
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain and Structure information: based on WT protein | Annotated databases | Deep learning-based pathogenicity predictions | Folding stability-based pathogenicity predictions | Sequence/structure-based pathogenicity predictions | Phase Separation | Evolutionary/physical properties | Molecular Dynamics-based analysis | DOI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | IUPred2 | ANCHOR2 | AlphaFold | MobiDB | PhosphoSitePlus | ClinVar | gnomAD | ESM1b | AlphaMissense | FoldX | Rosetta | Foldetta | PremPS | REVEL | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PSMutPred | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | |||||||||||||||||||||||||||||||||||||||||||||
| Score | Prediction | Score | Prediction | pLDDT | disorder | disorder | LTP | HTP | KL | PTM | Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | IP RF | SP RF | Prediction | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.1123G>T | G375W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G375W is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33438028‑G‑T). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of evidence, including the high‑accuracy tools, points to a pathogenic effect for G375W. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.604312 | Disordered | 0.428340 | Uncertain | 0.301 | 0.836 | 0.625 | 6-33438028-G-T | -9.654 | Likely Pathogenic | 0.464 | Ambiguous | Likely Benign | 4.33 | Destabilizing | 2.0 | 7.01 | Destabilizing | 5.67 | Destabilizing | 0.22 | Likely Benign | 0.450 | Likely Benign | -1.26 | Neutral | 0.992 | Probably Damaging | 0.869 | Possibly Damaging | 1.31 | Pathogenic | 0.01 | Affected | 4.32 | 12 | 0.0992 | 0.4368 | -2 | -7 | -0.5 | 129.16 | |||||||||||||||||||||||||||
| c.1141G>T | G381W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G381W is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33438046‑G‑T). Prediction tools that agree on a benign effect include premPS, PROVEAN, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments give AlphaMissense‑Optimized a benign call, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) a pathogenic call, and Foldetta (combining FoldX‑MD and Rosetta outputs) a pathogenic call. No prediction or folding result is missing or inconclusive. Overall, the majority of tools and the high‑accuracy methods indicate a pathogenic impact. Thus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.724957 | Disordered | 0.431692 | Uncertain | 0.301 | 0.951 | 0.750 | 6-33438046-G-T | 1 | 6.24e-7 | -10.173 | Likely Pathogenic | 0.438 | Ambiguous | Likely Benign | 8.81 | Destabilizing | 2.0 | 3.17 | Destabilizing | 5.99 | Destabilizing | 0.02 | Likely Benign | 0.576 | Likely Pathogenic | -1.04 | Neutral | 0.996 | Probably Damaging | 0.920 | Probably Damaging | 1.31 | Pathogenic | 0.01 | Affected | 4.32 | 9 | 0.0993 | 0.4000 | -2 | -7 | -0.5 | 129.16 | |||||||||||||||||||||||||
| c.1147G>T | G383W 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G383W is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33438052‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as benign, and Foldetta as pathogenic. Because the majority of conventional predictors favor pathogenicity while the high‑accuracy subset is split, the overall evidence leans toward a pathogenic interpretation. This conclusion does not conflict with the ClinVar uncertain status, which reflects the current lack of definitive clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.728858 | Disordered | 0.429104 | Uncertain | 0.296 | 0.949 | 0.750 | Uncertain | 1 | 6-33438052-G-T | 1 | 6.22e-7 | -10.161 | Likely Pathogenic | 0.439 | Ambiguous | Likely Benign | 5.81 | Destabilizing | 3.6 | 4.44 | Destabilizing | 5.13 | Destabilizing | 0.08 | Likely Benign | 0.469 | Likely Benign | -1.01 | Neutral | 0.959 | Probably Damaging | 0.704 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 4.32 | 7 | 0.0972 | 0.3785 | -2 | -7 | -0.5 | 129.16 | |||||||||||||||||||||||
| c.31G>T | G11W 2D AIThe SynGAP1 missense variant G11W is catalogued in gnomAD (ID 6‑33420295‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | 6-33420295-G-T | -5.819 | Likely Benign | 0.403 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -0.67 | Neutral | 0.959 | Probably Damaging | 0.318 | Benign | 3.87 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0747 | 0.4731 | -2 | -7 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||||||||
| c.3346G>T | G1116W 2D AIThe SynGAP1 missense variant G1116W is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443898‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta’s protein‑folding stability result is unavailable. Taken together, the majority of reliable predictors and the consensus high‑accuracy tools indicate a benign effect. This conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | 6-33443898-G-T | -9.448 | Likely Pathogenic | 0.356 | Ambiguous | Likely Benign | 0.405 | Likely Benign | -1.27 | Neutral | 0.996 | Probably Damaging | 0.946 | Probably Damaging | 4.04 | Benign | 0.01 | Affected | 4.32 | 2 | 0.0776 | 0.4046 | -2 | -7 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||
| c.3355G>T | G1119W 2D AIThe SynGAP1 missense variant G1119W is reported in gnomAD (ID 6‑33443907‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | 6-33443907-G-T | -10.904 | Likely Pathogenic | 0.336 | Likely Benign | Likely Benign | 0.386 | Likely Benign | -1.19 | Neutral | 0.997 | Probably Damaging | 0.949 | Probably Damaging | 3.90 | Benign | 0.02 | Affected | 4.32 | 2 | 0.0948 | 0.4059 | -2 | -7 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||||||||
| c.3379G>T | G1127W 2D AIThe SynGAP1 missense variant G1127W is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443931‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta’s protein‑folding stability result is unavailable. Overall, the balance of evidence, particularly from the high‑accuracy tools, indicates that the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | 6-33443931-G-T | 3 | 2.01e-6 | -9.850 | Likely Pathogenic | 0.418 | Ambiguous | Likely Benign | 0.394 | Likely Benign | -1.36 | Neutral | 0.983 | Probably Damaging | 0.665 | Possibly Damaging | 4.79 | Benign | 0.03 | Affected | 4.32 | 4 | 0.0782 | 0.4032 | -2 | -7 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||
| c.1003C>T | R335C 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant R335C is listed in ClinVar with an uncertain significance (ClinVar ID 2835865.0) and is present in gnomAD (ID 6‑33437908‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL and premPS, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive are AlphaMissense‑Optimized, FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect. This conclusion aligns with the ClinVar designation of uncertain significance, which does not contradict the prediction that the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.305330 | Structured | 0.331028 | Uncertain | 0.483 | 0.428 | 0.500 | Uncertain | 1 | 6-33437908-C-T | 1 | 6.20e-7 | -14.354 | Likely Pathogenic | 0.938 | Likely Pathogenic | Ambiguous | 0.53 | Ambiguous | 0.1 | 0.85 | Ambiguous | 0.69 | Ambiguous | 0.46 | Likely Benign | 0.277 | Likely Benign | -5.69 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.67 | Pathogenic | 0.01 | Affected | 3.38 | 22 | 0.2882 | 0.3290 | -3 | -4 | 7.0 | -53.05 | ||||||||||||||||||||||
| c.1190G>T | C397F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397F is reported in gnomAD (6-33438095-G-T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, Rosetta, and the protein‑folding stability method Foldetta. FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as benign, while Foldetta indicates a pathogenic effect. Because the majority of tools (nine benign vs. four pathogenic) lean toward a benign outcome and the high‑accuracy consensus is split, the variant is most likely benign. This assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | 6-33438095-G-T | 1 | 6.20e-7 | -6.571 | Likely Benign | 0.194 | Likely Benign | Likely Benign | 1.31 | Ambiguous | 1.7 | 3.61 | Destabilizing | 2.46 | Destabilizing | 0.12 | Likely Benign | 0.493 | Likely Benign | -1.95 | Neutral | 0.952 | Possibly Damaging | 0.497 | Possibly Damaging | 4.65 | Benign | 0.07 | Tolerated | 3.41 | 15 | 0.1426 | 0.5231 | -2 | -4 | 0.3 | 44.04 | ||||||||||||||||||||||||
| c.1427T>G | F476C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F476C is catalogued in gnomAD (ID 6‑33438459‑T‑G) but has no ClinVar entry. Functional prediction tools split in two groups: benign predictions come from REVEL, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, seven tools predict pathogenicity versus six predicting benign, with no ClinVar evidence to contradict this assessment. Thus, the variant is most likely pathogenic based on the current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.257454 | Structured | 0.397815 | Uncertain | 0.821 | 0.250 | 0.000 | 6-33438459-T-G | -9.270 | Likely Pathogenic | 0.745 | Likely Pathogenic | Likely Benign | 2.05 | Destabilizing | 0.1 | 2.62 | Destabilizing | 2.34 | Destabilizing | -0.30 | Likely Benign | 0.280 | Likely Benign | 2.69 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.46 | Benign | 0.83 | Tolerated | 3.40 | 22 | 0.2051 | 0.1251 | -2 | -4 | -0.3 | -44.04 | |||||||||||||||||||||||||||
| c.1513T>G | Y505D 2D 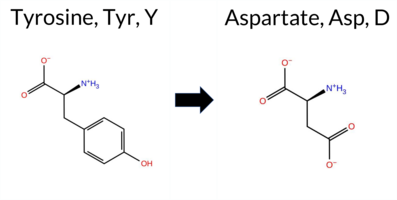 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y505D is listed in ClinVar as Pathogenic (ClinVar ID 3172759.0) and is not reported in gnomAD. Prediction tools that indicate a benign effect are limited to FATHMM, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support this view: AlphaMissense‑Optimized scores the variant as Pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a Likely Pathogenic verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as Pathogenic. Based on the overwhelming agreement among these predictions, the variant is most likely pathogenic, and this conclusion aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.292227 | Uncertain | 0.909 | 0.188 | 0.000 | Likely Pathogenic | 1 | -14.078 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 4.98 | Destabilizing | 0.1 | 4.72 | Destabilizing | 4.85 | Destabilizing | 2.49 | Destabilizing | 0.718 | Likely Pathogenic | -9.95 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.60 | Benign | 0.00 | Affected | 3.37 | 35 | 0.3940 | 0.0612 | -3 | -4 | -2.2 | -48.09 | |||||||||||||||||||||||||
| c.1543C>T | R515C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R515C is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33438786‑C‑T). Prediction tools that indicate a benign effect include only AlphaMissense‑Optimized. All other evaluated predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—classify the variant as pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No other high‑accuracy tools provide a definitive prediction. Based on the overall consensus, the variant is most likely pathogenic, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.055536 | Structured | 0.191256 | Uncertain | 0.924 | 0.275 | 0.000 | Uncertain | 1 | 6-33438786-C-T | 1 | 6.20e-7 | -10.973 | Likely Pathogenic | 0.628 | Likely Pathogenic | Likely Benign | 1.00 | Ambiguous | 0.0 | 1.13 | Ambiguous | 1.07 | Ambiguous | 0.72 | Ambiguous | 0.691 | Likely Pathogenic | -5.49 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.36 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.3055 | 0.1546 | -3 | -4 | 7.0 | -53.05 | ||||||||||||||||||||||
| c.1726T>C | C576R 2D 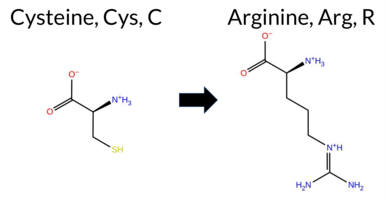 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C576R (ClinVar ID 2780076) is reported as Pathogenic and is not present in gnomAD. Prediction tools that classify the variant as benign include only FATHMM. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy assessments further support this: AlphaMissense‑Optimized returns a pathogenic score; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenicity. No predictions or stability results are missing or inconclusive. Based on the overwhelming agreement among predictive tools and the high‑accuracy methods, the variant is most likely pathogenic, consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.017684 | Uncertain | 0.913 | 0.245 | 0.000 | Pathogenic/Likely path. | 2 | -14.886 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 7.20 | Destabilizing | 1.0 | 4.09 | Destabilizing | 5.65 | Destabilizing | 1.64 | Destabilizing | 0.579 | Likely Pathogenic | -10.88 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.38 | Benign | 0.00 | Affected | 3.37 | 35 | 0.1887 | 0.1279 | -3 | -4 | -7.0 | 53.05 | |||||||||||||||||||||||||
| c.1790T>G | F597C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F597C is reported in gnomAD (variant ID 6-33440842‑T‑G) but has no ClinVar entry. Functional prediction tools uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool in the dataset predicts a benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Based on the unanimous pathogenic predictions and the absence of any benign calls, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | 6-33440842-T-G | 1 | 6.19e-7 | -12.099 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 3.77 | Destabilizing | 0.2 | 4.17 | Destabilizing | 3.97 | Destabilizing | 1.97 | Destabilizing | 0.953 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -2.19 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.2618 | 0.0783 | -2 | -4 | -0.3 | -44.04 | ||||||||||||||||||||||||
| c.1907T>G | F636C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F636C is reported in gnomAD (ID 6‑33440959‑T‑G) but has no ClinVar entry. Prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). Only FATHMM predicts a benign outcome; FoldX is uncertain and therefore not counted as evidence. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming majority of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | 6-33440959-T-G | 3 | 1.86e-6 | -13.287 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | 1.74 | Ambiguous | 0.1 | 2.65 | Destabilizing | 2.20 | Destabilizing | 1.22 | Destabilizing | 0.612 | Likely Pathogenic | -7.67 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.40 | Benign | 0.04 | Affected | 3.37 | 34 | 0.2301 | 0.0830 | -2 | -4 | -0.3 | -44.04 | ||||||||||||||||||||||||
| c.2428C>T | R810C 2D AIThe SynGAP1 missense variant R810C is listed in gnomAD (6‑33442980‑C‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.486429 | Structured | 0.851848 | Binding | 0.263 | 0.907 | 0.375 | 6-33442980-C-T | 2 | 1.24e-6 | -8.925 | Likely Pathogenic | 0.839 | Likely Pathogenic | Ambiguous | 0.245 | Likely Benign | -4.91 | Deleterious | 1.000 | Probably Damaging | 0.991 | Probably Damaging | 2.32 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3517 | 0.4263 | -3 | -4 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.2560C>T | R854C 2D AIThe SynGAP1 missense variant R854C is listed in ClinVar (ID 2896479) with an uncertain significance designation and is present in gnomAD (variant ID 6‑33443112‑C‑T). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT each predict a pathogenic impact. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign effect for R854C, which does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.488780 | Uncertain | 0.277 | 0.815 | 0.750 | Uncertain | 2 | 6-33443112-C-T | 3 | 1.86e-6 | -5.082 | Likely Benign | 0.170 | Likely Benign | Likely Benign | 0.174 | Likely Benign | -2.48 | Neutral | 1.000 | Probably Damaging | 0.947 | Probably Damaging | 4.05 | Benign | 0.01 | Affected | 3.88 | 3 | 0.3275 | 0.4217 | -3 | -4 | 7.0 | -53.05 | ||||||||||||||||||||||||||||||||
| c.3016T>G | Y1006D 2D AIThe SynGAP1 missense variant Y1006D is catalogued in gnomAD (variant ID 6‑33443568‑T‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM. Those that predict a pathogenic impact are PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” AlphaMissense‑Optimized is uncertain, and no Foldetta (FoldX‑MD/ Rosetta) stability data are available. Considering the high‑accuracy consensus, the variant is most likely benign; this assessment does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.930554 | Binding | 0.264 | 0.896 | 0.750 | 6-33443568-T-G | -5.296 | Likely Benign | 0.898 | Likely Pathogenic | Ambiguous | 0.196 | Likely Benign | -1.53 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.73 | Benign | 0.56 | Tolerated | 3.77 | 5 | 0.3776 | 0.1094 | -3 | -4 | -2.2 | -48.09 | ||||||||||||||||||||||||||||||||||||
| c.310C>T | R104C 2D AIThe SynGAP1 missense variant R104C has no ClinVar entry and is present in gnomAD (ID 6‑33432175‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | 6-33432175-C-T | 2 | 1.24e-6 | -5.716 | Likely Benign | 0.475 | Ambiguous | Likely Benign | 0.109 | Likely Benign | -1.41 | Neutral | 0.993 | Probably Damaging | 0.446 | Benign | 3.99 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2954 | 0.3292 | -3 | -4 | 7.0 | -53.05 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||||
| c.3307C>T | R1103C 2D AISynGAP1 missense variant R1103C is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33443859‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic). AlphaMissense‑Optimized reports a benign outcome, while Foldetta results are unavailable. Overall, the balance of evidence favors a pathogenic interpretation, which is in contrast to the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.945666 | Disordered | 0.957363 | Binding | 0.328 | 0.862 | 0.875 | Uncertain | 1 | 6-33443859-C-T | 6 | 3.92e-6 | -2.440 | Likely Benign | 0.246 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -3.01 | Deleterious | 0.996 | Probably Damaging | 0.787 | Possibly Damaging | 2.41 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.3376 | 0.4121 | -3 | -4 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.3826G>T | D1276Y 2D 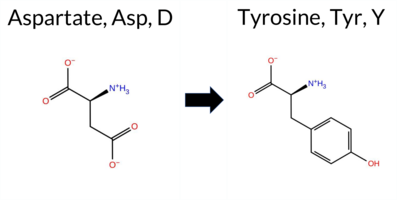 AIThe SynGAP1 missense variant D1276Y is catalogued in gnomAD (ID 6‑33447874‑G‑T) but has no ClinVar entry. Functional prediction tools fall into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus indicates a likely pathogenic effect; Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar status, as none is currently reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.666105 | Disordered | 0.802156 | Binding | 0.636 | 0.705 | 0.625 | 6-33447874-G-T | -1.558 | Likely Benign | 0.768 | Likely Pathogenic | Likely Benign | 0.325 | Likely Benign | -6.66 | Deleterious | 0.999 | Probably Damaging | 0.928 | Probably Damaging | 1.18 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.0385 | 0.5082 | -3 | -4 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||
| c.4024G>T | D1342Y 2D AIThe SynGAP1 missense variant D1342Y is reported in gnomAD (6‑33451898‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions are made by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.921076 | Disordered | 0.981682 | Binding | 0.316 | 0.678 | 0.875 | 6-33451898-G-T | -4.108 | Likely Benign | 0.398 | Ambiguous | Likely Benign | 0.095 | Likely Benign | -1.34 | Neutral | 0.939 | Possibly Damaging | 0.496 | Possibly Damaging | 3.98 | Benign | 0.01 | Affected | 4.32 | 4 | 0.0868 | 0.5377 | -3 | -4 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||
| c.469C>T | R157C 2D AIThe SynGAP1 missense variant R157C is listed in gnomAD (ID 6‑33432766‑C‑T) but has no ClinVar entry. Functional prediction tools split in two groups: benign predictions come from REVEL and FATHMM, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (which is “Likely Pathogenic”). AlphaMissense‑Optimized returns an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. High‑accuracy assessments therefore indicate a likely pathogenic consensus from SGM‑Consensus, an uncertain AlphaMissense‑Optimized score, and no Foldetta data. Overall, the majority of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.523978 | Binding | 0.306 | 0.777 | 0.375 | 6-33432766-C-T | 7 | 4.34e-6 | -11.524 | Likely Pathogenic | 0.880 | Likely Pathogenic | Ambiguous | 0.237 | Likely Benign | -4.02 | Deleterious | 1.000 | Probably Damaging | 0.990 | Probably Damaging | 3.77 | Benign | 0.00 | Affected | 3.74 | 4 | 0.3696 | 0.2092 | -3 | -4 | 7.0 | -53.05 | ||||||||||||||||||||||||||||||||||
| c.653T>G | F218C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F218C is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33435295‑T‑G). Prediction tools that agree on a benign effect include only FATHMM, whereas the majority of tools (REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default) predict a pathogenic impact. Results that are uncertain or unavailable are FoldX, ESM1b, AlphaMissense‑Optimized, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic prediction (2 pathogenic vs. 1 benign votes); and Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for F218C, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.281712 | Structured | 0.408725 | Uncertain | 0.848 | 0.272 | 0.000 | 6-33435295-T-G | 1 | 6.20e-7 | -7.234 | In-Between | 0.948 | Likely Pathogenic | Ambiguous | 1.49 | Ambiguous | 0.1 | 2.20 | Destabilizing | 1.85 | Ambiguous | 1.02 | Destabilizing | 0.744 | Likely Pathogenic | -4.92 | Deleterious | 0.994 | Probably Damaging | 0.667 | Possibly Damaging | 5.78 | Benign | 0.03 | Affected | 3.41 | 13 | 0.2330 | 0.1321 | -2 | -4 | -0.3 | -44.04 | |||||||||||||||||||||||||
| c.67G>T | D23Y 2D AIThe SynGAP1 D23Y missense variant is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33420331‑G‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic impact. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.557691 | Disordered | 0.440341 | Uncertain | 0.369 | 0.892 | 0.375 | 6-33420331-G-T | -4.191 | Likely Benign | 0.729 | Likely Pathogenic | Likely Benign | 0.142 | Likely Benign | -2.87 | Deleterious | 0.972 | Probably Damaging | 0.861 | Possibly Damaging | 3.46 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1013 | 0.7951 | -3 | -4 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||
| c.772C>T | R258C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R258C missense variant is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33437677‑C‑T). Prediction tools that agree on a benign effect include only FATHMM. All other evaluated predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—indicate a pathogenic or likely pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, which does not contradict its current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.295083 | Structured | 0.293667 | Uncertain | 0.894 | 0.260 | 0.250 | Uncertain | 1 | 6-33437677-C-T | 1 | 6.20e-7 | -10.285 | Likely Pathogenic | 0.790 | Likely Pathogenic | Ambiguous | 1.17 | Ambiguous | 0.4 | 1.76 | Ambiguous | 1.47 | Ambiguous | 0.87 | Ambiguous | 0.771 | Likely Pathogenic | -6.79 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 5.77 | Benign | 0.00 | Affected | 3.39 | 15 | 0.3307 | 0.3411 | -3 | -4 | 7.0 | -53.05 | ||||||||||||||||||||||
| c.961C>T | R321C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R321C is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33437866‑C‑T). Prediction tools that agree on a benign effect include REVEL, premPS, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Five tools (SGM‑Consensus, FoldX, Rosetta, AlphaMissense‑Default, and Foldetta) report uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of predictions (six out of eleven) support a pathogenic impact, while three support benign and five are inconclusive. Thus, the variant is most likely pathogenic based on current computational evidence, and this does not contradict its ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.175930 | Structured | 0.423273 | Uncertain | 0.931 | 0.297 | 0.125 | Conflicting | 2 | 6-33437866-C-T | 9 | 5.58e-6 | -10.025 | Likely Pathogenic | 0.387 | Ambiguous | Likely Benign | 0.57 | Ambiguous | 0.1 | 0.56 | Ambiguous | 0.57 | Ambiguous | 0.18 | Likely Benign | 0.495 | Likely Benign | -4.59 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.89 | Pathogenic | 0.01 | Affected | 3.38 | 23 | 0.3313 | 0.2516 | -3 | -4 | 7.0 | -53.05 | ||||||||||||||||||||||
| c.985C>T | R329C 2D 3DClick to see structure in 3D Viewer AISynGAP1 R329C is listed in ClinVar with an uncertain significance and is present in gnomAD (variant ID 6‑33437890‑C‑T). Functional prediction tools cluster into two groups: benign predictions from REVEL, FoldX, Rosetta, Foldetta, and FATHMM; pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta) as benign. Overall, the majority of conventional predictors lean toward pathogenicity, whereas the high‑accuracy Foldetta result is benign, leaving the variant’s effect ambiguous. Based on the prevailing predictions, the variant is most likely pathogenic, which contrasts with its ClinVar uncertain designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.384043 | Structured | 0.376086 | Uncertain | 0.887 | 0.479 | 0.250 | Uncertain | 1 | 6-33437890-C-T | 2 | 1.24e-6 | -9.433 | Likely Pathogenic | 0.865 | Likely Pathogenic | Ambiguous | 0.44 | Likely Benign | 0.1 | 0.40 | Likely Benign | 0.42 | Likely Benign | 0.69 | Ambiguous | 0.313 | Likely Benign | -5.70 | Deleterious | 0.999 | Probably Damaging | 0.825 | Possibly Damaging | 3.98 | Benign | 0.00 | Affected | 3.41 | 15 | 0.3553 | 0.2921 | -3 | -4 | 7.0 | -53.05 | ||||||||||||||||||||||
| c.100T>G | Y34D 2D AIThe SynGAP1 missense variant Y34D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized independently predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation; there is no contradictory ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.435847 | Uncertain | 0.303 | 0.855 | 0.375 | -2.653 | Likely Benign | 0.357 | Ambiguous | Likely Benign | 0.199 | Likely Benign | -1.20 | Neutral | 0.824 | Possibly Damaging | 0.828 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 0.4092 | 0.1322 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.1012G>T | D338Y 2D AIThe SynGAP1 D338Y missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools largely agree on a deleterious effect: premPS is the sole predictor labeling it benign, whereas the remaining seven tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—classify it as pathogenic. Predictions from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive. High‑accuracy assessments further support a pathogenic interpretation: the SGM Consensus (derived from a unanimous majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, AlphaMissense‑Optimized remains uncertain, and Foldetta is also uncertain. Overall, the preponderance of evidence indicates that D338Y is most likely pathogenic, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -14.190 | Likely Pathogenic | 0.945 | Likely Pathogenic | Ambiguous | 1.10 | Ambiguous | 1.3 | 0.91 | Ambiguous | 1.01 | Ambiguous | 0.22 | Likely Benign | 0.552 | Likely Pathogenic | -6.49 | Deleterious | 0.989 | Probably Damaging | 0.832 | Possibly Damaging | 1.66 | Pathogenic | 0.00 | Affected | 0.0687 | 0.5609 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1013A>T | D338V 2D 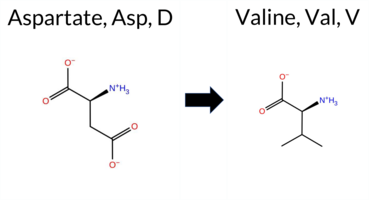 3DClick to see structure in 3D Viewer AIThe SynGAP1 D338V missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only premPS, whereas the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict pathogenicity. High‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain; the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. No prediction or folding‑stability result is missing or inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -11.494 | Likely Pathogenic | 0.927 | Likely Pathogenic | Ambiguous | 1.64 | Ambiguous | 0.2 | 1.08 | Ambiguous | 1.36 | Ambiguous | 0.23 | Likely Benign | 0.553 | Likely Pathogenic | -6.79 | Deleterious | 0.891 | Possibly Damaging | 0.492 | Possibly Damaging | 1.73 | Pathogenic | 0.01 | Affected | 0.0879 | 0.5745 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1021G>T | G341C 2D  AIThe SynGAP1 missense variant G341C has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include FoldX, Foldetta, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (likely benign). Tools that predict a pathogenic effect are REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; Rosetta remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall consensus of the available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | -6.156 | Likely Benign | 0.190 | Likely Benign | Likely Benign | 0.42 | Likely Benign | 1.2 | -0.92 | Ambiguous | -0.25 | Likely Benign | 0.24 | Likely Benign | 0.501 | Likely Pathogenic | -2.40 | Neutral | 0.997 | Probably Damaging | 0.870 | Possibly Damaging | 0.34 | Pathogenic | 0.01 | Affected | 0.1285 | 0.3650 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1022G>T | G341V 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant G341V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, polyPhen‑2 HumVar, and the SGM‑Consensus score (derived from a majority of benign calls among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as uncertain. No evidence from FoldX or premPS is available. Overall, the majority of predictions support a benign classification, and this is consistent with the lack of ClinVar annotation. Therefore, the variant is most likely benign and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | -5.371 | Likely Benign | 0.247 | Likely Benign | Likely Benign | 0.86 | Ambiguous | 0.3 | -2.24 | Stabilizing | -0.69 | Ambiguous | -0.50 | Ambiguous | 0.459 | Likely Benign | -2.29 | Neutral | 0.801 | Possibly Damaging | 0.192 | Benign | 0.42 | Pathogenic | 0.05 | Affected | 0.1019 | 0.3649 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1024T>G | Y342D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y342D is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include premPS and SIFT, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict it to be pathogenic. FoldX reports an uncertain effect, while Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta is pathogenic. Based on the preponderance of pathogenic predictions and the absence of benign evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.366687 | Structured | 0.408200 | Uncertain | 0.866 | 0.487 | 0.250 | -10.940 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 1.60 | Ambiguous | 0.1 | 2.70 | Destabilizing | 2.15 | Destabilizing | 0.13 | Likely Benign | 0.668 | Likely Pathogenic | -7.19 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.71 | Pathogenic | 0.07 | Tolerated | 0.3925 | 0.0862 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1028T>A | V343D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343D is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, PROVEAN, ESM1b, FATHMM, premPS, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. Rosetta and Foldetta, which evaluate protein‑folding stability, also predict a pathogenic outcome, while FoldX remains uncertain. No tool in the dataset predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta is pathogenic. Consequently, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -15.523 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.57 | Ambiguous | 0.2 | 3.40 | Destabilizing | 2.49 | Destabilizing | 1.73 | Destabilizing | 0.530 | Likely Pathogenic | -5.62 | Deleterious | 0.996 | Probably Damaging | 0.930 | Probably Damaging | 1.59 | Pathogenic | 0.00 | Affected | 0.1781 | 0.2261 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1028T>G | V343G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—classify the variant as pathogenic. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Thus, the preponderance of evidence indicates that V343G is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -11.332 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 2.49 | Destabilizing | 0.1 | 4.40 | Destabilizing | 3.45 | Destabilizing | 1.68 | Destabilizing | 0.421 | Likely Benign | -5.84 | Deleterious | 0.898 | Possibly Damaging | 0.996 | Probably Damaging | 1.60 | Pathogenic | 0.00 | Affected | 0.1982 | 0.3341 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1030G>T | G344C 2D AIThe SynGAP1 missense variant G344C is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic outcome include SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy methods specifically show pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No tool predicts a benign effect. Based on the unanimous pathogenic predictions and the absence of any ClinVar or gnomAD evidence, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.264545 | Structured | 0.368110 | Uncertain | 0.913 | 0.485 | 0.250 | -12.880 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 8.52 | Destabilizing | 2.9 | 7.52 | Destabilizing | 8.02 | Destabilizing | 0.59 | Ambiguous | 0.794 | Likely Pathogenic | -8.02 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -0.47 | Pathogenic | 0.01 | Affected | 0.1184 | 0.4788 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1031G>T | G344V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G344V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while the only inconclusive result is premPS, which is listed as uncertain. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic status; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports pathogenic. Consequently, the variant is most likely pathogenic, and this prediction is consistent with the absence of a ClinVar entry (no contradiction). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.264545 | Structured | 0.368110 | Uncertain | 0.913 | 0.485 | 0.250 | -14.913 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 13.33 | Destabilizing | 1.5 | 14.51 | Destabilizing | 13.92 | Destabilizing | 0.53 | Ambiguous | 0.889 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -0.48 | Pathogenic | 0.03 | Affected | 0.1244 | 0.4744 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1034T>C | L345P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L345P is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.260850 | Structured | 0.354989 | Uncertain | 0.936 | 0.478 | 0.125 | -10.994 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.33 | Destabilizing | 0.2 | 3.26 | Destabilizing | 3.30 | Destabilizing | 1.27 | Destabilizing | 0.335 | Likely Benign | -4.80 | Deleterious | 0.998 | Probably Damaging | 0.889 | Possibly Damaging | 1.70 | Pathogenic | 0.02 | Affected | 0.3581 | 0.1342 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1037T>G | V346G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V346G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). All available in‑silico predictors classify it as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a pathogenic verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports pathogenic. With all evidence converging on a deleterious impact and no ClinVar annotation to contradict, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.260850 | Structured | 0.350921 | Uncertain | 0.949 | 0.461 | 0.000 | -12.779 | Likely Pathogenic | 0.969 | Likely Pathogenic | Likely Pathogenic | 4.24 | Destabilizing | 0.2 | 4.62 | Destabilizing | 4.43 | Destabilizing | 2.15 | Destabilizing | 0.667 | Likely Pathogenic | -6.03 | Deleterious | 0.991 | Probably Damaging | 0.999 | Probably Damaging | 1.50 | Pathogenic | 0.00 | Affected | 0.2378 | 0.2522 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1043T>G | V348G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V348G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods reinforce this assessment: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates a pathogenic effect. No prediction or stability result is missing or inconclusive. Based on the overwhelming agreement among pathogenic predictions and the lack of a benign consensus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.170161 | Structured | 0.346556 | Uncertain | 0.951 | 0.414 | 0.000 | -12.793 | Likely Pathogenic | 0.964 | Likely Pathogenic | Likely Pathogenic | 3.97 | Destabilizing | 0.2 | 5.70 | Destabilizing | 4.84 | Destabilizing | 2.23 | Destabilizing | 0.419 | Likely Benign | -6.18 | Deleterious | 0.637 | Possibly Damaging | 0.989 | Probably Damaging | 1.56 | Pathogenic | 0.00 | Affected | 0.2229 | 0.2082 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1046C>T | P349L 2D  AIThe SynGAP1 missense variant P349L is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and AlphaMissense‑Optimized, whereas the majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy methods give conflicting results: AlphaMissense‑Optimized reports a benign outcome, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Other stability‑based predictors (FoldX, Rosetta, premPS) are also inconclusive. Overall, the preponderance of evidence from the consensus of multiple in‑silico tools points to a pathogenic effect for P349L. This prediction does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.167087 | Structured | 0.348607 | Uncertain | 0.947 | 0.396 | 0.000 | -11.734 | Likely Pathogenic | 0.650 | Likely Pathogenic | Likely Benign | 0.70 | Ambiguous | 0.6 | 1.17 | Ambiguous | 0.94 | Ambiguous | 0.57 | Ambiguous | 0.326 | Likely Benign | -8.04 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 1.51 | Pathogenic | 0.00 | Affected | 0.2222 | 0.6867 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1049T>G | V350G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V350G lies in the C2 domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, polyPhen‑2 HumDiv, and AlphaMissense‑Optimized. The remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict it to be pathogenic. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All predictions are available and consistent. Based on the preponderance of pathogenic calls, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -12.267 | Likely Pathogenic | 0.597 | Likely Pathogenic | Likely Benign | 2.57 | Destabilizing | 0.0 | 4.25 | Destabilizing | 3.41 | Destabilizing | 1.93 | Destabilizing | 0.330 | Likely Benign | -5.46 | Deleterious | 0.435 | Benign | 0.920 | Probably Damaging | 1.61 | Pathogenic | 0.02 | Affected | 0.2316 | 0.2522 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.104T>A | V35D 2D AIThe SynGAP1 missense variant V35D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the overall assessment. Overall, the majority of evidence points to the variant being most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -4.232 | Likely Benign | 0.321 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -0.42 | Neutral | 0.824 | Possibly Damaging | 0.828 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.1540 | 0.1547 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.104T>G | V35G 2D AIThe SynGAP1 missense variant V35G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus also predicts benign; Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because the variant is not currently catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -3.614 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.143 | Likely Benign | 0.66 | Neutral | 0.693 | Possibly Damaging | 0.824 | Possibly Damaging | 4.41 | Benign | 0.00 | Affected | 0.1514 | 0.2618 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.1058T>C | L353P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L353P is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a strong bias toward pathogenicity: REVEL predicts benign, whereas FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all predict pathogenic. Two tools report uncertainty: ESM1b and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Overall, the majority of evidence points to a pathogenic impact, which is consistent with the ClinVar designation of uncertain significance but leans toward pathogenicity rather than benign. Thus, the variant is most likely pathogenic, and this prediction does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.137348 | Structured | 0.373584 | Uncertain | 0.926 | 0.315 | 0.000 | Uncertain | 1 | -7.913 | In-Between | 0.936 | Likely Pathogenic | Ambiguous | 4.63 | Destabilizing | 0.1 | 10.19 | Destabilizing | 7.41 | Destabilizing | 2.17 | Destabilizing | 0.464 | Likely Benign | -3.70 | Deleterious | 0.947 | Possibly Damaging | 0.454 | Possibly Damaging | 1.29 | Pathogenic | 0.02 | Affected | 3.37 | 25 | 0.3582 | 0.1645 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||
| c.1064G>T | G355V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G355V is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain or unavailable results are reported for FoldX, premPS, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, whereas the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect. Foldetta’s stability prediction is unavailable. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.388832 | Uncertain | 0.810 | 0.354 | 0.125 | -10.111 | Likely Pathogenic | 0.675 | Likely Pathogenic | Likely Benign | 1.78 | Ambiguous | 0.6 | 0.14 | Likely Benign | 0.96 | Ambiguous | 0.53 | Ambiguous | 0.346 | Likely Benign | -7.59 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.78 | Pathogenic | 0.04 | Affected | 0.1221 | 0.4685 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1066C>T | R356C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R356C is listed in ClinVar as Benign (ClinVar ID 469145.0) and is present in gnomAD (ID 6‑33437971‑C‑T). Functional prediction tools cluster into two groups: benign predictions from REVEL and AlphaMissense‑Optimized, and pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. Uncertain results are reported by FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as inconclusive. Overall, the majority of evidence points to a pathogenic effect, contradicting the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.219301 | Structured | 0.395028 | Uncertain | 0.802 | 0.373 | 0.250 | Likely Benign | 1 | 6-33437971-C-T | 5 | 3.10e-6 | -11.827 | Likely Pathogenic | 0.774 | Likely Pathogenic | Likely Benign | 0.76 | Ambiguous | 0.0 | 1.19 | Ambiguous | 0.98 | Ambiguous | 0.84 | Ambiguous | 0.312 | Likely Benign | -7.12 | Deleterious | 1.000 | Probably Damaging | 0.990 | Probably Damaging | 1.67 | Pathogenic | 0.00 | Affected | 3.39 | 22 | 0.3238 | 0.3618 | -4 | -3 | 7.0 | -53.05 | 212.3 | 91.0 | -0.1 | 0.3 | -0.3 | 0.1 | X | Potentially Pathogenic | Arg356 is located in a loop that includes a short helical section and connects two anti-parallel β sheet strands (res. Gly341-Pro349, res. Thr359-Pro364). In the WT simulations, the guanidinium group of Arg356 alternately forms salt bridges with the carboxylate groups of the GAP domain residues, Glu446 and Glu698. Arg356 also forms hydrogen bonds with the hydroxyl group of the GAP domain residue Thr691 and interacts with Met409 at the C2-GAP interface.In the variant simulations, the Cys356 mutation fails to maintain any of the Arg356 interactions and only occasionally forms weak hydrogen bonds with nearby C2 domain residues (e.g., Gln407). Although no negative structural effects are observed during the simulations, Arg356 is located at the C2 and GAP domain interface, making the residue swap potentially detrimental to the tertiary structure assembly. | |||||||||||||
| c.1067G>T | R356L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R356L is not reported in ClinVar and is present in gnomAD (ID 6‑33437972‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, FoldX, and Foldetta, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive are AlphaMissense‑Optimized, Rosetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the preponderance of evidence points to a pathogenic effect for R356L, and this conclusion does not contradict ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.219301 | Structured | 0.395028 | Uncertain | 0.802 | 0.373 | 0.250 | 6-33437972-G-T | -13.957 | Likely Pathogenic | 0.911 | Likely Pathogenic | Ambiguous | -0.04 | Likely Benign | 0.1 | -0.57 | Ambiguous | -0.31 | Likely Benign | 0.68 | Ambiguous | 0.412 | Likely Benign | -6.20 | Deleterious | 0.993 | Probably Damaging | 0.982 | Probably Damaging | 1.69 | Pathogenic | 0.02 | Affected | 3.39 | 22 | 0.2110 | 0.5242 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||
| c.1070A>T | H357L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 H357L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv, while polyPhen‑2 HumVar and ESM1b are benign or uncertain, respectively. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign (3 benign vs 1 pathogenic), and Foldetta also predicts benign. No predictions are missing or inconclusive. Overall, the variant is most likely benign based on the majority of computational evidence, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.203355 | Structured | 0.399052 | Uncertain | 0.861 | 0.413 | 0.250 | -7.281 | In-Between | 0.140 | Likely Benign | Likely Benign | -0.18 | Likely Benign | 0.1 | 0.14 | Likely Benign | -0.02 | Likely Benign | 0.10 | Likely Benign | 0.203 | Likely Benign | -3.39 | Deleterious | 0.704 | Possibly Damaging | 0.169 | Benign | 4.20 | Benign | 0.25 | Tolerated | 0.1084 | 0.5971 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||
| c.107A>T | H36L 2D AIThe SynGAP1 missense variant H36L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign classification. AlphaMissense‑Optimized also reports benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of evidence points to a benign effect. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -2.403 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -1.73 | Neutral | 0.010 | Benign | 0.011 | Benign | 4.19 | Benign | 0.00 | Affected | 0.1310 | 0.6111 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.1084T>A | W362R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W362R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify it as pathogenic. Benign predictions are absent; all evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—return pathogenic or likely pathogenic calls. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports pathogenic. Consequently, the variant is most likely pathogenic, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.430310 | Uncertain | 0.957 | 0.552 | 0.125 | -14.004 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.64 | Destabilizing | 0.3 | 3.90 | Destabilizing | 3.27 | Destabilizing | 1.10 | Destabilizing | 0.706 | Likely Pathogenic | -12.87 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 3.39 | 24 | 0.4811 | 0.0571 | 2 | -3 | -3.6 | -30.03 | 287.5 | -34.1 | -0.2 | 0.1 | -0.6 | 0.2 | X | X | X | Potentially Pathogenic | The indole ring of Trp362, located on the surface of an anti-parallel β sheet (res. Thr359-Pro364) in the C2 domain, stacks with nearby residues (e.g., Arg401, Arg272). In the variant simulations, the guanidinium group of the introduced residue Arg362 forms a salt bridge with the carboxylate group of Glu273 and, like Trp362, stacks with other arginine residues (e.g., Arg401, Arg272). This residue is at both the C2-membrane interface and the C2-RasGTPase interface, so the residue swap could potentially affect both interactions. However, these phenomena cannot be addressed using solvent-only simulations. Notably, Arg272, which stacks with both the non-mutated Trp362 and the mutated Arg362, forms a salt bridge directly with Asp105 of Ras in the WT simulations. Therefore, the residue swap could affect the C2 domain stability, the SynGAP-membrane association, and the SynGAP-Ras association. | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||
| c.1084T>C | W362R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W362R (ClinVar ID 41461.0) is listed as Pathogenic and is not reported in gnomAD. All available in silico predictors classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments concur: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) reports Pathogenic. Thus, the variant is most likely pathogenic, and this prediction aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.430310 | Uncertain | 0.957 | 0.552 | 0.125 | Pathogenic | 2 | -14.004 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.64 | Destabilizing | 0.3 | 3.90 | Destabilizing | 3.27 | Destabilizing | 1.10 | Destabilizing | 0.706 | Likely Pathogenic | -12.87 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 3.39 | 24 | 0.4811 | 0.0571 | 2 | -3 | -3.6 | -30.03 | 287.5 | -34.1 | -0.2 | 0.1 | -0.6 | 0.2 | X | X | X | Potentially Pathogenic | The indole ring of Trp362, located on the surface of an anti-parallel β sheet (res. Thr359-Pro364) in the C2 domain, stacks with nearby residues (e.g., Arg401, Arg272). In the variant simulations, the guanidinium group of the introduced residue Arg362 forms a salt bridge with the carboxylate group of Glu273 and, like Trp362, stacks with other arginine residues (e.g., Arg401, Arg272). This residue is at both the C2-membrane interface and the C2-RasGTPase interface, so the residue swap could potentially affect both interactions. However, these phenomena cannot be addressed using solvent-only simulations. Notably, Arg272, which stacks with both the non-mutated Trp362 and the mutated Arg362, forms a salt bridge directly with Asp105 of Ras in the WT simulations. Therefore, the residue swap could affect the C2 domain stability, the SynGAP-membrane association, and the SynGAP-Ras association. | 10.1016/j.ajhg.2020.11.011 | |||||||||||||
| c.1085G>C | W362S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W362S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as pathogenic, while premPS remains uncertain. High‑accuracy assessments corroborate this trend: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports likely pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta outputs) also indicates pathogenic. No tool predicts a benign outcome. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.430310 | Uncertain | 0.957 | 0.552 | 0.125 | -13.228 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 4.08 | Destabilizing | 0.0 | 4.55 | Destabilizing | 4.32 | Destabilizing | 0.95 | Ambiguous | 0.625 | Likely Pathogenic | -12.87 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.31 | Pathogenic | 0.00 | Affected | 0.5106 | 0.1215 | Weaken | -2 | -3 | 0.1 | -99.14 | ||||||||||||||||||||||||||||
| c.1087T>G | Y363D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y363D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are none; all evaluated algorithms predict a deleterious impact. Pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions or stability results are missing or inconclusive. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.435392 | Uncertain | 0.954 | 0.586 | 0.125 | -13.840 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.86 | Destabilizing | 0.2 | 3.03 | Destabilizing | 2.95 | Destabilizing | 1.73 | Destabilizing | 0.635 | Likely Pathogenic | -8.94 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.54 | Pathogenic | 0.00 | Affected | 0.4720 | 0.1853 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1091C>T | P364L 2D 3DClick to see structure in 3D Viewer AISynGAP1 P364L is reported in gnomAD (ID 6‑33437996‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, AlphaMissense‑Optimized, and Foldetta; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Three tools—FoldX, Rosetta, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign. Overall, the balance of evidence slightly favors a benign effect, and this conclusion does not contradict any ClinVar classification because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.390993 | Structured | 0.439474 | Uncertain | 0.942 | 0.590 | 0.250 | 6-33437996-C-T | -10.620 | Likely Pathogenic | 0.457 | Ambiguous | Likely Benign | 0.88 | Ambiguous | 0.9 | -0.73 | Ambiguous | 0.08 | Likely Benign | 0.31 | Likely Benign | 0.387 | Likely Benign | -7.78 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 1.54 | Pathogenic | 0.18 | Tolerated | 3.39 | 20 | 0.2200 | 0.6207 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||
| c.1094T>G | V365G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V365G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity unanimously classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign effect. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. All available evidence points to a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.414856 | Structured | 0.441505 | Uncertain | 0.923 | 0.608 | 0.250 | -13.020 | Likely Pathogenic | 0.944 | Likely Pathogenic | Ambiguous | 4.18 | Destabilizing | 0.2 | 4.83 | Destabilizing | 4.51 | Destabilizing | 2.18 | Destabilizing | 0.576 | Likely Pathogenic | -5.88 | Deleterious | 0.688 | Possibly Damaging | 0.989 | Probably Damaging | 1.66 | Pathogenic | 0.00 | Affected | 0.1835 | 0.2717 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1100T>C | L367P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L367P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on benign impact include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict pathogenicity are FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, SIFT, and FATHMM; premPS remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic effect. Overall, the majority of predictions lean toward a benign effect, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.370445 | Structured | 0.441805 | Uncertain | 0.790 | 0.657 | 0.250 | -2.418 | Likely Benign | 0.160 | Likely Benign | Likely Benign | 2.13 | Destabilizing | 0.4 | 4.05 | Destabilizing | 3.09 | Destabilizing | 0.72 | Ambiguous | 0.212 | Likely Benign | -0.50 | Neutral | 0.627 | Possibly Damaging | 0.196 | Benign | 1.72 | Pathogenic | 0.02 | Affected | 0.3946 | 0.1874 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1103C>T | P368L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P368L is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33438008‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Predictions that are uncertain or inconclusive are FoldX, Rosetta, premPS, AlphaMissense‑Default, and Foldetta. High‑accuracy assessments give AlphaMissense‑Optimized a benign score, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain. Based on the overall distribution of predictions, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.363090 | Structured | 0.439989 | Uncertain | 0.580 | 0.677 | 0.250 | 6-33438008-C-T | 1 | 6.33e-7 | -6.520 | Likely Benign | 0.444 | Ambiguous | Likely Benign | 1.52 | Ambiguous | 0.7 | 1.15 | Ambiguous | 1.34 | Ambiguous | 0.52 | Ambiguous | 0.248 | Likely Benign | -6.61 | Deleterious | 0.991 | Probably Damaging | 0.831 | Possibly Damaging | 1.77 | Pathogenic | 0.00 | Affected | 3.42 | 19 | 0.2336 | 0.7125 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||
| c.1108G>T | G370C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370C has no ClinVar entry and is not reported in gnomAD. Functional prediction tools fall into two groups: benign predictions come from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from REVEL, FoldX, Rosetta, Foldetta, and FATHMM. ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. No prediction or stability result is missing. Overall, the majority of tools predict a benign effect, and the high‑accuracy consensus also leans benign, while only one high‑accuracy method (Foldetta) suggests pathogenicity. Thus, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -7.071 | In-Between | 0.119 | Likely Benign | Likely Benign | 3.01 | Destabilizing | 2.1 | 2.03 | Destabilizing | 2.52 | Destabilizing | 0.29 | Likely Benign | 0.511 | Likely Pathogenic | -1.00 | Neutral | 0.353 | Benign | 0.010 | Benign | 1.32 | Pathogenic | 0.06 | Tolerated | 0.1245 | 0.4412 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1109G>T | G370V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster into two groups: benign (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic (FoldX, Rosetta, Foldetta, and FATHMM). High‑accuracy assessments further refine this picture: AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic outcome. Overall, the majority of evidence points toward a benign impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -5.328 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 4.98 | Destabilizing | 3.8 | 5.61 | Destabilizing | 5.30 | Destabilizing | -0.43 | Likely Benign | 0.427 | Likely Benign | 0.03 | Neutral | 0.000 | Benign | 0.000 | Benign | 1.32 | Pathogenic | 0.29 | Tolerated | 0.1306 | 0.4198 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.110C>A | S37Y 2D  AIThe SynGAP1 missense variant S37Y is listed in gnomAD (ID 6‑33423519‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and Foldetta (FoldX‑MD/Rosetta stability assessment) has no available result. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta data is missing. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.433492 | Uncertain | 0.317 | 0.806 | 0.500 | 6-33423519-C-A | 1 | 6.20e-7 | -4.447 | Likely Benign | 0.370 | Ambiguous | Likely Benign | 0.132 | Likely Benign | -1.61 | Neutral | 0.880 | Possibly Damaging | 0.888 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1247 | 0.5256 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||
| c.1117G>A | G373R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G373R is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33438022‑G‑A). Prediction tools that classify the variant as benign include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Optimized, and premPS. Those that predict pathogenicity are REVEL, FoldX, Foldetta, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as pathogenic. No prediction or folding result is missing or inconclusive. Overall, the majority of tools (six versus five) and the consensus of high‑accuracy methods lean toward a benign effect. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.529623 | Disordered | 0.429267 | Uncertain | 0.295 | 0.799 | 0.625 | 6-33438022-G-A | 1 | 6.28e-7 | -7.878 | In-Between | 0.653 | Likely Pathogenic | Likely Benign | 4.28 | Destabilizing | 3.5 | 0.14 | Likely Benign | 2.21 | Destabilizing | 0.21 | Likely Benign | 0.510 | Likely Pathogenic | -0.64 | Neutral | 0.001 | Benign | 0.000 | Benign | 3.90 | Benign | 0.01 | Affected | 3.53 | 16 | 0.1089 | 0.4524 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||
| c.1117G>C | G373R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G373R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, FoldX, Foldetta, SIFT, and AlphaMissense‑Default; ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as pathogenic. Overall, the majority of tools (seven benign vs five pathogenic) lean toward a benign interpretation, and this does not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.529623 | Disordered | 0.429267 | Uncertain | 0.295 | 0.799 | 0.625 | -7.878 | In-Between | 0.653 | Likely Pathogenic | Likely Benign | 4.28 | Destabilizing | 3.5 | 0.14 | Likely Benign | 2.21 | Destabilizing | 0.21 | Likely Benign | 0.522 | Likely Pathogenic | -0.64 | Neutral | 0.001 | Benign | 0.000 | Benign | 3.90 | Benign | 0.01 | Affected | 3.53 | 16 | 0.1089 | 0.4524 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||
| c.1118G>T | G373V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G373V is listed in ClinVar with an uncertain significance and is present in gnomAD (variant ID 6‑33438023‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are FoldX, Foldetta, and SIFT, while Rosetta is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as pathogenic. Overall, the majority of predictions support a benign impact, and this consensus does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.529623 | Disordered | 0.429267 | Uncertain | 0.295 | 0.799 | 0.625 | Uncertain | 1 | 6-33438023-G-T | 6 | 5.03e-6 | -6.062 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 5.32 | Destabilizing | 3.2 | 0.82 | Ambiguous | 3.07 | Destabilizing | 0.09 | Likely Benign | 0.428 | Likely Benign | -0.98 | Neutral | 0.007 | Benign | 0.001 | Benign | 3.90 | Benign | 0.00 | Affected | 3.53 | 16 | 0.1424 | 0.4004 | -1 | -3 | 4.6 | 42.08 | 207.6 | -68.1 | 1.9 | 1.1 | -0.6 | 0.1 | Uncertain | Gly373 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364, res. Ala399-Ile411). Because the Ω loop is assumed to directly interact with the membrane, it moves arbitrarily throughout the WT solvent simulations. The Ω loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play major roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are observed in the variant simulations, Val373 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. However, since the effect on the Gly-rich Ω loop dynamics can only be studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1124G>T | G375V 2D AIThe SynGAP1 missense variant G375V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions (premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic predictions (REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, and FATHMM). High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also benign, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a pathogenic effect. No prediction is missing or inconclusive. Overall, the majority of tools and the consensus score suggest a benign effect, but the Foldetta result introduces uncertainty. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.604312 | Disordered | 0.428340 | Uncertain | 0.301 | 0.836 | 0.625 | -6.149 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 3.93 | Destabilizing | 3.5 | 7.55 | Destabilizing | 5.74 | Destabilizing | 0.02 | Likely Benign | 0.547 | Likely Pathogenic | -0.92 | Neutral | 0.845 | Possibly Damaging | 0.186 | Benign | 1.32 | Pathogenic | 0.06 | Tolerated | 0.1653 | 0.4193 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1126G>T | G376C 2D AISynGAP1 missense variant G376C is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from Rosetta, premPS, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls come from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Two tools report uncertainty: Foldetta and ESM1b. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign verdict; Foldetta remains uncertain. Overall, the majority of conventional predictors lean toward pathogenicity, whereas the most accurate methods favor a benign effect. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | Uncertain | 1 | -7.686 | In-Between | 0.125 | Likely Benign | Likely Benign | 2.56 | Destabilizing | 0.5 | 0.22 | Likely Benign | 1.39 | Ambiguous | 0.16 | Likely Benign | 0.560 | Likely Pathogenic | -1.15 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1476 | 0.3929 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||
| c.1127G>T | G376V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G376V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions arise from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The consensus score from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, matching the majority of individual benign calls. High‑accuracy methods give mixed results: AlphaMissense‑Optimized predicts benign, SGM Consensus predicts benign, whereas Foldetta (integrating FoldX‑MD and Rosetta) predicts pathogenic. No prediction is missing or inconclusive. Overall, the balance of evidence leans toward a benign effect; this is consistent with the lack of ClinVar annotation and gnomAD presence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | -6.242 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 4.84 | Destabilizing | 0.8 | -0.81 | Ambiguous | 2.02 | Destabilizing | -0.18 | Likely Benign | 0.541 | Likely Pathogenic | -0.66 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1594 | 0.3525 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1132G>T | G378C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G378C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of tools (8/12) predict pathogenicity, and the high‑accuracy consensus leans toward benign only for AlphaMissense‑Optimized and SGM Consensus, while Foldetta indicates instability. Thus, the variant is most likely pathogenic based on the preponderance of evidence, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.767246 | Disordered | 0.432858 | Uncertain | 0.341 | 0.915 | 0.625 | -7.981 | In-Between | 0.203 | Likely Benign | Likely Benign | 7.63 | Destabilizing | 2.7 | 12.14 | Destabilizing | 9.89 | Destabilizing | 0.26 | Likely Benign | 0.635 | Likely Pathogenic | -1.18 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1595 | 0.4240 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1136C>G | S379W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S379W is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33438041‑C‑G). Prediction tools that indicate a benign effect include premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic impact comprise REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus as benign. Because the majority of conventional tools favor pathogenicity while the high‑accuracy subset is split, the overall evidence leans toward a pathogenic effect. This conclusion does not contradict the ClinVar uncertain status, which remains unresolved. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.728858 | Disordered | 0.433206 | Uncertain | 0.327 | 0.931 | 0.625 | Uncertain | 1 | 6-33438041-C-G | -8.898 | Likely Pathogenic | 0.388 | Ambiguous | Likely Benign | 4.32 | Destabilizing | 3.4 | 3.56 | Destabilizing | 3.94 | Destabilizing | 0.16 | Likely Benign | 0.520 | Likely Pathogenic | -1.02 | Neutral | 0.998 | Probably Damaging | 0.844 | Possibly Damaging | 3.82 | Benign | 0.01 | Affected | 4.32 | 11 | 0.1196 | 0.6070 | -2 | -3 | -0.1 | 99.14 | 271.3 | -75.7 | 1.4 | 1.0 | 0.6 | 0.5 | Uncertain | Ser379 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364, res. Ala399-Ile411). Because the Ω loop is assumed to directly interact with the membrane, it moves arbitrarily throughout the WT solvent simulations. The Ω loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play major roles in protein functions that require flexibility, and thus hydrophobic residues like tryptophan are rarely tolerated. Although no major negative structural effects are observed in the variant simulations, Trp379 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. However, since the effect on Gly-rich Ω loop dynamics can only be studied through the SynGAP-membrane complex, no definite conclusions can be drawn | |||||||||||||||||
| c.1139G>T | G380V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G380V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions are reported by REVEL, FoldX, polyPhen‑2 HumDiv, and SIFT; Rosetta is uncertain. The SGM‑Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority of the four high‑accuracy tools) is benign, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a pathogenic impact. Overall, the majority of evidence points to a benign effect, and this does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.724957 | Disordered | 0.432982 | Uncertain | 0.316 | 0.939 | 0.750 | -6.234 | Likely Benign | 0.172 | Likely Benign | Likely Benign | 7.60 | Destabilizing | 2.5 | 1.75 | Ambiguous | 4.68 | Destabilizing | -0.17 | Likely Benign | 0.574 | Likely Pathogenic | -0.70 | Neutral | 0.816 | Possibly Damaging | 0.210 | Benign | 2.54 | Benign | 0.02 | Affected | 0.1618 | 0.3708 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.113C>T | P38L 2D AIThe SynGAP1 missense variant P38L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (gnomAD ID 6‑33423522‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.433285 | Uncertain | 0.344 | 0.791 | 0.375 | Conflicting | 4 | 6-33423522-C-T | 8 | 4.96e-6 | -2.469 | Likely Benign | 0.197 | Likely Benign | Likely Benign | 0.141 | Likely Benign | -2.56 | Deleterious | 0.983 | Probably Damaging | 0.931 | Probably Damaging | 4.02 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2432 | 0.7057 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||
| c.1141G>A | G381R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G381R is listed in ClinVar with an uncertain significance (ClinVar ID 4801336) and is present in gnomAD (variant ID 6‑33438046‑G‑A). Prediction tools that classify the variant as benign include premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that predict pathogenicity are REVEL, FoldX, Rosetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as benign, while both the SGM‑Consensus and Foldetta (combining FoldX‑MD and Rosetta outputs) predict pathogenicity. Overall, the preponderance of evidence points to a pathogenic effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.724957 | Disordered | 0.431692 | Uncertain | 0.301 | 0.951 | 0.750 | Uncertain | 1 | 6-33438046-G-A | -8.990 | Likely Pathogenic | 0.652 | Likely Pathogenic | Likely Benign | 5.60 | Destabilizing | 0.9 | 2.80 | Destabilizing | 4.20 | Destabilizing | 0.20 | Likely Benign | 0.589 | Likely Pathogenic | -0.82 | Neutral | 0.985 | Probably Damaging | 0.795 | Possibly Damaging | 1.32 | Pathogenic | 0.08 | Tolerated | 4.32 | 9 | 0.1339 | 0.3945 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||
| c.1141G>C | G381R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G381R is catalogued in gnomAD (ID 6‑33438046‑G‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized; pathogenic predictions arise from SGM‑Consensus, REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized reports a benign outcome, whereas the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta) both predict pathogenicity. Overall, the majority of evidence indicates a pathogenic impact for G381R, and this conclusion is not contradicted by ClinVar status, which currently lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.724957 | Disordered | 0.431692 | Uncertain | 0.301 | 0.951 | 0.750 | 6-33438046-G-C | 2 | 1.25e-6 | -8.990 | Likely Pathogenic | 0.652 | Likely Pathogenic | Likely Benign | 5.60 | Destabilizing | 0.9 | 2.80 | Destabilizing | 4.20 | Destabilizing | 0.20 | Likely Benign | 0.589 | Likely Pathogenic | -0.82 | Neutral | 0.985 | Probably Damaging | 0.795 | Possibly Damaging | 1.32 | Pathogenic | 0.08 | Tolerated | 4.32 | 9 | 0.1339 | 0.3945 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||
| c.1142G>T | G381V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G381V is listed in ClinVar with an uncertain significance (ClinVar ID 1940172.0) and is present in the gnomAD database (6‑33438047‑G‑T). Functional prediction tools that report a benign effect include premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, FoldX, Rosetta, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a majority‑benign vote and is reported as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of predictions lean toward a benign impact, and this is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.724957 | Disordered | 0.431692 | Uncertain | 0.301 | 0.951 | 0.750 | Uncertain | 1 | 6-33438047-G-T | 2 | 1.25e-6 | -5.967 | Likely Benign | 0.146 | Likely Benign | Likely Benign | 7.16 | Destabilizing | 1.0 | 4.10 | Destabilizing | 5.63 | Destabilizing | -0.32 | Likely Benign | 0.618 | Likely Pathogenic | -0.95 | Neutral | 0.386 | Benign | 0.157 | Benign | 1.32 | Pathogenic | 0.10 | Tolerated | 4.32 | 9 | 0.1621 | 0.3902 | -1 | -3 | 4.6 | 42.08 | 214.6 | -68.8 | 0.3 | 0.7 | -0.5 | 0.3 | Uncertain | Gly381 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364, res. Ala399-Ile411). Because the Ω loop is assumed to directly interact with the membrane, it moves arbitrarily throughout the WT solvent simulations. The Ω loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play major roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are observed in the variant simulations, Val381 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. However, since the effects on Gly-rich Ω loop dynamics can only be well studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1145G>T | G382V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G382V has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a split opinion: benign predictions come from PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions are returned by REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM, while premPS is uncertain. High‑accuracy assessments give a mixed signal: AlphaMissense‑Optimized predicts benign, SGM‑Consensus indicates likely benign, but Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of tools lean toward pathogenicity, and the high‑accuracy Foldetta result supports this. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429690 | Uncertain | 0.315 | 0.951 | 0.750 | -5.988 | Likely Benign | 0.155 | Likely Benign | Likely Benign | 6.40 | Destabilizing | 1.6 | 9.28 | Destabilizing | 7.84 | Destabilizing | -0.53 | Ambiguous | 0.571 | Likely Pathogenic | -0.54 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 1.32 | Pathogenic | 0.03 | Affected | 0.1631 | 0.3708 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1148G>T | G383V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G383V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect are FoldX, Rosetta, polyPhen‑2 HumDiv, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of predictions lean toward a benign impact, and this conclusion does not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429104 | Uncertain | 0.296 | 0.949 | 0.750 | -5.769 | Likely Benign | 0.145 | Likely Benign | Likely Benign | 5.13 | Destabilizing | 2.1 | 4.06 | Destabilizing | 4.60 | Destabilizing | -0.26 | Likely Benign | 0.406 | Likely Benign | -0.72 | Neutral | 0.668 | Possibly Damaging | 0.207 | Benign | 4.12 | Benign | 0.01 | Affected | 0.1597 | 0.3493 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1150G>T | G384C 2D AIThe SynGAP1 missense variant G384C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic; and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split and is treated as unavailable. Overall, the majority of predictions (seven pathogenic vs. five benign) and the pathogenic Foldetta result indicate that the variant is most likely pathogenic, with no contradiction to ClinVar status because the variant is not yet classified there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.728858 | Disordered | 0.427831 | Uncertain | 0.323 | 0.934 | 0.750 | -8.363 | Likely Pathogenic | 0.135 | Likely Benign | Likely Benign | 2.41 | Destabilizing | 2.0 | 5.50 | Destabilizing | 3.96 | Destabilizing | 0.17 | Likely Benign | 0.456 | Likely Benign | -1.07 | Neutral | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1530 | 0.4240 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1154C>G | S385W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S385W is listed in ClinVar as Benign (ClinVar ID 218691.0) and is present in gnomAD (ID 6‑33438059‑C‑G). Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM Consensus as Benign, and Foldetta as Uncertain. Taken together, the majority of evidence points to a benign impact, which aligns with the ClinVar classification and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.733139 | Disordered | 0.425480 | Uncertain | 0.341 | 0.925 | 0.750 | Benign | 1 | 6-33438059-C-G | -9.353 | Likely Pathogenic | 0.362 | Ambiguous | Likely Benign | 0.53 | Ambiguous | 0.2 | 0.69 | Ambiguous | 0.61 | Ambiguous | 0.00 | Likely Benign | 0.373 | Likely Benign | -0.84 | Neutral | 0.986 | Probably Damaging | 0.968 | Probably Damaging | 4.63 | Benign | 0.00 | Affected | 4.32 | 3 | 0.1272 | 0.6670 | -2 | -3 | -0.1 | 99.14 | 260.4 | -71.2 | 0.5 | 1.3 | 0.7 | 0.4 | Uncertain | Ser385 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364, res. Ala399-Ile411). Because the Ω loop is assumed to directly interact with the membrane, it moves arbitrarily throughout the WT solvent simulations. The Ω loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play major roles in protein functions that require flexibility, and thus hydrophobic residues like tryptophan are rarely tolerated. Although no major negative structural effects are observed in the variant simulations, Trp385 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. However, since the effects on Gly-rich Ω loop dynamics can only be studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | 10.1016/j.ajhg.2020.11.011 | ||||||||||||||||
