
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain and Structure information: based on WT protein | Annotated databases | Deep learning-based pathogenicity predictions | Folding stability-based pathogenicity predictions | Sequence/structure-based pathogenicity predictions | Phase Separation | Evolutionary/physical properties | Molecular Dynamics-based analysis | DOI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | IUPred2 | ANCHOR2 | AlphaFold | MobiDB | PhosphoSitePlus | ClinVar | gnomAD | ESM1b | AlphaMissense | FoldX | Rosetta | Foldetta | PremPS | REVEL | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PSMutPred | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | |||||||||||||||||||||||||||||||||||||||||||||
| Score | Prediction | Score | Prediction | pLDDT | disorder | disorder | LTP | HTP | KL | PTM | Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | IP RF | SP RF | Prediction | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.913A>T | T305S 2D 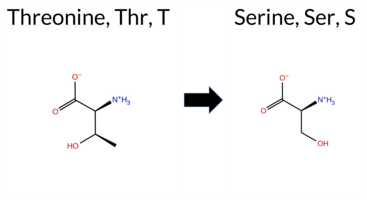 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T305S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions that are inconclusive or uncertain are provided by Rosetta, Foldetta, and premPS. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign,” and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.359901 | Structured | 0.299706 | Uncertain | 0.872 | 0.274 | 0.125 | -2.899 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.45 | Likely Benign | 0.4 | 1.21 | Ambiguous | 0.83 | Ambiguous | 0.55 | Ambiguous | 0.135 | Likely Benign | -0.60 | Neutral | 0.760 | Possibly Damaging | 0.484 | Possibly Damaging | 1.86 | Pathogenic | 0.54 | Tolerated | 0.3579 | 0.4496 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.914C>G | T305S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T305S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions that are inconclusive or uncertain are provided by Rosetta, Foldetta, and premPS. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign,” and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.359901 | Structured | 0.299706 | Uncertain | 0.872 | 0.274 | 0.125 | -2.899 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.45 | Likely Benign | 0.4 | 1.21 | Ambiguous | 0.83 | Ambiguous | 0.55 | Ambiguous | 0.104 | Likely Benign | -0.60 | Neutral | 0.760 | Possibly Damaging | 0.484 | Possibly Damaging | 1.86 | Pathogenic | 0.54 | Tolerated | 0.3579 | 0.4496 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.931C>T | H311Y 2D 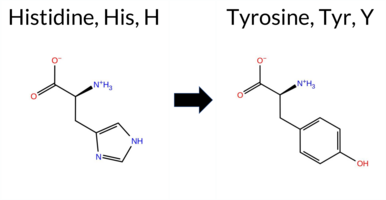 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H311Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, ESM1b, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Tools with uncertain or inconclusive results—Rosetta, Foldetta, and AlphaMissense‑Default—are treated as unavailable for pathogenicity assessment. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta remains uncertain. Overall, the predictions are split, with five tools favoring benign, five favoring pathogenic, and three inconclusive. Based on the available evidence, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.229226 | Structured | 0.354792 | Uncertain | 0.902 | 0.314 | 0.125 | -4.513 | Likely Benign | 0.342 | Ambiguous | Likely Benign | -0.06 | Likely Benign | 0.2 | -1.58 | Ambiguous | -0.82 | Ambiguous | 0.18 | Likely Benign | 0.456 | Likely Benign | -3.80 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.86 | Pathogenic | 0.02 | Affected | 0.0965 | 0.4180 | 0 | 2 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||
| c.971G>T | R324L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R324L is catalogued in gnomAD (6-33437876‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, and SIFT; pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome, and Foldetta (integrating FoldX‑MD and Rosetta outputs) reports a benign stability change. Overall, the majority of evidence points toward a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.257454 | Structured | 0.426893 | Uncertain | 0.954 | 0.397 | 0.000 | 6-33437876-G-T | 1 | 6.20e-7 | -10.328 | Likely Pathogenic | 0.575 | Likely Pathogenic | Likely Benign | -0.28 | Likely Benign | 0.0 | -0.08 | Likely Benign | -0.18 | Likely Benign | 0.29 | Likely Benign | 0.489 | Likely Benign | -2.20 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.86 | Pathogenic | 0.63 | Tolerated | 3.39 | 22 | 0.2310 | 0.5607 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||
| c.991T>A | S331T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S331T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while FoldX and premPS are inconclusive (Uncertain). High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Taken together, the majority of evidence indicates that S331T is most likely benign, and this conclusion does not contradict the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.433034 | Structured | 0.346458 | Uncertain | 0.658 | 0.475 | 0.250 | -2.474 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.60 | Ambiguous | 0.2 | -0.10 | Likely Benign | 0.25 | Likely Benign | -0.72 | Ambiguous | 0.095 | Likely Benign | 1.13 | Neutral | 0.003 | Benign | 0.002 | Benign | 1.86 | Pathogenic | 0.98 | Tolerated | 0.1505 | 0.4552 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1011G>C | K337N 2D 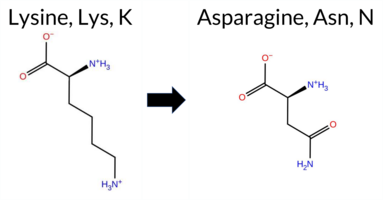 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K337N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, premPS, and SIFT. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions (seven pathogenic vs. five benign) and the high‑accuracy consensus lean toward a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.348540 | Uncertain | 0.449 | 0.438 | 0.500 | -13.095 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.12 | Likely Benign | 0.1 | 0.36 | Likely Benign | 0.24 | Likely Benign | -0.02 | Likely Benign | 0.280 | Likely Benign | -4.38 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.87 | Pathogenic | 0.11 | Tolerated | 0.2945 | 0.1315 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1011G>T | K337N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K337N is not reported in ClinVar and has no entries in gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, Rosetta, premPS, and SIFT, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. With seven tools supporting pathogenicity versus five supporting benign, the overall prediction leans toward pathogenic. No ClinVar entry contradicts this assessment, and the variant is absent from gnomAD, so the pathogenic prediction is not challenged by population data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.348540 | Uncertain | 0.449 | 0.438 | 0.500 | -13.095 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.12 | Likely Benign | 0.1 | 0.36 | Likely Benign | 0.24 | Likely Benign | -0.02 | Likely Benign | 0.280 | Likely Benign | -4.38 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.87 | Pathogenic | 0.11 | Tolerated | 0.2945 | 0.1315 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.3166G>A | G1056S 2D  AIThe SynGAP1 missense variant G1056S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.868632 | Binding | 0.402 | 0.935 | 0.875 | -5.252 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.339 | Likely Benign | -0.28 | Neutral | 0.451 | Benign | 0.149 | Benign | 1.87 | Pathogenic | 0.55 | Tolerated | 0.2497 | 0.5702 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3763A>C | K1255Q 2D 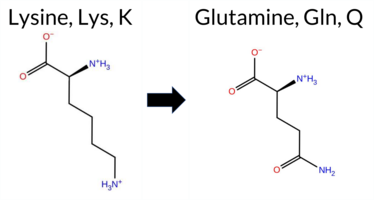 AIThe SynGAP1 missense variant K1255Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. In contrast, the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate pathogenicity. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus also reports a likely pathogenic classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect for K1255Q, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.637480 | Disordered | 0.417615 | Uncertain | 0.880 | 0.563 | 0.625 | -12.680 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.282 | Likely Benign | -3.19 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.87 | Pathogenic | 0.00 | Affected | 0.3625 | 0.1102 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||||||||||
