 0.983887 | Binding | 0.307 | 0.900 | 0.750 | -7.333 | In-Between | 0.370 | Ambiguous | Likely Benign | 0.138 | Likely Benign | -2.61 | Deleterious | 0.898 | Possibly Damaging | 0.477 | Possibly Damaging | 3.81 | Benign | 0.00 | Affected | 0.0949 | 0.5536 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3236G>A | S1079N 2D AIThe SynGAP1 missense variant S1079N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority of the high‑accuracy tools) also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of predictions indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983887 | Binding | 0.307 | 0.900 | 0.750 | -4.989 | Likely Benign | 0.505 | Ambiguous | Likely Benign | 0.026 | Likely Benign | -0.97 | Neutral | 0.001 | Benign | 0.001 | Benign | 3.93 | Benign | 0.00 | Affected | 0.1225 | 0.4657 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3236G>C | S1079T 2D AIThe SynGAP1 missense variant S1079T is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly suggests the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for S1079T. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983887 | Binding | 0.307 | 0.900 | 0.750 | -3.225 | Likely Benign | 0.163 | Likely Benign | Likely Benign | 0.023 | Likely Benign | -1.28 | Neutral | 0.001 | Benign | 0.003 | Benign | 3.91 | Benign | 0.00 | Affected | 0.1283 | 0.5920 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3236G>T | S1079I 2D AIThe SynGAP1 missense variant S1079I is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.919029 | Disordered | 0.983887 | Binding | 0.307 | 0.900 | 0.750 | -4.732 | Likely Benign | 0.688 | Likely Pathogenic | Likely Benign | 0.093 | Likely Benign | -2.86 | Deleterious | 0.078 | Benign | 0.025 | Benign | 3.83 | Benign | 0.00 | Affected | 0.0921 | 0.4775 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3237C>A | S1079R 2D AIThe SynGAP1 missense variant S1079R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (gnomAD ID 6‑33443789‑C‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Those that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is inconclusive. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments are limited: AlphaMissense‑Optimized remains uncertain, SGM‑Consensus is benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983887 | Binding | 0.307 | 0.900 | 0.750 | Uncertain | 1 | 6-33443789-C-A | 4 | 2.51e-6 | -4.579 | Likely Benign | 0.955 | Likely Pathogenic | Ambiguous | 0.123 | Likely Benign | -1.81 | Neutral | 0.177 | Benign | 0.075 | Benign | 3.86 | Benign | 0.00 | Affected | 3.77 | 5 | 0.0811 | 0.4028 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||
| c.3237C>G | S1079R 2D AIThe SynGAP1 missense variant S1079R is listed in ClinVar (ID 1047537.0) as Benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and no Foldetta (FoldX‑MD/Rosetta stability) result is available. High‑accuracy assessments therefore show a benign consensus (SGM‑Consensus) with one uncertain AlphaMissense‑Optimized prediction and no destabilizing Foldetta evidence. Overall, the majority of predictions support a benign classification, which is consistent with the ClinVar status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983887 | Binding | 0.307 | 0.900 | 0.750 | Benign | 1 | -4.579 | Likely Benign | 0.955 | Likely Pathogenic | Ambiguous | 0.124 | Likely Benign | -1.81 | Neutral | 0.177 | Benign | 0.075 | Benign | 3.86 | Benign | 0.00 | Affected | 3.77 | 5 | 0.0811 | 0.4028 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||
| c.3238G>A | A1080T 2D AIThe SynGAP1 missense variant A1080T (ClinVar ID 1473274.0) is listed as “Uncertain” in ClinVar and is present in gnomAD (ID 6‑33443790‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly suggests the variant is most likely benign, which does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.981457 | Binding | 0.303 | 0.900 | 0.750 | Conflicting | 2 | 6-33443790-G-A | 17 | 1.06e-5 | -3.928 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.144 | Likely Benign | -0.19 | Neutral | 0.253 | Benign | 0.042 | Benign | 4.10 | Benign | 0.60 | Tolerated | 3.77 | 5 | 0.1564 | 0.7103 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||
| c.3238G>C | A1080P 2D AIThe SynGAP1 missense variant A1080P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for A1080P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.981457 | Binding | 0.303 | 0.900 | 0.750 | -2.429 | Likely Benign | 0.254 | Likely Benign | Likely Benign | 0.170 | Likely Benign | -1.15 | Neutral | 0.996 | Probably Damaging | 0.833 | Possibly Damaging | 3.96 | Benign | 0.02 | Affected | 0.1884 | 0.5324 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3238G>T | A1080S 2D AIThe SynGAP1 missense variant A1080S is listed in ClinVar (ID 2703014.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443790‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign effect, and this conclusion does not contradict the ClinVar designation, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.981457 | Binding | 0.303 | 0.900 | 0.750 | Uncertain | 1 | 6-33443790-G-T | 1 | 6.26e-7 | -3.277 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.103 | Likely Benign | 0.01 | Neutral | 0.702 | Possibly Damaging | 0.346 | Benign | 4.16 | Benign | 0.08 | Tolerated | 3.77 | 5 | 0.2498 | 0.5915 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||||
| c.3239C>A | A1080E 2D  AIThe SynGAP1 missense variant A1080E is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33443791‑C‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM. Tools that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. High‑accuracy evidence therefore points to a benign or uncertain impact: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus is benign, and Foldetta data are missing. Overall, the balance of predictions leans toward a benign effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.981457 | Binding | 0.303 | 0.900 | 0.750 | 6-33443791-C-A | -3.672 | Likely Benign | 0.855 | Likely Pathogenic | Ambiguous | 0.090 | Likely Benign | -1.50 | Neutral | 0.901 | Possibly Damaging | 0.540 | Possibly Damaging | 4.00 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1394 | 0.2437 | -1 | 0 | -5.3 | 58.04 | ||||||||||||||||||||||||||||||||||||
| c.3239C>G | A1080G 2D AIThe SynGAP1 missense variant A1080G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable. Overall, the consensus of the majority of prediction algorithms and the high‑accuracy tools points to a benign effect for A1080G, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.981457 | Binding | 0.303 | 0.900 | 0.750 | -3.515 | Likely Benign | 0.213 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -0.80 | Neutral | 0.901 | Possibly Damaging | 0.355 | Benign | 4.00 | Benign | 0.04 | Affected | 0.2153 | 0.4958 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3239C>T | A1080V 2D AIThe SynGAP1 missense variant A1080V is listed in gnomAD (ID 6‑33443791‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; no Foldetta stability data are available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.981457 | Binding | 0.303 | 0.900 | 0.750 | 6-33443791-C-T | -4.087 | Likely Benign | 0.229 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -1.06 | Neutral | 0.481 | Possibly Damaging | 0.144 | Benign | 3.99 | Benign | 0.04 | Affected | 3.77 | 5 | 0.1298 | 0.6056 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||||
| c.323A>C | K108T 2D AIThe SynGAP1 missense variant K108T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is not available for this variant. Overall, the majority of evidence points toward a benign impact, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.626927 | Disordered | 0.673331 | Binding | 0.338 | 0.858 | 0.875 | -2.941 | Likely Benign | 0.855 | Likely Pathogenic | Ambiguous | 0.156 | Likely Benign | -1.48 | Neutral | 0.998 | Probably Damaging | 0.981 | Probably Damaging | 4.08 | Benign | 0.03 | Affected | 0.2179 | 0.3538 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||||||||||||
| c.323A>G | K108R 2D AIThe SynGAP1 missense variant K108R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33432188‑A‑G). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign, and the SGM‑Consensus itself is benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.626927 | Disordered | 0.673331 | Binding | 0.338 | 0.858 | 0.875 | Uncertain | 1 | 6-33432188-A-G | 6 | 3.72e-6 | -2.892 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.184 | Likely Benign | 0.37 | Neutral | 0.993 | Probably Damaging | 0.956 | Probably Damaging | 4.22 | Benign | 1.00 | Tolerated | 3.61 | 5 | 0.5286 | 0.1229 | Weaken | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||
| c.323A>T | K108M 2D AIThe SynGAP1 K108M missense variant has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which collectively suggest a likely benign outcome. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and Foldetta stability analysis is unavailable. Overall, the balance of evidence leans toward a benign interpretation, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.626927 | Disordered | 0.673331 | Binding | 0.338 | 0.858 | 0.875 | -3.863 | Likely Benign | 0.909 | Likely Pathogenic | Ambiguous | 0.216 | Likely Benign | -1.64 | Neutral | 0.999 | Probably Damaging | 0.990 | Probably Damaging | 4.03 | Benign | 0.01 | Affected | 0.1271 | 0.4144 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||||||||||||
| c.3241G>A | A1081T 2D AIThe SynGAP1 missense variant A1081T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.979759 | Binding | 0.288 | 0.895 | 0.750 | -3.887 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.07 | Neutral | 0.440 | Benign | 0.184 | Benign | 4.01 | Benign | 0.15 | Tolerated | 0.1567 | 0.6112 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3241G>C | A1081P 2D AIThe SynGAP1 missense variant A1081P is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.979759 | Binding | 0.288 | 0.895 | 0.750 | -2.967 | Likely Benign | 0.178 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -1.07 | Neutral | 0.005 | Benign | 0.010 | Benign | 4.00 | Benign | 0.14 | Tolerated | 0.1878 | 0.4540 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3241G>T | A1081S 2D AIThe SynGAP1 missense variant A1081S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.979759 | Binding | 0.288 | 0.895 | 0.750 | -3.536 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -0.29 | Neutral | 0.021 | Benign | 0.031 | Benign | 4.02 | Benign | 0.23 | Tolerated | 0.2268 | 0.4915 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3242C>A | A1081D 2D AIThe SynGAP1 missense variant A1081D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) which classifies the variant as Likely Benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is Uncertain, and the Foldetta protein‑folding stability assessment is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.979759 | Binding | 0.288 | 0.895 | 0.750 | -4.603 | Likely Benign | 0.892 | Likely Pathogenic | Ambiguous | 0.095 | Likely Benign | -1.84 | Neutral | 0.611 | Possibly Damaging | 0.404 | Benign | 3.97 | Benign | 0.04 | Affected | 0.2069 | 0.2600 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.3242C>G | A1081G 2D AIThe missense variant A1081G in SynGAP1 has no entry in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as likely benign. Foldetta results are unavailable. Overall, the computational evidence strongly supports a benign classification, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.979759 | Binding | 0.288 | 0.895 | 0.750 | -3.174 | Likely Benign | 0.191 | Likely Benign | Likely Benign | 0.033 | Likely Benign | -1.43 | Neutral | 0.393 | Benign | 0.184 | Benign | 3.99 | Benign | 0.23 | Tolerated | 0.1751 | 0.4348 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3242C>T | A1081V 2D AIThe SynGAP1 missense variant A1081V is reported in gnomAD (ID 6‑33443794‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, creating a single discordant signal. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that A1081V is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.979759 | Binding | 0.288 | 0.895 | 0.750 | 6-33443794-C-T | -3.973 | Likely Benign | 0.172 | Likely Benign | Likely Benign | 0.036 | Likely Benign | -1.32 | Neutral | 0.611 | Possibly Damaging | 0.399 | Benign | 4.04 | Benign | 0.37 | Tolerated | 3.77 | 5 | 0.1353 | 0.4884 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||||
| c.3244C>A | Q1082K 2D AIThe SynGAP1 missense variant Q1082K is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all score benign, and AlphaMissense‑Optimized also predicts benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. No tools predict pathogenicity, and AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is Likely Benign, while Foldetta results are unavailable. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.979325 | Binding | 0.347 | 0.896 | 0.875 | -4.488 | Likely Benign | 0.460 | Ambiguous | Likely Benign | 0.087 | Likely Benign | -1.13 | Neutral | 0.224 | Benign | 0.058 | Benign | 4.19 | Benign | 0.12 | Tolerated | 0.2008 | 0.4972 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3244C>G | Q1082E 2D AIThe SynGAP1 missense variant Q1082E is reported in gnomAD (ID 6‑33443796‑C‑G) and has no ClinVar entry. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Based on the unanimous benign predictions and lack of ClinVar pathogenic annotation, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.979325 | Binding | 0.347 | 0.896 | 0.875 | 6-33443796-C-G | -3.437 | Likely Benign | 0.267 | Likely Benign | Likely Benign | 0.090 | Likely Benign | -0.87 | Neutral | 0.112 | Benign | 0.026 | Benign | 4.19 | Benign | 0.07 | Tolerated | 3.77 | 5 | 0.1485 | 0.3227 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||||||||
| c.3245A>C | Q1082P 2D AIThe SynGAP1 missense variant Q1082P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.979325 | Binding | 0.347 | 0.896 | 0.875 | -2.141 | Likely Benign | 0.064 | Likely Benign | Likely Benign | 0.287 | Likely Benign | -0.71 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.16 | Benign | 0.05 | Affected | 0.2121 | 0.6144 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3245A>G | Q1082R 2D AIThe SynGAP1 missense variant Q1082R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.979325 | Binding | 0.347 | 0.896 | 0.875 | -3.584 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.076 | Likely Benign | -0.96 | Neutral | 0.224 | Benign | 0.058 | Benign | 4.14 | Benign | 0.10 | Tolerated | 0.1590 | 0.3014 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3245A>T | Q1082L 2D AIThe SynGAP1 missense variant Q1082L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.979325 | Binding | 0.347 | 0.896 | 0.875 | -3.284 | Likely Benign | 0.171 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -1.30 | Neutral | 0.224 | Benign | 0.058 | Benign | 4.12 | Benign | 1.00 | Tolerated | 0.0979 | 0.6824 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3246G>C | Q1082H 2D AIThe SynGAP1 missense variant Q1082H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.979325 | Binding | 0.347 | 0.896 | 0.875 | -4.307 | Likely Benign | 0.273 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -1.27 | Neutral | 0.002 | Benign | 0.002 | Benign | 4.11 | Benign | 0.03 | Affected | 3.77 | 5 | 0.1580 | 0.4779 | 0 | 3 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||
| c.3246G>T | Q1082H 2D AIThe SynGAP1 missense variant Q1082H is listed in gnomAD (ID 6‑33443798‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or tolerated outcomes, while only SIFT predicts a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.979325 | Binding | 0.347 | 0.896 | 0.875 | 6-33443798-G-T | -4.307 | Likely Benign | 0.273 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -1.27 | Neutral | 0.002 | Benign | 0.002 | Benign | 4.11 | Benign | 0.03 | Affected | 3.77 | 5 | 0.1580 | 0.4779 | 0 | 3 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||||
| c.3247A>C | K1083Q 2D AIThe SynGAP1 missense variant K1083Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions come from polyPhen‑2 HumDiv and HumVar. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign effect. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools points to a benign impact for K1083Q, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | -2.214 | Likely Benign | 0.390 | Ambiguous | Likely Benign | 0.099 | Likely Benign | -0.50 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 4.06 | Benign | 0.37 | Tolerated | 0.4821 | 0.1647 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3247A>G | K1083E 2D AIThe SynGAP1 missense variant K1083E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is not available for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | -3.538 | Likely Benign | 0.864 | Likely Pathogenic | Ambiguous | 0.133 | Likely Benign | -0.78 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 4.09 | Benign | 0.27 | Tolerated | 0.4135 | 0.1717 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3248A>C | K1083T 2D AIThe SynGAP1 missense variant K1083T is reported in gnomAD (ID 6‑33443800‑A‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | 6-33443800-A-C | 2 | 1.26e-6 | -2.870 | Likely Benign | 0.690 | Likely Pathogenic | Likely Benign | 0.233 | Likely Benign | -0.76 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 4.05 | Benign | 0.31 | Tolerated | 3.77 | 5 | 0.2192 | 0.4150 | -1 | 0 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||||||
| c.3248A>G | K1083R 2D AIThe SynGAP1 missense variant K1083R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | -2.212 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.119 | Likely Benign | -0.53 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 4.06 | Benign | 0.84 | Tolerated | 0.5053 | 0.1903 | Weaken | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||||||
| c.3248A>T | K1083I 2D  AIThe SynGAP1 missense variant K1083I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | -3.207 | Likely Benign | 0.836 | Likely Pathogenic | Ambiguous | 0.239 | Likely Benign | -1.65 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 4.02 | Benign | 0.30 | Tolerated | 0.1394 | 0.3978 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||||||||||||
| c.3249A>C | K1083N 2D AIThe SynGAP1 missense variant K1083N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | -4.088 | Likely Benign | 0.873 | Likely Pathogenic | Ambiguous | 0.053 | Likely Benign | -0.83 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 4.04 | Benign | 0.21 | Tolerated | 0.3939 | 0.2234 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3249A>T | K1083N 2D AIThe SynGAP1 missense variant K1083N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | -4.088 | Likely Benign | 0.873 | Likely Pathogenic | Ambiguous | 0.053 | Likely Benign | -0.83 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 4.04 | Benign | 0.21 | Tolerated | 0.3939 | 0.2234 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.324G>C | K108N 2D AISynGAP1 missense variant K108N is reported in gnomAD (6‑33432189‑G‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign calls from REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign); pathogenic calls from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicting pathogenic, whereas the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign; Foldetta results are unavailable. Consequently, the evidence is evenly split, with no single prediction dominating. The variant is therefore not clearly benign or pathogenic based on current computational data, and this lack of consensus does not contradict any ClinVar classification, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.626927 | Disordered | 0.673331 | Binding | 0.338 | 0.858 | 0.875 | 6-33432189-G-C | 1 | 6.20e-7 | -3.015 | Likely Benign | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.068 | Likely Benign | -1.35 | Neutral | 0.998 | Probably Damaging | 0.981 | Probably Damaging | 4.07 | Benign | 0.03 | Affected | 3.61 | 5 | 0.3904 | 0.1820 | 0 | 1 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||||||
| c.324G>T | K108N 2D AIThe SynGAP1 missense variant K108N is not reported in ClinVar and has no gnomAD entry. Consensus predictions from multiple in‑silico tools are split: benign calls come from REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) which labels the variant as Likely Benign. Pathogenic calls arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized, the latter two high‑accuracy predictors both flagging the variant as Pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not return a result for this variant, so its stability impact is unavailable. Overall, the majority of high‑confidence tools (AlphaMissense‑Optimized and the SGM‑Consensus) disagree, with AlphaMissense‑Optimized indicating pathogenicity while the consensus suggests benign. Because ClinVar contains no classification, there is no contradiction; the variant is most likely pathogenic based on the most reliable predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.626927 | Disordered | 0.673331 | Binding | 0.338 | 0.858 | 0.875 | -3.015 | Likely Benign | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.068 | Likely Benign | -1.35 | Neutral | 0.998 | Probably Damaging | 0.981 | Probably Damaging | 4.07 | Benign | 0.03 | Affected | 3.61 | 5 | 0.3904 | 0.1820 | 0 | 1 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||
| c.3250C>A | P1084T 2D AIThe SynGAP1 missense variant P1084T is reported in ClinVar as “Not submitted” and is present in gnomAD (ID 6‑33443802‑C‑A). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it as pathogenic, but this is the sole discordant call. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign; Foldetta results are not available. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently contains no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | 6-33443802-C-A | -4.665 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.127 | Likely Benign | -2.14 | Neutral | 0.025 | Benign | 0.012 | Benign | 4.11 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1546 | 0.6793 | -1 | 0 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||
| c.3250C>G | P1084A 2D AIThe SynGAP1 missense variant P1084A is listed in ClinVar (ID 2827308.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which itself is “Likely Benign”). In contrast, PROVEAN and polyPhen‑2 HumDiv predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | Uncertain | 1 | -3.928 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -2.54 | Deleterious | 0.649 | Possibly Damaging | 0.157 | Benign | 4.05 | Benign | 0.35 | Tolerated | 3.77 | 5 | 0.3164 | 0.5784 | -1 | 1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||
| c.3250C>T | P1084S 2D AIThe SynGAP1 missense variant P1084S is reported in gnomAD (variant ID 6‑33443802‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | 6-33443802-C-T | 1 | 6.31e-7 | -3.987 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.086 | Likely Benign | -2.24 | Neutral | 0.481 | Possibly Damaging | 0.157 | Benign | 4.03 | Benign | 0.03 | Affected | 3.77 | 5 | 0.3102 | 0.6215 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.3251C>A | P1084H 2D AIThe SynGAP1 missense variant P1084H is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443803‑C‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which reports “Likely Benign”). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar designation of uncertainty rather than a definitive pathogenic claim. Thus, the variant is most likely benign, and its prediction profile does not contradict the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | Uncertain | 1 | 6-33443803-C-A | 1 | 6.31e-7 | -4.125 | Likely Benign | 0.323 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -3.16 | Deleterious | 0.997 | Probably Damaging | 0.840 | Possibly Damaging | 3.96 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1751 | 0.5523 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||
| c.3251C>G | P1084R 2D AIThe SynGAP1 missense variant P1084R is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443803‑C‑G). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome (2 benign vs 1 pathogenic vote). Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for P1084R, and this conclusion does not contradict the ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | 6-33443803-C-G | 4 | 2.52e-6 | -4.171 | Likely Benign | 0.562 | Ambiguous | Likely Benign | 0.153 | Likely Benign | -2.87 | Deleterious | 0.970 | Probably Damaging | 0.637 | Possibly Damaging | 3.99 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1370 | 0.4153 | -2 | 0 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||
| c.3251C>T | P1084L 2D AIThe SynGAP1 missense variant P1084L is reported in gnomAD (ID 6‑33443803‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | 6-33443803-C-T | 1 | 6.31e-7 | -4.547 | Likely Benign | 0.175 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -3.33 | Deleterious | 0.649 | Possibly Damaging | 0.157 | Benign | 4.00 | Benign | 0.01 | Affected | 3.77 | 5 | 0.2218 | 0.6470 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.3253C>G | R1085G 2D AIThe SynGAP1 missense variant R1085G is reported in gnomAD (ID 6‑33443805‑C‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions from REVEL, ESM1b, and FATHMM; pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. A high‑accuracy consensus (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a 2‑to‑2 split, leaving the consensus inconclusive. No Foldetta stability assessment is available. Overall, the majority of evidence (five pathogenic versus three benign) points to a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.978838 | Binding | 0.270 | 0.888 | 1.000 | 6-33443805-C-G | -4.225 | Likely Benign | 0.846 | Likely Pathogenic | Ambiguous | 0.179 | Likely Benign | -2.79 | Deleterious | 0.997 | Probably Damaging | 0.993 | Probably Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.3310 | 0.3693 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||
| c.3253C>T | R1085W 2D AISynGAP1 missense variant R1085W is listed in ClinVar as Uncertain and is present in gnomAD (ID 6‑33443805‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized reports an uncertain outcome. The high‑accuracy consensus (SGM) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields no majority and is therefore unavailable, and Foldetta stability analysis is also unavailable. Overall, the majority of evidence points to a pathogenic effect, which contrasts with the ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.978838 | Binding | 0.270 | 0.888 | 1.000 | Uncertain | 2 | 6-33443805-C-T | 2 | 1.26e-6 | -6.339 | Likely Benign | 0.821 | Likely Pathogenic | Ambiguous | 0.202 | Likely Benign | -3.15 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.70 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1238 | 0.3700 | -3 | 2 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||
| c.3254G>A | R1085Q 2D  AIThe SynGAP1 missense variant R1085Q (ClinVar ID 1729448.0) is listed as ClinVar status Uncertain and is present in gnomAD (6‑33443806‑G‑A). Functional prediction tools show a split opinion: benign predictions come from REVEL, PROVEAN, ESM1b, and FATHMM, whereas pathogenic predictions are reported by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also favors benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, which is consistent with the ClinVar uncertain designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.978838 | Binding | 0.270 | 0.888 | 1.000 | Uncertain | 1 | 6-33443806-G-A | 5 | 3.16e-6 | -3.843 | Likely Benign | 0.589 | Likely Pathogenic | Likely Benign | 0.224 | Likely Benign | -1.43 | Neutral | 0.998 | Probably Damaging | 0.988 | Probably Damaging | 2.73 | Benign | 0.02 | Affected | 3.77 | 5 | 0.2962 | 0.2751 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||
| c.3254G>C | R1085P 2D AIThe SynGAP1 missense variant R1085P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2). Foldetta results are unavailable. Overall, the majority of standard predictors (five pathogenic vs. four benign) lean toward a pathogenic interpretation, but the high‑accuracy AlphaMissense‑Optimized prediction and the inconclusive SGM Consensus temper this view. The variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.978838 | Binding | 0.270 | 0.888 | 1.000 | -2.527 | Likely Benign | 0.759 | Likely Pathogenic | Likely Benign | 0.260 | Likely Benign | -2.55 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.71 | Benign | 0.01 | Affected | 0.1988 | 0.4524 | 0 | -2 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||||||||||
| c.3254G>T | R1085L 2D AIThe SynGAP1 missense variant R1085L is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R1085L, and this conclusion does not contradict the ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.978838 | Binding | 0.270 | 0.888 | 1.000 | -3.674 | Likely Benign | 0.734 | Likely Pathogenic | Likely Benign | 0.243 | Likely Benign | -2.38 | Neutral | 0.997 | Probably Damaging | 0.993 | Probably Damaging | 2.72 | Benign | 0.01 | Affected | 0.1909 | 0.4568 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3256C>A | P1086T 2D AIThe SynGAP1 missense variant P1086T is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443808‑C‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic impact. The variant is most likely pathogenic based on the available computational evidence, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.849326 | Disordered | 0.977190 | Binding | 0.393 | 0.885 | 1.000 | 6-33443808-C-A | -4.181 | Likely Benign | 0.568 | Likely Pathogenic | Likely Benign | 0.229 | Likely Benign | -2.97 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.75 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1343 | 0.5848 | -1 | 0 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||