| c.1159G>T | G387C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G387C is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33438064‑G‑T). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of high‑confidence tools lean toward a benign interpretation, and this does not contradict the ClinVar status, which has no pathogenic classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.642678 | Disordered | 0.422910 | Uncertain | 0.293 | 0.861 | 0.750 | 6-33438064-G-T | 1 | 6.21e-7 | -7.609 | In-Between | 0.146 | Likely Benign | Likely Benign | 2.88 | Destabilizing | 0.6 | 2.34 | Destabilizing | 2.61 | Destabilizing | -0.03 | Likely Benign | 0.430 | Likely Benign | -0.58 | Neutral | 0.859 | Possibly Damaging | 0.346 | Benign | 1.32 | Pathogenic | 0.01 | Affected | 4.32 | 3 | 0.1589 | 0.4240 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||
| c.115T>G | Y39D 2D AIThe SynGAP1 missense variant Y39D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for Y39D, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.432876 | Uncertain | 0.343 | 0.787 | 0.375 | -2.338 | Likely Benign | 0.333 | Likely Benign | Likely Benign | 0.166 | Likely Benign | -1.86 | Neutral | 0.824 | Possibly Damaging | 0.828 | Possibly Damaging | 3.98 | Benign | 0.00 | Affected | 0.4099 | 0.0903 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.1160G>T | G387V 2D 3DClick to see structure in 3D Viewer AISynGAP1 G387V is listed in ClinVar with an uncertain significance and is present in gnomAD (variant ID 6-33438065-G-T). Functional prediction tools that report a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, SIFT, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as likely benign, while the Foldetta stability assessment (combining FoldX‑MD and Rosetta) indicates a pathogenic change. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as pathogenic. Overall, the majority of predictions favor a benign impact, and this consensus does not contradict the ClinVar uncertain status; thus the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.642678 | Disordered | 0.422910 | Uncertain | 0.293 | 0.861 | 0.750 | Uncertain | 1 | 6-33438065-G-T | 22 | 1.37e-5 | -6.199 | Likely Benign | 0.153 | Likely Benign | Likely Benign | 5.13 | Destabilizing | 1.8 | 6.44 | Destabilizing | 5.79 | Destabilizing | -0.33 | Likely Benign | 0.390 | Likely Benign | -0.54 | Neutral | 0.069 | Benign | 0.077 | Benign | 1.32 | Pathogenic | 0.01 | Affected | 4.32 | 3 | 0.1586 | 0.3708 | -1 | -3 | 4.6 | 42.08 | 207.7 | -68.4 | -0.7 | 0.8 | -0.5 | 0.1 | Uncertain | Gly387 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364 and res. Ala399-Ile411). The Ω loop is assumed to directly interact with the membrane, and it is observed to move arbitrarily throughout the WT solvent simulations. This loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play significant roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are visualized in the variant’s simulations, Val387 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. Since the effects on the Gly-rich Ω loop dynamics can only be well studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1162G>T | G388C 2D 3DClick to see structure in 3D Viewer AISynGAP1 G388C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, and PROVEAN, while pathogenic predictions arise from REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized predicts benign, Foldetta predicts pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Because the majority of evidence points to pathogenicity and there is no ClinVar entry to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.736850 | Disordered | 0.420316 | Uncertain | 0.319 | 0.827 | 0.750 | -8.088 | Likely Pathogenic | 0.121 | Likely Benign | Likely Benign | 4.01 | Destabilizing | 3.7 | 4.95 | Destabilizing | 4.48 | Destabilizing | 0.03 | Likely Benign | 0.603 | Likely Pathogenic | -0.98 | Neutral | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1497 | 0.4112 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1166C>T | S389L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S389L is catalogued in gnomAD (6‑33438071‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, Foldetta, and the SGM‑Consensus score (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments are consistent with the benign consensus: AlphaMissense‑Optimized reports Benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Benign. No prediction or folding stability result is missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.703578 | Disordered | 0.417444 | Uncertain | 0.306 | 0.803 | 0.875 | 6-33438071-C-T | 7 | 4.40e-6 | -6.040 | Likely Benign | 0.165 | Likely Benign | Likely Benign | 0.04 | Likely Benign | 0.1 | 0.69 | Ambiguous | 0.37 | Likely Benign | -0.23 | Likely Benign | 0.456 | Likely Benign | -0.75 | Neutral | 0.462 | Possibly Damaging | 0.693 | Possibly Damaging | 5.05 | Benign | 0.01 | Affected | 4.32 | 8 | 0.1844 | 0.5871 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||
| c.1169G>T | G390V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G390V has no ClinVar entry and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from REVEL, FoldX, Rosetta, Foldetta, SIFT, and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default (benign), ESM1b (uncertain), FATHMM (pathogenic), and PROVEAN (benign), also yields a benign verdict; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, indicates pathogenic. With two high‑accuracy tools supporting benign and one supporting pathogenic, the overall evidence leans toward a benign effect. This conclusion does not contradict ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.626927 | Disordered | 0.413274 | Uncertain | 0.304 | 0.763 | 0.875 | -7.137 | In-Between | 0.146 | Likely Benign | Likely Benign | 3.74 | Destabilizing | 0.6 | 4.88 | Destabilizing | 4.31 | Destabilizing | -0.16 | Likely Benign | 0.509 | Likely Pathogenic | -0.88 | Neutral | 0.002 | Benign | 0.002 | Benign | 1.32 | Pathogenic | 0.01 | Affected | 0.1537 | 0.4004 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||
| c.1171G>T | G391C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G391C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, and PROVEAN, whereas the remaining tools—REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—predict it to be pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence points to a pathogenic effect. This prediction does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | -8.596 | Likely Pathogenic | 0.123 | Likely Benign | Likely Benign | 2.65 | Destabilizing | 0.7 | 5.03 | Destabilizing | 3.84 | Destabilizing | 0.11 | Likely Benign | 0.640 | Likely Pathogenic | -1.29 | Neutral | 1.000 | Probably Damaging | 0.970 | Probably Damaging | 1.32 | Pathogenic | 0.03 | Affected | 0.1483 | 0.4225 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1172G>T | G391V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G391V is listed in ClinVar as Benign (ClinVar ID 1014488.0) and is present in gnomAD (variant ID 6‑33438077‑G‑T). Prediction tools that classify the variant as benign include premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus. Tools that predict pathogenicity are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. With two high‑accuracy tools supporting benign and one supporting pathogenic, the overall prediction leans toward a benign effect. This conclusion aligns with the ClinVar benign classification, so there is no contradiction with the existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | Likely Benign | 1 | 6-33438077-G-T | 3 | 1.86e-6 | -6.642 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 4.23 | Destabilizing | 1.3 | 4.81 | Destabilizing | 4.52 | Destabilizing | -0.11 | Likely Benign | 0.595 | Likely Pathogenic | -0.98 | Neutral | 0.994 | Probably Damaging | 0.887 | Possibly Damaging | 1.32 | Pathogenic | 0.10 | Tolerated | 3.69 | 8 | 0.1621 | 0.3821 | -1 | -3 | 4.6 | 42.08 | 228.6 | -69.0 | 0.0 | 0.8 | -0.5 | 0.3 | Uncertain | Gly387 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364 and res. Ala399-Ile411). The Ω loop is assumed to directly interact with the membrane, and it is observed to move arbitrarily throughout the WT solvent simulations. This loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play significant roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are visualized in the variant’s simulations, Val391 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. Since the effects on the Gly-rich Ω loop dynamics can only be well studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1177G>T | G393C 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G393C is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Rosetta, premPS, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus (likely pathogenic), REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain (no definitive stability change). The majority of evidence points toward a pathogenic effect, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -8.854 | Likely Pathogenic | 0.181 | Likely Benign | Likely Benign | 2.99 | Destabilizing | 0.9 | -0.26 | Likely Benign | 1.37 | Ambiguous | 0.43 | Likely Benign | 0.769 | Likely Pathogenic | -3.05 | Deleterious | 0.999 | Probably Damaging | 0.936 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1593 | 0.4408 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1178G>T | G393V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G393V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Rosetta is uncertain and is treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while Foldetta predicts pathogenic. The SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑vs‑2 split. Overall, the majority of evidence (8 pathogenic vs. 4 benign) points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -6.358 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 5.56 | Destabilizing | 2.3 | -0.72 | Ambiguous | 2.42 | Destabilizing | -0.01 | Likely Benign | 0.639 | Likely Pathogenic | -2.69 | Deleterious | 0.982 | Probably Damaging | 0.648 | Possibly Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1712 | 0.4144 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||
| c.1181A>T | K394I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 K394I missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include premPS, polyPhen‑2 HumVar, and FATHMM, while a majority (seven) predict pathogenicity: SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) remains Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. No evidence from these tools contradicts the ClinVar status, which is absent. Overall, the preponderance of pathogenic predictions suggests the variant is most likely pathogenic, with no conflict from ClinVar data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.505461 | Disordered | 0.399336 | Uncertain | 0.387 | 0.634 | 0.625 | -9.244 | Likely Pathogenic | 0.876 | Likely Pathogenic | Ambiguous | 0.78 | Ambiguous | 0.2 | 1.10 | Ambiguous | 0.94 | Ambiguous | 0.19 | Likely Benign | 0.519 | Likely Pathogenic | -3.96 | Deleterious | 0.700 | Possibly Damaging | 0.403 | Benign | 4.59 | Benign | 0.00 | Affected | 0.1728 | 0.4123 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1184G>T | G395V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G395V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only FoldX and SIFT predict pathogenic. The SGM‑Consensus score, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and the Foldetta stability analysis is uncertain. Taken together, the preponderance of evidence points to a benign impact for G395V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.513880 | Disordered | 0.396199 | Uncertain | 0.474 | 0.601 | 0.500 | -5.617 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 2.83 | Destabilizing | 0.7 | -0.37 | Likely Benign | 1.23 | Ambiguous | 0.08 | Likely Benign | 0.288 | Likely Benign | -1.49 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.21 | Benign | 0.03 | Affected | 0.1290 | 0.4183 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1186G>T | G396C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G396C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar; premPS is uncertain. The high‑accuracy consensus methods give a mixed signal: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of individual predictors and the SGM‑Consensus lean toward a benign interpretation, and the two high‑accuracy tools that are available also favor benign over pathogenic. Therefore, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.394626 | Uncertain | 0.640 | 0.584 | 0.500 | -5.459 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 2.15 | Destabilizing | 0.7 | 2.52 | Destabilizing | 2.34 | Destabilizing | 0.59 | Ambiguous | 0.411 | Likely Benign | -3.00 | Deleterious | 0.983 | Probably Damaging | 0.533 | Possibly Damaging | 3.89 | Benign | 0.08 | Tolerated | 0.1181 | 0.4541 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1189T>C | C397R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Rosetta, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and AlphaMissense‑Default. The remaining tools (FoldX, premPS, ESM1b) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign (2 benign vs. 1 pathogenic, 1 uncertain), and Foldetta predicts a benign impact on protein folding stability. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | -7.094 | In-Between | 0.708 | Likely Pathogenic | Likely Benign | 0.55 | Ambiguous | 0.8 | 0.40 | Likely Benign | 0.48 | Likely Benign | 0.96 | Ambiguous | 0.514 | Likely Pathogenic | -1.46 | Neutral | 0.480 | Possibly Damaging | 0.226 | Benign | 4.65 | Benign | 0.11 | Tolerated | 0.1780 | 0.1853 | -4 | -3 | -7.0 | 53.05 | ||||||||||||||||||||||||||||||
| c.1189T>G | C397G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach consensus all indicate a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the variant as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies it as “Likely Benign.” No tool predicts pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. No pathogenic predictions are present. Therefore, based on the available computational evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | 1.244 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.68 | Ambiguous | 0.2 | 1.42 | Ambiguous | 1.05 | Ambiguous | 0.04 | Likely Benign | 0.272 | Likely Benign | 1.51 | Neutral | 0.276 | Benign | 0.066 | Benign | 5.93 | Benign | 0.60 | Tolerated | 0.3678 | 0.3729 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.118G>T | D40Y 2D AIThe SynGAP1 D40Y missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus indicates Likely Benign, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for D40Y, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -4.313 | Likely Benign | 0.483 | Ambiguous | Likely Benign | 0.182 | Likely Benign | -1.72 | Neutral | 0.388 | Benign | 0.328 | Benign | 3.98 | Benign | 0.00 | Affected | 0.1173 | 0.7918 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.1193C>T | P398L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant P398L (ClinVar ID 2415189.0) is listed as Uncertain in ClinVar and is present in gnomAD (ID 6‑33438098‑C‑T). Functional prediction tools that agree on a benign effect include Foldetta, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, and SIFT. Predictions that are uncertain or inconclusive are FoldX, Rosetta, premPS, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Based on the available predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.436924 | Structured | 0.401041 | Uncertain | 0.891 | 0.525 | 0.250 | Uncertain | 1 | 6-33438098-C-T | 8 | 4.96e-6 | -7.518 | In-Between | 0.547 | Ambiguous | Likely Benign | 1.48 | Ambiguous | 0.2 | -0.54 | Ambiguous | 0.47 | Likely Benign | 0.62 | Ambiguous | 0.599 | Likely Pathogenic | -7.10 | Deleterious | 0.961 | Probably Damaging | 0.256 | Benign | 5.72 | Benign | 0.01 | Affected | 3.40 | 16 | 0.2248 | 0.7157 | -3 | -3 | 5.4 | 16.04 | 245.8 | -68.6 | -0.1 | 0.0 | -0.3 | 0.2 | X | Potentially Pathogenic | Pro398 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364 and res. Ala399-Ile411). The Ω loop is assumed to directly interact with the membrane, and it is observed to move arbitrarily throughout the WT solvent simulations. Although the residue swap does not influence the nearby secondary structure elements, proline is often found at the ends of β sheets due to its disfavored status during folding.Additionally, the Ω loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone. Ω loops are known to play significant roles in protein functions that require flexibility, and thus hydrophobic residues like leucine are rarely tolerated. Although no negative structural effects are visualized in the variant’s simulations, Leu398 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. Since the effects on the Gly-rich Ω loop dynamics can only be well studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1199T>G | V400G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V400G missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are polyPhen‑2 HumDiv and FATHMM; all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.398279 | Structured | 0.415488 | Uncertain | 0.951 | 0.451 | 0.000 | -11.508 | Likely Pathogenic | 0.969 | Likely Pathogenic | Likely Pathogenic | 4.84 | Destabilizing | 0.1 | 5.40 | Destabilizing | 5.12 | Destabilizing | 2.40 | Destabilizing | 0.819 | Likely Pathogenic | -5.70 | Deleterious | 0.335 | Benign | 0.920 | Probably Damaging | 5.27 | Benign | 0.00 | Affected | 0.2296 | 0.2522 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.119A>T | D40V 2D AIThe SynGAP1 missense variant D40V is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -3.350 | Likely Benign | 0.431 | Ambiguous | Likely Benign | 0.222 | Likely Benign | -1.16 | Neutral | 0.028 | Benign | 0.088 | Benign | 4.00 | Benign | 0.00 | Affected | 0.1679 | 0.8357 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.11C>A | S4Y 2D AIThe SynGAP1 missense variant S4Y is reported in gnomAD (ID 6‑33420275‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign; Foldetta results are unavailable. Taken together, the majority of evidence points to a benign impact. There is no ClinVar classification to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | 6-33420275-C-A | -5.156 | Likely Benign | 0.209 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -0.34 | Neutral | 0.880 | Possibly Damaging | 0.608 | Possibly Damaging | 4.12 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0748 | 0.6181 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||
| c.1202G>T | R401L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R401L is not reported in ClinVar and is present in gnomAD (ID 6‑33438107‑G‑T). Prediction tools that classify the variant as benign include Rosetta, FATHMM, and premPS, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) indicate a pathogenic effect. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. No evidence from FoldX alone is available. Based on the preponderance of pathogenic predictions and the corroborating high‑accuracy tools, R401L is most likely pathogenic; this assessment does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.314870 | Structured | 0.424277 | Uncertain | 0.961 | 0.419 | 0.000 | 6-33438107-G-T | 1 | 6.20e-7 | -12.280 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | -1.52 | Ambiguous | 0.1 | -0.23 | Likely Benign | -0.88 | Ambiguous | 0.22 | Likely Benign | 0.858 | Likely Pathogenic | -6.42 | Deleterious | 0.997 | Probably Damaging | 0.987 | Probably Damaging | 5.44 | Benign | 0.02 | Affected | 3.38 | 27 | 0.1972 | 0.4263 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||
| c.1205T>C | L402P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L402P is not reported in ClinVar and is absent from gnomAD. Consensus among in‑silico predictors is overwhelmingly pathogenic: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a deleterious effect, whereas only FATHMM predicts a benign outcome. High‑accuracy tools reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic effect; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Taken together, the evidence strongly supports a pathogenic classification, and this assessment does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.243554 | Structured | 0.431978 | Uncertain | 0.961 | 0.383 | 0.000 | -12.030 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 7.40 | Destabilizing | 0.1 | 5.82 | Destabilizing | 6.61 | Destabilizing | 2.22 | Destabilizing | 0.616 | Likely Pathogenic | -4.80 | Deleterious | 1.000 | Probably Damaging | 0.980 | Probably Damaging | 3.68 | Benign | 0.01 | Affected | 0.3628 | 0.1874 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1208A>T | K403I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K403I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, premPS, and FATHMM, while the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions (nine pathogenic vs. five benign) and the high‑accuracy tools lean toward a pathogenic effect. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.275179 | Structured | 0.424920 | Uncertain | 0.960 | 0.372 | 0.000 | -15.239 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.17 | Likely Benign | 0.1 | -0.46 | Likely Benign | -0.15 | Likely Benign | 0.36 | Likely Benign | 0.519 | Likely Pathogenic | -7.27 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.70 | Benign | 0.00 | Affected | 0.1223 | 0.4141 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1213C>T | R405C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R405C missense variant is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33438118‑C‑T). Prediction tools that indicate a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas a majority of tools (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as likely pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) remains uncertain. Overall, the balance of evidence points to a pathogenic effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.404888 | Uncertain | 0.949 | 0.315 | 0.000 | Conflicting | 3 | 6-33438118-C-T | 6 | 3.72e-6 | -9.206 | Likely Pathogenic | 0.713 | Likely Pathogenic | Likely Benign | 0.72 | Ambiguous | 0.1 | 1.51 | Ambiguous | 1.12 | Ambiguous | 1.21 | Destabilizing | 0.427 | Likely Benign | -7.27 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.61 | Benign | 0.02 | Affected | 3.38 | 28 | 0.3227 | 0.3964 | -4 | -3 | 7.0 | -53.05 | 221.3 | 82.6 | -0.1 | 0.0 | -0.2 | 0.3 | X | X | Potentially Pathogenic | The guanidinium group of Arg405, located in an anti-parallel β sheet strand of the C2 domain (res. Ala399-Ile411), forms a salt bridge with the carboxylate group of the Glu446 side chain from an opposing α helix (res. Val441-Ser457) in the GAP domain. The positively charged Arg405 side chain also stacks with the aromatic ring of the Phe358 side chain from a loop preceding the β strand (res. Thr359-Thr366), which could assist in maintaining the anti-parallel strand arrangement.In the variant simulations, the thiol-containing side chain of Cys405 is neutral and smaller compared to the arginine side chain. The lack of Arg405-Phe358 stacking affects the loop structure, causing it to assume a β strand form—an effect that could be exacerbated during protein folding. Moreover, the inability of Cys405 to form a salt bridge with Glu446 could affect the tertiary structure assembly, although this is not apparent based on the variant simulations. | ||||||||||||
| c.1216T>G | Y406D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y406D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and FATHMM. In contrast, the majority of tools predict a pathogenic impact: FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score is “Likely Pathogenic,” while premPS is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the preponderance of evidence, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status (which is currently unreported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.179055 | Structured | 0.393707 | Uncertain | 0.946 | 0.291 | 0.000 | -14.832 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.82 | Destabilizing | 0.3 | 4.28 | Destabilizing | 4.05 | Destabilizing | 0.98 | Ambiguous | 0.347 | Likely Benign | -7.64 | Deleterious | 0.999 | Probably Damaging | 0.950 | Probably Damaging | 3.77 | Benign | 0.01 | Affected | 0.4439 | 0.0903 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.121C>T | R41C 2D AIThe SynGAP1 missense variant R41C is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33423530‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar) and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and no result is available from Foldetta (protein‑folding stability). Taken together, the majority of evidence points to a benign impact for R41C, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.311707 | Structured | 0.431757 | Uncertain | 0.344 | 0.765 | 0.375 | Conflicting | 3 | 6-33423530-C-T | 7 | 4.34e-6 | -4.745 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -1.10 | Neutral | 0.976 | Probably Damaging | 0.919 | Probably Damaging | 4.13 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3485 | 0.4520 | -4 | -3 | 7.0 | -53.05 | ||||||||||||||||||||||||||||||||
| c.1232T>A | I411N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I411N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.339366 | Uncertain | 0.927 | 0.198 | 0.000 | -14.426 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 3.00 | Destabilizing | 0.1 | 3.71 | Destabilizing | 3.36 | Destabilizing | 2.16 | Destabilizing | 0.556 | Likely Pathogenic | -6.42 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 0.0738 | 0.0700 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1238C>T | P413L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P413L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM. Those that predict a pathogenic effect comprise SGM‑Consensus, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Rosetta and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.332472 | Uncertain | 0.927 | 0.201 | 0.000 | -12.735 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.61 | Destabilizing | 0.4 | 1.34 | Ambiguous | 1.98 | Ambiguous | 0.30 | Likely Benign | 0.461 | Likely Benign | -9.21 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.19 | Benign | 0.00 | Affected | 0.2056 | 0.5614 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1247T>C | L416P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L416P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions from REVEL, SIFT, and FATHMM; pathogenic predictions from the remaining 13 tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen2_HumDiv, polyPhen2_HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, and the SGM Consensus). High‑accuracy methods specifically indicate pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No predictions are inconclusive. Based on the overall consensus of the majority of tools and the high‑accuracy methods, the variant is most likely pathogenic, which is consistent with the lack of ClinVar reporting and gnomAD absence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.336105 | Uncertain | 0.935 | 0.227 | 0.000 | -8.859 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 2.59 | Destabilizing | 0.1 | 6.23 | Destabilizing | 4.41 | Destabilizing | 1.27 | Destabilizing | 0.284 | Likely Benign | -3.56 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.35 | Benign | 0.21 | Tolerated | 0.3589 | 0.1353 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1249T>G | Y417D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y417D is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.346865 | Uncertain | 0.958 | 0.250 | 0.000 | -14.955 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.13 | Destabilizing | 0.1 | 5.37 | Destabilizing | 5.25 | Destabilizing | 2.43 | Destabilizing | 0.688 | Likely Pathogenic | -9.55 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.96 | Benign | 0.00 | Affected | 0.4388 | 0.0400 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1253A>T | K418I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K418I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM. In contrast, the majority of tools predict a pathogenic outcome: SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta provide uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as Uncertain. Overall, the preponderance of evidence points to a pathogenic effect for K418I, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.360134 | Uncertain | 0.948 | 0.263 | 0.000 | -14.895 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.51 | Ambiguous | 0.0 | 0.64 | Ambiguous | 0.58 | Ambiguous | 0.27 | Likely Benign | 0.428 | Likely Benign | -7.27 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.32 | Benign | 0.01 | Affected | 0.1297 | 0.2432 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.125C>T | P42L 2D AIThe SynGAP1 missense variant P42L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | -3.781 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -1.87 | Neutral | 0.909 | Possibly Damaging | 0.927 | Probably Damaging | 4.19 | Benign | 0.00 | Affected | 0.2312 | 0.5560 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.1267T>G | Y423D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y423D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, while the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.421885 | Uncertain | 0.975 | 0.242 | 0.000 | -13.279 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 4.67 | Destabilizing | 0.1 | 4.79 | Destabilizing | 4.73 | Destabilizing | 1.78 | Destabilizing | 0.553 | Likely Pathogenic | -8.28 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.38 | Benign | 0.04 | Affected | 0.3262 | 0.0412 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1271T>A | V424D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V424D is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors indicates a pathogenic effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, whereas only FATHMM predicts a benign outcome. High‑accuracy tools reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels it likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. No prediction is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -15.531 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.60 | Destabilizing | 0.1 | 3.50 | Destabilizing | 3.55 | Destabilizing | 2.13 | Destabilizing | 0.615 | Likely Pathogenic | -5.87 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.1536 | 0.0845 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1271T>G | V424G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V424G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM. All other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic consensus (3 pathogenic vs. 1 benign); and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. No predictions are missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -13.697 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 3.90 | Destabilizing | 0.1 | 4.84 | Destabilizing | 4.37 | Destabilizing | 2.44 | Destabilizing | 0.622 | Likely Pathogenic | -6.26 | Deleterious | 0.994 | Probably Damaging | 1.000 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.1396 | 0.2029 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1277A>T | N426I 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant N426I has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and the protein‑folding stability method Foldetta. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicting pathogenicity, and Foldetta indicating a benign folding stability outcome. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.394941 | Uncertain | 0.959 | 0.287 | 0.000 | -10.158 | Likely Pathogenic | 0.570 | Likely Pathogenic | Likely Benign | 0.67 | Ambiguous | 0.0 | 0.14 | Likely Benign | 0.41 | Likely Benign | 0.23 | Likely Benign | 0.216 | Likely Benign | -5.71 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 3.31 | Benign | 0.09 | Tolerated | 0.0656 | 0.3244 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.127G>T | G43C 2D AIThe SynGAP1 missense variant G43C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431462 | Uncertain | 0.396 | 0.762 | 0.375 | -4.599 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.90 | Neutral | 0.987 | Probably Damaging | 0.750 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.1445 | 0.3438 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.1280A>T | H427L 2D 3DClick to see structure in 3D Viewer AISynGAP1 H427L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: nine tools (REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Optimized) predict a benign effect, while five tools (SGM‑Consensus, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default) predict pathogenicity. High‑accuracy methods provide a more focused view: AlphaMissense‑Optimized indicates a benign outcome; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, remains pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. Taken together, the majority of evidence supports a benign impact for H427L, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.081712 | Structured | 0.394261 | Uncertain | 0.962 | 0.287 | 0.000 | -9.691 | Likely Pathogenic | 0.755 | Likely Pathogenic | Likely Benign | -0.17 | Likely Benign | 0.0 | 0.05 | Likely Benign | -0.06 | Likely Benign | 0.31 | Likely Benign | 0.272 | Likely Benign | -5.38 | Deleterious | 0.299 | Benign | 0.033 | Benign | 3.42 | Benign | 0.01 | Affected | 0.0942 | 0.4953 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.1282T>G | Y428D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Y428D is not reported in ClinVar and is absent from gnomAD. Across a broad panel of in silico predictors, 15 tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) uniformly predict a deleterious effect, whereas only FATHMM classifies it as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote) is likely pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic effect. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.389652 | Uncertain | 0.965 | 0.292 | 0.000 | -13.473 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.24 | Destabilizing | 0.0 | 4.19 | Destabilizing | 3.72 | Destabilizing | 1.64 | Destabilizing | 0.554 | Likely Pathogenic | -9.55 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.50 | Benign | 0.01 | Affected | 0.4458 | 0.1003 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1285C>T | R429W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R429W is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33438190‑C‑T). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b; premPS and AlphaMissense‑Default are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of predictions lean toward a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.390504 | Uncertain | 0.959 | 0.290 | 0.000 | Conflicting | 5 | 6-33438190-C-T | 65 | 4.03e-5 | -10.666 | Likely Pathogenic | 0.500 | Ambiguous | Likely Benign | 0.31 | Likely Benign | 0.1 | -0.13 | Likely Benign | 0.09 | Likely Benign | 0.52 | Ambiguous | 0.282 | Likely Benign | -3.19 | Deleterious | 1.000 | Probably Damaging | 0.990 | Probably Damaging | 3.41 | Benign | 0.03 | Affected | 3.38 | 25 | 0.1232 | 0.3422 | 2 | -3 | 3.6 | 30.03 | 252.3 | 45.5 | 0.0 | 0.0 | 0.2 | 0.1 | X | Potentially Pathogenic | The guanidinium group of Arg429, located in an α helix (res. Met414-Glu436), either forms a salt bridge with the carboxylate group of an acidic residue (Asp474, Asp467) or a H-bond with the hydroxyl group of Ser471 in an opposing α helix (res. Ala461-Phe476). In the variant simulations, the indole ring of the Trp429 side chain cannot form ionic interactions with the acidic residues. Although it forms a H-bond with Ser471, the bonding is not as strong as that of arginine. The residue swap could affect the tertiary structure assembly during folding; however, no large-scale negative effects were seen during the simulations. | ||||||||||||||
| c.128G>T | G43V 2D AIThe SynGAP1 missense variant G43V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431462 | Uncertain | 0.396 | 0.762 | 0.375 | -3.745 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -0.79 | Neutral | 0.320 | Benign | 0.140 | Benign | 4.28 | Benign | 0.00 | Affected | 0.1156 | 0.3120 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.1292T>C | L431P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L431P (ClinVar ID 661045.0) is reported as Pathogenic and is not present in gnomAD. Prediction tools that classify the variant as benign include only FATHMM. All other evaluated tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Pathogenic. Based on the overwhelming consensus of pathogenic predictions and the ClinVar designation, the variant is most likely pathogenic, with no contradiction to its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.094817 | Structured | 0.374755 | Uncertain | 0.959 | 0.300 | 0.000 | Likely Pathogenic | 1 | -14.222 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 6.78 | Destabilizing | 0.3 | 11.59 | Destabilizing | 9.19 | Destabilizing | 2.29 | Destabilizing | 0.659 | Likely Pathogenic | -6.39 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.91 | Benign | 0.05 | Affected | 3.37 | 29 | 0.3484 | 0.2163 | -3 | -3 | -5.4 | -16.04 | 222.4 | 62.8 | 0.1 | 0.0 | 0.1 | 0.0 | X | Potentially Pathogenic | The iso-butyl side chain of Leu431, located in an α helix (res. Met414-Glu436), packs against other hydrophobic residues in an interhelix space (e.g., Val434, Leu435, Leu696, Leu711) in the WT simulations. While the backbone amide group of Leu431 forms an H-bond with the carbonyl group of His427, the cyclic five-membered pyrrolidine ring of Pro431, lacking the necessary amide group, cannot do the same. Thus, although the cyclic five-membered pyrrolidine ring of Pro431 packs almost as favorably as the side chain of Leu431 in the hydrophobic niche, the residue swap causes the α helix to partially unfold in the variant simulations. | ||||||||||||||||
| c.1294T>C | C432R 2D 3DClick to see structure in 3D Viewer AIClinVar has no entry for this SynGAP1 missense variant, and it is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM. The majority of algorithms predict a pathogenic impact: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). FoldX and Foldetta report uncertain results. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, while Foldetta remains inconclusive. Overall, the consensus of the available predictions indicates that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.362533 | Uncertain | 0.960 | 0.285 | 0.000 | -13.858 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.03 | Ambiguous | 0.2 | 2.05 | Destabilizing | 1.54 | Ambiguous | 1.73 | Destabilizing | 0.690 | Likely Pathogenic | -11.46 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.45 | Benign | 0.01 | Affected | 0.1359 | 0.1766 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.1294T>G | C432G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C432G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM. Those that predict a pathogenic effect comprise SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Predictions that are inconclusive or uncertain are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points to a pathogenic impact. This conclusion is consistent with the absence of a ClinVar classification; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.362533 | Uncertain | 0.960 | 0.285 | 0.000 | -10.247 | Likely Pathogenic | 0.831 | Likely Pathogenic | Ambiguous | 1.37 | Ambiguous | 0.0 | 2.25 | Destabilizing | 1.81 | Ambiguous | 1.60 | Destabilizing | 0.631 | Likely Pathogenic | -11.46 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.45 | Benign | 0.03 | Affected | 0.2796 | 0.2130 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1301T>A | V434D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign are SIFT and FATHMM; all other evaluated algorithms—including REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, and ESM1b—predict it to be pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized returns a pathogenic score; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among pathogenic predictors and the corroborating high‑accuracy tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | -14.765 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 3.26 | Destabilizing | 0.0 | 3.33 | Destabilizing | 3.30 | Destabilizing | 1.78 | Destabilizing | 0.592 | Likely Pathogenic | -5.18 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.38 | Benign | 0.13 | Tolerated | 0.1307 | 0.0741 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1301T>G | V434G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are SIFT and FATHMM, while the remaining tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default) all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as pathogenic. Taken together, the overwhelming majority of evidence indicates a deleterious effect. Therefore, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | -12.519 | Likely Pathogenic | 0.809 | Likely Pathogenic | Ambiguous | 3.08 | Destabilizing | 0.0 | 4.37 | Destabilizing | 3.73 | Destabilizing | 1.91 | Destabilizing | 0.564 | Likely Pathogenic | -5.16 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.39 | Benign | 0.07 | Tolerated | 0.1758 | 0.2408 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.130T>A | W44R 2D AIThe SynGAP1 W44R missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of tools (seven pathogenic vs. three benign) indicate a pathogenic impact. Thus, the variant is most likely pathogenic, and this prediction does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -4.850 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.291 | Likely Benign | -5.06 | Deleterious | 0.943 | Possibly Damaging | 0.888 | Possibly Damaging | 3.16 | Benign | 0.00 | Affected | 0.4268 | 0.0749 | 2 | -3 | -3.6 | -30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.130T>C | W44R 2D AISynGAP1 W44R is not reported in ClinVar and is absent from gnomAD. Consensus from standard predictors shows a split: benign calls come from REVEL, ESM1b, and FATHMM, while pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessment focuses on AlphaMissense‑Optimized, which predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive, and Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -4.850 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.291 | Likely Benign | -5.06 | Deleterious | 0.943 | Possibly Damaging | 0.888 | Possibly Damaging | 3.16 | Benign | 0.00 | Affected | 0.4268 | 0.0749 | 2 | -3 | -3.6 | -30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.1310C>T | P437L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P437L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. Predictions that are inconclusive or uncertain are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts a likely pathogenic outcome, while AlphaMissense‑Optimized and Foldetta are uncertain. Taken together, the majority of evidence points toward a pathogenic impact for P437L, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.306196 | Uncertain | 0.921 | 0.298 | 0.000 | -13.554 | Likely Pathogenic | 0.787 | Likely Pathogenic | Ambiguous | 1.10 | Ambiguous | 0.0 | -3.52 | Stabilizing | -1.21 | Ambiguous | 0.35 | Likely Benign | 0.324 | Likely Benign | -8.48 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.67 | Benign | 0.04 | Affected | 0.2228 | 0.6354 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1316T>C | L439P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L439P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a deleterious effect: pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a benign outcome. High‑accuracy methods reinforce the pathogenic view: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.281542 | Uncertain | 0.942 | 0.265 | 0.000 | -9.929 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 6.30 | Destabilizing | 0.2 | 7.62 | Destabilizing | 6.96 | Destabilizing | 1.28 | Destabilizing | 0.539 | Likely Pathogenic | -5.72 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.21 | Benign | 0.02 | Affected | 0.3426 | 0.1192 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1319A>T | N440I 2D AISynGAP1 missense variant N440I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. Uncertain results are reported by FoldX, Rosetta, and Foldetta. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as likely pathogenic; Foldetta remains inconclusive. Overall, the balance of evidence favors a pathogenic interpretation, and this conclusion does not contradict the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.267204 | Uncertain | 0.929 | 0.245 | 0.000 | -10.365 | Likely Pathogenic | 0.778 | Likely Pathogenic | Likely Benign | 0.97 | Ambiguous | 0.9 | 1.10 | Ambiguous | 1.04 | Ambiguous | 0.10 | Likely Benign | 0.100 | Likely Benign | -4.07 | Deleterious | 0.322 | Benign | 0.109 | Benign | 3.47 | Benign | 0.03 | Affected | 0.0554 | 0.3772 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.131G>C | W44S 2D AIThe SynGAP1 missense variant W44S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default) predict a pathogenic impact; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, the balance of evidence favors a pathogenic classification, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -5.113 | Likely Benign | 0.921 | Likely Pathogenic | Ambiguous | 0.275 | Likely Benign | -4.68 | Deleterious | 0.824 | Possibly Damaging | 0.775 | Possibly Damaging | 3.16 | Benign | 0.00 | Affected | 0.4139 | 0.2371 | -2 | -3 | 0.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.1322T>A | V441D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V441D is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and Foldetta, whereas a majority of tools (SGM Consensus, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. FoldX and Rosetta are inconclusive, and AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show that the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenicity, while Foldetta predicts benign stability. Overall, the balance of evidence leans toward pathogenicity, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -15.392 | Likely Pathogenic | 0.934 | Likely Pathogenic | Ambiguous | -0.57 | Ambiguous | 0.1 | 0.56 | Ambiguous | -0.01 | Likely Benign | 1.15 | Destabilizing | 0.308 | Likely Benign | -6.07 | Deleterious | 1.000 | Probably Damaging | 0.959 | Probably Damaging | 3.38 | Benign | 0.10 | Tolerated | 0.1232 | 0.0698 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1322T>G | V441G 2D 3DClick to see structure in 3D Viewer AISynGAP1 variant V441G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into three groups: benign predictions come from REVEL, FoldX, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; the remaining tools (Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized remains benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) favors pathogenic, and Foldetta is inconclusive. Taken together, the majority of evidence points to a benign effect, but the SGM Consensus and several individual pathogenic predictors introduce uncertainty. Therefore, the variant is most likely benign based on the overall prediction landscape, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -12.380 | Likely Pathogenic | 0.478 | Ambiguous | Likely Benign | 0.18 | Likely Benign | 0.0 | 1.25 | Ambiguous | 0.72 | Ambiguous | 0.95 | Ambiguous | 0.273 | Likely Benign | -5.88 | Deleterious | 0.841 | Possibly Damaging | 0.997 | Probably Damaging | 3.41 | Benign | 0.24 | Tolerated | 0.1664 | 0.1715 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1325A>T | K442I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K442I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy assessments are: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of predictions, including the two high‑accuracy pathogenic calls, indicate a pathogenic impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.170161 | Structured | 0.255766 | Uncertain | 0.912 | 0.225 | 0.000 | -14.921 | Likely Pathogenic | 0.965 | Likely Pathogenic | Likely Pathogenic | 0.16 | Likely Benign | 0.1 | -0.16 | Likely Benign | 0.00 | Likely Benign | 0.30 | Likely Benign | 0.350 | Likely Benign | -6.91 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.37 | Benign | 0.02 | Affected | 0.0943 | 0.3073 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1327G>T | G443C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The remaining tools are inconclusive: Foldetta is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta remains uncertain. Overall, the consensus of the majority of evidence points to a benign impact for G443C, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -4.308 | Likely Benign | 0.208 | Likely Benign | Likely Benign | -0.06 | Likely Benign | 0.0 | -0.95 | Ambiguous | -0.51 | Ambiguous | -0.10 | Likely Benign | 0.260 | Likely Benign | -2.94 | Deleterious | 0.977 | Probably Damaging | 0.504 | Possibly Damaging | 3.37 | Benign | 0.16 | Tolerated | 0.1258 | 0.2782 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1328G>T | G443V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Overall, the majority of evidence points to a benign impact for G443V, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -4.130 | Likely Benign | 0.274 | Likely Benign | Likely Benign | 0.17 | Likely Benign | 0.2 | -2.19 | Stabilizing | -1.01 | Ambiguous | 0.21 | Likely Benign | 0.099 | Likely Benign | -2.90 | Deleterious | 0.585 | Possibly Damaging | 0.195 | Benign | 3.36 | Benign | 0.12 | Tolerated | 0.1089 | 0.3375 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1340T>A | V447D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant V447D lies in the GAP domain. ClinVar has no entry for this change, and it is absent from gnomAD. Prediction tools that agree on benign impact are REVEL and FATHMM, whereas the remaining predictors—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classify the variant as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta also reports a pathogenic effect. Overall, the evidence points to a pathogenic effect for V447D, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.283801 | Uncertain | 0.970 | 0.243 | 0.000 | -16.643 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.34 | Destabilizing | 0.1 | 3.59 | Destabilizing | 3.97 | Destabilizing | 2.29 | Destabilizing | 0.491 | Likely Benign | -5.33 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.30 | Benign | 0.01 | Affected | 0.1424 | 0.0541 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1340T>G | V447G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V447G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and FATHMM, while the remaining tools—FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. No prediction or stability result is missing or inconclusive. Overall, the preponderance of evidence indicates that V447G is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.250310 | Structured | 0.283801 | Uncertain | 0.970 | 0.243 | 0.000 | -13.648 | Likely Pathogenic | 0.861 | Likely Pathogenic | Ambiguous | 3.81 | Destabilizing | 0.1 | 4.62 | Destabilizing | 4.22 | Destabilizing | 2.28 | Destabilizing | 0.499 | Likely Benign | -5.43 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.1863 | 0.2240 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.134A>T | N45I 2D AIThe SynGAP1 missense variant N45I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.401658 | Structured | 0.431853 | Uncertain | 0.498 | 0.741 | 0.375 | -4.063 | Likely Benign | 0.568 | Likely Pathogenic | Likely Benign | 0.147 | Likely Benign | -1.32 | Neutral | 0.943 | Possibly Damaging | 0.924 | Probably Damaging | 4.04 | Benign | 0.00 | Affected | 0.0861 | 0.7406 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.1352T>C | L451P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L451P is reported in ClinVar as Pathogenic (ClinVar ID 3064222.0) and is not found in gnomAD. Prediction tools that assess functional impact uniformly classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on these predictions, the variant is most likely pathogenic, and this conclusion aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.281712 | Structured | 0.314017 | Uncertain | 0.978 | 0.232 | 0.000 | Likely Pathogenic | 1 | -14.549 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.92 | Destabilizing | 0.2 | 8.57 | Destabilizing | 7.75 | Destabilizing | 2.58 | Destabilizing | 0.750 | Likely Pathogenic | -6.81 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.43 | Pathogenic | 0.00 | Affected | 3.37 | 34 | 0.2823 | 0.1221 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||
| c.1355T>A | V452D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V452D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, while all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy predictors, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.335645 | Structured | 0.315167 | Uncertain | 0.970 | 0.229 | 0.000 | -15.793 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.92 | Destabilizing | 0.1 | 3.37 | Destabilizing | 3.65 | Destabilizing | 2.52 | Destabilizing | 0.630 | Likely Pathogenic | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.16 | Benign | 0.00 | Affected | 0.1320 | 0.0610 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1355T>G | V452G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V452G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. AlphaMissense‑Optimized remains uncertain. Overall, the preponderance of evidence indicates that V452G is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.335645 | Structured | 0.315167 | Uncertain | 0.970 | 0.229 | 0.000 | -13.410 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 3.70 | Destabilizing | 0.1 | 3.37 | Destabilizing | 3.54 | Destabilizing | 2.46 | Destabilizing | 0.570 | Likely Pathogenic | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.17 | Benign | 0.00 | Affected | 0.1948 | 0.2567 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1358A>T | H453L 2D 3DClick to see structure in 3D Viewer AISynGAP1 H453L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, premPS, SIFT, FATHMM, and the protein‑folding stability method Foldetta; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments indicate that AlphaMissense‑Optimized is uncertain, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta predicts benign. Overall, the predictions are split, with a slight edge toward pathogenicity from the consensus and high‑accuracy tools, but the presence of several benign calls and the uncertainty of AlphaMissense‑Optimized temper this conclusion. Thus, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.352862 | Structured | 0.316097 | Uncertain | 0.946 | 0.200 | 0.000 | -10.438 | Likely Pathogenic | 0.834 | Likely Pathogenic | Ambiguous | -0.59 | Ambiguous | 0.1 | 0.04 | Likely Benign | -0.28 | Likely Benign | 0.31 | Likely Benign | 0.413 | Likely Benign | -10.87 | Deleterious | 0.998 | Probably Damaging | 0.973 | Probably Damaging | 3.45 | Benign | 0.11 | Tolerated | 0.0841 | 0.4964 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.1361T>A | I454N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I454N resides in the GAP domain. ClinVar has no entry for this variant, and it is absent from gnomAD. Prediction tools that agree on benign impact are REVEL and FATHMM, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict pathogenicity. High‑accuracy methods further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic, and Foldetta predicts a destabilizing, pathogenic change. Overall, the evidence strongly favors a pathogenic classification, and this assessment does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.312811 | Uncertain | 0.965 | 0.182 | 0.000 | -14.246 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.24 | Destabilizing | 0.1 | 3.56 | Destabilizing | 3.90 | Destabilizing | 2.44 | Destabilizing | 0.497 | Likely Benign | -6.82 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.25 | Benign | 0.00 | Affected | 0.0750 | 0.0412 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1364T>C | L455P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L455P is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.310377 | Uncertain | 0.963 | 0.168 | 0.000 | -14.150 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 7.45 | Destabilizing | 0.2 | 11.99 | Destabilizing | 9.72 | Destabilizing | 2.19 | Destabilizing | 0.695 | Likely Pathogenic | -6.72 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.23 | Benign | 0.00 | Affected | 0.3211 | 0.1221 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1375G>T | G459C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G459C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the majority of algorithms (SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. Uncertain or inconclusive results come from Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Taken together, the preponderance of evidence points to a pathogenic effect for G459C, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.289888 | Uncertain | 0.903 | 0.150 | 0.125 | -12.109 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 2.46 | Destabilizing | 0.2 | 1.49 | Ambiguous | 1.98 | Ambiguous | 0.65 | Ambiguous | 0.586 | Likely Pathogenic | -8.46 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.00 | Benign | 0.00 | Affected | 0.0960 | 0.4103 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1376G>T | G459V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G459V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact; premPS is inconclusive and therefore not counted. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the consensus of these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for G459V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.289888 | Uncertain | 0.903 | 0.150 | 0.125 | -13.606 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.79 | Destabilizing | 0.1 | 5.01 | Destabilizing | 4.40 | Destabilizing | 0.71 | Ambiguous | 0.605 | Likely Pathogenic | -8.46 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.99 | Benign | 0.00 | Affected | 0.0997 | 0.4240 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.137C>T | P46L 2D AIThe SynGAP1 missense variant P46L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for P46L, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.390993 | Structured | 0.433588 | Uncertain | 0.549 | 0.741 | 0.375 | -5.135 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 0.076 | Likely Benign | -1.14 | Neutral | 0.909 | Possibly Damaging | 0.927 | Probably Damaging | 4.12 | Benign | 0.00 | Affected | 0.2469 | 0.6633 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.1387G>T | D463Y 2D 3DClick to see structure in 3D Viewer AIClinVar reports no entry for this SynGAP1 D463Y variant, and it is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Uncertain results come from AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of conventional tools lean toward pathogenicity, while the high‑accuracy Foldetta suggests benign. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | -14.387 | Likely Pathogenic | 0.904 | Likely Pathogenic | Ambiguous | -0.14 | Likely Benign | 0.1 | 0.77 | Ambiguous | 0.32 | Likely Benign | 0.07 | Likely Benign | 0.399 | Likely Benign | -7.95 | Deleterious | 0.998 | Probably Damaging | 0.904 | Possibly Damaging | 3.35 | Benign | 0.14 | Tolerated | 0.0540 | 0.6213 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1388A>T | D463V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D463V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, premPS, SIFT, and FATHMM, whereas those that agree on a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Uncertain predictions come from Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points toward a pathogenic impact for D463V, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | -12.374 | Likely Pathogenic | 0.880 | Likely Pathogenic | Ambiguous | 0.23 | Likely Benign | 0.1 | 0.98 | Ambiguous | 0.61 | Ambiguous | 0.33 | Likely Benign | 0.521 | Likely Pathogenic | -7.95 | Deleterious | 0.973 | Probably Damaging | 0.658 | Possibly Damaging | 3.31 | Benign | 0.09 | Tolerated | 0.0713 | 0.5526 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1394T>A | L465H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L465H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic. Based on the consensus of all available predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.346032 | Structured | 0.319240 | Uncertain | 0.956 | 0.202 | 0.000 | -16.751 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.15 | Destabilizing | 0.4 | 2.29 | Destabilizing | 2.72 | Destabilizing | 2.54 | Destabilizing | 0.764 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.1289 | 0.1089 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1394T>C | L465P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L465P is listed in ClinVar as Pathogenic (ClinVar ID 1067821.0) and is not reported in gnomAD. Prediction tools that assess functional impact uniformly classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this conclusion aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.346032 | Structured | 0.319240 | Uncertain | 0.956 | 0.202 | 0.000 | Likely Pathogenic | 1 | -14.824 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 7.18 | Destabilizing | 0.3 | 10.85 | Destabilizing | 9.02 | Destabilizing | 2.73 | Destabilizing | 0.778 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 3.37 | 34 | 0.3387 | 0.1582 | -3 | -3 | -5.4 | -16.04 | 211.1 | 65.9 | 0.1 | 0.0 | -0.2 | 0.0 | X | Potentially Pathogenic | The iso-butyl side chain of Leu465, located in the middle of an α helix (res. Ala461–Phe476), packs with hydrophobic residues (e.g., Phe464, Met468, Tyr497, Ile494) in an inter-helix space formed with two other α helices (res. Ala461–Phe476 and res. Thr488-Gly502). In the variant simulations, the cyclic five-membered pyrrolidine ring of Pro465 is not as optimal as the side chain of Leu465 for filling the three α helix hydrophobic niche. Although the residue swap does not cause a large-scale conformational shift during the simulations, the H-bond between the backbone amide group of Leu465 and the backbone carbonyl group of Ala461 is lost. This, in turn, breaks the continuity of the α helix secondary structure element. | ||||||||||||||||
| c.1399G>T | D467Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D467Y is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from FoldX, Foldetta, premPS, and SIFT, whereas pathogenic calls are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy methods give a clearer picture: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. No evidence from Rosetta is available due to its uncertain status. Overall, the majority of high‑confidence predictions lean toward pathogenicity, and this does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.329932 | Uncertain | 0.940 | 0.246 | 0.000 | -14.373 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.36 | Likely Benign | 0.1 | -0.60 | Ambiguous | -0.12 | Likely Benign | 0.07 | Likely Benign | 0.846 | Likely Pathogenic | -8.70 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.28 | Pathogenic | 0.07 | Tolerated | 0.0517 | 0.5521 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.13C>G | R5G 2D  AIThe SynGAP1 missense variant R5G is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.547847 | Binding | 0.363 | 0.920 | 0.750 | Uncertain | 1 | -3.639 | Likely Benign | 0.150 | Likely Benign | Likely Benign | 0.169 | Likely Benign | -0.16 | Neutral | 0.013 | Benign | 0.003 | Benign | 4.12 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3760 | 0.4332 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||
| c.1400A>T | D467V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D467V missense variant is not reported in ClinVar or gnomAD. Prediction tools cluster into two groups: benign predictions come from Rosetta and premPS, while the majority—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—label the change as pathogenic. Two tools, FoldX and Foldetta, give uncertain results. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus indicates likely pathogenic, and Foldetta remains uncertain. Overall, the consensus of high‑confidence predictors points to a pathogenic effect, and this conclusion is consistent with the absence of any ClinVar annotation or gnomAD observation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.329932 | Uncertain | 0.940 | 0.246 | 0.000 | -15.041 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 1.24 | Ambiguous | 0.1 | 0.08 | Likely Benign | 0.66 | Ambiguous | 0.02 | Likely Benign | 0.893 | Likely Pathogenic | -8.70 | Deleterious | 0.997 | Probably Damaging | 0.997 | Probably Damaging | -1.28 | Pathogenic | 0.02 | Affected | 0.0644 | 0.5274 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1418T>G | V473G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V473G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, while the remaining eleven tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) uniformly predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) votes pathogenic, and Foldetta also predicts pathogenic. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic based on the collective evidence, and this conclusion does not contradict any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.191378 | Structured | 0.362529 | Uncertain | 0.884 | 0.239 | 0.000 | -14.782 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 3.45 | Destabilizing | 0.0 | 3.40 | Destabilizing | 3.43 | Destabilizing | 2.69 | Destabilizing | 0.683 | Likely Pathogenic | -6.91 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.13 | Benign | 0.00 | Affected | 0.1885 | 0.2567 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1420G>T | D474Y 2D AIThe SynGAP1 missense variant D474Y is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include FoldX, Foldetta, and premPS, whereas the majority of tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as benign. Overall, the preponderance of evidence points to a pathogenic effect for D474Y. This conclusion does not contradict ClinVar status, as the variant is currently unreported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.373433 | Uncertain | 0.864 | 0.255 | 0.000 | -14.647 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.27 | Likely Benign | 0.5 | -0.74 | Ambiguous | -0.24 | Likely Benign | 0.20 | Likely Benign | 0.864 | Likely Pathogenic | -7.72 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.30 | Pathogenic | 0.01 | Affected | 0.0493 | 0.4237 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1421A>T | D474V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D474V missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that indicate a benign effect include Rosetta, Foldetta, and premPS, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as benign. Overall, the balance of evidence favors a pathogenic classification, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.373433 | Uncertain | 0.864 | 0.255 | 0.000 | -12.999 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.77 | Ambiguous | 0.0 | 0.18 | Likely Benign | 0.48 | Likely Benign | 0.18 | Likely Benign | 0.866 | Likely Pathogenic | -7.69 | Deleterious | 0.998 | Probably Damaging | 0.997 | Probably Damaging | -1.30 | Pathogenic | 0.04 | Affected | 0.0694 | 0.4510 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1423C>T | R475W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R475W is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33438455‑C‑T). Prediction tools that agree on a benign effect include only Foldetta, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus) uniformly predict a pathogenic impact; FoldX, Rosetta, and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic effect, which does not contradict the ClinVar “Uncertain” classification but suggests that the variant is more likely pathogenic rather than benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.382696 | Uncertain | 0.852 | 0.261 | 0.000 | Uncertain | 1 | 6-33438455-C-T | 1 | 6.20e-7 | -13.235 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 1.44 | Ambiguous | 0.4 | -0.92 | Ambiguous | 0.26 | Likely Benign | 0.56 | Ambiguous | 0.725 | Likely Pathogenic | -7.56 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | -1.45 | Pathogenic | 0.00 | Affected | 3.39 | 28 | 0.1231 | 0.2785 | 2 | -3 | 3.6 | 30.03 | 266.9 | 39.6 | 0.0 | 0.0 | 0.0 | 0.1 | X | X | X | Potentially Pathogenic | In the WT simulations, the guanidinium group of Arg475, located near the end of an α-helix (res. Ala461-Phe476), stacks with the phenyl ring of Phe476 and forms a salt bridge with Glu472. Additionally, Arg475 occasionally forms another salt bridge with the carboxylate group of Glu486 on the α-α loop connecting the two α-helices (res. Ala461-Phe476 and Leu489-Glu519) at the GAP-Ras interface. Therefore, Arg475 potentially plays a key role in positioning the loop by interacting with Glu486, which is necessary for the positioning of the “arginine finger” (Arg485) and, ultimately, for RasGTPase activation.In the variant simulations, Trp475 moves and stacks with Arg479 on the proceeding α-α loop, disrupting the terminal end of the α-helix. Lastly, the potential effect of the residue swap on the SynGAP-Ras complex formation or GTPase activation cannot be fully addressed using the SynGAP solvent-only simulations. | |||||||||||
| c.1435C>G | R479G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R479G is reported in gnomAD (ID 6-33438467-C-G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise AlphaMissense‑Default, ESM1b, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, Rosetta, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). FoldX and Foldetta, which assess protein‑folding stability, returned uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM Consensus as pathogenic, and Foldetta as inconclusive. Overall, the majority of computational evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Therefore, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.216401 | Structured | 0.419256 | Uncertain | 0.820 | 0.249 | 0.000 | 6-33438467-C-G | 1 | 6.20e-7 | -8.600 | Likely Pathogenic | 0.624 | Likely Pathogenic | Likely Benign | 0.97 | Ambiguous | 0.0 | 2.51 | Destabilizing | 1.74 | Ambiguous | 0.71 | Ambiguous | 0.228 | Likely Benign | -2.82 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.43 | Benign | 0.42 | Tolerated | 3.39 | 32 | 0.2911 | 0.2707 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1442A>T | H481L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H481L missense variant is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default; FoldX is uncertain and therefore not counted. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or stability result is missing or inconclusive. Overall, the majority of tools (seven benign vs five pathogenic) lean toward a benign interpretation, and this does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign, though a subset of high‑accuracy predictors suggest pathogenicity, indicating some uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.257454 | Structured | 0.430977 | Uncertain | 0.764 | 0.247 | 0.000 | -9.097 | Likely Pathogenic | 0.587 | Likely Pathogenic | Likely Benign | -0.58 | Ambiguous | 0.1 | 0.15 | Likely Benign | -0.22 | Likely Benign | 0.29 | Likely Benign | 0.349 | Likely Benign | -5.91 | Deleterious | 0.995 | Probably Damaging | 0.986 | Probably Damaging | 3.41 | Benign | 0.48 | Tolerated | 0.0661 | 0.4678 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.1445T>A | L482H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L482H is not reported in ClinVar (ClinVar status: None) and has no entries in gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are absent; all available pathogenic predictors (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus vote (Likely Pathogenic) uniformly indicate a deleterious impact. Uncertain predictions come from FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as Uncertain. No evidence suggests a benign outcome. Consequently, the variant is most likely pathogenic based on the consensus of pathogenic predictions, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.426236 | Uncertain | 0.795 | 0.248 | 0.000 | -13.825 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 1.49 | Ambiguous | 0.0 | 1.44 | Ambiguous | 1.47 | Ambiguous | 1.64 | Destabilizing | 0.886 | Likely Pathogenic | -6.91 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.28 | Pathogenic | 0.00 | Affected | 0.1000 | 0.0879 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1445T>C | L482P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L482P is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify the substitution as pathogenic: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a pathogenic impact. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.426236 | Uncertain | 0.795 | 0.248 | 0.000 | -12.866 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.59 | Destabilizing | 0.1 | 10.87 | Destabilizing | 7.23 | Destabilizing | 1.55 | Destabilizing | 0.896 | Likely Pathogenic | -6.91 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.30 | Pathogenic | 0.01 | Affected | 0.2756 | 0.0825 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1448T>A | I483K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I483K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.415850 | Uncertain | 0.798 | 0.254 | 0.000 | -18.260 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.53 | Destabilizing | 0.1 | 5.15 | Destabilizing | 4.34 | Destabilizing | 2.01 | Destabilizing | 0.585 | Likely Pathogenic | -6.50 | Deleterious | 0.962 | Probably Damaging | 0.991 | Probably Damaging | 3.14 | Benign | 0.00 | Affected | 0.0868 | 0.0670 | -2 | -3 | -8.4 | 15.01 | |||||||||||||||||||||||||||||
| c.1448T>G | I483R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I483R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.415850 | Uncertain | 0.798 | 0.254 | 0.000 | -17.066 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 5.14 | Destabilizing | 0.4 | 4.09 | Destabilizing | 4.62 | Destabilizing | 1.97 | Destabilizing | 0.595 | Likely Pathogenic | -6.50 | Deleterious | 0.997 | Probably Damaging | 0.991 | Probably Damaging | 3.14 | Benign | 0.00 | Affected | 0.1120 | 0.0870 | -2 | -3 | -9.0 | 43.03 | |||||||||||||||||||||||||||||