| c.805A>G | I269V 2D 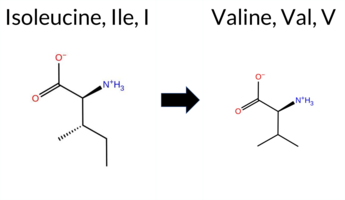 3DClick to see structure in 3D Viewer AIThe SynGAP1 I269V missense variant has no ClinVar record (ClinVar ID None) and is not reported in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Predictions that are uncertain or inconclusive are FoldX, Foldetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of tools lean toward a benign interpretation, but the high‑accuracy consensus indicates a pathogenic signal, leaving the variant’s clinical significance uncertain. This assessment does not contradict any existing ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.216401 | Structured | 0.343787 | Uncertain | 0.937 | 0.244 | 0.125 | -8.748 | Likely Pathogenic | 0.344 | Ambiguous | Likely Benign | 0.95 | Ambiguous | 0.0 | 0.49 | Likely Benign | 0.72 | Ambiguous | 0.71 | Ambiguous | 0.393 | Likely Benign | -0.72 | Neutral | 0.958 | Probably Damaging | 0.970 | Probably Damaging | 1.87 | Pathogenic | 0.10 | Tolerated | 0.0844 | 0.2859 | 4 | 3 | -0.3 | -14.03 | ||||||||||||||||||||||||||||||
| c.826C>A | P276T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 P276T missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Uncertain or inconclusive results come from Rosetta, Foldetta, and premPS. High‑accuracy methods give the following: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign, two pathogenic), and Foldetta is uncertain. Overall, the majority of tools (six pathogenic vs. four benign) indicate a pathogenic impact. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.037156 | Structured | 0.338937 | Uncertain | 0.724 | 0.230 | 0.250 | -5.793 | Likely Benign | 0.146 | Likely Benign | Likely Benign | 2.61 | Destabilizing | 0.1 | 0.75 | Ambiguous | 1.68 | Ambiguous | 0.63 | Ambiguous | 0.293 | Likely Benign | -3.53 | Deleterious | 0.961 | Probably Damaging | 0.721 | Possibly Damaging | 1.87 | Pathogenic | 0.03 | Affected | 0.1601 | 0.4676 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||
| c.827C>T | P276L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 P276L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, premPS, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. FoldX, Rosetta, and Foldetta provide uncertain or unavailable stability results and are therefore not considered evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta remains unavailable. Overall, the predictions are mixed, with an equal split between benign and pathogenic calls, and the high‑accuracy tools do not yield a definitive verdict. Consequently, the variant is most likely benign based on the current evidence, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.037156 | Structured | 0.338937 | Uncertain | 0.724 | 0.230 | 0.250 | -6.687 | Likely Benign | 0.196 | Likely Benign | Likely Benign | 1.64 | Ambiguous | 0.1 | 0.87 | Ambiguous | 1.26 | Ambiguous | 0.33 | Likely Benign | 0.439 | Likely Benign | -4.92 | Deleterious | 0.961 | Probably Damaging | 0.655 | Possibly Damaging | 1.87 | Pathogenic | 0.01 | Affected | 0.2179 | 0.5650 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||
| c.932A>C | H311P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H311P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign outcome; the remaining predictions are uncertain (FoldX, Foldetta, premPS, AlphaMissense‑Optimized). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Taken together, the majority of evidence points to a deleterious impact on protein function. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for H311P. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.229226 | Structured | 0.354792 | Uncertain | 0.902 | 0.314 | 0.125 | -10.454 | Likely Pathogenic | 0.869 | Likely Pathogenic | Ambiguous | 1.57 | Ambiguous | 0.6 | 2.39 | Destabilizing | 1.98 | Ambiguous | 0.74 | Ambiguous | 0.724 | Likely Pathogenic | -7.76 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.87 | Pathogenic | 0.01 | Affected | 0.2173 | 0.4172 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||
| c.932A>T | H311L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 H311L missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include AlphaMissense‑Optimized, Foldetta, premPS, and Rosetta. Tools that predict a pathogenic outcome are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labeling it likely pathogenic, and Foldetta predicting a benign effect. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.229226 | Structured | 0.354792 | Uncertain | 0.902 | 0.314 | 0.125 | -10.119 | Likely Pathogenic | 0.575 | Likely Pathogenic | Likely Benign | -0.53 | Ambiguous | 0.0 | -0.04 | Likely Benign | -0.29 | Likely Benign | 0.43 | Likely Benign | 0.663 | Likely Pathogenic | -7.99 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.87 | Pathogenic | 0.02 | Affected | 0.0961 | 0.5292 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.952C>T | P318S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P318S is present in gnomAD (variant ID 6‑33437857‑C‑T) but has no ClinVar entry. Functional prediction tools uniformly indicate a deleterious effect. Pathogenic predictions come from SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain predictions come from Rosetta and Foldetta. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is likely pathogenic, and Foldetta remains uncertain. Taken together, the overwhelming majority of evidence supports a pathogenic classification, and this conclusion is consistent with the absence of a ClinVar record rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.111485 | Structured | 0.400936 | Uncertain | 0.858 | 0.234 | 0.000 | 6-33437857-C-T | 1 | 6.19e-7 | -9.954 | Likely Pathogenic | 0.956 | Likely Pathogenic | Likely Pathogenic | 2.22 | Destabilizing | 0.1 | 1.71 | Ambiguous | 1.97 | Ambiguous | 1.00 | Destabilizing | 0.626 | Likely Pathogenic | -7.05 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.87 | Pathogenic | 0.03 | Affected | 3.38 | 23 | 0.3692 | 0.5653 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||
| c.992C>T | S331L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S331L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign or likely benign. Only FATHMM predicts pathogenicity, while FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the consensus of the majority of tools supports a benign classification, and this is consistent with the lack of ClinVar evidence; there is no contradiction with ClinVar status. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.433034 | Structured | 0.346458 | Uncertain | 0.658 | 0.475 | 0.250 | -6.570 | Likely Benign | 0.219 | Likely Benign | Likely Benign | 0.54 | Ambiguous | 0.0 | 1.06 | Ambiguous | 0.80 | Ambiguous | 0.07 | Likely Benign | 0.090 | Likely Benign | -1.72 | Neutral | 0.270 | Benign | 0.136 | Benign | 1.87 | Pathogenic | 0.58 | Tolerated | 0.0953 | 0.4124 | -3 | -2 | 4.6 | 26.08 | |||||||||||||||||||||||||||||
| c.1051G>T | A351S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A351S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only FATHMM predicts a pathogenic outcome. High‑accuracy methods—AlphaMissense‑Optimized, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (integrating FoldX‑MD and Rosetta)—all report benign or likely benign. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely benign; this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.216401 | Structured | 0.362025 | Uncertain | 0.925 | 0.342 | 0.000 | -4.823 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.24 | Likely Benign | 0.0 | 0.25 | Likely Benign | 0.25 | Likely Benign | 0.27 | Likely Benign | 0.016 | Likely Benign | -1.42 | Neutral | 0.080 | Benign | 0.023 | Benign | 1.88 | Pathogenic | 0.13 | Tolerated | 0.2558 | 0.5334 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.3763A>G | K1255E 2D  AIThe SynGAP1 missense variant K1255E is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are not available. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.637480 | Disordered | 0.417615 | Uncertain | 0.880 | 0.563 | 0.625 | -15.072 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.308 | Likely Benign | -3.12 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.88 | Pathogenic | 0.00 | Affected | 0.3094 | 0.0877 | 0 | 1 | 0.4 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.805A>C | I269L 2D 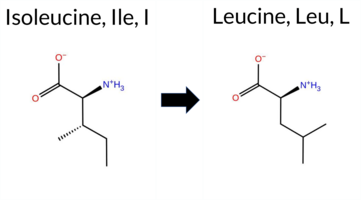 3DClick to see structure in 3D Viewer AIThe SynGAP1 I269L missense variant is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors shows a predominance of benign calls: REVEL, FoldX, Rosetta, premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized all predict benign. Pathogenicity is suggested by polyPhen‑2 (HumDiv and HumVar) and FATHMM, while ESM1b and AlphaMissense‑Default remain uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, Foldetta predicts benign stability, and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to an equal split between benign and pathogenic signals. Overall, the balance of evidence favors a benign effect for I269L, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.216401 | Structured | 0.343787 | Uncertain | 0.937 | 0.244 | 0.125 | -7.588 | In-Between | 0.470 | Ambiguous | Likely Benign | 0.00 | Likely Benign | 0.0 | 0.19 | Likely Benign | 0.10 | Likely Benign | 0.47 | Likely Benign | 0.348 | Likely Benign | -1.47 | Neutral | 0.981 | Probably Damaging | 0.970 | Probably Damaging | 1.88 | Pathogenic | 0.28 | Tolerated | 0.0662 | 0.2817 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.805A>T | I269L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I269L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two consensus groups: eight tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, AlphaMissense‑Optimized) predict a benign effect, while three tools (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM) predict pathogenicity. Two tools (ESM1b, AlphaMissense‑Default) return uncertain results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized classifies the variant as benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, also predicts a benign impact. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields no clear majority and is therefore unavailable as evidence. Overall, the preponderance of evidence supports a benign classification, which is consistent with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.216401 | Structured | 0.343787 | Uncertain | 0.937 | 0.244 | 0.125 | -7.588 | In-Between | 0.470 | Ambiguous | Likely Benign | 0.00 | Likely Benign | 0.0 | 0.19 | Likely Benign | 0.10 | Likely Benign | 0.47 | Likely Benign | 0.348 | Likely Benign | -1.47 | Neutral | 0.981 | Probably Damaging | 0.970 | Probably Damaging | 1.88 | Pathogenic | 0.28 | Tolerated | 0.0662 | 0.2817 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.815G>A | R272Q 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant R272Q is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33437720‑G‑A). Prediction tools that classify the variant as benign include REVEL, Rosetta, Foldetta, AlphaMissense‑Default, AlphaMissense‑Optimized, and PROVEAN. Those that predict pathogenicity are premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. The high‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive; and Foldetta predicts benign. With the majority of high‑accuracy tools supporting a benign effect, the variant is most likely benign, which does not contradict its current ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.071867 | Structured | 0.425620 | Uncertain | 0.925 | 0.215 | 0.125 | Uncertain | 2 | 6-33437720-G-A | 14 | 8.67e-6 | -9.559 | Likely Pathogenic | 0.286 | Likely Benign | Likely Benign | 0.73 | Ambiguous | 0.1 | 0.15 | Likely Benign | 0.44 | Likely Benign | 1.00 | Destabilizing | 0.321 | Likely Benign | -1.81 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.88 | Pathogenic | 0.03 | Affected | 3.38 | 19 | 0.2966 | 0.1973 | 1 | 1 | 1.0 | -28.06 | 255.7 | 52.9 | 0.0 | 0.0 | -0.2 | 0.1 | X | Uncertain | The guanidinium group of Arg272, located at the end of an anti-parallel β sheet strand (res. Arg259-Arg272), is stably maintained in an upright and outward position via stacking with the indole ring of the Trp362 side chain in another β strand (res. Thr359-Pro364). In the WT simulations, Arg272 forms hydrogen bonds with the glycine-rich Ω loop residues (res. Val365-Pro398, e.g., Gly380) and creates a salt bridge with the carboxylate group of the Asp304 side chain.In the variant simulations, the carboxamide group of the Gln272 side chain does not stack with the indole ring of Trp362 as stably as the guanidinium group of Arg272 in the WT. Consequently, the Gln272 side chain is freer to interact with the loop residues than Arg272, potentially negatively affecting the dynamic SynGAP-membrane association. Additionally, Arg272 faces the RasGTPase interface, so the residue swap could impact the SynGAP-Ras complex formation and GTPase activation. | ||||||||||||||
| c.835C>T | R279W 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant R279W is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a strong bias toward pathogenicity: benign predictions are limited to REVEL, whereas pathogenic predictions are made by FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive include Foldetta, AlphaMissense‑Optimized, Rosetta, and premPS. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta is uncertain. Overall, the preponderance of evidence indicates that R279W is most likely pathogenic, a conclusion that contradicts the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.309382 | Uncertain | 0.887 | 0.257 | 0.125 | Conflicting | 2 | -11.417 | Likely Pathogenic | 0.942 | Likely Pathogenic | Ambiguous | 2.00 | Destabilizing | 0.8 | 1.47 | Ambiguous | 1.74 | Ambiguous | 0.80 | Ambiguous | 0.485 | Likely Benign | -6.29 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.88 | Pathogenic | 0.00 | Affected | 3.39 | 18 | 0.1200 | 0.2519 | 2 | -3 | 3.6 | 30.03 | 270.0 | 38.3 | 0.1 | 0.0 | 0.3 | 0.0 | Uncertain | The guanidinium group of Arg279, located at the beginning of an anti-parallel β sheet strand (res. Arg279-Leu286), can form hydrogen bond with the backbone carbonyl groups of nearby loop residues (e.g., Ser296, Ser331, and As332) and form salt bridges with the carboxylate groups of Asp330 and Asp332. In the WT simulations, Arg279 sporadically forms a salt bridge even with the carboxylate group of Glu613, loosely connecting the C2 domain and GAP domain. Meanwhile, the indole ring of the Trp279 side chain is unable to hydrogen bond with the loop residues in the variant simulations. The lack of hydrogen bond or salt bridge formation with the loop residues could be significant, as Arg279 and the loops face the polar head group region of the membrane. Thus, although Trp279 could interact with the membrane surface as a “lipid anchor,” any changes to the wider loop dynamics could still adversely affect the formation of a stable SynGAP-membrane association. However, no definite conclusions on the effect of the residue swap on the SynGAP-membrane association can be drawn from solvent-only simulations. | |||||||||||||||||
| c.850C>A | L284I 2D 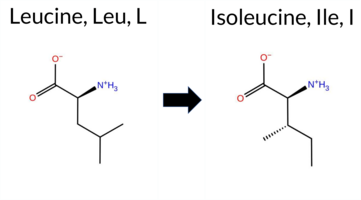 3DClick to see structure in 3D Viewer AIThe SynGAP1 L284I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No evidence from FoldX or Rosetta alone is conclusive. Overall, the majority of predictions, including the high‑accuracy methods, support a benign classification. This consensus does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.094817 | Structured | 0.371601 | Uncertain | 0.950 | 0.255 | 0.000 | -5.853 | Likely Benign | 0.293 | Likely Benign | Likely Benign | 0.50 | Ambiguous | 0.0 | 1.22 | Ambiguous | 0.86 | Ambiguous | 0.49 | Likely Benign | 0.364 | Likely Benign | -1.51 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.88 | Pathogenic | 0.13 | Tolerated | 0.0738 | 0.3006 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.854G>A | C285Y 2D 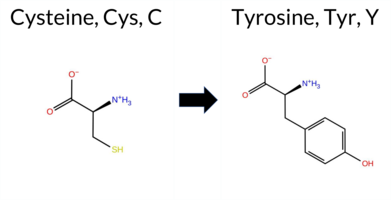 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C285Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and Rosetta. Those that agree on a pathogenic effect include SGM‑Consensus, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive are Foldetta, premPS, ESM1b, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact for C285Y. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.144935 | Structured | 0.375400 | Uncertain | 0.946 | 0.250 | 0.000 | -7.210 | In-Between | 0.895 | Likely Pathogenic | Ambiguous | 3.66 | Destabilizing | 1.1 | -2.33 | Stabilizing | 0.67 | Ambiguous | 0.53 | Ambiguous | 0.484 | Likely Benign | -8.67 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.88 | Pathogenic | 0.18 | Tolerated | 0.1413 | 0.3689 | 0 | -2 | -3.8 | 60.04 | |||||||||||||||||||||||||||||
| c.884C>T | T295I 2D 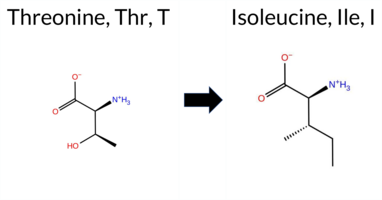 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T295I is reported in gnomAD (ID 6‑33437789‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two consensus groups: benign predictions come from FoldX and Foldetta, while pathogenic predictions are supported by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results are reported by AlphaMissense‑Optimized, Rosetta, and premPS and are treated as inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points toward a pathogenic effect, with only a minority of tools indicating benign or uncertain outcomes. This prediction does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.401658 | Structured | 0.295548 | Uncertain | 0.881 | 0.288 | 0.125 | 6-33437789-C-T | 4 | 2.48e-6 | -9.330 | Likely Pathogenic | 0.892 | Likely Pathogenic | Ambiguous | 0.21 | Likely Benign | 0.2 | 0.55 | Ambiguous | 0.38 | Likely Benign | 0.58 | Ambiguous | 0.607 | Likely Pathogenic | -4.87 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.88 | Pathogenic | 0.04 | Affected | 3.38 | 23 | 0.1025 | 0.5599 | -1 | 0 | 5.2 | 12.05 | ||||||||||||||||||||||||