| c.3256C>G | P1086A 2D AIThe SynGAP1 missense variant P1086A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign, and Foldetta data are unavailable. Taken together, the balance of evidence points to a benign effect for P1086A. This conclusion does not conflict with the ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.849326 | Disordered | 0.977190 | Binding | 0.393 | 0.885 | 1.000 | -4.317 | Likely Benign | 0.372 | Ambiguous | Likely Benign | 0.206 | Likely Benign | -2.98 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.78 | Benign | 0.00 | Affected | 0.3084 | 0.4463 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3256C>T | P1086S 2D AIThe SynGAP1 missense variant P1086S is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443808‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.849326 | Disordered | 0.977190 | Binding | 0.393 | 0.885 | 1.000 | 6-33443808-C-T | -3.165 | Likely Benign | 0.604 | Likely Pathogenic | Likely Benign | 0.212 | Likely Benign | -3.04 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.78 | Benign | 0.00 | Affected | 3.77 | 5 | 0.3067 | 0.4894 | -1 | 1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||
| c.3257C>A | P1086Q 2D AIThe SynGAP1 missense variant P1086Q is not reported in ClinVar (ClinVar status: None) but is present in gnomAD (ID 6‑33443809‑C‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs. two benign votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. four benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.849326 | Disordered | 0.977190 | Binding | 0.393 | 0.885 | 1.000 | 6-33443809-C-A | -4.668 | Likely Benign | 0.652 | Likely Pathogenic | Likely Benign | 0.185 | Likely Benign | -2.92 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.77 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1285 | 0.4784 | -1 | 0 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||
| c.3257C>G | P1086R 2D AIThe SynGAP1 missense variant P1086R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two pathogenic vs. two benign votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, more tools (five) predict pathogenicity than benign (three), and the high‑accuracy consensus is inconclusive. Therefore, the variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar status because the variant is not yet reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.849326 | Disordered | 0.977190 | Binding | 0.393 | 0.885 | 1.000 | -5.190 | Likely Benign | 0.848 | Likely Pathogenic | Ambiguous | 0.205 | Likely Benign | -3.42 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.86 | Benign | 0.00 | Affected | 0.1319 | 0.3551 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||||
| c.3257C>T | P1086L 2D AIThe SynGAP1 missense variant P1086L is not reported in ClinVar (ClinVar status: not present) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which simply indicates the variant has not yet been reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.849326 | Disordered | 0.977190 | Binding | 0.393 | 0.885 | 1.000 | -4.694 | Likely Benign | 0.607 | Likely Pathogenic | Likely Benign | 0.166 | Likely Benign | -3.57 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.73 | Benign | 0.00 | Affected | 0.2055 | 0.6326 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3259T>A | S1087T 2D AIThe SynGAP1 missense variant S1087T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to the variant being most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | -4.455 | Likely Benign | 0.128 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -1.01 | Neutral | 0.790 | Possibly Damaging | 0.266 | Benign | 2.66 | Benign | 0.25 | Tolerated | 0.1333 | 0.6503 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3259T>C | S1087P 2D AIThe SynGAP1 missense variant S1087P is reported in gnomAD (ID 6‑33443811‑T‑C) and has no ClinVar entry. All available in silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Based on the unanimous benign predictions and lack of ClinVar pathogenic annotation, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | 6-33443811-T-C | 1 | 6.34e-7 | -2.946 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.135 | Likely Benign | -1.92 | Neutral | 0.006 | Benign | 0.008 | Benign | 2.56 | Benign | 0.12 | Tolerated | 3.77 | 5 | 0.1803 | 0.5700 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||
| c.3259T>G | S1087A 2D AIThe SynGAP1 missense variant S1087A is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Based on the consensus of all available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | -3.649 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.057 | Likely Benign | -1.03 | Neutral | 0.447 | Benign | 0.139 | Benign | 2.64 | Benign | 0.34 | Tolerated | 0.4464 | 0.5405 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.325A>C | S109R 2D AIThe SynGAP1 missense variant S109R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM, whereas pathogenic calls come from SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments highlight that AlphaMissense‑Optimized predicts pathogenicity, whereas the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts benign impact; Foldetta data are unavailable. Overall, the balance of evidence leans toward a benign effect for S109R, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.669335 | Binding | 0.328 | 0.864 | 0.750 | -4.830 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.225 | Likely Benign | -1.80 | Neutral | 0.002 | Benign | 0.001 | Benign | 3.48 | Benign | 0.00 | Affected | 0.0884 | 0.2700 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.325A>G | S109G 2D AIThe SynGAP1 missense variant S109G is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority of the four high‑accuracy tools) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.669335 | Binding | 0.328 | 0.864 | 0.750 | -3.918 | Likely Benign | 0.549 | Ambiguous | Likely Benign | 0.132 | Likely Benign | -1.95 | Neutral | 0.378 | Benign | 0.067 | Benign | 3.51 | Benign | 0.00 | Affected | 0.2686 | 0.3779 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.325A>T | S109C 2D AIThe SynGAP1 missense variant S109C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus (majority vote) also as Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.669335 | Binding | 0.328 | 0.864 | 0.750 | -6.268 | Likely Benign | 0.761 | Likely Pathogenic | Likely Benign | 0.217 | Likely Benign | -2.19 | Neutral | 0.983 | Probably Damaging | 0.431 | Benign | 3.46 | Benign | 0.00 | Affected | 0.1084 | 0.5354 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.3260C>A | S1087Y 2D AIThe SynGAP1 missense variant S1087Y is catalogued in gnomAD (ID 6‑33443812‑C‑A) and has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as benign, and no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | 6-33443812-C-A | -4.194 | Likely Benign | 0.587 | Likely Pathogenic | Likely Benign | 0.107 | Likely Benign | -2.41 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.56 | Benign | 0.02 | Affected | 3.77 | 5 | 0.0671 | 0.5845 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||
| c.3260C>G | S1087C 2D AIThe SynGAP1 missense variant S1087C is catalogued in gnomAD (ID 6‑33443812‑C‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact, while ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | 6-33443812-C-G | 1 | 6.34e-7 | -7.369 | In-Between | 0.194 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -2.22 | Neutral | 0.997 | Probably Damaging | 0.840 | Possibly Damaging | 2.55 | Benign | 0.05 | Affected | 3.77 | 5 | 0.0979 | 0.6118 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||
| c.3260C>T | S1087F 2D AISynGAP1 missense variant S1087F is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of reliable predictors and the two high‑accuracy tools suggest a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | Uncertain | 1 | -3.843 | Likely Benign | 0.497 | Ambiguous | Likely Benign | 0.105 | Likely Benign | -2.75 | Deleterious | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.56 | Benign | 0.03 | Affected | 3.77 | 5 | 0.0645 | 0.6029 | -2 | -3 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||
| c.3262A>C | S1088R 2D AIThe SynGAP1 missense variant S1088R is not reported in ClinVar and has no gnomAD entry. Prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, and FATHMM, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as Likely Benign, reflecting the majority of benign calls. High‑accuracy assessments further support this: AlphaMissense‑Optimized labels the variant as Pathogenic, but the SGM‑Consensus (majority vote) remains Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence leans toward a benign effect; this conclusion does not conflict with ClinVar, which contains no entry for S1088R. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.975261 | Binding | 0.336 | 0.889 | 1.000 | -4.588 | Likely Benign | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.209 | Likely Benign | -1.96 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1190 | 0.4502 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.3262A>G | S1088G 2D AIThe SynGAP1 missense variant S1088G is listed in ClinVar (ID 2742833.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as Likely Benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this is consistent with the ClinVar “Uncertain” status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.975261 | Binding | 0.336 | 0.889 | 1.000 | Uncertain | 1 | -5.034 | Likely Benign | 0.285 | Likely Benign | Likely Benign | 0.163 | Likely Benign | -1.83 | Neutral | 0.979 | Probably Damaging | 0.973 | Probably Damaging | 2.63 | Benign | 0.03 | Affected | 3.77 | 5 | 0.2541 | 0.5170 | 0 | 1 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||
| c.3262A>T | S1088C 2D AIThe SynGAP1 missense variant S1088C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar) and SIFT. Two tools, AlphaMissense‑Default and ESM1b, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it contains two benign and two uncertain calls, and Foldetta data are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.910643 | Disordered | 0.975261 | Binding | 0.336 | 0.889 | 1.000 | -7.532 | In-Between | 0.547 | Ambiguous | Likely Benign | 0.212 | Likely Benign | -2.33 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.59 | Benign | 0.01 | Affected | 0.1571 | 0.6166 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3263G>A | S1088N 2D AIThe SynGAP1 missense variant S1088N has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, while the SGM‑Consensus (derived from the four high‑accuracy tools) indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence leans toward a benign impact, and this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.975261 | Binding | 0.336 | 0.889 | 1.000 | -5.227 | Likely Benign | 0.791 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.25 | Neutral | 0.991 | Probably Damaging | 0.982 | Probably Damaging | 2.69 | Benign | 0.02 | Affected | 0.1940 | 0.5197 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3263G>C | S1088T 2D AIThe SynGAP1 missense variant S1088T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.975261 | Binding | 0.336 | 0.889 | 1.000 | -4.569 | Likely Benign | 0.295 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -1.32 | Neutral | 0.979 | Probably Damaging | 0.973 | Probably Damaging | 2.71 | Benign | 0.04 | Affected | 0.2048 | 0.6514 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3263G>T | S1088I 2D AIThe SynGAP1 missense variant S1088I is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33443815‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. AlphaMissense‑Optimized is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the balance of evidence, the variant is most likely benign; this assessment does not contradict ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.975261 | Binding | 0.336 | 0.889 | 1.000 | 6-33443815-G-T | -4.893 | Likely Benign | 0.891 | Likely Pathogenic | Ambiguous | 0.288 | Likely Benign | -2.05 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.62 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1322 | 0.5712 | -2 | -1 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||
| c.3264C>A | S1088R 2D AIThe SynGAP1 missense variant S1088R is not reported in ClinVar and is present in gnomAD (ID 6‑33443816‑C‑A). Prediction tools that classify the variant as benign include REVEL, PROVEAN, ESM1b, and FATHMM, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized predict it to be pathogenic. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains benign; Foldetta results are unavailable. Overall, the balance of evidence—five pathogenic versus four benign predictions—suggests the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for S1088R. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.975261 | Binding | 0.336 | 0.889 | 1.000 | 6-33443816-C-A | -4.588 | Likely Benign | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.181 | Likely Benign | -1.96 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1190 | 0.4502 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||
| c.3264C>G | S1088R 2D AIThe SynGAP1 missense variant S1088R has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, whereas the SGM‑Consensus remains Benign; Foldetta results are unavailable. Overall, the balance of evidence slightly favors a pathogenic interpretation, with no conflict with ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.975261 | Binding | 0.336 | 0.889 | 1.000 | -4.588 | Likely Benign | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.181 | Likely Benign | -1.96 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1190 | 0.4502 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.3265G>A | G1089R 2D AIThe SynGAP1 missense variant G1089R is catalogued in gnomAD (ID 6‑33443817‑G‑A) but has no ClinVar entry. Functional prediction tools split in a 6‑to‑3 ratio: benign calls come from REVEL, polyPhen‑2 HumVar, and ESM1b, while pathogenic calls come from PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. AlphaMissense‑Optimized yields an Uncertain result, and no Foldetta stability assessment is available. Overall, the majority of high‑confidence predictors lean toward pathogenicity, and this assessment does not conflict with ClinVar status, which is currently unreported. Thus, the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | 6-33443817-G-A | 1 | 6.35e-7 | -4.757 | Likely Benign | 0.897 | Likely Pathogenic | Ambiguous | 0.222 | Likely Benign | -3.13 | Deleterious | 0.896 | Possibly Damaging | 0.325 | Benign | 2.42 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.0934 | 0.4415 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||
| c.3265G>C | G1089R 2D AIThe SynGAP1 missense variant G1089R is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 HumVar, and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. Grouping by consensus, seven tools predict pathogenicity and three predict benign, giving a net pathogenic signal. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic. Foldetta stability analysis is unavailable. Overall, the evidence points to the variant being most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -4.757 | Likely Benign | 0.897 | Likely Pathogenic | Ambiguous | 0.228 | Likely Benign | -3.13 | Deleterious | 0.896 | Possibly Damaging | 0.325 | Benign | 2.42 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.0934 | 0.4415 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3265G>T | G1089W 2D AIThe SynGAP1 missense variant G1089W is not reported in ClinVar and has no allele in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the consensus SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments give an uncertain result from AlphaMissense‑Optimized, a Likely Pathogenic verdict from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and no available Foldetta stability data. Overall, the majority of evidence points to a deleterious effect, so the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -6.561 | Likely Benign | 0.863 | Likely Pathogenic | Ambiguous | 0.236 | Likely Benign | -3.45 | Deleterious | 1.000 | Probably Damaging | 0.988 | Probably Damaging | 2.37 | Pathogenic | 0.00 | Affected | 0.0840 | 0.4610 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.3266G>A | G1089E 2D AIThe SynGAP1 missense variant G1089E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas a majority (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default) predict a pathogenic impact; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a 2‑vs‑2 split, and Foldetta results are unavailable. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -3.233 | Likely Benign | 0.926 | Likely Pathogenic | Ambiguous | 0.170 | Likely Benign | -2.85 | Deleterious | 0.992 | Probably Damaging | 0.834 | Possibly Damaging | 2.59 | Benign | 0.01 | Affected | 0.1460 | 0.4387 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3266G>C | G1089A 2D AIThe SynGAP1 missense variant G1089A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation—there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -2.864 | Likely Benign | 0.224 | Likely Benign | Likely Benign | 0.142 | Likely Benign | -1.73 | Neutral | 0.186 | Benign | 0.055 | Benign | 2.45 | Pathogenic | 0.02 | Affected | 0.3475 | 0.5331 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3266G>T | G1089V 2D AIThe SynGAP1 missense variant G1089V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact for G1089V. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -4.809 | Likely Benign | 0.527 | Ambiguous | Likely Benign | 0.182 | Likely Benign | -2.81 | Deleterious | 0.984 | Probably Damaging | 0.722 | Possibly Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.1326 | 0.4413 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3268A>C | N1090H 2D AIThe SynGAP1 missense variant N1090H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -3.744 | Likely Benign | 0.447 | Ambiguous | Likely Benign | 0.094 | Likely Benign | -1.42 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.67 | Benign | 0.10 | Tolerated | 0.1604 | 0.7550 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||||||||||
| c.3268A>G | N1090D 2D AIThe SynGAP1 missense variant N1090D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and no Foldetta stability data is available. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -3.354 | Likely Benign | 0.827 | Likely Pathogenic | Ambiguous | 0.066 | Likely Benign | -1.39 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.70 | Benign | 0.47 | Tolerated | 0.1994 | 0.4375 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3268A>T | N1090Y 2D AIThe SynGAP1 missense variant N1090Y is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign, reflecting the majority of benign calls. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect for the variant, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -4.744 | Likely Benign | 0.651 | Likely Pathogenic | Likely Benign | 0.139 | Likely Benign | -2.26 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.66 | Benign | 0.05 | Affected | 0.0661 | 0.5998 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3269A>C | N1090T 2D AIThe SynGAP1 missense variant N1090T is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign, while the majority of high‑accuracy predictors (AlphaMissense‑Optimized and the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also support a benign classification. In contrast, polyPhen‑2 (both HumDiv and HumVar tracks) predict a pathogenic impact, but these are the only tools in disagreement. The AlphaMissense‑Default score is uncertain, and no Foldetta stability assessment is available. Overall, the preponderance of evidence points to a benign effect for the variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -3.478 | Likely Benign | 0.542 | Ambiguous | Likely Benign | 0.129 | Likely Benign | -0.78 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.77 | Benign | 0.58 | Tolerated | 0.1451 | 0.7930 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||||||||||
| c.3269A>G | N1090S 2D AIThe SynGAP1 missense variant N1090S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -2.451 | Likely Benign | 0.228 | Likely Benign | Likely Benign | 0.135 | Likely Benign | -0.49 | Neutral | 0.997 | Probably Damaging | 0.983 | Probably Damaging | 2.97 | Benign | 0.38 | Tolerated | 0.3980 | 0.7620 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3269A>T | N1090I 2D AIThe SynGAP1 missense variant N1090I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the balance of evidence from both general and high‑accuracy predictors points to a benign classification, and this conclusion does not contradict the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -4.356 | Likely Benign | 0.765 | Likely Pathogenic | Likely Benign | 0.173 | Likely Benign | -2.14 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.67 | Benign | 0.02 | Affected | 0.0781 | 0.6279 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.326G>A | S109N 2D AIThe SynGAP1 missense variant S109N is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM, while pathogenic calls come from polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to a likely benign verdict. High‑accuracy assessments further support this: AlphaMissense‑Optimized is uncertain, and Foldetta data are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.669335 | Binding | 0.328 | 0.864 | 0.750 | -5.509 | Likely Benign | 0.863 | Likely Pathogenic | Ambiguous | 0.116 | Likely Benign | -1.45 | Neutral | 0.596 | Possibly Damaging | 0.074 | Benign | 3.49 | Benign | 0.00 | Affected | 0.1350 | 0.3717 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.326G>C | S109T 2D AIThe SynGAP1 missense variant S109T is catalogued in gnomAD (6‑33432191‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all report benign or likely benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments confirm the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus, derived from the majority of the high‑accuracy predictors, is benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S109T is most likely benign, and this assessment does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.669335 | Binding | 0.328 | 0.864 | 0.750 | 6-33432191-G-C | 2 | 1.24e-6 | -4.065 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.090 | Likely Benign | -1.19 | Neutral | 0.231 | Benign | 0.050 | Benign | 3.59 | Benign | 0.00 | Affected | 3.61 | 5 | 0.1519 | 0.5212 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||
| c.326G>T | S109I 2D AIThe SynGAP1 missense variant S109I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized remains uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a 2‑vs‑2 tie, and Foldetta data are unavailable. Overall, the majority of evidence (five benign vs. three pathogenic predictions) points toward a benign impact. This conclusion does not contradict ClinVar, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.622677 | Disordered | 0.669335 | Binding | 0.328 | 0.864 | 0.750 | -5.195 | Likely Benign | 0.927 | Likely Pathogenic | Ambiguous | 0.200 | Likely Benign | -2.56 | Deleterious | 0.267 | Benign | 0.039 | Benign | 3.47 | Benign | 0.00 | Affected | 0.0910 | 0.4930 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3270T>A | N1090K 2D AIThe SynGAP1 missense variant N1090K is reported in ClinVar as “None” and is present in gnomAD (ID 6‑33443822‑T‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) and the consensus result lean toward a benign interpretation. This conclusion does not contradict ClinVar, which currently has no pathogenic classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | 6-33443822-T-A | 2 | 1.28e-6 | -3.423 | Likely Benign | 0.963 | Likely Pathogenic | Likely Pathogenic | 0.053 | Likely Benign | -1.52 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.73 | Benign | 0.18 | Tolerated | 3.77 | 5 | 0.2147 | 0.6121 | 0 | 1 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||||||
| c.3270T>G | N1090K 2D AIThe SynGAP1 missense variant N1090K has no ClinVar entry and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains Likely Benign; Foldetta results are unavailable. The overall balance of evidence leans toward a benign interpretation, and this is consistent with the lack of a ClinVar classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -3.423 | Likely Benign | 0.963 | Likely Pathogenic | Likely Pathogenic | 0.053 | Likely Benign | -1.52 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.73 | Benign | 0.18 | Tolerated | 3.77 | 5 | 0.2147 | 0.6121 | 0 | 1 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||
| c.3271C>A | L1091I 2D AIThe SynGAP1 missense variant L1091I is reported in gnomAD (ID 6‑33443823‑C‑A) but has no ClinVar entry. Across the available in‑silico predictors, every tool classified the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all returned benign scores. No tool predicted pathogenicity, so the pathogenic‑prediction group is empty. High‑accuracy assessments reinforce this benign view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available for this variant. Overall, the computational evidence strongly supports a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.924947 | Disordered | 0.984454 | Binding | 0.376 | 0.889 | 1.000 | 6-33443823-C-A | -4.304 | Likely Benign | 0.273 | Likely Benign | Likely Benign | 0.057 | Likely Benign | -0.66 | Neutral | 0.186 | Benign | 0.055 | Benign | 2.55 | Benign | 0.13 | Tolerated | 3.77 | 5 | 0.1073 | 0.3911 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||||||||
| c.3271C>G | L1091V 2D AIThe SynGAP1 missense variant L1091V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also reports Likely Benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is consistent with the lack of ClinVar evidence, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.924947 | Disordered | 0.984454 | Binding | 0.376 | 0.889 | 1.000 | -4.587 | Likely Benign | 0.328 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.62 | Neutral | 0.779 | Possibly Damaging | 0.211 | Benign | 2.57 | Benign | 0.08 | Tolerated | 0.1560 | 0.3561 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3272T>A | L1091Q 2D AIThe SynGAP1 missense variant L1091Q is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments are less definitive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs 2 benign), and Foldetta results are unavailable. Overall, the majority of available predictions (five pathogenic vs three benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.924947 | Disordered | 0.984454 | Binding | 0.376 | 0.889 | 1.000 | -4.381 | Likely Benign | 0.854 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.32 | Neutral | 0.997 | Probably Damaging | 0.939 | Probably Damaging | 2.47 | Pathogenic | 0.02 | Affected | 0.1192 | 0.1514 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3272T>C | L1091P 2D AIThe SynGAP1 missense variant L1091P is listed in gnomAD (ID 6‑33443824‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, and ESM1b, while pathogenic predictions are reported by polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized returns an uncertain result, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, leaving it inconclusive. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of tools (five pathogenic vs three benign) predict a deleterious effect, and the high‑accuracy AlphaMissense‑Optimized score is uncertain. Consequently, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.924947 | Disordered | 0.984454 | Binding | 0.376 | 0.889 | 1.000 | 6-33443824-T-C | -4.139 | Likely Benign | 0.871 | Likely Pathogenic | Ambiguous | 0.180 | Likely Benign | -1.25 | Neutral | 0.997 | Probably Damaging | 0.939 | Probably Damaging | 2.45 | Pathogenic | 0.04 | Affected | 3.77 | 5 | 0.3199 | 0.1918 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||