| c.1453C>G | R485G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R485G is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (gnomAD ID: 6‑33438485‑C‑G). Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tools predict a benign outcome. Uncertain or inconclusive predictions come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Uncertain. Overall, the evidence strongly favors a pathogenic classification, and this conclusion does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.377409 | Uncertain | 0.805 | 0.246 | 0.125 | 6-33438485-C-G | 1 | 6.20e-7 | -15.777 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.84 | Ambiguous | 0.1 | 1.60 | Ambiguous | 1.22 | Ambiguous | 0.98 | Ambiguous | 0.631 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.3140 | 0.2678 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1453C>T | R485C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R485C (gnomAD ID 6‑33438485‑C‑T) is listed in ClinVar with an uncertain significance. Functional prediction tools largely disagree: benign calls come from Rosetta and premPS, whereas pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus is labeled likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) remains uncertain. With the majority of evidence pointing to pathogenicity and no contradictory data from ClinVar, the variant is most likely pathogenic, although ClinVar has not yet reached a definitive classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.377409 | Uncertain | 0.805 | 0.246 | 0.125 | Uncertain | 2 | 6-33438485-C-T | 9 | 5.58e-6 | -14.294 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 1.00 | Ambiguous | 0.1 | 0.26 | Likely Benign | 0.63 | Ambiguous | 0.44 | Likely Benign | 0.597 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.90 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.3350 | 0.2762 | -4 | -3 | 7.0 | -53.05 | 225.5 | 99.6 | -0.1 | 0.0 | -0.3 | 0.2 | X | Uncertain | The guanidinium group of Arg485 is located in a short helical structure (res. Glu480-Leu482) within an α-α loop connecting the two α-helices (res. Ala461-Phe476 and Leu489-Glu519) at the GAP-Ras interface. The side chain of Arg485 acts as the “arginine finger” of SynGAP, playing a crucial role in Ras-GTPase activation. Consequently, the residue swap inhibits the conversion of GTP to GDP at the enzyme’s active site. Although no negative effects on the protein structure are observed during the simulations, no definite conclusions can be drawn due to the critical role of Arg485 in GTPase activation. | |||||||||||||
| c.145T>C | C49R 2D AIThe SynGAP1 missense variant C49R has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain and no Foldetta (FoldX‑MD/Rosetta) result is available. Taken together, the preponderance of evidence points to a pathogenic effect, and this conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.209395 | Structured | 0.445316 | Uncertain | 0.541 | 0.704 | 0.000 | -10.000 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 0.320 | Likely Benign | -3.53 | Deleterious | 0.676 | Possibly Damaging | 0.761 | Possibly Damaging | 3.95 | Benign | 0.00 | Affected | 0.1605 | 0.2120 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||||||||||||
| c.145T>G | C49G 2D AIThe SynGAP1 missense variant C49G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic versus four benign) lean toward a pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant has no existing ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.209395 | Structured | 0.445316 | Uncertain | 0.541 | 0.704 | 0.000 | -6.464 | Likely Benign | 0.674 | Likely Pathogenic | Likely Benign | 0.351 | Likely Benign | -3.64 | Deleterious | 0.462 | Possibly Damaging | 0.599 | Possibly Damaging | 3.87 | Benign | 0.00 | Affected | 0.2419 | 0.2528 | -3 | -3 | -2.9 | -46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.1460A>T | N487I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N487I has no ClinVar entry and is not reported in gnomAD. Functional prediction tools largely disagree, but the majority indicate a deleterious effect. Benign predictions come from Rosetta, premPS, and FATHMM, whereas pathogenic predictions are reported by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are provided by FoldX and Foldetta. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also predicts pathogenic; Foldetta remains inconclusive. Overall, the preponderance of evidence supports a pathogenic classification for N487I, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.209395 | Structured | 0.338511 | Uncertain | 0.890 | 0.243 | 0.125 | -16.592 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.71 | Ambiguous | 0.1 | 0.13 | Likely Benign | 0.92 | Ambiguous | 0.33 | Likely Benign | 0.591 | Likely Pathogenic | -8.95 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.67 | Benign | 0.00 | Affected | 0.0633 | 0.3531 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.1466T>A | L489H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L489H is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic. No tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a destabilizing, pathogenic effect. All available predictions are concordant and supportive. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.326126 | Uncertain | 0.949 | 0.234 | 0.125 | -15.946 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.12 | Destabilizing | 0.2 | 2.13 | Destabilizing | 2.63 | Destabilizing | 1.99 | Destabilizing | 0.919 | Likely Pathogenic | -6.74 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.60 | Pathogenic | 0.00 | Affected | 0.1132 | 0.0871 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1466T>C | L489P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L489P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All evaluated in‑silico predictors classify the change as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. Overall, the variant is most likely pathogenic based on the consensus of predictive tools, a conclusion that contradicts the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.326126 | Uncertain | 0.949 | 0.234 | 0.125 | Conflicting | 2 | -13.520 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.50 | Destabilizing | 0.1 | 4.69 | Destabilizing | 3.60 | Destabilizing | 1.73 | Destabilizing | 0.939 | Likely Pathogenic | -6.74 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.56 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.3673 | 0.1474 | -3 | -3 | -5.4 | -16.04 | 209.9 | 61.9 | 0.1 | 0.0 | 0.6 | 0.1 | X | Potentially Pathogenic | The iso-butyl side chain of Leu489, located in the α-helix (res. Leu489-Glu519) within an inter-helix space of four helices (res. Ala461-Phe476, res. Val441-Ser457, and res. Met414-Glu436), packs with hydrophobic residues (e.g., Cys432, Ala448, Lys444, Ala493, Val447, Met468). In the variant simulations, Pro489 is located near the beginning of the α-helix, so the residue swap with Leu489 does not affect the continuity of the secondary structure element. However, the side chain of proline is not as optimal as that of leucine for maintaining hydrophobic packing with nearby residues (e.g., Ala448, Lys444). Additionally, the consistently maintained hydrogen bond interaction between the backbone amide group of Leu489 and the carbonyl of Glu436 is lost due to the residue swap, potentially affecting the tertiary structure integrity. | ||||||||||||||||
| c.1475A>T | K492I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K492I is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions from premPS and FATHMM; pathogenic predictions from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results come from FoldX, Rosetta, and Foldetta. High‑accuracy methods give a pathogenic verdict: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also predicts pathogenic (3/4 votes). Foldetta remains inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.327121 | Uncertain | 0.947 | 0.192 | 0.000 | -18.661 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | -0.54 | Ambiguous | 0.1 | -0.59 | Ambiguous | -0.57 | Ambiguous | 0.49 | Likely Benign | 0.645 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.94 | Benign | 0.00 | Affected | 0.0875 | 0.2810 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1481T>A | I494K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I494K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity uniformly classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool in the dataset predicts a benign outcome. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.353330 | Uncertain | 0.941 | 0.157 | 0.000 | -15.950 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 4.23 | Destabilizing | 0.2 | 5.33 | Destabilizing | 4.78 | Destabilizing | 1.89 | Destabilizing | 0.925 | Likely Pathogenic | -6.40 | Deleterious | 0.987 | Probably Damaging | 0.937 | Probably Damaging | -1.41 | Pathogenic | 0.00 | Affected | 0.1015 | 0.0870 | -2 | -3 | -8.4 | 15.01 | |||||||||||||||||||||||||||||
| c.1481T>G | I494R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I494R is listed in ClinVar as Pathogenic (ClinVar ID 1685460.0) and is not reported in gnomAD. Prediction tools that assess functional impact all converge on a pathogenic outcome: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate pathogenicity. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Thus, the variant is most likely pathogenic, and this prediction aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.353330 | Uncertain | 0.941 | 0.157 | 0.000 | Likely Pathogenic | 1 | -15.758 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 6.71 | Destabilizing | 0.3 | 3.40 | Destabilizing | 5.06 | Destabilizing | 2.19 | Destabilizing | 0.911 | Likely Pathogenic | -6.43 | Deleterious | 0.999 | Probably Damaging | 0.957 | Probably Damaging | -1.41 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.1180 | 0.0870 | -2 | -3 | -9.0 | 43.03 | 273.9 | -59.8 | 0.0 | 0.0 | 0.0 | 0.1 | X | X | X | X | Potentially Pathogenic | The sec-butyl side chain of Ile494, located in an α-helix (res. Leu489-Glu519), packs against hydrophobic residues (e.g., Phe484, Leu465, Trp572, Ala493, Met468) in an inter-helix space (res. Leu489-Glu519 and res. Ala461-Phe476). In the variant simulations, the bulkier and positively charged residue, Arg494, weakens the integrity of the opposing helix. Additionally, the bulkier Arg494 stacks with Phe484, causing the α-helices to move farther apart to accommodate it. This mutation could have substantial negative effects due to the fundamental role of hydrophobic packing, which is disrupted by Arg494 during protein folding. | |||||||||||||
| c.1489T>G | Y497D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y497D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic. No tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a destabilizing, pathogenic effect. All available predictions are concordant and indicate a likely pathogenic impact. Thus, based on the collective computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.092881 | Structured | 0.393608 | Uncertain | 0.950 | 0.181 | 0.000 | -12.128 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 4.70 | Destabilizing | 0.2 | 4.29 | Destabilizing | 4.50 | Destabilizing | 2.36 | Destabilizing | 0.918 | Likely Pathogenic | -9.81 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.67 | Pathogenic | 0.01 | Affected | 0.3621 | 0.0612 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1495A>G | R499G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R499G is listed in gnomAD (variant ID 6‑33438527‑A‑G) but has no ClinVar entry. Prediction tools that assess the variant’s effect on protein function overwhelmingly indicate a deleterious outcome: REVEL, FoldX‑MD (Rosetta component), Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify it as pathogenic. Only FoldX (overall) and AlphaMissense‑Optimized report uncertain results, which are treated as unavailable evidence. No tool predicts a benign effect. High‑accuracy assessments specifically show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as pathogenic. Taken together, the preponderance of evidence supports a pathogenic classification for R499G, and this conclusion is not contradicted by any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.071867 | Structured | 0.386723 | Uncertain | 0.899 | 0.146 | 0.000 | 6-33438527-A-G | 3 | 1.86e-6 | -9.960 | Likely Pathogenic | 0.789 | Likely Pathogenic | Ambiguous | 1.55 | Ambiguous | 0.1 | 2.75 | Destabilizing | 2.15 | Destabilizing | 1.41 | Destabilizing | 0.681 | Likely Pathogenic | -4.50 | Deleterious | 0.958 | Probably Damaging | 0.769 | Possibly Damaging | -1.47 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.2888 | 0.1798 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1496G>T | R499I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R499I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely indicate a deleterious effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all classify the change as pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports it as likely pathogenic. No tool predicts a benign outcome; the remaining predictions (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) are uncertain or inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Taken together, the overwhelming majority of evidence supports a pathogenic effect, and this is consistent with the absence of a ClinVar entry; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.071867 | Structured | 0.386723 | Uncertain | 0.899 | 0.146 | 0.000 | -9.069 | Likely Pathogenic | 0.861 | Likely Pathogenic | Ambiguous | 1.53 | Ambiguous | 0.1 | 0.61 | Ambiguous | 1.07 | Ambiguous | 0.57 | Ambiguous | 0.713 | Likely Pathogenic | -5.50 | Deleterious | 0.998 | Probably Damaging | 0.922 | Probably Damaging | -1.47 | Pathogenic | 0.00 | Affected | 0.1401 | 0.2146 | -2 | -3 | 9.0 | -43.03 | |||||||||||||||||||||||||||||
| c.1499T>C | L500P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L500P is listed in ClinVar (ID 2708686.0) as Pathogenic and is not reported in gnomAD. All available in‑silico predictors classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Thus, the variant is most likely pathogenic, and this prediction aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.066181 | Structured | 0.382942 | Uncertain | 0.893 | 0.150 | 0.000 | Pathogenic | 1 | -15.898 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 5.91 | Destabilizing | 0.3 | 8.90 | Destabilizing | 7.41 | Destabilizing | 1.92 | Destabilizing | 0.894 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.37 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.3103 | 0.0625 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||
| c.149T>A | I50N 2D AIThe SynGAP1 missense variant I50N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, whereas the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as benign. Foldetta results are unavailable. Overall, the predictions are mixed, with an equal split between benign and pathogenic calls, but the most reliable single‑tool prediction (AlphaMissense‑Optimized) and the majority of individual tools lean toward pathogenicity. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.295083 | Structured | 0.449965 | Uncertain | 0.545 | 0.708 | 0.000 | -7.091 | In-Between | 0.962 | Likely Pathogenic | Likely Pathogenic | 0.135 | Likely Benign | -2.37 | Neutral | 0.842 | Possibly Damaging | 0.272 | Benign | 3.73 | Benign | 0.00 | Affected | 0.0824 | 0.0412 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.1502T>A | I501N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I501N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM, while a majority of tools (SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. Uncertain results come from AlphaMissense‑Optimized, Foldetta, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as inconclusive, whereas the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; Foldetta likewise remains inconclusive. Overall, the balance of evidence favors a pathogenic classification, and this conclusion does not contradict any existing ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.079919 | Structured | 0.366596 | Uncertain | 0.886 | 0.153 | 0.000 | -11.105 | Likely Pathogenic | 0.854 | Likely Pathogenic | Ambiguous | 2.22 | Destabilizing | 0.0 | 1.70 | Ambiguous | 1.96 | Ambiguous | 1.90 | Destabilizing | 0.445 | Likely Benign | -6.13 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.37 | Benign | 0.01 | Affected | 0.0825 | 0.0270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1504G>T | G502C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G502C lies in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect include only premPS. The majority of tools predict a pathogenic effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. Tools with uncertain or inconclusive outputs are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a unanimous majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as uncertain. Overall, the preponderance of evidence points to a pathogenic impact for G502C, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.083462 | Structured | 0.340113 | Uncertain | 0.882 | 0.152 | 0.000 | -12.086 | Likely Pathogenic | 0.907 | Likely Pathogenic | Ambiguous | 1.02 | Ambiguous | 0.5 | 1.55 | Ambiguous | 1.29 | Ambiguous | 0.30 | Likely Benign | 0.845 | Likely Pathogenic | -8.65 | Deleterious | 1.000 | Probably Damaging | 0.988 | Probably Damaging | -1.67 | Pathogenic | 0.00 | Affected | 0.1279 | 0.2691 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1505G>T | G502V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G502V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: all evaluated algorithms except premPS (which predicts benign) classify the substitution as pathogenic or likely pathogenic. The consensus of high‑accuracy predictors is consistent: AlphaMissense‑Optimized reports a pathogenic effect; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a destabilizing, pathogenic outcome. In contrast, premPS is the sole tool suggesting a benign impact. Overall, the overwhelming majority of evidence points to a pathogenic effect for G502V, and this conclusion does not conflict with the absence of a ClinVar classification. Thus, the variant is most likely pathogenic, and this assessment is not contradicted by ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.083462 | Structured | 0.340113 | Uncertain | 0.882 | 0.152 | 0.000 | -15.278 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 3.56 | Destabilizing | 1.0 | 5.50 | Destabilizing | 4.53 | Destabilizing | 0.43 | Likely Benign | 0.917 | Likely Pathogenic | -8.65 | Deleterious | 0.999 | Probably Damaging | 0.944 | Probably Damaging | -1.67 | Pathogenic | 0.00 | Affected | 0.1406 | 0.3387 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1511A>T | K504I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K504I is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess the variant’s effect fall into two groups: benign predictions include REVEL, FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized; pathogenic predictions include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus as pathogenic. Because the majority of tools (seven) predict pathogenicity while three high‑accuracy methods provide conflicting evidence, the overall prediction leans toward pathogenic. This assessment does not contradict ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.028107 | Structured | 0.304984 | Uncertain | 0.850 | 0.189 | 0.000 | -12.597 | Likely Pathogenic | 0.727 | Likely Pathogenic | Likely Benign | -0.04 | Likely Benign | 0.3 | -0.36 | Likely Benign | -0.20 | Likely Benign | 0.40 | Likely Benign | 0.491 | Likely Benign | -7.35 | Deleterious | 0.996 | Probably Damaging | 0.993 | Probably Damaging | -1.49 | Pathogenic | 0.02 | Affected | 0.0769 | 0.2713 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1517T>A | L506H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L506H is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.034884 | Structured | 0.279180 | Uncertain | 0.924 | 0.196 | 0.000 | -12.999 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 4.45 | Destabilizing | 0.3 | 3.47 | Destabilizing | 3.96 | Destabilizing | 2.18 | Destabilizing | 0.758 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.54 | Pathogenic | 0.00 | Affected | 0.0919 | 0.0488 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1517T>C | L506P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L506P is listed in ClinVar (ID 975474.0) as Pathogenic and is not reported in gnomAD. All available in‑silico predictors classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is Pathogenic. Based on the unanimous computational evidence, the variant is most likely pathogenic, which aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.034884 | Structured | 0.279180 | Uncertain | 0.924 | 0.196 | 0.000 | Likely Pathogenic | 1 | -12.088 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 5.48 | Destabilizing | 0.7 | 10.19 | Destabilizing | 7.84 | Destabilizing | 2.50 | Destabilizing | 0.737 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.55 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.3047 | 0.0625 | -3 | -3 | -5.4 | -16.04 | 182.6 | 64.9 | 0.1 | 0.0 | 0.2 | 0.1 | X | Potentially Pathogenic | Leu506 is located in the middle of an α-helix (res. Gly502-Tyr518) within the inter-helix space of two helices (res. Gly502-Tyr518 and res. Glu582-Met603). In the WT simulations, the iso-butyl side chain of Leu506 hydrophobically packs with residues in the inter-helix space (e.g., Ile510, Phe597, Leu598, Ala601). In the variant simulations, the cyclic five-membered pyrrolidine ring of Pro506 is not as optimal as Leu506 for hydrophobic packing with nearby residues. Additionally, Pro506 cannot maintain the hydrogen bond with the backbone oxygen of Gly502 as Leu506 does in the WT, which disrupts the secondary structure element. | ||||||||||||||||
| c.1522G>T | D508Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D508Y missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. FoldX and Rosetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labeling it likely pathogenic, and Foldetta predicting a benign outcome. Overall, the majority of evidence (seven pathogenic vs. five benign) points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.019401 | Structured | 0.255890 | Uncertain | 0.890 | 0.228 | 0.000 | -12.917 | Likely Pathogenic | 0.739 | Likely Pathogenic | Likely Benign | -0.53 | Ambiguous | 0.2 | 0.72 | Ambiguous | 0.10 | Likely Benign | 0.06 | Likely Benign | 0.363 | Likely Benign | -8.37 | Deleterious | 1.000 | Probably Damaging | 0.965 | Probably Damaging | 3.26 | Benign | 0.01 | Affected | 0.0908 | 0.4736 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1523A>T | D508V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D508V missense change is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and Foldetta. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (a folding‑stability method combining FoldX‑MD and Rosetta outputs) as benign. With seven benign versus six pathogenic calls and two of the three high‑accuracy tools supporting benign, the variant is most likely benign. This conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.019401 | Structured | 0.255890 | Uncertain | 0.890 | 0.228 | 0.000 | -13.529 | Likely Pathogenic | 0.691 | Likely Pathogenic | Likely Benign | 0.07 | Likely Benign | 0.1 | 0.66 | Ambiguous | 0.37 | Likely Benign | 0.07 | Likely Benign | 0.381 | Likely Benign | -8.37 | Deleterious | 0.985 | Probably Damaging | 0.895 | Possibly Damaging | 3.27 | Benign | 0.08 | Tolerated | 0.1050 | 0.4604 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1529T>A | I510N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I510N is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a pathogenic or likely pathogenic outcome. No tool in the dataset predicts a benign effect. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic impact. Based on the uniform predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.025762 | Structured | 0.250630 | Uncertain | 0.945 | 0.273 | 0.000 | -12.784 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 3.09 | Destabilizing | 0.1 | 3.00 | Destabilizing | 3.05 | Destabilizing | 2.08 | Destabilizing | 0.925 | Likely Pathogenic | -5.62 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.45 | Pathogenic | 0.00 | Affected | 0.0761 | 0.0270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.152T>A | I51N 2D AIThe SynGAP1 missense variant I51N is not reported in ClinVar (ClinVar status: not reported) and is absent from gnomAD (gnomAD: not present). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method, has no available output for this variant. Consequently, the evidence is split evenly between benign and pathogenic predictions, with no decisive support from the most accurate methods. The variant is therefore inconclusive; it is not contradicted by any ClinVar record. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.291804 | Structured | 0.454181 | Uncertain | 0.606 | 0.710 | 0.000 | -9.287 | Likely Pathogenic | 0.909 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.77 | Neutral | 0.704 | Possibly Damaging | 0.272 | Benign | 4.13 | Benign | 0.00 | Affected | 0.1005 | 0.0769 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.1532G>T | G511V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G511V is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, which currently has no classification for G511V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.244404 | Uncertain | 0.924 | 0.287 | 0.000 | -11.738 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 3.47 | Destabilizing | 0.2 | 0.79 | Ambiguous | 2.13 | Destabilizing | 0.65 | Ambiguous | 0.540 | Likely Pathogenic | -8.71 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.16 | Benign | 0.01 | Affected | 0.1557 | 0.3019 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1541T>A | I514N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I514N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the overwhelming consensus of pathogenic predictions and the lack of benign evidence, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.049374 | Structured | 0.221408 | Uncertain | 0.948 | 0.266 | 0.000 | -13.869 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.41 | Destabilizing | 0.3 | 2.41 | Destabilizing | 2.91 | Destabilizing | 2.61 | Destabilizing | 0.582 | Likely Pathogenic | -6.86 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.82 | Benign | 0.00 | Affected | 0.0770 | 0.0142 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1550T>C | L517P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L517P is not reported in ClinVar and is absent from gnomAD. Prediction tools uniformly indicate a deleterious effect: pathogenic predictions are returned by REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the change as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels it likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a pathogenic effect. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.147645 | Uncertain | 0.938 | 0.296 | 0.000 | -15.546 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 5.88 | Destabilizing | 0.8 | 7.96 | Destabilizing | 6.92 | Destabilizing | 1.92 | Destabilizing | 0.937 | Likely Pathogenic | -6.70 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.50 | Pathogenic | 0.00 | Affected | 0.3570 | 0.1963 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1552T>G | Y518D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y518D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all predict pathogenicity, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.139895 | Structured | 0.126970 | Uncertain | 0.897 | 0.321 | 0.000 | -12.247 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 3.92 | Destabilizing | 0.9 | 2.68 | Destabilizing | 3.30 | Destabilizing | 1.36 | Destabilizing | 0.619 | Likely Pathogenic | -9.41 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.40 | Benign | 0.03 | Affected | 0.3682 | 0.0212 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1559C>T | S520F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S520F is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools that classify the variant as benign include Rosetta, Foldetta, and premPS. Those that predict pathogenicity are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX gives an uncertain result. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, whereas Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts a benign impact. Overall, the majority of evidence points to a pathogenic effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.094817 | Structured | 0.084894 | Uncertain | 0.887 | 0.337 | 0.000 | Uncertain | 1 | -12.541 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | -1.20 | Ambiguous | 0.4 | 0.39 | Likely Benign | -0.41 | Likely Benign | 0.25 | Likely Benign | 0.833 | Likely Pathogenic | -5.57 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | -1.36 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.0668 | 0.4779 | -2 | -3 | 3.6 | 60.10 | |||||||||||||||||||||||||
| c.155C>G | S52W 2D AIThe SynGAP1 missense variant S52W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, whereas those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign). AlphaMissense‑Optimized is uncertain, and Foldetta results are unavailable. Overall, more tools (five) predict pathogenicity than benign (three), and no high‑accuracy consensus or folding‑stability evidence contradicts this trend. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not conflict with the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.291804 | Structured | 0.457753 | Uncertain | 0.499 | 0.677 | 0.000 | -8.649 | Likely Pathogenic | 0.909 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.85 | Neutral | 0.986 | Probably Damaging | 0.968 | Probably Damaging | 4.05 | Benign | 0.00 | Affected | 0.0580 | 0.6254 | -2 | -3 | -0.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.1568A>T | N523I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N523I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, premPS, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools report uncertain results: Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—indicates pathogenicity. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains inconclusive. Overall, the preponderance of evidence (eight pathogenic versus three benign predictions) suggests that the variant is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.069024 | Structured | 0.033426 | Uncertain | 0.883 | 0.383 | 0.125 | -12.862 | Likely Pathogenic | 0.761 | Likely Pathogenic | Likely Benign | 0.26 | Likely Benign | 0.2 | -1.53 | Ambiguous | -0.64 | Ambiguous | 0.33 | Likely Benign | 0.726 | Likely Pathogenic | -8.18 | Deleterious | 0.989 | Probably Damaging | 0.946 | Probably Damaging | -1.42 | Pathogenic | 0.00 | Affected | 0.0627 | 0.3714 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.1570T>C | C524R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C524R is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Across the evaluated in‑silico tools, all pathogenic‑predicating algorithms—REVEL, SGM‑Consensus, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—return a pathogenic or likely pathogenic verdict. The only tool with an inconclusive result is FoldX, which is treated as unavailable. High‑accuracy predictors reinforce this assessment: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, also predicts pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.134866 | Structured | 0.024729 | Uncertain | 0.916 | 0.385 | 0.125 | -14.337 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | -0.53 | Ambiguous | 0.5 | 9.04 | Destabilizing | 4.26 | Destabilizing | 1.62 | Destabilizing | 0.917 | Likely Pathogenic | -11.93 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.40 | Pathogenic | 0.00 | Affected | 0.2031 | 0.1463 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.1570T>G | C524G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change at residue 524 (C524G) is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while FoldX and Foldetta are uncertain. No tool predicts a benign effect. High‑accuracy assessments further support a damaging outcome: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, remains uncertain. Overall, the evidence strongly favors a pathogenic impact for C524G, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.134866 | Structured | 0.024729 | Uncertain | 0.916 | 0.385 | 0.125 | -13.597 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 1.07 | Ambiguous | 0.2 | 2.00 | Destabilizing | 1.54 | Ambiguous | 1.66 | Destabilizing | 0.924 | Likely Pathogenic | -11.93 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.40 | Pathogenic | 0.00 | Affected | 0.3579 | 0.2745 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1577T>G | V526G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V526G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on pathogenicity include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; no tool predicts it benign. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as pathogenic. All available evidence points to a deleterious effect. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.139895 | Structured | 0.023118 | Uncertain | 0.943 | 0.403 | 0.000 | -15.811 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 3.21 | Destabilizing | 0.3 | 4.79 | Destabilizing | 4.00 | Destabilizing | 2.19 | Destabilizing | 0.935 | Likely Pathogenic | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.49 | Pathogenic | 0.00 | Affected | 0.1636 | 0.1837 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1579G>T | D527Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant D527Y is listed in ClinVar with an uncertain significance (ClinVar ID 1698369.0) and is not reported in gnomAD. Functional prediction tools cluster into two groups: the single benign prediction from premPS versus a consensus of pathogenic predictions from the remaining 12 tools (REVEL, SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. Protein‑stability calculations from FoldX and Rosetta are also uncertain. Overall, the preponderance of evidence indicates that D527Y is most likely pathogenic, which does not contradict the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.139895 | Structured | 0.021908 | Uncertain | 0.913 | 0.408 | 0.000 | Uncertain | 1 | -15.386 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | -0.77 | Ambiguous | 0.2 | 1.89 | Ambiguous | 0.56 | Ambiguous | -0.14 | Likely Benign | 0.905 | Likely Pathogenic | -8.79 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -2.41 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.0554 | 0.4229 | -4 | -3 | 2.2 | 48.09 | 270.9 | -45.7 | 0.1 | 0.1 | -0.1 | 0.0 | X | Potentially Pathogenic | Asp527 is located on an α-α loop between the two α-helices (res. Gly502-Tyr518 and Ala533-Val560). In the WT simulations, the carboxylate group of the Asp527 side chain forms hydrogen bonds with the backbone atoms of loop residues (e.g., Ile529, Lys530) facing the membrane surface. In the variant simulations, Tyr527 is a bulkier residue that faces away from the loop and stacks with Phe646 in a nearby α-helix (res. Ser614-Ser668). Regardless, no negative structural effects are observed during the variant simulations. However, due to its location near the SynGAP-membrane interface, the effect of the residue swap cannot be fully addressed using the SynGAP solvent-only simulations. | ||||||||||||||||
| c.1580A>T | D527V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D527V missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that are uncertain (FoldX, Rosetta, Foldetta, premPS) provide no definitive evidence. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, while Foldetta remains uncertain. Overall, the majority of reliable predictors classify the variant as pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists. Thus, based on current computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.139895 | Structured | 0.021908 | Uncertain | 0.913 | 0.408 | 0.000 | -16.844 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.71 | Ambiguous | 0.7 | 0.84 | Ambiguous | 0.78 | Ambiguous | -0.55 | Ambiguous | 0.938 | Likely Pathogenic | -8.78 | Deleterious | 0.998 | Probably Damaging | 0.997 | Probably Damaging | -2.40 | Pathogenic | 0.00 | Affected | 0.0659 | 0.3984 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1583C>T | P528L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P528L missense variant is listed in ClinVar with an Uncertain significance and is not reported in gnomAD. Prediction tools that classify the variant as benign include premPS. All other evaluated algorithms—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—predict it to be pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized reports an uncertain outcome; the SGM‑Consensus, which aggregates the four high‑confidence predictors, indicates a likely pathogenic effect; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also yields an uncertain result. Taken together, the overwhelming majority of tools support a pathogenic interpretation. Therefore, the variant is most likely pathogenic, a conclusion that does not contradict its ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.225814 | Structured | 0.020396 | Uncertain | 0.909 | 0.403 | 0.000 | Uncertain | 1 | -13.752 | Likely Pathogenic | 0.853 | Likely Pathogenic | Ambiguous | 1.31 | Ambiguous | 0.1 | 0.61 | Ambiguous | 0.96 | Ambiguous | 0.19 | Likely Benign | 0.555 | Likely Pathogenic | -9.65 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.48 | Pathogenic | 0.00 | Affected | 0.1975 | 0.5574 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||
| c.1586T>A | I529N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I529N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign) all predict a neutral impact. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores the variant as benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a benign effect. Taken together, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.318242 | Structured | 0.019545 | Uncertain | 0.901 | 0.403 | 0.000 | -4.478 | Likely Benign | 0.283 | Likely Benign | Likely Benign | 0.12 | Likely Benign | 0.2 | -0.05 | Likely Benign | 0.04 | Likely Benign | 0.46 | Likely Benign | 0.378 | Likely Benign | -0.31 | Neutral | 0.997 | Probably Damaging | 0.952 | Probably Damaging | -1.24 | Pathogenic | 0.20 | Tolerated | 0.0722 | 0.0700 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.158G>T | G53V 2D AIThe SynGAP1 missense variant G53V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM. Tools that agree on a pathogenic effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign). Foldetta results are unavailable. Overall, the majority of predictions (six pathogenic vs. three benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.268042 | Structured | 0.460894 | Uncertain | 0.386 | 0.666 | 0.000 | -8.308 | Likely Pathogenic | 0.959 | Likely Pathogenic | Likely Pathogenic | 0.238 | Likely Benign | -1.71 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 4.11 | Benign | 0.00 | Affected | 0.1193 | 0.4833 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.1591T>C | C531R 2D AIThe SynGAP1 missense variant C531R is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, and polyPhen‑2 HumVar, whereas the remaining tools—REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar classification because the variant is currently unreported in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.281712 | Structured | 0.017941 | Uncertain | 0.878 | 0.401 | 0.000 | -12.600 | Likely Pathogenic | 0.966 | Likely Pathogenic | Likely Pathogenic | 0.31 | Likely Benign | 2.0 | -0.27 | Likely Benign | 0.02 | Likely Benign | 1.40 | Destabilizing | 0.619 | Likely Pathogenic | -9.85 | Deleterious | 0.929 | Possibly Damaging | 0.385 | Benign | -1.24 | Pathogenic | 0.00 | Affected | 0.1322 | 0.1566 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.1591T>G | C531G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C531G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX and AlphaMissense‑Optimized, while the majority of other in silico predictors (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) indicate a pathogenic impact. Uncertain results come from Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as inconclusive. Overall, the balance of evidence leans toward pathogenicity, with no ClinVar entry to contradict this assessment. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.281712 | Structured | 0.017941 | Uncertain | 0.878 | 0.401 | 0.000 | -10.037 | Likely Pathogenic | 0.577 | Likely Pathogenic | Likely Benign | 0.47 | Likely Benign | 0.3 | 1.49 | Ambiguous | 0.98 | Ambiguous | 1.42 | Destabilizing | 0.545 | Likely Pathogenic | -9.76 | Deleterious | 0.985 | Probably Damaging | 0.832 | Possibly Damaging | -1.24 | Pathogenic | 0.00 | Affected | 0.2653 | 0.2297 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1616A>T | H539L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H539L is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include Rosetta and premPS, whereas the majority of tools predict a pathogenic impact: REVEL, SIFT, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized is pathogenic, the SGM Consensus is likely pathogenic, and Foldetta’s stability prediction is uncertain. No evidence from the available data contradicts the ClinVar status, which is currently unreported. Overall, the preponderance of computational evidence indicates that H539L is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.031398 | Uncertain | 0.948 | 0.360 | 0.000 | -14.161 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | -1.76 | Ambiguous | 0.1 | -0.17 | Likely Benign | -0.97 | Ambiguous | 0.35 | Likely Benign | 0.879 | Likely Pathogenic | -10.17 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | -1.29 | Pathogenic | 0.01 | Affected | 0.0758 | 0.3680 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.161A>T | N54I 2D AIThe SynGAP1 missense variant N54I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward pathogenicity. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.196879 | Structured | 0.464669 | Uncertain | 0.504 | 0.659 | 0.000 | -9.919 | Likely Pathogenic | 0.890 | Likely Pathogenic | Ambiguous | 0.201 | Likely Benign | -1.70 | Neutral | 0.943 | Possibly Damaging | 0.924 | Probably Damaging | 4.15 | Benign | 0.00 | Affected | 0.0637 | 0.6793 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.1625A>T | N542I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N542I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls from FoldX, Rosetta, Foldetta, and premPS, whereas pathogenic calls come from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus. High‑accuracy assessments give AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta) as benign. Thus, the majority of evidence points to a deleterious effect, with only a minority of tools predicting benign stability. The variant is most likely pathogenic, and this assessment does not contradict any ClinVar classification because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.053060 | Structured | 0.026143 | Uncertain | 0.953 | 0.331 | 0.000 | -14.975 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.14 | Likely Benign | 0.4 | -0.38 | Likely Benign | -0.12 | Likely Benign | 0.42 | Likely Benign | 0.829 | Likely Pathogenic | -7.99 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.37 | Pathogenic | 0.02 | Affected | 0.0638 | 0.5427 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.1628T>C | L543P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L543P (ClinVar ID 4540554) is classified as Pathogenic in ClinVar and is not reported in gnomAD. Prediction tools that assess functional impact uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. No tool in the dataset predicts a benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is Pathogenic. All available predictions and stability analyses support a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion is consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.020918 | Uncertain | 0.963 | 0.314 | 0.000 | Likely Pathogenic | 1 | -15.958 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 8.56 | Destabilizing | 0.6 | 13.44 | Destabilizing | 11.00 | Destabilizing | 1.54 | Destabilizing | 0.770 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.89 | Pathogenic | 0.00 | Affected | 0.3457 | 0.0992 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||
| c.1636T>C | C546R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C546R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include only SIFT, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX and Foldetta return uncertain results and are therefore not considered evidence for either side. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as Uncertain. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.027463 | Structured | 0.009041 | Uncertain | 0.960 | 0.288 | 0.000 | -15.237 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | -0.70 | Ambiguous | 0.3 | 2.04 | Destabilizing | 0.67 | Ambiguous | 1.70 | Destabilizing | 0.894 | Likely Pathogenic | -9.73 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.26 | Pathogenic | 0.06 | Tolerated | 0.1808 | 0.1685 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.1636T>G | C546G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant C546G is not reported in ClinVar and is present in gnomAD (ID 6‑33438879‑T‑G). Prediction tools that indicate a benign effect include only SIFT, whereas the remaining tools—REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus—predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta is inconclusive and therefore not considered evidence. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.027463 | Structured | 0.009041 | Uncertain | 0.960 | 0.288 | 0.000 | 6-33438879-T-G | 1 | 6.20e-7 | -14.026 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 1.55 | Ambiguous | 0.0 | 2.43 | Destabilizing | 1.99 | Ambiguous | 1.53 | Destabilizing | 0.877 | Likely Pathogenic | -9.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.26 | Pathogenic | 0.06 | Tolerated | 3.37 | 35 | 0.3135 | 0.2513 | -3 | -3 | -2.9 | -46.09 | ||||||||||||||||||||||||
| c.1639T>C | C547R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C547R is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while no tool in the dataset predicts a benign outcome. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Based on the unanimous computational evidence, the variant is most likely pathogenic, a conclusion that contradicts the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.007912 | Uncertain | 0.971 | 0.275 | 0.000 | Uncertain | 1 | -16.967 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 7.76 | Destabilizing | 0.8 | 5.83 | Destabilizing | 6.80 | Destabilizing | 1.69 | Destabilizing | 0.900 | Likely Pathogenic | -11.60 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.33 | Pathogenic | 0.02 | Affected | 3.37 | 35 | 0.1802 | 0.1408 | -4 | -3 | -7.0 | 53.05 | 267.4 | -90.3 | 0.0 | 0.0 | -0.1 | 0.1 | X | X | X | X | Potentially Pathogenic | Cys547 is located in an α-helix (res. Ala533-Val560). The thiol side chain of Cys is situated in a hydrophobic inter-helix space, where it packs hydrophobically with other residues such as Ile626, Leu551, and Phe652. Additionally, the thiol side chain of Cys547 weakly hydrogen bonds with the carbonyl group of Leu543 in the same α-helix. In the variant simulations, the bulkier, positively charged guanidinium group of Arg547 must rotate out of the hydrophobic space. Consequently, it forms ionic interactions with the carboxylate groups of Glu548 in the same helix and Glu656 in the neighboring α-helix (res. Glu666-Asp644). This causes the two helices to slightly separate, significantly affecting the secondary structure integrity of the latter helix. These negative structural effects could be more pronounced during protein folding and are likely to be undermined in the MD simulations. | |||||||||||||
| c.1639T>G | C547G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C547G is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenic. No tool predicts a benign outcome. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments further support a harmful impact: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a pathogenic effect. Taken together, the evidence overwhelmingly points to a pathogenic classification for this variant, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.007912 | Uncertain | 0.971 | 0.275 | 0.000 | -14.182 | Likely Pathogenic | 0.923 | Likely Pathogenic | Ambiguous | 2.21 | Destabilizing | 0.0 | 2.99 | Destabilizing | 2.60 | Destabilizing | 1.83 | Destabilizing | 0.902 | Likely Pathogenic | -11.60 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.30 | Pathogenic | 0.05 | Affected | 0.3201 | 0.2502 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1646T>C | L549S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L549S is catalogued in gnomAD (ID 6‑33438889‑T‑C) but has no ClinVar entry. Functional prediction tools largely converge on a deleterious effect: SIFT reports a benign change, whereas the remaining 10 evaluated algorithms (SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. The high‑accuracy predictors reinforce this trend: AlphaMissense‑Optimized returns a pathogenic score, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive. No evidence from FoldX or Rosetta alone is available. Consequently, the variant is most likely pathogenic, and this assessment does not conflict with ClinVar, which contains no classification for this allele. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.025762 | Structured | 0.007921 | Uncertain | 0.955 | 0.281 | 0.000 | 6-33438889-T-C | 1 | 6.20e-7 | -13.018 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.76 | Ambiguous | 0.4 | 1.03 | Ambiguous | 1.40 | Ambiguous | 1.01 | Destabilizing | 0.801 | Likely Pathogenic | -4.85 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.14 | Pathogenic | 0.34 | Tolerated | 3.37 | 35 | 0.2965 | 0.0305 | -2 | -3 | -4.6 | -26.08 | ||||||||||||||||||||||||
| c.1652T>C | L551P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L551P (ClinVar ID 547942.0) is classified as Pathogenic in ClinVar and is not reported in gnomAD. Prediction tools that assess functional impact uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. No tool in the dataset predicts a benign outcome. High‑accuracy assessments further support this: AlphaMissense‑Optimized is Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is Pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this conclusion aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009977 | Structured | 0.006653 | Uncertain | 0.960 | 0.254 | 0.000 | Likely Pathogenic | 1 | -14.620 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 6.66 | Destabilizing | 0.1 | 6.58 | Destabilizing | 6.62 | Destabilizing | 2.66 | Destabilizing | 0.953 | Likely Pathogenic | -4.70 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.60 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.3377 | 0.1353 | -3 | -3 | -5.4 | -16.04 | 208.6 | 60.9 | 0.1 | 0.0 | -0.3 | 0.0 | X | Potentially Pathogenic | L551 is located on an α-helix (res. Ala533-Val560). The iso-butyl side chain of Leu551 hydrophobically packs with nearby hydrophobic residues such as Cys547, Phe652, Leu633, and Ile630 in the inter-helix space. In the variant simulations, the pyrrolidine side chain of Pro551 is not as optimal as leucine for hydrophobic packing with the nearby residues. Moreover, Pro551 lacks the amide group, and thus, it cannot form a hydrogen bond with the backbone carbonyl group of Cys547, which disrupts the continuity of the secondary structure element. | ||||||||||||||||
| c.1654T>C | C552R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C552R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only SIFT, whereas the remaining tools (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) yields an uncertain result, which is treated as unavailable evidence. Overall, the preponderance of predictions supports a pathogenic classification, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.013265 | Structured | 0.005714 | Uncertain | 0.955 | 0.256 | 0.000 | -14.315 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | -1.18 | Ambiguous | 0.1 | -0.51 | Ambiguous | -0.85 | Ambiguous | 1.10 | Destabilizing | 0.809 | Likely Pathogenic | -9.93 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.19 | Pathogenic | 0.48 | Tolerated | 0.1449 | 0.1366 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.1654T>G | C552G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C552G is not reported in ClinVar and is absent from gnomAD. In silico predictors that classify the change as benign include FoldX, Rosetta, Foldetta, and SIFT, whereas the majority of tools predict it to be pathogenic: SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy subset shows AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. With six pathogenic versus four benign predictions overall, the evidence leans toward a deleterious effect. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.013265 | Structured | 0.005714 | Uncertain | 0.955 | 0.256 | 0.000 | -11.570 | Likely Pathogenic | 0.965 | Likely Pathogenic | Likely Pathogenic | 0.27 | Likely Benign | 0.0 | 0.29 | Likely Benign | 0.28 | Likely Benign | 1.06 | Destabilizing | 0.745 | Likely Pathogenic | -10.20 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.21 | Pathogenic | 0.35 | Tolerated | 0.2338 | 0.1930 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1661T>G | V554G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V554G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, while the remaining eleven tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as pathogenic. No predictions are missing or inconclusive. Overall, the variant is most likely pathogenic based on the collective evidence, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.020522 | Structured | 0.007349 | Uncertain | 0.955 | 0.226 | 0.000 | -13.879 | Likely Pathogenic | 0.942 | Likely Pathogenic | Ambiguous | 3.31 | Destabilizing | 0.1 | 4.13 | Destabilizing | 3.72 | Destabilizing | 2.40 | Destabilizing | 0.594 | Likely Pathogenic | -6.96 | Deleterious | 0.997 | Probably Damaging | 1.000 | Probably Damaging | 3.21 | Benign | 0.00 | Affected | 0.1619 | 0.1548 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1664T>A | V555D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V555D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are none, while a majority of algorithms predict a pathogenic impact: REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta provide uncertain results. High‑accuracy methods reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta remains uncertain. Overall, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.013265 | Structured | 0.008218 | Uncertain | 0.943 | 0.225 | 0.000 | -16.413 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 1.84 | Ambiguous | 0.1 | 1.70 | Ambiguous | 1.77 | Ambiguous | 1.25 | Destabilizing | 0.896 | Likely Pathogenic | -5.71 | Deleterious | 1.000 | Probably Damaging | 0.987 | Probably Damaging | -1.39 | Pathogenic | 0.00 | Affected | 0.1411 | 0.0541 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1664T>G | V555G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V555G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on pathogenicity include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; no tool predicts it benign. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as pathogenic. No predictions are inconclusive or missing. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.013265 | Structured | 0.008218 | Uncertain | 0.943 | 0.225 | 0.000 | -13.327 | Likely Pathogenic | 0.899 | Likely Pathogenic | Ambiguous | 2.07 | Destabilizing | 0.1 | 2.22 | Destabilizing | 2.15 | Destabilizing | 1.07 | Destabilizing | 0.798 | Likely Pathogenic | -6.35 | Deleterious | 0.984 | Probably Damaging | 1.000 | Probably Damaging | -1.39 | Pathogenic | 0.01 | Affected | 0.1994 | 0.1677 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1673A>T | H558L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H558L missense variant has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, ESM1b, and FATHMM; FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. With eight benign versus five pathogenic predictions and two high‑accuracy benign calls, the variant is most likely benign. This conclusion is not contradicted by ClinVar, which contains no entry for H558L. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.033407 | Structured | 0.011039 | Uncertain | 0.897 | 0.200 | 0.000 | -13.563 | Likely Pathogenic | 0.250 | Likely Benign | Likely Benign | -0.52 | Ambiguous | 0.0 | 0.21 | Likely Benign | -0.16 | Likely Benign | 0.30 | Likely Benign | 0.509 | Likely Pathogenic | -5.40 | Deleterious | 0.001 | Benign | 0.005 | Benign | -0.98 | Pathogenic | 0.29 | Tolerated | 0.1002 | 0.3141 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.1675T>C | C559R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C559R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, Rosetta, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Uncertain or inconclusive results come from FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of tools (six pathogenic vs five benign) lean toward a pathogenic interpretation, and this does not contradict any ClinVar status because the variant is not yet classified in ClinVar. Thus, the variant is most likely pathogenic based on current predictive evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.016021 | Structured | 0.010460 | Uncertain | 0.842 | 0.204 | 0.000 | -9.509 | Likely Pathogenic | 0.779 | Likely Pathogenic | Likely Benign | -1.03 | Ambiguous | 0.1 | -0.38 | Likely Benign | -0.71 | Ambiguous | 0.79 | Ambiguous | 0.498 | Likely Benign | -8.58 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.48 | Benign | 0.46 | Tolerated | 0.2498 | 0.1720 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.1675T>G | C559G 2D 3DClick to see structure in 3D Viewer AISynGAP1 C559G is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include FoldX, Foldetta, SIFT, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Uncertain calls from Rosetta and premPS are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta as benign. Overall, the majority of predictions (seven pathogenic vs five benign) and the high‑accuracy consensus lean toward a pathogenic effect. This conclusion does not contradict ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.016021 | Structured | 0.010460 | Uncertain | 0.842 | 0.204 | 0.000 | -12.342 | Likely Pathogenic | 0.581 | Likely Pathogenic | Likely Benign | 0.12 | Likely Benign | 0.1 | 0.53 | Ambiguous | 0.33 | Likely Benign | 0.76 | Ambiguous | 0.547 | Likely Pathogenic | -8.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.50 | Benign | 0.37 | Tolerated | 0.2454 | 0.2001 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1679T>G | V560G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V560G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only SIFT, whereas the majority of tools (REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. Uncertain results come from FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, AlphaMissense‑Optimized is uncertain, and Foldetta is uncertain. Overall, the preponderance of evidence indicates that V560G is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.021381 | Structured | 0.013872 | Uncertain | 0.853 | 0.204 | 0.000 | -12.485 | Likely Pathogenic | 0.799 | Likely Pathogenic | Ambiguous | 0.66 | Ambiguous | 0.1 | 2.12 | Destabilizing | 1.39 | Ambiguous | 1.80 | Destabilizing | 0.753 | Likely Pathogenic | -5.87 | Deleterious | 0.981 | Probably Damaging | 1.000 | Probably Damaging | -1.25 | Pathogenic | 0.19 | Tolerated | 0.1738 | 0.2036 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.167T>C | L56P 2D AIThe SynGAP1 missense variant L56P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of computational evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.342579 | Structured | 0.476218 | Uncertain | 0.495 | 0.657 | 0.000 | -9.991 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.307 | Likely Benign | -2.62 | Deleterious | 0.943 | Possibly Damaging | 0.944 | Probably Damaging | 3.77 | Benign | 0.00 | Affected | 0.3806 | 0.1342 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.1685C>T | P562L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant P562L is listed in ClinVar (ID 41462) as Pathogenic and is present in gnomAD (variant ID 6‑33440737‑C‑T). Prediction tools that indicate a benign effect include Rosetta and premPS, whereas the remaining tools—REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is Uncertain. Overall, the preponderance of evidence points to a pathogenic effect for P562L, which is consistent with its ClinVar classification and does not contradict the reported status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.022306 | Structured | 0.023606 | Uncertain | 0.893 | 0.200 | 0.000 | Pathogenic/Likely path. | 12 | 6-33440737-C-T | -13.438 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 3.54 | Destabilizing | 0.8 | 0.17 | Likely Benign | 1.86 | Ambiguous | -0.14 | Likely Benign | 0.829 | Likely Pathogenic | -9.95 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 0.58 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.2250 | 0.4510 | -3 | -3 | 5.4 | 16.04 | 228.8 | -68.5 | -0.1 | 0.0 | 0.1 | 0.2 | X | Potentially Pathogenic | Pro562 is located on an α-α loop between two α-helices (res. Ala533-Val560 and res. Arg563-Glu578). The cyclic pyrrolidine side chain of Pro562 hydrophobically packs with other residues in the inter-helix space, such as Leu565, Ile501, and Phe561. In the variant simulations, Leu562 packs more favorably with the nearby hydrophobic residues, and the backbone amide group of Leu562 (absent in proline) does not form any intra-protein hydrogen bonds. However, prolines are well-suited for unstructured regions like loops, and thus, Pro562 in the WT is necessary at the end of the helix to induce a tight turn during folding. Although no negative structural effects are observed during the simulations, the residue swap could potentially cause extensive damage to the protein structure during folding. | 10.1016/j.ajhg.2020.11.011 | ||||||||||||||
| c.1687A>T | R563W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R563W is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show that AlphaMissense‑Optimized is inconclusive, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also inconclusive. No evidence from the uncertain tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) supports either outcome. Overall, the balance of evidence favors a pathogenic classification for R563W, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.039760 | Structured | 0.031987 | Uncertain | 0.876 | 0.209 | 0.000 | -12.454 | Likely Pathogenic | 0.854 | Likely Pathogenic | Ambiguous | 1.25 | Ambiguous | 0.1 | 0.79 | Ambiguous | 1.02 | Ambiguous | 0.34 | Likely Benign | 0.302 | Likely Benign | -6.75 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.42 | Benign | 0.01 | Affected | 0.1378 | 0.2088 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||
| c.1694T>C | L565P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L565P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity largely agree: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect, while only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. No prediction or folding‑stability result is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.034884 | Structured | 0.045819 | Uncertain | 0.922 | 0.205 | 0.000 | -13.369 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.59 | Destabilizing | 0.4 | 6.82 | Destabilizing | 5.21 | Destabilizing | 2.33 | Destabilizing | 0.546 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.74 | Benign | 0.00 | Affected | 0.3825 | 0.0877 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1694T>G | L565R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L565R is not reported in ClinVar (ClinVar status: not listed) but is present in the gnomAD database (gnomAD ID: 6‑33440746‑T‑G). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, Foldetta, and the SGM Consensus—consistently predict a pathogenic impact. The Rosetta stability assessment is inconclusive and is therefore treated as unavailable. High‑accuracy methods all support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar evidence (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.034884 | Structured | 0.045819 | Uncertain | 0.922 | 0.205 | 0.000 | 6-33440746-T-G | -16.070 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.71 | Destabilizing | 0.1 | 1.88 | Ambiguous | 3.30 | Destabilizing | 2.66 | Destabilizing | 0.547 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.74 | Benign | 0.00 | Affected | 3.37 | 35 | 0.1373 | 0.0615 | -2 | -3 | -8.3 | 43.03 | ||||||||||||||||||||||||||
| c.170T>A | L57H 2D AIThe SynGAP1 missense variant L57H is not reported in ClinVar and has no entry in gnomAD. Prediction tools show a split: benign calls come from REVEL, PROVEAN, ESM1b, and FATHMM, whereas pathogenic calls come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments further indicate that AlphaMissense‑Optimized is Uncertain, whereas the SGM‑Consensus remains Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of high‑confidence tools and the consensus score favor a benign effect, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.254060 | Structured | 0.481044 | Uncertain | 0.554 | 0.642 | 0.000 | -6.251 | Likely Benign | 0.796 | Likely Pathogenic | Ambiguous | 0.173 | Likely Benign | -1.58 | Neutral | 0.984 | Probably Damaging | 0.971 | Probably Damaging | 3.90 | Benign | 0.00 | Affected | 0.1045 | 0.0611 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.170T>C | L57P 2D AIThe SynGAP1 missense variant L57P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM. Tools that agree on a pathogenic effect include polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (six pathogenic vs. three benign) indicate that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.254060 | Structured | 0.481044 | Uncertain | 0.554 | 0.642 | 0.000 | -10.724 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.242 | Likely Benign | -1.77 | Neutral | 0.943 | Possibly Damaging | 0.944 | Probably Damaging | 3.90 | Benign | 0.00 | Affected | 0.3651 | 0.1703 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.1712C>G | S571W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S571W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that provide a clear verdict overwhelmingly classify the substitution as pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict deleterious effects. No tool in the dataset returned a benign classification; the only non‑conclusive results come from FoldX, Rosetta, Foldetta, and premPS, which are listed as uncertain. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta’s stability analysis is inconclusive. Based on the aggregate predictions, the variant is most likely pathogenic, and this assessment is consistent with the absence of a ClinVar entry (no contradictory status). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.069024 | Structured | 0.045569 | Uncertain | 0.928 | 0.270 | 0.000 | -14.025 | Likely Pathogenic | 0.961 | Likely Pathogenic | Likely Pathogenic | -1.13 | Ambiguous | 0.1 | -1.44 | Ambiguous | -1.29 | Ambiguous | 0.67 | Ambiguous | 0.867 | Likely Pathogenic | -6.64 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.32 | Pathogenic | 0.00 | Affected | 0.0648 | 0.3809 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1712C>T | S571L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S571L is listed in ClinVar with an uncertain significance (ClinVar ID 3897075) and is present in gnomAD (ID 6‑33440764‑C‑T). Prediction tools that indicate a benign effect include premPS and AlphaMissense‑Optimized, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) all predict a pathogenic outcome. The high‑accuracy AlphaMissense‑Optimized score is benign, whereas the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—supports pathogenicity. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for S571L, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.069024 | Structured | 0.045569 | Uncertain | 0.928 | 0.270 | 0.000 | Uncertain | 2 | 6-33440764-C-T | 1 | 6.23e-7 | -11.651 | Likely Pathogenic | 0.660 | Likely Pathogenic | Likely Benign | -1.53 | Ambiguous | 0.1 | -1.05 | Ambiguous | -1.29 | Ambiguous | 0.27 | Likely Benign | 0.841 | Likely Pathogenic | -5.61 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | -1.25 | Pathogenic | 0.04 | Affected | 3.37 | 35 | 0.0959 | 0.3918 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||
| c.1714T>A | W572R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W572R is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while no tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.150080 | Structured | 0.039626 | Uncertain | 0.935 | 0.256 | 0.000 | -17.511 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 4.84 | Destabilizing | 0.1 | 6.19 | Destabilizing | 5.52 | Destabilizing | 1.79 | Destabilizing | 0.894 | Likely Pathogenic | -12.81 | Deleterious | -1.25 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.4059 | 0.0212 | 2 | -3 | -3.6 | -30.03 | 312.6 | -37.6 | 0.0 | 0.0 | -1.0 | 0.0 | X | X | Potentially Pathogenic | The indole ring of Trp572, located in an α-helix (res. Arg563-Glu578), lies in a hydrophobic inter-helix space, where it makes extensive hydrophobic interactions with nearby residues such as Met470, Phe569, Leu588, and Ile589. The guanidinium group of Arg572 is similarly sized to the tryptophan it replaced; however, it is also positively charged. In the variant simulations, Arg572 forms hydrogen bonds with other residues in the inter-helix space, such as Ser592 and the backbone carbonyl atom of Leu465. Additionally, Arg572 hydrophobically packs its carbon chain with surrounding residues such as Phe569 and Ile589.However, the introduced residue arginine is too hydrophilic and charged for the hydrophobic space, disrupting the hydrophobic packing of the inter-helix space. Indeed, in the second simulation, Arg572 successfully escapes the hydrophobic niche completely, causing the whole protein to partially unfold.Overall, the residue swap is highly likely to cause critical protein folding problems, as evidenced by the effects seen in the variant simulations. | |||||||||||||||||||||
| c.1714T>C | W572R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W572R is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate pathogenicity, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic.” No tool in the dataset predicts a benign outcome. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the unanimous computational evidence, the variant is most likely pathogenic, which is consistent with its ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.150080 | Structured | 0.039626 | Uncertain | 0.935 | 0.256 | 0.000 | Not provided | 1 | -17.511 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 4.84 | Destabilizing | 0.1 | 6.19 | Destabilizing | 5.52 | Destabilizing | 1.79 | Destabilizing | 0.894 | Likely Pathogenic | -12.81 | Deleterious | -1.25 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.4059 | 0.0212 | 2 | -3 | -3.6 | -30.03 | 312.6 | -37.6 | 0.0 | 0.0 | -1.0 | 0.0 | X | X | Potentially Pathogenic | The indole ring of Trp572, located in an α-helix (res. Arg563-Glu578), lies in a hydrophobic inter-helix space, where it makes extensive hydrophobic interactions with nearby residues such as Met470, Phe569, Leu588, and Ile589. The guanidinium group of Arg572 is similarly sized to the tryptophan it replaced; however, it is also positively charged. In the variant simulations, Arg572 forms hydrogen bonds with other residues in the inter-helix space, such as Ser592 and the backbone carbonyl atom of Leu465. Additionally, Arg572 hydrophobically packs its carbon chain with surrounding residues such as Phe569 and Ile589.However, the introduced residue arginine is too hydrophilic and charged for the hydrophobic space, disrupting the hydrophobic packing of the inter-helix space. Indeed, in the second simulation, Arg572 successfully escapes the hydrophobic niche completely, causing the whole protein to partially unfold.Overall, the residue swap is highly likely to cause critical protein folding problems, as evidenced by the effects seen in the variant simulations. | |||||||||||||||||||
| c.1715G>C | W572S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W572S is listed in ClinVar as Pathogenic (ClinVar ID 1069317.0) and is not reported in gnomAD. All available in silico predictors classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Therefore, the variant is most likely pathogenic, and this prediction aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.150080 | Structured | 0.039626 | Uncertain | 0.935 | 0.256 | 0.000 | Pathogenic | 1 | -17.461 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.78 | Destabilizing | 0.2 | 3.37 | Destabilizing | 4.58 | Destabilizing | 1.79 | Destabilizing | 0.775 | Likely Pathogenic | -12.74 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.24 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.3958 | 0.0857 | -2 | -3 | 0.1 | -99.14 | 235.1 | 76.6 | 0.0 | 0.0 | -0.4 | 0.1 | X | Potentially Pathogenic | The introduced residue Ser572, located in an α-helix (res. Arg563-Glu578), is considerably smaller than the tryptophan it replaced. The indole ring of the Trp572 side chain lies in a hydrophobic inter-helix space, where it makes extensive hydrophobic interactions with nearby residues such as Met470, Phe569, Leu588, and Ile589. In the variant simulations, all these favorable packing interactions are completely removed, as the introduced residue Ser572 is too hydrophilic or small to fill the hydrophobic niche occupied by the indole ring. Moreover, the hydroxyl group of Ser572 forms hydrogen bonds with the carbonyl groups of Glu567 and Val568 within the same α-helix, potentially lowering its integrity. Overall, the residue swap is highly likely to cause critical protein folding problems that are underestimated based on the effects seen in the variant simulations. | ||||||||||||||||
| c.1717C>T | R573W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R573W (ClinVar ID 421734) is classified as Pathogenic in ClinVar and is not reported in gnomAD. Benign predictions: none. Pathogenic predictions: REVEL, FoldX, PROVEAN, polyPhen2_HumDiv, polyPhen2_HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized. Uncertain predictions: Rosetta, Foldetta, premPS. High‑accuracy tools: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; Foldetta is Uncertain. Overall, the variant is most likely pathogenic, in agreement with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.134866 | Structured | 0.032433 | Uncertain | 0.934 | 0.235 | 0.000 | Pathogenic/Likely path. | 8 | -14.078 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 2.37 | Destabilizing | 0.7 | 0.57 | Ambiguous | 1.47 | Ambiguous | 0.88 | Ambiguous | 0.758 | Likely Pathogenic | -6.94 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | -1.48 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.1179 | 0.2643 | 2 | -3 | 3.6 | 30.03 | 257.6 | 39.0 | 0.1 | 0.0 | 0.2 | 0.0 | X | X | Potentially Pathogenic | The guanidinium group of Arg573, located in an α-helix (res. Arg563-Glu578), forms a salt bridge with the carboxylate groups of Glu582 and/or Asp586 from a nearby α-helix (res. Glu582-Met603) in the WT simulations. Additionally, the Arg573 side chain stacks planarly with the aromatic phenol ring of Tyr665 and hydrogen bonds with the hydroxyl group of Ser668 from another α-helix (res. Ser641-Ser668). In the variant simulations, the indole ring of the Trp573 side chain is unable to maintain the same level of coordination as the positively charged Arg573 side chain. Indeed, Trp573 is seen hydrogen bonding only briefly with the carboxylate group of Glu582. Consequently, the integrity of the opposing α-helix end (res. Glu582-Met603) is weakened. Overall, the residue swap has the potential to substantially affect the tertiary structure assembly during the protein folding process. | |||||||||||||||