| c.926G>A | G309D 2D 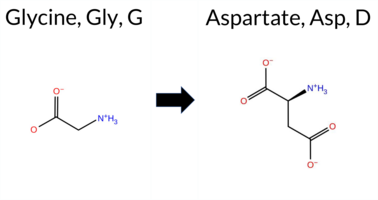 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G309D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions are provided by SIFT and Rosetta, whereas the remaining 12 tools (REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, SGM‑Consensus) are pathogenic. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta is inconclusive. Taken together, the majority of evidence supports a pathogenic effect. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.275179 | Structured | 0.338439 | Uncertain | 0.882 | 0.342 | 0.125 | -15.264 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.89 | Destabilizing | 0.5 | -0.03 | Likely Benign | 1.43 | Ambiguous | 0.75 | Ambiguous | 0.523 | Likely Pathogenic | -6.43 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.88 | Pathogenic | 0.06 | Tolerated | 0.1889 | 0.2678 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.937G>C | E313Q 2D 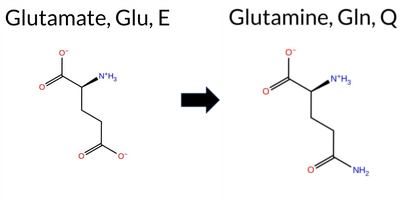 3DClick to see structure in 3D Viewer AISynGAP1 E313Q is listed in ClinVar with an Uncertain significance status and is not reported in gnomAD. Prediction tools that classify the variant as benign include FoldX, Foldetta, premPS, PROVEAN, and AlphaMissense‑Optimized. Those that predict pathogenicity are REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized indicates a benign effect, Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign stability, whereas the SGM‑Consensus remains pathogenic. Overall, the balance of evidence leans toward pathogenicity, and this assessment does not contradict the ClinVar designation of Uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.170161 | Structured | 0.366526 | Uncertain | 0.898 | 0.304 | 0.125 | Uncertain | 1 | -11.420 | Likely Pathogenic | 0.629 | Likely Pathogenic | Likely Benign | 0.19 | Likely Benign | 0.1 | 0.55 | Ambiguous | 0.37 | Likely Benign | 0.50 | Likely Benign | 0.505 | Likely Pathogenic | -2.42 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.88 | Pathogenic | 0.05 | Affected | 0.1523 | 0.7396 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||
| c.938A>C | E313A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E313A is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only AlphaMissense‑Optimized, whereas the remaining pathogenic‑or‑likely‑pathogenic predictors are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, Foldetta, premPS) returned uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect. Thus, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.170161 | Structured | 0.366526 | Uncertain | 0.898 | 0.304 | 0.125 | -14.591 | Likely Pathogenic | 0.747 | Likely Pathogenic | Likely Benign | 1.07 | Ambiguous | 0.3 | 0.97 | Ambiguous | 1.02 | Ambiguous | 0.62 | Ambiguous | 0.680 | Likely Pathogenic | -4.88 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.88 | Pathogenic | 0.02 | Affected | 0.3416 | 0.6743 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.939G>C | E313D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E313D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and SIFT, whereas a majority of tools predict a pathogenic impact: polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicating a likely pathogenic effect, and Foldetta yielding an uncertain stability change. Overall, the balance of evidence points to a pathogenic interpretation; this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.170161 | Structured | 0.366526 | Uncertain | 0.898 | 0.304 | 0.125 | -8.309 | Likely Pathogenic | 0.636 | Likely Pathogenic | Likely Benign | 0.87 | Ambiguous | 0.0 | 0.96 | Ambiguous | 0.92 | Ambiguous | 0.60 | Ambiguous | 0.346 | Likely Benign | -1.66 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.88 | Pathogenic | 0.14 | Tolerated | 0.1970 | 0.4640 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.939G>T | E313D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E313D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and SIFT, whereas a majority of tools predict a pathogenic impact: polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicating a likely pathogenic outcome, and Foldetta yielding an uncertain stability change. Overall, the balance of evidence points to a pathogenic effect for E313D, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.170161 | Structured | 0.366526 | Uncertain | 0.898 | 0.304 | 0.125 | -8.309 | Likely Pathogenic | 0.636 | Likely Pathogenic | Likely Benign | 0.87 | Ambiguous | 0.0 | 0.96 | Ambiguous | 0.92 | Ambiguous | 0.60 | Ambiguous | 0.346 | Likely Benign | -1.66 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.88 | Pathogenic | 0.14 | Tolerated | 0.1970 | 0.4640 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.956C>T | A319V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 A319V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. FoldX, Rosetta, and Foldetta give uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta is also inconclusive. Overall, the majority of evidence points to a benign effect, and this does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.410405 | Uncertain | 0.879 | 0.254 | 0.125 | -8.179 | Likely Pathogenic | 0.141 | Likely Benign | Likely Benign | 0.58 | Ambiguous | 0.4 | 0.63 | Ambiguous | 0.61 | Ambiguous | 0.23 | Likely Benign | 0.329 | Likely Benign | -2.32 | Neutral | 0.989 | Probably Damaging | 0.824 | Possibly Damaging | 1.88 | Pathogenic | 0.10 | Tolerated | 0.1094 | 0.6390 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||
| c.1003C>A | R335S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R335S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, premPS, and SIFT; pathogenic predictions from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. No evidence from FoldX or Rosetta alone is available. Based on the preponderance of pathogenic predictions and the high‑accuracy tools, the variant is most likely pathogenic, which is consistent with the absence of ClinVar reporting and gnomAD data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.305330 | Structured | 0.331028 | Uncertain | 0.483 | 0.428 | 0.500 | -9.286 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.68 | Ambiguous | 0.1 | 0.72 | Ambiguous | 0.70 | Ambiguous | 0.14 | Likely Benign | 0.184 | Likely Benign | -3.30 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.89 | Pathogenic | 0.11 | Tolerated | 0.2598 | 0.4005 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||
| c.1042G>C | V348L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V348L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No other high‑confidence tools provide conflicting evidence. Based on the preponderance of predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.170161 | Structured | 0.346556 | Uncertain | 0.951 | 0.414 | 0.000 | -4.842 | Likely Benign | 0.270 | Likely Benign | Likely Benign | -0.58 | Ambiguous | 0.0 | 0.04 | Likely Benign | -0.27 | Likely Benign | 0.24 | Likely Benign | 0.072 | Likely Benign | -0.75 | Neutral | 0.264 | Benign | 0.030 | Benign | 1.89 | Pathogenic | 0.62 | Tolerated | 0.1088 | 0.4953 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1042G>T | V348L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V348L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No other high‑confidence tools provide conflicting evidence. Based on the preponderance of predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.170161 | Structured | 0.346556 | Uncertain | 0.951 | 0.414 | 0.000 | -4.842 | Likely Benign | 0.270 | Likely Benign | Likely Benign | -0.58 | Ambiguous | 0.0 | 0.04 | Likely Benign | -0.27 | Likely Benign | 0.24 | Likely Benign | 0.072 | Likely Benign | -0.75 | Neutral | 0.264 | Benign | 0.030 | Benign | 1.89 | Pathogenic | 0.62 | Tolerated | 0.1088 | 0.4953 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1628T>A | L543Q 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L543Q is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. No tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a destabilizing, pathogenic effect. All available predictions are concordant and supportive. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.020918 | Uncertain | 0.963 | 0.314 | 0.000 | -14.851 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.03 | Destabilizing | 0.2 | 3.48 | Destabilizing | 3.26 | Destabilizing | 2.25 | Destabilizing | 0.746 | Likely Pathogenic | -5.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.89 | Pathogenic | 0.00 | Affected | 0.0983 | 0.0488 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1628T>C | L543P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L543P (ClinVar ID 4540554) is classified as Pathogenic in ClinVar and is not reported in gnomAD. Prediction tools that assess functional impact uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. No tool in the dataset predicts a benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is Pathogenic. All available predictions and stability analyses support a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion is consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.020918 | Uncertain | 0.963 | 0.314 | 0.000 | Likely Pathogenic | 1 | -15.958 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 8.56 | Destabilizing | 0.6 | 13.44 | Destabilizing | 11.00 | Destabilizing | 1.54 | Destabilizing | 0.770 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.89 | Pathogenic | 0.00 | Affected | 0.3457 | 0.0992 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||