| c.3272T>G | L1091R 2D AIThe SynGAP1 missense variant L1091R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of consensus tools (five pathogenic vs. three benign) suggest a pathogenic impact. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.924947 | Disordered | 0.984454 | Binding | 0.376 | 0.889 | 1.000 | -3.662 | Likely Benign | 0.896 | Likely Pathogenic | Ambiguous | 0.191 | Likely Benign | -1.51 | Neutral | 0.997 | Probably Damaging | 0.939 | Probably Damaging | 2.47 | Pathogenic | 0.02 | Affected | 0.1274 | 0.1357 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3274T>A | L1092M 2D AIThe SynGAP1 missense variant L1092M is not reported in ClinVar and has no entries in gnomAD, indicating it has not been catalogued in these databases. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the substitution as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The AlphaMissense‑Default tool remains uncertain, and no Foldetta stability assessment is available. High‑accuracy predictions from AlphaMissense‑Optimized and the SGM‑Consensus both support a benign outcome, whereas the absence of a Foldetta result precludes a stability‑based conclusion. Overall, the majority of evidence points to a benign effect for the variant, and this assessment does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.924947 | Disordered | 0.985431 | Binding | 0.385 | 0.890 | 1.000 | -5.348 | Likely Benign | 0.383 | Ambiguous | Likely Benign | 0.083 | Likely Benign | -0.15 | Neutral | 0.986 | Probably Damaging | 0.875 | Possibly Damaging | 2.66 | Benign | 0.21 | Tolerated | 0.0919 | 0.4479 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3274T>G | L1092V 2D AIThe SynGAP1 missense variant L1092V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The high‑accuracy consensus (SGM‑Consensus) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.924947 | Disordered | 0.985431 | Binding | 0.385 | 0.890 | 1.000 | -4.906 | Likely Benign | 0.451 | Ambiguous | Likely Benign | 0.034 | Likely Benign | -0.36 | Neutral | 0.051 | Benign | 0.037 | Benign | 2.79 | Benign | 0.37 | Tolerated | 0.1658 | 0.3912 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3275T>C | L1092S 2D AIThe SynGAP1 missense variant L1092S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for this variant. This conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.924947 | Disordered | 0.985431 | Binding | 0.385 | 0.890 | 1.000 | -3.649 | Likely Benign | 0.900 | Likely Pathogenic | Ambiguous | 0.121 | Likely Benign | -0.42 | Neutral | 0.986 | Probably Damaging | 0.823 | Possibly Damaging | 2.68 | Benign | 0.25 | Tolerated | 0.3086 | 0.1119 | -3 | -2 | -4.6 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3275T>G | L1092W 2D AIThe SynGAP1 missense variant L1092W is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, while those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. Two tools give uncertain results: ESM1b and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta is unavailable. Overall, the majority of conventional tools predict pathogenicity, but the high‑accuracy consensus leans toward a benign interpretation. Thus, the variant is most likely benign based on the most reliable predictions, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.924947 | Disordered | 0.985431 | Binding | 0.385 | 0.890 | 1.000 | -7.014 | In-Between | 0.804 | Likely Pathogenic | Ambiguous | 0.120 | Likely Benign | -2.00 | Neutral | 0.999 | Probably Damaging | 0.968 | Probably Damaging | 2.58 | Benign | 0.03 | Affected | 0.0787 | 0.4033 | -2 | -2 | -4.7 | 73.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3276G>C | L1092F 2D AIThe SynGAP1 missense variant L1092F is reported in gnomAD (6‑33443828‑G‑C) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.924947 | Disordered | 0.985431 | Binding | 0.385 | 0.890 | 1.000 | 6-33443828-G-C | 1 | 6.40e-7 | -5.010 | Likely Benign | 0.623 | Likely Pathogenic | Likely Benign | 0.067 | Likely Benign | -1.40 | Neutral | 0.986 | Probably Damaging | 0.823 | Possibly Damaging | 2.63 | Benign | 0.18 | Tolerated | 3.77 | 5 | 0.0772 | 0.4060 | 0 | 2 | -1.0 | 34.02 | ||||||||||||||||||||||||||||||||||
| c.3276G>T | L1092F 2D AIThe SynGAP1 missense variant L1092F is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools show a split: benign calls from REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also leans benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the lack of ClinVar annotation and gnomAD absence. Therefore, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.924947 | Disordered | 0.985431 | Binding | 0.385 | 0.890 | 1.000 | -5.010 | Likely Benign | 0.623 | Likely Pathogenic | Likely Benign | 0.067 | Likely Benign | -1.40 | Neutral | 0.986 | Probably Damaging | 0.823 | Possibly Damaging | 2.63 | Benign | 0.18 | Tolerated | 3.77 | 5 | 0.0772 | 0.4060 | 0 | 2 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||
| c.3277C>A | Q1093K 2D AIThe SynGAP1 missense variant Q1093K is reported in gnomAD (variant ID 6‑33443829‑C‑A) but has no ClinVar entry. Functional prediction tools uniformly favor a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the change as benign, and AlphaMissense‑Optimized also predicts benign. No tool predicts pathogenicity; AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983312 | Binding | 0.351 | 0.886 | 1.000 | 6-33443829-C-A | -3.919 | Likely Benign | 0.558 | Ambiguous | Likely Benign | 0.061 | Likely Benign | -0.92 | Neutral | 0.224 | Benign | 0.091 | Benign | 2.78 | Benign | 0.06 | Tolerated | 3.77 | 5 | 0.1926 | 0.5861 | 1 | 1 | -0.4 | 0.04 | ||||||||||||||||||||||||||||||||||||
| c.3277C>G | Q1093E 2D AIThe SynGAP1 missense change Q1093E is not reported in ClinVar and is absent from gnomAD. In silico assessment shows unanimous benign predictions: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. No tool predicts pathogenicity. High‑accuracy consensus methods reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” The protein‑folding stability predictor Foldetta was not available for this variant. Overall, the evidence strongly supports a benign effect, and this conclusion is consistent with the lack of ClinVar annotation. Thus, the variant is most likely benign and does not contradict existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983312 | Binding | 0.351 | 0.886 | 1.000 | -3.104 | Likely Benign | 0.265 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.59 | Neutral | 0.112 | Benign | 0.041 | Benign | 2.78 | Benign | 0.07 | Tolerated | 0.1514 | 0.3717 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3278A>C | Q1093P 2D AIThe SynGAP1 missense variant Q1093P is not reported in ClinVar and is absent from gnomAD. Consensus from most in silico predictors classifies it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic effect. High‑accuracy assessments reinforce the benign interpretation: AlphaMissense‑Optimized scores benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely benign, while Foldetta stability analysis is unavailable. Overall, the evidence points to a benign effect for Q1093P, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983312 | Binding | 0.351 | 0.886 | 1.000 | -2.613 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.137 | Likely Benign | -0.96 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.69 | Benign | 0.05 | Affected | 0.2059 | 0.5956 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3278A>G | Q1093R 2D AIThe SynGAP1 missense variant Q1093R is reported in gnomAD (ID 6‑33443830‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic prediction comes from SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983312 | Binding | 0.351 | 0.886 | 1.000 | 6-33443830-A-G | 1 | 6.40e-7 | -3.681 | Likely Benign | 0.483 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.06 | Neutral | 0.224 | Benign | 0.091 | Benign | 2.73 | Benign | 0.04 | Affected | 3.77 | 5 | 0.1537 | 0.3904 | 1 | 1 | -1.0 | 28.06 | ||||||||||||||||||||||||||||||||||
| c.3278A>T | Q1093L 2D AIThe SynGAP1 missense variant Q1093L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983312 | Binding | 0.351 | 0.886 | 1.000 | -3.242 | Likely Benign | 0.165 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -1.10 | Neutral | 0.224 | Benign | 0.091 | Benign | 2.72 | Benign | 0.03 | Affected | 0.0899 | 0.6837 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3279G>C | Q1093H 2D AIThe SynGAP1 missense variant Q1093H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983312 | Binding | 0.351 | 0.886 | 1.000 | -4.003 | Likely Benign | 0.337 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -1.14 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.69 | Benign | 0.02 | Affected | 0.1600 | 0.5269 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3279G>T | Q1093H 2D AIThe SynGAP1 missense variant Q1093H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983312 | Binding | 0.351 | 0.886 | 1.000 | -4.003 | Likely Benign | 0.337 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -1.14 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.69 | Benign | 0.02 | Affected | 0.1600 | 0.5269 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.327T>A | S109R 2D AIThe SynGAP1 missense variant S109R has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) remains benign; Foldetta stability analysis is unavailable. Overall, the majority of predictions lean toward a benign impact, and this is not contradicted by any ClinVar status. Thus, the variant is most likely benign based on the current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.669335 | Binding | 0.328 | 0.864 | 0.750 | -4.830 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.198 | Likely Benign | -1.80 | Neutral | 0.002 | Benign | 0.001 | Benign | 3.48 | Benign | 0.00 | Affected | 0.0884 | 0.2700 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.327T>G | S109R 2D AIThe SynGAP1 missense variant S109R has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign). Tools that predict pathogenicity are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods give mixed results: AlphaMissense‑Optimized reports a pathogenic effect, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome; Foldetta stability analysis is unavailable. Overall, the majority of predictions lean toward a benign impact, with one high‑accuracy tool suggesting pathogenicity, and there is no ClinVar status to contradict these findings. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.669335 | Binding | 0.328 | 0.864 | 0.750 | -4.830 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.198 | Likely Benign | -1.80 | Neutral | 0.002 | Benign | 0.001 | Benign | 3.48 | Benign | 0.00 | Affected | 0.0884 | 0.2700 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3280T>A | S1094T 2D AIThe SynGAP1 missense variant S1094T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.981352 | Binding | 0.358 | 0.877 | 1.000 | -4.009 | Likely Benign | 0.194 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.57 | Neutral | 0.790 | Possibly Damaging | 0.433 | Benign | 2.59 | Benign | 0.16 | Tolerated | 0.1874 | 0.6902 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3280T>C | S1094P 2D AIThe SynGAP1 missense variant S1094P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S1094P, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.981352 | Binding | 0.358 | 0.877 | 1.000 | -2.430 | Likely Benign | 0.285 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -1.31 | Neutral | 0.990 | Probably Damaging | 0.798 | Possibly Damaging | 2.46 | Pathogenic | 0.13 | Tolerated | 0.2322 | 0.6099 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3280T>G | S1094A 2D AIThe SynGAP1 missense variant S1094A is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity, so the pathogenic‑prediction group is empty. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.981352 | Binding | 0.358 | 0.877 | 1.000 | -3.595 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -0.78 | Neutral | 0.447 | Benign | 0.252 | Benign | 2.66 | Benign | 1.00 | Tolerated | 0.4692 | 0.5403 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3281C>A | S1094Y 2D AIThe SynGAP1 missense variant S1094Y is not reported in ClinVar (status: none) but is present in gnomAD (ID 6‑33443833‑C‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic). Foldetta, a protein‑folding‑stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions therefore show a benign call from AlphaMissense‑Optimized, an inconclusive SGM Consensus, and no Foldetta data. Overall, the balance of evidence leans toward a pathogenic interpretation (five pathogenic versus four benign calls), and this does not contradict the ClinVar status, which currently has no assertion for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.938133 | Disordered | 0.981352 | Binding | 0.358 | 0.877 | 1.000 | 6-33443833-C-A | -5.609 | Likely Benign | 0.631 | Likely Pathogenic | Likely Benign | 0.155 | Likely Benign | -2.07 | Neutral | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 2.45 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.1007 | 0.5877 | -2 | -3 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||
| c.3281C>G | S1094C 2D AIThe SynGAP1 missense variant S1094C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Two tools—AlphaMissense‑Default and ESM1b—return uncertain results. High‑accuracy assessments are limited: AlphaMissense‑Optimized indicates a benign effect; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields no clear consensus and is treated as unavailable; Foldetta data are not provided, so its stability prediction is also unavailable. Overall, the balance of evidence favors a pathogenic interpretation, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.938133 | Disordered | 0.981352 | Binding | 0.358 | 0.877 | 1.000 | -7.143 | In-Between | 0.425 | Ambiguous | Likely Benign | 0.131 | Likely Benign | -1.81 | Neutral | 0.997 | Probably Damaging | 0.946 | Probably Damaging | 2.45 | Pathogenic | 0.05 | Affected | 0.1403 | 0.6309 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3281C>T | S1094F 2D AIThe SynGAP1 missense variant S1094F is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive. Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the majority of conventional predictors lean toward pathogenicity, but the single high‑accuracy tool predicts benign and the consensus is unresolved. Thus, the variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.938133 | Disordered | 0.981352 | Binding | 0.358 | 0.877 | 1.000 | -5.655 | Likely Benign | 0.666 | Likely Pathogenic | Likely Benign | 0.150 | Likely Benign | -2.17 | Neutral | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 2.45 | Pathogenic | 0.02 | Affected | 0.0925 | 0.5851 | -3 | -2 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||||||
| c.3283C>A | P1095T 2D AIThe SynGAP1 missense variant P1095T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.979251 | Binding | 0.387 | 0.870 | 1.000 | -4.706 | Likely Benign | 0.195 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -1.42 | Neutral | 0.872 | Possibly Damaging | 0.399 | Benign | 2.77 | Benign | 0.13 | Tolerated | 0.1685 | 0.6677 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3283C>G | P1095A 2D AIThe SynGAP1 missense variant P1095A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.979251 | Binding | 0.387 | 0.870 | 1.000 | -4.555 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.80 | Neutral | 0.580 | Possibly Damaging | 0.242 | Benign | 2.79 | Benign | 0.18 | Tolerated | 0.3122 | 0.5703 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3283C>T | P1095S 2D AIThe SynGAP1 missense variant P1095S is reported in gnomAD (ID 6‑33443835‑C‑T) and has no ClinVar entry. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Based on the unanimous benign predictions and the absence of any pathogenic signal, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.979251 | Binding | 0.387 | 0.870 | 1.000 | 6-33443835-C-T | 7 | 4.51e-6 | -3.819 | Likely Benign | 0.182 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -0.64 | Neutral | 0.207 | Benign | 0.072 | Benign | 2.80 | Benign | 1.00 | Tolerated | 3.77 | 5 | 0.3150 | 0.5963 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.3284C>A | P1095Q 2D AIThe SynGAP1 missense variant P1095Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, but this is the sole discordant call. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.979251 | Binding | 0.387 | 0.870 | 1.000 | -4.171 | Likely Benign | 0.295 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -1.64 | Neutral | 0.922 | Possibly Damaging | 0.441 | Benign | 2.85 | Benign | 0.45 | Tolerated | 0.1573 | 0.5519 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3284C>G | P1095R 2D AIThe SynGAP1 missense variant P1095R is reported in ClinVar as “Not submitted” and is not present in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, all of which classify the change as benign or tolerant. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. AlphaMissense‑Default remains uncertain, and no Foldetta stability assessment is available. High‑accuracy consensus methods reinforce the benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” and Foldetta data are missing. Overall, the majority of reliable predictors and consensus analyses indicate that P1095R is most likely benign, and this conclusion does not contradict the ClinVar status, which currently contains no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.979251 | Binding | 0.387 | 0.870 | 1.000 | -5.180 | Likely Benign | 0.553 | Ambiguous | Likely Benign | 0.091 | Likely Benign | -1.93 | Neutral | 0.922 | Possibly Damaging | 0.528 | Possibly Damaging | 2.77 | Benign | 0.04 | Affected | 0.1513 | 0.4576 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3284C>T | P1095L 2D AIThe SynGAP1 missense variant P1095L is catalogued in gnomAD (ID 6‑33443836‑C‑T) but has no ClinVar record. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, and Foldetta data are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.979251 | Binding | 0.387 | 0.870 | 1.000 | 6-33443836-C-T | 1 | 6.44e-7 | -4.697 | Likely Benign | 0.363 | Ambiguous | Likely Benign | 0.126 | Likely Benign | -2.78 | Deleterious | 0.960 | Probably Damaging | 0.604 | Possibly Damaging | 2.74 | Benign | 0.03 | Affected | 3.77 | 5 | 0.2199 | 0.6347 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||
| c.3286G>A | E1096K 2D  AIThe SynGAP1 missense variant E1096K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus remains Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.976475 | Binding | 0.308 | 0.858 | 1.000 | -4.148 | Likely Benign | 0.845 | Likely Pathogenic | Ambiguous | 0.097 | Likely Benign | -1.44 | Neutral | 0.872 | Possibly Damaging | 0.478 | Possibly Damaging | 2.75 | Benign | 0.15 | Tolerated | 0.2440 | 0.7533 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3286G>C | E1096Q 2D  AIThe SynGAP1 missense variant E1096Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (both HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools suggests that E1096Q is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.976475 | Binding | 0.308 | 0.858 | 1.000 | -3.134 | Likely Benign | 0.462 | Ambiguous | Likely Benign | 0.142 | Likely Benign | -1.08 | Neutral | 0.954 | Possibly Damaging | 0.654 | Possibly Damaging | 2.73 | Benign | 0.29 | Tolerated | 0.1527 | 0.7459 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3287A>C | E1096A 2D  AIThe SynGAP1 missense variant E1096A is listed in ClinVar (ID 2579889.0) with an uncertain significance annotation and is not reported in gnomAD. Consensus from multiple in‑silico predictors shows a predominance of benign calls: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only polyPhen‑2 HumDiv assigns a pathogenic label, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign; Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the aggregate evidence points to a benign effect, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.976475 | Binding | 0.308 | 0.858 | 1.000 | Uncertain | 1 | -4.504 | Likely Benign | 0.510 | Ambiguous | Likely Benign | 0.164 | Likely Benign | -1.37 | Neutral | 0.626 | Possibly Damaging | 0.184 | Benign | 2.77 | Benign | 0.16 | Tolerated | 3.77 | 5 | 0.3805 | 0.7569 | -1 | 0 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||
| c.3287A>G | E1096G 2D  AIThe SynGAP1 missense variant E1096G is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors shows a split: polyPhen‑2 HumDiv and HumVar classify it as pathogenic, whereas REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized predict a benign effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome, and AlphaMissense‑Optimized itself is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑accuracy tools (AlphaMissense‑Optimized and SGM‑Consensus) indicate a benign impact, and no evidence contradicts this assessment with ClinVar data, which is currently lacking. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.976475 | Binding | 0.308 | 0.858 | 1.000 | -2.749 | Likely Benign | 0.536 | Ambiguous | Likely Benign | 0.118 | Likely Benign | -2.14 | Neutral | 0.872 | Possibly Damaging | 0.478 | Possibly Damaging | 2.70 | Benign | 0.06 | Tolerated | 0.2789 | 0.6220 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3287A>T | E1096V 2D  AIThe SynGAP1 missense variant E1096V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.976475 | Binding | 0.308 | 0.858 | 1.000 | -3.588 | Likely Benign | 0.650 | Likely Pathogenic | Likely Benign | 0.138 | Likely Benign | -1.06 | Neutral | 0.043 | Benign | 0.017 | Benign | 2.81 | Benign | 0.03 | Affected | 0.1082 | 0.7596 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3288G>C | E1096D 2D  AIThe SynGAP1 missense variant E1096D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign interpretation, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.976475 | Binding | 0.308 | 0.858 | 1.000 | -3.818 | Likely Benign | 0.192 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.40 | Neutral | 0.115 | Benign | 0.052 | Benign | 2.71 | Benign | 0.38 | Tolerated | 0.2034 | 0.4994 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3288G>T | E1096D 2D AIThe SynGAP1 missense variant E1096D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign classification, and this conclusion is not contradicted by any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.976475 | Binding | 0.308 | 0.858 | 1.000 | -3.818 | Likely Benign | 0.192 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.40 | Neutral | 0.115 | Benign | 0.052 | Benign | 2.71 | Benign | 0.38 | Tolerated | 0.2034 | 0.4994 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3289C>A | P1097T 2D AIThe SynGAP1 missense variant P1097T is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the computational evidence strongly supports a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.974957 | Binding | 0.384 | 0.858 | 1.000 | -5.000 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -1.38 | Neutral | 0.009 | Benign | 0.013 | Benign | 2.61 | Benign | 0.21 | Tolerated | 0.1751 | 0.6567 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3289C>G | P1097A 2D AIThe SynGAP1 missense variant P1097A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts pathogenicity. The high‑accuracy consensus (SGM‑Consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the unanimous benign predictions and the lack of any pathogenic evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.974957 | Binding | 0.384 | 0.858 | 1.000 | -4.315 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -1.77 | Neutral | 0.245 | Benign | 0.140 | Benign | 2.71 | Benign | 0.26 | Tolerated | 0.3199 | 0.5381 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3289C>T | P1097S 2D AIThe SynGAP1 missense variant P1097S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.974957 | Binding | 0.384 | 0.858 | 1.000 | -3.748 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -1.04 | Neutral | 0.025 | Benign | 0.023 | Benign | 2.62 | Benign | 0.70 | Tolerated | 0.3332 | 0.5691 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.328G>A | V110I 2D AIThe SynGAP1 missense variant V110I is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -4.409 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.10 | Neutral | 0.012 | Benign | 0.006 | Benign | 4.26 | Benign | 0.52 | Tolerated | 0.0875 | 0.4485 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.328G>C | V110L 2D AIThe SynGAP1 missense variant V110L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence overwhelmingly supports a benign impact for V110L, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -3.024 | Likely Benign | 0.357 | Ambiguous | Likely Benign | 0.058 | Likely Benign | -0.71 | Neutral | 0.158 | Benign | 0.025 | Benign | 4.20 | Benign | 0.50 | Tolerated | 0.1165 | 0.5145 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.328G>T | V110F 2D AIThe SynGAP1 missense variant V110F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, SGM‑Consensus is Likely Benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -4.872 | Likely Benign | 0.397 | Ambiguous | Likely Benign | 0.042 | Likely Benign | -1.63 | Neutral | 0.006 | Benign | 0.003 | Benign | 4.13 | Benign | 0.01 | Affected | 0.0794 | 0.4027 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.3290C>A | P1097Q 2D AIThe SynGAP1 missense variant P1097Q is reported in gnomAD (ID 6‑33443842‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.974957 | Binding | 0.384 | 0.858 | 1.000 | 6-33443842-C-A | -4.765 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -1.10 | Neutral | 0.918 | Possibly Damaging | 0.604 | Possibly Damaging | 2.58 | Benign | 0.12 | Tolerated | 3.77 | 5 | 0.1613 | 0.5136 | -1 | 0 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||
| c.3290C>G | P1097R 2D AIThe SynGAP1 missense variant P1097R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the preponderance of benign predictions and the lack of pathogenic evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.974957 | Binding | 0.384 | 0.858 | 1.000 | -4.938 | Likely Benign | 0.422 | Ambiguous | Likely Benign | 0.079 | Likely Benign | -1.75 | Neutral | 0.918 | Possibly Damaging | 0.525 | Possibly Damaging | 2.58 | Benign | 0.07 | Tolerated | 0.1517 | 0.4027 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3290C>T | P1097L 2D AIThe SynGAP1 missense variant P1097L is listed in ClinVar as Benign (ClinVar ID 2060978.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as benign; Foldetta results are unavailable. Overall, the majority of evidence supports a benign impact, and this conclusion is consistent with the ClinVar designation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.974957 | Binding | 0.384 | 0.858 | 1.000 | Benign | 1 | -4.410 | Likely Benign | 0.145 | Likely Benign | Likely Benign | 0.131 | Likely Benign | -2.07 | Neutral | 0.611 | Possibly Damaging | 0.198 | Benign | 2.64 | Benign | 0.05 | Affected | 3.77 | 5 | 0.2349 | 0.6356 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||
| c.3292A>C | S1098R 2D AIThe SynGAP1 missense variant S1098R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign, reflecting the majority of benign predictions. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, and the SGM‑Consensus (majority vote) also indicates Benign. Foldetta results are not available, so no stability evidence is considered. Overall, the majority of computational evidence supports a benign impact for S1098R, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | -4.583 | Likely Benign | 0.775 | Likely Pathogenic | Likely Benign | 0.127 | Likely Benign | -1.00 | Neutral | 0.586 | Possibly Damaging | 0.223 | Benign | 2.72 | Benign | 0.16 | Tolerated | 0.1053 | 0.4012 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3292A>G | S1098G 2D AIThe SynGAP1 missense variant S1098G is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the collective predictions strongly support a benign impact, and this conclusion is consistent with the lack of a ClinVar pathogenic classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | -3.494 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.39 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.72 | Benign | 0.57 | Tolerated | 0.2634 | 0.5206 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3292A>T | S1098C 2D AIThe SynGAP1 missense variant S1098C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus likewise indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | -6.553 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -1.46 | Neutral | 0.938 | Possibly Damaging | 0.665 | Possibly Damaging | 2.65 | Benign | 0.12 | Tolerated | 0.1249 | 0.6233 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.3293G>A | S1098N 2D AIThe SynGAP1 missense variant S1098N is listed in ClinVar (ID 864704.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33443845‑G‑A). All evaluated in‑silico predictors agree on a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the computational evidence strongly supports a benign classification, which is consistent with the ClinVar “Uncertain” status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | Conflicting | 2 | 6-33443845-G-A | 6 | 3.89e-6 | -5.120 | Likely Benign | 0.156 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.58 | Neutral | 0.369 | Benign | 0.120 | Benign | 2.76 | Benign | 0.36 | Tolerated | 3.77 | 5 | 0.1665 | 0.5145 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||
| c.3293G>C | S1098T 2D AIThe SynGAP1 missense variant S1098T is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | -4.144 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -0.18 | Neutral | 0.224 | Benign | 0.120 | Benign | 2.76 | Benign | 0.88 | Tolerated | 0.1706 | 0.6653 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3293G>T | S1098I 2D AIThe SynGAP1 missense variant S1098I is catalogued in gnomAD (ID 6‑33443845‑G‑T) and has no ClinVar entry. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | 6-33443845-G-T | -4.497 | Likely Benign | 0.182 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -0.92 | Neutral | 0.259 | Benign | 0.066 | Benign | 2.67 | Benign | 0.15 | Tolerated | 3.77 | 5 | 0.1219 | 0.5601 | -2 | -1 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||