| c.1721T>C | L574P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L574P is not reported in ClinVar and is present in gnomAD (6-33440773‑T‑C). Prediction tools that indicate a benign effect include REVEL, PROVEAN, and SIFT, whereas the majority of tools predict a pathogenic impact: FoldX, Rosetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, Foldetta, and the SGM Consensus score (likely pathogenic). High‑accuracy methods specifically give a pathogenic verdict: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.083462 | Structured | 0.026427 | Uncertain | 0.927 | 0.246 | 0.000 | 6-33440773-T-C | -13.394 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 2.95 | Destabilizing | 0.5 | 9.19 | Destabilizing | 6.07 | Destabilizing | 0.59 | Ambiguous | 0.442 | Likely Benign | -1.05 | Neutral | 1.000 | Probably Damaging | 0.971 | Probably Damaging | -1.29 | Pathogenic | 0.26 | Tolerated | 3.38 | 32 | 0.3811 | 0.0953 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||
| c.1723C>T | R575C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R575C is listed in ClinVar with an “Uncertain” status (ClinVar ID 537013.0) and is present in gnomAD (ID 6‑33440775‑C‑T). Prediction tools that indicate a benign effect include only AlphaMissense‑Optimized. All other evaluated algorithms predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Four tools (FoldX, Rosetta, Foldetta, premPS) returned uncertain results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the majority of predictions support a pathogenic effect. Thus, the variant is most likely pathogenic, which is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.021362 | Uncertain | 0.916 | 0.259 | 0.000 | Conflicting | 3 | 6-33440775-C-T | 23 | 1.43e-5 | -11.179 | Likely Pathogenic | 0.630 | Likely Pathogenic | Likely Benign | 1.39 | Ambiguous | 0.2 | 0.50 | Ambiguous | 0.95 | Ambiguous | 0.73 | Ambiguous | 0.715 | Likely Pathogenic | -5.43 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.30 | Pathogenic | 0.02 | Affected | 3.37 | 35 | 0.2969 | 0.1692 | -4 | -3 | 7.0 | -53.05 | 227.7 | 99.2 | 0.0 | 0.0 | 0.0 | 0.1 | X | Potentially Pathogenic | The guanidinium group of Arg575, located in an α-helix (res. Arg563-Glu578), forms salt bridges with the carboxylate groups of Asp463 and Asp467, and it also hydrogen bonds with the hydroxyl group of Ser466 on an opposing α-helix (res. Ala461-Phe476) in the WT simulations. In the variant simulations, the thiol group of the Cys575 side chain, which is neither positively charged nor particularly hydrophilic, packs against the hydrophobic Met470 on an opposing α-helix (res. Ala461-Arg475). Additionally, although the thiol group is not an effective hydrogen bonder, the Cys575 side chain rotates to hydrogen bond with the backbone carbonyl group of Ser571 in the same α-helix, which could theoretically lower the helix integrity. Overall, the residue swap has the potential to substantially affect the tertiary structure assembly during the protein folding process. | |||||||||||||
| c.1726T>G | C576G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C576G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from REVEL, FoldX (uncertain), Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a benign outcome. When high‑accuracy methods are considered, AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labels it likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) also predicts pathogenicity. No prediction is inconclusive or missing. Consequently, the variant is most likely pathogenic based on the consensus of available computational evidence, and this assessment does not contradict any ClinVar annotation (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.017684 | Uncertain | 0.913 | 0.245 | 0.000 | -14.809 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 1.66 | Ambiguous | 0.0 | 2.53 | Destabilizing | 2.10 | Destabilizing | 1.67 | Destabilizing | 0.586 | Likely Pathogenic | -10.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.38 | Benign | 0.00 | Affected | 0.3269 | 0.2574 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1735C>G | R579G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R579G is reported in gnomAD (ID 6‑33440787‑C‑G) and has no ClinVar entry. Prediction tools that assess pathogenicity uniformly favor a deleterious effect: SGM‑Consensus (Likely Pathogenic), REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenic. No tool in the dataset reports a benign outcome; the only uncertain calls are from FoldX, AlphaMissense‑Optimized, and Foldetta. High‑accuracy assessments further support pathogenicity: the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic, while AlphaMissense‑Optimized and Foldetta remain uncertain. Consequently, the collective evidence indicates that R579G is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.053060 | Structured | 0.022872 | Uncertain | 0.877 | 0.244 | 0.000 | 6-33440787-C-G | 1 | 6.20e-7 | -14.298 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 1.43 | Ambiguous | 0.0 | 2.36 | Destabilizing | 1.90 | Ambiguous | 1.32 | Destabilizing | 0.680 | Likely Pathogenic | -5.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.40 | Pathogenic | 0.01 | Affected | 3.37 | 34 | 0.3078 | 0.2554 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1738G>T | G580C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G580C is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a pathogenic outcome include REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools with uncertain or inconclusive results are Rosetta and premPS. High‑accuracy methods all support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No tool predicts benign. **Based on the consensus of available predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status (which is currently unreported).** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.025952 | Uncertain | 0.853 | 0.236 | 0.000 | -11.608 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 2.94 | Destabilizing | 0.1 | 1.18 | Ambiguous | 2.06 | Destabilizing | 0.60 | Ambiguous | 0.755 | Likely Pathogenic | -8.66 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.18 | Pathogenic | 0.01 | Affected | 0.1422 | 0.2146 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1739G>T | G580V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G580V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess functional impact uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. The only inconclusive result is premPS, which is listed as uncertain. No tool predicts a benign effect. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenic. Based on the unanimous pathogenic predictions and the absence of any benign calls, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.025952 | Uncertain | 0.853 | 0.236 | 0.000 | -13.705 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 4.10 | Destabilizing | 0.1 | 3.89 | Destabilizing | 4.00 | Destabilizing | 0.79 | Ambiguous | 0.750 | Likely Pathogenic | -8.66 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.18 | Pathogenic | 0.04 | Affected | 0.1270 | 0.2909 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1741C>T | R581W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R581W is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include only Rosetta, whereas the remaining pathogenic‑predicating tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—consistently classify the variant as deleterious. Uncertain or inconclusive results come from FoldX, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain”; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Pathogenic”; and Foldetta remains “Uncertain.” Overall, the preponderance of evidence points to a pathogenic impact, which contrasts with the ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.029544 | Uncertain | 0.829 | 0.236 | 0.000 | Uncertain | 2 | -12.855 | Likely Pathogenic | 0.920 | Likely Pathogenic | Ambiguous | 1.32 | Ambiguous | 0.1 | -0.32 | Likely Benign | 0.50 | Ambiguous | 0.68 | Ambiguous | 0.678 | Likely Pathogenic | -6.79 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | -1.37 | Pathogenic | 0.01 | Affected | 3.37 | 34 | 0.1142 | 0.3043 | 2 | -3 | 3.6 | 30.03 | 257.8 | 36.0 | 0.1 | 0.1 | 0.1 | 0.3 | X | X | Potentially Pathogenic | Arg581 is located on a short α-α loop between two α helices (res. Arg563-Glu578 and res. Glu582-Ser604). In the WT simulations, the guanidinium group of Arg581 forms salt bridges with the carboxylate groups of Asp583 within the same helix, as well as with Glu478 and/or Glu480 in a slightly α-helical loop (res. Glu478-Thr488) preceding another α helix (res. Ala461-Phe476).In the variant simulations, the neutral indole ring of the Trp581 side chain cannot form any of these salt bridges. Instead, it packs hydrophobically against Met477 and Ile587 without forming any direct hydrogen bonds. The tendency of the loop (res. Asp477-Thr488) to acquire an α-helical structure seems to marginally increase, potentially due to Trp581's inability to coordinate stable hydrogen bonds with the loop residues (e.g., Glu478-Arg581 salt bridge). Additionally, the residue swap could weaken the tertiary structure assembly and negatively affect the overall protein folding process. | |||||||||||||||
| c.1742G>T | R581L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R581L is not reported in ClinVar and is present in gnomAD (ID 6‑33440794‑G‑T). Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, premPS, and SIFT, whereas tools that predict pathogenicity are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy AlphaMissense‑Optimized score is pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenicity. In contrast, the Foldetta stability assessment, which integrates FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the majority of computational evidence supports a pathogenic classification for R581L, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.029544 | Uncertain | 0.829 | 0.236 | 0.000 | 6-33440794-G-T | 3 | 1.86e-6 | -10.134 | Likely Pathogenic | 0.958 | Likely Pathogenic | Likely Pathogenic | 0.29 | Likely Benign | 0.1 | -0.20 | Likely Benign | 0.05 | Likely Benign | 0.45 | Likely Benign | 0.654 | Likely Pathogenic | -5.93 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.33 | Pathogenic | 0.08 | Tolerated | 3.37 | 34 | 0.1550 | 0.3483 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||
| c.1747G>T | D583Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D583Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect are Foldetta and premPS, whereas the remaining tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict a pathogenic outcome; FoldX and Rosetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | -12.187 | Likely Pathogenic | 0.970 | Likely Pathogenic | Likely Pathogenic | 0.75 | Ambiguous | 0.2 | -1.10 | Ambiguous | -0.18 | Likely Benign | 0.10 | Likely Benign | 0.760 | Likely Pathogenic | -8.50 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.45 | Pathogenic | 0.01 | Affected | 0.0537 | 0.3868 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1748A>T | D583V 2D 3DClick to see structure in 3D Viewer AISynGAP1 D583V is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Rosetta, Foldetta, premPS, and SIFT, whereas pathogenic predictions are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX and ESM1b give uncertain results. High‑accuracy methods give a split view: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) predicts benign. Overall, the majority of tools support a pathogenic effect, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | -7.796 | In-Between | 0.973 | Likely Pathogenic | Likely Pathogenic | 1.20 | Ambiguous | 0.2 | -0.31 | Likely Benign | 0.45 | Likely Benign | 0.12 | Likely Benign | 0.839 | Likely Pathogenic | -8.63 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | -1.40 | Pathogenic | 0.08 | Tolerated | 0.0778 | 0.4090 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1751T>A | I584N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I584N is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic. No tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a pathogenic impact. All available predictions are concordant and supportive. Based on these computational assessments, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.059222 | Structured | 0.046673 | Uncertain | 0.846 | 0.244 | 0.000 | -13.153 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 2.70 | Destabilizing | 0.1 | 2.13 | Destabilizing | 2.42 | Destabilizing | 2.08 | Destabilizing | 0.706 | Likely Pathogenic | -6.57 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.18 | Pathogenic | 0.01 | Affected | 0.0693 | 0.0470 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1756G>T | D586Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D586Y missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that indicate a benign effect include FoldX, SIFT, and premPS, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic outcome. Rosetta and Foldetta, which assess protein‑folding stability, return uncertain results and are therefore not considered evidence for either direction. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also as pathogenic, and Foldetta as uncertain. Overall, the preponderance of pathogenic predictions (13 vs. 3 benign) suggests that D586Y is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -12.916 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 0.37 | Likely Benign | 0.4 | 1.09 | Ambiguous | 0.73 | Ambiguous | -0.49 | Likely Benign | 0.712 | Likely Pathogenic | -5.38 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.16 | Pathogenic | 0.41 | Tolerated | 0.0623 | 0.5634 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1757A>T | D586V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D586V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, premPS, and SIFT. Tools that predict pathogenicity include SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions (nine pathogenic vs. five benign) indicate a pathogenic effect. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -12.409 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.40 | Likely Benign | 0.2 | 0.03 | Likely Benign | 0.22 | Likely Benign | 0.18 | Likely Benign | 0.801 | Likely Pathogenic | -5.58 | Deleterious | 0.998 | Probably Damaging | 0.999 | Probably Damaging | -1.23 | Pathogenic | 0.24 | Tolerated | 0.0826 | 0.5458 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1759A>G | R587G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R587G is not reported in ClinVar and is present in gnomAD (ID 6‑33440811‑A‑G). Prediction tools that agree on a benign effect include SIFT and AlphaMissense‑Optimized, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) classify the change as pathogenic. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized predicts benign, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—returns pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the preponderance of evidence from multiple independent predictors indicates that R587G is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | 6-33440811-A-G | 2 | 1.24e-6 | -13.780 | Likely Pathogenic | 0.780 | Likely Pathogenic | Likely Benign | 1.55 | Ambiguous | 0.2 | 2.43 | Destabilizing | 1.99 | Ambiguous | 1.55 | Destabilizing | 0.578 | Likely Pathogenic | -6.07 | Deleterious | 1.000 | Probably Damaging | 0.972 | Probably Damaging | -1.28 | Pathogenic | 0.07 | Tolerated | 3.37 | 35 | 0.3183 | 0.3401 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1763T>A | L588H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L588H is listed in ClinVar (ID 422233.0) as Pathogenic and is not reported in gnomAD. All available in silico predictors classify the change as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Pathogenic. Thus, the variant is most likely pathogenic, and this prediction aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.082229 | Uncertain | 0.887 | 0.214 | 0.000 | Likely Pathogenic | 1 | -16.947 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.20 | Destabilizing | 0.2 | 3.69 | Destabilizing | 3.95 | Destabilizing | 2.26 | Destabilizing | 0.939 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.42 | Pathogenic | 0.00 | Affected | 3.38 | 34 | 0.0980 | 0.0456 | -2 | -3 | -7.0 | 23.98 | 214.3 | 20.9 | 0.0 | 0.0 | 0.0 | 0.2 | X | X | X | Potentially Pathogenic | The isobutyl group of the Leu588 side chain, located in an α helix (res. Glu582-Met603), packs against hydrophobic residues in the inter-helix hydrophobic space (e.g., Ile584, Trp572, Phe484, Met470, Val473, Ile483).In the variant simulations, the imidazole ring of His588 is aromatic but contains polar delta and epsilon nitrogen atoms that are not suited for the hydrophobic niche. The protonated epsilon nitrogen forms a hydrogen bond with the backbone carbonyl group of Ala469, which can disrupt the continuity of the opposing α helix (res. Phe476-Lys460).While the residue swap could affect the tertiary assembly and the underlying protein folding process, it is difficult to determine if the mutation would be tolerated based solely on the variant simulations. | ||||||||||||||
| c.1763T>C | L588P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L588P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All available in silico predictors classify the change as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.082229 | Uncertain | 0.887 | 0.214 | 0.000 | Uncertain | 2 | -14.771 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 5.61 | Destabilizing | 0.5 | 12.91 | Destabilizing | 9.26 | Destabilizing | 2.33 | Destabilizing | 0.932 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.42 | Pathogenic | 0.00 | Affected | 3.38 | 34 | 0.3533 | 0.0992 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||
| c.1766T>A | I589N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I589N is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. All available predictions are consistent and conclusive. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.018415 | Structured | 0.084536 | Uncertain | 0.927 | 0.214 | 0.000 | -15.539 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.72 | Destabilizing | 0.2 | 3.13 | Destabilizing | 3.43 | Destabilizing | 2.69 | Destabilizing | 0.930 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.99 | Pathogenic | 0.00 | Affected | 0.0968 | 0.0742 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.176T>C | L59P 2D AIThe SynGAP1 missense variant L59P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.212910 | Structured | 0.484882 | Uncertain | 0.510 | 0.668 | 0.000 | -7.076 | In-Between | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.362 | Likely Benign | -2.74 | Deleterious | 0.943 | Possibly Damaging | 0.944 | Probably Damaging | 3.25 | Benign | 0.00 | Affected | 0.3787 | 0.1382 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.1778T>A | L593H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L593H is listed in ClinVar with an uncertain significance and is not present in gnomAD. In silico predictors that classify the variant as benign include only FATHMM. All other evaluated tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic effect. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No prediction or stability result is missing or inconclusive. Overall, the variant is most likely pathogenic based on the consensus of predictions, and this assessment does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.110534 | Uncertain | 0.941 | 0.151 | 0.000 | Uncertain | 1 | -16.504 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.52 | Destabilizing | 0.2 | 2.32 | Destabilizing | 2.42 | Destabilizing | 2.75 | Destabilizing | 0.812 | Likely Pathogenic | -6.77 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.77 | Benign | 0.00 | Affected | 3.37 | 35 | 0.1101 | 0.0541 | -2 | -3 | -7.0 | 23.98 | 222.0 | 20.7 | 0.0 | 0.0 | 0.2 | 0.0 | X | X | Potentially Pathogenic | The iso-propyl side chain of Leu593, located in an α helix (res. Glu582-Met603), packs favourably with multiple hydrophobic residues in the inter-helix space (e.g., Leu598, Ile589, Phe594, Phe561).In the variant simulations, His593 retains a similar packing arrangement via its aromatic imidazole ring. However, the polar nitrogen atoms introduce hydrogen bond donors and acceptors into the previously hydrophobic space. The epsilon protonated nitrogen of His593 forms a stable hydrogen bond with the phenol group of the Tyr505 side chain in an α helix (res. Gln503-Tyr518).While the residue swap could affect the tertiary assembly and the underlying protein folding process, it is difficult to determine if the mutation would be tolerated based solely on the variant simulations. | |||||||||||||||
| c.1778T>C | L593P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L593P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic outcome. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, a conclusion that contradicts its current ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.110534 | Uncertain | 0.941 | 0.151 | 0.000 | Uncertain | 1 | -13.961 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.75 | Destabilizing | 0.9 | 10.77 | Destabilizing | 8.26 | Destabilizing | 2.43 | Destabilizing | 0.777 | Likely Pathogenic | -6.77 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.77 | Benign | 0.00 | Affected | 0.3783 | 0.1274 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||
| c.1784T>C | L595P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L595P is listed in ClinVar with an “Uncertain” status (ClinVar ID 3172762.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.015344 | Structured | 0.128444 | Uncertain | 0.920 | 0.150 | 0.000 | Uncertain | 1 | -11.856 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.09 | Destabilizing | 0.8 | 5.88 | Destabilizing | 3.99 | Destabilizing | 1.78 | Destabilizing | 0.747 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.72 | Benign | 0.00 | Affected | 3.37 | 35 | 0.3336 | 0.1713 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||
| c.1786C>T | R596C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R596C is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33440838‑C‑T). Prediction tools that indicate a benign effect include only premPS. All other evaluated algorithms—REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—classify the variant as pathogenic or likely pathogenic, while Rosetta remains inconclusive. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. **Thus, the variant is most likely pathogenic based on the collective predictions, which does not contradict the ClinVar uncertain status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.017797 | Structured | 0.135423 | Uncertain | 0.918 | 0.134 | 0.000 | Conflicting | 2 | 6-33440838-C-T | 6 | 3.72e-6 | -10.805 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | 2.94 | Destabilizing | 0.0 | 1.49 | Ambiguous | 2.22 | Destabilizing | -0.03 | Likely Benign | 0.633 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.3429 | 0.2211 | -4 | -3 | 7.0 | -53.05 | 230.7 | 97.9 | -0.1 | 0.0 | -0.3 | 0.4 | X | X | Potentially Pathogenic | The guanidinium group of Arg596, located in an α helix (res. Glu582-Met603), forms a salt bridge with the carboxylate group of Glu495 from another α helix (res. Leu489-Glu519). In the WT simulations, the side chain of Arg596 hydrogen bonds with the backbone carbonyl groups of Asn487, Glu486, Arg485, and Phe484. Additionally, Arg596 can hydrogen bond with the carboxamide group of the Asn487 side chain on an opposing loop that links two α helices (res. Ala461-Arg475, res. Leu489-Glu519).In the variant simulations, the thiol group of the Cys596 side chain is unable to form salt bridges or any of the hydrogen bonds that the Arg596 side chain can. Thus, the residue swap could affect the tertiary structure assembly more profoundly than observed in the simulations. Notably, Arg596 plays a key role in positioning the aforementioned loop, which is crucial for the placement of the “arginine finger” or the Arg485 side chain during RasGTPase activation. | ||||||||||||
| c.178G>T | D60Y 2D AIThe SynGAP1 D60Y missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the D60Y variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -7.748 | In-Between | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.221 | Likely Benign | -2.60 | Deleterious | 0.972 | Probably Damaging | 0.969 | Probably Damaging | 3.90 | Benign | 0.00 | Affected | 0.0517 | 0.7790 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||||
| c.1793T>A | L598H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L598H is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized returns a pathogenic score; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.007259 | Structured | 0.147872 | Uncertain | 0.953 | 0.154 | 0.000 | -17.210 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 2.52 | Destabilizing | 0.2 | 2.37 | Destabilizing | 2.45 | Destabilizing | 2.24 | Destabilizing | 0.648 | Likely Pathogenic | -6.87 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.10 | Benign | 0.00 | Affected | 0.1060 | 0.0879 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1793T>C | L598P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L598P is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: pathogenic predictions come from SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a benign outcome. High‑accuracy methods reinforce the pathogenic view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates it is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also classifies it as pathogenic. No prediction or stability result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.007259 | Structured | 0.147872 | Uncertain | 0.953 | 0.154 | 0.000 | -12.621 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 8.31 | Destabilizing | 0.5 | 12.04 | Destabilizing | 10.18 | Destabilizing | 2.02 | Destabilizing | 0.705 | Likely Pathogenic | -6.87 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.10 | Benign | 0.00 | Affected | 0.2742 | 0.1192 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1795T>C | C599R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C599R is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a deleterious effect: REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, PROVEAN, ESM1b, FATHMM, premPS, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic, while FoldX reports an uncertain outcome. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic status; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009865 | Structured | 0.151725 | Uncertain | 0.960 | 0.151 | 0.000 | -15.968 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.00 | Ambiguous | 0.3 | 3.29 | Destabilizing | 2.15 | Destabilizing | 1.54 | Destabilizing | 0.909 | Likely Pathogenic | -11.95 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.50 | Pathogenic | 0.00 | Affected | 0.1519 | 0.1566 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.1795T>G | C599G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a deleterious effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts a benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the evidence strongly favors a pathogenic effect, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009865 | Structured | 0.151725 | Uncertain | 0.960 | 0.151 | 0.000 | -13.342 | Likely Pathogenic | 0.949 | Likely Pathogenic | Ambiguous | 1.40 | Ambiguous | 0.0 | 0.90 | Ambiguous | 1.15 | Ambiguous | 1.43 | Destabilizing | 0.923 | Likely Pathogenic | -11.95 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.50 | Pathogenic | 0.03 | Affected | 0.2444 | 0.2130 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1799C>T | P600L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P600L is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include Rosetta and premPS, whereas the majority of tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. FoldX and Foldetta give uncertain results. High‑accuracy methods specifically show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta remains inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for P600L, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.162960 | Uncertain | 0.947 | 0.147 | 0.000 | -13.209 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.36 | Ambiguous | 0.1 | -3.58 | Stabilizing | -1.11 | Ambiguous | -0.49 | Likely Benign | 0.734 | Likely Pathogenic | -9.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.35 | Pathogenic | 0.00 | Affected | 0.2391 | 0.5555 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.179A>T | D60V 2D AIThe SynGAP1 D60V missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the majority of evaluated predictors (7 of 10) indicate pathogenicity, while only three suggest benignity. Therefore, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -6.576 | Likely Benign | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.254 | Likely Benign | -2.72 | Deleterious | 0.972 | Probably Damaging | 0.954 | Probably Damaging | 3.91 | Benign | 0.00 | Affected | 0.0765 | 0.7962 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.1805T>A | I602N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I602N is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All available evidence points to a damaging impact. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010221 | Structured | 0.186541 | Uncertain | 0.963 | 0.171 | 0.000 | -16.033 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 2.94 | Destabilizing | 0.2 | 3.34 | Destabilizing | 3.14 | Destabilizing | 2.31 | Destabilizing | 0.880 | Likely Pathogenic | -6.89 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -2.01 | Pathogenic | 0.00 | Affected | 0.0718 | 0.0700 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1811C>G | S604W 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S604W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Benign predictions are provided by Rosetta, premPS, and FATHMM. Stability‑based methods give mixed results: FoldX is uncertain, while Foldetta is also uncertain. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors pathogenic. Foldetta remains inconclusive. Overall, the consensus of the majority of tools indicates a pathogenic effect. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.192527 | Uncertain | 0.911 | 0.195 | 0.000 | -19.117 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | -1.23 | Ambiguous | 0.1 | -2.05 | Stabilizing | -1.64 | Ambiguous | -0.19 | Likely Benign | 0.645 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.07 | Benign | 0.00 | Affected | 0.0818 | 0.4602 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1814C>T | P605L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P605L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only premPS. All other evaluated tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, AlphaMissense‑Default, AlphaMissense‑Optimized, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, indicates a pathogenic effect. Based on the overwhelming agreement among these predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.023087 | Structured | 0.192737 | Uncertain | 0.929 | 0.231 | 0.000 | -12.114 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.65 | Destabilizing | 1.1 | 2.74 | Destabilizing | 2.70 | Destabilizing | -0.10 | Likely Benign | 0.814 | Likely Pathogenic | -9.95 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 0.69 | Pathogenic | 0.00 | Affected | 0.2232 | 0.6158 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1820T>A | L607H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L607H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on pathogenicity include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a benign effect. Uncertain predictions come from FoldX, Rosetta, and Foldetta. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta’s stability analysis is inconclusive. Taken together, the overwhelming majority of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.194229 | Uncertain | 0.869 | 0.250 | 0.000 | -14.775 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.76 | Ambiguous | 0.1 | 1.88 | Ambiguous | 1.32 | Ambiguous | 1.38 | Destabilizing | 0.906 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.48 | Pathogenic | 0.01 | Affected | 0.1199 | 0.0541 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1820T>C | L607P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L607P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while the SGM‑Consensus score is “Likely Pathogenic.” No tool in the dataset predicts a benign outcome; the only inconclusive result is FoldX, which is listed as uncertain and therefore does not influence the overall assessment. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. Consequently, the variant is most likely pathogenic based on the available predictions, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.194229 | Uncertain | 0.869 | 0.250 | 0.000 | -14.059 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 1.11 | Ambiguous | 0.7 | 6.93 | Destabilizing | 4.02 | Destabilizing | 1.29 | Destabilizing | 0.922 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.54 | Pathogenic | 0.00 | Affected | 0.3710 | 0.1274 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1826G>T | G609V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G609V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from premPS and AlphaMissense‑Optimized, whereas the remaining 10 tools (SGM Consensus, REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM) all predict pathogenicity. High‑accuracy assessments further highlight the discordance: AlphaMissense‑Optimized reports a benign effect, whereas the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta outputs) both indicate pathogenicity. With the majority of evidence pointing to deleterious impact, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.203786 | Uncertain | 0.851 | 0.252 | 0.000 | -11.049 | Likely Pathogenic | 0.374 | Ambiguous | Likely Benign | 4.17 | Destabilizing | 0.3 | 3.77 | Destabilizing | 3.97 | Destabilizing | 0.34 | Likely Benign | 0.734 | Likely Pathogenic | -4.47 | Deleterious | 0.974 | Probably Damaging | 0.818 | Possibly Damaging | -1.48 | Pathogenic | 0.02 | Affected | 0.1269 | 0.3317 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1829T>A | L610H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L610H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic. No tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a destabilizing, pathogenic effect. All available predictions are concordant and supportive. Based on these computational assessments, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.209504 | Uncertain | 0.888 | 0.253 | 0.000 | -14.573 | Likely Pathogenic | 0.960 | Likely Pathogenic | Likely Pathogenic | 4.83 | Destabilizing | 0.5 | 4.98 | Destabilizing | 4.91 | Destabilizing | 2.03 | Destabilizing | 0.927 | Likely Pathogenic | -6.91 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.59 | Pathogenic | 0.00 | Affected | 0.1067 | 0.0741 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1829T>C | L610P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L610P is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, every tool examined (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) classifies the substitution as pathogenic; no tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a pathogenic effect. Because all evidence points to a deleterious impact and there is no ClinVar entry to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.209504 | Uncertain | 0.888 | 0.253 | 0.000 | -14.863 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 6.02 | Destabilizing | 0.2 | 8.15 | Destabilizing | 7.09 | Destabilizing | 1.99 | Destabilizing | 0.934 | Likely Pathogenic | -6.91 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.59 | Pathogenic | 0.00 | Affected | 0.3599 | 0.1113 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1840T>G | Y614D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y614D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM predicts it to be benign, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify it as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.182134 | Uncertain | 0.852 | 0.254 | 0.000 | -15.073 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 2.14 | Destabilizing | 0.6 | 4.16 | Destabilizing | 3.15 | Destabilizing | 1.69 | Destabilizing | 0.597 | Likely Pathogenic | -9.83 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.40 | Benign | 0.02 | Affected | 0.4013 | 0.0573 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1841A>C | Y614S 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant Y614S is not reported in ClinVar and is present in gnomAD (ID 6‑33440893‑A‑C). Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas the majority of algorithms—AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, PROVEAN, polyPhen‑2 (HumDiv and HumVar), premPS, Rosetta, Foldetta, and the SGM Consensus—indicate pathogenicity; FoldX remains uncertain. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized scores the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labels it likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic effect. Overall, the preponderance of evidence points to a pathogenic classification, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.182134 | Uncertain | 0.852 | 0.254 | 0.000 | 6-33440893-A-C | 1 | 6.20e-7 | -12.709 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.74 | Ambiguous | 0.4 | 3.25 | Destabilizing | 2.50 | Destabilizing | 2.05 | Destabilizing | 0.482 | Likely Benign | -8.83 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.48 | Benign | 0.09 | Tolerated | 3.37 | 35 | 0.4155 | 0.1220 | -2 | -3 | 0.5 | -76.10 | ||||||||||||||||||||||||
| c.1844C>T | P615L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P615L is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS and Rosetta, whereas the remaining tools—REVEL, FoldX, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, PROVEAN, and the SGM Consensus—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenicity, and Foldetta yields an uncertain result. Taken together, the overwhelming majority of computational evidence indicates that P615L is likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.179032 | Uncertain | 0.879 | 0.255 | 0.000 | -11.884 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.03 | Destabilizing | 0.4 | -0.01 | Likely Benign | 1.01 | Ambiguous | 0.50 | Likely Benign | 0.699 | Likely Pathogenic | -9.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.13 | Pathogenic | 0.04 | Affected | 0.1949 | 0.4692 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1846G>T | D616Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D616Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM, whereas a majority of tools predict a pathogenic outcome: SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as likely pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No evidence from FoldX or Rosetta alone is conclusive. Overall, the balance of evidence favors a pathogenic interpretation, and this conclusion does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | -12.638 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 1.70 | Ambiguous | 0.3 | 1.32 | Ambiguous | 1.51 | Ambiguous | 0.35 | Likely Benign | 0.374 | Likely Benign | -7.43 | Deleterious | 0.999 | Probably Damaging | 0.970 | Probably Damaging | 3.28 | Benign | 0.01 | Affected | 0.0465 | 0.4069 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1847A>T | D616V 2D 3DClick to see structure in 3D Viewer AISynGAP1 D616V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, premPS, and FATHMM, while pathogenic calls are made by FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus. Uncertain results are reported by Rosetta and AlphaMissense‑Optimized. High‑accuracy assessments give a pathogenic signal: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of evidence, including the high‑accuracy tools, supports a pathogenic effect for D616V. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | -13.992 | Likely Pathogenic | 0.919 | Likely Pathogenic | Ambiguous | 2.41 | Destabilizing | 0.2 | 1.95 | Ambiguous | 2.18 | Destabilizing | 0.36 | Likely Benign | 0.268 | Likely Benign | -7.36 | Deleterious | 0.972 | Probably Damaging | 0.682 | Possibly Damaging | 3.26 | Benign | 0.00 | Affected | 0.0699 | 0.4393 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.184G>T | D62Y 2D AIThe SynGAP1 missense variant D62Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -6.313 | Likely Benign | 0.569 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | -2.17 | Neutral | 0.388 | Benign | 0.328 | Benign | 4.03 | Benign | 0.00 | Affected | 0.0657 | 0.5588 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.185A>T | D62V 2D AIThe SynGAP1 missense variant D62V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate a benign outcome, while the sole pathogenic signal comes from SIFT. AlphaMissense‑Default remains uncertain, and no Foldetta stability assessment is available. High‑accuracy methods reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports Likely Benign, and Foldetta data are missing. Taken together, the preponderance of evidence supports a benign classification for D62V, and this assessment does not conflict with the absence of a ClinVar entry. Therefore, the variant is most likely benign, and this conclusion does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -4.417 | Likely Benign | 0.489 | Ambiguous | Likely Benign | 0.118 | Likely Benign | -2.04 | Neutral | 0.028 | Benign | 0.088 | Benign | 4.04 | Benign | 0.00 | Affected | 0.1039 | 0.6129 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.1861C>G | R621G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R621G is reported in gnomAD (ID 6‑33440913‑C‑G) but has no ClinVar entry. In silico predictors largely agree on a deleterious effect: pathogenic calls come from REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The only benign prediction is from FATHMM; FoldX and Foldetta give uncertain results. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence indicates the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.084420 | Uncertain | 0.945 | 0.216 | 0.000 | 6-33440913-C-G | 1 | 6.20e-7 | -16.611 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.18 | Ambiguous | 0.3 | 2.07 | Destabilizing | 1.63 | Ambiguous | 1.17 | Destabilizing | 0.558 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.84 | Benign | 0.01 | Affected | 3.37 | 35 | 0.3109 | 0.2651 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1868T>A | L623H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L623H is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while no tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized indicates pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No contradictory evidence is present. Based on the unanimous computational predictions, the variant is most likely pathogenic, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.060667 | Uncertain | 0.962 | 0.211 | 0.000 | -14.631 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.76 | Destabilizing | 0.3 | 2.22 | Destabilizing | 2.49 | Destabilizing | 2.40 | Destabilizing | 0.793 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.55 | Pathogenic | 0.00 | Affected | 0.1099 | 0.0541 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1868T>C | L623P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L623P is not reported in ClinVar and is absent from gnomAD. Prediction tools uniformly indicate a deleterious effect: pathogenic predictions are returned by REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the change as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels it likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a pathogenic effect. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.060667 | Uncertain | 0.962 | 0.211 | 0.000 | -12.385 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.70 | Destabilizing | 0.2 | 11.18 | Destabilizing | 8.94 | Destabilizing | 2.26 | Destabilizing | 0.788 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.56 | Pathogenic | 0.00 | Affected | 0.3961 | 0.1474 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1874T>A | L625H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L625H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.045896 | Uncertain | 0.966 | 0.215 | 0.000 | -14.264 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.85 | Destabilizing | 0.3 | 2.96 | Destabilizing | 2.91 | Destabilizing | 2.02 | Destabilizing | 0.738 | Likely Pathogenic | -6.87 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.98 | Benign | 0.00 | Affected | 0.0954 | 0.0541 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1874T>C | L625P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L625P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy methods give consistent results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.045896 | Uncertain | 0.966 | 0.215 | 0.000 | -14.819 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 5.47 | Destabilizing | 0.6 | 12.49 | Destabilizing | 8.98 | Destabilizing | 1.98 | Destabilizing | 0.746 | Likely Pathogenic | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.98 | Benign | 0.00 | Affected | 0.3294 | 0.1313 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1877T>A | I626N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I626N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods further support this: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the unanimous agreement of the majority of tools and the corroborating high‑accuracy predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.040732 | Uncertain | 0.970 | 0.223 | 0.000 | -15.240 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.49 | Destabilizing | 0.1 | 3.56 | Destabilizing | 3.53 | Destabilizing | 2.36 | Destabilizing | 0.621 | Likely Pathogenic | -6.41 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.03 | Benign | 0.00 | Affected | 0.0880 | 0.0270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1886T>A | V629D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V629D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all return pathogenic or likely pathogenic calls, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized indicates pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) predicts pathogenic. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -17.143 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 3.85 | Destabilizing | 0.1 | 3.82 | Destabilizing | 3.84 | Destabilizing | 2.17 | Destabilizing | 0.713 | Likely Pathogenic | -6.37 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.06 | Benign | 0.00 | Affected | 0.1284 | 0.0541 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1886T>G | V629G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V629G missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining eleven tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) uniformly predict a pathogenic outcome; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. Taken together, the overwhelming majority of evidence indicates that V629G is likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -13.150 | Likely Pathogenic | 0.871 | Likely Pathogenic | Ambiguous | 3.81 | Destabilizing | 0.0 | 4.52 | Destabilizing | 4.17 | Destabilizing | 1.94 | Destabilizing | 0.678 | Likely Pathogenic | -6.47 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.07 | Benign | 0.00 | Affected | 0.1903 | 0.2240 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1889T>A | I630N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I630N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic. Based on the consensus of all available predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.036106 | Uncertain | 0.966 | 0.236 | 0.000 | -14.259 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | 2.96 | Destabilizing | 0.0 | 2.55 | Destabilizing | 2.76 | Destabilizing | 2.39 | Destabilizing | 0.825 | Likely Pathogenic | -4.86 | Deleterious | 1.000 | Probably Damaging | 0.994 | Probably Damaging | -1.49 | Pathogenic | 0.00 | Affected | 0.0853 | 0.0270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1895A>T | N632I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N632I is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include premPS and Foldetta, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus (likely pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, remains likely pathogenic; and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, indicates a benign effect. Overall, the preponderance of evidence points to a pathogenic classification for N632I, and this conclusion does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.041437 | Uncertain | 0.938 | 0.254 | 0.000 | -12.620 | Likely Pathogenic | 0.881 | Likely Pathogenic | Ambiguous | 1.33 | Ambiguous | 0.3 | -1.24 | Ambiguous | 0.05 | Likely Benign | 0.20 | Likely Benign | 0.839 | Likely Pathogenic | -7.76 | Deleterious | 0.987 | Probably Damaging | 0.887 | Possibly Damaging | -1.56 | Pathogenic | 0.02 | Affected | 0.0712 | 0.5973 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.1898T>C | L633P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L633P (ClinVar ID 858973.0) is listed as Pathogenic and is not reported in gnomAD. Prediction tools that classify the variant as benign include only FATHMM. All other evaluated tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized scores it as Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts Pathogenic. Based on the overwhelming consensus of pathogenic predictions and the ClinVar designation, the variant is most likely pathogenic, with no contradiction to its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.045407 | Uncertain | 0.952 | 0.252 | 0.000 | Pathogenic/Likely path. | 2 | -15.669 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.60 | Destabilizing | 0.2 | 10.15 | Destabilizing | 8.38 | Destabilizing | 2.42 | Destabilizing | 0.693 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.70 | Benign | 0.00 | Affected | 3.37 | 34 | 0.3528 | 0.0953 | -3 | -3 | -5.4 | -16.04 | 193.2 | 65.1 | 0.0 | 0.0 | 0.1 | 0.0 | X | Potentially Pathogenic | The iso-butyl side chain of Leu633, located in the middle of an α helix (res. Glu617-Asn635), packs hydrophobically with nearby residues (e.g., Leu653, Val629, Leu551) in the WT simulations.In the variant simulations, the pyrrolidine side chain of Pro633 is not as optimal for hydrophobic packing as Leu633 in the WT. Additionally, proline lacks a free backbone amide group, so Pro633 cannot form a hydrogen bond with the backbone carbonyl group of Val629, which disrupts the continuity of the secondary structure element. | ||||||||||||||||
| c.1904A>T | N635I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N635I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX is uncertain. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts a benign impact. Overall, the majority of tools lean toward a pathogenic interpretation, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.039760 | Structured | 0.060246 | Uncertain | 0.900 | 0.252 | 0.000 | -15.012 | Likely Pathogenic | 0.608 | Likely Pathogenic | Likely Benign | 0.94 | Ambiguous | 0.1 | -0.05 | Likely Benign | 0.45 | Likely Benign | -0.35 | Likely Benign | 0.363 | Likely Benign | -8.56 | Deleterious | 0.980 | Probably Damaging | 0.889 | Possibly Damaging | 2.88 | Benign | 0.00 | Affected | 0.0736 | 0.3776 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.191T>A | I64K 2D AIThe SynGAP1 missense variant I64K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains benign; Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, and this conclusion does not contradict the ClinVar status, which contains no report for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | -3.206 | Likely Benign | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.159 | Likely Benign | -0.47 | Neutral | 0.334 | Benign | 0.029 | Benign | 4.07 | Benign | 0.00 | Affected | 0.0878 | 0.0740 | -2 | -3 | -8.4 | 15.01 | |||||||||||||||||||||||||||||||||||||||
| c.191T>G | I64R 2D AIThe SynGAP1 missense variant I64R is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which classifies the variant as “Likely Benign.” Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the Foldetta protein‑folding stability assessment is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar assertion, so there is no contradiction with existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | -2.108 | Likely Benign | 0.936 | Likely Pathogenic | Ambiguous | 0.165 | Likely Benign | -0.54 | Neutral | 0.842 | Possibly Damaging | 0.068 | Benign | 4.05 | Benign | 0.00 | Affected | 0.1103 | 0.0940 | -2 | -3 | -9.0 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.1930G>T | D644Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 D644Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, premPS, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default; the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports it as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. The remaining tools, FoldX, REVEL, premPS, polyPhen‑2 HumVar, and FATHMM, support a benign effect, while PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default support a pathogenic effect. Overall, the majority of evidence leans toward pathogenicity, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -10.143 | Likely Pathogenic | 0.721 | Likely Pathogenic | Likely Benign | 0.03 | Likely Benign | 0.1 | -1.18 | Ambiguous | -0.58 | Ambiguous | -0.04 | Likely Benign | 0.318 | Likely Benign | -4.93 | Deleterious | 0.968 | Probably Damaging | 0.311 | Benign | 3.44 | Benign | 0.02 | Affected | 0.0679 | 0.6094 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1931A>T | D644V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D644V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -6.230 | Likely Benign | 0.636 | Likely Pathogenic | Likely Benign | 0.50 | Ambiguous | 0.0 | -0.35 | Likely Benign | 0.08 | Likely Benign | -0.03 | Likely Benign | 0.347 | Likely Benign | -4.96 | Deleterious | 0.198 | Benign | 0.052 | Benign | 3.49 | Benign | 0.11 | Tolerated | 0.0951 | 0.6267 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||
| c.1937T>C | L646P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L646P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.303751 | Uncertain | 0.941 | 0.344 | 0.000 | -12.956 | Likely Pathogenic | 0.970 | Likely Pathogenic | Likely Pathogenic | 7.34 | Destabilizing | 0.5 | 12.08 | Destabilizing | 9.71 | Destabilizing | 2.72 | Destabilizing | 0.604 | Likely Pathogenic | -4.55 | Deleterious | 0.999 | Probably Damaging | 0.926 | Probably Damaging | 3.17 | Benign | 0.00 | Affected | 0.3481 | 0.1874 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1939G>T | G647C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G647C is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, the majority (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus “Likely Benign”) uniformly predict a benign effect, whereas only SIFT classifies the change as pathogenic. High‑accuracy tools give consistent benign results: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a benign stability change. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -2.955 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.41 | Likely Benign | 0.0 | -0.20 | Likely Benign | 0.11 | Likely Benign | 0.05 | Likely Benign | 0.112 | Likely Benign | -1.63 | Neutral | 0.003 | Benign | 0.004 | Benign | 3.44 | Benign | 0.02 | Affected | 0.1194 | 0.3250 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1940G>T | G647V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G647V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that assess sequence conservation and structural impact uniformly classify the substitution as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a tolerated change. No tool predicts pathogenicity; only FoldX and Rosetta report uncertain stability effects, which are treated as unavailable evidence. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a benign effect. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -4.713 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.85 | Ambiguous | 0.1 | -0.59 | Ambiguous | 0.13 | Likely Benign | -0.13 | Likely Benign | 0.115 | Likely Benign | -1.77 | Neutral | 0.178 | Benign | 0.073 | Benign | 3.52 | Benign | 0.12 | Tolerated | 0.1318 | 0.3946 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1949A>T | N650I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N650I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, Rosetta, Foldetta, premPS, and FATHMM; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX reports an uncertain outcome. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the balance of evidence favors a pathogenic effect for the variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.086953 | Structured | 0.361944 | Uncertain | 0.961 | 0.357 | 0.000 | -15.940 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.50 | Ambiguous | 0.2 | 0.21 | Likely Benign | 0.36 | Likely Benign | 0.21 | Likely Benign | 0.485 | Likely Benign | -8.97 | Deleterious | 0.999 | Probably Damaging | 0.955 | Probably Damaging | 3.02 | Benign | 0.00 | Affected | 0.0907 | 0.4339 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.194A>T | H65L 2D AIThe SynGAP1 H65L missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” AlphaMissense‑Optimized returns an uncertain result, and Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.476188 | Uncertain | 0.458 | 0.758 | 0.125 | -1.889 | Likely Benign | 0.836 | Likely Pathogenic | Ambiguous | 0.159 | Likely Benign | -1.65 | Neutral | 0.462 | Possibly Damaging | 0.227 | Benign | 4.22 | Benign | 0.00 | Affected | 0.0718 | 0.4760 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.1958T>C | L653P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L653P is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.049374 | Structured | 0.335213 | Uncertain | 0.963 | 0.332 | 0.000 | -17.675 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 6.02 | Destabilizing | 0.8 | 9.20 | Destabilizing | 7.61 | Destabilizing | 2.56 | Destabilizing | 0.700 | Likely Pathogenic | -6.51 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.09 | Benign | 0.00 | Affected | 0.3251 | 0.1874 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1964T>C | L655P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L655P is catalogued in gnomAD (ID 6‑33441223‑T‑C) but has no ClinVar entry. Functional prediction tools show a split: benign calls come from REVEL, PROVEAN, SIFT, and FATHMM, whereas pathogenic calls are made by Rosetta, Foldetta, both polyPhen‑2 versions, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX and premPS are uncertain. High‑accuracy assessments give AlphaMissense‑Optimized as pathogenic, Foldetta as pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Overall, the majority of evidence points to a pathogenic effect. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.015344 | Structured | 0.268808 | Uncertain | 0.961 | 0.274 | 0.000 | 6-33441223-T-C | 1 | 6.20e-7 | -9.771 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 1.33 | Ambiguous | 0.5 | 6.52 | Destabilizing | 3.93 | Destabilizing | 0.57 | Ambiguous | 0.126 | Likely Benign | -1.36 | Neutral | 0.981 | Probably Damaging | 0.772 | Possibly Damaging | 3.47 | Benign | 0.32 | Tolerated | 3.39 | 24 | 0.3598 | 0.1274 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||
| c.1969T>A | W657R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W657R is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, SIFT, and FATHMM, whereas pathogenic calls are made by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus score, which is labeled Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, and the Foldetta stability analysis is inconclusive. Overall, the majority of evidence points to a pathogenic impact for W657R, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | -13.391 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 1.56 | Ambiguous | 0.2 | 1.64 | Ambiguous | 1.60 | Ambiguous | 1.29 | Destabilizing | 0.461 | Likely Benign | -11.96 | Deleterious | 0.999 | Probably Damaging | 0.964 | Probably Damaging | 3.48 | Benign | 0.07 | Tolerated | 0.4684 | 0.0000 | 2 | -3 | -3.6 | -30.03 | |||||||||||||||||||||||||||||
| c.1969T>C | W657R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W657R is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, while pathogenic predictions are made by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority of the four high‑accuracy tools) is pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive. FoldX and Rosetta individually report uncertain effects on protein stability. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | -13.391 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 1.56 | Ambiguous | 0.2 | 1.64 | Ambiguous | 1.60 | Ambiguous | 1.29 | Destabilizing | 0.461 | Likely Benign | -11.96 | Deleterious | 0.999 | Probably Damaging | 0.964 | Probably Damaging | 3.48 | Benign | 0.07 | Tolerated | 0.4684 | 0.0000 | 2 | -3 | -3.6 | -30.03 | |||||||||||||||||||||||||||||
| c.1970G>C | W657S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W657S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools (SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta) predict a pathogenic impact; Rosetta remains uncertain. High‑accuracy methods all support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | -11.817 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 2.27 | Destabilizing | 0.2 | 1.87 | Ambiguous | 2.07 | Destabilizing | 1.26 | Destabilizing | 0.334 | Likely Benign | -11.82 | Deleterious | 0.999 | Probably Damaging | 0.947 | Probably Damaging | 3.52 | Benign | 0.09 | Tolerated | 0.4299 | 0.0489 | -2 | -3 | 0.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1972G>T | G658C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G658C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. One tool, ESM1b, yields an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign (2 benign vs. 1 pathogenic, 1 uncertain), and Foldetta predicts a benign impact on protein stability. Overall, the majority of evidence points to a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.029376 | Structured | 0.180299 | Uncertain | 0.942 | 0.251 | 0.000 | -7.577 | In-Between | 0.170 | Likely Benign | Likely Benign | 0.05 | Likely Benign | 0.0 | 0.04 | Likely Benign | 0.05 | Likely Benign | 0.46 | Likely Benign | 0.146 | Likely Benign | -3.49 | Deleterious | 0.989 | Probably Damaging | 0.544 | Possibly Damaging | 3.37 | Benign | 0.04 | Affected | 0.1509 | 0.3141 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1973G>T | G658V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G658V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv; Rosetta’s output is uncertain and therefore not used as evidence. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta) as Benign. Taken together, the majority of reliable predictors indicate a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.029376 | Structured | 0.180299 | Uncertain | 0.942 | 0.251 | 0.000 | -6.815 | Likely Benign | 0.166 | Likely Benign | Likely Benign | -0.07 | Likely Benign | 0.0 | -0.71 | Ambiguous | -0.39 | Likely Benign | 0.34 | Likely Benign | 0.117 | Likely Benign | -3.23 | Deleterious | 0.841 | Possibly Damaging | 0.264 | Benign | 3.38 | Benign | 0.13 | Tolerated | 0.1337 | 0.3330 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.197C>T | P66L 2D AIThe SynGAP1 missense variant P66L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized calling the variant pathogenic, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome; Foldetta results are not available. Overall, the predictions are split evenly between benign and pathogenic, with high‑accuracy tools providing opposing conclusions. Consequently, the variant’s impact remains uncertain and does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.394753 | Structured | 0.474132 | Uncertain | 0.455 | 0.762 | 0.125 | -2.437 | Likely Benign | 0.972 | Likely Pathogenic | Likely Pathogenic | 0.194 | Likely Benign | -2.48 | Neutral | 0.909 | Possibly Damaging | 0.713 | Possibly Damaging | 3.92 | Benign | 0.00 | Affected | 0.2412 | 0.6687 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.1993T>G | Y665D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y665D is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as Uncertain. No evidence from these tools contradicts the ClinVar status, which is currently unreported. Overall, the balance of evidence—seven pathogenic versus four benign predictions, with a pathogenic consensus from SGM‑Consensus—suggests the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.086641 | Uncertain | 0.922 | 0.361 | 0.000 | -10.101 | Likely Pathogenic | 0.737 | Likely Pathogenic | Likely Benign | 1.15 | Ambiguous | 0.2 | 1.30 | Ambiguous | 1.23 | Ambiguous | 0.15 | Likely Benign | 0.227 | Likely Benign | -3.67 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.75 | Benign | 1.00 | Tolerated | 0.3838 | 0.0573 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.2000T>A | I667N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I667N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.083597 | Uncertain | 0.927 | 0.379 | 0.000 | -15.730 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 2.89 | Destabilizing | 0.2 | 2.84 | Destabilizing | 2.87 | Destabilizing | 2.56 | Destabilizing | 0.572 | Likely Pathogenic | -6.73 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.95 | Benign | 0.00 | Affected | 0.0841 | 0.0470 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.2006A>T | N669I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N669I is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect include premPS and FATHMM, whereas the remaining ten tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the majority‑vote SGM‑Consensus—predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain (treated as unavailable), SGM‑Consensus as likely pathogenic, and Foldetta as uncertain (also treated as unavailable). The overall consensus of the available predictions leans strongly toward pathogenicity, and this conclusion does not conflict with the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.086615 | Uncertain | 0.872 | 0.380 | 0.000 | -13.324 | Likely Pathogenic | 0.862 | Likely Pathogenic | Ambiguous | 0.84 | Ambiguous | 0.0 | 1.09 | Ambiguous | 0.97 | Ambiguous | 0.31 | Likely Benign | 0.517 | Likely Pathogenic | -8.18 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.0749 | 0.4697 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.2009T>C | L670P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L670P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. Stability‑based methods are inconclusive: FoldX, Rosetta, and the combined Foldetta output are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar assertion; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.161087 | Structured | 0.090855 | Uncertain | 0.812 | 0.385 | 0.000 | -4.916 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.56 | Ambiguous | 0.3 | 1.83 | Ambiguous | 1.20 | Ambiguous | -0.25 | Likely Benign | 0.211 | Likely Benign | 2.42 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.41 | Benign | 0.26 | Tolerated | 0.3590 | 0.1474 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.200T>C | L67P 2D AIThe SynGAP1 missense variant L67P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are unavailable. Overall, the majority of evidence points toward a benign impact. The variant’s predicted benign status does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.473668 | Uncertain | 0.428 | 0.761 | 0.125 | -2.852 | Likely Benign | 0.945 | Likely Pathogenic | Ambiguous | 0.150 | Likely Benign | -0.80 | Neutral | 0.943 | Possibly Damaging | 0.766 | Possibly Damaging | 4.07 | Benign | 0.00 | Affected | 0.3052 | 0.1382 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2011G>T | D671Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D671Y missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split consensus: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy methods give a mixed signal: AlphaMissense‑Optimized and the SGM‑Consensus both indicate pathogenicity, while Foldetta predicts a benign effect on protein stability. Because the majority of standard predictors lean toward pathogenicity and the two high‑confidence tools also favor a deleterious outcome, the variant is most likely pathogenic. This assessment does not contradict ClinVar, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | -13.107 | Likely Pathogenic | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.31 | Likely Benign | 0.1 | 0.05 | Likely Benign | 0.18 | Likely Benign | 0.11 | Likely Benign | 0.385 | Likely Benign | -6.05 | Deleterious | 1.000 | Probably Damaging | 0.961 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 0.0564 | 0.6376 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.2012A>T | D671V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D671V missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus methods give a pathogenic signal: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. Overall, the majority of predictions (seven pathogenic vs. three benign) support a pathogenic classification. There is no ClinVar entry to contradict this assessment, so the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | -12.376 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 0.63 | Ambiguous | 0.1 | 0.51 | Ambiguous | 0.57 | Ambiguous | 0.14 | Likely Benign | 0.379 | Likely Benign | -6.08 | Deleterious | 0.975 | Probably Damaging | 0.885 | Possibly Damaging | 3.31 | Benign | 0.01 | Affected | 0.0820 | 0.6651 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.2018T>C | L673P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L673P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. All other evaluated tools—SGM‑Consensus, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized indicates benign, but the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts Pathogenic. Taken together, the overwhelming majority of predictions, including the high‑accuracy tools, classify the variant as pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.104692 | Uncertain | 0.545 | 0.369 | 0.000 | -12.160 | Likely Pathogenic | 0.622 | Likely Pathogenic | Likely Benign | 2.41 | Destabilizing | 0.3 | 1.63 | Ambiguous | 2.02 | Destabilizing | 1.17 | Destabilizing | 0.207 | Likely Benign | -4.10 | Deleterious | 1.000 | Probably Damaging | 0.978 | Probably Damaging | 3.37 | Benign | 0.02 | Affected | 0.3552 | 0.1474 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2024A>T | N675I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N675I is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools cluster into two groups: benign predictions come from REVEL, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX and Rosetta give uncertain results and are treated as unavailable. High‑accuracy methods give mixed outcomes: AlphaMissense‑Optimized predicts benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta predicts benign. Overall, the majority of tools (7/12) indicate pathogenicity, while 5/12 indicate benign. Thus, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.111024 | Uncertain | 0.513 | 0.333 | 0.000 | -13.254 | Likely Pathogenic | 0.574 | Likely Pathogenic | Likely Benign | 1.00 | Ambiguous | 0.1 | -0.97 | Ambiguous | 0.02 | Likely Benign | 0.41 | Likely Benign | 0.338 | Likely Benign | -7.37 | Deleterious | 0.999 | Probably Damaging | 0.955 | Probably Damaging | 3.37 | Benign | 0.00 | Affected | 0.0620 | 0.7188 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.203T>C | L68P 2D AIThe SynGAP1 missense variant L68P is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign). Tools that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions lean toward pathogenic, but the consensus from SGM and the high‑accuracy AlphaMissense‑Optimized are contradictory, leaving the variant’s clinical significance uncertain. No conflict exists with ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.470567 | Uncertain | 0.405 | 0.768 | 0.250 | -2.122 | Likely Benign | 0.956 | Likely Pathogenic | Likely Pathogenic | 0.143 | Likely Benign | -0.57 | Neutral | 0.943 | Possibly Damaging | 0.766 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 0.2827 | 0.1382 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2041G>C | G681R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G681R is listed in gnomAD (6-33441300-G-C) but has no ClinVar entry. In silico predictors largely converge on a deleterious effect: benign calls are limited to FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—report pathogenicity. premPS is inconclusive. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates pathogenic folding instability. No prediction tool suggests a benign outcome, and the variant’s presence in gnomAD does not alter the consensus. Thus, the variant is most likely pathogenic, with no conflict with ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | 6-33441300-G-C | 1 | 6.20e-7 | -12.170 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 2.25 | Destabilizing | 1.7 | 5.46 | Destabilizing | 3.86 | Destabilizing | 0.99 | Ambiguous | 0.556 | Likely Pathogenic | -7.98 | Deleterious | 0.999 | Probably Damaging | 0.928 | Probably Damaging | 3.42 | Benign | 0.00 | Affected | 3.43 | 14 | 0.1022 | 0.3879 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||