| c.1628T>G | L543R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L543R is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic. No tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a pathogenic impact. All available predictions are concordant and supportive. Based on these computational assessments, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.020918 | Uncertain | 0.963 | 0.314 | 0.000 | -18.563 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.47 | Destabilizing | 1.2 | 8.02 | Destabilizing | 5.75 | Destabilizing | 1.64 | Destabilizing | 0.739 | Likely Pathogenic | -5.97 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.89 | Pathogenic | 0.00 | Affected | 0.1229 | 0.0488 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.827C>G | P276R 2D 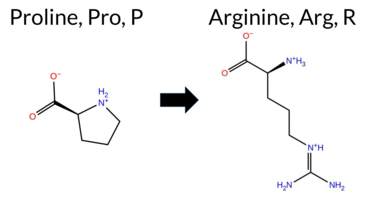 3DClick to see structure in 3D Viewer AIThe SynGAP1 P276R missense variant is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (gnomAD ID 6‑33437732‑C‑G). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. Stability‑based predictors (FoldX, Rosetta, premPS, Foldetta) are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic classification; this conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.037156 | Structured | 0.338937 | Uncertain | 0.724 | 0.230 | 0.250 | 6-33437732-C-G | 7 | 4.34e-6 | -10.983 | Likely Pathogenic | 0.714 | Likely Pathogenic | Likely Benign | 1.78 | Ambiguous | 0.2 | 1.02 | Ambiguous | 1.40 | Ambiguous | 0.78 | Ambiguous | 0.498 | Likely Benign | -4.52 | Deleterious | 0.994 | Probably Damaging | 0.892 | Possibly Damaging | 1.89 | Pathogenic | 0.01 | Affected | 3.38 | 19 | 0.1445 | 0.2828 | -2 | 0 | -2.9 | 59.07 | ||||||||||||||||||||||||
| c.931C>A | H311N 2D 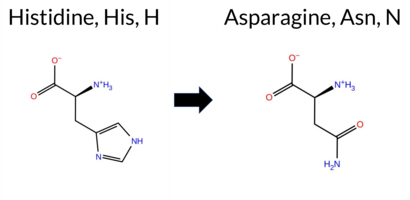 3DClick to see structure in 3D Viewer AIThe SynGAP1 H311N missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, SIFT, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain or inconclusive results are reported for FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as uncertain. Overall, the majority of evaluated tools predict pathogenicity, suggesting that H311N is most likely pathogenic. This prediction does not contradict ClinVar status, as no ClinVar classification is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.229226 | Structured | 0.354792 | Uncertain | 0.902 | 0.314 | 0.125 | -9.545 | Likely Pathogenic | 0.661 | Likely Pathogenic | Likely Benign | 0.82 | Ambiguous | 0.1 | 1.12 | Ambiguous | 0.97 | Ambiguous | 0.72 | Ambiguous | 0.475 | Likely Benign | -5.35 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.89 | Pathogenic | 0.06 | Tolerated | 0.1566 | 0.2380 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||
| c.956C>G | A319G 2D 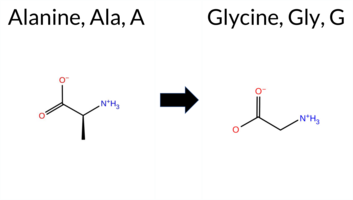 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A319G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all predict benign or likely benign. In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM predict pathogenicity, while Rosetta remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports benign. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.179055 | Structured | 0.410405 | Uncertain | 0.879 | 0.254 | 0.125 | -6.505 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.11 | Likely Benign | 0.4 | 0.56 | Ambiguous | 0.34 | Likely Benign | 0.39 | Likely Benign | 0.219 | Likely Benign | -1.83 | Neutral | 0.994 | Probably Damaging | 0.900 | Possibly Damaging | 1.89 | Pathogenic | 0.09 | Tolerated | 0.2345 | 0.4532 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.959T>G | V320G 2D 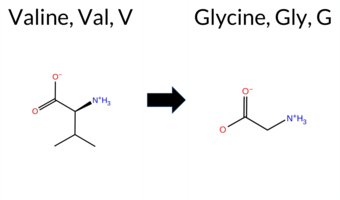 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V320G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools cluster into two agreement groups: the single benign prediction comes from REVEL, while the pathogenic group includes FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: Rosetta and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain; the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—returns pathogenic; and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, also predicts pathogenic. Overall, the preponderance of evidence indicates that V320G is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.185198 | Structured | 0.419626 | Uncertain | 0.905 | 0.266 | 0.125 | -9.043 | Likely Pathogenic | 0.816 | Likely Pathogenic | Ambiguous | 2.15 | Destabilizing | 1.1 | 1.87 | Ambiguous | 2.01 | Destabilizing | 1.48 | Destabilizing | 0.438 | Likely Benign | -5.74 | Deleterious | 0.958 | Probably Damaging | 0.999 | Probably Damaging | 1.89 | Pathogenic | 0.02 | Affected | 0.1688 | 0.1949 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.961C>T | R321C 2D 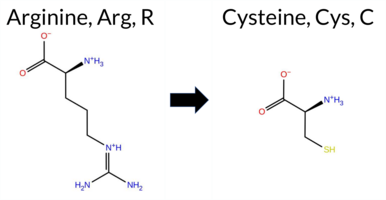 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R321C is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33437866‑C‑T). Prediction tools that agree on a benign effect include REVEL, premPS, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Five tools (SGM‑Consensus, FoldX, Rosetta, AlphaMissense‑Default, and Foldetta) report uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of predictions (six out of eleven) support a pathogenic impact, while three support benign and five are inconclusive. Thus, the variant is most likely pathogenic based on current computational evidence, and this does not contradict its ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.175930 | Structured | 0.423273 | Uncertain | 0.931 | 0.297 | 0.125 | Conflicting | 2 | 6-33437866-C-T | 9 | 5.58e-6 | -10.025 | Likely Pathogenic | 0.387 | Ambiguous | Likely Benign | 0.57 | Ambiguous | 0.1 | 0.56 | Ambiguous | 0.57 | Ambiguous | 0.18 | Likely Benign | 0.495 | Likely Benign | -4.59 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.89 | Pathogenic | 0.01 | Affected | 3.38 | 23 | 0.3313 | 0.2516 | -3 | -4 | 7.0 | -53.05 | ||||||||||||||||||||||
| c.991T>G | S331A 2D 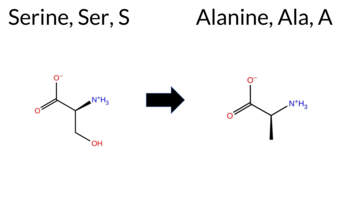 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S331A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only FATHMM predicts pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a benign majority vote (3 benign vs. 1 pathogenic). High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote) is benign; and Foldetta, combining FoldX‑MD and Rosetta outputs, is benign. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.433034 | Structured | 0.346458 | Uncertain | 0.658 | 0.475 | 0.250 | -2.199 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.1 | -0.08 | Likely Benign | 0.07 | Likely Benign | -0.15 | Likely Benign | 0.071 | Likely Benign | -0.44 | Neutral | 0.139 | Benign | 0.060 | Benign | 1.89 | Pathogenic | 0.67 | Tolerated | 0.5314 | 0.3008 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||