| c.3294T>A | S1098R 2D AIThe SynGAP1 missense variant S1098R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments therefore support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is benign, and the protein‑folding stability method Foldetta is not available for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | -4.583 | Likely Benign | 0.775 | Likely Pathogenic | Likely Benign | 0.133 | Likely Benign | -1.00 | Neutral | 0.586 | Possibly Damaging | 0.223 | Benign | 2.72 | Benign | 0.16 | Tolerated | 0.1053 | 0.4012 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3294T>G | S1098R 2D AIThe SynGAP1 missense variant S1098R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments therefore support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is benign, and the protein‑folding stability method Foldetta is not available for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | -4.583 | Likely Benign | 0.775 | Likely Pathogenic | Likely Benign | 0.134 | Likely Benign | -1.00 | Neutral | 0.586 | Possibly Damaging | 0.223 | Benign | 2.72 | Benign | 0.16 | Tolerated | 0.1053 | 0.4012 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3295T>A | Y1099N 2D  AIThe SynGAP1 missense variant Y1099N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | -4.329 | Likely Benign | 0.269 | Likely Benign | Likely Benign | 0.143 | Likely Benign | -1.01 | Neutral | 0.818 | Possibly Damaging | 0.360 | Benign | 2.83 | Benign | 0.16 | Tolerated | 0.2121 | 0.0935 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3295T>C | Y1099H 2D  AIThe SynGAP1 missense variant Y1099H is reported in gnomAD (variant ID 6-33443847‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | 6-33443847-T-C | 1 | 6.49e-7 | -3.620 | Likely Benign | 0.267 | Likely Benign | Likely Benign | 0.123 | Likely Benign | -0.50 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.76 | Benign | 0.18 | Tolerated | 3.77 | 5 | 0.2453 | 0.0735 | 2 | 0 | -1.9 | -26.03 | ||||||||||||||||||||||||||||||||||
| c.3295T>G | Y1099D 2D  AIThe SynGAP1 missense variant Y1099D is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all score the change as tolerated or benign, while only polyPhen‑2 HumDiv flags it as pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is Likely Benign, and Foldetta results are unavailable. Taken together, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar assertion, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | -4.193 | Likely Benign | 0.429 | Ambiguous | Likely Benign | 0.130 | Likely Benign | -1.08 | Neutral | 0.818 | Possibly Damaging | 0.435 | Benign | 2.79 | Benign | 0.13 | Tolerated | 0.3819 | 0.0935 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.3296A>C | Y1099S 2D  AIThe SynGAP1 missense variant Y1099S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | -1.775 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.156 | Likely Benign | -0.23 | Neutral | 0.149 | Benign | 0.026 | Benign | 2.90 | Benign | 0.62 | Tolerated | 0.4564 | 0.2298 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||||||||||||
| c.3296A>G | Y1099C 2D  AIThe SynGAP1 missense variant Y1099C is reported in gnomAD (variant ID 6‑33443848‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | 6-33443848-A-G | 3 | 1.95e-6 | -5.670 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -1.13 | Neutral | 0.997 | Probably Damaging | 0.840 | Possibly Damaging | 2.73 | Benign | 0.14 | Tolerated | 3.77 | 5 | 0.3008 | 0.2382 | -2 | 0 | 3.8 | -60.04 | ||||||||||||||||||||||||||||||||||
| c.3296A>T | Y1099F 2D  AIThe SynGAP1 missense variant Y1099F is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | -3.651 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.85 | Neutral | 0.006 | Benign | 0.008 | Benign | 2.75 | Benign | 0.27 | Tolerated | 0.2453 | 0.2780 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3298G>A | G1100S 2D AIThe SynGAP1 missense variant G1100S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for G1100S, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -2.898 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -0.65 | Neutral | 0.943 | Possibly Damaging | 0.595 | Possibly Damaging | 2.10 | Pathogenic | 0.28 | Tolerated | 0.2501 | 0.5699 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3298G>C | G1100R 2D AIThe SynGAP1 missense variant G1100R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of available predictions lean toward pathogenicity, and this does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -2.923 | Likely Benign | 0.603 | Likely Pathogenic | Likely Benign | 0.176 | Likely Benign | -2.05 | Neutral | 0.992 | Probably Damaging | 0.906 | Possibly Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.0986 | 0.5217 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3298G>T | G1100C 2D AIThe SynGAP1 missense variant G1100C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -6.488 | Likely Benign | 0.150 | Likely Benign | Likely Benign | 0.174 | Likely Benign | -2.25 | Neutral | 0.999 | Probably Damaging | 0.950 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.1354 | 0.4768 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3299G>A | G1100D 2D AIThe SynGAP1 missense variant G1100D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -5.235 | Likely Benign | 0.577 | Likely Pathogenic | Likely Benign | 0.141 | Likely Benign | -1.67 | Neutral | 0.049 | Benign | 0.030 | Benign | 1.93 | Pathogenic | 0.01 | Affected | 0.1924 | 0.3043 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3299G>C | G1100A 2D AIThe SynGAP1 missense variant G1100A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign based on current computational evidence, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -2.862 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -0.95 | Neutral | 0.943 | Possibly Damaging | 0.667 | Possibly Damaging | 2.01 | Pathogenic | 0.05 | Affected | 0.3442 | 0.5154 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3299G>T | G1100V 2D AIThe SynGAP1 missense variant G1100V is reported in gnomAD (variant ID 6‑33443851‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign); pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the balance of evidence favors a benign classification for G1100V, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | 6-33443851-G-T | 1 | 6.51e-7 | -2.362 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.198 | Likely Benign | -2.43 | Neutral | 0.992 | Probably Damaging | 0.906 | Possibly Damaging | 1.93 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1297 | 0.4036 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||
| c.329T>A | V110D 2D AIThe SynGAP1 missense variant V110D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic vs. two benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated tools (seven pathogenic vs. three benign) indicate a pathogenic effect. This prediction is consistent with the lack of ClinVar reporting and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -4.536 | Likely Benign | 0.970 | Likely Pathogenic | Likely Pathogenic | 0.189 | Likely Benign | -2.84 | Deleterious | 0.978 | Probably Damaging | 0.500 | Possibly Damaging | 4.04 | Benign | 0.00 | Affected | 0.1849 | 0.1115 | -2 | -3 | -7.7 | 15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.329T>C | V110A 2D AIThe SynGAP1 missense variant V110A is reported as “Likely Benign” in ClinVar and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the consensus of the available predictions points to a benign impact for V110A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -2.971 | Likely Benign | 0.717 | Likely Pathogenic | Likely Benign | 0.075 | Likely Benign | -1.35 | Neutral | 0.462 | Possibly Damaging | 0.122 | Benign | 4.14 | Benign | 0.13 | Tolerated | 0.3196 | 0.2872 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.329T>G | V110G 2D AIThe SynGAP1 missense variant V110G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence points toward a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -4.012 | Likely Benign | 0.724 | Likely Pathogenic | Likely Benign | 0.162 | Likely Benign | -2.87 | Deleterious | 0.377 | Benign | 0.928 | Probably Damaging | 4.06 | Benign | 0.00 | Affected | 0.2302 | 0.2671 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.32G>A | G11E 2D AIThe SynGAP1 missense variant G11E is reported in gnomAD (variant ID 6-33420296‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus itself is likely benign. Foldetta results are not available, so they do not influence the assessment. Overall, the preponderance of evidence points to a benign variant, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | 6-33420296-G-A | 5 | 3.24e-6 | -4.206 | Likely Benign | 0.219 | Likely Benign | Likely Benign | 0.109 | Likely Benign | 0.09 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.95 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1444 | 0.4628 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.32G>C | G11A 2D AIThe SynGAP1 missense variant G11A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | -3.611 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -0.28 | Neutral | 0.105 | Benign | 0.007 | Benign | 4.00 | Benign | 0.00 | Affected | 0.4045 | 0.5440 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.32G>T | G11V 2D AIThe SynGAP1 missense variant G11V is reported in gnomAD (variant ID 6‑33420296‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that G11V is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | 6-33420296-G-T | -4.079 | Likely Benign | 0.160 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -0.38 | Neutral | 0.668 | Possibly Damaging | 0.049 | Benign | 3.93 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1292 | 0.4522 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||
| c.3301C>A | P1101T 2D AIThe SynGAP1 missense variant P1101T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.968967 | Binding | 0.457 | 0.861 | 0.875 | -4.161 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.126 | Likely Benign | -1.60 | Neutral | 0.115 | Benign | 0.031 | Benign | 4.22 | Benign | 0.04 | Affected | 0.1729 | 0.5860 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3301C>G | P1101A 2D AIThe SynGAP1 missense variant P1101A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess pathogenicity uniformly predict a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Consequently, the variant is most likely benign based on the collective predictions, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.968967 | Binding | 0.457 | 0.861 | 0.875 | -3.825 | Likely Benign | 0.057 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -1.34 | Neutral | 0.010 | Benign | 0.010 | Benign | 4.26 | Benign | 0.08 | Tolerated | 0.3154 | 0.5218 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3301C>T | P1101S 2D AIThe SynGAP1 missense variant P1101S is reported in gnomAD (variant ID 6‑33443853‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, creating a single discordant call. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.968967 | Binding | 0.457 | 0.861 | 0.875 | 6-33443853-C-T | 1 | 6.52e-7 | -3.845 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -1.31 | Neutral | 0.626 | Possibly Damaging | 0.255 | Benign | 4.25 | Benign | 0.07 | Tolerated | 3.77 | 5 | 0.3265 | 0.5400 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.3302C>A | P1101H 2D AIThe SynGAP1 missense variant P1101H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect for P1101H, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.968967 | Binding | 0.457 | 0.861 | 0.875 | -5.370 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.167 | Likely Benign | -1.87 | Neutral | 0.996 | Probably Damaging | 0.864 | Possibly Damaging | 4.18 | Benign | 0.02 | Affected | 0.2046 | 0.4597 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3302C>G | P1101R 2D AIThe SynGAP1 missense variant P1101R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy methods further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of evidence points to a benign effect, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.968967 | Binding | 0.457 | 0.861 | 0.875 | -4.772 | Likely Benign | 0.227 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -1.28 | Neutral | 0.960 | Probably Damaging | 0.761 | Possibly Damaging | 4.27 | Benign | 0.36 | Tolerated | 0.1497 | 0.4644 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3302C>T | P1101L 2D AIThe SynGAP1 missense variant P1101L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for P1101L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.968967 | Binding | 0.457 | 0.861 | 0.875 | -4.335 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -2.19 | Neutral | 0.770 | Possibly Damaging | 0.255 | Benign | 4.27 | Benign | 0.04 | Affected | 0.2310 | 0.6050 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3304G>A | A1102T 2D AIThe SynGAP1 missense variant A1102T is listed in ClinVar (ID 1422927) as Benign and is present in gnomAD (6‑33443856‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence supports a benign classification, which is consistent with the ClinVar status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | Benign | 1 | 6-33443856-G-A | 11 | 7.17e-6 | -3.540 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.30 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.32 | Pathogenic | 0.95 | Tolerated | 3.77 | 5 | 0.1755 | 0.7699 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||
| c.3304G>C | A1102P 2D AIThe SynGAP1 missense variant A1102P is listed in ClinVar (ID 2789225.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | Uncertain | 1 | -5.120 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -0.97 | Neutral | 0.000 | Benign | 0.002 | Benign | 2.26 | Pathogenic | 0.13 | Tolerated | 3.77 | 5 | 0.1978 | 0.5919 | -1 | 1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||
| c.3304G>T | A1102S 2D AIThe SynGAP1 missense variant A1102S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign classification, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | -2.900 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.041 | Likely Benign | 0.05 | Neutral | 0.019 | Benign | 0.032 | Benign | 2.57 | Benign | 0.59 | Tolerated | 0.2666 | 0.6501 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3305C>A | A1102D 2D AIThe SynGAP1 missense variant A1102D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | -4.647 | Likely Benign | 0.388 | Ambiguous | Likely Benign | 0.081 | Likely Benign | -1.38 | Neutral | 0.033 | Benign | 0.028 | Benign | 2.27 | Pathogenic | 0.12 | Tolerated | 0.2045 | 0.2984 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3305C>G | A1102G 2D AIThe SynGAP1 A1102G missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The high‑accuracy consensus (SGM‑Consensus) classifies the variant as “Likely Benign,” and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | -3.070 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.060 | Likely Benign | 0.39 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.30 | Pathogenic | 0.14 | Tolerated | 0.1947 | 0.4958 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3305C>T | A1102V 2D AIThe SynGAP1 missense variant A1102V is listed in ClinVar (ID 2846719.0) as Benign and is present in gnomAD (variant ID 6‑33443857‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion aligns with the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | Benign | 1 | 6-33443857-C-T | -2.440 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -1.27 | Neutral | 0.017 | Benign | 0.028 | Benign | 2.29 | Pathogenic | 0.12 | Tolerated | 3.77 | 5 | 0.1333 | 0.6264 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||
| c.3307C>A | R1103S 2D AIThe SynGAP1 missense variant R1103S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, and Foldetta results are unavailable. Overall, the majority of evidence (six benign versus two pathogenic predictions) indicates that R1103S is most likely benign, and this conclusion does not contradict any ClinVar annotation because no ClinVar claim exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.945666 | Disordered | 0.957363 | Binding | 0.328 | 0.862 | 0.875 | -3.611 | Likely Benign | 0.464 | Ambiguous | Likely Benign | 0.111 | Likely Benign | -1.94 | Neutral | 0.511 | Possibly Damaging | 0.187 | Benign | 2.48 | Pathogenic | 0.28 | Tolerated | 0.3025 | 0.4224 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3307C>G | R1103G 2D AIThe SynGAP1 missense variant R1103G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the balance of evidence—five benign predictions versus four pathogenic, with a benign high‑accuracy tool and no conflicting ClinVar annotation—suggests that the variant is most likely benign. This conclusion does not contradict any ClinVar status, as the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.945666 | Disordered | 0.957363 | Binding | 0.328 | 0.862 | 0.875 | -3.516 | Likely Benign | 0.221 | Likely Benign | Likely Benign | 0.132 | Likely Benign | -2.65 | Deleterious | 0.911 | Possibly Damaging | 0.308 | Benign | 2.44 | Pathogenic | 0.03 | Affected | 0.3429 | 0.4077 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3307C>T | R1103C 2D AISynGAP1 missense variant R1103C is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33443859‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic). AlphaMissense‑Optimized reports a benign outcome, while Foldetta results are unavailable. Overall, the balance of evidence favors a pathogenic interpretation, which is in contrast to the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.945666 | Disordered | 0.957363 | Binding | 0.328 | 0.862 | 0.875 | Uncertain | 1 | 6-33443859-C-T | 6 | 3.92e-6 | -2.440 | Likely Benign | 0.246 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -3.01 | Deleterious | 0.996 | Probably Damaging | 0.787 | Possibly Damaging | 2.41 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.3376 | 0.4121 | -3 | -4 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.3308G>A | R1103H 2D AIThe SynGAP1 missense variant R1103H is listed in ClinVar (ID 577408.0) as benign and is present in gnomAD (variant ID 6‑33443860‑G‑A). Prediction tools that classify the variant as benign include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as benign, while Foldetta results are unavailable. Overall, the majority of predictions support a benign impact, and this conclusion aligns with the ClinVar benign classification, indicating no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.957363 | Binding | 0.328 | 0.862 | 0.875 | Benign/Likely benign | 3 | 6-33443860-G-A | 31 | 2.03e-5 | -3.622 | Likely Benign | 0.156 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -1.97 | Neutral | 0.996 | Probably Damaging | 0.733 | Possibly Damaging | 2.49 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.2977 | 0.2380 | 2 | 0 | 1.3 | -19.05 | ||||||||||||||||||||||||||||||||
| c.3308G>C | R1103P 2D AIThe SynGAP1 missense variant R1103P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of predictions (six benign vs. four pathogenic) support a benign classification. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.957363 | Binding | 0.328 | 0.862 | 0.875 | -2.149 | Likely Benign | 0.229 | Likely Benign | Likely Benign | 0.098 | Likely Benign | -2.48 | Neutral | 0.969 | Probably Damaging | 0.659 | Possibly Damaging | 2.43 | Pathogenic | 0.02 | Affected | 0.2288 | 0.5109 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3308G>T | R1103L 2D AIThe SynGAP1 missense variant R1103L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443860‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” outcome (3 benign vs. 1 pathogenic votes). High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.957363 | Binding | 0.328 | 0.862 | 0.875 | Uncertain | 1 | 6-33443860-G-T | -2.330 | Likely Benign | 0.205 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -2.35 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.44 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.2098 | 0.5181 | -3 | -2 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||
| c.3310C>A | P1104T 2D AIThe SynGAP1 missense variant P1104T is reported in gnomAD (variant ID 6‑33443862‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.936162 | Disordered | 0.954801 | Binding | 0.440 | 0.863 | 0.875 | 6-33443862-C-A | -3.995 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -0.14 | Neutral | 0.770 | Possibly Damaging | 0.481 | Possibly Damaging | 2.76 | Benign | 0.09 | Tolerated | 3.77 | 5 | 0.1549 | 0.6700 | -1 | 0 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||
| c.3310C>G | P1104A 2D AIThe SynGAP1 missense variant P1104A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.936162 | Disordered | 0.954801 | Binding | 0.440 | 0.863 | 0.875 | -3.677 | Likely Benign | 0.059 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.54 | Neutral | 0.409 | Benign | 0.184 | Benign | 2.74 | Benign | 0.22 | Tolerated | 0.3289 | 0.5315 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3310C>T | P1104S 2D AIThe SynGAP1 missense variant P1104S is listed in ClinVar (ID 2912797.0) as Benign and is present in gnomAD (variant ID 6‑33443862‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign, and AlphaMissense‑Optimized also reports Benign. Foldetta results are not available. Overall, the majority of computational evidence supports a benign classification, which is consistent with the ClinVar status. Thus, the variant is most likely benign and does not contradict the ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.936162 | Disordered | 0.954801 | Binding | 0.440 | 0.863 | 0.875 | Benign | 1 | 6-33443862-C-T | 1 | 6.54e-7 | -2.330 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.30 | Neutral | 0.770 | Possibly Damaging | 0.404 | Benign | 2.77 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.3271 | 0.5746 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||
| c.3311C>A | P1104Q 2D AIThe SynGAP1 missense variant P1104Q is reported in gnomAD (ID 6‑33443863‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign likelihood. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.936162 | Disordered | 0.954801 | Binding | 0.440 | 0.863 | 0.875 | 6-33443863-C-A | -3.161 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -0.64 | Neutral | 0.986 | Probably Damaging | 0.825 | Possibly Damaging | 2.68 | Benign | 0.06 | Tolerated | 3.77 | 5 | 0.1498 | 0.5063 | -1 | 0 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||
| c.3311C>G | P1104R 2D AIThe SynGAP1 missense variant P1104R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus likewise indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.936162 | Disordered | 0.954801 | Binding | 0.440 | 0.863 | 0.875 | -3.864 | Likely Benign | 0.328 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -0.64 | Neutral | 0.986 | Probably Damaging | 0.761 | Possibly Damaging | 2.68 | Benign | 0.06 | Tolerated | 0.1387 | 0.3703 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3311C>T | P1104L 2D AIThe SynGAP1 missense variant P1104L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also predicts Likely Benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy predictors, points to a benign impact. This conclusion is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.936162 | Disordered | 0.954801 | Binding | 0.440 | 0.863 | 0.875 | -3.846 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -0.33 | Neutral | 0.626 | Possibly Damaging | 0.168 | Benign | 2.81 | Benign | 1.00 | Tolerated | 0.2264 | 0.6795 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3313C>G | R1105G 2D AIThe SynGAP1 missense variant R1105G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Consensus from standard in silico predictors shows a split: benign calls come from REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, polyPhen‑2 HumDiv, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessment gives a benign prediction from AlphaMissense‑Optimized, a pathogenic consensus from the SGM method (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and no result from Foldetta, so its stability impact is unavailable. Overall, the majority of tools lean toward a benign effect, but the high‑accuracy consensus is conflicted. Thus, the variant is most likely benign based on the bulk of predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.901269 | Disordered | 0.954396 | Binding | 0.330 | 0.863 | 0.875 | -4.900 | Likely Benign | 0.438 | Ambiguous | Likely Benign | 0.146 | Likely Benign | -3.48 | Deleterious | 0.677 | Possibly Damaging | 0.168 | Benign | 2.45 | Pathogenic | 0.09 | Tolerated | 0.3293 | 0.4269 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3313C>T | R1105W 2D AIThe SynGAP1 missense variant R1105W is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33443865‑C‑T). Prediction tools show mixed results: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The AlphaMissense‑Default tool remains uncertain. A consensus analysis (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a pathogenic majority. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, whereas the SGM Consensus predicts pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for R1105W, which does not conflict with the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.901269 | Disordered | 0.954396 | Binding | 0.330 | 0.863 | 0.875 | Uncertain | 1 | 6-33443865-C-T | 6 | 3.93e-6 | -6.911 | Likely Benign | 0.488 | Ambiguous | Likely Benign | 0.133 | Likely Benign | -4.34 | Deleterious | 0.999 | Probably Damaging | 0.696 | Possibly Damaging | 2.42 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.1154 | 0.4117 | -3 | 2 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||
| c.3314G>A | R1105Q 2D AIThe SynGAP1 missense variant R1105Q is listed in ClinVar (ID 1803693.0) with an uncertain significance status and is present in gnomAD (variant ID 6‑33443866‑G‑A). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or tolerated outcomes. Only polyPhen‑2 HumDiv predicts a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, which is consistent with its ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.901269 | Disordered | 0.954396 | Binding | 0.330 | 0.863 | 0.875 | Uncertain | 2 | 6-33443866-G-A | 3 | 1.96e-6 | -3.666 | Likely Benign | 0.216 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -1.21 | Neutral | 0.958 | Probably Damaging | 0.194 | Benign | 2.50 | Benign | 0.16 | Tolerated | 3.77 | 5 | 0.2942 | 0.3174 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||
| c.3314G>C | R1105P 2D AIThe SynGAP1 missense variant R1105P is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leans pathogenic. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of predictions (six benign vs. two pathogenic) suggest a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current predictive evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.901269 | Disordered | 0.954396 | Binding | 0.330 | 0.863 | 0.875 | -3.325 | Likely Benign | 0.425 | Ambiguous | Likely Benign | 0.149 | Likely Benign | -3.22 | Deleterious | 0.007 | Benign | 0.006 | Benign | 2.44 | Pathogenic | 0.08 | Tolerated | 0.2031 | 0.5101 | 0 | -2 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||||||||||
| c.3314G>T | R1105L 2D AIThe SynGAP1 missense variant R1105L is not reported in ClinVar and is present in gnomAD (ID 6‑33443866‑G‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized classifies the variant as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels it pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not provide a result for this residue, so its stability impact is unavailable. Overall, the balance of evidence leans toward a benign effect, with no conflict with ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.901269 | Disordered | 0.954396 | Binding | 0.330 | 0.863 | 0.875 | 6-33443866-G-T | -4.031 | Likely Benign | 0.459 | Ambiguous | Likely Benign | 0.125 | Likely Benign | -3.51 | Deleterious | 0.677 | Possibly Damaging | 0.168 | Benign | 2.46 | Pathogenic | 0.19 | Tolerated | 3.77 | 5 | 0.1734 | 0.5373 | -2 | -3 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||
| c.3316C>A | Q1106K 2D AIThe SynGAP1 missense variant Q1106K is catalogued in gnomAD (ID 6‑33443868‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized; pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because the votes are split (two benign, one pathogenic, one uncertain). Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence favors a benign effect, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.885302 | Disordered | 0.952043 | Binding | 0.382 | 0.870 | 0.875 | 6-33443868-C-A | -3.368 | Likely Benign | 0.527 | Ambiguous | Likely Benign | 0.115 | Likely Benign | -2.49 | Neutral | 0.963 | Probably Damaging | 0.959 | Probably Damaging | 1.82 | Pathogenic | 0.16 | Tolerated | 3.77 | 5 | 0.1827 | 0.4800 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||