| c.2041G>T | G681C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G681C is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all classify it as pathogenic, while only FATHMM predicts a benign outcome. Uncertain calls come from FoldX, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, Foldetta (combining FoldX‑MD and Rosetta) is pathogenic, and AlphaMissense‑Optimized remains inconclusive. Overall, the evidence strongly favors a pathogenic interpretation, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | -12.374 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 1.89 | Ambiguous | 1.3 | 2.63 | Destabilizing | 2.26 | Destabilizing | 0.66 | Ambiguous | 0.554 | Likely Pathogenic | -8.98 | Deleterious | 1.000 | Probably Damaging | 0.959 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.1194 | 0.3886 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.2042G>T | G681V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G681V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the majority of algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—classify the change as pathogenic. High‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain; SGM‑Consensus predicts a likely pathogenic effect; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic outcome. No other high‑confidence predictions are available. Taken together, the consensus of pathogenic predictions outweighs the single benign call, indicating that G681V is most likely pathogenic. This assessment is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | -14.043 | Likely Pathogenic | 0.953 | Likely Pathogenic | Ambiguous | 3.21 | Destabilizing | 2.0 | 6.12 | Destabilizing | 4.67 | Destabilizing | 0.64 | Ambiguous | 0.572 | Likely Pathogenic | -8.98 | Deleterious | 0.999 | Probably Damaging | 0.928 | Probably Damaging | 3.33 | Benign | 0.01 | Affected | 0.1350 | 0.3840 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2044T>G | Y682D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y682D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, whereas SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. The high‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a pathogenic effect. With no ClinVar assertion to oppose these findings, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.141467 | Uncertain | 0.758 | 0.328 | 0.000 | -15.094 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 2.81 | Destabilizing | 0.4 | 3.06 | Destabilizing | 2.94 | Destabilizing | 0.96 | Ambiguous | 0.639 | Likely Pathogenic | -9.60 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.32 | Benign | 0.01 | Affected | 0.4191 | 0.0760 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.2048T>A | I683N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I683N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all predict pathogenicity, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized returns a pathogenic score, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) also indicates pathogenicity. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.143268 | Uncertain | 0.848 | 0.314 | 0.000 | -12.120 | Likely Pathogenic | 0.974 | Likely Pathogenic | Likely Pathogenic | 2.18 | Destabilizing | 0.1 | 2.08 | Destabilizing | 2.13 | Destabilizing | 1.46 | Destabilizing | 0.546 | Likely Pathogenic | -6.87 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 3.26 | Benign | 0.01 | Affected | 0.0978 | 0.1133 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.2050G>T | D684Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess the variant’s effect largely agree on a deleterious outcome: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. Only premPS and FATHMM predict a benign effect. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict the absence of ClinVar reporting. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | -15.224 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.65 | Destabilizing | 1.5 | 2.12 | Destabilizing | 2.89 | Destabilizing | -0.06 | Likely Benign | 0.600 | Likely Pathogenic | -8.98 | Deleterious | 1.000 | Probably Damaging | 0.963 | Probably Damaging | 3.44 | Benign | 0.00 | Affected | 0.0575 | 0.6564 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.2051A>T | D684V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while only premPS and FATHMM predict a benign outcome. High‑accuracy assessments reinforce the pathogenic interpretation: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No evidence suggests a benign effect, and the lack of ClinVar annotation means there is no conflicting clinical classification. Therefore, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | -16.128 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.86 | Destabilizing | 1.1 | 2.06 | Destabilizing | 2.96 | Destabilizing | 0.07 | Likely Benign | 0.601 | Likely Pathogenic | -8.98 | Deleterious | 0.901 | Possibly Damaging | 0.480 | Possibly Damaging | 3.44 | Benign | 0.00 | Affected | 0.0775 | 0.6209 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.2056G>T | G686C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G686C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from FoldX, Rosetta, FATHMM, and the protein‑folding stability method Foldetta; pathogenic predictions arise from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, with the SGM‑Consensus score labeling the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as Pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.177104 | Uncertain | 0.919 | 0.268 | 0.000 | -12.790 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.25 | Likely Benign | 0.2 | 0.41 | Likely Benign | 0.33 | Likely Benign | 0.93 | Ambiguous | 0.553 | Likely Pathogenic | -8.14 | Deleterious | 1.000 | Probably Damaging | 0.988 | Probably Damaging | 3.31 | Benign | 0.02 | Affected | 0.1209 | 0.3246 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.2057G>T | G686V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G686V has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on benign impact are Rosetta and FATHMM, while the majority of other in silico predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score (Likely Pathogenic) indicate a pathogenic effect. Uncertain results come from FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for G686V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.177104 | Uncertain | 0.919 | 0.268 | 0.000 | -13.751 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.89 | Ambiguous | 0.5 | 0.21 | Likely Benign | 0.55 | Ambiguous | 0.74 | Ambiguous | 0.570 | Likely Pathogenic | -8.08 | Deleterious | 1.000 | Probably Damaging | 0.979 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.1059 | 0.3646 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2066T>A | L689H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L689H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods further support this: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.227227 | Uncertain | 0.963 | 0.248 | 0.000 | -14.659 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.40 | Destabilizing | 0.1 | 2.50 | Destabilizing | 2.95 | Destabilizing | 2.21 | Destabilizing | 0.532 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.14 | Benign | 0.00 | Affected | 0.1013 | 0.0456 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2066T>C | L689P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L689P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.227227 | Uncertain | 0.963 | 0.248 | 0.000 | -17.900 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.76 | Destabilizing | 0.2 | 13.35 | Destabilizing | 10.06 | Destabilizing | 2.29 | Destabilizing | 0.631 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.17 | Benign | 0.00 | Affected | 0.3668 | 0.1353 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.206T>A | I69N 2D AIThe SynGAP1 missense variant I69N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | -3.220 | Likely Benign | 0.631 | Likely Pathogenic | Likely Benign | 0.110 | Likely Benign | -0.90 | Neutral | 0.943 | Possibly Damaging | 0.781 | Possibly Damaging | 4.10 | Benign | 0.00 | Affected | 0.0859 | 0.0412 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2075T>C | L692P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L692P is listed in ClinVar with an “Uncertain” status (ClinVar ID 847082.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, which does not contradict its current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.064632 | Structured | 0.295225 | Uncertain | 0.966 | 0.243 | 0.000 | Uncertain | 1 | -16.447 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 9.19 | Destabilizing | 0.1 | 13.20 | Destabilizing | 11.20 | Destabilizing | 1.69 | Destabilizing | 0.668 | Likely Pathogenic | -6.98 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.06 | Benign | 0.00 | Affected | 3.42 | 17 | 0.3642 | 0.1025 | -3 | -3 | -5.4 | -16.04 | 186.2 | 62.8 | -0.2 | 0.1 | -0.7 | 0.3 | X | Potentially Pathogenic | The isobutyl side chain of Leu692, located in the middle of an α-helix (res. Leu685-Gln702), engages in hydrophobic packing with nearby residues (e.g., Leu441, Leu431, Leu696) in the inter-helix space. Prolines lack a free amide group necessary for hydrogen bonding with the carbonyl group of Glu688 in the same manner as Leu692 in the WT. Consequently, the residue swap with proline disrupts the continuity of the secondary structure element in the variant simulations. Additionally, the side chain of Pro692 is not as optimal as Leu692 for hydrophobic packing in the inter-helix space. | ||||||||||||||||
| c.2078A>T | H693L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant H693L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from Foldetta, premPS, polyPhen‑2 HumVar, and FATHMM, while pathogenic predictions are made by REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Uncertain results are reported by FoldX and Rosetta. High‑accuracy assessments indicate AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus also leans pathogenic, whereas Foldetta predicts benign stability. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.073402 | Structured | 0.323991 | Uncertain | 0.964 | 0.260 | 0.000 | -14.006 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | -0.53 | Ambiguous | 0.1 | 0.92 | Ambiguous | 0.20 | Likely Benign | -0.29 | Likely Benign | 0.573 | Likely Pathogenic | -10.96 | Deleterious | 0.979 | Probably Damaging | 0.390 | Benign | 3.18 | Benign | 0.01 | Affected | 0.0824 | 0.4675 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.2084T>C | L695P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L695P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.373419 | Uncertain | 0.942 | 0.258 | 0.000 | -17.496 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.63 | Destabilizing | 0.2 | 4.73 | Destabilizing | 4.68 | Destabilizing | 2.11 | Destabilizing | 0.612 | Likely Pathogenic | -6.85 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.16 | Benign | 0.00 | Affected | 0.3044 | 0.0992 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2087T>A | L696H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L696H is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: all available predictors except FATHMM classify the variant as pathogenic (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized). Only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Taken together, the overwhelming majority of computational evidence supports a pathogenic classification, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.390093 | Uncertain | 0.962 | 0.267 | 0.000 | -17.042 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.56 | Destabilizing | 0.0 | 2.55 | Destabilizing | 2.56 | Destabilizing | 2.07 | Destabilizing | 0.569 | Likely Pathogenic | -6.58 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.00 | Benign | 0.00 | Affected | 0.1009 | 0.0488 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2087T>C | L696P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L696P is listed in ClinVar as Pathogenic (ClinVar ID 1699350.0) and is not reported in gnomAD. Prediction tools that classify the variant as benign include only FATHMM; all other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—report it as pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a pathogenic effect. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote) is pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a destabilizing, pathogenic outcome. Taken together, the overwhelming majority of predictions and the high‑accuracy tools classify the variant as pathogenic, fully consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.390093 | Uncertain | 0.962 | 0.267 | 0.000 | Likely Pathogenic | 1 | -16.926 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.66 | Destabilizing | 0.2 | 10.84 | Destabilizing | 8.75 | Destabilizing | 2.13 | Destabilizing | 0.678 | Likely Pathogenic | -6.58 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.00 | Benign | 0.00 | Affected | 3.46 | 13 | 0.3065 | 0.1995 | -3 | -3 | -5.4 | -16.04 | 180.6 | 65.9 | 0.1 | 0.0 | -0.6 | 0.1 | X | Potentially Pathogenic | The isobutyl side chain of Leu696, located in the middle of an α-helix (res. Leu685-Gln702), engages in hydrophobic packing with nearby residues (e.g., Leu441, Leu431, Leu692, Leu714) in the inter-helix space. Prolines lack a free amide group necessary for hydrogen bonding with the carbonyl group of Leu692 in the same manner as Leu696 in the WT. Consequently, the residue swap with proline disrupts the continuity of the secondary structure element in the variant simulations. Additionally, the side chain of Pro696 is not as optimal as Leu696 for hydrophobic packing in the inter-helix space. | ||||||||||||||||
| c.2089T>A | W697R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W697R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict a pathogenic effect: SGM‑Consensus, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, PROVEAN, AlphaMissense‑Default, and premPS. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence from multiple pathogenic‑predicting tools indicates that W697R is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.400169 | Uncertain | 0.945 | 0.297 | 0.000 | -10.020 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 1.14 | Ambiguous | 0.1 | 1.18 | Ambiguous | 1.16 | Ambiguous | 1.25 | Destabilizing | 0.401 | Likely Benign | -9.50 | Deleterious | 1.000 | Probably Damaging | 0.994 | Probably Damaging | 3.45 | Benign | 0.02 | Affected | 3.46 | 13 | 0.3944 | 0.0612 | 2 | -3 | -3.6 | -30.03 | 254.4 | -41.2 | 0.0 | 0.0 | -0.7 | 0.0 | X | Potentially Benign | The indole ring of Trp697, located on the outer surface of an α-helix (res. Leu685-Val699), is not involved in any long-lasting interactions in the WT simulations. In the variant simulations, the positively charged guanidinium side chain of Arg697 occasionally forms hydrogen bonds with nearby residues, such as Ser722 and Asn719. However, similar to Trp697 in the WT, Arg697 does not form any long-lasting interactions and thus does not induce any negative structural effects in the simulations. | ||||||||||||||||||
| c.2089T>C | W697R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W697R is listed in ClinVar as Benign (ClinVar ID 703213.0) and is present in the gnomAD database (gnomAD ID 6‑33441348‑T‑C). Functional prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict a pathogenic impact: premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence from multiple pathogenic‑predicting tools suggests that the variant is most likely pathogenic, which contradicts its current ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.400169 | Uncertain | 0.945 | 0.297 | 0.000 | Likely Benign | 1 | 6-33441348-T-C | 1 | 6.20e-7 | -10.020 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 1.14 | Ambiguous | 0.1 | 1.18 | Ambiguous | 1.16 | Ambiguous | 1.25 | Destabilizing | 0.401 | Likely Benign | -9.50 | Deleterious | 1.000 | Probably Damaging | 0.994 | Probably Damaging | 3.45 | Benign | 0.02 | Affected | 3.46 | 13 | 0.3944 | 0.0612 | 2 | -3 | -3.6 | -30.03 | 254.4 | -41.2 | 0.0 | 0.0 | -0.7 | 0.0 | X | Potentially Benign | The indole ring of Trp697, located on the outer surface of an α-helix (res. Leu685-Val699), is not involved in any long-lasting interactions in the WT simulations. In the variant simulations, the positively charged guanidinium side chain of Arg697 occasionally forms hydrogen bonds with nearby residues, such as Ser722 and Asn719. However, similar to Trp697 in the WT, Arg697 does not form any long-lasting interactions and thus does not induce any negative structural effects in the simulations. | |||||||||||||
| c.208C>G | R70G 2D AIThe SynGAP1 missense variant R70G is listed in gnomAD (ID 6‑33425816‑C‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar) and SIFT. AlphaMissense‑Default is uncertain, and Foldetta (FoldX‑MD/Rosetta stability assessment) has no result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also indicates a likely benign outcome; Foldetta data are unavailable. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.458981 | Uncertain | 0.392 | 0.793 | 0.375 | 6-33425816-C-G | 1 | 6.20e-7 | -3.555 | Likely Benign | 0.430 | Ambiguous | Likely Benign | 0.095 | Likely Benign | -0.61 | Neutral | 0.962 | Probably Damaging | 0.726 | Possibly Damaging | 4.11 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3078 | 0.3194 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||
| c.208C>T | R70W 2D AIThe SynGAP1 missense variant R70W is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.458981 | Uncertain | 0.392 | 0.793 | 0.375 | -3.558 | Likely Benign | 0.588 | Likely Pathogenic | Likely Benign | 0.148 | Likely Benign | -1.83 | Neutral | 0.999 | Probably Damaging | 0.876 | Possibly Damaging | 4.06 | Benign | 0.00 | Affected | 0.1061 | 0.4028 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2090G>C | W697S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W697S is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are premPS, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as unavailable due to uncertainty. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.400169 | Uncertain | 0.945 | 0.297 | 0.000 | -4.900 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 1.90 | Ambiguous | 0.1 | 1.58 | Ambiguous | 1.74 | Ambiguous | 1.13 | Destabilizing | 0.322 | Likely Benign | -8.89 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 3.53 | Benign | 0.13 | Tolerated | 0.3738 | 0.1089 | -2 | -3 | 0.1 | -99.14 | ||||||||||||||||||||||||||||||
| c.2096T>G | V699G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V699G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include FATHMM and AlphaMissense‑Optimized, while the remaining tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b) uniformly predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic verdict; and Foldetta also predicts pathogenic. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.069024 | Structured | 0.432975 | Uncertain | 0.935 | 0.315 | 0.000 | -11.912 | Likely Pathogenic | 0.481 | Ambiguous | Likely Benign | 2.22 | Destabilizing | 0.0 | 3.25 | Destabilizing | 2.74 | Destabilizing | 1.77 | Destabilizing | 0.514 | Likely Pathogenic | -5.11 | Deleterious | 0.972 | Probably Damaging | 0.999 | Probably Damaging | 3.37 | Benign | 0.01 | Affected | 0.1830 | 0.2240 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.2099T>C | L700P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L700P is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while only FATHMM predicts it benign. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Pathogenic” verdict (3 pathogenic vs. 1 benign). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.416255 | Uncertain | 0.927 | 0.331 | 0.000 | -13.092 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 7.29 | Destabilizing | 0.5 | 12.85 | Destabilizing | 10.07 | Destabilizing | 1.84 | Destabilizing | 0.541 | Likely Pathogenic | -4.31 | Deleterious | 0.999 | Probably Damaging | 0.966 | Probably Damaging | 3.30 | Benign | 0.01 | Affected | 0.3603 | 0.1025 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2102C>T | P701L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P701L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and ESM1b. The remaining tools (FoldX, Rosetta, AlphaMissense‑Default) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of predictions (7 benign vs. 3 pathogenic) and the two high‑accuracy benign calls suggest that the variant is most likely benign. This conclusion does not contradict any ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.404318 | Uncertain | 0.918 | 0.345 | 0.000 | -10.185 | Likely Pathogenic | 0.515 | Ambiguous | Likely Benign | 1.15 | Ambiguous | 0.0 | -0.68 | Ambiguous | 0.24 | Likely Benign | 0.12 | Likely Benign | 0.116 | Likely Benign | -3.04 | Deleterious | 0.642 | Possibly Damaging | 0.087 | Benign | 3.50 | Benign | 0.09 | Tolerated | 0.2018 | 0.5546 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||
| c.2108T>A | L703H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L703H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from REVEL and FATHMM, whereas pathogenic predictions are made by FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts Pathogenic; the SGM‑Consensus itself is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts Pathogenic. Taken together, the overwhelming majority of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.144935 | Structured | 0.388282 | Uncertain | 0.929 | 0.353 | 0.000 | -12.886 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 2.52 | Destabilizing | 0.0 | 2.29 | Destabilizing | 2.41 | Destabilizing | 1.75 | Destabilizing | 0.420 | Likely Benign | -5.71 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.09 | Benign | 0.00 | Affected | 0.1038 | 0.0288 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2108T>C | L703P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L703P is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic, while only FATHMM predicts it benign. The SGM‑Consensus, which is a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Pathogenic” result (3 pathogenic vs. 1 benign). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; SGM‑Consensus is likely pathogenic; and Foldetta, integrating FoldX‑MD and Rosetta outputs, is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.144935 | Structured | 0.388282 | Uncertain | 0.929 | 0.353 | 0.000 | -13.766 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 4.44 | Destabilizing | 0.1 | 9.38 | Destabilizing | 6.91 | Destabilizing | 1.54 | Destabilizing | 0.559 | Likely Pathogenic | -5.70 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.11 | Benign | 0.00 | Affected | 0.3698 | 0.1186 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.211G>T | D71Y 2D AIThe SynGAP1 missense variant D71Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -4.090 | Likely Benign | 0.740 | Likely Pathogenic | Likely Benign | 0.188 | Likely Benign | -2.49 | Neutral | 0.842 | Possibly Damaging | 0.189 | Benign | 4.00 | Benign | 0.00 | Affected | 0.0584 | 0.6086 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2123T>A | L708H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L708H is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default; the SGM Consensus score is also labeled Likely Pathogenic. Stability‑based methods FoldX and Rosetta return uncertain results, and Foldetta likewise is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the majority of conventional predictors lean toward pathogenicity, whereas a few high‑accuracy tools suggest benign or are inconclusive. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | -8.059 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | 1.84 | Ambiguous | 0.1 | 1.55 | Ambiguous | 1.70 | Ambiguous | 1.44 | Destabilizing | 0.243 | Likely Benign | -4.68 | Deleterious | 1.000 | Probably Damaging | 0.981 | Probably Damaging | 3.25 | Benign | 0.02 | Affected | 0.0824 | 0.0288 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2123T>C | L708P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L708P is not reported in ClinVar and is absent from gnomAD. Consensus among most in‑silico predictors is strongly pathogenic: FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a deleterious effect. Only REVEL and FATHMM predict a benign outcome, while AlphaMissense‑Optimized is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is inconclusive, SGM Consensus reports “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic change. Taken together, the preponderance of evidence points to a pathogenic impact for L708P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | -10.268 | Likely Pathogenic | 0.932 | Likely Pathogenic | Ambiguous | 4.29 | Destabilizing | 0.1 | 6.43 | Destabilizing | 5.36 | Destabilizing | 1.62 | Destabilizing | 0.298 | Likely Benign | -4.47 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 3.39 | Benign | 0.04 | Affected | 0.2800 | 0.0825 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2126T>C | L709P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L709P is not reported in ClinVar and is absent from gnomAD. Benign predictions come from REVEL, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from FoldX, Rosetta, Foldetta, polyPhen2_HumDiv, polyPhen2_HumVar, and AlphaMissense‑Default. High‑accuracy tools give mixed results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, FATHMM, PROVEAN) also predicts benign; Foldetta predicts pathogenic. Because the majority of standard predictors and the SGM Consensus favor benign, the variant is most likely benign, and this is consistent with the lack of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.243554 | Structured | 0.365830 | Uncertain | 0.934 | 0.379 | 0.000 | -7.517 | In-Between | 0.767 | Likely Pathogenic | Likely Benign | 2.81 | Destabilizing | 0.0 | 5.65 | Destabilizing | 4.23 | Destabilizing | 0.31 | Likely Benign | 0.167 | Likely Benign | -1.93 | Neutral | 1.000 | Probably Damaging | 0.975 | Probably Damaging | 3.47 | Benign | 0.37 | Tolerated | 0.3004 | 0.0825 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||
| c.212A>T | D71V 2D AIThe SynGAP1 D71V missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to a likely benign classification (3 benign vs. 1 pathogenic votes). High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -3.549 | Likely Benign | 0.780 | Likely Pathogenic | Likely Benign | 0.183 | Likely Benign | -2.28 | Neutral | 0.334 | Benign | 0.060 | Benign | 4.01 | Benign | 0.00 | Affected | 0.0771 | 0.6197 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2132T>C | L711P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L711P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and FATHMM; all other evaluated algorithms—including FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.308712 | Structured | 0.377436 | Uncertain | 0.950 | 0.364 | 0.000 | -12.128 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 5.70 | Destabilizing | 0.1 | 8.15 | Destabilizing | 6.93 | Destabilizing | 2.09 | Destabilizing | 0.375 | Likely Benign | -6.59 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.28 | Benign | 0.00 | Affected | 0.3300 | 0.0986 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2134G>T | G712C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G712C is catalogued in gnomAD (6‑33441599‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the majority of algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | 6-33441599-G-T | 1 | 6.20e-7 | -11.376 | Likely Pathogenic | 0.829 | Likely Pathogenic | Ambiguous | 2.54 | Destabilizing | 0.0 | 5.72 | Destabilizing | 4.13 | Destabilizing | 0.56 | Ambiguous | 0.516 | Likely Pathogenic | -7.75 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.31 | Benign | 0.00 | Affected | 3.50 | 9 | 0.1513 | 0.3947 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||
| c.2135G>T | G712V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G712V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, while the majority of tools (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic impact. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | -10.466 | Likely Pathogenic | 0.877 | Likely Pathogenic | Ambiguous | 3.79 | Destabilizing | 0.0 | 6.18 | Destabilizing | 4.99 | Destabilizing | 0.79 | Ambiguous | 0.410 | Likely Benign | -7.55 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.32 | Benign | 0.00 | Affected | 0.1395 | 0.3712 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2138C>T | P713L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P713L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact; AlphaMissense‑Optimized and premPS are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the balance of evidence favors a pathogenic classification, and this conclusion does not contradict the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.393235 | Uncertain | 0.961 | 0.371 | 0.000 | -11.323 | Likely Pathogenic | 0.850 | Likely Pathogenic | Ambiguous | 0.18 | Likely Benign | 0.1 | -0.03 | Likely Benign | 0.08 | Likely Benign | 0.55 | Ambiguous | 0.324 | Likely Benign | -8.60 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.1993 | 0.5261 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.2141T>C | L714P 2D AIThe SynGAP1 missense variant L714P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the preponderance of evidence from these tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.402311 | Uncertain | 0.961 | 0.369 | 0.000 | -12.562 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 7.48 | Destabilizing | 1.9 | 13.54 | Destabilizing | 10.51 | Destabilizing | 2.26 | Destabilizing | 0.441 | Likely Benign | -6.44 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.08 | Benign | 0.00 | Affected | 0.3572 | 0.1253 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2144C>T | P715L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P715L is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL, Rosetta, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX and premPS give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. Because the majority of consensus and individual predictors indicate pathogenicity, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for P715L. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.243554 | Structured | 0.409757 | Uncertain | 0.956 | 0.362 | 0.000 | -12.207 | Likely Pathogenic | 0.764 | Likely Pathogenic | Likely Benign | 1.43 | Ambiguous | 0.1 | 0.06 | Likely Benign | 0.75 | Ambiguous | 0.58 | Ambiguous | 0.318 | Likely Benign | -9.10 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.39 | Benign | 0.01 | Affected | 0.1944 | 0.5105 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.2150T>A | L717H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L717H occurs in the GAP domain. ClinVar has no entry for this variant, and it is not present in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM; all other evaluated algorithms (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact, and the SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions or stability results are missing or inconclusive. Based on the overwhelming majority of computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.429342 | Uncertain | 0.969 | 0.397 | 0.000 | -11.107 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 2.27 | Destabilizing | 0.1 | 2.04 | Destabilizing | 2.16 | Destabilizing | 1.05 | Destabilizing | 0.317 | Likely Benign | -5.23 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 0.0918 | 0.0558 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2150T>C | L717P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L717P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from REVEL and FATHMM, while the remaining 13 tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus) all classify the variant as pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts a pathogenic effect. No predictions are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.429342 | Uncertain | 0.969 | 0.397 | 0.000 | -10.214 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.96 | Destabilizing | 0.2 | 6.56 | Destabilizing | 5.26 | Destabilizing | 1.66 | Destabilizing | 0.447 | Likely Benign | -5.32 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.28 | Benign | 0.00 | Affected | 0.3065 | 0.1053 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2153T>A | L718H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L718H is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools cluster into two groups: the single benign predictor REVEL, and a consensus of pathogenic predictions from FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are inconclusive or missing. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.438417 | Uncertain | 0.966 | 0.385 | 0.000 | -14.923 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.84 | Destabilizing | 0.1 | 2.68 | Destabilizing | 3.26 | Destabilizing | 2.69 | Destabilizing | 0.460 | Likely Benign | -6.64 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.1020 | 0.0526 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2153T>C | L718P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L718P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity are unanimous: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool in the dataset predicts a benign effect, so the benign‑prediction group is empty. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Consequently, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.438417 | Uncertain | 0.966 | 0.385 | 0.000 | -15.643 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.56 | Destabilizing | 0.1 | 9.69 | Destabilizing | 8.13 | Destabilizing | 2.35 | Destabilizing | 0.660 | Likely Pathogenic | -6.64 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.3854 | 0.1221 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2156A>T | N719I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Two tools report uncertainty: Rosetta and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of predictions lean toward a benign impact, with no conflict with ClinVar status. Thus, the variant is most likely benign based on the current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -10.794 | Likely Pathogenic | 0.399 | Ambiguous | Likely Benign | -0.19 | Likely Benign | 0.0 | -0.74 | Ambiguous | -0.47 | Likely Benign | 0.40 | Likely Benign | 0.146 | Likely Benign | -4.88 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.71 | Benign | 0.10 | Tolerated | 0.0408 | 0.4493 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||
| c.2158G>T | D720Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D720Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and premPS, while pathogenic predictions are made by SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Predictions from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive. The high‑accuracy consensus (SGM Consensus) classifies the variant as Likely Pathogenic, and Foldetta likewise yields an uncertain stability change. AlphaMissense‑Optimized remains inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -14.771 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | -0.74 | Ambiguous | 0.1 | -1.38 | Ambiguous | -1.06 | Ambiguous | 0.20 | Likely Benign | 0.499 | Likely Benign | -7.05 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.11 | Pathogenic | 0.00 | Affected | 0.0626 | 0.5973 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.2159A>T | D720V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D720V has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, Foldetta, and premPS, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -12.730 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 0.08 | Likely Benign | 0.0 | -0.78 | Ambiguous | -0.35 | Likely Benign | 0.20 | Likely Benign | 0.437 | Likely Benign | -7.18 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 2.12 | Pathogenic | 0.00 | Affected | 0.0822 | 0.5708 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.2162T>A | I721N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -14.905 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.82 | Destabilizing | 0.0 | 3.21 | Destabilizing | 3.02 | Destabilizing | 2.10 | Destabilizing | 0.425 | Likely Benign | -6.30 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.19 | Pathogenic | 0.00 | Affected | 0.0737 | 0.0340 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.2174T>C | L725P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L725P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. **Based on the collective predictions, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.455613 | Uncertain | 0.911 | 0.491 | 0.625 | -15.390 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 4.91 | Destabilizing | 0.1 | 9.02 | Destabilizing | 6.97 | Destabilizing | 1.82 | Destabilizing | 0.396 | Likely Benign | -6.08 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.3796 | 0.1664 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||
| c.2176A>T | R726W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R726W has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a split: benign calls from REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further reveal AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. No prediction or stability result is missing or inconclusive. Overall, the majority of tools (seven) predict a benign effect, but the SGM‑Consensus and several high‑accuracy methods indicate pathogenicity, leaving the variant’s clinical significance uncertain. The predictions do not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.521092 | Disordered | 0.449098 | Uncertain | 0.888 | 0.513 | 0.625 | -10.091 | Likely Pathogenic | 0.580 | Likely Pathogenic | Likely Benign | 0.46 | Likely Benign | 0.1 | 0.46 | Likely Benign | 0.46 | Likely Benign | 0.15 | Likely Benign | 0.217 | Likely Benign | -3.72 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.57 | Benign | 0.01 | Affected | 0.1161 | 0.4252 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||
| c.217A>T | R73W 2D AIThe SynGAP1 missense variant R73W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.453164 | Uncertain | 0.332 | 0.826 | 0.375 | -5.874 | Likely Benign | 0.318 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -1.96 | Neutral | 0.962 | Probably Damaging | 0.274 | Benign | 3.98 | Benign | 0.00 | Affected | 0.1505 | 0.4035 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2180A>T | N727I 2D 3DClick to see structure in 3D Viewer AISynGAP1 N727I is not reported in ClinVar and is absent from gnomAD. Benign predictions come from REVEL, FoldX, premPS, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen2_HumDiv, polyPhen2_HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Foldetta and Rosetta provide inconclusive results. High‑accuracy tools give a mixed picture: AlphaMissense‑Optimized predicts benign, SGM‑Consensus predicts likely pathogenic, and Foldetta is uncertain. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -10.230 | Likely Pathogenic | 0.577 | Likely Pathogenic | Likely Benign | 0.17 | Likely Benign | 0.1 | 0.90 | Ambiguous | 0.54 | Ambiguous | 0.43 | Likely Benign | 0.319 | Likely Benign | -5.93 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.13 | Pathogenic | 0.03 | Affected | 0.0666 | 0.5917 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||
| c.2183C>T | P728L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P728L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, and premPS, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact; FoldX and AlphaMissense‑Optimized are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the preponderance of evidence from standard and high‑accuracy predictors points to a pathogenic effect for P728L. This conclusion is not contradicted by any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.632174 | Disordered | 0.434760 | Uncertain | 0.725 | 0.567 | 0.625 | -11.125 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 0.79 | Ambiguous | 0.0 | 0.15 | Likely Benign | 0.47 | Likely Benign | 0.20 | Likely Benign | 0.402 | Likely Benign | -8.27 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 0.66 | Pathogenic | 0.00 | Affected | 0.2321 | 0.4713 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||
| c.2189T>A | I730N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I730N is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar) and SIFT. The remaining tools—premPS, ESM1b, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -7.987 | In-Between | 0.369 | Ambiguous | Likely Benign | 0.09 | Likely Benign | 0.2 | 0.04 | Likely Benign | 0.07 | Likely Benign | 0.70 | Ambiguous | 0.080 | Likely Benign | -1.60 | Neutral | 1.000 | Probably Damaging | 0.986 | Probably Damaging | 3.46 | Benign | 0.02 | Affected | 0.0969 | 0.0533 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||
| c.2194A>G | R732G 2D AIThe SynGAP1 R732G missense variant is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (ID 6‑33441659‑A‑G). Prediction tools that agree on a benign effect include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs. 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of standard predictors lean toward pathogenicity, while the most accurate single predictor (AlphaMissense‑Optimized) suggests a benign outcome. Given the lack of ClinVar evidence, there is no contradiction; the variant is most likely pathogenic based on the collective predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | 6-33441659-A-G | 1 | 6.20e-7 | -9.348 | Likely Pathogenic | 0.295 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -2.98 | Deleterious | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.56 | Benign | 0.03 | Affected | 3.59 | 7 | 0.3080 | 0.2942 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||
| c.2194A>T | R732W 2D AIThe SynGAP1 missense variant R732W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, five tools predict pathogenicity versus four predicting benign, with no ClinVar evidence to contradict these computational findings. Thus, the variant is most likely pathogenic based on the current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | -11.976 | Likely Pathogenic | 0.252 | Likely Benign | Likely Benign | 0.161 | Likely Benign | -3.57 | Deleterious | 1.000 | Probably Damaging | 0.973 | Probably Damaging | 2.53 | Benign | 0.01 | Affected | 0.1332 | 0.2748 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2201C>T | P734L 2D AIThe SynGAP1 missense variant P734L is reported in gnomAD (variant ID 6‑33441666‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, creating a single discordant call. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.411273 | Uncertain | 0.368 | 0.721 | 0.875 | 6-33441666-C-T | 3 | 1.86e-6 | -3.472 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -2.11 | Neutral | 0.897 | Possibly Damaging | 0.330 | Benign | 2.69 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.2390 | 0.5059 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.2206C>T | R736C 2D AISynGAP1 missense variant R736C is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33441671‑C‑T). Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized scores benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also returns benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence indicates a benign effect, which does not conflict with the ClinVar uncertain designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | Conflicting | 3 | 6-33441671-C-T | 8 | 4.96e-6 | -7.113 | In-Between | 0.120 | Likely Benign | Likely Benign | 0.190 | Likely Benign | -2.06 | Neutral | 0.999 | Probably Damaging | 0.825 | Possibly Damaging | 2.48 | Pathogenic | 0.00 | Affected | 4.07 | 3 | 0.3740 | 0.1691 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.2218C>T | R740W 2D AIThe SynGAP1 missense variant R740W is listed in ClinVar with an uncertain significance and is present in the gnomAD database (ID 6‑33441683‑C‑T). Prediction tools that classify the variant as benign include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while those that predict pathogenicity are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized predicting a benign effect; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two benign vs. two pathogenic calls) and is treated as unavailable, and no Foldetta stability data are reported. Overall, the majority of conventional tools (five pathogenic vs. four benign) suggest a pathogenic impact, whereas the single high‑accuracy tool indicates benign. Thus, the variant is most likely pathogenic based on the aggregate predictions, and this assessment does not contradict the ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.771762 | Disordered | 0.475392 | Uncertain | 0.269 | 0.849 | 0.875 | Uncertain | 2 | 6-33441683-C-T | 6 | 3.72e-6 | -8.561 | Likely Pathogenic | 0.168 | Likely Benign | Likely Benign | 0.180 | Likely Benign | -3.09 | Deleterious | 1.000 | Probably Damaging | 0.938 | Probably Damaging | 2.52 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1566 | 0.3375 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||
| c.2222C>T | P741L 2D AIThe SynGAP1 missense variant P741L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.493550 | Uncertain | 0.354 | 0.859 | 0.875 | -4.850 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.63 | Neutral | 0.001 | Benign | 0.003 | Benign | 2.84 | Benign | 0.03 | Affected | 0.1978 | 0.5780 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2228C>T | P743L 2D AIThe SynGAP1 missense variant P743L is listed in gnomAD (ID 6‑33441693‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.526809 | Binding | 0.317 | 0.862 | 0.875 | 6-33441693-C-T | 1 | 6.19e-7 | -4.838 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.112 | Likely Benign | -2.21 | Neutral | 0.801 | Possibly Damaging | 0.192 | Benign | 2.73 | Benign | 0.00 | Affected | 4.32 | 2 | 0.2166 | 0.5533 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.2234C>T | P745L 2D AIThe SynGAP1 missense variant P745L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus (majority vote) as Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | -6.303 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.211 | Likely Benign | -3.79 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.53 | Benign | 0.01 | Affected | 0.2205 | 0.5188 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2237T>G | V746G 2D AIThe SynGAP1 missense variant V746G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized scores benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, V746G is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.808535 | Disordered | 0.576597 | Binding | 0.336 | 0.867 | 0.875 | -2.971 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -0.89 | Neutral | 0.136 | Benign | 0.270 | Benign | 2.74 | Benign | 0.02 | Affected | 0.1897 | 0.2550 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2240T>G | V747G 2D AIThe SynGAP1 missense variant V747G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as benign, and Foldetta results are unavailable. Overall, the consensus of the available predictions indicates that V747G is most likely benign, and this conclusion does not contradict any ClinVar status because the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | -3.883 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -1.22 | Neutral | 0.589 | Possibly Damaging | 0.917 | Probably Damaging | 2.56 | Benign | 0.00 | Affected | 0.1960 | 0.2374 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2243T>C | L748P 2D AIThe SynGAP1 missense variant L748P is reported in gnomAD (ID 6‑33441708‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions are made by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments reinforce the benign view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.611637 | Binding | 0.339 | 0.863 | 0.750 | 6-33441708-T-C | 1 | 6.20e-7 | -2.732 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.69 | Neutral | 0.912 | Possibly Damaging | 0.611 | Possibly Damaging | 2.69 | Benign | 0.02 | Affected | 4.32 | 2 | 0.3503 | 0.1399 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||
| c.2245C>T | R749W 2D AIThe SynGAP1 missense variant R749W is listed in ClinVar as benign and is observed in gnomAD (ID 6‑33441710‑C‑T). Prediction tools that classify the variant as benign include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also returns benign, and Foldetta stability analysis is unavailable. Overall, the majority of evidence, especially from high‑confidence methods, supports a benign effect. This consensus aligns with the ClinVar designation, so there is no contradiction between the predictions and the reported clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.675549 | Disordered | 0.626050 | Binding | 0.337 | 0.860 | 0.625 | Likely Benign | 1 | 6-33441710-C-T | 3 | 1.86e-6 | -7.647 | In-Between | 0.338 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -2.62 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.59 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1215 | 0.4088 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||
| c.2248G>A | G750R 2D AIThe SynGAP1 missense variant G750R is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33441713‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs four benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | 6-33441713-G-A | 17 | 1.05e-5 | -3.861 | Likely Benign | 0.619 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -2.09 | Neutral | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.45 | Pathogenic | 0.00 | Affected | 3.99 | 5 | 0.0960 | 0.4015 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.2248G>C | G750R 2D AIThe SynGAP1 missense variant G750R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments are mixed: AlphaMissense‑Optimized predicts benign, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2), and Foldetta data are unavailable. Overall, the majority of standard predictors (5 pathogenic vs 4 benign) lean toward pathogenicity, and no ClinVar annotation contradicts this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -3.861 | Likely Benign | 0.619 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -2.09 | Neutral | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.45 | Pathogenic | 0.00 | Affected | 3.99 | 5 | 0.0960 | 0.4015 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2249G>T | G750V 2D AIThe SynGAP1 missense variant G750V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions therefore show AlphaMissense‑Optimized as benign, while the remaining high‑confidence tools (PROVEAN, polyPhen‑2, SIFT, FATHMM) all indicate pathogenicity. Overall, the majority of evidence (five pathogenic vs four benign) points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -4.512 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -2.54 | Deleterious | 0.984 | Probably Damaging | 0.850 | Possibly Damaging | 2.45 | Pathogenic | 0.01 | Affected | 0.1270 | 0.4212 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2252C>T | P751L 2D AIThe SynGAP1 missense variant P751L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.667683 | Binding | 0.386 | 0.866 | 0.625 | -3.558 | Likely Benign | 0.143 | Likely Benign | Likely Benign | 0.207 | Likely Benign | -1.62 | Neutral | 0.316 | Benign | 0.062 | Benign | 2.94 | Benign | 0.24 | Tolerated | 0.2318 | 0.6451 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2255C>G | S752W 2D AIThe SynGAP1 missense variant S752W is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas seven tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic, and the Foldetta stability analysis is unavailable. Overall, the majority of predictions lean toward pathogenicity, and this conclusion is not contradicted by ClinVar status (which is absent). Thus, the variant is most likely pathogenic based on the collective evidence, despite the single benign prediction from AlphaMissense‑Optimized. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.690594 | Binding | 0.365 | 0.877 | 0.625 | -6.771 | Likely Benign | 0.565 | Likely Pathogenic | Likely Benign | 0.341 | Likely Benign | -3.54 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 1.49 | Pathogenic | 0.00 | Affected | 0.0837 | 0.6241 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2269G>T | G757C 2D AIThe SynGAP1 missense variant G757C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.830995 | Binding | 0.310 | 0.869 | 0.375 | -6.652 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.179 | Likely Benign | -1.74 | Neutral | 0.997 | Probably Damaging | 0.870 | Possibly Damaging | 2.65 | Benign | 0.04 | Affected | 0.1302 | 0.3630 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2270G>T | G757V 2D AIThe SynGAP1 missense variant G757V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.830995 | Binding | 0.310 | 0.869 | 0.375 | -4.840 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -1.47 | Neutral | 0.801 | Possibly Damaging | 0.494 | Possibly Damaging | 2.68 | Benign | 0.07 | Tolerated | 0.1089 | 0.3512 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2272T>G | Y758D 2D AIThe SynGAP1 missense variant Y758D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized independently predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.856063 | Binding | 0.289 | 0.871 | 0.375 | -3.688 | Likely Benign | 0.481 | Ambiguous | Likely Benign | 0.160 | Likely Benign | -1.89 | Neutral | 0.973 | Probably Damaging | 0.796 | Possibly Damaging | 2.71 | Benign | 0.02 | Affected | 0.4647 | 0.0331 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2273A>C | Y758S 2D AIThe SynGAP1 missense variant Y758S is reported in gnomAD (variant ID 6‑33441738‑A‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment does not contradict any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.856063 | Binding | 0.289 | 0.871 | 0.375 | 6-33441738-A-C | 1 | 6.20e-7 | -1.912 | Likely Benign | 0.215 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -0.54 | Neutral | 0.912 | Possibly Damaging | 0.629 | Possibly Damaging | 2.75 | Benign | 0.06 | Tolerated | 3.99 | 5 | 0.5377 | 0.1663 | Weaken | -2 | -3 | 0.5 | -76.10 | |||||||||||||||||||||||||||||||||
| c.227C>T | S76F 2D AIThe SynGAP1 missense variant S76F is reported in ClinVar as “Not submitted” and is present in gnomAD (variant ID 6-33425835-C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.444487 | Uncertain | 0.279 | 0.826 | 0.500 | 6-33425835-C-T | 4 | 2.48e-6 | -4.557 | Likely Benign | 0.197 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -2.40 | Neutral | 0.972 | Probably Damaging | 0.831 | Possibly Damaging | 3.71 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0501 | 0.4887 | -2 | -3 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||
| c.2284G>T | D762Y 2D AIThe SynGAP1 D762Y variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas a larger group predicts a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus methods give the following results: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic”; Foldetta (a protein‑folding stability approach combining FoldX‑MD and Rosetta) has no available output for this variant. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -6.959 | Likely Benign | 0.905 | Likely Pathogenic | Ambiguous | 0.219 | Likely Benign | -3.24 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 2.07 | Pathogenic | 0.01 | Affected | 0.0700 | 0.7929 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2285A>T | D762V 2D AIThe SynGAP1 D762V missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b. Tools that agree on a pathogenic effect include polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returned an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of consensus tools predict a pathogenic impact, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -6.122 | Likely Benign | 0.949 | Likely Pathogenic | Ambiguous | 0.229 | Likely Benign | -1.98 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.08 | Pathogenic | 0.01 | Affected | 0.1105 | 0.8785 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.2288T>A | L763H 2D AIThe SynGAP1 missense variant L763H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on benign include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the predictions are mixed, with a slight tilt toward pathogenicity (5 pathogenic vs. 4 benign). Thus, the variant is most likely pathogenic according to the aggregate predictions, and this assessment does not contradict ClinVar, which currently has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -6.681 | Likely Benign | 0.640 | Likely Pathogenic | Likely Benign | 0.180 | Likely Benign | -0.79 | Neutral | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 2.36 | Pathogenic | 0.03 | Affected | 0.1021 | 0.0887 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2288T>C | L763P 2D AIThe SynGAP1 missense variant L763P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain. For high‑accuracy assessment, AlphaMissense‑Optimized predicts benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic, with one uncertain). Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -5.802 | Likely Benign | 0.550 | Ambiguous | Likely Benign | 0.158 | Likely Benign | -0.89 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.36 | Pathogenic | 0.12 | Tolerated | 0.3920 | 0.1182 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2291A>T | N764I 2D AIThe SynGAP1 missense variant N764I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, more tools predict pathogenicity (five) than benignity (three), and no ClinVar evidence contradicts this assessment. Thus, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.919527 | Binding | 0.305 | 0.861 | 0.250 | -6.879 | Likely Benign | 0.883 | Likely Pathogenic | Ambiguous | 0.115 | Likely Benign | -2.58 | Deleterious | 0.906 | Possibly Damaging | 0.679 | Possibly Damaging | 2.58 | Benign | 0.00 | Affected | 0.0581 | 0.4483 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.2297C>A | S766Y 2D AIThe SynGAP1 missense variant S766Y is reported in gnomAD (ID 6‑33442455‑C‑A) but has no ClinVar entry. Functional prediction tools show mixed results: benign calls come from REVEL, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts benign, but the SGM‑Consensus (majority vote) predicts pathogenic, and no Foldetta stability data are available. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.923125 | Binding | 0.338 | 0.874 | 0.250 | 6-33442455-C-A | -8.636 | Likely Pathogenic | 0.641 | Likely Pathogenic | Likely Benign | 0.222 | Likely Benign | -2.67 | Deleterious | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 3.64 | 6 | 0.0794 | 0.5609 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||
| c.2297C>T | S766F 2D AIThe SynGAP1 missense variant S766F is listed in gnomAD (ID 6‑33442455‑C‑T) but has no ClinVar entry. Functional prediction tools show a split verdict: benign calls come from REVEL, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments further reveal AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; Foldetta results are unavailable. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.923125 | Binding | 0.338 | 0.874 | 0.250 | 6-33442455-C-T | -8.944 | Likely Pathogenic | 0.709 | Likely Pathogenic | Likely Benign | 0.233 | Likely Benign | -2.87 | Deleterious | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 4.08 | Benign | 0.00 | Affected | 3.64 | 6 | 0.0770 | 0.5887 | -2 | -3 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||
| c.2300T>A | I767N 2D AIThe SynGAP1 missense variant I767N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | -5.117 | Likely Benign | 0.541 | Ambiguous | Likely Benign | 0.122 | Likely Benign | -0.16 | Neutral | 0.977 | Probably Damaging | 0.632 | Possibly Damaging | 4.04 | Benign | 0.06 | Tolerated | 0.1039 | 0.1012 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2302G>T | D768Y 2D AIThe SynGAP1 missense variant D768Y is listed in ClinVar with status “Uncertain” (ClinVar ID 1061652.0) and is present in gnomAD (variant ID 6‑33442460‑G‑T). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain,” SGM‑Consensus as “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic impact, which does not contradict the ClinVar designation of uncertainty. Thus, based on current predictions, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | Uncertain | 1 | 6-33442460-G-T | -9.866 | Likely Pathogenic | 0.824 | Likely Pathogenic | Ambiguous | 0.234 | Likely Benign | -2.86 | Deleterious | 0.989 | Probably Damaging | 0.806 | Possibly Damaging | 4.01 | Benign | 0.07 | Tolerated | 3.64 | 6 | 0.0581 | 0.7525 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||
| c.2303A>T | D768V 2D AIThe SynGAP1 D768V variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Based on the preponderance of pathogenic predictions and the SGM‑Consensus result, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | -9.528 | Likely Pathogenic | 0.880 | Likely Pathogenic | Ambiguous | 0.164 | Likely Benign | -2.62 | Deleterious | 0.611 | Possibly Damaging | 0.140 | Benign | 4.04 | Benign | 0.02 | Affected | 0.0802 | 0.8019 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2306T>A | L769H 2D AIThe SynGAP1 missense variant L769H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, all of which classify the variant as benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, all of which report the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | -6.038 | Likely Benign | 0.422 | Ambiguous | Likely Benign | 0.235 | Likely Benign | -1.96 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 0.1016 | 0.0828 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2306T>C | L769P 2D AIThe SynGAP1 missense variant L769P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | -6.261 | Likely Benign | 0.338 | Likely Benign | Likely Benign | 0.200 | Likely Benign | -1.75 | Neutral | 0.925 | Possibly Damaging | 0.427 | Benign | 3.90 | Benign | 0.00 | Affected | 0.3573 | 0.1323 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2324G>T | R775L 2D AIThe SynGAP1 missense variant R775L (ClinVar ID 4327035) is present in gnomAD (ID 6‑33442482‑G‑T). Prediction tools that agree on benign include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those predicting pathogenic are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also indicates likely benign; Foldetta results are unavailable. Overall, the consensus of computational evidence points to a benign effect, consistent with the ClinVar annotation and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.895337 | Binding | 0.320 | 0.896 | 0.250 | 1 | 6-33442482-G-T | -5.951 | Likely Benign | 0.598 | Likely Pathogenic | Likely Benign | 0.124 | Likely Benign | -1.86 | Neutral | 0.933 | Possibly Damaging | 0.871 | Possibly Damaging | 4.13 | Benign | 0.06 | Tolerated | 3.64 | 6 | 0.1665 | 0.5089 | -2 | -3 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||
| c.2326G>T | G776C 2D AIThe SynGAP1 missense variant G776C is not reported in ClinVar but is present in gnomAD (ID 6‑33442484‑G‑T). Prediction tools cluster into benign (REVEL, FATHMM, AlphaMissense‑Optimized) and pathogenic (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT). Two tools (ESM1b, AlphaMissense‑Default) return uncertain results. High‑accuracy assessments are limited: AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta data are unavailable. Overall, the majority of conventional predictors indicate pathogenicity, whereas the single high‑accuracy tool suggests benign. Given the preponderance of pathogenic predictions and the absence of a ClinVar entry, the variant is most likely pathogenic and does not contradict any existing ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.377384 | Structured | 0.886983 | Binding | 0.296 | 0.888 | 0.250 | 6-33442484-G-T | -7.974 | In-Between | 0.380 | Ambiguous | Likely Benign | 0.181 | Likely Benign | -2.59 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 4.15 | Benign | 0.01 | Affected | 3.64 | 6 | 0.1235 | 0.4403 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||
| c.232C>T | R78C 2D AIThe SynGAP1 missense variant R78C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R78C, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.448183 | Uncertain | 0.304 | 0.866 | 0.500 | -6.079 | Likely Benign | 0.467 | Ambiguous | Likely Benign | 0.114 | Likely Benign | -2.13 | Neutral | 0.991 | Probably Damaging | 0.194 | Benign | 3.80 | Benign | 0.00 | Affected | 0.3255 | 0.2804 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||||||||
| c.2330T>A | L777H 2D AIThe SynGAP1 missense variant L777H has no ClinVar record and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors a benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence—including the consensus of high‑accuracy tools—suggests that the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | -6.719 | Likely Benign | 0.492 | Ambiguous | Likely Benign | 0.230 | Likely Benign | -2.51 | Deleterious | 0.997 | Probably Damaging | 0.993 | Probably Damaging | 3.95 | Benign | 0.00 | Affected | 0.1052 | 0.0972 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2330T>C | L777P 2D AIThe SynGAP1 missense variant L777P is catalogued in gnomAD (6‑33442488‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions are reported by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus also indicates a likely benign outcome. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for the variant, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | 6-33442488-T-C | -5.807 | Likely Benign | 0.361 | Ambiguous | Likely Benign | 0.255 | Likely Benign | -2.13 | Neutral | 0.991 | Probably Damaging | 0.985 | Probably Damaging | 3.94 | Benign | 0.00 | Affected | 3.64 | 6 | 0.3732 | 0.1664 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||
| c.2344G>T | D782Y 2D AIThe SynGAP1 missense variant D782Y is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools largely converge on a deleterious effect: pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default, while only REVEL predicts a benign outcome. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is classified as Likely Pathogenic. Foldetta, a protein‑folding stability predictor that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -8.785 | Likely Pathogenic | 0.919 | Likely Pathogenic | Ambiguous | 0.382 | Likely Benign | -3.75 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.0559 | 0.6202 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2345A>T | D782V 2D AIThe SynGAP1 missense variant D782V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (which reports “Likely Pathogenic”). The high‑accuracy AlphaMissense‑Optimized tool yields an uncertain result, and the Foldetta stability assessment is unavailable. Overall, the consensus of the available predictions strongly favors a pathogenic effect for D782V. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -8.250 | Likely Pathogenic | 0.931 | Likely Pathogenic | Ambiguous | 0.462 | Likely Benign | -3.59 | Deleterious | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.0803 | 0.6477 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2353C>T | R785C 2D AIThe SynGAP1 R785C missense variant is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33442905‑C‑T). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—indicates a likely pathogenic outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points toward a pathogenic impact, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | Uncertain | 1 | 6-33442905-C-T | 29 | 1.80e-5 | -5.887 | Likely Benign | 0.662 | Likely Pathogenic | Likely Benign | 0.126 | Likely Benign | -5.06 | Deleterious | 0.144 | Benign | 0.046 | Benign | 2.22 | Pathogenic | 0.00 | Affected | 3.64 | 6 | 0.3775 | 0.3530 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||
| c.2357T>A | L786H 2D AIThe SynGAP1 missense variant L786H is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. Grouping by agreement, two tools predict benign, seven predict pathogenic, and AlphaMissense‑Optimized remains uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is inconclusive, but the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | -5.892 | Likely Benign | 0.836 | Likely Pathogenic | Ambiguous | 0.168 | Likely Benign | -3.72 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.79 | Pathogenic | 0.00 | Affected | 0.1309 | 0.1214 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||
| c.2357T>C | L786P 2D AISynGAP1 missense variant L786P is reported in gnomAD (ID 6‑33442909‑T‑C) but has no ClinVar entry. Functional prediction tools show discordant results: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further highlight this split: AlphaMissense‑Optimized reports a benign effect, SGM‑Consensus confirms a likely pathogenic outcome, and Foldetta results are unavailable. Overall, the majority of conventional tools and the SGM‑Consensus support a pathogenic interpretation, while one high‑accuracy tool suggests benign. No ClinVar status is present, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | 6-33442909-T-C | -3.217 | Likely Benign | 0.656 | Likely Pathogenic | Likely Benign | 0.219 | Likely Benign | -4.06 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 1.79 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3343 | 0.1814 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.2360C>T | P787L 2D AIThe SynGAP1 missense variant P787L is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that classify the variant as benign include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default—predict it to be pathogenic. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; Foldetta results are unavailable. Overall, the majority of predictions (seven pathogenic vs. three benign) lean toward pathogenicity, and this conclusion is not contradicted by any ClinVar annotation. Thus, the variant is most likely pathogenic based on the current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.901269 | Disordered | 0.613211 | Binding | 0.377 | 0.899 | 0.750 | -3.924 | Likely Benign | 0.747 | Likely Pathogenic | Likely Benign | 0.256 | Likely Benign | -5.89 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.45 | Pathogenic | 0.01 | Affected | 0.2254 | 0.6034 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2366C>T | P789L 2D AIThe SynGAP1 P789L missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence (five pathogenic vs. three benign predictions, with the SGM Consensus supporting pathogenicity) indicates that P789L is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -4.623 | Likely Benign | 0.457 | Ambiguous | Likely Benign | 0.303 | Likely Benign | -5.91 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1958 | 0.4866 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.236A>T | N79I 2D AIThe SynGAP1 missense variant N79I is listed in ClinVar (ID 4759645) with an uncertain significance status and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 HumDiv and SIFT predict pathogenicity. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | Uncertain | 1 | -3.958 | Likely Benign | 0.337 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -1.42 | Neutral | 0.939 | Possibly Damaging | 0.080 | Benign | 4.14 | Benign | 0.00 | Affected | 0.0572 | 0.4924 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||
| c.2381C>T | P794L 2D AIThe SynGAP1 missense variant P794L is listed in ClinVar as Benign (ClinVar ID 859213.0) and is present in the gnomAD database (gnomAD ID 6‑33442933‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as benign, while Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the consensus of available predictions indicates that P794L is most likely benign, and this conclusion is consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.979741 | Disordered | 0.408951 | Uncertain | 0.550 | 0.898 | 0.875 | Benign/Likely benign | 2 | 6-33442933-C-T | 73 | 4.52e-5 | -3.808 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.80 | Neutral | 0.761 | Possibly Damaging | 0.321 | Benign | 4.24 | Benign | 0.03 | Affected | 4.07 | 3 | 0.2417 | 0.6733 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||
| c.2384C>T | P795L 2D AIThe SynGAP1 missense variant P795L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a “Likely Benign” status. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) confirms a benign outcome. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that P795L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.972450 | Disordered | 0.410339 | Uncertain | 0.457 | 0.903 | 0.875 | -2.677 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.45 | Neutral | 0.016 | Benign | 0.010 | Benign | 4.27 | Benign | 0.04 | Affected | 0.2372 | 0.6499 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2387C>T | P796L 2D AIThe SynGAP1 P796L missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is also benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.936162 | Disordered | 0.426363 | Uncertain | 0.427 | 0.900 | 0.875 | -5.442 | Likely Benign | 0.069 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -1.06 | Neutral | 0.325 | Benign | 0.182 | Benign | 4.24 | Benign | 0.01 | Affected | 0.2328 | 0.5712 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2390C>T | P797L 2D AIThe SynGAP1 missense variant P797L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.926919 | Disordered | 0.449970 | Uncertain | 0.561 | 0.902 | 0.875 | -5.631 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.033 | Likely Benign | -0.66 | Neutral | 0.818 | Possibly Damaging | 0.637 | Possibly Damaging | 4.23 | Benign | 0.40 | Tolerated | 0.2550 | 0.6538 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2393C>T | P798L 2D AIThe SynGAP1 missense variant P798L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33442945‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign; a Foldetta stability prediction is not available. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.871313 | Disordered | 0.492709 | Uncertain | 0.426 | 0.899 | 0.875 | Uncertain | 2 | 6-33442945-C-T | 6 | 3.72e-6 | -5.640 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -0.86 | Neutral | 0.981 | Probably Damaging | 0.631 | Possibly Damaging | 4.21 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2039 | 0.5555 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||
| c.2396C>T | P799L 2D AIThe SynGAP1 missense variant P799L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for P799L, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.871313 | Disordered | 0.537892 | Binding | 0.400 | 0.894 | 0.750 | -5.296 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -0.93 | Neutral | 0.905 | Possibly Damaging | 0.670 | Possibly Damaging | 4.27 | Benign | 0.00 | Affected | 0.2078 | 0.6285 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2398G>T | G800C 2D AIThe SynGAP1 missense variant G800C is not reported in ClinVar and has no allele in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM‑Consensus also indicates a benign outcome; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of a ClinVar pathogenic annotation. Based on the aggregate predictions, the variant is most likely benign, and this is consistent with the lack of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.852992 | Disordered | 0.588350 | Binding | 0.303 | 0.884 | 0.625 | -7.074 | In-Between | 0.129 | Likely Benign | Likely Benign | 0.144 | Likely Benign | -1.53 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 2.70 | Benign | 0.15 | Tolerated | 0.1102 | 0.4189 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||
| c.2399G>T | G800V 2D AIThe SynGAP1 missense variant G800V is predicted to be benign by all evaluated in‑silico tools. ClinVar contains no entry for this variant, and it is not reported in gnomAD. Consensus predictions from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a likely benign outcome. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a pathogenic ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.852992 | Disordered | 0.588350 | Binding | 0.303 | 0.884 | 0.625 | -4.890 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.24 | Neutral | 0.451 | Benign | 0.157 | Benign | 2.73 | Benign | 0.25 | Tolerated | 0.1130 | 0.4198 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||
| c.239A>T | K80I 2D AIThe SynGAP1 missense variant K80I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized remains uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta data are unavailable. Consequently, the variant’s predicted impact is ambiguous, with an equal split between benign and pathogenic signals and no ClinVar entry to contradict the computational assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.637480 | Disordered | 0.477530 | Uncertain | 0.331 | 0.873 | 0.500 | -5.320 | Likely Benign | 0.947 | Likely Pathogenic | Ambiguous | 0.083 | Likely Benign | -2.54 | Deleterious | 0.939 | Possibly Damaging | 0.164 | Benign | 3.86 | Benign | 0.00 | Affected | 0.1095 | 0.2981 | -2 | -3 | 8.4 | -15.01 | ||||||||||||||||||||||||||||||||||||||||