| c.998A>T | K333I 2D 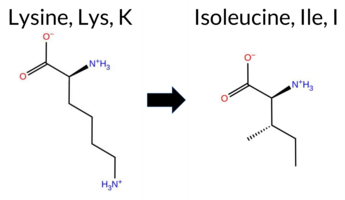 3DClick to see structure in 3D Viewer AIThe SynGAP1 K333I missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX and premPS, whereas the majority of tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; Rosetta and Foldetta give uncertain results. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.311707 | Structured | 0.330781 | Uncertain | 0.537 | 0.447 | 0.500 | -14.517 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.30 | Likely Benign | 0.0 | 0.95 | Ambiguous | 0.63 | Ambiguous | 0.43 | Likely Benign | 0.544 | Likely Pathogenic | -6.49 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.89 | Pathogenic | 0.03 | Affected | 0.0931 | 0.3521 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1015A>C | K339Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K339Q missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, and Foldetta. Those that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or folding stability result is missing or inconclusive. Overall, the majority of evaluated tools (8 pathogenic vs. 4 benign) indicate a pathogenic effect. This conclusion is not contradicted by ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -10.952 | Likely Pathogenic | 0.863 | Likely Pathogenic | Ambiguous | 0.06 | Likely Benign | 0.0 | -0.50 | Ambiguous | -0.22 | Likely Benign | -0.02 | Likely Benign | 0.458 | Likely Benign | -3.06 | Deleterious | 0.982 | Probably Damaging | 0.824 | Possibly Damaging | 1.90 | Pathogenic | 0.04 | Affected | 0.4041 | 0.1012 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1453C>T | R485C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R485C (gnomAD ID 6‑33438485‑C‑T) is listed in ClinVar with an uncertain significance. Functional prediction tools largely disagree: benign calls come from Rosetta and premPS, whereas pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus is labeled likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) remains uncertain. With the majority of evidence pointing to pathogenicity and no contradictory data from ClinVar, the variant is most likely pathogenic, although ClinVar has not yet reached a definitive classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.377409 | Uncertain | 0.805 | 0.246 | 0.125 | Uncertain | 2 | 6-33438485-C-T | 9 | 5.58e-6 | -14.294 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 1.00 | Ambiguous | 0.1 | 0.26 | Likely Benign | 0.63 | Ambiguous | 0.44 | Likely Benign | 0.597 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.90 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.3350 | 0.2762 | -4 | -3 | 7.0 | -53.05 | 225.5 | 99.6 | -0.1 | 0.0 | -0.3 | 0.2 | X | Uncertain | The guanidinium group of Arg485 is located in a short helical structure (res. Glu480-Leu482) within an α-α loop connecting the two α-helices (res. Ala461-Phe476 and Leu489-Glu519) at the GAP-Ras interface. The side chain of Arg485 acts as the “arginine finger” of SynGAP, playing a crucial role in Ras-GTPase activation. Consequently, the residue swap inhibits the conversion of GTP to GDP at the enzyme’s active site. Although no negative effects on the protein structure are observed during the simulations, no definite conclusions can be drawn due to the critical role of Arg485 in GTPase activation. | |||||||||||||
| c.794A>G | K265R 2D 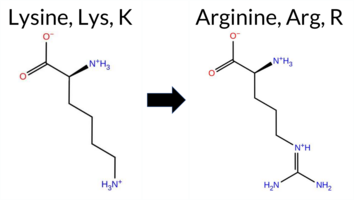 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K265R is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster into two groups: benign predictions include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports benign. No prediction or stability result is missing or inconclusive. Based on the aggregate evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.209395 | Structured | 0.309758 | Uncertain | 0.936 | 0.275 | 0.000 | -3.366 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.04 | Likely Benign | 0.1 | -0.44 | Likely Benign | -0.20 | Likely Benign | 0.30 | Likely Benign | 0.223 | Likely Benign | -1.08 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.90 | Pathogenic | 0.36 | Tolerated | 0.4713 | 0.0976 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.801G>C | W267C 2D 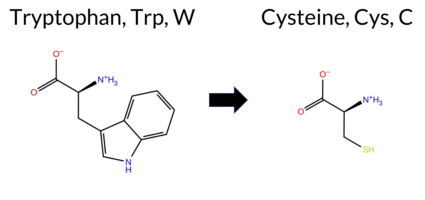 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W267C is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as pathogenic: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Thus, the variant is most likely pathogenic based on the consensus of all predictions, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.298060 | Uncertain | 0.943 | 0.274 | 0.000 | -12.351 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.81 | Destabilizing | 0.2 | 2.20 | Destabilizing | 2.51 | Destabilizing | 1.00 | Destabilizing | 0.705 | Likely Pathogenic | -11.95 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.90 | Pathogenic | 0.01 | Affected | 0.3573 | 0.1700 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||||
| c.801G>T | W267C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W267C is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as pathogenic: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Thus, the variant is most likely pathogenic based on the consensus of all predictions, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.298060 | Uncertain | 0.943 | 0.274 | 0.000 | -12.351 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.81 | Destabilizing | 0.2 | 2.20 | Destabilizing | 2.51 | Destabilizing | 1.00 | Destabilizing | 0.705 | Likely Pathogenic | -11.95 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.90 | Pathogenic | 0.01 | Affected | 0.3573 | 0.1700 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||||
| c.836G>C | R279P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R279P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess functional impact uniformly indicate a deleterious effect: REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as pathogenic. No tool predicts a benign effect. FoldX reports an uncertain outcome, but Rosetta, Foldetta, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all predict pathogenicity. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is pathogenic; the SGM‑Consensus, based on a majority vote of the aforementioned predictors, is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.309382 | Uncertain | 0.887 | 0.257 | 0.125 | -14.926 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 1.42 | Ambiguous | 0.9 | 4.27 | Destabilizing | 2.85 | Destabilizing | 1.21 | Destabilizing | 0.626 | Likely Pathogenic | -5.01 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.90 | Pathogenic | 0.02 | Affected | 0.2179 | 0.3281 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.853T>A | C285S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C285S has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include only SIFT, whereas a majority of the remaining predictors (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default) indicate a pathogenic impact. The remaining tools (FoldX, Rosetta, Foldetta, ESM1b, AlphaMissense‑Optimized) return uncertain results, which are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.144935 | Structured | 0.375400 | Uncertain | 0.946 | 0.250 | 0.000 | -7.149 | In-Between | 0.868 | Likely Pathogenic | Ambiguous | 0.73 | Ambiguous | 0.1 | 0.75 | Ambiguous | 0.74 | Ambiguous | 1.20 | Destabilizing | 0.507 | Likely Pathogenic | -8.09 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 1.90 | Pathogenic | 0.08 | Tolerated | 0.5210 | 0.2038 | Weaken | 0 | -1 | -3.3 | -16.06 | ||||||||||||||||||||||||||||
| c.854G>C | C285S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C285S is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and SIFT, whereas premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default all predict a pathogenic outcome. Predictions from FoldX, Rosetta, ESM1b, AlphaMissense‑Optimized, and Foldetta are inconclusive. High‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic classification; Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also uncertain. Overall, the preponderance of evidence points to a pathogenic effect for C285S, and this assessment does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.144935 | Structured | 0.375400 | Uncertain | 0.946 | 0.250 | 0.000 | -7.149 | In-Between | 0.868 | Likely Pathogenic | Ambiguous | 0.73 | Ambiguous | 0.1 | 0.75 | Ambiguous | 0.74 | Ambiguous | 1.20 | Destabilizing | 0.499 | Likely Benign | -8.09 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 1.90 | Pathogenic | 0.08 | Tolerated | 0.5210 | 0.2038 | Weaken | 0 | -1 | -3.3 | -16.06 | ||||||||||||||||||||||||||||