| c.3316C>G | Q1106E 2D AIThe SynGAP1 missense variant Q1106E is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, PolyPhen‑2 (HumDiv and HumVar) and FATHMM predict pathogenicity. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also likely benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for Q1106E, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.952043 | Binding | 0.382 | 0.870 | 0.875 | -5.074 | Likely Benign | 0.307 | Likely Benign | Likely Benign | 0.127 | Likely Benign | -1.60 | Neutral | 0.963 | Probably Damaging | 0.959 | Probably Damaging | 1.80 | Pathogenic | 0.15 | Tolerated | 0.1381 | 0.2663 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3317A>C | Q1106P 2D AIThe SynGAP1 missense variant Q1106P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the balance of evidence (5 benign vs 4 pathogenic predictions, with a benign high‑accuracy score and no contradictory ClinVar annotation) indicates that the variant is most likely benign. This conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.885302 | Disordered | 0.952043 | Binding | 0.382 | 0.870 | 0.875 | -3.807 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.162 | Likely Benign | -3.14 | Deleterious | 0.996 | Probably Damaging | 0.988 | Probably Damaging | 1.75 | Pathogenic | 0.31 | Tolerated | 0.2029 | 0.5781 | 0 | -1 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3317A>G | Q1106R 2D AIThe SynGAP1 missense variant Q1106R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it contains two benign, one pathogenic, and one uncertain call. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑confidence predictors (four benign vs. three pathogenic) lean toward a benign interpretation, and this assessment does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.885302 | Disordered | 0.952043 | Binding | 0.382 | 0.870 | 0.875 | -4.211 | Likely Benign | 0.475 | Ambiguous | Likely Benign | 0.133 | Likely Benign | -2.20 | Neutral | 0.985 | Probably Damaging | 0.973 | Probably Damaging | 1.80 | Pathogenic | 0.09 | Tolerated | 0.1412 | 0.2850 | 1 | 1 | -1.0 | 28.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3317A>T | Q1106L 2D AIThe SynGAP1 missense variant Q1106L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic impact. This assessment does not contradict ClinVar status, as the variant is currently unreported in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.885302 | Disordered | 0.952043 | Binding | 0.382 | 0.870 | 0.875 | -4.219 | Likely Benign | 0.171 | Likely Benign | Likely Benign | 0.169 | Likely Benign | -4.46 | Deleterious | 0.985 | Probably Damaging | 0.973 | Probably Damaging | 1.77 | Pathogenic | 0.05 | Affected | 0.0833 | 0.6282 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3318A>C | Q1106H 2D AIThe SynGAP1 missense variant Q1106H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.885302 | Disordered | 0.952043 | Binding | 0.382 | 0.870 | 0.875 | -4.893 | Likely Benign | 0.370 | Ambiguous | Likely Benign | 0.174 | Likely Benign | -3.03 | Deleterious | 0.996 | Probably Damaging | 0.992 | Probably Damaging | 1.75 | Pathogenic | 0.03 | Affected | 0.1453 | 0.4214 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3318A>T | Q1106H 2D AIThe SynGAP1 missense variant Q1106H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.885302 | Disordered | 0.952043 | Binding | 0.382 | 0.870 | 0.875 | -4.893 | Likely Benign | 0.370 | Ambiguous | Likely Benign | 0.174 | Likely Benign | -3.03 | Deleterious | 0.996 | Probably Damaging | 0.992 | Probably Damaging | 1.75 | Pathogenic | 0.03 | Affected | 0.1453 | 0.4214 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3319C>A | Q1107K 2D AIThe SynGAP1 missense variant Q1107K is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools largely support a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. Only polyPhen‑2 HumDiv predicts a pathogenic effect, and AlphaMissense‑Default remains uncertain. High‑accuracy assessments further reinforce the benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion is not in conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.951017 | Binding | 0.393 | 0.880 | 0.875 | -4.066 | Likely Benign | 0.400 | Ambiguous | Likely Benign | 0.095 | Likely Benign | -1.99 | Neutral | 0.920 | Possibly Damaging | 0.425 | Benign | 2.60 | Benign | 0.30 | Tolerated | 0.1805 | 0.5276 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3319C>G | Q1107E 2D AIThe SynGAP1 missense variant Q1107E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.951017 | Binding | 0.393 | 0.880 | 0.875 | -3.875 | Likely Benign | 0.231 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -1.54 | Neutral | 0.920 | Possibly Damaging | 0.425 | Benign | 2.59 | Benign | 0.02 | Affected | 0.1453 | 0.2932 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.331C>A | P111T 2D AIThe SynGAP1 missense variant P111T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.650020 | Binding | 0.438 | 0.858 | 0.750 | -2.800 | Likely Benign | 0.305 | Likely Benign | Likely Benign | 0.061 | Likely Benign | -1.44 | Neutral | 0.421 | Benign | 0.050 | Benign | 4.11 | Benign | 0.02 | Affected | 0.1650 | 0.5402 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.331C>G | P111A 2D AIThe SynGAP1 missense variant P111A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.650020 | Binding | 0.438 | 0.858 | 0.750 | -2.726 | Likely Benign | 0.163 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -1.42 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.26 | Benign | 0.04 | Affected | 0.3520 | 0.4825 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.331C>T | P111S 2D AIThe SynGAP1 missense variant P111S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods also support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the collective predictions strongly suggest that P111S is most likely benign, and this conclusion is not contradicted by any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.650020 | Binding | 0.438 | 0.858 | 0.750 | -2.773 | Likely Benign | 0.307 | Likely Benign | Likely Benign | 0.033 | Likely Benign | -1.03 | Neutral | 0.131 | Benign | 0.026 | Benign | 4.21 | Benign | 0.29 | Tolerated | 0.3515 | 0.4973 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3320A>C | Q1107P 2D AIThe SynGAP1 missense variant Q1107P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign effect for Q1107P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.951017 | Binding | 0.393 | 0.880 | 0.875 | -2.643 | Likely Benign | 0.047 | Likely Benign | Likely Benign | 0.135 | Likely Benign | -2.35 | Neutral | 0.965 | Probably Damaging | 0.611 | Possibly Damaging | 2.57 | Benign | 0.01 | Affected | 0.2109 | 0.5767 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3320A>G | Q1107R 2D AIThe SynGAP1 missense variant Q1107R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” AlphaMissense‑Optimized also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.951017 | Binding | 0.393 | 0.880 | 0.875 | -2.837 | Likely Benign | 0.394 | Ambiguous | Likely Benign | 0.126 | Likely Benign | -1.76 | Neutral | 0.965 | Probably Damaging | 0.425 | Benign | 2.55 | Benign | 0.04 | Affected | 0.1482 | 0.3319 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3320A>T | Q1107L 2D AIThe SynGAP1 missense variant Q1107L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.951017 | Binding | 0.393 | 0.880 | 0.875 | -3.785 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.119 | Likely Benign | -3.27 | Deleterious | 0.006 | Benign | 0.004 | Benign | 2.53 | Benign | 0.01 | Affected | 0.0820 | 0.6447 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3321G>C | Q1107H 2D AIThe SynGAP1 missense variant Q1107H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.951017 | Binding | 0.393 | 0.880 | 0.875 | -3.546 | Likely Benign | 0.260 | Likely Benign | Likely Benign | 0.136 | Likely Benign | -2.58 | Deleterious | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.51 | Benign | 0.01 | Affected | 0.1514 | 0.4683 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3321G>T | Q1107H 2D AIThe SynGAP1 missense variant Q1107H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, while Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.951017 | Binding | 0.393 | 0.880 | 0.875 | -3.546 | Likely Benign | 0.260 | Likely Benign | Likely Benign | 0.136 | Likely Benign | -2.58 | Deleterious | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.51 | Benign | 0.01 | Affected | 0.1514 | 0.4683 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3322A>C | S1108R 2D AISynGAP1 missense variant S1108R is not reported in ClinVar (status: None) and is absent from gnomAD (no entry). Prediction tools that classify the variant as benign include REVEL, polyPhen‑2 HumVar, ESM1b, and FATHMM. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The high‑accuracy consensus (SGM Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN is inconclusive, as it yields a 2‑vs‑2 split. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no available result for this variant. Overall, the computational evidence is mixed, with an equal number of benign and pathogenic calls and no high‑confidence consensus. Thus, the variant is most likely pathogenic based on the current predictions, and this assessment does not contradict the ClinVar status, which is unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | -5.878 | Likely Benign | 0.912 | Likely Pathogenic | Ambiguous | 0.130 | Likely Benign | -2.75 | Deleterious | 0.611 | Possibly Damaging | 0.329 | Benign | 2.54 | Benign | 0.04 | Affected | 0.0864 | 0.3492 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3322A>G | S1108G 2D AIThe SynGAP1 missense variant S1108G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs three pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Thus, the variant is most likely benign based on current predictive data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | -6.496 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.084 | Likely Benign | -2.59 | Deleterious | 0.568 | Possibly Damaging | 0.239 | Benign | 2.46 | Pathogenic | 0.16 | Tolerated | 0.2268 | 0.3975 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3322A>T | S1108C 2D AIThe SynGAP1 missense variant S1108C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is labeled Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains Likely Pathogenic; Foldetta results are unavailable. Overall, the majority of predictions (seven pathogenic vs. three benign) support a pathogenic classification, and this conclusion does not contradict any ClinVar status because none is available. Thus, the variant is most likely pathogenic based on the current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | -9.189 | Likely Pathogenic | 0.183 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -3.30 | Deleterious | 0.992 | Probably Damaging | 0.820 | Possibly Damaging | 2.42 | Pathogenic | 0.04 | Affected | 0.0992 | 0.5299 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.3323G>A | S1108N 2D AIThe SynGAP1 missense variant S1108N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S1108N, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | -6.488 | Likely Benign | 0.250 | Likely Benign | Likely Benign | 0.070 | Likely Benign | -2.02 | Neutral | 0.611 | Possibly Damaging | 0.239 | Benign | 2.47 | Pathogenic | 0.06 | Tolerated | 0.1283 | 0.3952 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3323G>C | S1108T 2D AIThe SynGAP1 missense variant S1108T is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | -5.710 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.49 | Neutral | 0.393 | Benign | 0.239 | Benign | 2.56 | Benign | 0.25 | Tolerated | 0.1304 | 0.5365 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3323G>T | S1108I 2D AIThe SynGAP1 missense variant S1108I is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33443875‑G‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no reported result for this variant. Overall, the balance of evidence (five benign versus four pathogenic predictions) suggests the variant is most likely benign, and this conclusion does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | Uncertain | 1 | 6-33443875-G-T | -3.666 | Likely Benign | 0.292 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -3.73 | Deleterious | 0.971 | Probably Damaging | 0.604 | Possibly Damaging | 2.44 | Pathogenic | 0.10 | Tolerated | 3.77 | 5 | 0.0949 | 0.4602 | -2 | -1 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||
| c.3324C>A | S1108R 2D AISynGAP1 missense variant S1108R has no ClinVar record and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumVar, ESM1b, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is a 2‑to‑2 split and therefore unavailable; Foldetta, which would combine FoldX‑MD and Rosetta outputs, has no reported result. Consequently, the evidence is evenly divided, leaving the variant’s functional impact uncertain. The predictions do not contradict any ClinVar status, as none is available. Overall, the variant is most likely of uncertain significance rather than definitively benign or pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | -5.878 | Likely Benign | 0.912 | Likely Pathogenic | Ambiguous | 0.109 | Likely Benign | -2.75 | Deleterious | 0.611 | Possibly Damaging | 0.329 | Benign | 2.54 | Benign | 0.04 | Affected | 0.0864 | 0.3492 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3324C>G | S1108R 2D AIThe SynGAP1 missense variant S1108R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic vs. two benign). Foldetta, which would assess protein‑folding stability, has no available result for this variant. Overall, the computational evidence is balanced, providing no clear bias toward benign or pathogenic. Thus, the variant’s likely impact remains uncertain, and there is no contradiction with the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | -5.878 | Likely Benign | 0.912 | Likely Pathogenic | Ambiguous | 0.108 | Likely Benign | -2.75 | Deleterious | 0.611 | Possibly Damaging | 0.329 | Benign | 2.54 | Benign | 0.04 | Affected | 0.0864 | 0.3492 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3325C>A | L1109I 2D AIThe SynGAP1 missense variant L1109I is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion is consistent with the lack of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | -5.475 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.40 | Neutral | 0.126 | Benign | 0.040 | Benign | 2.70 | Benign | 0.23 | Tolerated | 0.1087 | 0.4703 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3325C>G | L1109V 2D AIThe SynGAP1 missense variant L1109V is catalogued in gnomAD (ID 6‑33443877‑C‑G) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool reports a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the set predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a Likely Benign verdict. Foldetta results are not available for this variant. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | 6-33443877-C-G | -5.490 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -0.52 | Neutral | 0.001 | Benign | 0.005 | Benign | 2.72 | Benign | 0.19 | Tolerated | 4.32 | 2 | 0.1676 | 0.4353 | 1 | 2 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||
| c.3325C>T | L1109F 2D AIThe SynGAP1 missense variant L1109F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to the variant being most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | -3.459 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -1.04 | Neutral | 0.832 | Possibly Damaging | 0.324 | Benign | 2.74 | Benign | 0.12 | Tolerated | 0.0780 | 0.4540 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3326T>A | L1109H 2D AIThe SynGAP1 missense variant L1109H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | -4.353 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -0.56 | Neutral | 0.832 | Possibly Damaging | 0.499 | Possibly Damaging | 2.70 | Benign | 0.04 | Affected | 0.1250 | 0.1845 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3326T>C | L1109P 2D AIThe SynGAP1 missense variant L1109P is listed in ClinVar with an uncertain significance (ClinVar ID 1730257.0) and is not reported in gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized indicates a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign classification, which does not contradict the ClinVar uncertain status. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | Conflicting | 2 | -5.313 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.151 | Likely Benign | -0.52 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.65 | Benign | 0.07 | Tolerated | 4.32 | 2 | 0.3159 | 0.2330 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.3326T>G | L1109R 2D AIThe SynGAP1 missense variant L1109R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | -5.440 | Likely Benign | 0.408 | Ambiguous | Likely Benign | 0.139 | Likely Benign | -0.70 | Neutral | 0.586 | Possibly Damaging | 0.225 | Benign | 2.68 | Benign | 0.34 | Tolerated | 0.1352 | 0.1919 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3328A>C | S1110R 2D AIThe SynGAP1 missense variant S1110R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the balance of evidence (six benign versus three pathogenic predictions) indicates that the variant is most likely benign. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -5.075 | Likely Benign | 0.773 | Likely Pathogenic | Likely Benign | 0.065 | Likely Benign | -2.46 | Neutral | 0.144 | Benign | 0.042 | Benign | 2.31 | Pathogenic | 0.01 | Affected | 0.1057 | 0.3571 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3328A>G | S1110G 2D AIThe SynGAP1 missense variant S1110G is listed in ClinVar (ID 1722210.0) as benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion aligns with the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | Likely Benign | 1 | -4.674 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -2.26 | Neutral | 0.036 | Benign | 0.026 | Benign | 2.19 | Pathogenic | 0.08 | Tolerated | 4.32 | 2 | 0.2559 | 0.4806 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||
| c.3328A>T | S1110C 2D AIThe SynGAP1 missense variant S1110C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also resolves to benign (2 benign vs. 1 pathogenic vote). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy consensus—points to a benign impact. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -7.250 | In-Between | 0.096 | Likely Benign | Likely Benign | 0.027 | Likely Benign | -2.12 | Neutral | 0.898 | Possibly Damaging | 0.477 | Possibly Damaging | 2.16 | Pathogenic | 0.01 | Affected | 0.1214 | 0.5954 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3329G>A | S1110N 2D AIThe SynGAP1 missense variant S1110N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S1110N, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -5.028 | Likely Benign | 0.167 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -1.99 | Neutral | 0.144 | Benign | 0.078 | Benign | 2.20 | Pathogenic | 0.01 | Affected | 0.1542 | 0.4716 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3329G>C | S1110T 2D AIThe SynGAP1 missense variant S1110T is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the preponderance of benign predictions and the consensus from high‑accuracy tools, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -3.989 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.032 | Likely Benign | -1.76 | Neutral | 0.001 | Benign | 0.003 | Benign | 2.21 | Pathogenic | 0.04 | Affected | 0.1581 | 0.6224 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3329G>T | S1110I 2D AIThe SynGAP1 missense variant S1110I is catalogued in gnomAD (ID 6‑33443881‑G‑T) but has no ClinVar submission. Functional prediction tools show a split: six algorithms (REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a benign effect, whereas three (PROVEAN, SIFT, FATHMM) predict pathogenicity. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized classifies the variant as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic); Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no available result for this residue. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | 6-33443881-G-T | -6.124 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -2.99 | Deleterious | 0.007 | Benign | 0.003 | Benign | 2.17 | Pathogenic | 0.01 | Affected | 4.32 | 2 | 0.1203 | 0.4971 | -2 | -1 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||
| c.332C>A | P111Q 2D AIThe SynGAP1 missense variant P111Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments further support this view: AlphaMissense‑Optimized indicates benign, SGM‑Consensus confirms likely benign, and Foldetta data are unavailable. Taken together, the preponderance of evidence points to a benign effect for P111Q, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.650020 | Binding | 0.438 | 0.858 | 0.750 | -4.726 | Likely Benign | 0.543 | Ambiguous | Likely Benign | 0.079 | Likely Benign | -2.10 | Neutral | 0.421 | Benign | 0.054 | Benign | 4.06 | Benign | 0.00 | Affected | 0.1593 | 0.4232 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.332C>G | P111R 2D AIThe SynGAP1 missense variant P111R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.650020 | Binding | 0.438 | 0.858 | 0.750 | -4.811 | Likely Benign | 0.782 | Likely Pathogenic | Likely Benign | 0.100 | Likely Benign | -2.34 | Neutral | 0.421 | Benign | 0.075 | Benign | 4.06 | Benign | 0.00 | Affected | 0.1578 | 0.3015 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.332C>T | P111L 2D AIThe SynGAP1 missense variant P111L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that P111L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.707965 | Disordered | 0.650020 | Binding | 0.438 | 0.858 | 0.750 | -4.430 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.089 | Likely Benign | -2.81 | Deleterious | 0.421 | Benign | 0.055 | Benign | 4.06 | Benign | 0.00 | Affected | 0.2355 | 0.7085 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3330C>A | S1110R 2D AIThe SynGAP1 missense variant S1110R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs. three pathogenic predictions) points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -5.075 | Likely Benign | 0.773 | Likely Pathogenic | Likely Benign | 0.043 | Likely Benign | -2.46 | Neutral | 0.144 | Benign | 0.042 | Benign | 2.31 | Pathogenic | 0.01 | Affected | 0.1057 | 0.3571 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3330C>G | S1110R 2D AIThe SynGAP1 missense variant S1110R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs. three pathogenic predictions) points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -5.075 | Likely Benign | 0.773 | Likely Pathogenic | Likely Benign | 0.043 | Likely Benign | -2.46 | Neutral | 0.144 | Benign | 0.042 | Benign | 2.31 | Pathogenic | 0.01 | Affected | 0.1057 | 0.3571 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3331A>C | K1111Q 2D AIThe SynGAP1 missense variant K1111Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only polyPhen‑2 HumDiv predicts it as pathogenic. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely benign. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign; Foldetta results are not available. Overall, the consensus of available predictions indicates that K1111Q is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.921455 | Binding | 0.300 | 0.902 | 0.875 | -3.687 | Likely Benign | 0.261 | Likely Benign | Likely Benign | 0.036 | Likely Benign | -0.80 | Neutral | 0.666 | Possibly Damaging | 0.267 | Benign | 2.66 | Benign | 0.31 | Tolerated | 0.4577 | 0.1714 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3331A>G | K1111E 2D AIThe SynGAP1 missense variant K1111E is not reported in ClinVar and has no entry in gnomAD. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all score it as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also classifies it as likely benign. Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy tools further support a benign interpretation: AlphaMissense‑Optimized returns a benign prediction, while the SGM‑Consensus (majority vote) remains benign; a Foldetta stability assessment is unavailable. Taken together, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.921455 | Binding | 0.300 | 0.902 | 0.875 | -3.666 | Likely Benign | 0.565 | Likely Pathogenic | Likely Benign | 0.089 | Likely Benign | -0.86 | Neutral | 0.451 | Benign | 0.193 | Benign | 2.69 | Benign | 0.23 | Tolerated | 0.3846 | 0.1833 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3332A>C | K1111T 2D AIThe SynGAP1 missense variant K1111T is listed in ClinVar (ID 4746140) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443884‑A‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. No tool in the dataset predicts pathogenicity. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant, so its status is unavailable. Overall, the collective predictions point to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.921455 | Binding | 0.300 | 0.902 | 0.875 | Uncertain | 1 | 6-33443884-A-C | 2 | 1.32e-6 | -4.037 | Likely Benign | 0.519 | Ambiguous | Likely Benign | 0.080 | Likely Benign | -0.90 | Neutral | 0.292 | Benign | 0.110 | Benign | 2.64 | Benign | 0.28 | Tolerated | 4.32 | 2 | 0.2250 | 0.4425 | -1 | 0 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||||
| c.3332A>G | K1111R 2D AIThe SynGAP1 missense variant K1111R is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.921455 | Binding | 0.300 | 0.902 | 0.875 | -2.495 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -0.30 | Neutral | 0.006 | Benign | 0.006 | Benign | 2.74 | Benign | 0.71 | Tolerated | 0.4664 | 0.2031 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||||||||
| c.3332A>T | K1111M 2D AIThe SynGAP1 missense variant K1111M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Taken together, the majority of evidence points to a benign impact for K1111M. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.921455 | Binding | 0.300 | 0.902 | 0.875 | -5.579 | Likely Benign | 0.759 | Likely Pathogenic | Likely Benign | 0.071 | Likely Benign | -1.75 | Neutral | 0.072 | Benign | 0.029 | Benign | 2.59 | Benign | 0.12 | Tolerated | 0.1388 | 0.4701 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||||||||||||
| c.3333G>C | K1111N 2D AIThe SynGAP1 missense variant K1111N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and AlphaMissense‑Optimized is uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.921455 | Binding | 0.300 | 0.902 | 0.875 | -4.503 | Likely Benign | 0.833 | Likely Pathogenic | Ambiguous | 0.048 | Likely Benign | -0.77 | Neutral | 0.666 | Possibly Damaging | 0.211 | Benign | 2.64 | Benign | 0.15 | Tolerated | 0.3667 | 0.2350 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3333G>T | K1111N 2D AIThe SynGAP1 missense variant K1111N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and AlphaMissense‑Optimized is uncertain. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.921455 | Binding | 0.300 | 0.902 | 0.875 | -4.503 | Likely Benign | 0.833 | Likely Pathogenic | Ambiguous | 0.048 | Likely Benign | -0.77 | Neutral | 0.666 | Possibly Damaging | 0.211 | Benign | 2.64 | Benign | 0.15 | Tolerated | 0.3667 | 0.2350 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3334G>A | E1112K 2D AIThe SynGAP1 missense variant E1112K is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign. Only two tools predict pathogenicity: SIFT and AlphaMissense‑Default. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the majority of predictions, including the high‑accuracy tools, suggest the variant is most likely benign, and this is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.909381 | Binding | 0.335 | 0.902 | 0.875 | -3.772 | Likely Benign | 0.684 | Likely Pathogenic | Likely Benign | 0.210 | Likely Benign | 0.15 | Neutral | 0.245 | Benign | 0.096 | Benign | 2.82 | Benign | 0.02 | Affected | 0.3002 | 0.7704 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3334G>C | E1112Q 2D AIThe SynGAP1 missense variant E1112Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from multiple in silico predictors and high‑accuracy tools points to a benign impact for E1112Q, and this conclusion does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.909381 | Binding | 0.335 | 0.902 | 0.875 | -1.975 | Likely Benign | 0.364 | Ambiguous | Likely Benign | 0.102 | Likely Benign | -0.48 | Neutral | 0.611 | Possibly Damaging | 0.305 | Benign | 2.71 | Benign | 0.42 | Tolerated | 0.2031 | 0.7430 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3335A>C | E1112A 2D AIThe SynGAP1 missense variant E1112A is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.909381 | Binding | 0.335 | 0.902 | 0.875 | -3.227 | Likely Benign | 0.373 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -1.86 | Neutral | 0.393 | Benign | 0.131 | Benign | 2.71 | Benign | 0.02 | Affected | 0.3929 | 0.7528 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.3335A>G | E1112G 2D AIThe SynGAP1 missense variant E1112G is reported in gnomAD (ID 6‑33443887‑A‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN and SIFT. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yielding a “Likely Benign” classification. AlphaMissense‑Optimized independently predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.909381 | Binding | 0.335 | 0.902 | 0.875 | 6-33443887-A-G | 1 | 6.73e-7 | -3.459 | Likely Benign | 0.277 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -2.58 | Deleterious | 0.058 | Benign | 0.015 | Benign | 2.70 | Benign | 0.01 | Affected | 4.32 | 2 | 0.2795 | 0.6220 | -2 | 0 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||