| c.23T>A | I8N 2D AIThe SynGAP1 missense variant I8N is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools (polyPhen‑2 HumDiv and SIFT) predict pathogenicity, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the consensus of the majority of predictors, including the high‑accuracy methods, points to a benign impact. This conclusion is consistent with the absence of any ClinVar classification, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.543080 | Binding | 0.341 | 0.916 | 0.625 | -3.979 | Likely Benign | 0.163 | Likely Benign | Likely Benign | 0.120 | Likely Benign | 0.05 | Neutral | 0.561 | Possibly Damaging | 0.032 | Benign | 3.99 | Benign | 0.00 | Affected | 0.0803 | 0.0540 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2401G>T | G801C 2D AIThe SynGAP1 missense variant G801C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM‑Consensus also indicates a benign outcome; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.874069 | Disordered | 0.636323 | Binding | 0.320 | 0.892 | 0.625 | -7.542 | In-Between | 0.125 | Likely Benign | Likely Benign | 0.156 | Likely Benign | -1.83 | Neutral | 0.992 | Probably Damaging | 0.873 | Possibly Damaging | 2.67 | Benign | 0.06 | Tolerated | 0.1039 | 0.4021 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||
| c.2402G>T | G801V 2D AIThe SynGAP1 missense variant G801V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that G801V is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.874069 | Disordered | 0.636323 | Binding | 0.320 | 0.892 | 0.625 | -4.781 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.098 | Likely Benign | -1.64 | Neutral | 0.611 | Possibly Damaging | 0.272 | Benign | 2.70 | Benign | 0.11 | Tolerated | 0.1041 | 0.4230 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||
| c.2404G>T | G802C 2D AIThe SynGAP1 missense variant G802C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence—including the consensus of multiple independent predictors and the high‑accuracy tools—supports a benign classification for G802C. This conclusion is consistent with the lack of any ClinVar pathogenic annotation, so there is no contradiction with existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.894241 | Disordered | 0.681966 | Binding | 0.294 | 0.898 | 0.625 | -6.332 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.127 | Likely Benign | -1.60 | Neutral | 0.983 | Probably Damaging | 0.819 | Possibly Damaging | 2.65 | Benign | 0.00 | Affected | 0.1426 | 0.4126 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||
| c.2405G>T | G802V 2D AIThe SynGAP1 missense variant G802V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign. No Foldetta stability analysis is available for this variant. Overall, the consensus of the available predictions indicates that G802V is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.894241 | Disordered | 0.681966 | Binding | 0.294 | 0.898 | 0.625 | -3.871 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.49 | Neutral | 0.411 | Benign | 0.321 | Benign | 2.67 | Benign | 0.00 | Affected | 0.1345 | 0.3533 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||
| c.2408A>T | K803I 2D AIThe SynGAP1 K803I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. The high‑accuracy AlphaMissense‑Optimized assessment is uncertain, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently contains no classification for K803I. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.827927 | Disordered | 0.733908 | Binding | 0.349 | 0.900 | 0.625 | -5.207 | Likely Benign | 0.894 | Likely Pathogenic | Ambiguous | 0.196 | Likely Benign | -4.06 | Deleterious | 0.995 | Probably Damaging | 0.913 | Probably Damaging | 2.31 | Pathogenic | 0.00 | Affected | 0.1425 | 0.3889 | -2 | -3 | 8.4 | -15.01 | ||||||||||||||||||||||||||||||||||||||
| c.2410G>T | D804Y 2D AIThe SynGAP1 missense variant D804Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) all predict a pathogenic outcome. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, therefore classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; Foldetta results are unavailable. Overall, the majority of predictions support a pathogenic effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.801317 | Disordered | 0.786762 | Binding | 0.294 | 0.900 | 0.625 | -8.356 | Likely Pathogenic | 0.782 | Likely Pathogenic | Likely Benign | 0.357 | Likely Benign | -5.16 | Deleterious | 0.999 | Probably Damaging | 0.983 | Probably Damaging | 1.18 | Pathogenic | 0.01 | Affected | 0.0886 | 0.6653 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||
| c.2411A>T | D804V 2D AIThe SynGAP1 missense variant D804V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—predict a pathogenic impact. The AlphaMissense‑Optimized score is uncertain, providing no definitive evidence. High‑accuracy assessments show that the SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome; AlphaMissense‑Optimized remains inconclusive, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for D804V, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.801317 | Disordered | 0.786762 | Binding | 0.294 | 0.900 | 0.625 | -8.143 | Likely Pathogenic | 0.832 | Likely Pathogenic | Ambiguous | 0.402 | Likely Benign | -4.77 | Deleterious | 0.997 | Probably Damaging | 0.951 | Probably Damaging | 1.19 | Pathogenic | 0.01 | Affected | 0.1196 | 0.6981 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||
| c.2414T>C | L805P 2D AIThe SynGAP1 missense variant L805P is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments give a benign result from AlphaMissense‑Optimized, a pathogenic outcome from the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and no Foldetta data are available. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.775545 | Disordered | 0.827669 | Binding | 0.341 | 0.903 | 0.625 | Uncertain | 1 | -4.661 | Likely Benign | 0.444 | Ambiguous | Likely Benign | 0.272 | Likely Benign | -3.40 | Deleterious | 0.975 | Probably Damaging | 0.767 | Possibly Damaging | 2.36 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3128 | 0.1650 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.2417T>C | F806S 2D  AIThe SynGAP1 missense variant F806S is catalogued in gnomAD (ID 6‑33442969‑T‑C) but has no ClinVar entry. Functional prediction tools split into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, whereas the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because none is reported. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | 6-33442969-T-C | 1 | 6.20e-7 | -6.959 | Likely Benign | 0.911 | Likely Pathogenic | Ambiguous | 0.269 | Likely Benign | -4.13 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.21 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.5067 | 0.0391 | Weaken | -2 | -3 | -3.6 | -60.10 | ||||||||||||||||||||||||||||||||
| c.2419T>G | Y807D 2D AIThe SynGAP1 missense variant Y807D is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect are limited to REVEL, while the majority of algorithms (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. Uncertain calls come from ESM1b and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as pathogenic, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for Y807D, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.699094 | Disordered | 0.853760 | Binding | 0.336 | 0.901 | 0.500 | -7.598 | In-Between | 0.862 | Likely Pathogenic | Ambiguous | 0.186 | Likely Benign | -4.40 | Deleterious | 0.966 | Probably Damaging | 0.773 | Possibly Damaging | 2.42 | Pathogenic | 0.00 | Affected | 0.3944 | 0.0704 | -4 | -3 | -2.2 | -48.09 | ||||||||||||||||||||||||||||||||||||||
| c.2423T>G | V808G 2D AIThe SynGAP1 V808G missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of individual predictors (five benign vs. three pathogenic) support a benign classification, and this is not contradicted by any ClinVar annotation. Thus, based on the available predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.699094 | Disordered | 0.856438 | Binding | 0.289 | 0.903 | 0.500 | -6.635 | Likely Benign | 0.460 | Ambiguous | Likely Benign | 0.268 | Likely Benign | -2.94 | Deleterious | 0.069 | Benign | 0.135 | Benign | 2.27 | Pathogenic | 0.00 | Affected | 0.1925 | 0.3336 | -1 | -3 | -4.6 | -42.08 | |||||||||||||||||||||||||||||||||||||||
| c.242T>C | L81P 2D AIThe SynGAP1 missense variant L81P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is not available for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.632174 | Disordered | 0.502033 | Binding | 0.291 | 0.878 | 0.250 | -4.413 | Likely Benign | 0.827 | Likely Pathogenic | Ambiguous | 0.095 | Likely Benign | -1.51 | Neutral | 0.919 | Possibly Damaging | 0.226 | Benign | 3.92 | Benign | 0.00 | Affected | 0.3062 | 0.1818 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2432C>T | P811L 2D AIThe SynGAP1 P811L missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. AlphaMissense‑Default is uncertain. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, leans toward benign (2 benign vs. 1 pathogenic, 1 uncertain). AlphaMissense‑Optimized also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from high‑accuracy tools and consensus methods indicates that P811L is most likely benign. This assessment does not contradict ClinVar status, as the variant is not yet reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.411940 | Structured | 0.847064 | Binding | 0.382 | 0.910 | 0.250 | -6.184 | Likely Benign | 0.491 | Ambiguous | Likely Benign | 0.150 | Likely Benign | -3.98 | Deleterious | 0.982 | Probably Damaging | 0.824 | Possibly Damaging | 2.71 | Benign | 0.01 | Affected | 0.2327 | 0.6621 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2435C>T | P812L 2D AIThe SynGAP1 P812L missense variant is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (gnomAD ID: 6‑33442987‑C‑T). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic, and Foldetta’s protein‑folding stability analysis is unavailable. Overall, the majority of predictions lean toward pathogenicity, which is consistent with the lack of ClinVar reporting but does not contradict it. Thus, based on the available computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.414856 | Structured | 0.842442 | Binding | 0.388 | 0.901 | 0.125 | 6-33442987-C-T | 1 | 6.20e-7 | -7.121 | In-Between | 0.591 | Likely Pathogenic | Likely Benign | 0.172 | Likely Benign | -2.61 | Deleterious | 0.978 | Probably Damaging | 0.824 | Possibly Damaging | 2.76 | Benign | 0.01 | Affected | 4.32 | 4 | 0.2131 | 0.6634 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.2438T>C | L813P 2D AIThe SynGAP1 missense variant L813P is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign, while the majority of high‑accuracy predictors (AlphaMissense‑Optimized and the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also support a benign classification. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact, and AlphaMissense‑Default remains uncertain. No Foldetta stability assessment is available, so it does not influence the overall interpretation. Overall, the preponderance of evidence points to a benign effect for the variant, and this conclusion is not contradicted by any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.411940 | Structured | 0.838481 | Binding | 0.292 | 0.905 | 0.250 | -2.572 | Likely Benign | 0.554 | Ambiguous | Likely Benign | 0.122 | Likely Benign | -1.96 | Neutral | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 2.58 | Benign | 0.21 | Tolerated | 0.3564 | 0.1458 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2443C>T | R815C 2D AIThe SynGAP1 missense variant R815C is listed in ClinVar (ID 660618.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33442995‑C‑T). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized result is “Uncertain.” The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of predictions indicates a pathogenic effect, which does not contradict the ClinVar “Uncertain” classification but suggests that the variant is more likely pathogenic rather than benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.394753 | Structured | 0.780568 | Binding | 0.278 | 0.907 | 0.250 | Uncertain | 1 | 6-33442995-C-T | 5 | 3.10e-6 | -9.373 | Likely Pathogenic | 0.828 | Likely Pathogenic | Ambiguous | 0.174 | Likely Benign | -3.89 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.59 | Benign | 0.00 | Affected | 4.32 | 4 | 0.3389 | 0.3682 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||
| c.2444G>T | R815L 2D AISynGAP1 missense variant R815L is listed in ClinVar (ID 2505666.0) with an uncertain significance annotation and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and FATHMM, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of tools indicates a pathogenic effect, which contrasts with the ClinVar uncertain classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.394753 | Structured | 0.780568 | Binding | 0.278 | 0.907 | 0.250 | Uncertain | 1 | -8.546 | Likely Pathogenic | 0.865 | Likely Pathogenic | Ambiguous | 0.175 | Likely Benign | -3.06 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.63 | Benign | 0.03 | Affected | 4.32 | 4 | 0.1817 | 0.5132 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||
| c.2453C>T | P818L 2D AIThe SynGAP1 missense variant P818L is catalogued in gnomAD (ID 6‑33443005‑C‑T) but has no ClinVar entry. Functional prediction tools fall into two consensus groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). AlphaMissense‑Optimized returns an uncertain result. High‑accuracy assessments are limited: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus indicates “Likely Pathogenic,” and Foldetta data are unavailable. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Thus, the variant is most likely pathogenic based on current predictive tools. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.715889 | Binding | 0.371 | 0.893 | 0.625 | 6-33443005-C-T | 1 | 6.20e-7 | -6.064 | Likely Benign | 0.938 | Likely Pathogenic | Ambiguous | 0.285 | Likely Benign | -5.81 | Deleterious | 0.997 | Probably Damaging | 0.954 | Probably Damaging | 1.98 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.2351 | 0.6951 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.2458T>G | Y820D 2D AIThe SynGAP1 missense variant Y820D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. In contrast, a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all indicate likely pathogenicity. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools suggests that Y820D is most likely pathogenic, with no ClinVar status to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.733139 | Disordered | 0.695550 | Binding | 0.293 | 0.883 | 0.625 | -10.497 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 0.169 | Likely Benign | -2.77 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.73 | Benign | 0.08 | Tolerated | 0.4113 | 0.0704 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.245T>C | L82P 2D AIThe SynGAP1 missense variant L82P is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33425853‑T‑C). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors pathogenicity. Foldetta, a protein‑folding stability method, did not provide a result for this variant. Overall, the majority of evidence points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.637480 | Disordered | 0.517720 | Binding | 0.284 | 0.890 | 0.375 | 6-33425853-T-C | 1 | 6.20e-7 | -7.667 | In-Between | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.125 | Likely Benign | -2.73 | Deleterious | 0.939 | Possibly Damaging | 0.162 | Benign | 3.64 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3477 | 0.1118 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.2461T>C | C821R 2D AIThe SynGAP1 missense variant C821R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑accuracy predictors (six out of nine) indicate a pathogenic impact, while three suggest benign. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.745909 | Disordered | 0.672821 | Binding | 0.351 | 0.883 | 0.750 | -3.997 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.183 | Likely Benign | -2.93 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.69 | Benign | 0.03 | Affected | 0.1944 | 0.1746 | -4 | -3 | -7.0 | 53.05 | ||||||||||||||||||||||||||||||||||||||||
| c.2461T>G | C821G 2D AIThe SynGAP1 missense variant C821G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward pathogenicity. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.745909 | Disordered | 0.672821 | Binding | 0.351 | 0.883 | 0.750 | -4.613 | Likely Benign | 0.910 | Likely Pathogenic | Ambiguous | 0.187 | Likely Benign | -2.94 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.68 | Benign | 0.05 | Affected | 0.3287 | 0.2747 | -3 | -3 | -2.9 | -46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2474C>G | S825W 2D AIThe SynGAP1 missense variant S825W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess pathogenicity largely agree: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all predict a deleterious effect. Only REVEL classifies the variant as benign, representing the sole benign prediction. High‑accuracy methods further support a pathogenic interpretation: AlphaMissense‑Optimized returns a pathogenic score, and the SGM‑Consensus indicates “Likely Pathogenic.” Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the consensus of the majority of tools, including the high‑accuracy predictors, indicates that S825W is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.618614 | Binding | 0.384 | 0.886 | 0.750 | -8.396 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.316 | Likely Benign | -5.25 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.0818 | 0.6083 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2474C>T | S825L 2D AIThe SynGAP1 missense variant S825L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443026‑C‑T). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain,” and the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a pathogenic effect, which is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.618614 | Binding | 0.384 | 0.886 | 0.750 | Uncertain | 1 | 6-33443026-C-T | 1 | 6.20e-7 | -4.987 | Likely Benign | 0.910 | Likely Pathogenic | Ambiguous | 0.249 | Likely Benign | -4.30 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.94 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1252 | 0.5747 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||
| c.2476G>T | D826Y 2D AIThe SynGAP1 missense variant D826Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL classifies it as benign, whereas the remaining predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—label it pathogenic. The consensus score from the SGM framework, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM Consensus also indicates Likely Pathogenic; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a pathogenic effect for D826Y. Thus, the variant is most likely pathogenic, and this assessment does not contradict the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.666105 | Disordered | 0.627309 | Binding | 0.327 | 0.886 | 0.625 | -8.029 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.336 | Likely Benign | -4.25 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.47 | Pathogenic | 0.00 | Affected | 0.0683 | 0.7410 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2477A>T | D826V 2D AIThe SynGAP1 missense variant D826V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect for D826V, and this conclusion does not conflict with ClinVar, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.666105 | Disordered | 0.627309 | Binding | 0.327 | 0.886 | 0.625 | -6.918 | Likely Benign | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.428 | Likely Benign | -4.64 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.48 | Pathogenic | 0.00 | Affected | 0.1017 | 0.8023 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2480T>A | I827N 2D AIThe SynGAP1 missense variant I827N is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, whereas the SGM‑Consensus (majority vote) supports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the preponderance of evidence points to a benign effect; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.636272 | Binding | 0.383 | 0.884 | 0.625 | -4.860 | Likely Benign | 0.804 | Likely Pathogenic | Ambiguous | 0.114 | Likely Benign | -1.69 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.66 | Benign | 0.11 | Tolerated | 0.0876 | 0.0342 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2489C>T | P830L 2D AIThe SynGAP1 missense variant P830L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign. Foldetta results are unavailable. Overall, the majority of reliable predictors lean toward a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.590140 | Disordered | 0.618152 | Binding | 0.333 | 0.874 | 0.500 | -3.990 | Likely Benign | 0.362 | Ambiguous | Likely Benign | 0.269 | Likely Benign | -5.31 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.65 | Benign | 0.00 | Affected | 0.2138 | 0.6631 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.248G>T | R83I 2D AIThe SynGAP1 missense variant R83I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic vs. two benign). Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the preponderance of evidence (seven pathogenic vs. three benign predictions) indicates that R83I is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.637480 | Disordered | 0.522784 | Binding | 0.275 | 0.895 | 0.250 | -4.021 | Likely Benign | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.183 | Likely Benign | -3.15 | Deleterious | 0.972 | Probably Damaging | 0.766 | Possibly Damaging | 3.17 | Benign | 0.00 | Affected | 0.1450 | 0.3219 | -2 | -3 | 9.0 | -43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2504T>C | L835P 2D AIThe SynGAP1 missense variant L835P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the overall assessment. Overall, the majority of evidence points to the variant being most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.642742 | Binding | 0.319 | 0.863 | 0.125 | -3.024 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.144 | Likely Benign | 0.03 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.78 | Benign | 0.04 | Affected | 0.3455 | 0.1079 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.250C>T | R84C 2D AIThe SynGAP1 missense variant R84C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that R84C is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.666105 | Disordered | 0.529205 | Binding | 0.298 | 0.888 | 0.500 | -9.044 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.243 | Likely Benign | -3.25 | Deleterious | 0.999 | Probably Damaging | 0.876 | Possibly Damaging | 3.66 | Benign | 0.00 | Affected | 0.3281 | 0.3657 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||||||||
| c.2510T>A | V837D 2D AIThe SynGAP1 missense variant V837D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.626284 | Binding | 0.318 | 0.871 | 0.125 | -5.712 | Likely Benign | 0.785 | Likely Pathogenic | Ambiguous | 0.127 | Likely Benign | -0.51 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.61 | Benign | 0.85 | Tolerated | 0.1415 | 0.0902 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2510T>G | V837G 2D AIThe SynGAP1 missense variant V837G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are not available, so they do not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.626284 | Binding | 0.318 | 0.871 | 0.125 | -2.599 | Likely Benign | 0.315 | Likely Benign | Likely Benign | 0.187 | Likely Benign | 0.93 | Neutral | 0.997 | Probably Damaging | 0.999 | Probably Damaging | 3.18 | Benign | 1.00 | Tolerated | 0.2260 | 0.2422 | -1 | -3 | -4.6 | -42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2513A>T | N838I 2D AIThe SynGAP1 missense variant N838I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of algorithms predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for N838I. This conclusion is consistent with the absence of a ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.622677 | Disordered | 0.613320 | Binding | 0.276 | 0.861 | 0.250 | -8.061 | Likely Pathogenic | 0.890 | Likely Pathogenic | Ambiguous | 0.170 | Likely Benign | -4.44 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.63 | Benign | 0.01 | Affected | 0.0642 | 0.4900 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2522T>G | V841G 2D AIThe SynGAP1 missense variant V841G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are REVEL and FATHMM, while the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, whereas the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect for V841G, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.622677 | Disordered | 0.616495 | Binding | 0.261 | 0.873 | 0.125 | -10.054 | Likely Pathogenic | 0.913 | Likely Pathogenic | Ambiguous | 0.288 | Likely Benign | -4.11 | Deleterious | 0.997 | Probably Damaging | 0.999 | Probably Damaging | 2.51 | Benign | 0.00 | Affected | 0.2351 | 0.2422 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2531T>C | L844P 2D AIThe SynGAP1 missense variant L844P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Consensus from standard in‑silico predictors shows a split: benign calls come from REVEL and FATHMM, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; ESM1b remains uncertain. High‑accuracy assessment further supports a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to pathogenic. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence from both conventional and high‑accuracy tools indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.595080 | Disordered | 0.611301 | Binding | 0.304 | 0.835 | 0.375 | -7.425 | In-Between | 0.962 | Likely Pathogenic | Likely Pathogenic | 0.319 | Likely Benign | -3.18 | Deleterious | 0.986 | Probably Damaging | 0.876 | Possibly Damaging | 2.58 | Benign | 0.01 | Affected | 0.3528 | 0.1458 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2533G>T | D845Y 2D AIThe SynGAP1 missense variant D845Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic. Foldetta results are not available for this variant. Overall, the preponderance of evidence from multiple in silico predictors indicates that D845Y is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -9.917 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.384 | Likely Benign | -6.55 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.0590 | 0.6699 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2534A>T | D845V 2D AIThe SynGAP1 D845V missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL classifies it as benign, whereas the remaining 11 predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support a harmful outcome: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a pathogenic effect. This conclusion is consistent with the absence of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -8.914 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.426 | Likely Benign | -6.15 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.0871 | 0.7088 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2542G>T | G848C 2D AIThe SynGAP1 missense variant G848C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.563942 | Binding | 0.287 | 0.816 | 0.500 | -6.987 | Likely Benign | 0.248 | Likely Benign | Likely Benign | 0.237 | Likely Benign | -1.14 | Neutral | 0.998 | Probably Damaging | 0.922 | Probably Damaging | 2.55 | Benign | 0.01 | Affected | 0.1264 | 0.4400 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2545G>T | D849Y 2D AIThe SynGAP1 missense variant D849Y has no ClinVar entry and is not reported in gnomAD. Consensus from multiple in‑silico predictors shows a split: benign calls come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default remains uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” status. High‑accuracy tools further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Taken together, the majority of reliable predictors and consensus analyses indicate a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with existing clinical evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.554191 | Binding | 0.319 | 0.813 | 0.500 | -6.200 | Likely Benign | 0.383 | Ambiguous | Likely Benign | 0.150 | Likely Benign | -1.92 | Neutral | 0.971 | Probably Damaging | 0.773 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.0767 | 0.7690 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2546A>T | D849V 2D AIThe SynGAP1 missense variant D849V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of computational evidence points to a benign effect, and this is consistent with the lack of any ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.554191 | Binding | 0.319 | 0.813 | 0.500 | -3.819 | Likely Benign | 0.319 | Likely Benign | Likely Benign | 0.137 | Likely Benign | -2.10 | Neutral | 0.918 | Possibly Damaging | 0.481 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 0.1161 | 0.7963 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2549G>T | G850V 2D AIThe SynGAP1 missense variant G850V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools converge on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; no Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for G850V, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.648219 | Disordered | 0.540897 | Binding | 0.312 | 0.820 | 0.500 | -5.379 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.213 | Likely Benign | -1.74 | Neutral | 0.960 | Probably Damaging | 0.679 | Possibly Damaging | 4.21 | Benign | 0.02 | Affected | 0.1255 | 0.4077 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2552C>T | P851L 2D AIThe SynGAP1 missense variant P851L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.648219 | Disordered | 0.526893 | Binding | 0.347 | 0.819 | 0.625 | -3.907 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.149 | Likely Benign | -1.13 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 4.25 | Benign | 0.05 | Affected | 0.2129 | 0.7047 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2554G>T | G852C 2D AIThe SynGAP1 missense variant G852C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and Foldetta results are unavailable. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign,” and AlphaMissense‑Optimized independently predicts a benign outcome. With the majority of evidence pointing to a benign effect and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.506063 | Binding | 0.276 | 0.816 | 0.625 | -7.462 | In-Between | 0.107 | Likely Benign | Likely Benign | 0.132 | Likely Benign | -1.75 | Neutral | 0.992 | Probably Damaging | 0.873 | Possibly Damaging | 4.10 | Benign | 0.01 | Affected | 0.1278 | 0.4808 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2555G>T | G852V 2D AIThe SynGAP1 missense variant G852V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for G852V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.506063 | Binding | 0.276 | 0.816 | 0.625 | -5.629 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.30 | Neutral | 0.918 | Possibly Damaging | 0.697 | Possibly Damaging | 4.19 | Benign | 0.02 | Affected | 0.1205 | 0.4173 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2557G>T | G853C 2D AIThe SynGAP1 G853C missense variant has no ClinVar record and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.657645 | Disordered | 0.496246 | Uncertain | 0.284 | 0.815 | 0.625 | -7.021 | In-Between | 0.096 | Likely Benign | Likely Benign | 0.120 | Likely Benign | -1.65 | Neutral | 0.992 | Probably Damaging | 0.873 | Possibly Damaging | 4.10 | Benign | 0.00 | Affected | 0.1292 | 0.4944 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2558G>T | G853V 2D AIThe SynGAP1 missense variant G853V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for G853V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.657645 | Disordered | 0.496246 | Uncertain | 0.284 | 0.815 | 0.625 | -5.688 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.53 | Neutral | 0.611 | Possibly Damaging | 0.502 | Possibly Damaging | 4.14 | Benign | 0.01 | Affected | 0.1188 | 0.4609 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2564T>A | L855H 2D AIThe SynGAP1 missense variant L855H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.485558 | Uncertain | 0.285 | 0.823 | 0.625 | -3.224 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -2.07 | Neutral | 0.938 | Possibly Damaging | 0.690 | Possibly Damaging | 3.96 | Benign | 0.01 | Affected | 0.1137 | 0.1313 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2564T>C | L855P 2D AIThe SynGAP1 missense variant L855P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.485558 | Uncertain | 0.285 | 0.823 | 0.625 | -2.434 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -1.19 | Neutral | 0.586 | Possibly Damaging | 0.377 | Benign | 3.97 | Benign | 0.14 | Tolerated | 0.3633 | 0.1805 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2567A>T | N856I 2D AIThe SynGAP1 missense variant N856I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv and SIFT predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.477615 | Uncertain | 0.263 | 0.827 | 0.500 | -4.360 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.086 | Likely Benign | -2.30 | Neutral | 0.692 | Possibly Damaging | 0.202 | Benign | 4.08 | Benign | 0.04 | Affected | 0.0744 | 0.6453 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2579T>A | V860D 2D AIThe SynGAP1 missense variant V860D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.545602 | Disordered | 0.518121 | Binding | 0.269 | 0.803 | 0.250 | -4.310 | Likely Benign | 0.590 | Likely Pathogenic | Likely Benign | 0.164 | Likely Benign | -1.98 | Neutral | 0.971 | Probably Damaging | 0.690 | Possibly Damaging | 4.08 | Benign | 0.00 | Affected | 0.1382 | 0.1047 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2579T>G | V860G 2D AIThe SynGAP1 missense variant V860G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools cluster into two groups: the benign group includes REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; the pathogenic group contains only SIFT. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, the SGM Consensus (derived from the majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign, while Foldetta results are unavailable. Overall, the collective evidence indicates that V860G is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.545602 | Disordered | 0.518121 | Binding | 0.269 | 0.803 | 0.250 | -3.471 | Likely Benign | 0.173 | Likely Benign | Likely Benign | 0.198 | Likely Benign | -1.54 | Neutral | 0.012 | Benign | 0.009 | Benign | 4.11 | Benign | 0.00 | Affected | 0.2312 | 0.2547 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.257T>A | V86D 2D AIThe SynGAP1 missense variant V86D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on benign impact include REVEL, PROVEAN, and FATHMM, whereas those that agree on pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; ESM1b is uncertain. High‑accuracy assessment shows AlphaMissense‑Optimized predicts pathogenic, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leans benign (two benign, one pathogenic, one uncertain). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of conventional tools predict pathogenicity, whereas the high‑accuracy consensus suggests benign. Based on the aggregate predictions, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, which currently has no entry for V86D. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.685117 | Disordered | 0.552911 | Binding | 0.295 | 0.887 | 0.500 | -7.141 | In-Between | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.147 | Likely Benign | -2.38 | Neutral | 0.824 | Possibly Damaging | 0.485 | Possibly Damaging | 3.73 | Benign | 0.00 | Affected | 0.1590 | 0.1047 | -2 | -3 | -7.7 | 15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.257T>G | V86G 2D AIThe SynGAP1 missense variant V86G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split. Foldetta, which evaluates protein‑folding stability via FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence (five pathogenic versus four benign predictions) indicates that V86G is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.685117 | Disordered | 0.552911 | Binding | 0.295 | 0.887 | 0.500 | -4.212 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.130 | Likely Benign | -2.50 | Deleterious | 0.307 | Benign | 0.824 | Possibly Damaging | 3.74 | Benign | 0.00 | Affected | 0.2321 | 0.2547 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2582C>G | S861W 2D AIThe SynGAP1 missense variant S861W has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority of the four high‑accuracy tools) remains pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this assessment does not contradict any ClinVar status because none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.540903 | Binding | 0.285 | 0.797 | 0.250 | -8.538 | Likely Pathogenic | 0.585 | Likely Pathogenic | Likely Benign | 0.267 | Likely Benign | -3.13 | Deleterious | 0.999 | Probably Damaging | 0.975 | Probably Damaging | 3.89 | Benign | 0.01 | Affected | 0.0874 | 0.6247 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2585A>T | N862I 2D AIThe SynGAP1 missense variant N862I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b; AlphaMissense‑Default is uncertain. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a pathogenic majority (2 pathogenic vs. 1 benign, 1 uncertain). High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM Consensus as pathogenic, and Foldetta results are unavailable. Overall, the majority of predictions support a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.525368 | Disordered | 0.564559 | Binding | 0.257 | 0.791 | 0.250 | -8.702 | Likely Pathogenic | 0.561 | Ambiguous | Likely Benign | 0.195 | Likely Benign | -3.19 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 4.03 | Benign | 0.03 | Affected | 0.0844 | 0.6443 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.2588T>C | L863P 2D AIThe SynGAP1 missense variant L863P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.594839 | Binding | 0.267 | 0.795 | 0.250 | -4.760 | Likely Benign | 0.234 | Likely Benign | Likely Benign | 0.202 | Likely Benign | -0.78 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 4.01 | Benign | 0.12 | Tolerated | 0.3520 | 0.1458 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2597T>G | V866G 2D AIThe SynGAP1 missense variant V866G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact for V866G, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.638070 | Binding | 0.266 | 0.788 | 0.250 | -1.519 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -2.30 | Neutral | 0.912 | Possibly Damaging | 0.993 | Probably Damaging | 2.69 | Benign | 0.07 | Tolerated | 0.2548 | 0.2157 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2599G>A | G867R 2D AIThe SynGAP1 missense variant G867R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.657954 | Binding | 0.285 | 0.801 | 0.250 | -3.569 | Likely Benign | 0.755 | Likely Pathogenic | Likely Benign | 0.131 | Likely Benign | -1.76 | Neutral | 0.997 | Probably Damaging | 0.943 | Probably Damaging | 2.70 | Benign | 0.05 | Affected | 3.82 | 4 | 0.0886 | 0.4494 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.2599G>C | G867R 2D AIThe SynGAP1 missense variant G867R is catalogued in gnomAD (6‑33443151‑G‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score, whereas pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized reports a benign effect, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign status. No Foldetta stability analysis is available for this residue. Overall, the majority of evidence points to a benign effect for G867R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.657954 | Binding | 0.285 | 0.801 | 0.250 | 6-33443151-G-C | 1 | 6.20e-7 | -3.569 | Likely Benign | 0.755 | Likely Pathogenic | Likely Benign | 0.131 | Likely Benign | -1.76 | Neutral | 0.997 | Probably Damaging | 0.943 | Probably Damaging | 2.70 | Benign | 0.05 | Affected | 3.82 | 4 | 0.0886 | 0.4494 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||
| c.2600G>T | G867V 2D AIThe SynGAP1 missense variant G867V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Two tools—ESM1b and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split between benign and uncertain calls. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence (five benign predictions versus two pathogenic predictions, with no decisive high‑accuracy support for pathogenicity) indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.517562 | Disordered | 0.657954 | Binding | 0.285 | 0.801 | 0.250 | -7.049 | In-Between | 0.441 | Ambiguous | Likely Benign | 0.111 | Likely Benign | -2.23 | Neutral | 0.999 | Probably Damaging | 0.958 | Probably Damaging | 2.70 | Benign | 0.10 | Tolerated | 0.1125 | 0.4613 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2602G>T | D868Y 2D AIThe SynGAP1 missense variant D868Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default all indicate deleteriousness. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for D868Y. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.525368 | Disordered | 0.676362 | Binding | 0.262 | 0.815 | 0.250 | -6.031 | Likely Benign | 0.815 | Likely Pathogenic | Ambiguous | 0.256 | Likely Benign | -2.90 | Deleterious | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.47 | Pathogenic | 0.05 | Affected | 0.1115 | 0.6847 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2603A>T | D868V 2D AIThe SynGAP1 missense variant D868V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, and ESM1b, whereas those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.525368 | Disordered | 0.676362 | Binding | 0.262 | 0.815 | 0.250 | -5.200 | Likely Benign | 0.853 | Likely Pathogenic | Ambiguous | 0.182 | Likely Benign | -3.17 | Deleterious | 0.999 | Probably Damaging | 0.966 | Probably Damaging | 2.49 | Pathogenic | 0.25 | Tolerated | 0.1485 | 0.6753 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2606T>C | L869P 2D AIThe SynGAP1 missense variant L869P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification, and AlphaMissense‑Optimized independently predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy tools—supports a benign interpretation. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.688653 | Binding | 0.272 | 0.839 | 0.250 | -2.394 | Likely Benign | 0.523 | Ambiguous | Likely Benign | 0.174 | Likely Benign | -0.99 | Neutral | 0.990 | Probably Damaging | 0.900 | Possibly Damaging | 2.58 | Benign | 0.04 | Affected | 0.3687 | 0.1503 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2609T>C | L870P 2D AIThe SynGAP1 missense variant L870P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.483068 | Structured | 0.688079 | Binding | 0.274 | 0.850 | 0.125 | -4.462 | Likely Benign | 0.402 | Ambiguous | Likely Benign | 0.194 | Likely Benign | -1.92 | Neutral | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 2.59 | Benign | 0.13 | Tolerated | 0.3795 | 0.1864 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2612A>T | H871L 2D AIThe SynGAP1 missense variant H871L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification—there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.480142 | Structured | 0.679301 | Binding | 0.279 | 0.858 | 0.250 | -4.562 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.167 | Likely Benign | -2.16 | Neutral | 0.069 | Benign | 0.054 | Benign | 2.65 | Benign | 0.14 | Tolerated | 0.0953 | 0.4714 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2627C>T | S876L 2D AISynGAP1 variant S876L is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect; the SGM Consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive and therefore unavailable; Foldetta stability analysis is also unavailable. Overall, the majority of available predictions favor a benign impact, suggesting the variant is most likely benign. This conclusion does not conflict with the ClinVar uncertain status, which reflects the current lack of definitive evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.549308 | Disordered | 0.631130 | Binding | 0.280 | 0.872 | 0.250 | Uncertain | 2 | -5.856 | Likely Benign | 0.489 | Ambiguous | Likely Benign | 0.249 | Likely Benign | -3.56 | Deleterious | 0.998 | Probably Damaging | 0.992 | Probably Damaging | 2.57 | Benign | 0.05 | Affected | 3.77 | 5 | 0.1268 | 0.6053 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||||||
| c.2630T>C | L877P 2D AIThe SynGAP1 missense variant L877P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.653063 | Disordered | 0.634010 | Binding | 0.265 | 0.875 | 0.250 | -4.960 | Likely Benign | 0.312 | Likely Benign | Likely Benign | 0.154 | Likely Benign | -1.57 | Neutral | 0.986 | Probably Damaging | 0.876 | Possibly Damaging | 2.59 | Benign | 0.03 | Affected | 0.3899 | 0.1543 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.263T>G | V88G 2D AIThe SynGAP1 missense variant V88G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2) and Foldetta results are unavailable. Overall, the majority of predictions (5 benign vs. 4 pathogenic) and the lack of a ClinVar pathogenic classification suggest that V88G is most likely benign, with no contradiction to existing ClinVar data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.703578 | Disordered | 0.552910 | Binding | 0.323 | 0.870 | 0.500 | -8.588 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.084 | Likely Benign | -2.40 | Neutral | 0.024 | Benign | 0.208 | Benign | 3.68 | Benign | 0.00 | Affected | 0.2608 | 0.2609 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2645G>T | G882V 2D AIThe SynGAP1 missense variant G882V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation—there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.632888 | Binding | 0.306 | 0.878 | 0.250 | -5.893 | Likely Benign | 0.257 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -2.41 | Neutral | 0.224 | Benign | 0.066 | Benign | 2.06 | Pathogenic | 0.02 | Affected | 0.1178 | 0.4717 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2648T>C | L883P 2D AIThe SynGAP1 missense variant L883P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence supports a benign classification for this variant, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.641952 | Binding | 0.334 | 0.886 | 0.250 | -2.572 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -0.58 | Neutral | 0.934 | Possibly Damaging | 0.516 | Possibly Damaging | 2.62 | Benign | 0.21 | Tolerated | 0.3462 | 0.1658 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2654C>T | P885L 2D AIThe SynGAP1 missense variant P885L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.636133 | Binding | 0.344 | 0.917 | 0.250 | -4.352 | Likely Benign | 0.150 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -1.99 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.75 | Benign | 0.00 | Affected | 0.2138 | 0.6951 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2660C>T | P887L 2D AIThe SynGAP1 missense variant P887L is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.602269 | Binding | 0.348 | 0.925 | 0.500 | -4.008 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -1.72 | Neutral | 0.152 | Benign | 0.070 | Benign | 2.81 | Benign | 0.18 | Tolerated | 0.1800 | 0.5164 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2665G>A | G889R 2D AIThe SynGAP1 missense variant G889R is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (ID 6‑33443217‑G‑A). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized; pathogenic predictions come from SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs three pathogenic) supports a benign impact. This conclusion does not contradict ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | 6-33443217-G-A | 1 | 6.20e-7 | -3.357 | Likely Benign | 0.685 | Likely Pathogenic | Likely Benign | 0.070 | Likely Benign | -1.96 | Neutral | 0.027 | Benign | 0.009 | Benign | 2.38 | Pathogenic | 0.02 | Affected | 4.32 | 4 | 0.0925 | 0.4165 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.2665G>C | G889R 2D AIThe SynGAP1 missense variant G889R is not reported in ClinVar (ClinVar status: not present) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | -3.357 | Likely Benign | 0.685 | Likely Pathogenic | Likely Benign | 0.072 | Likely Benign | -1.96 | Neutral | 0.027 | Benign | 0.009 | Benign | 2.38 | Pathogenic | 0.02 | Affected | 4.32 | 4 | 0.0925 | 0.4165 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2666G>T | G889V 2D AIThe SynGAP1 missense variant G889V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for G889V, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | -5.781 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -2.11 | Neutral | 0.611 | Possibly Damaging | 0.243 | Benign | 2.39 | Pathogenic | 0.03 | Affected | 0.1198 | 0.4523 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2668C>T | R890C 2D AIThe SynGAP1 missense variant R890C is listed in ClinVar as benign and is present in gnomAD (6-33443220-C‑T). Functional prediction tools show mixed results: benign predictions come from REVEL, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions are reported by PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus also indicates benign; Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence leans toward a benign effect, which is consistent with the ClinVar classification and does not contradict the reported status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.720929 | Disordered | 0.531156 | Binding | 0.284 | 0.928 | 0.625 | Benign | 1 | 6-33443220-C-T | 9 | 5.58e-6 | -5.786 | Likely Benign | 0.402 | Ambiguous | Likely Benign | 0.200 | Likely Benign | -3.38 | Deleterious | 1.000 | Probably Damaging | 0.971 | Probably Damaging | 3.94 | Benign | 0.04 | Affected | 4.32 | 4 | 0.3626 | 0.2206 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.266C>T | P89L 2D AISynGAP1 missense variant P89L is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 HumVar, ESM1b, and FATHMM, while pathogenic calls are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts pathogenicity, whereas the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of tools favor a pathogenic effect, but the evidence is not unanimous. Therefore, the variant is most likely pathogenic according to the current predictions, and this assessment does not contradict its ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.703578 | Disordered | 0.545797 | Binding | 0.316 | 0.865 | 0.500 | Uncertain | 2 | -6.775 | Likely Benign | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.119 | Likely Benign | -3.29 | Deleterious | 0.889 | Possibly Damaging | 0.058 | Benign | 3.73 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2399 | 0.5638 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||
| c.2672T>A | L891H 2D AIThe SynGAP1 missense variant L891H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.505861 | Binding | 0.305 | 0.923 | 0.750 | -4.604 | Likely Benign | 0.320 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -1.79 | Neutral | 0.122 | Benign | 0.134 | Benign | 2.69 | Benign | 0.00 | Affected | 0.1099 | 0.1189 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2672T>C | L891P 2D AIThe SynGAP1 missense variant L891P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for L891P, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.505861 | Binding | 0.305 | 0.923 | 0.750 | -3.835 | Likely Benign | 0.169 | Likely Benign | Likely Benign | 0.126 | Likely Benign | -1.61 | Neutral | 0.990 | Probably Damaging | 0.900 | Possibly Damaging | 2.67 | Benign | 0.01 | Affected | 0.3594 | 0.1138 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2681G>T | G894V 2D AIThe SynGAP1 missense variant G894V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also leans benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no conflict with the ClinVar status. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.788093 | Disordered | 0.425700 | Uncertain | 0.310 | 0.925 | 0.750 | -5.047 | Likely Benign | 0.395 | Ambiguous | Likely Benign | 0.188 | Likely Benign | -2.61 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.64 | Benign | 0.02 | Affected | 0.1187 | 0.4091 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2686G>T | G896C 2D AIThe SynGAP1 missense variant G896C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.412816 | Uncertain | 0.314 | 0.923 | 0.625 | -5.607 | Likely Benign | 0.245 | Likely Benign | Likely Benign | 0.193 | Likely Benign | -2.59 | Deleterious | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.50 | Benign | 0.10 | Tolerated | 0.1270 | 0.4369 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2696T>A | I899N 2D AIThe SynGAP1 missense variant I899N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is likely benign, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for I899N, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.443727 | Uncertain | 0.292 | 0.928 | 0.375 | -3.328 | Likely Benign | 0.469 | Ambiguous | Likely Benign | 0.058 | Likely Benign | -0.68 | Neutral | 0.611 | Possibly Damaging | 0.140 | Benign | 2.76 | Benign | 0.00 | Affected | 0.0933 | 0.0470 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.269T>G | V90G 2D AIThe SynGAP1 missense variant V90G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign verdict. Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.570702 | Disordered | 0.542047 | Binding | 0.343 | 0.873 | 0.500 | -2.617 | Likely Benign | 0.358 | Ambiguous | Likely Benign | 0.094 | Likely Benign | -0.55 | Neutral | 0.024 | Benign | 0.208 | Benign | 4.00 | Benign | 0.00 | Affected | 0.2047 | 0.2936 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.26A>T | H9L 2D AIThe SynGAP1 missense variant H9L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.528099 | Binding | 0.394 | 0.916 | 0.750 | -2.603 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.151 | Likely Benign | 0.48 | Neutral | 0.024 | Benign | 0.002 | Benign | 4.25 | Benign | 0.00 | Affected | 0.1255 | 0.6285 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2707G>T | G903C 2D AIThe SynGAP1 missense variant G903C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain and does not influence the consensus. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, which would provide a protein‑folding stability estimate, is unavailable for this variant. Overall, the majority of evidence (five pathogenic versus three benign predictions) points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.680603 | Disordered | 0.549818 | Binding | 0.291 | 0.917 | 0.375 | -6.593 | Likely Benign | 0.471 | Ambiguous | Likely Benign | 0.151 | Likely Benign | -3.28 | Deleterious | 1.000 | Probably Damaging | 0.990 | Probably Damaging | 2.32 | Pathogenic | 0.02 | Affected | 0.1158 | 0.4420 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2708G>T | G903V 2D AIThe SynGAP1 missense variant G903V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, while Foldetta results are unavailable. Overall, the balance of evidence (five benign predictions versus three pathogenic predictions, with no conflicting ClinVar annotation) indicates that the variant is most likely benign. This conclusion does not contradict any ClinVar status, as the variant is not currently catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.680603 | Disordered | 0.549818 | Binding | 0.291 | 0.917 | 0.375 | -4.750 | Likely Benign | 0.491 | Ambiguous | Likely Benign | 0.168 | Likely Benign | -1.93 | Neutral | 0.997 | Probably Damaging | 0.959 | Probably Damaging | 2.44 | Pathogenic | 0.95 | Tolerated | 0.1269 | 0.4266 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2713C>T | R905C 2D AIThe SynGAP1 missense variant R905C (ClinVar ID 469152.0) is listed as Uncertain in ClinVar and is present in gnomAD (ID 6‑33443265‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of standard predictors indicate a pathogenic impact, whereas the high‑accuracy AlphaMissense‑Optimized tool suggests a benign effect. Consequently, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.671169 | Disordered | 0.618085 | Binding | 0.291 | 0.920 | 0.250 | Conflicting | 2 | 6-33443265-C-T | 15 | 9.31e-6 | -5.578 | Likely Benign | 0.723 | Likely Pathogenic | Likely Benign | 0.194 | Likely Benign | -3.14 | Deleterious | 1.000 | Probably Damaging | 0.980 | Probably Damaging | 2.57 | Benign | 0.01 | Affected | 3.77 | 5 | 0.3231 | 0.3642 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.2714G>T | R905L 2D AIThe SynGAP1 missense variant R905L is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (allele ID 6‑33443266‑G‑T). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of predictions leans toward a benign interpretation, and this conclusion does not contradict the ClinVar status, which currently has no assertion for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.671169 | Disordered | 0.618085 | Binding | 0.291 | 0.920 | 0.250 | 6-33443266-G-T | -3.284 | Likely Benign | 0.709 | Likely Pathogenic | Likely Benign | 0.242 | Likely Benign | -2.55 | Deleterious | 0.963 | Probably Damaging | 0.753 | Possibly Damaging | 2.61 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1676 | 0.4905 | -2 | -3 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||
| c.2717T>A | L906H 2D AIThe SynGAP1 missense variant L906H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs. two pathogenic votes) and Foldetta results are unavailable. Overall, the majority of standard predictors lean toward pathogenicity, but the most reliable high‑accuracy tool suggests a benign outcome and the consensus is unresolved. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.644316 | Binding | 0.315 | 0.920 | 0.250 | -4.367 | Likely Benign | 0.732 | Likely Pathogenic | Likely Benign | 0.124 | Likely Benign | -1.01 | Neutral | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.18 | Pathogenic | 0.02 | Affected | 0.1125 | 0.1073 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2717T>C | L906P 2D AIThe SynGAP1 missense variant L906P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome. Foldetta, which would provide a protein‑folding stability estimate, is unavailable for this variant. Overall, the preponderance of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.644316 | Binding | 0.315 | 0.920 | 0.250 | -3.662 | Likely Benign | 0.478 | Ambiguous | Likely Benign | 0.150 | Likely Benign | 0.08 | Neutral | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 2.18 | Pathogenic | 0.10 | Tolerated | 0.3430 | 0.1458 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2728G>T | G910C 2D AIThe SynGAP1 missense variant G910C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta data are unavailable. Overall, the majority of tools (five pathogenic vs four benign) lean toward a pathogenic interpretation, and the high‑accuracy benign prediction from AlphaMissense‑Optimized does not override this trend. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.762850 | Disordered | 0.707319 | Binding | 0.264 | 0.917 | 0.250 | -6.359 | Likely Benign | 0.723 | Likely Pathogenic | Likely Benign | 0.252 | Likely Benign | -3.11 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.69 | Benign | 0.01 | Affected | 0.1179 | 0.4194 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2732T>A | V911D 2D AIThe SynGAP1 missense variant V911D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of high‑confidence predictors indicate a benign impact, and this is consistent with the lack of ClinVar evidence. Therefore, the variant is most likely benign and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.685117 | Disordered | 0.724137 | Binding | 0.327 | 0.914 | 0.375 | -4.422 | Likely Benign | 0.624 | Likely Pathogenic | Likely Benign | 0.114 | Likely Benign | -0.41 | Neutral | 0.500 | Possibly Damaging | 0.157 | Benign | 2.71 | Benign | 0.05 | Affected | 0.1416 | 0.1289 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2732T>G | V911G 2D AIThe SynGAP1 missense variant V911G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). All available in silico predictors classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity, so the benign group includes every listed predictor, while the pathogenic group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.685117 | Disordered | 0.724137 | Binding | 0.327 | 0.914 | 0.375 | -2.754 | Likely Benign | 0.222 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -0.04 | Neutral | 0.004 | Benign | 0.003 | Benign | 2.74 | Benign | 0.16 | Tolerated | 0.2307 | 0.3360 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2740G>T | D914Y 2D AIThe SynGAP1 missense variant D914Y is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of tools (five pathogenic vs. four benign) and the lack of a benign consensus from the high‑accuracy methods suggest that D914Y is most likely pathogenic. This prediction does not contradict ClinVar status, as the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.699094 | Disordered | 0.785987 | Binding | 0.320 | 0.892 | 0.250 | -4.768 | Likely Benign | 0.740 | Likely Pathogenic | Likely Benign | 0.239 | Likely Benign | -2.65 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 2.62 | Benign | 0.01 | Affected | 0.0907 | 0.7583 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2743G>T | G915C 2D AIThe SynGAP1 missense variant G915C is listed in ClinVar with no submitted interpretation and is present in gnomAD (variant ID 6-33443295-G‑T). Functional prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.657645 | Disordered | 0.808641 | Binding | 0.302 | 0.880 | 0.375 | 6-33443295-G-T | 1 | 6.20e-7 | -6.446 | Likely Benign | 0.185 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -2.84 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 2.64 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1023 | 0.4471 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.2747T>A | V916D 2D AIThe SynGAP1 missense variant V916D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT uniformly predict a pathogenic outcome. AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus indicates Likely Benign, and Foldetta (which integrates FoldX‑MD and Rosetta outputs) is not available for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.835395 | Binding | 0.308 | 0.879 | 0.250 | -2.653 | Likely Benign | 0.392 | Ambiguous | Likely Benign | 0.139 | Likely Benign | -1.48 | Neutral | 0.975 | Probably Damaging | 0.767 | Possibly Damaging | 2.69 | Benign | 0.01 | Affected | 0.1580 | 0.1113 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2747T>G | V916G 2D AIThe SynGAP1 missense variant V916G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.642678 | Disordered | 0.835395 | Binding | 0.308 | 0.879 | 0.250 | -1.950 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -0.63 | Neutral | 0.666 | Possibly Damaging | 0.917 | Probably Damaging | 2.69 | Benign | 0.05 | Affected | 0.2420 | 0.2681 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.274G>A | G92R 2D AIThe SynGAP1 missense variant G92R is listed in gnomAD (6-33425882‑G‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain,” SGM‑Consensus as “Likely Benign,” and Foldetta results are unavailable. Taken together, the preponderance of evidence from multiple independent predictors and the consensus score points to a benign classification. This conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.537848 | Binding | 0.337 | 0.874 | 0.625 | 6-33425882-G-A | 1 | 6.20e-7 | -2.909 | Likely Benign | 0.876 | Likely Pathogenic | Ambiguous | 0.139 | Likely Benign | -2.38 | Neutral | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 4.01 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1041 | 0.4605 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||
| c.274G>C | G92R 2D AIThe SynGAP1 missense variant G92R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence tools and the consensus prediction lean toward a benign interpretation, with no conflict with ClinVar status. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.537848 | Binding | 0.337 | 0.874 | 0.625 | -2.909 | Likely Benign | 0.876 | Likely Pathogenic | Ambiguous | 0.139 | Likely Benign | -2.38 | Neutral | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 4.01 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1041 | 0.4605 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.2750C>T | P917L 2D AIThe SynGAP1 missense variant P917L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.863949 | Binding | 0.314 | 0.862 | 0.375 | -3.369 | Likely Benign | 0.172 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -2.35 | Neutral | 0.425 | Benign | 0.233 | Benign | 2.75 | Benign | 0.00 | Affected | 0.2001 | 0.6351 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.275G>T | G92V 2D AIThe SynGAP1 missense variant G92V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign impact for G92V, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.537848 | Binding | 0.337 | 0.874 | 0.625 | -4.627 | Likely Benign | 0.338 | Likely Benign | Likely Benign | 0.164 | Likely Benign | -2.62 | Deleterious | 0.999 | Probably Damaging | 0.972 | Probably Damaging | 4.00 | Benign | 0.00 | Affected | 0.1171 | 0.4363 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2762T>C | L921P 2D AIThe SynGAP1 missense variant L921P is recorded in gnomAD (ID 6‑33443314‑T‑C) but has no ClinVar submission. Functional prediction tools cluster into two groups: benign calls from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized; pathogenic calls from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because the votes are split (two benign, one pathogenic, one uncertain). Foldetta, a protein‑folding stability predictor that combines FoldX‑MD and Rosetta outputs, has no reported result for this variant. Consequently, the evidence is evenly divided, with high‑accuracy tools not providing a decisive verdict. The variant is therefore inconclusive; it does not contradict ClinVar status, which currently has no entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.608892 | Disordered | 0.943282 | Binding | 0.311 | 0.845 | 0.375 | 6-33443314-T-C | 1 | 6.20e-7 | -2.087 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.215 | Likely Benign | -1.83 | Neutral | 0.998 | Probably Damaging | 0.958 | Probably Damaging | 2.39 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3581 | 0.1343 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.2768T>A | I923N 2D AIThe SynGAP1 missense variant I923N (ClinVar ID 647043.0) is listed as “Uncertain” and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign” because the majority of its constituent tools (three benign, one pathogenic) favor a benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as benign, and the Foldetta stability analysis is unavailable. Overall, the collective predictions point to a benign impact, which does not contradict the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.562014 | Disordered | 0.964857 | Binding | 0.292 | 0.852 | 0.250 | Uncertain | 1 | -0.733 | Likely Benign | 0.712 | Likely Pathogenic | Likely Benign | 0.108 | Likely Benign | -1.16 | Neutral | 0.991 | Probably Damaging | 0.793 | Possibly Damaging | 2.70 | Benign | 0.13 | Tolerated | 3.77 | 5 | 0.1164 | 0.1073 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||
| c.2771C>T | P924L 2D AIThe SynGAP1 missense variant P924L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence indicates that P924L is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.521092 | Disordered | 0.971858 | Binding | 0.293 | 0.846 | 0.250 | -6.361 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.422 | Likely Benign | -6.89 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 0.66 | Pathogenic | 0.00 | Affected | 0.1771 | 0.4941 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2774T>A | L925H 2D AIThe SynGAP1 missense variant L925H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic.” No Foldetta stability result is available. Overall, the majority of evidence points to a pathogenic effect for L925H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.977963 | Binding | 0.290 | 0.852 | 0.125 | -3.369 | Likely Benign | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.475 | Likely Benign | -5.24 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.1219 | 0.1494 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2774T>C | L925P 2D AIThe SynGAP1 missense variant L925P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only ESM1b, whereas the remaining tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic. Foldetta results are not available for this variant. Overall, the preponderance of evidence from multiple in silico predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.977963 | Binding | 0.290 | 0.852 | 0.125 | -2.733 | Likely Benign | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.501 | Likely Pathogenic | -5.12 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.3083 | 0.1996 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.277C>G | R93G 2D AIThe SynGAP1 missense variant R93G is listed in ClinVar (ID 2504251.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (derived from the same set of predictors) labels it “Likely Benign”; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that R93G is most likely benign, which does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.549151 | Binding | 0.290 | 0.874 | 0.625 | Uncertain | 1 | -2.674 | Likely Benign | 0.400 | Ambiguous | Likely Benign | 0.093 | Likely Benign | -1.69 | Neutral | 0.103 | Benign | 0.019 | Benign | 3.99 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3809 | 0.3814 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||
| c.277C>T | R93W 2D AIThe SynGAP1 missense variant R93W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R93W, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.549151 | Binding | 0.290 | 0.874 | 0.625 | -5.652 | Likely Benign | 0.514 | Ambiguous | Likely Benign | 0.094 | Likely Benign | -2.22 | Neutral | 0.981 | Probably Damaging | 0.257 | Benign | 3.96 | Benign | 0.00 | Affected | 0.1413 | 0.3835 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2786A>T | N929I 2D AIThe SynGAP1 missense variant N929I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL classifies it as benign, whereas the remaining eight tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict pathogenicity. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus result is consistent. Foldetta, a protein‑folding stability method that combines FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not conflict with the current ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.986867 | Binding | 0.321 | 0.851 | 0.375 | -11.799 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.297 | Likely Benign | -6.82 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.45 | Pathogenic | 0.00 | Affected | 0.0780 | 0.6486 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2789C>T | P930L 2D AIThe SynGAP1 missense variant P930L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts Pathogenic, and the SGM‑Consensus also indicates Likely Pathogenic. Foldetta results are unavailable. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for P930L, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.988036 | Binding | 0.304 | 0.855 | 0.375 | -10.690 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.440 | Likely Benign | -7.25 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 0.66 | Pathogenic | 0.00 | Affected | 0.2275 | 0.6113 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2792T>A | L931H 2D AIThe SynGAP1 missense variant L931H has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. ESM1b and AlphaMissense‑Optimized are uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.549308 | Disordered | 0.989212 | Binding | 0.335 | 0.856 | 0.375 | -7.495 | In-Between | 0.954 | Likely Pathogenic | Ambiguous | 0.301 | Likely Benign | -3.76 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.38 | Pathogenic | 0.00 | Affected | 0.1198 | 0.1205 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2792T>C | L931P 2D AIThe SynGAP1 missense variant L931P is not reported in ClinVar and is absent from gnomAD. Prediction tools show a strong bias toward pathogenicity: SIFT, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, PROVEAN, and AlphaMissense‑Default all predict a deleterious effect, whereas only REVEL indicates a benign outcome. High‑accuracy assessments reinforce this trend: the SGM consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic, and AlphaMissense‑Optimized returns an uncertain result. Foldetta predictions are unavailable. Overall, the preponderance of evidence points to a pathogenic impact for L931P, and this conclusion is consistent with the absence of any ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.549308 | Disordered | 0.989212 | Binding | 0.335 | 0.856 | 0.375 | -8.624 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 0.376 | Likely Benign | -3.43 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.3203 | 0.1698 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2798A>T | H933L 2D AIThe SynGAP1 H933L missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.987531 | Binding | 0.305 | 0.862 | 0.625 | -0.858 | Likely Benign | 0.470 | Ambiguous | Likely Benign | 0.388 | Likely Benign | -6.26 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.38 | Pathogenic | 0.02 | Affected | 0.1003 | 0.5749 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2809G>T | D937Y 2D AIThe SynGAP1 missense variant D937Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic versus four benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic based on current computational evidence, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.819762 | Disordered | 0.963385 | Binding | 0.348 | 0.883 | 0.625 | -4.720 | Likely Benign | 0.711 | Likely Pathogenic | Likely Benign | 0.137 | Likely Benign | -2.52 | Deleterious | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.67 | Benign | 0.05 | Affected | 0.1305 | 0.7494 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2810A>T | D937V 2D AIThe SynGAP1 missense variant D937V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) support a benign classification. This consensus does not contradict ClinVar status, as no ClinVar entry exists for this variant. Thus, based on current computational evidence, the D937V variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.963385 | Binding | 0.348 | 0.883 | 0.625 | -3.418 | Likely Benign | 0.673 | Likely Pathogenic | Likely Benign | 0.141 | Likely Benign | -2.21 | Neutral | 1.000 | Probably Damaging | 0.977 | Probably Damaging | 2.70 | Benign | 0.02 | Affected | 0.1766 | 0.7408 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2813G>T | G938V 2D AIThe SynGAP1 missense variant G938V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence indicates that G938V is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.949795 | Binding | 0.318 | 0.883 | 0.625 | -6.112 | Likely Benign | 0.153 | Likely Benign | Likely Benign | 0.123 | Likely Benign | -0.81 | Neutral | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 2.83 | Benign | 0.31 | Tolerated | 0.1230 | 0.3811 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2816C>T | P939L 2D AIThe SynGAP1 missense variant P939L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta are unavailable. Overall, five tools predict pathogenicity versus four predicting benign, so the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -4.191 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.159 | Likely Benign | -3.69 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 0.2113 | 0.5142 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2818G>T | G940C 2D AIThe SynGAP1 missense variant G940C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this conclusion, so the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.920635 | Binding | 0.383 | 0.902 | 0.625 | -8.158 | Likely Pathogenic | 0.097 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -0.40 | Neutral | 0.996 | Probably Damaging | 0.905 | Possibly Damaging | 2.70 | Benign | 0.11 | Tolerated | 0.1307 | 0.4354 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2819G>T | G940V 2D AIThe SynGAP1 missense variant G940V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the consensus of available predictions points to a benign impact, and this is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.920635 | Binding | 0.383 | 0.902 | 0.625 | -6.252 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.110 | Likely Benign | 0.36 | Neutral | 0.626 | Possibly Damaging | 0.419 | Benign | 2.92 | Benign | 0.31 | Tolerated | 0.1222 | 0.3822 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.281C>T | P94L 2D AIThe SynGAP1 missense variant P94L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.570978 | Binding | 0.350 | 0.869 | 0.625 | -2.721 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -2.27 | Neutral | 0.198 | Benign | 0.017 | Benign | 4.13 | Benign | 0.00 | Affected | 0.2125 | 0.5862 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2822C>T | P941L 2D AIThe SynGAP1 missense variant P941L is listed in ClinVar (ID 3451960.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). The only tool that predicts a pathogenic outcome is SIFT. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates “Likely Benign.” No Foldetta (FoldX‑MD/Rosetta) stability result is available, so it does not influence the assessment. Overall, the majority of predictions, including the high‑accuracy tools, point to a benign effect, which is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.900790 | Binding | 0.403 | 0.906 | 0.625 | Uncertain | 2 | -5.692 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -0.44 | Neutral | 0.144 | Benign | 0.039 | Benign | 2.76 | Benign | 0.01 | Affected | 0.2196 | 0.5931 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||