| c.3335A>T | E1112V 2D AIThe SynGAP1 missense variant E1112V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) also as benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign impact for E1112V, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.909381 | Binding | 0.335 | 0.902 | 0.875 | -3.971 | Likely Benign | 0.579 | Likely Pathogenic | Likely Benign | 0.139 | Likely Benign | -2.28 | Neutral | 0.440 | Benign | 0.140 | Benign | 2.70 | Benign | 0.00 | Affected | 0.1481 | 0.7567 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3336G>C | E1112D 2D AIThe SynGAP1 missense variant E1112D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.909381 | Binding | 0.335 | 0.902 | 0.875 | -4.286 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -1.24 | Neutral | 0.005 | Benign | 0.005 | Benign | 2.69 | Benign | 0.06 | Tolerated | 4.32 | 2 | 0.2343 | 0.5553 | 2 | 3 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.3336G>T | E1112D 2D AIThe SynGAP1 missense variant E1112D is catalogued in gnomAD (variant ID 6-33443888‑G‑T) but has no ClinVar entry. Across the spectrum of in‑silico predictors, every tool examined—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classifies the substitution as benign. No pathogenic predictions are reported. Grouping by consensus, all listed tools fall into the benign category, with no tools indicating pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Based on the unanimous benign predictions and the absence of any ClinVar pathogenic classification, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.909381 | Binding | 0.335 | 0.902 | 0.875 | 6-33443888-G-T | -4.286 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -1.24 | Neutral | 0.005 | Benign | 0.005 | Benign | 2.69 | Benign | 0.06 | Tolerated | 4.32 | 2 | 0.2343 | 0.5553 | 2 | 3 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||
| c.3337G>A | G1113S 2D AIThe SynGAP1 missense variant G1113S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | -3.601 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.061 | Likely Benign | -0.51 | Neutral | 0.905 | Possibly Damaging | 0.538 | Possibly Damaging | 2.58 | Benign | 0.35 | Tolerated | 0.2499 | 0.5309 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3337G>C | G1113R 2D AIThe SynGAP1 missense variant G1113R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current predictive data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | -4.765 | Likely Benign | 0.618 | Likely Pathogenic | Likely Benign | 0.063 | Likely Benign | -1.54 | Neutral | 0.986 | Probably Damaging | 0.848 | Possibly Damaging | 2.65 | Benign | 0.64 | Tolerated | 0.0939 | 0.4426 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3337G>T | G1113C 2D AIThe SynGAP1 missense variant G1113C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. ESM1b is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | -7.917 | In-Between | 0.128 | Likely Benign | Likely Benign | 0.130 | Likely Benign | -1.64 | Neutral | 0.999 | Probably Damaging | 0.917 | Probably Damaging | 2.50 | Benign | 0.06 | Tolerated | 0.1325 | 0.4956 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3338G>A | G1113D 2D AIThe SynGAP1 missense variant G1113D is listed in ClinVar with an uncertain significance (ClinVar ID 2766136.0) and is present in gnomAD (variant ID 6‑33443890‑G‑A). Functional prediction tools that agree on a benign outcome include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—all of which classify the substitution as benign. AlphaMissense‑Optimized also predicts a benign effect, whereas AlphaMissense‑Default remains uncertain. No tool predicts a pathogenic effect. The high‑accuracy consensus methods reinforce this benign assessment: the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” and AlphaMissense‑Optimized also indicates benign. Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective evidence strongly supports a benign impact, and this conclusion does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | Uncertain | 1 | 6-33443890-G-A | -4.638 | Likely Benign | 0.354 | Ambiguous | Likely Benign | 0.061 | Likely Benign | -0.72 | Neutral | 0.029 | Benign | 0.017 | Benign | 2.58 | Benign | 0.34 | Tolerated | 4.32 | 2 | 0.1872 | 0.2452 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||
| c.3338G>C | G1113A 2D AIThe SynGAP1 missense variant G1113A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of tools, including the high‑accuracy predictors, indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | -4.714 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -1.15 | Neutral | 0.798 | Possibly Damaging | 0.433 | Benign | 2.58 | Benign | 0.50 | Tolerated | 0.3444 | 0.5342 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3338G>T | G1113V 2D AIThe SynGAP1 missense variant G1113V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | -5.708 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -1.98 | Neutral | 0.827 | Possibly Damaging | 0.456 | Possibly Damaging | 2.53 | Benign | 0.11 | Tolerated | 0.1333 | 0.4224 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3340A>C | S1114R 2D AIThe SynGAP1 missense variant S1114R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates a likely benign outcome. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence supports a benign classification, and this conclusion does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | -5.718 | Likely Benign | 0.696 | Likely Pathogenic | Likely Benign | 0.037 | Likely Benign | -1.52 | Neutral | 0.157 | Benign | 0.153 | Benign | 2.68 | Benign | 0.03 | Affected | 0.0999 | 0.3649 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3340A>G | S1114G 2D AIThe SynGAP1 missense variant S1114G is reported in gnomAD (ID 6‑33443892‑A‑G) but has no ClinVar entry. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | 6-33443892-A-G | -3.894 | Likely Benign | 0.064 | Likely Benign | Likely Benign | 0.080 | Likely Benign | -1.33 | Neutral | 0.000 | Benign | 0.000 | Benign | 2.75 | Benign | 0.08 | Tolerated | 4.32 | 2 | 0.2414 | 0.4914 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||||
| c.3340A>T | S1114C 2D AIThe SynGAP1 missense variant S1114C is reported in gnomAD (ID 6‑33443892‑A‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | 6-33443892-A-T | -8.600 | Likely Pathogenic | 0.091 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -1.77 | Neutral | 0.938 | Possibly Damaging | 0.552 | Possibly Damaging | 2.63 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1101 | 0.5675 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.3341G>A | S1114N 2D AIThe SynGAP1 missense variant S1114N is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool predicts pathogenicity. The high‑accuracy consensus, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), also reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | -6.089 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.51 | Neutral | 0.071 | Benign | 0.058 | Benign | 2.71 | Benign | 0.06 | Tolerated | 0.1416 | 0.4610 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3341G>C | S1114T 2D AIThe SynGAP1 missense variant S1114T is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | -5.919 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -0.87 | Neutral | 0.071 | Benign | 0.078 | Benign | 2.69 | Benign | 0.10 | Tolerated | 0.1424 | 0.6095 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3341G>T | S1114I 2D AIThe SynGAP1 missense variant S1114I is reported in gnomAD (ID 6‑33443893‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | 6-33443893-G-T | -6.718 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.023 | Likely Benign | -1.86 | Neutral | 0.570 | Possibly Damaging | 0.292 | Benign | 2.65 | Benign | 0.02 | Affected | 4.32 | 2 | 0.1159 | 0.5276 | -2 | -1 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||
| c.3342C>A | S1114R 2D AIThe SynGAP1 missense variant S1114R is not reported in ClinVar and has no entry in gnomAD. Prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. Only two tools predict pathogenicity: SIFT and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of predictions, including the high‑accuracy tools, suggest the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | -5.718 | Likely Benign | 0.696 | Likely Pathogenic | Likely Benign | 0.035 | Likely Benign | -1.52 | Neutral | 0.157 | Benign | 0.153 | Benign | 2.68 | Benign | 0.03 | Affected | 0.0999 | 0.3649 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3342C>G | S1114R 2D AIThe SynGAP1 missense variant S1114R is not reported in ClinVar and has no entry in gnomAD. Prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. Only two tools predict pathogenicity: SIFT and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of predictions, including the high‑accuracy tools, suggest the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | -5.718 | Likely Benign | 0.696 | Likely Pathogenic | Likely Benign | 0.035 | Likely Benign | -1.52 | Neutral | 0.157 | Benign | 0.153 | Benign | 2.68 | Benign | 0.03 | Affected | 0.0999 | 0.3649 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3343A>C | I1115L 2D AIThe SynGAP1 missense variant I1115L is listed in ClinVar (ID 4178654) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443895‑A‑C). All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic effect, so the pathogenic‑prediction group is empty. High‑accuracy assessments reinforce this benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the computational evidence strongly supports a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | Uncertain | 1 | 6-33443895-A-C | 1 | 7.56e-7 | -2.308 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.120 | Likely Benign | -0.46 | Neutral | 0.004 | Benign | 0.007 | Benign | 2.82 | Benign | 1.00 | Tolerated | 4.32 | 2 | 0.1154 | 0.4575 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||||
| c.3343A>G | I1115V 2D AIThe SynGAP1 missense variant I1115V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | -2.512 | Likely Benign | 0.060 | Likely Benign | Likely Benign | 0.104 | Likely Benign | 0.06 | Neutral | 0.002 | Benign | 0.007 | Benign | 2.76 | Benign | 0.68 | Tolerated | 0.1561 | 0.3871 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3343A>T | I1115F 2D AIThe SynGAP1 missense variant I1115F is reported in gnomAD (ID 6‑33443895‑A‑T) and has no ClinVar entry. All available in silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are not available. Based on the unanimous benign predictions and the lack of a ClinVar pathogenic classification, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | 6-33443895-A-T | -3.426 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.98 | Neutral | 0.230 | Benign | 0.098 | Benign | 2.71 | Benign | 0.19 | Tolerated | 4.32 | 2 | 0.0769 | 0.4338 | 0 | 1 | -1.7 | 34.02 | ||||||||||||||||||||||||||||||||||||
| c.3344T>A | I1115N 2D AIThe SynGAP1 missense variant I1115N is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | -4.018 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -0.60 | Neutral | 0.009 | Benign | 0.011 | Benign | 2.77 | Benign | 0.08 | Tolerated | 0.1299 | 0.1270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3344T>C | I1115T 2D AIThe SynGAP1 missense variant I1115T is listed in ClinVar (ID 130530) as Benign and is present in gnomAD (variant ID 6‑33443896‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts Benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly favors a benign impact, consistent with the ClinVar designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | Benign | 10 | 6-33443896-T-C | 20536 | 1.36e-2 | -2.670 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -0.04 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.76 | Benign | 0.23 | Tolerated | 4.32 | 2 | 0.1397 | 0.2252 | 0 | -1 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||
| c.3344T>G | I1115S 2D AIThe SynGAP1 missense variant I1115S is catalogued in gnomAD (ID 6‑33443896‑T‑G) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the variant as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions is benign, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | 6-33443896-T-G | -0.579 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.128 | Likely Benign | 0.67 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.88 | Benign | 0.14 | Tolerated | 4.32 | 2 | 0.2986 | 0.1453 | -2 | -1 | -5.3 | -26.08 | ||||||||||||||||||||||||||||||||||||
| c.3345T>G | I1115M 2D AIThe SynGAP1 missense variant I1115M is reported in gnomAD (variant ID 6‑33443897‑T‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, creating a single discordant call. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | 6-33443897-T-G | 4 | 2.77e-6 | -3.708 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -0.38 | Neutral | 0.512 | Possibly Damaging | 0.200 | Benign | 2.71 | Benign | 0.23 | Tolerated | 4.32 | 2 | 0.1030 | 0.4162 | 1 | 2 | -2.6 | 18.03 | ||||||||||||||||||||||||||||||||||
| c.3346G>A | G1116R 2D AIThe SynGAP1 missense variant G1116R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of computational evidence points to a benign effect for G1116R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | -6.379 | Likely Benign | 0.495 | Ambiguous | Likely Benign | 0.368 | Likely Benign | -0.60 | Neutral | 0.922 | Possibly Damaging | 0.657 | Possibly Damaging | 4.18 | Benign | 0.04 | Affected | 0.0925 | 0.4342 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3346G>C | G1116R 2D AIThe SynGAP1 missense variant G1116R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of computational evidence points to a benign effect for G1116R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | -6.379 | Likely Benign | 0.495 | Ambiguous | Likely Benign | 0.368 | Likely Benign | -0.60 | Neutral | 0.922 | Possibly Damaging | 0.657 | Possibly Damaging | 4.18 | Benign | 0.04 | Affected | 0.0925 | 0.4342 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3346G>T | G1116W 2D AIThe SynGAP1 missense variant G1116W is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443898‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta’s protein‑folding stability result is unavailable. Taken together, the majority of reliable predictors and the consensus high‑accuracy tools indicate a benign effect. This conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | 6-33443898-G-T | -9.448 | Likely Pathogenic | 0.356 | Ambiguous | Likely Benign | 0.405 | Likely Benign | -1.27 | Neutral | 0.996 | Probably Damaging | 0.946 | Probably Damaging | 4.04 | Benign | 0.01 | Affected | 4.32 | 2 | 0.0776 | 0.4046 | -2 | -7 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||
| c.3347G>A | G1116E 2D AIThe SynGAP1 missense variant G1116E is reported in gnomAD (variant ID 6-33443899‑G‑A) but has no ClinVar entry. Functional prediction tools uniformly favor a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while AlphaMissense‑Default remains uncertain. No tool predicts pathogenicity. The high‑accuracy consensus (SGM‑Consensus) also indicates a likely benign outcome, and AlphaMissense‑Optimized corroborates this. Foldetta, a protein‑folding stability predictor, was not available for this variant. Overall, the collective evidence strongly supports a benign impact, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | 6-33443899-G-A | -6.375 | Likely Benign | 0.375 | Ambiguous | Likely Benign | 0.345 | Likely Benign | -0.33 | Neutral | 0.043 | Benign | 0.022 | Benign | 4.06 | Benign | 0.07 | Tolerated | 4.32 | 2 | 0.1467 | 0.4069 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||||
| c.3347G>C | G1116A 2D AIThe SynGAP1 missense variant G1116A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | -5.753 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.288 | Likely Benign | -0.24 | Neutral | 0.010 | Benign | 0.022 | Benign | 4.08 | Benign | 0.24 | Tolerated | 0.3417 | 0.4944 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3347G>T | G1116V 2D AIThe SynGAP1 missense variant G1116V is reported in gnomAD (variant ID 6‑33443899‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, creating a single discordant signal. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | 6-33443899-G-T | -6.426 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.393 | Likely Benign | -0.79 | Neutral | 0.626 | Possibly Damaging | 0.375 | Benign | 4.06 | Benign | 0.06 | Tolerated | 4.32 | 2 | 0.1268 | 0.3494 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||
| c.3349G>A | G1117S 2D AIThe SynGAP1 missense variant G1117S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.853192 | Binding | 0.323 | 0.914 | 0.750 | -3.890 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.211 | Likely Benign | -0.37 | Neutral | 0.032 | Benign | 0.026 | Benign | 5.08 | Benign | 0.17 | Tolerated | 0.2572 | 0.5111 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3349G>C | G1117R 2D AIThe SynGAP1 missense variant G1117R is not reported in ClinVar and is absent from gnomAD. Consensus among most in silico predictors is benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized all classify the change as tolerated or benign. No tool predicts pathogenicity. Two predictors (ESM1b and AlphaMissense‑Default) return uncertain results, but these do not alter the overall benign consensus. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta data are unavailable. Consequently, the variant is most likely benign, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.853192 | Binding | 0.323 | 0.914 | 0.750 | -7.132 | In-Between | 0.519 | Ambiguous | Likely Benign | 0.253 | Likely Benign | -0.68 | Neutral | 0.006 | Benign | 0.008 | Benign | 4.62 | Benign | 0.08 | Tolerated | 0.0937 | 0.4542 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3349G>T | G1117C 2D AIThe SynGAP1 missense variant G1117C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.853192 | Binding | 0.323 | 0.914 | 0.750 | -9.045 | Likely Pathogenic | 0.112 | Likely Benign | Likely Benign | 0.359 | Likely Benign | -1.30 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 4.56 | Benign | 0.03 | Affected | 0.1347 | 0.4612 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.334G>A | G112R 2D AIThe SynGAP1 missense variant G112R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments are less definitive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of standard predictors lean toward a benign impact, and this is not contradicted by any ClinVar annotation. Thus, based on current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.728858 | Disordered | 0.640153 | Binding | 0.332 | 0.867 | 0.750 | -3.680 | Likely Benign | 0.866 | Likely Pathogenic | Ambiguous | 0.141 | Likely Benign | -3.31 | Deleterious | 0.002 | Benign | 0.004 | Benign | 3.94 | Benign | 0.00 | Affected | 0.0983 | 0.3978 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.334G>C | G112R 2D AIThe SynGAP1 missense variant G112R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments are less definitive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of standard predictors lean toward a benign impact, and this is not contradicted by any ClinVar annotation. Thus, based on current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.728858 | Disordered | 0.640153 | Binding | 0.332 | 0.867 | 0.750 | -3.680 | Likely Benign | 0.866 | Likely Pathogenic | Ambiguous | 0.142 | Likely Benign | -3.31 | Deleterious | 0.002 | Benign | 0.004 | Benign | 3.94 | Benign | 0.00 | Affected | 0.0983 | 0.3978 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.334G>T | G112W 2D AIThe SynGAP1 missense variant G112W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 pathogenic vs. 2 benign votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic interpretation. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.728858 | Disordered | 0.640153 | Binding | 0.332 | 0.867 | 0.750 | -6.382 | Likely Benign | 0.720 | Likely Pathogenic | Likely Benign | 0.190 | Likely Benign | -3.98 | Deleterious | 0.983 | Probably Damaging | 0.778 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 0.0631 | 0.4212 | -7 | -2 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||||||||||||
| c.3350G>A | G1117D 2D AIThe SynGAP1 missense variant G1117D is catalogued in gnomAD (6‑33443902‑G‑A) but has no ClinVar entry. In silico predictors that agree on benign impact include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic effect, while ESM1b and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it contains two benign and two uncertain calls, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar classification (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.853192 | Binding | 0.323 | 0.914 | 0.750 | 6-33443902-G-A | 1 | 6.61e-7 | -7.594 | In-Between | 0.358 | Ambiguous | Likely Benign | 0.315 | Likely Benign | -0.38 | Neutral | 0.666 | Possibly Damaging | 0.355 | Benign | 4.57 | Benign | 0.24 | Tolerated | 4.32 | 2 | 0.1920 | 0.2635 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||
| c.3350G>C | G1117A 2D AIThe SynGAP1 missense variant G1117A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.853192 | Binding | 0.323 | 0.914 | 0.750 | -6.514 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -0.41 | Neutral | 0.152 | Benign | 0.071 | Benign | 4.61 | Benign | 0.23 | Tolerated | 0.3545 | 0.4944 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3350G>T | G1117V 2D AIThe SynGAP1 missense variant G1117V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.853192 | Binding | 0.323 | 0.914 | 0.750 | -7.251 | In-Between | 0.083 | Likely Benign | Likely Benign | 0.284 | Likely Benign | -1.32 | Neutral | 0.011 | Benign | 0.014 | Benign | 4.57 | Benign | 0.04 | Affected | 0.1312 | 0.3680 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3352A>C | S1118R 2D AIThe SynGAP1 missense variant S1118R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S1118R is most likely benign, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | -2.670 | Likely Benign | 0.553 | Ambiguous | Likely Benign | 0.175 | Likely Benign | -0.74 | Neutral | 0.034 | Benign | 0.023 | Benign | 5.17 | Benign | 0.05 | Affected | 4.32 | 2 | 0.1314 | 0.3431 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.3352A>G | S1118G 2D AIThe SynGAP1 missense variant S1118G is reported in gnomAD (ID 6‑33443904‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the preponderance of evidence indicates that S1118G is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | 6-33443904-A-G | 1 | 6.98e-7 | -1.615 | Likely Benign | 0.057 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -0.61 | Neutral | 0.790 | Possibly Damaging | 0.433 | Benign | 5.82 | Benign | 0.08 | Tolerated | 4.32 | 2 | 0.2737 | 0.4657 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||
| c.3352A>T | S1118C 2D AIThe SynGAP1 missense variant S1118C is listed in gnomAD (ID 6‑33443904‑A‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, while ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | 6-33443904-A-T | -7.402 | In-Between | 0.096 | Likely Benign | Likely Benign | 0.311 | Likely Benign | -1.05 | Neutral | 0.997 | Probably Damaging | 0.889 | Possibly Damaging | 5.15 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1648 | 0.5695 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.3353G>A | S1118N 2D AIThe SynGAP1 missense variant S1118N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that the variant is most likely benign, and this assessment is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | -6.551 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.223 | Likely Benign | -0.53 | Neutral | 0.901 | Possibly Damaging | 0.433 | Benign | 5.26 | Benign | 0.06 | Tolerated | 0.1924 | 0.4215 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3353G>C | S1118T 2D AIThe SynGAP1 missense variant S1118T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | -6.063 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.196 | Likely Benign | -0.67 | Neutral | 0.790 | Possibly Damaging | 0.433 | Benign | 5.18 | Benign | 0.10 | Tolerated | 0.2066 | 0.5587 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3353G>T | S1118I 2D AIThe SynGAP1 missense variant S1118I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | -5.710 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 0.309 | Likely Benign | -1.09 | Neutral | 0.990 | Probably Damaging | 0.798 | Possibly Damaging | 5.16 | Benign | 0.01 | Affected | 0.1486 | 0.4580 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3354C>A | S1118R 2D AIThe SynGAP1 missense variant S1118R (ClinVar ID 2656489.0) is listed as ClinVar status Uncertain and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic effect, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar Uncertain designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | Uncertain | 1 | -2.670 | Likely Benign | 0.553 | Ambiguous | Likely Benign | 0.166 | Likely Benign | -0.74 | Neutral | 0.034 | Benign | 0.023 | Benign | 5.17 | Benign | 0.05 | Affected | 4.32 | 2 | 0.1314 | 0.3431 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||
| c.3354C>G | S1118R 2D AIThe SynGAP1 missense variant S1118R is listed in gnomAD (ID 6‑33443906‑C‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.828802 | Binding | 0.310 | 0.919 | 0.750 | 6-33443906-C-G | -2.670 | Likely Benign | 0.553 | Ambiguous | Likely Benign | 0.165 | Likely Benign | -0.74 | Neutral | 0.034 | Benign | 0.023 | Benign | 5.17 | Benign | 0.05 | Affected | 4.32 | 2 | 0.1314 | 0.3431 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||
| c.3355G>A | G1119R 2D AIThe SynGAP1 missense variant G1119R is listed in ClinVar as benign and is present in gnomAD (ID 6‑33443907‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and ESM1b predict a pathogenic impact. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, aligning with the ClinVar classification; there is no contradiction between the predictions and the reported ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | Benign | 1 | 6-33443907-G-A | 64 | 4.23e-5 | -8.489 | Likely Pathogenic | 0.473 | Ambiguous | Likely Benign | 0.303 | Likely Benign | 0.10 | Neutral | 0.969 | Probably Damaging | 0.462 | Possibly Damaging | 4.03 | Benign | 0.10 | Tolerated | 4.32 | 2 | 0.1112 | 0.4733 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||
| c.3355G>C | G1119R 2D AIThe SynGAP1 missense variant G1119R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. AlphaMissense‑Default is uncertain. High‑accuracy assessment shows AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | -8.489 | Likely Pathogenic | 0.473 | Ambiguous | Likely Benign | 0.289 | Likely Benign | 0.10 | Neutral | 0.969 | Probably Damaging | 0.462 | Possibly Damaging | 4.03 | Benign | 0.10 | Tolerated | 4.32 | 2 | 0.1112 | 0.4733 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3355G>T | G1119W 2D AIThe SynGAP1 missense variant G1119W is reported in gnomAD (ID 6‑33443907‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | 6-33443907-G-T | -10.904 | Likely Pathogenic | 0.336 | Likely Benign | Likely Benign | 0.386 | Likely Benign | -1.19 | Neutral | 0.997 | Probably Damaging | 0.949 | Probably Damaging | 3.90 | Benign | 0.02 | Affected | 4.32 | 2 | 0.0948 | 0.4059 | -2 | -7 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||||||||
| c.3356G>A | G1119E 2D AIThe SynGAP1 missense variant G1119E is reported in gnomAD (variant ID 6-33443908‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that G1119E is most likely benign, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | 6-33443908-G-A | 1 | 6.61e-7 | -9.151 | Likely Pathogenic | 0.338 | Likely Benign | Likely Benign | 0.284 | Likely Benign | -0.41 | Neutral | 0.005 | Benign | 0.013 | Benign | 3.93 | Benign | 0.12 | Tolerated | 4.32 | 2 | 0.1654 | 0.4259 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.3356G>C | G1119A 2D AIThe SynGAP1 missense variant G1119A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | -6.703 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.201 | Likely Benign | -0.33 | Neutral | 0.393 | Benign | 0.187 | Benign | 3.93 | Benign | 1.00 | Tolerated | 0.3591 | 0.4957 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3356G>T | G1119V 2D AIThe SynGAP1 missense variant G1119V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and polyPhen‑2 HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | -6.428 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.352 | Likely Benign | -0.94 | Neutral | 0.918 | Possibly Damaging | 0.604 | Possibly Damaging | 3.91 | Benign | 0.31 | Tolerated | 0.1493 | 0.3707 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3358G>A | G1120S 2D AIThe SynGAP1 missense variant G1120S is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions strongly suggests that G1120S is most likely benign, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.804931 | Binding | 0.335 | 0.925 | 0.875 | -4.959 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.286 | Likely Benign | -0.27 | Neutral | 0.451 | Benign | 0.209 | Benign | 3.69 | Benign | 0.77 | Tolerated | 0.2515 | 0.5311 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3358G>C | G1120R 2D AIThe SynGAP1 missense variant G1120R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign impact for G1120R, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.871313 | Disordered | 0.804931 | Binding | 0.335 | 0.925 | 0.875 | -8.784 | Likely Pathogenic | 0.565 | Likely Pathogenic | Likely Benign | 0.333 | Likely Benign | -0.77 | Neutral | 0.666 | Possibly Damaging | 0.221 | Benign | 3.60 | Benign | 0.05 | Affected | 0.0994 | 0.4142 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3358G>T | G1120C 2D AIThe SynGAP1 missense variant G1120C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” No Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for G1120C, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.804931 | Binding | 0.335 | 0.925 | 0.875 | -9.324 | Likely Pathogenic | 0.112 | Likely Benign | Likely Benign | 0.311 | Likely Benign | -1.32 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 3.60 | Benign | 0.03 | Affected | 0.1271 | 0.4227 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3359G>A | G1120D 2D AIThe SynGAP1 missense variant G1120D is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443911‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) resolves to a benign majority (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.871313 | Disordered | 0.804931 | Binding | 0.335 | 0.925 | 0.875 | 6-33443911-G-A | 4 | 2.65e-6 | -9.244 | Likely Pathogenic | 0.378 | Ambiguous | Likely Benign | 0.351 | Likely Benign | -0.82 | Neutral | 0.666 | Possibly Damaging | 0.355 | Benign | 3.60 | Benign | 0.19 | Tolerated | 3.77 | 5 | 0.1659 | 0.1835 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||