| c.2825C>T | P942L 2D AIThe SynGAP1 missense variant P942L is listed in ClinVar (ID 2851884.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443377‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.878102 | Binding | 0.365 | 0.915 | 0.625 | Uncertain | 1 | 6-33443377-C-T | 4 | 2.48e-6 | -5.063 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -2.00 | Neutral | 0.411 | Benign | 0.239 | Benign | 2.37 | Pathogenic | 0.00 | Affected | 4.32 | 4 | 0.2094 | 0.5507 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||
| c.2827G>C | G943R 2D AIThe SynGAP1 missense variant G943R is reported in gnomAD (variant ID 6-33443379‑G‑C) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity; only ESM1b is uncertain. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions strongly supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.973328 | Disordered | 0.860437 | Binding | 0.372 | 0.910 | 0.750 | 6-33443379-G-C | 1 | 6.20e-7 | -7.159 | In-Between | 0.335 | Likely Benign | Likely Benign | 0.308 | Likely Benign | 1.00 | Neutral | 0.411 | Benign | 0.062 | Benign | 2.86 | Benign | 0.94 | Tolerated | 4.32 | 4 | 0.0976 | 0.4933 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||
| c.2827G>T | G943C 2D AIThe SynGAP1 missense variant G943C is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which itself is “Likely Benign”). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b all predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence—including the consensus and high‑accuracy tools—points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.973328 | Disordered | 0.860437 | Binding | 0.372 | 0.910 | 0.750 | -9.822 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.356 | Likely Benign | -0.94 | Neutral | 0.938 | Possibly Damaging | 0.649 | Possibly Damaging | 2.84 | Benign | 0.10 | Tolerated | 0.1425 | 0.4439 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2828G>T | G943V 2D AIThe SynGAP1 missense variant G943V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. The only tool with an uncertain outcome is ESM1b, which does not alter the overall consensus. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign classification. High‑accuracy predictors corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Taken together, the evidence overwhelmingly supports a benign interpretation, and there is no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.973328 | Disordered | 0.860437 | Binding | 0.372 | 0.910 | 0.750 | -7.658 | In-Between | 0.092 | Likely Benign | Likely Benign | 0.265 | Likely Benign | -0.43 | Neutral | 0.224 | Benign | 0.062 | Benign | 2.85 | Benign | 0.16 | Tolerated | 0.1399 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2830G>T | G944C 2D AIThe SynGAP1 missense variant G944C is listed in ClinVar with no submitted interpretation and is present in gnomAD (variant ID 6‑33443382‑G‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote of the four contributing tools) also indicates Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which remains unclassified. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.852408 | Binding | 0.360 | 0.923 | 0.750 | 6-33443382-G-T | 1 | 6.20e-7 | -9.121 | Likely Pathogenic | 0.093 | Likely Benign | Likely Benign | 0.426 | Likely Benign | -1.79 | Neutral | 0.975 | Probably Damaging | 0.848 | Possibly Damaging | 3.70 | Benign | 0.00 | Affected | 4.32 | 4 | 0.1399 | 0.4439 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.2837G>T | G946V 2D AIThe SynGAP1 missense variant G946V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for G946V, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985417 | Disordered | 0.845792 | Binding | 0.357 | 0.920 | 0.750 | -7.528 | In-Between | 0.109 | Likely Benign | Likely Benign | 0.368 | Likely Benign | -0.30 | Neutral | 0.901 | Possibly Damaging | 0.516 | Possibly Damaging | 4.57 | Benign | 0.00 | Affected | 0.1524 | 0.3707 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2839G>A | G947R 2D AIThe SynGAP1 missense variant G947R is listed in gnomAD (6-33443391-G-A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Optimized) and pathogenic (SIFT). Two tools remain uncertain (ESM1b, AlphaMissense‑Default). High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—does not reach a definitive conclusion (two benign, two uncertain). Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no reported result for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988695 | Disordered | 0.850554 | Binding | 0.358 | 0.919 | 0.750 | 6-33443391-G-A | 6 | 3.72e-6 | -7.481 | In-Between | 0.343 | Ambiguous | Likely Benign | 0.273 | Likely Benign | -0.60 | Neutral | 0.411 | Benign | 0.140 | Benign | 4.94 | Benign | 0.04 | Affected | 4.32 | 4 | 0.1034 | 0.4733 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.2839G>C | G947R 2D AIThe SynGAP1 missense variant G947R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta stability analysis is unavailable. Overall, the balance of evidence favors a benign classification, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988695 | Disordered | 0.850554 | Binding | 0.358 | 0.919 | 0.750 | -7.481 | In-Between | 0.343 | Ambiguous | Likely Benign | 0.273 | Likely Benign | -0.60 | Neutral | 0.411 | Benign | 0.140 | Benign | 4.94 | Benign | 0.04 | Affected | 4.32 | 4 | 0.1034 | 0.4733 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2840G>T | G947V 2D AIThe SynGAP1 missense variant G947V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign, whereas polyPhen‑2 HumDiv and SIFT predict pathogenicity; ESM1b remains uncertain. High‑accuracy methods reinforce the benign assessment: AlphaMissense‑Optimized scores benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” and Foldetta data are not available. Overall, the preponderance of evidence points to a benign impact for G947V, and this conclusion is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988695 | Disordered | 0.850554 | Binding | 0.358 | 0.919 | 0.750 | -7.171 | In-Between | 0.105 | Likely Benign | Likely Benign | 0.296 | Likely Benign | -1.11 | Neutral | 0.586 | Possibly Damaging | 0.303 | Benign | 4.93 | Benign | 0.01 | Affected | 0.1443 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2842G>T | G948C 2D AIThe SynGAP1 missense variant G948C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The high‑accuracy consensus (SGM‑Consensus) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the majority of evidence points to a benign impact. The predictions do not contradict ClinVar status, as no ClinVar assertion exists for this variant. Thus, based on the available computational predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.862121 | Binding | 0.365 | 0.919 | 0.750 | -10.254 | Likely Pathogenic | 0.102 | Likely Benign | Likely Benign | 0.247 | Likely Benign | -0.77 | Neutral | 0.997 | Probably Damaging | 0.840 | Possibly Damaging | 4.50 | Benign | 0.04 | Affected | 0.1422 | 0.4439 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2843G>T | G948V 2D AIThe SynGAP1 missense variant G948V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that G948V is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.862121 | Binding | 0.365 | 0.919 | 0.750 | -6.541 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.256 | Likely Benign | -0.48 | Neutral | 0.901 | Possibly Damaging | 0.435 | Benign | 4.52 | Benign | 0.06 | Tolerated | 0.1464 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2845G>T | G949C 2D AIThe SynGAP1 missense variant G949C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. four benign) indicate that the variant is most likely pathogenic. This assessment does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | -10.580 | Likely Pathogenic | 0.091 | Likely Benign | Likely Benign | 0.345 | Likely Benign | -1.02 | Neutral | 1.000 | Probably Damaging | 0.975 | Probably Damaging | 2.21 | Pathogenic | 0.01 | Affected | 0.1480 | 0.4097 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2846G>T | G949V 2D AIThe SynGAP1 missense variant G949V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | -7.364 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.328 | Likely Benign | -0.55 | Neutral | 0.992 | Probably Damaging | 0.834 | Possibly Damaging | 2.21 | Pathogenic | 0.01 | Affected | 0.1493 | 0.3165 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2848G>T | G950C 2D AIThe SynGAP1 missense variant G950C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑confidence predictors (five pathogenic vs. four benign) indicate a pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -9.589 | Likely Pathogenic | 0.092 | Likely Benign | Likely Benign | 0.395 | Likely Benign | -0.35 | Neutral | 0.983 | Probably Damaging | 0.750 | Possibly Damaging | 2.26 | Pathogenic | 0.01 | Affected | 0.1406 | 0.4439 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2849G>T | G950V 2D AIThe SynGAP1 missense variant G950V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that G950V is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -7.796 | In-Between | 0.093 | Likely Benign | Likely Benign | 0.428 | Likely Benign | -0.91 | Neutral | 0.411 | Benign | 0.239 | Benign | 2.26 | Pathogenic | 0.01 | Affected | 0.1392 | 0.3507 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.284A>T | H95L 2D AIThe SynGAP1 missense variant H95L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta results are not available for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.590542 | Binding | 0.335 | 0.875 | 0.625 | -1.967 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -2.31 | Neutral | 0.084 | Benign | 0.007 | Benign | 4.17 | Benign | 0.00 | Affected | 0.1017 | 0.5074 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2852A>T | H951L 2D AIThe SynGAP1 missense variant H951L is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.901477 | Binding | 0.415 | 0.925 | 0.750 | -4.822 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.192 | Likely Benign | -0.99 | Neutral | 0.022 | Benign | 0.018 | Benign | 5.47 | Benign | 0.43 | Tolerated | 0.2224 | 0.4526 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2854G>T | G952C 2D AIThe SynGAP1 missense variant G952C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.910621 | Binding | 0.341 | 0.926 | 0.750 | -8.599 | Likely Pathogenic | 0.088 | Likely Benign | Likely Benign | 0.272 | Likely Benign | -0.18 | Neutral | 0.371 | Benign | 0.169 | Benign | 3.20 | Benign | 0.01 | Affected | 0.1458 | 0.4439 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2855G>T | G952V 2D AIThe SynGAP1 missense variant G952V is listed in ClinVar (ID 2055482.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that G952V is most likely benign, and this conclusion does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.910621 | Binding | 0.341 | 0.926 | 0.750 | Uncertain | 1 | -7.074 | In-Between | 0.078 | Likely Benign | Likely Benign | 0.231 | Likely Benign | -0.33 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.20 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1538 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||
| c.2858C>T | P953L 2D AIThe SynGAP1 missense variant P953L is reported in gnomAD (variant ID 6‑33443410‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, representing a single dissenting opinion. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign effect, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.920633 | Binding | 0.403 | 0.926 | 0.750 | 6-33443410-C-T | 11 | 6.82e-6 | -6.069 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -1.34 | Neutral | 0.611 | Possibly Damaging | 0.096 | Benign | 2.76 | Benign | 0.25 | Tolerated | 3.77 | 5 | 0.2725 | 0.5778 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.2861C>T | P954L 2D AIThe SynGAP1 missense variant P954L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984159 | Disordered | 0.932268 | Binding | 0.465 | 0.926 | 0.750 | -5.607 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.43 | Neutral | 0.977 | Probably Damaging | 0.812 | Possibly Damaging | 2.78 | Benign | 0.55 | Tolerated | 0.2346 | 0.5867 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.286G>T | G96C 2D AIThe SynGAP1 missense variant G96C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus methods give a benign verdict: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” No result is available from Foldetta, so its folding‑stability assessment is not considered. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of any ClinVar classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.657645 | Disordered | 0.599491 | Binding | 0.335 | 0.871 | 0.625 | -5.140 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.112 | Likely Benign | -1.87 | Neutral | 0.981 | Probably Damaging | 0.216 | Benign | 4.12 | Benign | 0.00 | Affected | 0.1645 | 0.4142 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2870A>T | H957L 2D AIThe SynGAP1 missense variant H957L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only FATHMM predicts a pathogenic outcome. When predictions are grouped by consensus, the benign group contains eight tools, whereas the pathogenic group contains only FATHMM. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a Likely Benign result. Foldetta, a protein‑folding stability method, has no available output for this variant. Overall, the preponderance of evidence indicates that H957L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.968874 | Binding | 0.362 | 0.915 | 0.750 | -6.598 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.296 | Likely Benign | -0.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 2.44 | Pathogenic | 0.09 | Tolerated | 0.1525 | 0.5516 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2873A>T | H958L 2D AIThe SynGAP1 missense variant H958L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only two tools, SIFT and ESM1b, predict a pathogenic outcome. When the high‑accuracy consensus is considered, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a likely benign verdict, and AlphaMissense‑Optimized also reports benign. Foldetta predictions are unavailable. Overall, the majority of evidence supports a benign interpretation, and this is consistent with the lack of ClinVar annotation. Thus, the variant is most likely benign and does not contradict existing ClinVar data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.976011 | Binding | 0.371 | 0.913 | 0.750 | -8.422 | Likely Pathogenic | 0.087 | Likely Benign | Likely Benign | 0.181 | Likely Benign | -1.28 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.25 | Benign | 0.03 | Affected | 0.1723 | 0.5338 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2876A>T | H959L 2D AIThe SynGAP1 missense variant H959L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that H959L is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.980566 | Binding | 0.333 | 0.905 | 0.750 | -8.515 | Likely Pathogenic | 0.100 | Likely Benign | Likely Benign | 0.236 | Likely Benign | -1.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.14 | Benign | 0.09 | Tolerated | 0.1698 | 0.5538 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2879A>T | H960L 2D AIThe SynGAP1 missense variant H960L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic prediction comes from ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987911 | Disordered | 0.983385 | Binding | 0.380 | 0.901 | 0.750 | -8.386 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.130 | Likely Benign | -1.30 | Neutral | 0.174 | Benign | 0.043 | Benign | 4.17 | Benign | 0.33 | Tolerated | 0.1465 | 0.5595 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.287G>T | G96V 2D AIThe SynGAP1 missense variant G96V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.657645 | Disordered | 0.599491 | Binding | 0.335 | 0.871 | 0.625 | -4.266 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -1.43 | Neutral | 0.334 | Benign | 0.029 | Benign | 4.18 | Benign | 0.00 | Affected | 0.1575 | 0.3833 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2882A>T | H961L 2D AIThe SynGAP1 missense variant H961L is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT and ESM1b predict pathogenicity, but these are outliers among the consensus. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, SGM‑Consensus is benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.989835 | Disordered | 0.984562 | Binding | 0.323 | 0.893 | 0.750 | -8.547 | Likely Pathogenic | 0.109 | Likely Benign | Likely Benign | 0.155 | Likely Benign | -1.21 | Neutral | 0.144 | Benign | 0.078 | Benign | 4.13 | Benign | 0.01 | Affected | 0.1465 | 0.5344 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2885A>T | H962L 2D AIThe SynGAP1 missense variant H962L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for H962L, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.984483 | Binding | 0.369 | 0.886 | 0.750 | -8.478 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.151 | Likely Benign | -1.49 | Neutral | 0.494 | Possibly Damaging | 0.170 | Benign | 4.15 | Benign | 0.03 | Affected | 0.1738 | 0.5487 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2888A>T | H963L 2D AIThe SynGAP1 missense variant H963L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates benign, and the SGM‑Consensus likewise suggests a benign effect; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for H963L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.983973 | Binding | 0.325 | 0.886 | 0.750 | -8.110 | Likely Pathogenic | 0.109 | Likely Benign | Likely Benign | 0.172 | Likely Benign | -1.58 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.13 | Benign | 0.43 | Tolerated | 0.1626 | 0.5680 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2891A>T | H964L 2D AIThe SynGAP1 missense variant H964L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.568 | In-Between | 0.092 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.20 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.15 | Benign | 0.02 | Affected | 0.1409 | 0.5195 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2894A>T | H965L 2D AIThe SynGAP1 missense variant H965L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect. Benign predictors include REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool in the dataset returned a pathogenic prediction. Consensus predictors such as SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classify the variant as Likely Benign. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus is Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -6.708 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -1.66 | Neutral | 0.033 | Benign | 0.018 | Benign | 4.06 | Benign | 1.00 | Tolerated | 0.1606 | 0.5338 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2897A>T | H966L 2D AIThe SynGAP1 missense variant H966L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. No pathogenic predictions are present among the evaluated algorithms. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) reports likely benign. Foldetta results are not available for this variant. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -6.119 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -1.74 | Neutral | 0.174 | Benign | 0.062 | Benign | 4.05 | Benign | 0.35 | Tolerated | 0.1514 | 0.5316 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2899C>G | R967G 2D AIThe SynGAP1 missense variant R967G is reported in gnomAD (ID 6‑33443451‑C‑G) and has no ClinVar entry. All evaluated in silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Based on the unanimous benign predictions and absence of a ClinVar pathogenic claim, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.974374 | Disordered | 0.969686 | Binding | 0.340 | 0.888 | 0.750 | 6-33443451-C-G | 1 | 6.20e-7 | -2.214 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.279 | Likely Benign | -0.93 | Neutral | 0.005 | Benign | 0.007 | Benign | 4.17 | Benign | 0.18 | Tolerated | 4.32 | 2 | 0.3187 | 0.4070 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||
| c.28C>T | R10W 2D AIThe SynGAP1 missense variant R10W (ClinVar ID 1254556) is listed as “Uncertain” in ClinVar and is present in gnomAD (ID 6‑33420292‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is not in conflict with the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.513657 | Binding | 0.330 | 0.915 | 0.625 | Uncertain | 2 | 6-33420292-C-T | 2 | 1.30e-6 | -5.707 | Likely Benign | 0.503 | Ambiguous | Likely Benign | 0.236 | Likely Benign | -0.31 | Neutral | 0.964 | Probably Damaging | 0.190 | Benign | 4.10 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1461 | 0.4542 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||
| c.2900G>T | R967L 2D AIThe SynGAP1 missense variant R967L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443452‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for R967L, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.974374 | Disordered | 0.969686 | Binding | 0.340 | 0.888 | 0.750 | Uncertain | 1 | 6-33443452-G-T | 1 | 6.20e-7 | -3.496 | Likely Benign | 0.164 | Likely Benign | Likely Benign | 0.123 | Likely Benign | -0.99 | Neutral | 0.959 | Probably Damaging | 0.586 | Possibly Damaging | 4.15 | Benign | 0.75 | Tolerated | 4.32 | 2 | 0.1964 | 0.5093 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||
| c.2902G>T | G968C 2D AIThe SynGAP1 missense variant G968C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -8.441 | Likely Pathogenic | 0.117 | Likely Benign | Likely Benign | 0.170 | Likely Benign | -0.99 | Neutral | 0.992 | Probably Damaging | 0.820 | Possibly Damaging | 4.12 | Benign | 0.07 | Tolerated | 0.1294 | 0.4768 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2903G>T | G968V 2D AIThe SynGAP1 missense variant G968V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -6.152 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.239 | Likely Benign | -1.08 | Neutral | 0.918 | Possibly Damaging | 0.525 | Possibly Damaging | 4.19 | Benign | 0.11 | Tolerated | 0.1252 | 0.4036 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2906G>T | G969V 2D AIThe SynGAP1 missense variant G969V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.956572 | Binding | 0.405 | 0.898 | 0.750 | -5.946 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.92 | Neutral | 0.761 | Possibly Damaging | 0.239 | Benign | 4.18 | Benign | 0.01 | Affected | 0.1497 | 0.3847 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2912C>T | P971L 2D AIThe SynGAP1 missense variant P971L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.951523 | Binding | 0.545 | 0.905 | 0.625 | -4.892 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -1.57 | Neutral | 0.144 | Benign | 0.026 | Benign | 3.93 | Benign | 0.00 | Affected | 0.2046 | 0.5985 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2915C>T | P972L 2D AIThe SynGAP1 missense variant P972L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.954150 | Binding | 0.472 | 0.904 | 0.625 | -4.399 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.020 | Likely Benign | -1.73 | Neutral | 0.036 | Benign | 0.026 | Benign | 4.24 | Benign | 0.02 | Affected | 0.2107 | 0.5447 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2918G>T | G973V 2D AIThe SynGAP1 missense variant G973V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. Grouping by consensus, the benign‑predicting tools outnumber the single pathogenic prediction. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign classification. Foldetta results are unavailable, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.959498 | Binding | 0.390 | 0.897 | 0.625 | -4.688 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -0.76 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.18 | Benign | 0.01 | Affected | 0.1392 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2920G>T | D974Y 2D AIThe SynGAP1 missense variant D974Y is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the majority of computational evidence points to a benign impact, which is consistent with the absence of ClinVar pathogenic classification and gnomAD observations. Thus, the variant is most likely benign, and this assessment does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -4.290 | Likely Benign | 0.384 | Ambiguous | Likely Benign | 0.130 | Likely Benign | -1.85 | Neutral | 0.716 | Possibly Damaging | 0.284 | Benign | 4.13 | Benign | 0.01 | Affected | 0.1072 | 0.6627 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2921A>T | D974V 2D AIThe SynGAP1 missense variant D974V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective predictions strongly suggest that D974V is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -3.629 | Likely Benign | 0.351 | Ambiguous | Likely Benign | 0.211 | Likely Benign | -1.49 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.16 | Benign | 0.01 | Affected | 0.1449 | 0.7286 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2933C>T | P978L 2D AIThe SynGAP1 missense variant P978L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.975775 | Binding | 0.425 | 0.892 | 0.625 | -4.621 | Likely Benign | 0.386 | Ambiguous | Likely Benign | 0.092 | Likely Benign | -2.08 | Neutral | 0.818 | Possibly Damaging | 0.378 | Benign | 4.15 | Benign | 0.01 | Affected | 0.2326 | 0.6997 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2939A>T | H980L 2D AIThe SynGAP1 missense variant H980L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.974598 | Binding | 0.309 | 0.892 | 0.625 | -1.984 | Likely Benign | 0.293 | Likely Benign | Likely Benign | 0.187 | Likely Benign | -1.98 | Neutral | 0.625 | Possibly Damaging | 0.265 | Benign | 4.16 | Benign | 0.00 | Affected | 0.1488 | 0.5538 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2941G>T | G981C 2D AIThe SynGAP1 missense variant G981C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for G981C, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.970320 | Binding | 0.275 | 0.897 | 0.625 | -5.646 | Likely Benign | 0.593 | Likely Pathogenic | Likely Benign | 0.228 | Likely Benign | -1.81 | Neutral | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 3.72 | Benign | 0.00 | Affected | 0.1398 | 0.4593 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2942G>T | G981V 2D AIThe SynGAP1 missense variant G981V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for G981V, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.970320 | Binding | 0.275 | 0.897 | 0.625 | -3.873 | Likely Benign | 0.714 | Likely Pathogenic | Likely Benign | 0.156 | Likely Benign | -2.10 | Neutral | 0.997 | Probably Damaging | 0.958 | Probably Damaging | 3.75 | Benign | 0.00 | Affected | 0.1322 | 0.3861 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2944T>G | Y982D 2D AIThe SynGAP1 missense variant Y982D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which collectively suggest a likely benign outcome. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and Foldetta (combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the balance of evidence leans toward a benign interpretation, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.966717 | Binding | 0.272 | 0.895 | 0.625 | -5.021 | Likely Benign | 0.952 | Likely Pathogenic | Ambiguous | 0.194 | Likely Benign | -1.29 | Neutral | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 3.87 | Benign | 0.00 | Affected | 0.3959 | 0.0545 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2959G>T | D987Y 2D AIThe SynGAP1 missense variant D987Y is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as likely pathogenic (3 pathogenic vs 1 benign). AlphaMissense‑Optimized returns an uncertain result, and Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -6.208 | Likely Benign | 0.831 | Likely Pathogenic | Ambiguous | 0.274 | Likely Benign | -4.41 | Deleterious | 0.999 | Probably Damaging | 0.966 | Probably Damaging | 2.33 | Pathogenic | 0.01 | Affected | 0.0632 | 0.6818 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2960A>T | D987V 2D AIThe SynGAP1 missense variant D987V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that D987V is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -4.647 | Likely Benign | 0.857 | Likely Pathogenic | Ambiguous | 0.389 | Likely Benign | -4.01 | Deleterious | 0.992 | Probably Damaging | 0.913 | Probably Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.0907 | 0.7202 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2963T>A | L988H 2D AIThe SynGAP1 missense variant L988H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2). Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs four benign) lean toward pathogenicity. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.918781 | Binding | 0.360 | 0.913 | 0.750 | -4.779 | Likely Benign | 0.733 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -2.70 | Deleterious | 0.998 | Probably Damaging | 0.947 | Probably Damaging | 2.62 | Benign | 0.00 | Affected | 0.1179 | 0.1414 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2963T>C | L988P 2D AIThe SynGAP1 missense variant L988P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward pathogenicity. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.918781 | Binding | 0.360 | 0.913 | 0.750 | -3.848 | Likely Benign | 0.703 | Likely Pathogenic | Likely Benign | 0.216 | Likely Benign | -2.96 | Deleterious | 0.977 | Probably Damaging | 0.900 | Possibly Damaging | 2.62 | Benign | 0.00 | Affected | 0.3282 | 0.1698 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2972G>T | G991V 2D AIThe SynGAP1 missense variant G991V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.911393 | Binding | 0.286 | 0.920 | 0.750 | -4.129 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -1.61 | Neutral | 0.440 | Benign | 0.253 | Benign | 4.16 | Benign | 0.01 | Affected | 0.1148 | 0.3961 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2975T>A | V992D 2D AIThe SynGAP1 missense variant V992D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only SIFT predicts pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates that V992D is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.921728 | Binding | 0.331 | 0.917 | 0.750 | -3.976 | Likely Benign | 0.306 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -1.24 | Neutral | 0.302 | Benign | 0.158 | Benign | 4.21 | Benign | 0.05 | Affected | 0.1645 | 0.1056 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2975T>G | V992G 2D AIThe SynGAP1 missense variant V992G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). All available in silico predictors classify the variant as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity, so the benign group includes every listed predictor, while the pathogenic group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.754692 | Disordered | 0.921728 | Binding | 0.331 | 0.917 | 0.750 | -1.906 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -0.28 | Neutral | 0.056 | Benign | 0.086 | Benign | 4.21 | Benign | 0.16 | Tolerated | 0.2114 | 0.2612 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2978C>T | P993L 2D AIThe SynGAP1 missense variant P993L is reported in ClinVar as “Not listed” and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which contains no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.923979 | Binding | 0.319 | 0.908 | 0.750 | -3.581 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.028 | Likely Benign | -1.37 | Neutral | 0.224 | Benign | 0.138 | Benign | 4.14 | Benign | 0.01 | Affected | 0.2299 | 0.6697 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2984C>T | P995L 2D AIThe SynGAP1 missense variant P995L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta results are not available for this variant. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.935305 | Binding | 0.338 | 0.902 | 0.750 | -4.948 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -1.03 | Neutral | 0.411 | Benign | 0.096 | Benign | 4.16 | Benign | 0.00 | Affected | 0.2294 | 0.6324 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2987C>T | P996L 2D AIThe SynGAP1 missense variant P996L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect; there is no conflict with ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.775545 | Disordered | 0.942262 | Binding | 0.312 | 0.900 | 0.750 | -5.302 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -1.65 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.25 | Benign | 0.02 | Affected | 0.2057 | 0.6631 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.298T>G | Y100D 2D AIThe SynGAP1 missense variant Y100D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -1.269 | Likely Benign | 0.215 | Likely Benign | Likely Benign | 0.233 | Likely Benign | -1.01 | Neutral | 0.003 | Benign | 0.003 | Benign | 4.22 | Benign | 0.00 | Affected | 0.4465 | 0.0373 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2999T>A | I1000N 2D AIThe SynGAP1 missense variant I1000N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | -5.246 | Likely Benign | 0.677 | Likely Pathogenic | Likely Benign | 0.145 | Likely Benign | -0.82 | Neutral | 0.995 | Probably Damaging | 0.913 | Probably Damaging | 2.72 | Benign | 0.16 | Tolerated | 0.1001 | 0.0900 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3002T>A | L1001H 2D AIThe SynGAP1 missense variant L1001H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | -3.119 | Likely Benign | 0.258 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -1.02 | Neutral | 0.997 | Probably Damaging | 0.870 | Possibly Damaging | 2.64 | Benign | 0.00 | Affected | 0.1141 | 0.0958 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3002T>C | L1001P 2D AIThe SynGAP1 missense variant L1001P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence—including high‑accuracy tools—points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | Uncertain | 1 | -3.071 | Likely Benign | 0.209 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -1.02 | Neutral | 0.966 | Probably Damaging | 0.690 | Possibly Damaging | 2.65 | Benign | 0.00 | Affected | 4.32 | 4 | 0.3322 | 0.0929 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.3005A>T | H1002L 2D AIThe SynGAP1 H1002L missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and the absence of the variant in population databases, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.953758 | Binding | 0.285 | 0.900 | 0.500 | -6.448 | Likely Benign | 0.556 | Ambiguous | Likely Benign | 0.157 | Likely Benign | -3.12 | Deleterious | 0.801 | Possibly Damaging | 0.602 | Possibly Damaging | 2.79 | Benign | 0.13 | Tolerated | 0.1296 | 0.5088 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3011A>T | H1004L 2D AIThe SynGAP1 missense variant H1004L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two pathogenic vs two benign votes). Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the balance of evidence leans toward a benign interpretation, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.808535 | Disordered | 0.943707 | Binding | 0.271 | 0.901 | 0.750 | -5.179 | Likely Benign | 0.716 | Likely Pathogenic | Likely Benign | 0.220 | Likely Benign | -3.14 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.75 | Benign | 0.55 | Tolerated | 0.1223 | 0.6026 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3022G>T | D1008Y 2D AIThe SynGAP1 missense variant D1008Y is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default) predict a pathogenic impact; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a tie and thus unavailable, and Foldetta results are not provided. Overall, the balance of evidence favors a pathogenic classification, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.694846 | Disordered | 0.919416 | Binding | 0.280 | 0.899 | 0.625 | -5.371 | Likely Benign | 0.928 | Likely Pathogenic | Ambiguous | 0.237 | Likely Benign | -3.71 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.62 | Benign | 0.00 | Affected | 0.1043 | 0.6293 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||||
| c.3023A>T | D1008V 2D AIThe SynGAP1 D1008V variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs. two benign votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, more tools (five) predict pathogenicity than benign (three), and the high‑accuracy assessments do not overturn this trend. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.694846 | Disordered | 0.919416 | Binding | 0.280 | 0.899 | 0.625 | -4.828 | Likely Benign | 0.944 | Likely Pathogenic | Ambiguous | 0.242 | Likely Benign | -3.61 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.64 | Benign | 0.01 | Affected | 0.1447 | 0.6608 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.3029T>C | F1010S 2D AIThe SynGAP1 missense variant F1010S is listed in gnomAD (ID 6‑33443581‑T‑C) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as benign; Foldetta results are unavailable. Overall, the balance of evidence, including the high‑accuracy benign predictions and the consensus benign call, indicates that the variant is most likely benign. This conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.741537 | Disordered | 0.912572 | Binding | 0.286 | 0.881 | 0.625 | 6-33443581-T-C | -1.722 | Likely Benign | 0.744 | Likely Pathogenic | Likely Benign | 0.153 | Likely Benign | -1.97 | Neutral | 0.994 | Probably Damaging | 0.892 | Possibly Damaging | 2.51 | Benign | 0.01 | Affected | 3.77 | 5 | 0.4196 | 0.0775 | -2 | -3 | -3.6 | -60.10 | ||||||||||||||||||||||||||||||||||||
| c.302A>T | H101L 2D AIThe SynGAP1 missense variant H101L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for H101L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.688356 | Binding | 0.370 | 0.884 | 0.625 | -1.376 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.33 | Neutral | 0.824 | Possibly Damaging | 0.840 | Possibly Damaging | 4.19 | Benign | 0.00 | Affected | 0.0924 | 0.4960 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3031G>A | G1011R 2D AIThe SynGAP1 missense variant G1011R is reported in gnomAD (variant ID 6‑33443583‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign, reflecting the majority of benign calls. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect for G1011R, and this conclusion does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.898380 | Binding | 0.332 | 0.869 | 0.625 | 6-33443583-G-A | -4.650 | Likely Benign | 0.609 | Likely Pathogenic | Likely Benign | 0.118 | Likely Benign | -0.79 | Neutral | 0.642 | Possibly Damaging | 0.494 | Possibly Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1041 | 0.4415 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||
| c.3031G>C | G1011R 2D AIThe SynGAP1 missense variant G1011R is not reported in ClinVar and is absent from gnomAD, so no population frequency or clinical assertion data are available. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.898380 | Binding | 0.332 | 0.869 | 0.625 | -4.650 | Likely Benign | 0.609 | Likely Pathogenic | Likely Benign | 0.118 | Likely Benign | -0.79 | Neutral | 0.642 | Possibly Damaging | 0.494 | Possibly Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1041 | 0.4415 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3032G>T | G1011V 2D AIThe SynGAP1 missense variant G1011V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.898380 | Binding | 0.332 | 0.869 | 0.625 | -4.883 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.133 | Likely Benign | -1.21 | Neutral | 0.473 | Possibly Damaging | 0.192 | Benign | 2.71 | Benign | 0.01 | Affected | 0.1352 | 0.3434 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3035C>T | P1012L 2D AIThe SynGAP1 missense variant P1012L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which contains no pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.894674 | Binding | 0.319 | 0.866 | 0.625 | -4.363 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.014 | Likely Benign | -0.90 | Neutral | 0.224 | Benign | 0.131 | Benign | 2.76 | Benign | 0.12 | Tolerated | 0.2155 | 0.6388 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3038C>T | S1013F 2D AIThe SynGAP1 missense variant S1013F is catalogued in gnomAD (ID 6‑33443590‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen2_HumVar, ESM1b, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen2_HumDiv, and SIFT. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign; the SGM Consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive due to mixed signals, and Foldetta results are unavailable. Overall, the balance of evidence, including the benign call from AlphaMissense‑Optimized, points to a likely benign effect. This conclusion does not conflict with ClinVar, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.823549 | Disordered | 0.899570 | Binding | 0.308 | 0.846 | 0.625 | 6-33443590-C-T | 1 | 6.20e-7 | -5.370 | Likely Benign | 0.353 | Ambiguous | Likely Benign | 0.057 | Likely Benign | -2.54 | Deleterious | 0.453 | Possibly Damaging | 0.272 | Benign | 2.65 | Benign | 0.03 | Affected | 3.77 | 5 | 0.0922 | 0.5803 | -2 | -3 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||
| c.3040G>T | G1014C 2D AIThe SynGAP1 G1014C missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.914808 | Binding | 0.293 | 0.835 | 0.625 | -7.424 | In-Between | 0.151 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -2.49 | Neutral | 0.997 | Probably Damaging | 0.889 | Possibly Damaging | 2.68 | Benign | 0.06 | Tolerated | 0.1401 | 0.4126 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3041G>T | G1014V 2D AIThe SynGAP1 missense variant G1014V is listed in ClinVar (ID 809922.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.914808 | Binding | 0.293 | 0.835 | 0.625 | Uncertain | 1 | -4.612 | Likely Benign | 0.181 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -2.47 | Neutral | 0.818 | Possibly Damaging | 0.377 | Benign | 2.72 | Benign | 0.06 | Tolerated | 3.77 | 5 | 0.1359 | 0.3533 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||
| c.3046G>T | D1016Y 2D AIThe SynGAP1 missense variant D1016Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all indicate pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. AlphaMissense‑Optimized returns an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. High‑accuracy assessment therefore points to a Likely Pathogenic classification from SGM‑Consensus, with AlphaMissense‑Optimized inconclusive and Foldetta missing. Based on the preponderance of pathogenic predictions and the lack of contradictory evidence from ClinVar, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.801317 | Disordered | 0.944705 | Binding | 0.323 | 0.811 | 0.625 | -4.432 | Likely Benign | 0.832 | Likely Pathogenic | Ambiguous | 0.350 | Likely Benign | -3.86 | Deleterious | 0.998 | Probably Damaging | 0.947 | Probably Damaging | 2.43 | Pathogenic | 0.00 | Affected | 0.1111 | 0.6531 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.3047A>T | D1016V 2D AIThe SynGAP1 D1016V missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments give an uncertain result from AlphaMissense‑Optimized, a Likely Pathogenic verdict from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and no available data from Foldetta. Overall, the majority of evidence points toward a deleterious effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.801317 | Disordered | 0.944705 | Binding | 0.323 | 0.811 | 0.625 | -3.208 | Likely Benign | 0.800 | Likely Pathogenic | Ambiguous | 0.362 | Likely Benign | -3.80 | Deleterious | 0.977 | Probably Damaging | 0.856 | Possibly Damaging | 2.45 | Pathogenic | 0.00 | Affected | 0.1424 | 0.7116 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3055C>T | R1019C 2D AIThe SynGAP1 missense variant R1019C is listed in ClinVar with an “Uncertain” status (ClinVar ID 1676922.0) and is present in gnomAD (ID 6‑33443607‑C‑T). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact; ESM1b remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote) remains pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.856457 | Disordered | 0.966400 | Binding | 0.315 | 0.794 | 0.500 | Conflicting | 2 | 6-33443607-C-T | 10 | 6.19e-6 | -7.386 | In-Between | 0.646 | Likely Pathogenic | Likely Benign | 0.168 | Likely Benign | -4.00 | Deleterious | 0.999 | Probably Damaging | 0.880 | Possibly Damaging | 2.36 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3016 | 0.3664 | -4 | -3 | 7.0 | -53.05 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||
| c.3056G>T | R1019L 2D AIThe SynGAP1 missense variant R1019L is listed in ClinVar with an “Uncertain” status (ClinVar ID 3364537.0) and is present in gnomAD (gnomAD ID 6‑33443608‑G‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote) remains pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a pathogenic impact, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.856457 | Disordered | 0.966400 | Binding | 0.315 | 0.794 | 0.500 | Uncertain | 1 | 6-33443608-G-T | 2 | 1.24e-6 | -5.194 | Likely Benign | 0.752 | Likely Pathogenic | Likely Benign | 0.110 | Likely Benign | -3.57 | Deleterious | 0.800 | Possibly Damaging | 0.573 | Possibly Damaging | 2.40 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1850 | 0.4886 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||
| c.3065T>A | L1022H 2D AIThe SynGAP1 missense variant L1022H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie (2 pathogenic, 2 benign) and is therefore inconclusive. Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the majority of conventional predictors lean toward pathogenicity, whereas the single high‑accuracy tool predicts benign and the consensus is unresolved. Thus, the variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.859585 | Disordered | 0.986981 | Binding | 0.339 | 0.752 | 0.500 | -2.473 | Likely Benign | 0.589 | Likely Pathogenic | Likely Benign | 0.140 | Likely Benign | -2.21 | Neutral | 0.999 | Probably Damaging | 0.944 | Probably Damaging | 2.49 | Pathogenic | 0.00 | Affected | 0.1165 | 0.1313 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3065T>C | L1022P 2D AIThe SynGAP1 missense variant L1022P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic, two benign), and Foldetta data are unavailable. Overall, the balance of evidence favors a pathogenic interpretation, and this assessment does not conflict with ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.859585 | Disordered | 0.986981 | Binding | 0.339 | 0.752 | 0.500 | -2.532 | Likely Benign | 0.643 | Likely Pathogenic | Likely Benign | 0.177 | Likely Benign | -2.22 | Neutral | 0.995 | Probably Damaging | 0.925 | Probably Damaging | 2.49 | Pathogenic | 0.01 | Affected | 0.3332 | 0.1589 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3071T>A | L1024H 2D AIThe SynGAP1 missense variant L1024H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized returns an Uncertain result, SGM‑Consensus indicates Likely Pathogenic, and Foldetta data are unavailable. Overall, the majority of evidence points toward a deleterious effect, suggesting the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -3.271 | Likely Benign | 0.868 | Likely Pathogenic | Ambiguous | 0.123 | Likely Benign | -3.09 | Deleterious | 1.000 | Probably Damaging | 0.981 | Probably Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.1198 | 0.1483 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3071T>C | L1024P 2D AIThe SynGAP1 missense variant L1024P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of standard prediction tools lean toward pathogenicity, while high‑accuracy methods are inconclusive. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -4.385 | Likely Benign | 0.730 | Likely Pathogenic | Likely Benign | 0.149 | Likely Benign | -2.42 | Neutral | 0.999 | Probably Damaging | 0.974 | Probably Damaging | 2.43 | Pathogenic | 0.03 | Affected | 0.3187 | 0.2033 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3076G>T | D1026Y 2D AIThe SynGAP1 missense variant D1026Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default all classify the variant as damaging. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -6.999 | Likely Benign | 0.892 | Likely Pathogenic | Ambiguous | 0.200 | Likely Benign | -3.08 | Deleterious | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 2.47 | Pathogenic | 0.00 | Affected | 0.0676 | 0.4936 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.3077A>T | D1026V 2D AIThe SynGAP1 missense variant D1026V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Tools that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and therefore also indicates a likely pathogenic outcome. AlphaMissense‑Optimized is uncertain, and no Foldetta stability result is available, so it does not contribute evidence. Overall, the majority of reliable predictors classify the variant as pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -5.871 | Likely Benign | 0.900 | Likely Pathogenic | Ambiguous | 0.144 | Likely Benign | -3.13 | Deleterious | 0.004 | Benign | 0.004 | Benign | 2.48 | Pathogenic | 0.00 | Affected | 0.0957 | 0.5236 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.307G>T | G103C 2D AIThe SynGAP1 missense variant G103C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect for G103C, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -5.681 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -1.12 | Neutral | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 4.17 | Benign | 0.00 | Affected | 0.1494 | 0.3767 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3080A>T | N1027I 2D AIThe SynGAP1 missense variant N1027I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -5.847 | Likely Benign | 0.751 | Likely Pathogenic | Likely Benign | 0.065 | Likely Benign | -2.36 | Neutral | 0.970 | Probably Damaging | 0.726 | Possibly Damaging | 2.71 | Benign | 0.02 | Affected | 0.0677 | 0.5724 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3083T>C | L1028P 2D AIThe SynGAP1 missense variant L1028P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Taken together, the majority of evidence points to a benign impact for this variant, and there is no ClinVar annotation to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -3.080 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 0.215 | Likely Benign | -0.42 | Neutral | 0.004 | Benign | 0.010 | Benign | 2.70 | Benign | 0.25 | Tolerated | 0.2846 | 0.1763 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3089A>T | H1030L 2D AIThe SynGAP1 missense variant H1030L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that H1030L is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -3.513 | Likely Benign | 0.270 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -2.35 | Neutral | 0.224 | Benign | 0.120 | Benign | 2.76 | Benign | 0.02 | Affected | 0.0968 | 0.5710 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.308G>T | G103V 2D AIThe SynGAP1 missense variant G103V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.584 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.126 | Likely Benign | -0.96 | Neutral | 0.820 | Possibly Damaging | 0.376 | Benign | 4.22 | Benign | 0.00 | Affected | 0.1420 | 0.3404 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3095T>C | L1032P 2D AIThe SynGAP1 missense variant L1032P has no ClinVar entry and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and the SGM‑Consensus score all classify the change as benign or likely benign. AlphaMissense‑Optimized also predicts a benign outcome, whereas SIFT uniquely flags the variant as pathogenic. The AlphaMissense‑Default assessment is uncertain, and no Foldetta stability data are available. High‑accuracy analyses reinforce the benign consensus: AlphaMissense‑Optimized reports a benign effect, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta results are missing, so they do not influence the interpretation. Overall, the majority of computational evidence supports a benign classification, which is consistent with the absence of a ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this conclusion does not contradict ClinVar status, as no ClinVar pathogenic claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -2.560 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.142 | Likely Benign | -0.71 | Neutral | 0.012 | Benign | 0.017 | Benign | 2.64 | Benign | 0.05 | Affected | 0.3024 | 0.2330 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3098C>A | S1033Y 2D AIThe SynGAP1 missense variant S1033Y is reported in gnomAD (ID 6‑33443650‑C‑A) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | 6-33443650-C-A | 1 | 6.19e-7 | -4.857 | Likely Benign | 0.564 | Ambiguous | Likely Benign | 0.034 | Likely Benign | -1.01 | Neutral | 0.021 | Benign | 0.008 | Benign | 2.69 | Benign | 0.04 | Affected | 3.77 | 5 | 0.0708 | 0.5262 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||
| c.3101C>T | P1034L 2D AIThe SynGAP1 missense variant P1034L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default is uncertain. The high‑accuracy AlphaMissense‑Optimized model classifies the variant as benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that P1034L is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -4.204 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.067 | Likely Benign | -3.24 | Deleterious | 0.001 | Benign | 0.005 | Benign | 2.53 | Benign | 0.01 | Affected | 0.2267 | 0.6937 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3104C>T | P1035L 2D AIThe SynGAP1 missense variant P1035L is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -5.694 | Likely Benign | 0.675 | Likely Pathogenic | Likely Benign | 0.071 | Likely Benign | -2.11 | Neutral | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 2.70 | Benign | 0.21 | Tolerated | 0.2444 | 0.7354 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3110T>A | I1037N 2D AIThe SynGAP1 missense variant I1037N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a Likely Benign classification, while AlphaMissense‑Optimized remains uncertain. Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -3.955 | Likely Benign | 0.832 | Likely Pathogenic | Ambiguous | 0.131 | Likely Benign | 1.56 | Neutral | 0.666 | Possibly Damaging | 0.211 | Benign | 2.81 | Benign | 0.34 | Tolerated | 0.1022 | 0.1140 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3116T>A | I1039N 2D AIThe SynGAP1 missense variant I1039N has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -3.469 | Likely Benign | 0.951 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.24 | Neutral | 0.011 | Benign | 0.010 | Benign | 2.67 | Benign | 0.02 | Affected | 0.1151 | 0.1311 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3118G>T | G1040C 2D AIThe SynGAP1 missense variant G1040C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all predict pathogenicity, while ESM1b and AlphaMissense‑Optimized predict a benign outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized reports a benign prediction, whereas the SGM‑Consensus remains Likely Pathogenic; the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence from multiple in silico tools indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -6.272 | Likely Benign | 0.620 | Likely Pathogenic | Likely Benign | 0.744 | Likely Pathogenic | -3.04 | Deleterious | 0.999 | Probably Damaging | 0.917 | Probably Damaging | -0.74 | Pathogenic | 0.00 | Affected | 0.1155 | 0.4556 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3119G>T | G1040V 2D AIThe SynGAP1 missense variant G1040V is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443671‑G‑T). Prediction tools that agree on a benign effect are ESM1b and AlphaMissense‑Optimized; those that predict a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta results are unavailable. Overall, the majority of predictions indicate a pathogenic impact, and this is not in conflict with the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | Uncertain | 1 | 6-33443671-G-T | 4 | 2.48e-6 | -3.453 | Likely Benign | 0.645 | Likely Pathogenic | Likely Benign | 0.774 | Likely Pathogenic | -2.89 | Deleterious | 0.827 | Possibly Damaging | 0.456 | Possibly Damaging | -0.74 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1239 | 0.4213 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||
| c.311G>T | R104L 2D AIThe SynGAP1 missense variant R104L is listed in ClinVar (ID 2746314.0) as Benign and is present in gnomAD (6‑33432176‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is benign, and the SGM‑Consensus (majority vote) is also benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the ClinVar benign classification and does not contradict the existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | Benign | 1 | 6-33432176-G-T | 1 | 6.20e-7 | -3.563 | Likely Benign | 0.578 | Likely Pathogenic | Likely Benign | 0.170 | Likely Benign | -1.38 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.05 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1681 | 0.4894 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||
| c.3122C>T | P1041L 2D AIThe SynGAP1 missense variant P1041L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also resolves to benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact. This conclusion does not contradict ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -4.901 | Likely Benign | 0.399 | Ambiguous | Likely Benign | 0.403 | Likely Benign | -3.14 | Deleterious | 0.905 | Possibly Damaging | 0.375 | Benign | 5.46 | Benign | 1.00 | Tolerated | 0.2357 | 0.6664 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3127A>T | R1043W 2D AIThe SynGAP1 missense variant R1043W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction; Foldetta results are unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -6.382 | Likely Benign | 0.523 | Ambiguous | Likely Benign | 0.444 | Likely Benign | -3.43 | Deleterious | 0.971 | Probably Damaging | 0.729 | Possibly Damaging | 5.38 | Benign | 0.00 | Affected | 0.1403 | 0.3696 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3131C>T | P1044L 2D AIThe SynGAP1 missense variant P1044L is not represented in ClinVar (no ClinVar ID) and is absent from gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic or likely pathogenic outcome. High‑accuracy assessments reinforce this benign prediction: AlphaMissense‑Optimized indicates benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.952126 | Binding | 0.331 | 0.855 | 0.750 | -4.327 | Likely Benign | 0.144 | Likely Benign | Likely Benign | 0.418 | Likely Benign | -1.64 | Neutral | 0.411 | Benign | 0.187 | Benign | 5.43 | Benign | 0.15 | Tolerated | 0.2264 | 0.6586 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3137C>T | P1046L 2D AIThe SynGAP1 missense variant P1046L is reported in gnomAD (ID 6‑33443689‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, SIFT and FATHMM predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors and the consensus analysis points to a benign classification. This conclusion is consistent with the absence of a ClinVar pathogenic report, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | 6-33443689-C-T | 1 | 6.20e-7 | -5.022 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -2.11 | Neutral | 0.001 | Benign | 0.005 | Benign | 2.35 | Pathogenic | 0.05 | Affected | 3.77 | 5 | 0.2036 | 0.6442 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.3140C>T | S1047L 2D AIThe SynGAP1 missense variant S1047L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443692‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta stability analysis is not available for this variant. Overall, the preponderance of computational evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.963420 | Disordered | 0.933764 | Binding | 0.409 | 0.909 | 0.750 | Uncertain | 1 | 6-33443692-C-T | 1 | 6.20e-7 | -4.062 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.032 | Likely Benign | -0.63 | Neutral | 0.144 | Benign | 0.058 | Benign | 2.60 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1575 | 0.5555 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||
| c.3142G>A | G1048R 2D AIThe SynGAP1 missense variant G1048R is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split: pathogenic calls come from REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar, whereas benign calls are made by PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a benign majority, and AlphaMissense‑Optimized itself predicts benign. The SGM‑Consensus, which aggregates these four predictors, reports a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar assertion; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | -4.305 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.503 | Likely Pathogenic | -0.54 | Neutral | 0.919 | Possibly Damaging | 0.728 | Possibly Damaging | 2.54 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0956 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3142G>C | G1048R 2D AIThe SynGAP1 missense variant G1048R is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM‑Consensus also indicates a likely benign outcome; the Foldetta protein‑folding stability assessment is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | Uncertain | 1 | -4.305 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.503 | Likely Pathogenic | -0.54 | Neutral | 0.919 | Possibly Damaging | 0.728 | Possibly Damaging | 2.54 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0956 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.3143G>T | G1048V 2D AIThe SynGAP1 missense variant G1048V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | -6.108 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.520 | Likely Pathogenic | -0.59 | Neutral | 0.958 | Probably Damaging | 0.787 | Possibly Damaging | 2.54 | Benign | 0.11 | Tolerated | 0.1312 | 0.3688 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3146C>T | P1049L 2D AIThe SynGAP1 missense variant P1049L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.917915 | Binding | 0.428 | 0.920 | 0.750 | -4.819 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -2.37 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.71 | Benign | 0.02 | Affected | 0.2267 | 0.5838 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3148G>A | G1050R 2D AIThe SynGAP1 missense variant G1050R is catalogued in gnomAD (ID 6‑33443700‑G‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign verdict, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | 6-33443700-G-A | 3 | 1.86e-6 | -6.637 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.045 | Likely Benign | -0.68 | Neutral | 0.009 | Benign | 0.008 | Benign | 2.48 | Pathogenic | 0.06 | Tolerated | 3.77 | 5 | 0.0939 | 0.4532 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.3148G>C | G1050R 2D AIThe SynGAP1 missense variant G1050R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority vote. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact; this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | -6.637 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.045 | Likely Benign | -0.68 | Neutral | 0.009 | Benign | 0.008 | Benign | 2.48 | Pathogenic | 0.06 | Tolerated | 3.77 | 5 | 0.0939 | 0.4532 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3149G>T | G1050V 2D AIThe SynGAP1 missense variant G1050V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no classification for G1050V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | -6.450 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.83 | Neutral | 0.126 | Benign | 0.096 | Benign | 2.49 | Pathogenic | 0.13 | Tolerated | 0.1260 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3151G>T | G1051C 2D AIThe SynGAP1 missense variant G1051C is listed in ClinVar as Pathogenic and is not reported in gnomAD. Functional prediction tools show a split assessment: benign calls come from REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls come from polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. High‑accuracy methods give a benign result from AlphaMissense‑Optimized; the SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is tied (2 benign vs. 2 pathogenic) and therefore inconclusive, and Foldetta’s stability prediction is unavailable. Overall, the majority of predictions lean toward a benign effect, which contradicts the ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.900141 | Binding | 0.358 | 0.936 | 0.875 | Likely Pathogenic | 1 | -9.050 | Likely Pathogenic | 0.122 | Likely Benign | Likely Benign | 0.497 | Likely Benign | -0.90 | Neutral | 0.971 | Probably Damaging | 0.750 | Possibly Damaging | -0.74 | Pathogenic | 0.10 | Tolerated | 3.77 | 5 | 0.1322 | 0.4612 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||
| c.3154G>A | G1052R 2D AISynGAP1 missense variant G1052R is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores the variant as benign, and the SGM consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar uncertain status but provides additional support toward a likely benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | Uncertain | 1 | -9.050 | Likely Pathogenic | 0.383 | Ambiguous | Likely Benign | 0.497 | Likely Benign | -0.41 | Neutral | 0.990 | Probably Damaging | 0.798 | Possibly Damaging | 3.90 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0976 | 0.4142 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||
| c.3154G>C | G1052R 2D AIThe SynGAP1 missense variant G1052R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and ESM1b predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | -9.050 | Likely Pathogenic | 0.383 | Ambiguous | Likely Benign | 0.497 | Likely Benign | -0.41 | Neutral | 0.990 | Probably Damaging | 0.798 | Possibly Damaging | 3.90 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0976 | 0.4142 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3155G>T | G1052V 2D AIThe SynGAP1 missense variant G1052V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign, while the high‑accuracy AlphaMissense‑Optimized score is benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign. In contrast, polyPhen‑2 HumDiv and HumVar both predict pathogenic, and ESM1b remains uncertain. No Foldetta stability assessment is available, so it does not influence the overall interpretation. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | -7.717 | In-Between | 0.094 | Likely Benign | Likely Benign | 0.452 | Likely Benign | -0.12 | Neutral | 0.901 | Possibly Damaging | 0.619 | Possibly Damaging | 3.90 | Benign | 0.19 | Tolerated | 0.1329 | 0.3499 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3160G>T | G1054C 2D AIThe SynGAP1 missense variant G1054C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | -9.548 | Likely Pathogenic | 0.118 | Likely Benign | Likely Benign | 0.297 | Likely Benign | -0.59 | Neutral | 0.999 | Probably Damaging | 0.907 | Possibly Damaging | 4.00 | Benign | 0.10 | Tolerated | 0.1413 | 0.4427 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3161G>T | G1054V 2D AIThe SynGAP1 missense variant G1054V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also reports likely benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign impact, and this is consistent with the lack of ClinVar evidence; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.878015 | Binding | 0.389 | 0.936 | 0.875 | -6.994 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.171 | Likely Benign | -0.22 | Neutral | 0.818 | Possibly Damaging | 0.221 | Benign | 4.01 | Benign | 0.18 | Tolerated | 0.1578 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3164G>T | G1055V 2D AIThe SynGAP1 missense variant G1055V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while ESM1b remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.982235 | Disordered | 0.872113 | Binding | 0.379 | 0.935 | 0.875 | -7.434 | In-Between | 0.114 | Likely Benign | Likely Benign | 0.399 | Likely Benign | 0.26 | Neutral | 0.818 | Possibly Damaging | 0.222 | Benign | 3.28 | Benign | 0.17 | Tolerated | 0.1399 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3166G>T | G1056C 2D AIThe SynGAP1 missense variant G1056C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign impact. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -9.974 | Likely Pathogenic | 0.122 | Likely Benign | Likely Benign | 0.432 | Likely Benign | -0.70 | Neutral | 0.994 | Probably Damaging | 0.777 | Possibly Damaging | 1.83 | Pathogenic | 0.06 | Tolerated | 0.1414 | 0.4439 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.3167G>T | G1056V 2D AIThe SynGAP1 missense variant G1056V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence (7 benign vs 2 pathogenic) supports a benign classification. This consensus does not contradict ClinVar status, which has no entry for this variant. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -8.130 | Likely Pathogenic | 0.097 | Likely Benign | Likely Benign | 0.448 | Likely Benign | -0.24 | Neutral | 0.292 | Benign | 0.110 | Benign | 1.83 | Pathogenic | 0.08 | Tolerated | 0.1488 | 0.3507 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.316A>G | R106G 2D AIThe SynGAP1 missense variant R106G is listed in gnomAD (ID 6‑33432181‑A‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Those that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar classification to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | 6-33432181-A-G | 1 | 6.20e-7 | -2.617 | Likely Benign | 0.835 | Likely Pathogenic | Ambiguous | 0.161 | Likely Benign | -2.21 | Neutral | 0.421 | Benign | 0.050 | Benign | 3.65 | Benign | 0.00 | Affected | 4.05 | 3 | 0.3680 | 0.3980 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||
| c.316A>T | R106W 2D AIThe SynGAP1 missense variant R106W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, more tools predict pathogenicity (5) than benign (3), and no ClinVar evidence contradicts this assessment. Thus, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | -5.350 | Likely Benign | 0.875 | Likely Pathogenic | Ambiguous | 0.240 | Likely Benign | -3.31 | Deleterious | 0.983 | Probably Damaging | 0.624 | Possibly Damaging | 3.62 | Benign | 0.00 | Affected | 0.1369 | 0.3995 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3172G>T | G1058C 2D AIThe SynGAP1 missense variant G1058C is reported in gnomAD (ID 6‑33443724‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and ESM1b. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of computational evidence points to a benign effect, and this is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.980739 | Disordered | 0.885724 | Binding | 0.407 | 0.929 | 0.875 | 6-33443724-G-T | 1 | 6.21e-7 | -9.384 | Likely Pathogenic | 0.132 | Likely Benign | Likely Benign | 0.264 | Likely Benign | -0.19 | Neutral | 0.600 | Possibly Damaging | 0.433 | Benign | 5.19 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1456 | 0.4627 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.3176G>T | G1059V 2D AIThe SynGAP1 missense variant G1059V is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score benign, while the majority‑vote SGM‑Consensus also classifies it as likely benign. Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy tools corroborate the benign prediction: AlphaMissense‑Optimized is benign and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely benign; Foldetta results are not available. Overall, the preponderance of evidence supports a benign classification for G1059V, and this assessment does not conflict with ClinVar, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.898939 | Binding | 0.399 | 0.926 | 0.875 | -7.242 | In-Between | 0.106 | Likely Benign | Likely Benign | 0.478 | Likely Benign | -0.82 | Neutral | 0.259 | Benign | 0.066 | Benign | 2.54 | Benign | 0.00 | Affected | 0.1462 | 0.3494 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3178G>T | G1060C 2D AIThe SynGAP1 missense variant G1060C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for G1060C, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | -9.630 | Likely Pathogenic | 0.116 | Likely Benign | Likely Benign | 0.363 | Likely Benign | -0.60 | Neutral | 0.999 | Probably Damaging | 0.917 | Probably Damaging | 2.63 | Benign | 0.12 | Tolerated | 0.1340 | 0.4227 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3179G>T | G1060V 2D AIThe SynGAP1 missense variant G1060V is listed in ClinVar as benign (ClinVar ID 1345112.0) and is observed in gnomAD (6‑33443731‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic effect. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as likely benign, and AlphaMissense‑Optimized also reports a benign outcome. No Foldetta stability assessment is available for this variant. Overall, the majority of evidence points to a benign impact, aligning with the ClinVar designation and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979242 | Disordered | 0.913048 | Binding | 0.407 | 0.928 | 0.875 | Benign | 1 | 6-33443731-G-T | 1 | 6.22e-7 | -6.966 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.369 | Likely Benign | -0.73 | Neutral | 0.986 | Probably Damaging | 0.728 | Possibly Damaging | 2.63 | Benign | 0.33 | Tolerated | 4.32 | 2 | 0.1453 | 0.3494 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||
| c.3181G>T | G1061C 2D AIThe SynGAP1 missense variant G1061C is listed in ClinVar (ID 536997.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33443733‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence (six benign vs. four pathogenic predictions) and the two high‑accuracy tools support a benign classification. This conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | Conflicting | 2 | 6-33443733-G-T | 6 | 3.73e-6 | -9.511 | Likely Pathogenic | 0.119 | Likely Benign | Likely Benign | 0.409 | Likely Benign | -1.46 | Neutral | 0.938 | Possibly Damaging | 0.665 | Possibly Damaging | 3.97 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1283 | 0.4227 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||