| c.3359G>C | G1120A 2D AIThe SynGAP1 missense variant G1120A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign likelihood. Foldetta results are unavailable. Overall, the consensus of available predictions indicates that G1120A is most likely benign, and this conclusion does not contradict any ClinVar status because none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.804931 | Binding | 0.335 | 0.925 | 0.875 | -7.192 | In-Between | 0.082 | Likely Benign | Likely Benign | 0.243 | Likely Benign | -0.39 | Neutral | 0.264 | Benign | 0.139 | Benign | 3.64 | Benign | 0.28 | Tolerated | 0.3435 | 0.4944 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3359G>T | G1120V 2D AIThe SynGAP1 missense variant G1120V is catalogued in gnomAD (6‑33443911‑G‑T) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the variant as benign, while ESM1b remains uncertain. No tool predicts pathogenicity. The high‑accuracy consensus, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), classifies the variant as “Likely Benign.” AlphaMissense‑Optimized also reports a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this assessment does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.804931 | Binding | 0.335 | 0.925 | 0.875 | 6-33443911-G-T | -7.659 | In-Between | 0.081 | Likely Benign | Likely Benign | 0.355 | Likely Benign | -1.20 | Neutral | 0.292 | Benign | 0.157 | Benign | 3.61 | Benign | 0.06 | Tolerated | 3.77 | 5 | 0.1360 | 0.3694 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||
| c.335G>A | G112E 2D AIThe SynGAP1 missense variant G112E is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split; Foldetta predictions are not available. Overall, the majority of evidence (five benign vs. three pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.728858 | Disordered | 0.640153 | Binding | 0.332 | 0.867 | 0.750 | -3.470 | Likely Benign | 0.818 | Likely Pathogenic | Ambiguous | 0.134 | Likely Benign | -3.30 | Deleterious | 0.421 | Benign | 0.146 | Benign | 3.96 | Benign | 0.00 | Affected | 0.1330 | 0.3814 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||||||||
| c.335G>C | G112A 2D AIThe SynGAP1 missense variant G112A is listed in ClinVar with an uncertain significance (ClinVar ID 1425533.0) and is present in gnomAD (6‑33432200‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. Foldetta results are unavailable. Overall, the majority of computational evidence indicates that the variant is most likely benign, which is consistent with its ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.640153 | Binding | 0.332 | 0.867 | 0.750 | Uncertain | 1 | 6-33432200-G-C | 15 | 9.30e-6 | -2.456 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -2.34 | Neutral | 0.231 | Benign | 0.054 | Benign | 4.07 | Benign | 0.00 | Affected | 3.61 | 5 | 0.3817 | 0.4429 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||
| c.335G>T | G112V 2D AIThe SynGAP1 missense variant G112V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.640153 | Binding | 0.332 | 0.867 | 0.750 | -2.411 | Likely Benign | 0.230 | Likely Benign | Likely Benign | 0.159 | Likely Benign | -3.39 | Deleterious | 0.421 | Benign | 0.108 | Benign | 3.96 | Benign | 0.00 | Affected | 0.1225 | 0.4218 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3361A>C | S1121R 2D AIThe SynGAP1 missense variant S1121R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign, reflecting the majority of benign predictions. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact for S1121R, and this conclusion is consistent with the lack of ClinVar reporting. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | -6.945 | Likely Benign | 0.597 | Likely Pathogenic | Likely Benign | 0.142 | Likely Benign | -0.34 | Neutral | 0.016 | Benign | 0.015 | Benign | 5.45 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1346 | 0.3431 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.3361A>G | S1121G 2D AIThe SynGAP1 missense variant S1121G is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443913‑A‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. Foldetta results are not available. Overall, the preponderance of evidence indicates that the variant is most likely benign, which does not contradict the current ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | Uncertain | 1 | 6-33443913-A-G | 1 | 7.00e-7 | -1.220 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -0.53 | Neutral | 0.003 | Benign | 0.004 | Benign | 6.63 | Benign | 0.00 | Affected | 3.77 | 5 | 0.2724 | 0.4657 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||
| c.3361A>T | S1121C 2D AIThe SynGAP1 missense variant S1121C is listed in gnomAD (ID 6‑33443913‑A‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | 6-33443913-A-T | -7.431 | In-Between | 0.094 | Likely Benign | Likely Benign | 0.354 | Likely Benign | -0.99 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 5.43 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1705 | 0.5691 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||
| c.3362G>A | S1121N 2D AIThe SynGAP1 missense variant S1121N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable. Overall, the consensus of the majority of predictors and the high‑accuracy tools points to a benign classification, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | -6.564 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.221 | Likely Benign | -0.12 | Neutral | 0.802 | Possibly Damaging | 0.266 | Benign | 5.50 | Benign | 0.00 | Affected | 0.1980 | 0.4211 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3362G>C | S1121T 2D AIThe SynGAP1 missense variant S1121T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is provided). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | -6.442 | Likely Benign | 0.069 | Likely Benign | Likely Benign | 0.105 | Likely Benign | -0.23 | Neutral | 0.011 | Benign | 0.026 | Benign | 5.45 | Benign | 0.00 | Affected | 0.2108 | 0.5582 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3362G>T | S1121I 2D AIThe SynGAP1 missense variant S1121I is listed in ClinVar (ID 4768628) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | Uncertain | 1 | -6.215 | Likely Benign | 0.147 | Likely Benign | Likely Benign | 0.455 | Likely Benign | -0.96 | Neutral | 0.875 | Possibly Damaging | 0.559 | Possibly Damaging | 5.44 | Benign | 0.00 | Affected | 0.1504 | 0.4776 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||
| c.3363C>A | S1121R 2D AIThe SynGAP1 missense variant S1121R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | -6.945 | Likely Benign | 0.597 | Likely Pathogenic | Likely Benign | 0.100 | Likely Benign | -0.34 | Neutral | 0.016 | Benign | 0.015 | Benign | 5.45 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1346 | 0.3431 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.3363C>G | S1121R 2D AIThe SynGAP1 missense variant S1121R is catalogued in gnomAD (ID 6‑33443915‑C‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all report benign. Only SIFT and AlphaMissense‑Default predict pathogenicity, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.810024 | Binding | 0.365 | 0.935 | 0.875 | 6-33443915-C-G | -6.945 | Likely Benign | 0.597 | Likely Pathogenic | Likely Benign | 0.101 | Likely Benign | -0.34 | Neutral | 0.016 | Benign | 0.015 | Benign | 5.45 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1346 | 0.3431 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||
| c.3364G>A | G1122S 2D AIThe SynGAP1 missense variant G1122S is listed in ClinVar (ID 643187) as Benign and is present in gnomAD (6‑33443916‑G‑A). Prediction tools that assess pathogenicity all converge on a benign outcome: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized indicates Benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports Likely Benign. The Foldetta stability analysis is unavailable for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, aligning with the ClinVar classification and showing no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.814918 | Binding | 0.357 | 0.932 | 0.875 | Benign/Likely benign | 3 | 6-33443916-G-A | 27 | 1.79e-5 | -4.880 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.189 | Likely Benign | -0.08 | Neutral | 0.022 | Benign | 0.006 | Benign | 4.89 | Benign | 0.92 | Tolerated | 3.77 | 5 | 0.2537 | 0.5111 | 1 | 0 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||||
| c.3364G>C | G1122R 2D AIThe SynGAP1 missense variant G1122R is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumVar, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, SIFT, and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence (six benign versus three pathogenic predictions, plus a benign consensus) indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.896620 | Disordered | 0.814918 | Binding | 0.357 | 0.932 | 0.875 | -9.063 | Likely Pathogenic | 0.507 | Ambiguous | Likely Benign | 0.319 | Likely Benign | -0.05 | Neutral | 0.639 | Possibly Damaging | 0.351 | Benign | 4.64 | Benign | 0.05 | Affected | 0.0976 | 0.4342 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3364G>T | G1122C 2D AIThe SynGAP1 missense variant G1122C is listed in ClinVar with no submitted interpretation and is present in gnomAD (variant ID 6‑33443916‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence—including the high‑confidence tools—supports a benign classification, and this conclusion does not contradict the ClinVar status, which currently has no pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.814918 | Binding | 0.357 | 0.932 | 0.875 | 6-33443916-G-T | -8.895 | Likely Pathogenic | 0.110 | Likely Benign | Likely Benign | 0.375 | Likely Benign | -1.30 | Neutral | 0.975 | Probably Damaging | 0.733 | Possibly Damaging | 4.48 | Benign | 0.08 | Tolerated | 3.77 | 5 | 0.1343 | 0.4227 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||
| c.3365G>A | G1122D 2D AIThe SynGAP1 missense variant G1122D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also favors benign (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that G1122D is most likely benign, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.896620 | Disordered | 0.814918 | Binding | 0.357 | 0.932 | 0.875 | -9.991 | Likely Pathogenic | 0.430 | Ambiguous | Likely Benign | 0.332 | Likely Benign | -0.49 | Neutral | 0.411 | Benign | 0.091 | Benign | 4.49 | Benign | 0.10 | Tolerated | 0.1746 | 0.2035 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3365G>C | G1122A 2D AIThe SynGAP1 missense variant G1122A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and there is no conflict with ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.814918 | Binding | 0.357 | 0.932 | 0.875 | -6.692 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.168 | Likely Benign | -0.52 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.52 | Benign | 0.62 | Tolerated | 0.3487 | 0.4944 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3365G>T | G1122V 2D AIThe SynGAP1 missense variant G1122V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) reports likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign, and this assessment is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.814918 | Binding | 0.357 | 0.932 | 0.875 | -6.398 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 0.271 | Likely Benign | -1.18 | Neutral | 0.059 | Benign | 0.025 | Benign | 4.49 | Benign | 0.10 | Tolerated | 0.1381 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3367G>A | G1123S 2D AIThe SynGAP1 missense variant G1123S is reported in gnomAD (variant ID 6-33443919-G-A) but has no ClinVar entry. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | 6-33443919-G-A | -4.918 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.291 | Likely Benign | -0.32 | Neutral | 0.292 | Benign | 0.157 | Benign | 4.66 | Benign | 0.57 | Tolerated | 3.77 | 5 | 0.2554 | 0.5111 | 0 | 1 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||||||||
| c.3367G>C | G1123R 2D AIThe SynGAP1 missense variant G1123R is not reported in ClinVar (no entry) and is absent from gnomAD. Consensus from routine in silico predictors shows a split: benign calls come from REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls arise from polyPhen‑2 HumDiv and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessment further supports a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign outcome, and no Foldetta stability data are available. Consequently, the overall evidence points to a benign effect for G1123R. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | -9.104 | Likely Pathogenic | 0.525 | Ambiguous | Likely Benign | 0.330 | Likely Benign | -0.46 | Neutral | 0.802 | Possibly Damaging | 0.435 | Benign | 4.34 | Benign | 0.57 | Tolerated | 0.0933 | 0.4342 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3367G>T | G1123C 2D AIThe SynGAP1 missense variant G1123C is listed in gnomAD (ID 6‑33443919‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and ESM1b predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are unavailable. Overall, the majority of evidence—including the consensus and high‑accuracy tools—supports a benign classification. This conclusion is not contradicted by ClinVar, which contains no record for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | 6-33443919-G-T | 1 | 6.64e-7 | -9.329 | Likely Pathogenic | 0.118 | Likely Benign | Likely Benign | 0.353 | Likely Benign | -1.17 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 4.34 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1348 | 0.4227 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.3368G>A | G1123D 2D AISynGAP1 missense variant G1123D is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33443920‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and ESM1b. AlphaMissense‑Default remains uncertain, and Foldetta stability analysis is unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus also benign, while Foldetta provides no result. Overall, the preponderance of evidence points to a benign effect, which does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | Uncertain | 2 | 6-33443920-G-A | 2 | 1.33e-6 | -10.321 | Likely Pathogenic | 0.405 | Ambiguous | Likely Benign | 0.360 | Likely Benign | -0.78 | Neutral | 0.500 | Possibly Damaging | 0.157 | Benign | 4.34 | Benign | 0.19 | Tolerated | 3.77 | 5 | 0.1792 | 0.2035 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||
| c.3368G>C | G1123A 2D AIThe SynGAP1 missense variant G1123A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated methods. High‑accuracy tools confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | -6.720 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.240 | Likely Benign | -0.55 | Neutral | 0.264 | Benign | 0.103 | Benign | 4.38 | Benign | 1.00 | Tolerated | 0.3544 | 0.4944 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3368G>T | G1123V 2D AIThe SynGAP1 missense variant G1123V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective predictions strongly suggest that G1123V is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | -7.129 | In-Between | 0.091 | Likely Benign | Likely Benign | 0.333 | Likely Benign | -1.03 | Neutral | 0.292 | Benign | 0.157 | Benign | 4.35 | Benign | 0.29 | Tolerated | 0.1362 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3370G>A | G1124R 2D AISynGAP1 missense variant G1124R is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33443922‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and ESM1b, while AlphaMissense‑Default remains uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to benign. High‑accuracy methods give AlphaMissense‑Optimized as benign; the SGM Consensus also supports benign. Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the ensemble of predictions leans toward a benign impact, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.837511 | Disordered | 0.833401 | Binding | 0.341 | 0.931 | 0.875 | Conflicting | 3 | 6-33443922-G-A | 24 | 1.60e-5 | -8.918 | Likely Pathogenic | 0.534 | Ambiguous | Likely Benign | 0.243 | Likely Benign | -0.58 | Neutral | 0.002 | Benign | 0.002 | Benign | 4.81 | Benign | 0.01 | Affected | 3.77 | 5 | 0.0935 | 0.4332 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||
| c.3370G>C | G1124R 2D AIThe SynGAP1 missense variant G1124R is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors shows a predominance of benign calls: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized all predict a benign effect, while SIFT and ESM1b predict pathogenicity. The AlphaMissense‑Default tool is uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized is benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also returns benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence points to a benign impact for G1124R, and this conclusion is consistent with the absence of any ClinVar annotation or gnomAD observation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.837511 | Disordered | 0.833401 | Binding | 0.341 | 0.931 | 0.875 | -8.918 | Likely Pathogenic | 0.534 | Ambiguous | Likely Benign | 0.239 | Likely Benign | -0.58 | Neutral | 0.002 | Benign | 0.002 | Benign | 4.81 | Benign | 0.01 | Affected | 3.77 | 5 | 0.0935 | 0.4332 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3371G>A | G1124E 2D AIThe SynGAP1 missense variant G1124E is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and ESM1b, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors a benign outcome (2 benign vs. 1 pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.837511 | Disordered | 0.833401 | Binding | 0.341 | 0.931 | 0.875 | -10.403 | Likely Pathogenic | 0.345 | Ambiguous | Likely Benign | 0.301 | Likely Benign | -0.84 | Neutral | 0.126 | Benign | 0.066 | Benign | 4.78 | Benign | 0.02 | Affected | 0.1405 | 0.4063 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3371G>C | G1124A 2D AIThe SynGAP1 missense variant G1124A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign,” and the Foldetta stability analysis is unavailable. Taken together, the overwhelming majority of computational evidence classifies G1124A as benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.833401 | Binding | 0.341 | 0.931 | 0.875 | -6.608 | Likely Benign | 0.084 | Likely Benign | Likely Benign | 0.199 | Likely Benign | -0.39 | Neutral | 0.059 | Benign | 0.041 | Benign | 4.81 | Benign | 0.02 | Affected | 0.3494 | 0.5138 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3371G>T | G1124V 2D AIThe SynGAP1 missense variant G1124V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of available predictions indicates that G1124V is most likely benign, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.833401 | Binding | 0.341 | 0.931 | 0.875 | -6.980 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.315 | Likely Benign | -0.96 | Neutral | 0.586 | Possibly Damaging | 0.172 | Benign | 4.75 | Benign | 0.00 | Affected | 0.1299 | 0.3888 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3373G>A | G1125R 2D AIThe SynGAP1 missense variant G1125R is catalogued in gnomAD (ID 6‑33443925‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. AlphaMissense‑Default remains uncertain. A high‑accuracy consensus (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a benign majority vote. AlphaMissense‑Optimized also reports benign. No Foldetta stability assessment is available. Collectively, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443925-G-A | -8.463 | Likely Pathogenic | 0.542 | Ambiguous | Likely Benign | 0.291 | Likely Benign | -0.54 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.56 | Benign | 0.04 | Affected | 3.77 | 5 | 0.0980 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3373G>C | G1125R 2D AIThe SynGAP1 missense variant G1125R is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443925‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta’s protein‑folding stability result is unavailable. Overall, the balance of evidence, particularly from the high‑accuracy tools, indicates that the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443925-G-C | 1 | 6.68e-7 | -8.463 | Likely Pathogenic | 0.542 | Ambiguous | Likely Benign | 0.290 | Likely Benign | -0.54 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.56 | Benign | 0.04 | Affected | 3.77 | 5 | 0.0980 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.3373G>T | G1125W 2D AIThe SynGAP1 missense variant G1125W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence, especially from the high‑accuracy tools, points to a benign classification. This conclusion is not contradicted by any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | -11.684 | Likely Pathogenic | 0.372 | Ambiguous | Likely Benign | 0.402 | Likely Benign | -1.10 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.50 | Benign | 0.00 | Affected | 0.0924 | 0.4046 | -7 | -2 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||||||||||||
| c.3374G>A | G1125E 2D AIThe SynGAP1 missense variant G1125E is not reported in ClinVar (ClinVar status: not reported) but is present in gnomAD (gnomAD ID 6‑33443926‑G‑A). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, AlphaMissense‑Optimized, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; AlphaMissense‑Default remains uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a benign outcome (2 benign vs. 1 pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM Consensus as benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443926-G-A | 11 | 7.34e-6 | -9.849 | Likely Pathogenic | 0.353 | Ambiguous | Likely Benign | 0.264 | Likely Benign | -0.32 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.58 | Benign | 0.09 | Tolerated | 3.77 | 5 | 0.1430 | 0.4063 | -2 | 0 | -3.1 | 72.06 | |||||||||||||||||||||||||||||||||||
| c.3374G>C | G1125A 2D AIThe SynGAP1 missense variant G1125A is listed in ClinVar with an “Uncertain” status and is present in gnomAD (6‑33443926‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (derived from the four high‑accuracy tools) is benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | Uncertain | 1 | 6-33443926-G-C | 1 | 6.68e-7 | -6.569 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.232 | Likely Benign | -0.60 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 4.60 | Benign | 0.11 | Tolerated | 3.77 | 5 | 0.3395 | 0.5138 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||
| c.3374G>T | G1125V 2D AIThe SynGAP1 missense variant G1125V is reported in gnomAD (variant ID 6-33443926-G-T) but has no ClinVar entry. Functional prediction tools cluster into two groups: six tools (REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score all predict a benign effect, whereas three tools (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT) predict pathogenicity. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443926-G-T | -6.776 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.320 | Likely Benign | -0.99 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.55 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1407 | 0.3688 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||
| c.3376G>A | G1126S 2D AIThe SynGAP1 missense variant G1126S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign impact for G1126S, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.837209 | Binding | 0.345 | 0.918 | 0.875 | -5.004 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.268 | Likely Benign | -0.28 | Neutral | 0.611 | Possibly Damaging | 0.171 | Benign | 4.78 | Benign | 0.74 | Tolerated | 0.2509 | 0.5300 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3376G>C | G1126R 2D AIThe SynGAP1 missense variant G1126R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Two tools—ESM1b and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is uncertain due to a 2‑to‑2 split between benign and uncertain calls; Foldetta’s protein‑folding stability analysis is unavailable. Overall, the balance of evidence leans toward a benign interpretation, and this conclusion does not contradict any ClinVar annotation because no ClinVar record exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.837209 | Binding | 0.345 | 0.918 | 0.875 | -7.760 | In-Between | 0.522 | Ambiguous | Likely Benign | 0.345 | Likely Benign | -0.56 | Neutral | 0.971 | Probably Damaging | 0.597 | Possibly Damaging | 4.77 | Benign | 0.01 | Affected | 0.0946 | 0.4532 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3376G>T | G1126C 2D AIThe SynGAP1 missense variant G1126C is listed in ClinVar (ID 469157.0) with an “Uncertain” status and is present in gnomAD (6‑33443928‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are SIFT and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.837209 | Binding | 0.345 | 0.918 | 0.875 | Uncertain | 1 | 6-33443928-G-T | 11 | 7.35e-6 | -9.389 | Likely Pathogenic | 0.113 | Likely Benign | Likely Benign | 0.449 | Likely Benign | -1.40 | Neutral | 0.005 | Benign | 0.005 | Benign | 4.74 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1326 | 0.4427 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||
| c.3377G>A | G1126D 2D AISynGAP1 missense variant G1126D is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split opinion: benign predictions come from REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The AlphaMissense‑Default result is uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.837209 | Binding | 0.345 | 0.918 | 0.875 | Uncertain | 1 | -8.888 | Likely Pathogenic | 0.432 | Ambiguous | Likely Benign | 0.376 | Likely Benign | -0.65 | Neutral | 0.906 | Possibly Damaging | 0.473 | Possibly Damaging | 4.82 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1725 | 0.2224 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||
| c.3377G>C | G1126A 2D AIThe SynGAP1 missense variant G1126A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.837209 | Binding | 0.345 | 0.918 | 0.875 | -6.402 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.228 | Likely Benign | -0.63 | Neutral | 0.124 | Benign | 0.061 | Benign | 4.83 | Benign | 0.56 | Tolerated | 0.3505 | 0.5333 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3377G>T | G1126V 2D AIThe SynGAP1 missense variant G1126V is listed in ClinVar with an uncertain significance and is present in the gnomAD database. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score all classify the change as benign. Only the SIFT algorithm predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized reports a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign, while Foldetta’s protein‑folding stability analysis is unavailable. Overall, the preponderance of evidence points to a benign variant, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.837209 | Binding | 0.345 | 0.918 | 0.875 | Uncertain | 1 | 6-33443929-G-T | -6.536 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.357 | Likely Benign | -1.20 | Neutral | 0.009 | Benign | 0.008 | Benign | 4.76 | Benign | 0.03 | Affected | 3.77 | 5 | 0.1366 | 0.3884 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||
| c.3379G>A | G1127R 2D AIThe SynGAP1 missense variant G1127R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443931‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Uncertain | 1 | 6-33443931-G-A | 2 | 1.34e-6 | -5.949 | Likely Benign | 0.629 | Likely Pathogenic | Likely Benign | 0.341 | Likely Benign | -0.87 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.86 | Benign | 0.12 | Tolerated | 4.32 | 4 | 0.0929 | 0.4532 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||
| c.3379G>C | G1127R 2D AIThe SynGAP1 missense variant G1127R is listed in ClinVar (ID 2967461.0) with an “Uncertain” clinical significance and is present in gnomAD (6‑33443931‑G‑C). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that G1127R is most likely benign, which does not contradict the ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Conflicting | 2 | 6-33443931-G-C | 16 | 1.07e-5 | -5.949 | Likely Benign | 0.629 | Likely Pathogenic | Likely Benign | 0.341 | Likely Benign | -0.87 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.86 | Benign | 0.12 | Tolerated | 4.32 | 4 | 0.0929 | 0.4532 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||
| c.3379G>T | G1127W 2D AIThe SynGAP1 missense variant G1127W is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443931‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta’s protein‑folding stability result is unavailable. Overall, the balance of evidence, particularly from the high‑accuracy tools, indicates that the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | 6-33443931-G-T | 3 | 2.01e-6 | -9.850 | Likely Pathogenic | 0.418 | Ambiguous | Likely Benign | 0.394 | Likely Benign | -1.36 | Neutral | 0.983 | Probably Damaging | 0.665 | Possibly Damaging | 4.79 | Benign | 0.03 | Affected | 4.32 | 4 | 0.0782 | 0.4032 | -2 | -7 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||