| c.3182G>T | G1061V 2D AIThe SynGAP1 missense variant G1061V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence indicates that G1061V is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.926729 | Binding | 0.394 | 0.923 | 0.875 | -6.709 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.307 | Likely Benign | -1.41 | Neutral | 0.224 | Benign | 0.066 | Benign | 3.98 | Benign | 0.00 | Affected | 0.1431 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3185G>T | G1062V 2D AIThe SynGAP1 missense variant G1062V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.936972 | Binding | 0.368 | 0.917 | 0.875 | -6.598 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.377 | Likely Benign | -0.78 | Neutral | 0.259 | Benign | 0.066 | Benign | 4.12 | Benign | 0.01 | Affected | 0.1441 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3187G>T | G1063C 2D AIThe SynGAP1 missense variant G1063C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which itself is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975134 | Disordered | 0.945784 | Binding | 0.394 | 0.913 | 0.875 | -8.315 | Likely Pathogenic | 0.106 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -1.07 | Neutral | 0.938 | Possibly Damaging | 0.477 | Possibly Damaging | 4.19 | Benign | 0.01 | Affected | 0.1440 | 0.4639 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3188G>T | G1063V 2D AIThe SynGAP1 missense variant G1063V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority of the high‑accuracy tools) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools indicates that G1063V is most likely benign, with no ClinVar status to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.975134 | Disordered | 0.945784 | Binding | 0.394 | 0.913 | 0.875 | -6.228 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.82 | Neutral | 0.004 | Benign | 0.002 | Benign | 4.29 | Benign | 0.03 | Affected | 0.1434 | 0.3707 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3194C>T | P1065L 2D AIThe SynGAP1 missense variant P1065L is listed in ClinVar as Benign and is present in gnomAD (ID 6‑33443746‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no reported result for this variant. Overall, the balance of evidence (5 benign vs. 4 pathogenic predictions) and the high‑accuracy benign call support a benign classification, aligning with the ClinVar status and indicating no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | Likely Benign | 1 | 6-33443746-C-T | 14 | 8.71e-6 | -5.085 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -2.94 | Deleterious | 0.950 | Possibly Damaging | 0.419 | Benign | 2.01 | Pathogenic | 0.00 | Affected | 4.32 | 2 | 0.2286 | 0.6922 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||
| c.3197C>T | P1066L 2D AIThe SynGAP1 missense variant P1066L is listed in ClinVar as a benign variant (ClinVar ID 951518.0) and is present in gnomAD (ID 6‑33443749‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, which is consistent with the ClinVar classification and does not contradict the reported status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.967676 | Disordered | 0.968838 | Binding | 0.403 | 0.913 | 0.875 | Likely Benign | 1 | 6-33443749-C-T | 14 | 8.71e-6 | -5.478 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -3.68 | Deleterious | 0.996 | Probably Damaging | 0.903 | Possibly Damaging | 2.72 | Benign | 0.00 | Affected | 4.32 | 2 | 0.2269 | 0.6780 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||
| c.319A>T | R107W 2D AIThe SynGAP1 missense variant R107W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two pathogenic vs two benign), and Foldetta results are unavailable. Overall, the majority of evidence (six pathogenic vs three benign) points to a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.663448 | Binding | 0.331 | 0.863 | 0.875 | -4.963 | Likely Benign | 0.965 | Likely Pathogenic | Likely Pathogenic | 0.328 | Likely Benign | -2.99 | Deleterious | 0.983 | Probably Damaging | 0.624 | Possibly Damaging | 2.95 | Benign | 0.00 | Affected | 0.1146 | 0.4228 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.31G>A | G11R 2D AIThe SynGAP1 missense variant G11R is catalogued in gnomAD (ID 6‑33420295‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | 6-33420295-G-A | -3.418 | Likely Benign | 0.428 | Ambiguous | Likely Benign | 0.102 | Likely Benign | -0.47 | Neutral | 0.498 | Possibly Damaging | 0.026 | Benign | 3.92 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1022 | 0.4596 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||
| c.31G>C | G11R 2D AIThe SynGAP1 missense variant G11R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | -3.418 | Likely Benign | 0.428 | Ambiguous | Likely Benign | 0.109 | Likely Benign | -0.47 | Neutral | 0.498 | Possibly Damaging | 0.026 | Benign | 3.92 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1022 | 0.4596 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3200C>T | P1067L 2D AIThe SynGAP1 missense variant P1067L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of predictions and the consensus analysis indicate a benign impact. This conclusion is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.966441 | Disordered | 0.975099 | Binding | 0.459 | 0.907 | 0.875 | -4.461 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.157 | Likely Benign | -3.01 | Deleterious | 0.951 | Possibly Damaging | 0.619 | Possibly Damaging | 2.76 | Benign | 0.01 | Affected | 0.2047 | 0.6198 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3208A>T | R1070W 2D AIThe SynGAP1 missense variant R1070W is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant as benign, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates it is likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this residue. Overall, the majority of tools and the SGM‑Consensus support a pathogenic effect, so the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.930790 | Disordered | 0.982693 | Binding | 0.297 | 0.906 | 0.875 | -8.063 | Likely Pathogenic | 0.743 | Likely Pathogenic | Likely Benign | 0.139 | Likely Benign | -3.42 | Deleterious | 0.999 | Probably Damaging | 0.956 | Probably Damaging | 3.72 | Benign | 0.00 | Affected | 0.1195 | 0.4293 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3211G>T | G1071C 2D AIThe SynGAP1 missense variant G1071C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence—including the consensus and high‑accuracy predictions—supports a benign classification, and this is consistent with the lack of ClinVar annotation. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983740 | Binding | 0.313 | 0.905 | 0.875 | -5.364 | Likely Benign | 0.305 | Likely Benign | Likely Benign | 0.182 | Likely Benign | -2.16 | Neutral | 0.997 | Probably Damaging | 0.889 | Possibly Damaging | 4.01 | Benign | 0.00 | Affected | 0.1323 | 0.4227 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3233T>G | V1078G 2D AIThe SynGAP1 missense variant V1078G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign (3 benign vs 1 pathogenic). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence indicates a benign impact. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.986989 | Binding | 0.294 | 0.898 | 0.750 | -3.270 | Likely Benign | 0.699 | Likely Pathogenic | Likely Benign | 0.168 | Likely Benign | -0.54 | Neutral | 0.157 | Benign | 0.292 | Benign | 3.85 | Benign | 0.00 | Affected | 0.2041 | 0.2693 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3248A>T | K1083I 2D AIThe SynGAP1 missense variant K1083I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | -3.207 | Likely Benign | 0.836 | Likely Pathogenic | Ambiguous | 0.239 | Likely Benign | -1.65 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 4.02 | Benign | 0.30 | Tolerated | 0.1394 | 0.3978 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||||||||||||
| c.3251C>T | P1084L 2D AIThe SynGAP1 missense variant P1084L is reported in gnomAD (ID 6‑33443803‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | 6-33443803-C-T | 1 | 6.31e-7 | -4.547 | Likely Benign | 0.175 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -3.33 | Deleterious | 0.649 | Possibly Damaging | 0.157 | Benign | 4.00 | Benign | 0.01 | Affected | 3.77 | 5 | 0.2218 | 0.6470 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.3253C>G | R1085G 2D AIThe SynGAP1 missense variant R1085G is reported in gnomAD (ID 6‑33443805‑C‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions from REVEL, ESM1b, and FATHMM; pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. A high‑accuracy consensus (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a 2‑to‑2 split, leaving the consensus inconclusive. No Foldetta stability assessment is available. Overall, the majority of evidence (five pathogenic versus three benign) points to a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.978838 | Binding | 0.270 | 0.888 | 1.000 | 6-33443805-C-G | -4.225 | Likely Benign | 0.846 | Likely Pathogenic | Ambiguous | 0.179 | Likely Benign | -2.79 | Deleterious | 0.997 | Probably Damaging | 0.993 | Probably Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.3310 | 0.3693 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||
| c.3257C>T | P1086L 2D AIThe SynGAP1 missense variant P1086L is not reported in ClinVar (ClinVar status: not present) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which simply indicates the variant has not yet been reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.849326 | Disordered | 0.977190 | Binding | 0.393 | 0.885 | 1.000 | -4.694 | Likely Benign | 0.607 | Likely Pathogenic | Likely Benign | 0.166 | Likely Benign | -3.57 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.73 | Benign | 0.00 | Affected | 0.2055 | 0.6326 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3260C>A | S1087Y 2D AIThe SynGAP1 missense variant S1087Y is catalogued in gnomAD (ID 6‑33443812‑C‑A) and has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as benign, and no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | 6-33443812-C-A | -4.194 | Likely Benign | 0.587 | Likely Pathogenic | Likely Benign | 0.107 | Likely Benign | -2.41 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.56 | Benign | 0.02 | Affected | 3.77 | 5 | 0.0671 | 0.5845 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||
| c.3260C>T | S1087F 2D AISynGAP1 missense variant S1087F is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of reliable predictors and the two high‑accuracy tools suggest a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | Uncertain | 1 | -3.843 | Likely Benign | 0.497 | Ambiguous | Likely Benign | 0.105 | Likely Benign | -2.75 | Deleterious | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.56 | Benign | 0.03 | Affected | 3.77 | 5 | 0.0645 | 0.6029 | -2 | -3 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||
| c.3265G>A | G1089R 2D AIThe SynGAP1 missense variant G1089R is catalogued in gnomAD (ID 6‑33443817‑G‑A) but has no ClinVar entry. Functional prediction tools split in a 6‑to‑3 ratio: benign calls come from REVEL, polyPhen‑2 HumVar, and ESM1b, while pathogenic calls come from PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. AlphaMissense‑Optimized yields an Uncertain result, and no Foldetta stability assessment is available. Overall, the majority of high‑confidence predictors lean toward pathogenicity, and this assessment does not conflict with ClinVar status, which is currently unreported. Thus, the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | 6-33443817-G-A | 1 | 6.35e-7 | -4.757 | Likely Benign | 0.897 | Likely Pathogenic | Ambiguous | 0.222 | Likely Benign | -3.13 | Deleterious | 0.896 | Possibly Damaging | 0.325 | Benign | 2.42 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.0934 | 0.4415 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||
| c.3265G>C | G1089R 2D AIThe SynGAP1 missense variant G1089R is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 HumVar, and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. Grouping by consensus, seven tools predict pathogenicity and three predict benign, giving a net pathogenic signal. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic. Foldetta stability analysis is unavailable. Overall, the evidence points to the variant being most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -4.757 | Likely Benign | 0.897 | Likely Pathogenic | Ambiguous | 0.228 | Likely Benign | -3.13 | Deleterious | 0.896 | Possibly Damaging | 0.325 | Benign | 2.42 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.0934 | 0.4415 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3266G>T | G1089V 2D AIThe SynGAP1 missense variant G1089V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact for G1089V. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -4.809 | Likely Benign | 0.527 | Ambiguous | Likely Benign | 0.182 | Likely Benign | -2.81 | Deleterious | 0.984 | Probably Damaging | 0.722 | Possibly Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.1326 | 0.4413 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3269A>T | N1090I 2D AIThe SynGAP1 missense variant N1090I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the balance of evidence from both general and high‑accuracy predictors points to a benign classification, and this conclusion does not contradict the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -4.356 | Likely Benign | 0.765 | Likely Pathogenic | Likely Benign | 0.173 | Likely Benign | -2.14 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.67 | Benign | 0.02 | Affected | 0.0781 | 0.6279 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3272T>C | L1091P 2D AIThe SynGAP1 missense variant L1091P is listed in gnomAD (ID 6‑33443824‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, and ESM1b, while pathogenic predictions are reported by polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized returns an uncertain result, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, leaving it inconclusive. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of tools (five pathogenic vs three benign) predict a deleterious effect, and the high‑accuracy AlphaMissense‑Optimized score is uncertain. Consequently, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.924947 | Disordered | 0.984454 | Binding | 0.376 | 0.889 | 1.000 | 6-33443824-T-C | -4.139 | Likely Benign | 0.871 | Likely Pathogenic | Ambiguous | 0.180 | Likely Benign | -1.25 | Neutral | 0.997 | Probably Damaging | 0.939 | Probably Damaging | 2.45 | Pathogenic | 0.04 | Affected | 3.77 | 5 | 0.3199 | 0.1918 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||
| c.3281C>A | S1094Y 2D AIThe SynGAP1 missense variant S1094Y is not reported in ClinVar (status: none) but is present in gnomAD (ID 6‑33443833‑C‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic). Foldetta, a protein‑folding‑stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions therefore show a benign call from AlphaMissense‑Optimized, an inconclusive SGM Consensus, and no Foldetta data. Overall, the balance of evidence leans toward a pathogenic interpretation (five pathogenic versus four benign calls), and this does not contradict the ClinVar status, which currently has no assertion for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.938133 | Disordered | 0.981352 | Binding | 0.358 | 0.877 | 1.000 | 6-33443833-C-A | -5.609 | Likely Benign | 0.631 | Likely Pathogenic | Likely Benign | 0.155 | Likely Benign | -2.07 | Neutral | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 2.45 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.1007 | 0.5877 | -2 | -3 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||
| c.3284C>T | P1095L 2D AIThe SynGAP1 missense variant P1095L is catalogued in gnomAD (ID 6‑33443836‑C‑T) but has no ClinVar record. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, and Foldetta data are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.979251 | Binding | 0.387 | 0.870 | 1.000 | 6-33443836-C-T | 1 | 6.44e-7 | -4.697 | Likely Benign | 0.363 | Ambiguous | Likely Benign | 0.126 | Likely Benign | -2.78 | Deleterious | 0.960 | Probably Damaging | 0.604 | Possibly Damaging | 2.74 | Benign | 0.03 | Affected | 3.77 | 5 | 0.2199 | 0.6347 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||
| c.3290C>T | P1097L 2D AIThe SynGAP1 missense variant P1097L is listed in ClinVar as Benign (ClinVar ID 2060978.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as benign; Foldetta results are unavailable. Overall, the majority of evidence supports a benign impact, and this conclusion is consistent with the ClinVar designation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.974957 | Binding | 0.384 | 0.858 | 1.000 | Benign | 1 | -4.410 | Likely Benign | 0.145 | Likely Benign | Likely Benign | 0.131 | Likely Benign | -2.07 | Neutral | 0.611 | Possibly Damaging | 0.198 | Benign | 2.64 | Benign | 0.05 | Affected | 3.77 | 5 | 0.2349 | 0.6356 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||
| c.3295T>G | Y1099D 2D AIThe SynGAP1 missense variant Y1099D is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all score the change as tolerated or benign, while only polyPhen‑2 HumDiv flags it as pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is Likely Benign, and Foldetta results are unavailable. Taken together, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar assertion, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | -4.193 | Likely Benign | 0.429 | Ambiguous | Likely Benign | 0.130 | Likely Benign | -1.08 | Neutral | 0.818 | Possibly Damaging | 0.435 | Benign | 2.79 | Benign | 0.13 | Tolerated | 0.3819 | 0.0935 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.3298G>T | G1100C 2D AIThe SynGAP1 missense variant G1100C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -6.488 | Likely Benign | 0.150 | Likely Benign | Likely Benign | 0.174 | Likely Benign | -2.25 | Neutral | 0.999 | Probably Damaging | 0.950 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.1354 | 0.4768 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.329T>A | V110D 2D AIThe SynGAP1 missense variant V110D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic vs. two benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated tools (seven pathogenic vs. three benign) indicate a pathogenic effect. This prediction is consistent with the lack of ClinVar reporting and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -4.536 | Likely Benign | 0.970 | Likely Pathogenic | Likely Pathogenic | 0.189 | Likely Benign | -2.84 | Deleterious | 0.978 | Probably Damaging | 0.500 | Possibly Damaging | 4.04 | Benign | 0.00 | Affected | 0.1849 | 0.1115 | -2 | -3 | -7.7 | 15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.329T>G | V110G 2D AIThe SynGAP1 missense variant V110G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence points toward a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -4.012 | Likely Benign | 0.724 | Likely Pathogenic | Likely Benign | 0.162 | Likely Benign | -2.87 | Deleterious | 0.377 | Benign | 0.928 | Probably Damaging | 4.06 | Benign | 0.00 | Affected | 0.2302 | 0.2671 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3302C>T | P1101L 2D AIThe SynGAP1 missense variant P1101L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for P1101L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.968967 | Binding | 0.457 | 0.861 | 0.875 | -4.335 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -2.19 | Neutral | 0.770 | Possibly Damaging | 0.255 | Benign | 4.27 | Benign | 0.04 | Affected | 0.2310 | 0.6050 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3311C>T | P1104L 2D AIThe SynGAP1 missense variant P1104L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also predicts Likely Benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy predictors, points to a benign impact. This conclusion is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.936162 | Disordered | 0.954801 | Binding | 0.440 | 0.863 | 0.875 | -3.846 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -0.33 | Neutral | 0.626 | Possibly Damaging | 0.168 | Benign | 2.81 | Benign | 1.00 | Tolerated | 0.2264 | 0.6795 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3314G>T | R1105L 2D AIThe SynGAP1 missense variant R1105L is not reported in ClinVar and is present in gnomAD (ID 6‑33443866‑G‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized classifies the variant as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels it pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not provide a result for this residue, so its stability impact is unavailable. Overall, the balance of evidence leans toward a benign effect, with no conflict with ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.901269 | Disordered | 0.954396 | Binding | 0.330 | 0.863 | 0.875 | 6-33443866-G-T | -4.031 | Likely Benign | 0.459 | Ambiguous | Likely Benign | 0.125 | Likely Benign | -3.51 | Deleterious | 0.677 | Possibly Damaging | 0.168 | Benign | 2.46 | Pathogenic | 0.19 | Tolerated | 3.77 | 5 | 0.1734 | 0.5373 | -2 | -3 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||
| c.3326T>A | L1109H 2D AIThe SynGAP1 missense variant L1109H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | -4.353 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -0.56 | Neutral | 0.832 | Possibly Damaging | 0.499 | Possibly Damaging | 2.70 | Benign | 0.04 | Affected | 0.1250 | 0.1845 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3326T>C | L1109P 2D AIThe SynGAP1 missense variant L1109P is listed in ClinVar with an uncertain significance (ClinVar ID 1730257.0) and is not reported in gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized indicates a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign classification, which does not contradict the ClinVar uncertain status. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | Conflicting | 2 | -5.313 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.151 | Likely Benign | -0.52 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.65 | Benign | 0.07 | Tolerated | 4.32 | 2 | 0.3159 | 0.2330 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.332C>T | P111L 2D AIThe SynGAP1 missense variant P111L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that P111L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.707965 | Disordered | 0.650020 | Binding | 0.438 | 0.858 | 0.750 | -4.430 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.089 | Likely Benign | -2.81 | Deleterious | 0.421 | Benign | 0.055 | Benign | 4.06 | Benign | 0.00 | Affected | 0.2355 | 0.7085 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3337G>T | G1113C 2D AIThe SynGAP1 missense variant G1113C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. ESM1b is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | -7.917 | In-Between | 0.128 | Likely Benign | Likely Benign | 0.130 | Likely Benign | -1.64 | Neutral | 0.999 | Probably Damaging | 0.917 | Probably Damaging | 2.50 | Benign | 0.06 | Tolerated | 0.1325 | 0.4956 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3338G>T | G1113V 2D AIThe SynGAP1 missense variant G1113V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.900456 | Binding | 0.327 | 0.910 | 0.875 | -5.708 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -1.98 | Neutral | 0.827 | Possibly Damaging | 0.456 | Possibly Damaging | 2.53 | Benign | 0.11 | Tolerated | 0.1333 | 0.4224 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3344T>A | I1115N 2D AIThe SynGAP1 missense variant I1115N is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | -4.018 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -0.60 | Neutral | 0.009 | Benign | 0.011 | Benign | 2.77 | Benign | 0.08 | Tolerated | 0.1299 | 0.1270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3349G>T | G1117C 2D AIThe SynGAP1 missense variant G1117C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.853192 | Binding | 0.323 | 0.914 | 0.750 | -9.045 | Likely Pathogenic | 0.112 | Likely Benign | Likely Benign | 0.359 | Likely Benign | -1.30 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 4.56 | Benign | 0.03 | Affected | 0.1347 | 0.4612 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3350G>T | G1117V 2D AIThe SynGAP1 missense variant G1117V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.853192 | Binding | 0.323 | 0.914 | 0.750 | -7.251 | In-Between | 0.083 | Likely Benign | Likely Benign | 0.284 | Likely Benign | -1.32 | Neutral | 0.011 | Benign | 0.014 | Benign | 4.57 | Benign | 0.04 | Affected | 0.1312 | 0.3680 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3356G>T | G1119V 2D AIThe SynGAP1 missense variant G1119V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and polyPhen‑2 HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | -6.428 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.352 | Likely Benign | -0.94 | Neutral | 0.918 | Possibly Damaging | 0.604 | Possibly Damaging | 3.91 | Benign | 0.31 | Tolerated | 0.1493 | 0.3707 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3358G>T | G1120C 2D AIThe SynGAP1 missense variant G1120C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” No Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for G1120C, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.804931 | Binding | 0.335 | 0.925 | 0.875 | -9.324 | Likely Pathogenic | 0.112 | Likely Benign | Likely Benign | 0.311 | Likely Benign | -1.32 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 3.60 | Benign | 0.03 | Affected | 0.1271 | 0.4227 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.335G>T | G112V 2D AIThe SynGAP1 missense variant G112V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.640153 | Binding | 0.332 | 0.867 | 0.750 | -2.411 | Likely Benign | 0.230 | Likely Benign | Likely Benign | 0.159 | Likely Benign | -3.39 | Deleterious | 0.421 | Benign | 0.108 | Benign | 3.96 | Benign | 0.00 | Affected | 0.1225 | 0.4218 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3364G>T | G1122C 2D AIThe SynGAP1 missense variant G1122C is listed in ClinVar with no submitted interpretation and is present in gnomAD (variant ID 6‑33443916‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence—including the high‑confidence tools—supports a benign classification, and this conclusion does not contradict the ClinVar status, which currently has no pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.814918 | Binding | 0.357 | 0.932 | 0.875 | 6-33443916-G-T | -8.895 | Likely Pathogenic | 0.110 | Likely Benign | Likely Benign | 0.375 | Likely Benign | -1.30 | Neutral | 0.975 | Probably Damaging | 0.733 | Possibly Damaging | 4.48 | Benign | 0.08 | Tolerated | 3.77 | 5 | 0.1343 | 0.4227 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||
| c.3365G>T | G1122V 2D AIThe SynGAP1 missense variant G1122V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) reports likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign, and this assessment is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.814918 | Binding | 0.357 | 0.932 | 0.875 | -6.398 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 0.271 | Likely Benign | -1.18 | Neutral | 0.059 | Benign | 0.025 | Benign | 4.49 | Benign | 0.10 | Tolerated | 0.1381 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3367G>T | G1123C 2D AIThe SynGAP1 missense variant G1123C is listed in gnomAD (ID 6‑33443919‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and ESM1b predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are unavailable. Overall, the majority of evidence—including the consensus and high‑accuracy tools—supports a benign classification. This conclusion is not contradicted by ClinVar, which contains no record for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | 6-33443919-G-T | 1 | 6.64e-7 | -9.329 | Likely Pathogenic | 0.118 | Likely Benign | Likely Benign | 0.353 | Likely Benign | -1.17 | Neutral | 0.994 | Probably Damaging | 0.840 | Possibly Damaging | 4.34 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1348 | 0.4227 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.3368G>T | G1123V 2D AIThe SynGAP1 missense variant G1123V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective predictions strongly suggest that G1123V is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.827246 | Binding | 0.346 | 0.934 | 0.875 | -7.129 | In-Between | 0.091 | Likely Benign | Likely Benign | 0.333 | Likely Benign | -1.03 | Neutral | 0.292 | Benign | 0.157 | Benign | 4.35 | Benign | 0.29 | Tolerated | 0.1362 | 0.3694 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3371G>T | G1124V 2D AIThe SynGAP1 missense variant G1124V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of available predictions indicates that G1124V is most likely benign, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.833401 | Binding | 0.341 | 0.931 | 0.875 | -6.980 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.315 | Likely Benign | -0.96 | Neutral | 0.586 | Possibly Damaging | 0.172 | Benign | 4.75 | Benign | 0.00 | Affected | 0.1299 | 0.3888 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3373G>A | G1125R 2D AIThe SynGAP1 missense variant G1125R is catalogued in gnomAD (ID 6‑33443925‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. AlphaMissense‑Default remains uncertain. A high‑accuracy consensus (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a benign majority vote. AlphaMissense‑Optimized also reports benign. No Foldetta stability assessment is available. Collectively, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443925-G-A | -8.463 | Likely Pathogenic | 0.542 | Ambiguous | Likely Benign | 0.291 | Likely Benign | -0.54 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.56 | Benign | 0.04 | Affected | 3.77 | 5 | 0.0980 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3373G>C | G1125R 2D AIThe SynGAP1 missense variant G1125R is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443925‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta’s protein‑folding stability result is unavailable. Overall, the balance of evidence, particularly from the high‑accuracy tools, indicates that the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443925-G-C | 1 | 6.68e-7 | -8.463 | Likely Pathogenic | 0.542 | Ambiguous | Likely Benign | 0.290 | Likely Benign | -0.54 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.56 | Benign | 0.04 | Affected | 3.77 | 5 | 0.0980 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.3376G>T | G1126C 2D AIThe SynGAP1 missense variant G1126C is listed in ClinVar (ID 469157.0) with an “Uncertain” status and is present in gnomAD (6‑33443928‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are SIFT and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.837209 | Binding | 0.345 | 0.918 | 0.875 | Uncertain | 1 | 6-33443928-G-T | 11 | 7.35e-6 | -9.389 | Likely Pathogenic | 0.113 | Likely Benign | Likely Benign | 0.449 | Likely Benign | -1.40 | Neutral | 0.005 | Benign | 0.005 | Benign | 4.74 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1326 | 0.4427 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||
| c.3377G>T | G1126V 2D AIThe SynGAP1 missense variant G1126V is listed in ClinVar with an uncertain significance and is present in the gnomAD database. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score all classify the change as benign. Only the SIFT algorithm predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized reports a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign, while Foldetta’s protein‑folding stability analysis is unavailable. Overall, the preponderance of evidence points to a benign variant, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.837209 | Binding | 0.345 | 0.918 | 0.875 | Uncertain | 1 | 6-33443929-G-T | -6.536 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.357 | Likely Benign | -1.20 | Neutral | 0.009 | Benign | 0.008 | Benign | 4.76 | Benign | 0.03 | Affected | 3.77 | 5 | 0.1366 | 0.3884 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||
| c.3379G>A | G1127R 2D AIThe SynGAP1 missense variant G1127R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443931‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Uncertain | 1 | 6-33443931-G-A | 2 | 1.34e-6 | -5.949 | Likely Benign | 0.629 | Likely Pathogenic | Likely Benign | 0.341 | Likely Benign | -0.87 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.86 | Benign | 0.12 | Tolerated | 4.32 | 4 | 0.0929 | 0.4532 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||
| c.3379G>C | G1127R 2D AIThe SynGAP1 missense variant G1127R is listed in ClinVar (ID 2967461.0) with an “Uncertain” clinical significance and is present in gnomAD (6‑33443931‑G‑C). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that G1127R is most likely benign, which does not contradict the ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Conflicting | 2 | 6-33443931-G-C | 16 | 1.07e-5 | -5.949 | Likely Benign | 0.629 | Likely Pathogenic | Likely Benign | 0.341 | Likely Benign | -0.87 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.86 | Benign | 0.12 | Tolerated | 4.32 | 4 | 0.0929 | 0.4532 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||
| c.3380G>T | G1127V 2D AIThe SynGAP1 missense variant G1127V is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443932‑G‑T). All available in silico predictors classify the change as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic prediction. Grouping by agreement, the benign‑predicting tools comprise the entire set, while no pathogenic predictions exist. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Uncertain | 1 | 6-33443932-G-T | 1 | 6.69e-7 | -6.097 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.230 | Likely Benign | -1.01 | Neutral | 0.004 | Benign | 0.005 | Benign | 4.81 | Benign | 0.17 | Tolerated | 4.32 | 4 | 0.1281 | 0.3669 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||
| c.3386T>C | L1129P 2D AIThe SynGAP1 missense variant L1129P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence—including high‑accuracy tools—points to a benign effect, and this conclusion does not contradict the current ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.876543 | Binding | 0.339 | 0.909 | 0.875 | Uncertain | 2 | -2.991 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 0.432 | Likely Benign | 0.27 | Neutral | 0.971 | Probably Damaging | 0.773 | Possibly Damaging | 5.44 | Benign | 0.00 | Affected | 4.32 | 4 | 0.3017 | 0.2231 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.338G>T | G113V 2D AIThe SynGAP1 missense variant G113V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.639486 | Binding | 0.350 | 0.870 | 0.750 | -3.752 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -1.77 | Neutral | 0.838 | Possibly Damaging | 0.145 | Benign | 4.18 | Benign | 0.05 | Affected | 0.1316 | 0.4002 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3392C>T | P1131L 2D AIThe SynGAP1 missense variant P1131L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy AlphaMissense‑Optimized tool classifies the variant as benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic, with one uncertain). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.919029 | Disordered | 0.855155 | Binding | 0.360 | 0.899 | 0.750 | -5.267 | Likely Benign | 0.420 | Ambiguous | Likely Benign | 0.293 | Likely Benign | -3.62 | Deleterious | 0.002 | Benign | 0.005 | Benign | 5.26 | Benign | 0.00 | Affected | 0.2043 | 0.6998 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3398T>A | I1133N 2D AIThe SynGAP1 missense variant I1133N is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Pathogenicity is suggested only by polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates Likely Benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign effect for I1133N, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | -5.887 | Likely Benign | 0.520 | Ambiguous | Likely Benign | 0.290 | Likely Benign | -1.07 | Neutral | 0.453 | Possibly Damaging | 0.162 | Benign | 5.51 | Benign | 0.02 | Affected | 0.0961 | 0.1140 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3410A>T | H1137L 2D AIThe SynGAP1 missense variant H1137L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the preponderance of benign predictions and the consensus from high‑accuracy methods, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.756488 | Binding | 0.314 | 0.879 | 0.875 | -2.215 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.359 | Likely Benign | -2.77 | Deleterious | 0.802 | Possibly Damaging | 0.534 | Possibly Damaging | 5.30 | Benign | 0.00 | Affected | 0.1199 | 0.6082 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3413C>A | S1138Y 2D AISynGAP1 missense variant S1138Y is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33444448‑C‑A). Prediction tools that agree on benign impact include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. AlphaMissense‑Default remains uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is ambiguous but leans toward benign (2 benign vs 1 pathogenic, 1 uncertain). High‑accuracy methods further support a benign interpretation: AlphaMissense‑Optimized predicts benign, while Foldetta results are unavailable. Overall, the balance of evidence, especially from high‑accuracy tools, suggests the variant is most likely benign, which does not contradict the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.891961 | Disordered | 0.738250 | Binding | 0.346 | 0.869 | 1.000 | Uncertain | 2 | 6-33444448-C-A | 3 | 1.86e-6 | -6.610 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.391 | Likely Benign | -2.51 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 5.41 | Benign | 0.05 | Affected | 4.32 | 4 | 0.1034 | 0.5449 | -2 | -3 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||
| c.3413C>T | S1138F 2D AIThe SynGAP1 missense variant S1138F is not reported in ClinVar and is present in gnomAD (ID 6‑33444448‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. The high‑accuracy AlphaMissense‑Optimized score is benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default (uncertain), ESM1b (benign), FATHMM (benign), and PROVEAN (pathogenic), resolves to benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates a benign effect. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.891961 | Disordered | 0.738250 | Binding | 0.346 | 0.869 | 1.000 | 6-33444448-C-T | 1 | 6.20e-7 | -6.298 | Likely Benign | 0.379 | Ambiguous | Likely Benign | 0.407 | Likely Benign | -2.88 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 5.42 | Benign | 0.03 | Affected | 4.32 | 4 | 0.0991 | 0.5623 | -2 | -3 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||
| c.3422C>T | P1141L 2D AIThe SynGAP1 missense variant P1141L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (two pathogenic versus one benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that P1141L is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.930790 | Disordered | 0.716087 | Binding | 0.364 | 0.852 | 1.000 | -3.817 | Likely Benign | 0.352 | Ambiguous | Likely Benign | 0.110 | Likely Benign | -4.64 | Deleterious | 0.954 | Possibly Damaging | 0.759 | Possibly Damaging | 0.98 | Pathogenic | 0.00 | Affected | 0.2147 | 0.6099 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3434A>T | N1145I 2D AIThe SynGAP1 missense variant N1145I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools cluster into two groups: pathogenic predictions come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; benign predictions come from ESM1b, FATHMM, and AlphaMissense‑Optimized. AlphaMissense‑Default is uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, while Foldetta results are unavailable. Overall, the majority of conventional predictors indicate pathogenicity, whereas the high‑accuracy subset leans benign. Based on the aggregate evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.722723 | Binding | 0.284 | 0.850 | 1.000 | -3.172 | Likely Benign | 0.378 | Ambiguous | Likely Benign | 0.504 | Likely Pathogenic | -4.19 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 5.41 | Benign | 0.00 | Affected | 0.0720 | 0.6145 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.3437C>T | P1146L 2D AISynGAP1 missense variant P1146L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas a separate group predicts pathogenicity: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also favors a benign outcome, while Foldetta results are unavailable. Overall, the majority of conventional predictors indicate pathogenicity, but the most accurate tools lean benign. Thus, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.919029 | Disordered | 0.732173 | Binding | 0.415 | 0.837 | 1.000 | -2.182 | Likely Benign | 0.483 | Ambiguous | Likely Benign | 0.564 | Likely Pathogenic | -5.25 | Deleterious | 0.992 | Probably Damaging | 0.912 | Probably Damaging | 5.51 | Benign | 0.00 | Affected | 0.2151 | 0.6446 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3446C>T | P1149L 2D AIThe SynGAP1 missense variant P1149L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for P1149L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.786938 | Binding | 0.424 | 0.837 | 0.625 | -3.438 | Likely Benign | 0.318 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -1.90 | Neutral | 0.818 | Possibly Damaging | 0.381 | Benign | 2.67 | Benign | 0.01 | Affected | 0.2235 | 0.5918 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3457C>T | R1153W 2D AIThe SynGAP1 missense variant R1153W is listed in ClinVar (ID 521099.0) with an uncertain significance designation and is present in the gnomAD database (variant ID 6‑33444492‑C‑T). Functional prediction tools cluster into two groups: benign predictions from REVEL and ESM1b, and pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus method SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely pathogenic. AlphaMissense‑Optimized independently predicts pathogenicity. No Foldetta stability assessment is available for this residue. Taken together, the majority of evidence points to a pathogenic effect, which is in contrast to the ClinVar uncertain classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.762850 | Disordered | 0.820118 | Binding | 0.361 | 0.848 | 0.625 | Uncertain | 2 | 6-33444492-C-T | 2 | 1.24e-6 | -5.812 | Likely Benign | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.317 | Likely Benign | -5.88 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.46 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1205 | 0.3340 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||
| c.3464T>G | V1155G 2D AIThe SynGAP1 missense variant V1155G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods give mixed results: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (benign), FATHMM (benign), and PROVEAN (benign), reports a benign outcome; Foldetta’s stability assessment is unavailable. Overall, the majority of conventional tools lean toward pathogenicity, whereas the high‑accuracy consensus leans benign, leaving the functional impact uncertain. The variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.855718 | Binding | 0.335 | 0.857 | 0.500 | -3.018 | Likely Benign | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.209 | Likely Benign | -1.91 | Neutral | 0.997 | Probably Damaging | 0.999 | Probably Damaging | 2.59 | Benign | 0.05 | Affected | 0.2211 | 0.2374 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3469T>A | W1157R 2D AIThe SynGAP1 missense variant W1157R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only ESM1b, whereas all other evaluated algorithms—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the preponderance of evidence indicates that W1157R is most likely pathogenic, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.694846 | Disordered | 0.877471 | Binding | 0.364 | 0.861 | 0.375 | -2.440 | Likely Benign | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.549 | Likely Pathogenic | -10.19 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.06 | Pathogenic | 0.00 | Affected | 0.4129 | 0.0612 | 2 | -3 | -3.6 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3469T>C | W1157R 2D AIThe SynGAP1 missense variant W1157R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only ESM1b, whereas all other evaluated algorithms—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the preponderance of evidence indicates that W1157R is most likely pathogenic, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.694846 | Disordered | 0.877471 | Binding | 0.364 | 0.861 | 0.375 | -2.440 | Likely Benign | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.549 | Likely Pathogenic | -10.19 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.06 | Pathogenic | 0.00 | Affected | 0.4129 | 0.0612 | 2 | -3 | -3.6 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.346T>G | Y116D 2D AIThe SynGAP1 missense variant Y116D is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions strongly suggests that Y116D is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.670235 | Binding | 0.381 | 0.878 | 0.625 | 0.542 | Likely Benign | 0.524 | Ambiguous | Likely Benign | 0.302 | Likely Benign | -0.14 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.24 | Benign | 0.83 | Tolerated | 0.4603 | 0.0545 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.3470G>C | W1157S 2D AIThe SynGAP1 missense variant W1157S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence indicates that W1157S is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.694846 | Disordered | 0.877471 | Binding | 0.364 | 0.861 | 0.375 | -1.158 | Likely Benign | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.394 | Likely Benign | -9.96 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.06 | Pathogenic | 0.00 | Affected | 0.4772 | 0.1227 | -2 | -3 | 0.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3473T>A | V1158D 2D AIThe SynGAP1 missense variant V1158D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence indicates that V1158D is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.877504 | Binding | 0.369 | 0.847 | 0.250 | -4.076 | Likely Benign | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.486 | Likely Benign | -4.56 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.1502 | 0.1820 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3473T>G | V1158G 2D AIThe SynGAP1 missense variant V1158G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic outcome. High‑accuracy assessments reinforce this pattern: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a pathogenic verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that V1158G is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.877504 | Binding | 0.369 | 0.847 | 0.250 | -3.058 | Likely Benign | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.433 | Likely Benign | -4.62 | Deleterious | 0.997 | Probably Damaging | 0.999 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.1870 | 0.3289 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3479A>T | N1160I 2D AIThe SynGAP1 missense variant N1160I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from multiple in silico tools indicates that the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.585406 | Disordered | 0.861611 | Binding | 0.361 | 0.836 | 0.375 | -4.996 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.440 | Likely Benign | -5.35 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.79 | Pathogenic | 0.01 | Affected | 0.0618 | 0.5903 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3485C>T | P1162L 2D AIThe SynGAP1 missense variant P1162L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, and FATHMM. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence (five pathogenic vs. four benign predictions) indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.858809 | Binding | 0.366 | 0.823 | 0.375 | -3.370 | Likely Benign | 0.962 | Likely Pathogenic | Likely Pathogenic | 0.209 | Likely Benign | -3.48 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.68 | Benign | 0.06 | Tolerated | 0.2153 | 0.7372 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3488A>T | H1163L 2D AIThe SynGAP1 missense variant H1163L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign) and Foldetta results are unavailable. Overall, the majority of predictions (5 pathogenic vs. 4 benign) lean toward a pathogenic impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.858469 | Binding | 0.328 | 0.825 | 0.375 | -2.401 | Likely Benign | 0.707 | Likely Pathogenic | Likely Benign | 0.568 | Likely Pathogenic | -3.15 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 5.55 | Benign | 0.10 | Tolerated | 0.0938 | 0.5356 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3491T>C | L1164P 2D AIThe SynGAP1 missense variant L1164P has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic impact are REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the majority of tools (seven pathogenic vs. three benign) predict a deleterious effect, indicating that the variant is most likely pathogenic. This prediction does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.853935 | Binding | 0.325 | 0.815 | 0.375 | -3.928 | Likely Benign | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.573 | Likely Pathogenic | -2.16 | Neutral | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 5.41 | Benign | 0.04 | Affected | 0.3173 | 0.1414 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3494C>G | S1165W 2D AIThe SynGAP1 missense variant S1165W has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign); pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome; Foldetta stability analysis is unavailable. Overall, the evidence is split, with an equal number of tools supporting benign versus pathogenic effects. Consequently, the variant is most likely pathogenic based on the current computational predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.835017 | Binding | 0.308 | 0.807 | 0.375 | -6.419 | Likely Benign | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.195 | Likely Benign | -2.29 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.54 | Benign | 0.02 | Affected | 0.0671 | 0.4807 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3499G>T | D1167Y 2D AIThe SynGAP1 missense variant D1167Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a 2‑to‑2 split and is therefore inconclusive. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑confidence predictors (six pathogenic vs. three benign) indicate that D1167Y is most likely pathogenic. This assessment does not contradict any ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.783999 | Binding | 0.336 | 0.798 | 0.500 | -4.050 | Likely Benign | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.281 | Likely Benign | -2.46 | Neutral | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.0640 | 0.7307 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||||
| c.3500A>T | D1167V 2D AIThe SynGAP1 missense variant D1167V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Based on the preponderance of pathogenic predictions and the high‑accuracy tool results, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.783999 | Binding | 0.336 | 0.798 | 0.500 | -2.750 | Likely Benign | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.288 | Likely Benign | -3.34 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.26 | Pathogenic | 0.00 | Affected | 0.0946 | 0.7515 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3503T>A | I1168N 2D AIThe SynGAP1 missense variant I1168N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote) which labels the variant as Likely Benign. In contrast, tools that predict a pathogenic effect are PolyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further show AlphaMissense‑Optimized as Pathogenic, while the SGM‑Consensus remains Likely Benign; Foldetta results are unavailable. Overall, the evidence is split evenly between benign and pathogenic predictions, with no consensus from the most accurate methods. Consequently, the variant’s pathogenicity is uncertain and does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.763262 | Binding | 0.423 | 0.796 | 0.500 | -4.269 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.455 | Likely Benign | -1.79 | Neutral | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 5.53 | Benign | 0.01 | Affected | 0.0943 | 0.0969 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3515A>T | H1172L 2D AIThe SynGAP1 missense variant H1172L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for H1172L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.626927 | Disordered | 0.673805 | Binding | 0.465 | 0.758 | 0.625 | -0.545 | Likely Benign | 0.446 | Ambiguous | Likely Benign | 0.426 | Likely Benign | -2.30 | Neutral | 0.451 | Benign | 0.265 | Benign | 5.47 | Benign | 0.01 | Affected | 0.0815 | 0.5285 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||
| c.3518T>A | I1173N 2D AIThe SynGAP1 missense variant I1173N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also leans benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Taken together, the preponderance of evidence points to a benign impact for I1173N, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.501700 | Disordered | 0.653145 | Binding | 0.521 | 0.756 | 0.375 | -4.130 | Likely Benign | 0.691 | Likely Pathogenic | Likely Benign | 0.443 | Likely Benign | -1.46 | Neutral | 0.925 | Possibly Damaging | 0.611 | Possibly Damaging | 5.34 | Benign | 0.02 | Affected | 0.0920 | 0.0142 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3532T>G | Y1178D 2D AIThe SynGAP1 missense variant Y1178D is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. AlphaMissense‑Optimized yields an Uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. Overall, the balance of evidence from high‑accuracy tools and consensus predictions leans toward a benign classification. This conclusion is not contradicted by ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.568433 | Binding | 0.554 | 0.688 | 0.250 | -1.250 | Likely Benign | 0.801 | Likely Pathogenic | Ambiguous | 0.434 | Likely Benign | -1.33 | Neutral | 0.995 | Probably Damaging | 0.846 | Possibly Damaging | 5.55 | Benign | 0.07 | Tolerated | 0.4562 | 0.0173 | -4 | -3 | -2.2 | -48.09 | ||||||||||||||||||||||||||||||||||||||
| c.3539T>A | L1180H 2D AIThe SynGAP1 missense variant L1180H is not reported in ClinVar and has no gnomAD allele, so its population frequency is unknown. Functional prediction tools show a split: benign calls come from REVEL, PROVEAN, ESM1b, and FATHMM, whereas pathogenic calls come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments highlight AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus leans benign; Foldetta results are not available. Overall, five of nine individual predictors favor pathogenicity, four favor benign, and the consensus tool suggests benign. Thus, the variant is most likely pathogenic based on the preponderance of high‑confidence predictions, and this assessment is not contradicted by ClinVar, which contains no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.626927 | Disordered | 0.559845 | Binding | 0.591 | 0.672 | 0.250 | -5.621 | Likely Benign | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.213 | Likely Benign | -0.22 | Neutral | 0.987 | Probably Damaging | 0.865 | Possibly Damaging | 2.65 | Benign | 0.00 | Affected | 0.0991 | 0.0860 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||
| c.3539T>C | L1180P 2D AISynGAP1 missense variant L1180P is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, whereas the SGM‑Consensus remains likely benign; Foldetta results are unavailable. Overall, the balance of evidence, including the consensus prediction and the higher number of benign calls, suggests the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.626927 | Disordered | 0.559845 | Binding | 0.591 | 0.672 | 0.250 | -4.564 | Likely Benign | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.213 | Likely Benign | -1.37 | Neutral | 0.992 | Probably Damaging | 0.930 | Probably Damaging | 2.65 | Benign | 0.00 | Affected | 0.3556 | 0.1155 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||
| c.3547T>G | Y1183D 2D AISynGAP1 missense variant Y1183D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools show mixed results: benign calls come from REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while pathogenic calls come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, a majority‑vote model of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts the variant as likely benign. High‑accuracy assessments further indicate AlphaMissense‑Optimized as pathogenic, whereas Foldetta (FoldX‑MD/Rosetta stability analysis) is not available for this residue. Overall, the balance of evidence leans toward a benign effect, and this assessment does not conflict with ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.566480 | Disordered | 0.527818 | Binding | 0.523 | 0.652 | 0.500 | -2.718 | Likely Benign | 0.972 | Likely Pathogenic | Likely Pathogenic | 0.097 | Likely Benign | -1.10 | Neutral | 0.986 | Probably Damaging | 0.787 | Possibly Damaging | 2.83 | Benign | 0.61 | Tolerated | 0.4602 | 0.0243 | -4 | -3 | -2.2 | -48.09 | ||||||||||||||||||||||||||||||||||||||
| c.3557C>G | S1186W 2D AIThe SynGAP1 missense variant S1186W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b remains uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic)—also favors pathogenic. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that S1186W is most likely pathogenic, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.562014 | Disordered | 0.506433 | Binding | 0.634 | 0.636 | 0.625 | -7.814 | In-Between | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.214 | Likely Benign | -3.43 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.62 | Benign | 0.01 | Affected | 0.0556 | 0.4158 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3562G>T | D1188Y 2D AIThe SynGAP1 D1188Y missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evaluated tools (seven pathogenic versus three benign) indicate a pathogenic impact. This prediction aligns with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.476583 | Structured | 0.484322 | Uncertain | 0.687 | 0.626 | 0.625 | -6.482 | Likely Benign | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.490 | Likely Benign | -4.49 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 5.40 | Benign | 0.00 | Affected | 0.0450 | 0.4714 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.3563A>T | D1188V 2D AIThe SynGAP1 D1188V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the balance of evidence (seven pathogenic versus three benign predictions) indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.476583 | Structured | 0.484322 | Uncertain | 0.687 | 0.626 | 0.625 | -4.482 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.479 | Likely Benign | -4.13 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 5.49 | Benign | 0.00 | Affected | 0.0592 | 0.4651 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3575T>C | L1192P 2D AIThe SynGAP1 missense variant L1192P is not reported in ClinVar and has no gnomAD entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, whereas the SGM‑Consensus remains benign; Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.575842 | Disordered | 0.441757 | Uncertain | 0.762 | 0.609 | 0.625 | -4.610 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.181 | Likely Benign | -1.94 | Neutral | 0.997 | Probably Damaging | 0.959 | Probably Damaging | 2.65 | Benign | 0.05 | Affected | 0.3515 | 0.1053 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||
| c.3577G>T | D1193Y 2D AIThe SynGAP1 missense variant D1193Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the overall pattern of predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.585406 | Disordered | 0.433390 | Uncertain | 0.807 | 0.600 | 0.375 | -8.233 | Likely Pathogenic | 0.883 | Likely Pathogenic | Ambiguous | 0.484 | Likely Benign | -2.94 | Deleterious | 0.992 | Probably Damaging | 0.947 | Probably Damaging | 5.50 | Benign | 0.00 | Affected | 0.0477 | 0.4111 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||
| c.3578A>T | D1193V 2D AIThe SynGAP1 D1193V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default, while only FATHMM predicts a benign outcome. ESM1b and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (two pathogenic vs. one benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.585406 | Disordered | 0.433390 | Uncertain | 0.807 | 0.600 | 0.375 | -7.297 | In-Between | 0.855 | Likely Pathogenic | Ambiguous | 0.526 | Likely Pathogenic | -2.92 | Deleterious | 0.977 | Probably Damaging | 0.856 | Possibly Damaging | 5.51 | Benign | 0.00 | Affected | 0.0752 | 0.4174 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3580A>T | R1194W 2D AIThe SynGAP1 missense variant R1194W is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, whereas the remaining methods—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (majority vote) confirms a likely pathogenic status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to the variant being most likely pathogenic, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.685117 | Disordered | 0.425297 | Uncertain | 0.796 | 0.602 | 0.375 | -9.583 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.514 | Likely Pathogenic | -2.98 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 5.41 | Benign | 0.00 | Affected | 0.1252 | 0.3162 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||
| c.3584T>G | V1195G 2D AIThe SynGAP1 missense variant V1195G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. Tools that agree on a benign effect are ESM1b and FATHMM. AlphaMissense‑Optimized is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive because it yields a 2‑to‑2 split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, six of the eight evaluated tools predict pathogenicity while only two predict benign, and no high‑accuracy consensus or folding‑stability evidence contradicts this. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not conflict with the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.604312 | Disordered | 0.434133 | Uncertain | 0.842 | 0.603 | 0.250 | -5.463 | Likely Benign | 0.881 | Likely Pathogenic | Ambiguous | 0.586 | Likely Pathogenic | -2.81 | Deleterious | 0.998 | Probably Damaging | 1.000 | Probably Damaging | 5.55 | Benign | 0.01 | Affected | 0.2005 | 0.2100 | -1 | -3 | -4.6 | -42.08 | |||||||||||||||||||||||||||||||||||||||
| c.358G>T | G120C 2D AIThe SynGAP1 missense variant G120C is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.659993 | Binding | 0.359 | 0.887 | 0.750 | -5.979 | Likely Benign | 0.179 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.73 | Neutral | 0.410 | Benign | 0.146 | Benign | 4.20 | Benign | 0.10 | Tolerated | 0.1376 | 0.4225 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3592T>G | Y1198D 2D AIThe SynGAP1 missense variant Y1198D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and SIFT, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely pathogenic effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools suggests that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.626927 | Disordered | 0.439379 | Uncertain | 0.853 | 0.593 | 0.250 | -14.709 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.325 | Likely Benign | -6.61 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.44 | Pathogenic | 0.18 | Tolerated | 0.4310 | 0.0373 | -4 | -3 | -2.2 | -48.09 | ||||||||||||||||||||||||||||||||||||||
| c.359G>T | G120V 2D AIThe SynGAP1 missense variant G120V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is not contradicted by any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.659993 | Binding | 0.359 | 0.887 | 0.750 | -4.571 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.036 | Likely Benign | -0.68 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.28 | Benign | 0.19 | Tolerated | 0.1321 | 0.3883 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3605T>A | I1202N 2D AIThe SynGAP1 missense variant I1202N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote) is likely pathogenic. Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, with no ClinVar status to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.529623 | Disordered | 0.510422 | Binding | 0.874 | 0.593 | 0.250 | -10.922 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.303 | Likely Benign | -5.65 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.79 | Pathogenic | 0.00 | Affected | 0.1014 | 0.0270 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3608A>T | H1203L 2D AIThe SynGAP1 missense variant H1203L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.618285 | Disordered | 0.527023 | Binding | 0.892 | 0.589 | 0.250 | -4.179 | Likely Benign | 0.153 | Likely Benign | Likely Benign | 0.361 | Likely Benign | -2.23 | Neutral | 0.473 | Possibly Damaging | 0.265 | Benign | 5.53 | Benign | 0.21 | Tolerated | 0.0682 | 0.2951 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||
| c.3611C>T | S1204L 2D AIThe SynGAP1 missense variant S1204L is reported in gnomAD (variant ID 6‑33446603‑C‑T) and has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; Foldetta results are not available. Based on the collective predictions, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.595080 | Disordered | 0.541098 | Binding | 0.887 | 0.587 | 0.375 | 6-33446603-C-T | 1 | 6.20e-7 | -4.672 | Likely Benign | 0.325 | Likely Benign | Likely Benign | 0.375 | Likely Benign | -0.86 | Neutral | 0.035 | Benign | 0.028 | Benign | 5.35 | Benign | 0.32 | Tolerated | 3.77 | 5 | 0.1000 | 0.4082 | -2 | -3 | 4.6 | 26.08 | |||||||||||||||||||||||||||||||||
| c.3614T>C | L1205P 2D AIThe SynGAP1 missense variant L1205P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All available in silico predictors classify the change as pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly indicates that the variant is pathogenic, which contradicts the current ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.595080 | Disordered | 0.552471 | Binding | 0.880 | 0.576 | 0.375 | Uncertain | 1 | -16.878 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.536 | Likely Pathogenic | -5.91 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.45 | Pathogenic | 0.00 | Affected | 0.3559 | 0.1053 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||
| c.3617A>T | K1206I 2D AIThe SynGAP1 missense variant K1206I is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are not available. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for K1206I. This conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.585406 | Disordered | 0.555819 | Binding | 0.893 | 0.569 | 0.375 | -13.526 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.302 | Likely Benign | -5.65 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.37 | Pathogenic | 0.01 | Affected | 0.0816 | 0.3521 | -2 | -3 | 8.4 | -15.01 | ||||||||||||||||||||||||||||||||||||||
| c.3626T>C | L1209P 2D AIThe SynGAP1 missense variant L1209P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as pathogenic; Foldetta results are unavailable. Based on the overwhelming agreement among pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.595080 | Disordered | 0.583711 | Binding | 0.899 | 0.574 | 0.375 | -17.259 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.448 | Likely Benign | -5.92 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.45 | Pathogenic | 0.00 | Affected | 0.3646 | 0.1053 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||
| c.3629A>T | H1210L 2D AIThe SynGAP1 missense variant H1210L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools indicates that H1210L is most likely benign, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.599170 | Disordered | 0.587579 | Binding | 0.900 | 0.567 | 0.375 | -4.018 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.168 | Likely Benign | -2.90 | Deleterious | 0.000 | Benign | 0.002 | Benign | 2.70 | Benign | 0.04 | Affected | 0.0673 | 0.4282 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||
| c.3638A>T | N1213I 2D AIThe SynGAP1 missense variant N1213I is not reported in ClinVar and is absent from gnomAD. Prediction tools show a split opinion: benign calls come from REVEL, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further highlight this discordance: AlphaMissense‑Optimized predicts a benign effect, whereas the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome. No Foldetta stability analysis is available for this residue. Overall, the preponderance of evidence points to a pathogenic effect for N1213I, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.580690 | Disordered | 0.521638 | Binding | 0.888 | 0.561 | 0.500 | -10.798 | Likely Pathogenic | 0.743 | Likely Pathogenic | Likely Benign | 0.093 | Likely Benign | -3.10 | Deleterious | 0.996 | Probably Damaging | 0.930 | Probably Damaging | 2.71 | Benign | 0.03 | Affected | 0.0437 | 0.4407 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3640C>G | R1214G 2D AIThe SynGAP1 missense variant R1214G is listed in gnomAD (6‑33446632‑C‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect are REVEL and AlphaMissense‑Optimized; those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable. Overall, the majority of tools predict a pathogenic impact, and this conclusion is not contradicted by any ClinVar status (none). Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.497853 | Structured | 0.506868 | Binding | 0.903 | 0.566 | 0.375 | 6-33446632-C-G | 1 | 6.20e-7 | -9.697 | Likely Pathogenic | 0.725 | Likely Pathogenic | Likely Benign | 0.131 | Likely Benign | -4.41 | Deleterious | 0.992 | Probably Damaging | 0.828 | Possibly Damaging | 2.48 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.3229 | 0.2361 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||
| c.3640C>T | R1214W 2D AIThe SynGAP1 missense variant R1214W is listed in ClinVar with an uncertain significance (ClinVar ID 1476244.0) and is present in the gnomAD database (gnomAD ID 6‑33446632‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL and AlphaMissense‑Optimized, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized indicates a benign effect, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as likely pathogenic. No Foldetta stability analysis is available for this residue. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not conflict with the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.497853 | Structured | 0.506868 | Binding | 0.903 | 0.566 | 0.375 | Uncertain | 1 | 6-33446632-C-T | 2 | 1.24e-6 | -8.799 | Likely Pathogenic | 0.710 | Likely Pathogenic | Likely Benign | 0.143 | Likely Benign | -4.95 | Deleterious | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.45 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1186 | 0.2367 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||
| c.3647T>C | L1216P 2D AIThe SynGAP1 missense variant L1216P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool predicts a benign outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as likely pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions strongly suggests that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.580690 | Disordered | 0.504713 | Binding | 0.863 | 0.563 | 0.250 | -16.029 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.524 | Likely Pathogenic | -5.28 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.13 | Pathogenic | 0.00 | Affected | 0.3281 | 0.1048 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||
| c.3655T>G | Y1219D 2D AIThe SynGAP1 missense variant Y1219D is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta results are not available for this variant. Based on the preponderance of pathogenic predictions, Y1219D is most likely pathogenic, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.613573 | Disordered | 0.474748 | Uncertain | 0.855 | 0.557 | 0.375 | -15.860 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.443 | Likely Benign | -7.11 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.4549 | 0.0573 | -4 | -3 | -2.2 | -48.09 | ||||||||||||||||||||||||||||||||||||||
| c.365C>T | P122L 2D AIThe SynGAP1 missense variant P122L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar, while pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.672358 | Binding | 0.400 | 0.887 | 0.750 | -3.810 | Likely Benign | 0.181 | Likely Benign | Likely Benign | 0.167 | Likely Benign | -2.92 | Deleterious | 0.906 | Possibly Damaging | 0.420 | Benign | 4.16 | Benign | 0.05 | Affected | 0.2657 | 0.6362 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3661C>T | R1221W 2D AIThe SynGAP1 missense variant R1221W is listed in ClinVar with an uncertain significance (ClinVar ID 1050818.0) and is present in the gnomAD database (gnomAD ID 6‑33446653‑C‑T). Functional prediction tools show a split assessment: benign predictions come from REVEL and FATHMM, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy analyses further refine the picture: AlphaMissense‑Optimized classifies the variant as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—deems it likely pathogenic. No Foldetta stability assessment is available for this residue. Overall, the majority of computational evidence points toward a pathogenic effect, which is consistent with the ClinVar designation of uncertain significance rather than a definitive benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.690604 | Disordered | 0.430363 | Uncertain | 0.906 | 0.539 | 0.375 | Conflicting | 3 | 6-33446653-C-T | 1 | 6.20e-7 | -10.938 | Likely Pathogenic | 0.651 | Likely Pathogenic | Likely Benign | 0.174 | Likely Benign | -4.57 | Deleterious | 1.000 | Probably Damaging | 0.987 | Probably Damaging | 2.50 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1242 | 0.2585 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||
| c.3664A>T | R1222W 2D AIThe SynGAP1 missense variant R1222W is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments show that the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Pathogenic, while AlphaMissense‑Optimized yields an Uncertain result and Foldetta data are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for R1222W. This conclusion is not contradicted by ClinVar status, which currently contains no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.703578 | Disordered | 0.423869 | Uncertain | 0.895 | 0.541 | 0.250 | -11.933 | Likely Pathogenic | 0.945 | Likely Pathogenic | Ambiguous | 0.260 | Likely Benign | -6.00 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.46 | Pathogenic | 0.03 | Affected | 0.0964 | 0.2761 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||
| c.3668T>C | L1223P 2D AIThe SynGAP1 missense variant L1223P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool predicts a benign outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus agrees. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the concordant pathogenic predictions and the lack of benign evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.608892 | Disordered | 0.436267 | Uncertain | 0.868 | 0.540 | 0.375 | -14.728 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.500 | Likely Pathogenic | -5.17 | Deleterious | 1.000 | Probably Damaging | 0.990 | Probably Damaging | 1.45 | Pathogenic | 0.01 | Affected | 0.3413 | 0.2352 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||
| c.3671T>C | L1224P 2D AIThe SynGAP1 missense variant L1224P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). In silico predictors that agree on a benign effect include REVEL and SIFT, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of predictions indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.525368 | Disordered | 0.441554 | Uncertain | 0.871 | 0.543 | 0.500 | -11.727 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.149 | Likely Benign | -3.18 | Deleterious | 0.998 | Probably Damaging | 0.948 | Probably Damaging | 2.36 | Pathogenic | 0.07 | Tolerated | 0.3618 | 0.1053 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||
| c.3671T>G | L1224R 2D AIThe SynGAP1 missense variant L1224R is listed in ClinVar with no assertion (ClinVar status: None) and is present in the gnomAD database (ID 6‑33446663‑T‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; ESM1b remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which currently contains no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.525368 | Disordered | 0.441554 | Uncertain | 0.871 | 0.543 | 0.500 | 6-33446663-T-G | 1 | 6.20e-7 | -7.504 | In-Between | 0.220 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -1.85 | Neutral | 0.989 | Probably Damaging | 0.900 | Possibly Damaging | 2.42 | Pathogenic | 0.33 | Tolerated | 3.77 | 5 | 0.1120 | 0.0558 | -2 | -3 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||
Found 8751 rows. Show 800 rows per page. Page 1/11 | Next »