| c.337G>A | G113R 2D AIThe SynGAP1 missense variant G113R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default, while AlphaMissense‑Optimized returns an uncertain result. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.639486 | Binding | 0.350 | 0.870 | 0.750 | -3.904 | Likely Benign | 0.791 | Likely Pathogenic | Ambiguous | 0.087 | Likely Benign | -1.50 | Neutral | 0.267 | Benign | 0.080 | Benign | 4.16 | Benign | 0.03 | Affected | 0.1025 | 0.4138 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.337G>C | G113R 2D AIThe SynGAP1 missense variant G113R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM all classify it as benign. In contrast, SIFT and AlphaMissense‑Default predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized score is uncertain, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as likely benign. Protein‑folding stability analysis via Foldetta is unavailable for this residue. Overall, the preponderance of evidence points to a benign effect, and this assessment does not conflict with the absence of a ClinVar classification. Thus, the variant is most likely benign, and this conclusion is consistent with the lack of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.639486 | Binding | 0.350 | 0.870 | 0.750 | -3.904 | Likely Benign | 0.791 | Likely Pathogenic | Ambiguous | 0.088 | Likely Benign | -1.50 | Neutral | 0.267 | Benign | 0.080 | Benign | 4.16 | Benign | 0.03 | Affected | 0.1025 | 0.4138 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.337G>T | G113W 2D AIThe SynGAP1 missense variant G113W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence (six benign versus four pathogenic predictions) supports a benign classification. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.639486 | Binding | 0.350 | 0.870 | 0.750 | -5.635 | Likely Benign | 0.620 | Likely Pathogenic | Likely Benign | 0.162 | Likely Benign | -2.44 | Neutral | 0.983 | Probably Damaging | 0.717 | Possibly Damaging | 4.10 | Benign | 0.01 | Affected | 0.0700 | 0.4462 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.3380G>A | G1127E 2D AIThe SynGAP1 missense variant G1127E is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the remaining tools (ESM1b and AlphaMissense‑Default) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a tie between benign and uncertain calls. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the evidence strongly points to a benign effect, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | -7.359 | In-Between | 0.422 | Ambiguous | Likely Benign | 0.314 | Likely Benign | -0.56 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.88 | Benign | 0.15 | Tolerated | 0.1395 | 0.4244 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3380G>C | G1127A 2D AIThe SynGAP1 missense variant G1127A is listed in ClinVar (ID 426748.0) with an uncertain significance status and is present in gnomAD (variant ID 6‑33443932‑G‑C). Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the variant as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the evidence strongly supports a benign classification, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Conflicting | 4 | 6-33443932-G-C | 4 | 2.68e-6 | -5.949 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.164 | Likely Benign | -0.43 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.83 | Benign | 1.00 | Tolerated | 4.32 | 4 | 0.3420 | 0.4933 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||
| c.3380G>T | G1127V 2D AIThe SynGAP1 missense variant G1127V is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443932‑G‑T). All available in silico predictors classify the change as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic prediction. Grouping by agreement, the benign‑predicting tools comprise the entire set, while no pathogenic predictions exist. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Uncertain | 1 | 6-33443932-G-T | 1 | 6.69e-7 | -6.097 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.230 | Likely Benign | -1.01 | Neutral | 0.004 | Benign | 0.005 | Benign | 4.81 | Benign | 0.17 | Tolerated | 4.32 | 4 | 0.1281 | 0.3669 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||
| c.3382G>A | G1128R 2D AIThe SynGAP1 missense variant G1128R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.865136 | Binding | 0.309 | 0.911 | 0.875 | -5.009 | Likely Benign | 0.692 | Likely Pathogenic | Likely Benign | 0.396 | Likely Benign | -0.79 | Neutral | 0.846 | Possibly Damaging | 0.346 | Benign | 4.38 | Benign | 0.12 | Tolerated | 0.0950 | 0.4725 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3382G>C | G1128R 2D AIThe SynGAP1 missense variant G1128R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.865136 | Binding | 0.309 | 0.911 | 0.875 | -5.009 | Likely Benign | 0.692 | Likely Pathogenic | Likely Benign | 0.397 | Likely Benign | -0.79 | Neutral | 0.846 | Possibly Damaging | 0.346 | Benign | 4.38 | Benign | 0.12 | Tolerated | 0.0950 | 0.4725 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3382G>T | G1128W 2D AIThe SynGAP1 missense variant G1128W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields an inconclusive result (two benign, two uncertain), and Foldetta data are unavailable. Overall, the balance of evidence favors a benign classification. This conclusion does not contradict ClinVar status, as the variant has no existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.896620 | Disordered | 0.865136 | Binding | 0.309 | 0.911 | 0.875 | -7.341 | In-Between | 0.402 | Ambiguous | Likely Benign | 0.464 | Likely Benign | -1.36 | Neutral | 0.011 | Benign | 0.008 | Benign | 4.36 | Benign | 0.02 | Affected | 0.0796 | 0.4540 | -7 | -2 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||||||||||||
| c.3383G>A | G1128E 2D AIThe SynGAP1 missense variant G1128E is present in gnomAD (6‑33443935‑G‑A) and has no ClinVar entry. In silico predictors that agree on a benign effect include REVEL, PROVEAN, PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as likely benign. High‑accuracy tools reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is a high‑accuracy majority vote. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its stability impact remains unknown. Overall, the computational evidence overwhelmingly supports a benign classification, and this is consistent with the absence of a pathogenic ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.865136 | Binding | 0.309 | 0.911 | 0.875 | 6-33443935-G-A | 1 | 6.70e-7 | -5.580 | Likely Benign | 0.452 | Ambiguous | Likely Benign | 0.235 | Likely Benign | -0.24 | Neutral | 0.002 | Benign | 0.008 | Benign | 4.43 | Benign | 1.00 | Tolerated | 4.32 | 4 | 0.1610 | 0.4497 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.3383G>C | G1128A 2D AIThe SynGAP1 missense variant G1128A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.865136 | Binding | 0.309 | 0.911 | 0.875 | -5.015 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.209 | Likely Benign | -0.26 | Neutral | 0.009 | Benign | 0.008 | Benign | 4.47 | Benign | 0.46 | Tolerated | 0.3588 | 0.5252 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3383G>T | G1128V 2D AIThe SynGAP1 missense variant G1128V is reported in gnomAD (variant ID 6‑33443935‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, creating a single discordant call. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.865136 | Binding | 0.309 | 0.911 | 0.875 | 6-33443935-G-T | -5.111 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.399 | Likely Benign | -0.69 | Neutral | 0.611 | Possibly Damaging | 0.185 | Benign | 4.39 | Benign | 0.09 | Tolerated | 4.32 | 4 | 0.1358 | 0.3922 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||
| c.3385C>A | L1129M 2D AIThe SynGAP1 missense variant L1129M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect for L1129M, and this conclusion does not conflict with ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.876543 | Binding | 0.339 | 0.909 | 0.875 | -4.233 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.295 | Likely Benign | -0.59 | Neutral | 0.918 | Possibly Damaging | 0.697 | Possibly Damaging | 5.43 | Benign | 0.00 | Affected | 0.1100 | 0.4236 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3385C>G | L1129V 2D AIThe SynGAP1 missense variant L1129V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.876543 | Binding | 0.339 | 0.909 | 0.875 | -3.595 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.262 | Likely Benign | -0.87 | Neutral | 0.393 | Benign | 0.187 | Benign | 5.46 | Benign | 0.00 | Affected | 0.1814 | 0.3804 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3386T>A | L1129Q 2D AIThe SynGAP1 missense variant L1129Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.876543 | Binding | 0.339 | 0.909 | 0.875 | -3.684 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.453 | Likely Benign | -1.63 | Neutral | 0.846 | Possibly Damaging | 0.525 | Possibly Damaging | 5.49 | Benign | 0.00 | Affected | 0.1278 | 0.1436 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3386T>C | L1129P 2D AIThe SynGAP1 missense variant L1129P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence—including high‑accuracy tools—points to a benign effect, and this conclusion does not contradict the current ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.876543 | Binding | 0.339 | 0.909 | 0.875 | Uncertain | 2 | -2.991 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 0.432 | Likely Benign | 0.27 | Neutral | 0.971 | Probably Damaging | 0.773 | Possibly Damaging | 5.44 | Benign | 0.00 | Affected | 4.32 | 4 | 0.3017 | 0.2231 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.3386T>G | L1129R 2D AIThe SynGAP1 missense variant L1129R is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.876543 | Binding | 0.339 | 0.909 | 0.875 | -2.613 | Likely Benign | 0.442 | Ambiguous | Likely Benign | 0.376 | Likely Benign | -1.68 | Neutral | 0.005 | Benign | 0.007 | Benign | 5.48 | Benign | 0.00 | Affected | 0.1334 | 0.1636 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3388A>C | K1130Q 2D AIThe SynGAP1 missense variant K1130Q is reported in gnomAD (ID 6‑33443940‑A‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.863782 | Binding | 0.350 | 0.904 | 0.750 | 6-33443940-A-C | -3.548 | Likely Benign | 0.529 | Ambiguous | Likely Benign | 0.337 | Likely Benign | -1.25 | Neutral | 0.818 | Possibly Damaging | 0.355 | Benign | 5.44 | Benign | 0.00 | Affected | 4.32 | 4 | 0.4964 | 0.1756 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||||||||
| c.3388A>G | K1130E 2D AIThe SynGAP1 missense variant K1130E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a Likely Benign classification, while AlphaMissense‑Optimized remains Uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of tools and the SGM consensus support a benign impact, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.863782 | Binding | 0.350 | 0.904 | 0.750 | -4.998 | Likely Benign | 0.946 | Likely Pathogenic | Ambiguous | 0.422 | Likely Benign | -1.23 | Neutral | 0.649 | Possibly Damaging | 0.266 | Benign | 5.45 | Benign | 0.00 | Affected | 0.4251 | 0.1876 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3389A>C | K1130T 2D AIThe SynGAP1 missense variant K1130T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) which classifies it as Likely Benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is Uncertain, and the Foldetta protein‑folding stability assessment is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.863782 | Binding | 0.350 | 0.904 | 0.750 | -3.550 | Likely Benign | 0.829 | Likely Pathogenic | Ambiguous | 0.397 | Likely Benign | -1.38 | Neutral | 0.481 | Possibly Damaging | 0.157 | Benign | 5.47 | Benign | 0.00 | Affected | 0.2572 | 0.4349 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||||||||||||
| c.3389A>G | K1130R 2D AIThe SynGAP1 missense variant K1130R is reported in gnomAD (variant ID 6‑33443941‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.863782 | Binding | 0.350 | 0.904 | 0.750 | 6-33443941-A-G | -2.163 | Likely Benign | 0.155 | Likely Benign | Likely Benign | 0.187 | Likely Benign | -0.94 | Neutral | 0.014 | Benign | 0.008 | Benign | 5.44 | Benign | 0.00 | Affected | 4.32 | 4 | 0.5096 | 0.2074 | Weaken | 2 | 3 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||||
| c.3389A>T | K1130M 2D AIThe SynGAP1 K1130M missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which collectively suggest a likely benign outcome. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. Overall, the balance of evidence leans toward a benign classification, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for K1130M. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.863782 | Binding | 0.350 | 0.904 | 0.750 | -4.844 | Likely Benign | 0.858 | Likely Pathogenic | Ambiguous | 0.476 | Likely Benign | -1.62 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 5.42 | Benign | 0.00 | Affected | 0.1703 | 0.4407 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||||||||||||
| c.338G>A | G113E 2D AIThe SynGAP1 missense variant G113E is not reported in ClinVar (ClinVar ID: None) but is present in gnomAD (ID 6‑33432203‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) also as benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.639486 | Binding | 0.350 | 0.870 | 0.750 | 6-33432203-G-A | 1 | 6.20e-7 | -3.169 | Likely Benign | 0.669 | Likely Pathogenic | Likely Benign | 0.092 | Likely Benign | -0.34 | Neutral | 0.028 | Benign | 0.006 | Benign | 4.21 | Benign | 0.05 | Affected | 3.61 | 5 | 0.1464 | 0.4035 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.338G>C | G113A 2D AIThe SynGAP1 missense variant G113A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign. Foldetta results are not available for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.639486 | Binding | 0.350 | 0.870 | 0.750 | -3.552 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.61 | Neutral | 0.131 | Benign | 0.039 | Benign | 4.20 | Benign | 0.22 | Tolerated | 0.4059 | 0.4684 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.338G>T | G113V 2D AIThe SynGAP1 missense variant G113V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.639486 | Binding | 0.350 | 0.870 | 0.750 | -3.752 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -1.77 | Neutral | 0.838 | Possibly Damaging | 0.145 | Benign | 4.18 | Benign | 0.05 | Affected | 0.1316 | 0.4002 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3390G>C | K1130N 2D AIThe SynGAP1 missense variant K1130N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) which classifies it as Likely Benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. Overall, the majority of evidence points to a benign impact. The variant’s predicted benign status does not contradict ClinVar, as no ClinVar entry exists. Thus, based on current computational predictions, the K1130N variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.863782 | Binding | 0.350 | 0.904 | 0.750 | -4.822 | Likely Benign | 0.946 | Likely Pathogenic | Ambiguous | 0.336 | Likely Benign | -1.02 | Neutral | 0.818 | Possibly Damaging | 0.287 | Benign | 5.43 | Benign | 0.00 | Affected | 0.4119 | 0.2393 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3390G>T | K1130N 2D AIThe SynGAP1 missense variant K1130N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) which classifies it as Likely Benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. Overall, the majority of evidence points to a benign impact. The variant’s predicted benign status does not contradict ClinVar, as no ClinVar entry exists. Thus, based on current computational predictions, the K1130N variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.863782 | Binding | 0.350 | 0.904 | 0.750 | -4.822 | Likely Benign | 0.946 | Likely Pathogenic | Ambiguous | 0.336 | Likely Benign | -1.02 | Neutral | 0.818 | Possibly Damaging | 0.287 | Benign | 5.43 | Benign | 0.00 | Affected | 0.4119 | 0.2393 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3391C>A | P1131T 2D AIThe SynGAP1 missense variant P1131T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.855155 | Binding | 0.360 | 0.899 | 0.750 | -4.766 | Likely Benign | 0.219 | Likely Benign | Likely Benign | 0.313 | Likely Benign | -2.93 | Deleterious | 0.245 | Benign | 0.096 | Benign | 5.30 | Benign | 0.00 | Affected | 0.1353 | 0.6315 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3391C>G | P1131A 2D AIThe SynGAP1 missense variant P1131A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.855155 | Binding | 0.360 | 0.899 | 0.750 | -4.785 | Likely Benign | 0.171 | Likely Benign | Likely Benign | 0.207 | Likely Benign | -2.88 | Deleterious | 0.009 | Benign | 0.008 | Benign | 5.36 | Benign | 0.00 | Affected | 0.2977 | 0.5501 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3391C>T | P1131S 2D AIThe SynGAP1 missense variant P1131S is reported in gnomAD (variant ID 6‑33443943‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this is not contradictory to ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.855155 | Binding | 0.360 | 0.899 | 0.750 | 6-33443943-C-T | 1 | 6.72e-7 | -4.089 | Likely Benign | 0.246 | Likely Benign | Likely Benign | 0.209 | Likely Benign | -2.62 | Deleterious | 0.025 | Benign | 0.015 | Benign | 5.54 | Benign | 0.00 | Affected | 4.32 | 4 | 0.2936 | 0.5936 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.3392C>A | P1131H 2D AIThe SynGAP1 missense variant P1131H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the balance of evidence leans toward a benign classification, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.919029 | Disordered | 0.855155 | Binding | 0.360 | 0.899 | 0.750 | -5.207 | Likely Benign | 0.454 | Ambiguous | Likely Benign | 0.357 | Likely Benign | -3.50 | Deleterious | 0.971 | Probably Damaging | 0.750 | Possibly Damaging | 5.24 | Benign | 0.00 | Affected | 0.1519 | 0.5546 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3392C>G | P1131R 2D AIThe SynGAP1 P1131R missense variant has no ClinVar record and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta data are unavailable. With five benign versus four pathogenic calls and a benign result from the most accurate tool, the variant is most likely benign. This assessment does not contradict ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.919029 | Disordered | 0.855155 | Binding | 0.360 | 0.899 | 0.750 | -3.792 | Likely Benign | 0.702 | Likely Pathogenic | Likely Benign | 0.416 | Likely Benign | -2.58 | Deleterious | 0.918 | Possibly Damaging | 0.420 | Benign | 5.26 | Benign | 0.00 | Affected | 0.1252 | 0.4010 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||||
| c.3392C>T | P1131L 2D AIThe SynGAP1 missense variant P1131L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy AlphaMissense‑Optimized tool classifies the variant as benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic, with one uncertain). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.919029 | Disordered | 0.855155 | Binding | 0.360 | 0.899 | 0.750 | -5.267 | Likely Benign | 0.420 | Ambiguous | Likely Benign | 0.293 | Likely Benign | -3.62 | Deleterious | 0.002 | Benign | 0.005 | Benign | 5.26 | Benign | 0.00 | Affected | 0.2043 | 0.6998 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3394T>A | S1132T 2D AIThe SynGAP1 missense variant S1132T is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification—there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.845506 | Binding | 0.289 | 0.894 | 0.750 | -3.700 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.190 | Likely Benign | -0.71 | Neutral | 0.011 | Benign | 0.017 | Benign | 5.45 | Benign | 0.50 | Tolerated | 0.1552 | 0.6309 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3394T>C | S1132P 2D AIThe SynGAP1 missense variant S1132P is listed in ClinVar with an uncertain significance (ClinVar ID 1341927.0) and is present in the gnomAD database (gnomAD ID 6‑33443946‑T‑C). All available in‑silico predictors uniformly classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or likely benign outcomes. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the computational evidence strongly supports a benign effect, which is consistent with the ClinVar uncertain classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.845506 | Binding | 0.289 | 0.894 | 0.750 | Conflicting | 3 | 6-33443946-T-C | 1 | 6.74e-7 | -1.423 | Likely Benign | 0.144 | Likely Benign | Likely Benign | 0.301 | Likely Benign | 0.38 | Neutral | 0.003 | Benign | 0.006 | Benign | 5.40 | Benign | 0.28 | Tolerated | 4.32 | 4 | 0.2081 | 0.5700 | 1 | -1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||
| c.3394T>G | S1132A 2D AIThe SynGAP1 missense variant S1132A is not reported in ClinVar and is absent from gnomAD, indicating no documented population frequency. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.845506 | Binding | 0.289 | 0.894 | 0.750 | -3.401 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.213 | Likely Benign | -0.60 | Neutral | 0.061 | Benign | 0.013 | Benign | 5.47 | Benign | 0.55 | Tolerated | 0.4416 | 0.5683 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3395C>A | S1132Y 2D AIThe SynGAP1 missense variant S1132Y is listed in ClinVar as a benign alteration (ClinVar ID 845357.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence supports a benign classification, which aligns with the ClinVar status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.845506 | Binding | 0.289 | 0.894 | 0.750 | Likely Benign | 1 | -5.894 | Likely Benign | 0.392 | Ambiguous | Likely Benign | 0.401 | Likely Benign | -1.76 | Neutral | 0.500 | Possibly Damaging | 0.208 | Benign | 5.40 | Benign | 0.09 | Tolerated | 4.32 | 4 | 0.0889 | 0.5233 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||
| c.3395C>G | S1132C 2D AIThe SynGAP1 missense variant S1132C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S1132C, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.845506 | Binding | 0.289 | 0.894 | 0.750 | -6.668 | Likely Benign | 0.142 | Likely Benign | Likely Benign | 0.318 | Likely Benign | -1.76 | Neutral | 0.977 | Probably Damaging | 0.777 | Possibly Damaging | 5.39 | Benign | 0.06 | Tolerated | 0.1199 | 0.5888 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.3395C>T | S1132F 2D AIThe SynGAP1 missense variant S1132F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.845506 | Binding | 0.289 | 0.894 | 0.750 | -5.458 | Likely Benign | 0.385 | Ambiguous | Likely Benign | 0.310 | Likely Benign | -2.02 | Neutral | 0.006 | Benign | 0.006 | Benign | 5.41 | Benign | 0.05 | Affected | 0.0921 | 0.5506 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.3397A>C | I1133L 2D AIThe SynGAP1 missense variant I1133L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that I1133L is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | -1.147 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.202 | Likely Benign | -0.26 | Neutral | 0.000 | Benign | 0.001 | Benign | 5.48 | Benign | 0.23 | Tolerated | 0.0911 | 0.4302 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3397A>G | I1133V 2D AIThe SynGAP1 missense variant I1133V is listed in ClinVar as Benign (ClinVar ID 999690.0) and is present in the gnomAD database (gnomAD ID 6‑33443949‑A‑G). All evaluated in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign. Foldetta results are unavailable. Consequently, the variant is most likely benign, and this prediction aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | Benign | 1 | 6-33443949-A-G | 22 | 1.48e-5 | -3.362 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.180 | Likely Benign | 0.06 | Neutral | 0.007 | Benign | 0.007 | Benign | 5.47 | Benign | 0.58 | Tolerated | 4.32 | 3 | 0.1190 | 0.3985 | 4 | 3 | -0.3 | -14.03 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||
| c.3397A>T | I1133F 2D AIThe SynGAP1 missense variant I1133F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | -2.941 | Likely Benign | 0.164 | Likely Benign | Likely Benign | 0.272 | Likely Benign | -1.21 | Neutral | 0.290 | Benign | 0.124 | Benign | 5.45 | Benign | 0.05 | Affected | 0.0598 | 0.3522 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3398T>A | I1133N 2D AIThe SynGAP1 missense variant I1133N is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Pathogenicity is suggested only by polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates Likely Benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign effect for I1133N, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | -5.887 | Likely Benign | 0.520 | Ambiguous | Likely Benign | 0.290 | Likely Benign | -1.07 | Neutral | 0.453 | Possibly Damaging | 0.162 | Benign | 5.51 | Benign | 0.02 | Affected | 0.0961 | 0.1140 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3398T>C | I1133T 2D AIThe SynGAP1 missense variant I1133T has no ClinVar entry and is present in gnomAD (ID 6‑33443950‑T‑C). Consensus among most tools is benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict a benign effect, while AlphaMissense‑Default remains uncertain and no tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign,” and Foldetta’s protein‑folding stability analysis is unavailable. Overall, the evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | 6-33443950-T-C | -4.522 | Likely Benign | 0.563 | Ambiguous | Likely Benign | 0.275 | Likely Benign | -0.30 | Neutral | 0.026 | Benign | 0.030 | Benign | 5.50 | Benign | 0.18 | Tolerated | 4.32 | 3 | 0.1007 | 0.1975 | -1 | 0 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||||||
| c.3398T>G | I1133S 2D AIThe SynGAP1 missense variant I1133S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective evidence strongly supports a benign impact for I1133S, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | -4.034 | Likely Benign | 0.492 | Ambiguous | Likely Benign | 0.186 | Likely Benign | -0.60 | Neutral | 0.007 | Benign | 0.016 | Benign | 5.70 | Benign | 0.08 | Tolerated | 0.2762 | 0.1110 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3399C>G | I1133M 2D AIThe SynGAP1 missense variant I1133M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | -3.409 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.223 | Likely Benign | -0.21 | Neutral | 0.016 | Benign | 0.009 | Benign | 5.45 | Benign | 0.06 | Tolerated | 0.0764 | 0.3685 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3400A>C | T1134P 2D AIThe SynGAP1 missense variant T1134P is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.813034 | Binding | 0.335 | 0.885 | 0.875 | -2.993 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.164 | Likely Benign | -0.57 | Neutral | 0.013 | Benign | 0.022 | Benign | 5.41 | Benign | 0.07 | Tolerated | 0.2001 | 0.5165 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3400A>G | T1134A 2D AIThe SynGAP1 missense variant T1134A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.813034 | Binding | 0.335 | 0.885 | 0.875 | -3.912 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.172 | Likely Benign | -0.71 | Neutral | 0.036 | Benign | 0.026 | Benign | 5.70 | Benign | 0.31 | Tolerated | 0.3756 | 0.4424 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3400A>T | T1134S 2D AIThe SynGAP1 missense variant T1134S is not reported in ClinVar and is absent from gnomAD, indicating no documented population frequency. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool predicts pathogenicity. The high‑accuracy consensus, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), also reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.813034 | Binding | 0.335 | 0.885 | 0.875 | -3.015 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.220 | Likely Benign | -0.42 | Neutral | 0.007 | Benign | 0.009 | Benign | 5.47 | Benign | 0.09 | Tolerated | 0.3189 | 0.4666 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3401C>A | T1134N 2D AIThe SynGAP1 missense variant T1134N is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that T1134N is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.813034 | Binding | 0.335 | 0.885 | 0.875 | -4.368 | Likely Benign | 0.176 | Likely Benign | Likely Benign | 0.193 | Likely Benign | -0.34 | Neutral | 0.001 | Benign | 0.001 | Benign | 5.42 | Benign | 0.04 | Affected | 0.1386 | 0.4870 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||||||||||||
| c.3401C>G | T1134S 2D AIThe SynGAP1 missense variant T1134S is not reported in ClinVar and is absent from gnomAD, indicating no documented population frequency. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool predicts pathogenicity. The high‑accuracy consensus, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), also reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.813034 | Binding | 0.335 | 0.885 | 0.875 | -3.015 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.228 | Likely Benign | -0.42 | Neutral | 0.007 | Benign | 0.009 | Benign | 5.47 | Benign | 0.09 | Tolerated | 0.3189 | 0.4666 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3401C>T | T1134I 2D AIThe SynGAP1 missense variant T1134I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.813034 | Binding | 0.335 | 0.885 | 0.875 | -4.072 | Likely Benign | 0.416 | Ambiguous | Likely Benign | 0.242 | Likely Benign | -1.93 | Neutral | 0.453 | Possibly Damaging | 0.162 | Benign | 5.52 | Benign | 0.44 | Tolerated | 0.1071 | 0.5447 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||||||||||||
Found 8751 rows. Show 800 rows per page. Page 4/11 « Previous | Next »