| c.883A>C | T295P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T295P is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect include FoldX and AlphaMissense‑Optimized, whereas the remaining evaluated algorithms (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) all predict a pathogenic outcome. High‑accuracy assessments further show that AlphaMissense‑Optimized classifies the variant as benign, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it as likely pathogenic, and Foldetta reports an uncertain stability change. With the majority of evidence pointing to deleterious effects, the variant is most likely pathogenic, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.401658 | Structured | 0.295548 | Uncertain | 0.881 | 0.288 | 0.125 | -9.941 | Likely Pathogenic | 0.688 | Likely Pathogenic | Likely Benign | 0.26 | Likely Benign | 0.2 | 3.30 | Destabilizing | 1.78 | Ambiguous | 0.60 | Ambiguous | 0.531 | Likely Pathogenic | -4.65 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.90 | Pathogenic | 0.02 | Affected | 0.2442 | 0.5327 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.937G>A | E313K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E313K is listed in ClinVar as Benign (ClinVar ID 3695040.0) and is not reported in gnomAD. Prediction tools that report a benign effect are absent; all available predictors that provide a definitive call classify the variant as pathogenic. These include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts Pathogenic, the SGM‑Consensus indicates Likely Pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) is Uncertain. Based on the overwhelming pathogenic predictions, the variant is most likely pathogenic, which contradicts its ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.170161 | Structured | 0.366526 | Uncertain | 0.898 | 0.304 | 0.125 | Likely Benign | 1 | -12.902 | Likely Pathogenic | 0.959 | Likely Pathogenic | Likely Pathogenic | 0.64 | Ambiguous | 0.6 | 1.40 | Ambiguous | 1.02 | Ambiguous | 0.75 | Ambiguous | 0.575 | Likely Pathogenic | -3.31 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 1.90 | Pathogenic | 0.02 | Affected | 0.2540 | 0.7708 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||
| c.944A>T | N315I 2D  3DClick to see structure in 3D Viewer AISynGAP1 variant N315I is not reported in ClinVar and is absent from gnomAD. In silico predictors that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, and AlphaMissense‑Optimized. Predictors that agree on a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. The high‑accuracy AlphaMissense‑Optimized score is benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; Foldetta, which integrates FoldX‑MD (uncertain) and Rosetta (benign), is considered unavailable. Overall, the balance of evidence leans toward pathogenicity, and this assessment does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -9.666 | Likely Pathogenic | 0.500 | Ambiguous | Likely Benign | -0.72 | Ambiguous | 0.4 | -0.17 | Likely Benign | -0.45 | Likely Benign | 0.36 | Likely Benign | 0.496 | Likely Benign | -5.19 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.90 | Pathogenic | 0.43 | Tolerated | 0.0763 | 0.7235 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.964G>C | A322P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A322P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, and SIFT, while pathogenic calls are made by SGM‑Consensus, Rosetta, Foldetta, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: FoldX and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar, which has no entry for this variant. Thus, the variant is most likely pathogenic, with no ClinVar evidence to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.175930 | Structured | 0.425745 | Uncertain | 0.938 | 0.334 | 0.000 | -10.558 | Likely Pathogenic | 0.878 | Likely Pathogenic | Ambiguous | 1.26 | Ambiguous | 1.3 | 5.43 | Destabilizing | 3.35 | Destabilizing | 0.40 | Likely Benign | 0.343 | Likely Benign | -1.72 | Neutral | 0.784 | Possibly Damaging | 0.390 | Benign | 1.90 | Pathogenic | 0.18 | Tolerated | 0.2064 | 0.5400 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1017G>C | K339N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K339N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and premPS. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the majority of evidence points to a pathogenic impact for K339N, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -11.117 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.20 | Likely Benign | 0.0 | 0.35 | Likely Benign | 0.28 | Likely Benign | 0.00 | Likely Benign | 0.410 | Likely Benign | -3.88 | Deleterious | 0.991 | Probably Damaging | 0.864 | Possibly Damaging | 1.91 | Pathogenic | 0.02 | Affected | 0.3224 | 0.1158 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1017G>T | K339N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K339N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and premPS. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta stability outputs) predicts a benign effect. Overall, the majority of evidence points toward a pathogenic impact for K339N, and this conclusion does not contradict any ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -11.117 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.20 | Likely Benign | 0.0 | 0.35 | Likely Benign | 0.28 | Likely Benign | 0.00 | Likely Benign | 0.410 | Likely Benign | -3.88 | Deleterious | 0.991 | Probably Damaging | 0.864 | Possibly Damaging | 1.91 | Pathogenic | 0.02 | Affected | 0.3224 | 0.1158 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1018G>C | A340P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A340P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, Foldetta, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The high‑accuracy predictors—AlphaMissense‑Optimized, the SGM‑Consensus, and Foldetta—each report a benign outcome. No prediction or folding‑stability result is missing or inconclusive; all available evidence points to a benign impact. Consequently, the variant is most likely benign based on the aggregate predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | -4.983 | Likely Benign | 0.264 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.5 | -0.58 | Ambiguous | -0.13 | Likely Benign | 0.46 | Likely Benign | 0.279 | Likely Benign | -1.48 | Neutral | 0.891 | Possibly Damaging | 0.575 | Possibly Damaging | 1.91 | Pathogenic | 0.25 | Tolerated | 0.2178 | 0.5468 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1019C>A | A340E 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant A340E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports a likely pathogenic outcome, while the protein‑folding stability method Foldetta is uncertain. AlphaMissense‑Optimized also yields an uncertain result. Overall, the majority of evidence points toward a pathogenic impact, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | -8.225 | Likely Pathogenic | 0.803 | Likely Pathogenic | Ambiguous | 0.63 | Ambiguous | 0.4 | 1.55 | Ambiguous | 1.09 | Ambiguous | 0.06 | Likely Benign | 0.138 | Likely Benign | -0.33 | Neutral | 0.625 | Possibly Damaging | 0.252 | Benign | 1.91 | Pathogenic | 0.39 | Tolerated | 0.1418 | 0.2160 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||
| c.2344G>T | D782Y 2D  AIThe SynGAP1 missense variant D782Y is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools largely converge on a deleterious effect: pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default, while only REVEL predicts a benign outcome. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is classified as Likely Pathogenic. Foldetta, a protein‑folding stability predictor that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -8.785 | Likely Pathogenic | 0.919 | Likely Pathogenic | Ambiguous | 0.382 | Likely Benign | -3.75 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.0559 | 0.6202 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2467A>T | S823C 2D  AIThe SynGAP1 missense variant S823C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. Two tools give uncertain results: ESM1b and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Pathogenic, while AlphaMissense‑Optimized remains uncertain. No Foldetta stability prediction is available, so it does not contribute to the assessment. Overall, the preponderance of evidence points to a pathogenic effect for S823C, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.685117 | Disordered | 0.627336 | Binding | 0.358 | 0.884 | 0.750 | -7.881 | In-Between | 0.911 | Likely Pathogenic | Ambiguous | 0.332 | Likely Benign | -3.80 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.1019 | 0.6137 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2474C>G | S825W 2D  AIThe SynGAP1 missense variant S825W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess pathogenicity largely agree: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all predict a deleterious effect. Only REVEL classifies the variant as benign, representing the sole benign prediction. High‑accuracy methods further support a pathogenic interpretation: AlphaMissense‑Optimized returns a pathogenic score, and the SGM‑Consensus indicates “Likely Pathogenic.” Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the consensus of the majority of tools, including the high‑accuracy predictors, indicates that S825W is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.618614 | Binding | 0.384 | 0.886 | 0.750 | -8.396 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.316 | Likely Benign | -5.25 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.0818 | 0.6083 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2533G>T | D845Y 2D AIThe SynGAP1 missense variant D845Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic. Foldetta results are not available for this variant. Overall, the preponderance of evidence from multiple in silico predictors indicates that D845Y is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -9.917 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.384 | Likely Benign | -6.55 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.0590 | 0.6699 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2534A>T | D845V 2D  AIThe SynGAP1 D845V missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL classifies it as benign, whereas the remaining 11 predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support a harmful outcome: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a pathogenic effect. This conclusion is consistent with the absence of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -8.914 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.426 | Likely Benign | -6.15 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.0871 | 0.7088 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3298G>T | G1100C 2D  AIThe SynGAP1 missense variant G1100C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -6.488 | Likely Benign | 0.150 | Likely Benign | Likely Benign | 0.174 | Likely Benign | -2.25 | Neutral | 0.999 | Probably Damaging | 0.950 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.1354 | 0.4768 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.794A>C | K265T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 K265T missense variant is reported in gnomAD (ID 6‑33437699‑A‑C) but has no ClinVar entry. Prediction tools that classify the variant as benign include REVEL, SIFT, and Rosetta. Those that predict pathogenicity are SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Four tools give uncertain results: FoldX, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.209395 | Structured | 0.309758 | Uncertain | 0.936 | 0.275 | 0.000 | 6-33437699-A-C | 1 | 6.20e-7 | -9.425 | Likely Pathogenic | 0.839 | Likely Pathogenic | Ambiguous | 0.99 | Ambiguous | 0.1 | 0.37 | Likely Benign | 0.68 | Ambiguous | 0.83 | Ambiguous | 0.441 | Likely Benign | -3.75 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.91 | Pathogenic | 0.07 | Tolerated | 3.38 | 18 | 0.2042 | 0.3178 | -1 | 0 | 3.2 | -27.07 | ||||||||||||||||||||||||
| c.836G>T | R279L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R279L is reported in gnomAD (ID 6‑33437741‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, and premPS. Those that predict a pathogenic effect comprise SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a unanimous majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a pathogenic impact. This conclusion is not contradicted by ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.309382 | Uncertain | 0.887 | 0.257 | 0.125 | 6-33437741-G-T | 1 | 6.20e-7 | -12.390 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 0.01 | Likely Benign | 0.2 | 0.14 | Likely Benign | 0.08 | Likely Benign | 0.39 | Likely Benign | 0.576 | Likely Pathogenic | -5.37 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.91 | Pathogenic | 0.03 | Affected | 3.39 | 18 | 0.1682 | 0.3266 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||
| c.887C>G | S296C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S296C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as benign. No predictions or stability results are missing or inconclusive. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.444081 | Structured | 0.282669 | Uncertain | 0.887 | 0.284 | 0.250 | -7.842 | In-Between | 0.286 | Likely Benign | Likely Benign | 0.46 | Likely Benign | 0.1 | -0.11 | Likely Benign | 0.18 | Likely Benign | 0.09 | Likely Benign | 0.361 | Likely Benign | -1.46 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.91 | Pathogenic | 0.05 | Affected | 0.1052 | 0.6052 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||
| c.893C>T | P298L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P298L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The remaining tools—FoldX, Rosetta, Foldetta, and ESM1b—return uncertain or inconclusive results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign outcome; and Foldetta’s stability prediction is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.328603 | Structured | 0.268765 | Uncertain | 0.860 | 0.283 | 0.500 | -7.334 | In-Between | 0.107 | Likely Benign | Likely Benign | 0.60 | Ambiguous | 0.2 | 1.53 | Ambiguous | 1.07 | Ambiguous | -0.16 | Likely Benign | 0.267 | Likely Benign | -0.82 | Neutral | 0.885 | Possibly Damaging | 0.589 | Possibly Damaging | 1.91 | Pathogenic | 0.21 | Tolerated | 0.2137 | 0.6795 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||
| c.943A>C | N315H 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant N315H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate benign or likely benign. In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM predict a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign stability. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -6.374 | Likely Benign | 0.156 | Likely Benign | Likely Benign | 0.29 | Likely Benign | 0.2 | 0.22 | Likely Benign | 0.26 | Likely Benign | 0.28 | Likely Benign | 0.258 | Likely Benign | -2.15 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.91 | Pathogenic | 0.76 | Tolerated | 0.1608 | 0.7296 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.952C>G | P318A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 P318A missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign outcome. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenicity, while AlphaMissense‑Optimized remains uncertain and Foldetta is also uncertain. Taken together, the overwhelming majority of evidence points to a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.111485 | Structured | 0.400936 | Uncertain | 0.858 | 0.234 | 0.000 | -9.642 | Likely Pathogenic | 0.872 | Likely Pathogenic | Ambiguous | 1.90 | Ambiguous | 0.2 | 1.69 | Ambiguous | 1.80 | Ambiguous | 0.94 | Ambiguous | 0.546 | Likely Pathogenic | -7.12 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.91 | Pathogenic | 0.04 | Affected | 0.3760 | 0.5585 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.997A>C | K333Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 K333Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, and SIFT, while those that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). Tools with inconclusive results are AlphaMissense‑Optimized, Foldetta, premPS, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as benign (combining FoldX‑MD and Rosetta outputs). Overall, the majority of evidence points toward a pathogenic classification, which does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.311707 | Structured | 0.330781 | Uncertain | 0.537 | 0.447 | 0.500 | -11.647 | Likely Pathogenic | 0.866 | Likely Pathogenic | Ambiguous | 0.00 | Likely Benign | 0.1 | 0.51 | Ambiguous | 0.26 | Likely Benign | 0.76 | Ambiguous | 0.444 | Likely Benign | -3.11 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.91 | Pathogenic | 0.08 | Tolerated | 0.4015 | 0.1219 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.997A>G | K333E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K333E missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, and SIFT. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a pathogenic impact for K333E. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.311707 | Structured | 0.330781 | Uncertain | 0.537 | 0.447 | 0.500 | -14.059 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | -0.06 | Likely Benign | 0.2 | 0.30 | Likely Benign | 0.12 | Likely Benign | 0.89 | Ambiguous | 0.488 | Likely Benign | -3.21 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.91 | Pathogenic | 0.09 | Tolerated | 0.3429 | 0.1039 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.999A>C | K333N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K333N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split opinion: benign predictions come from REVEL, FoldX, Rosetta, and SIFT, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). The high‑accuracy subset yields contrasting results: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus also indicates Likely Pathogenic, while Foldetta (combining FoldX‑MD and Rosetta stability outputs) predicts benign. premPS is inconclusive. Overall, the majority of tools lean toward pathogenicity, and the high‑accuracy predictions are not uniformly supportive of benign status. Therefore, K333N is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.311707 | Structured | 0.330781 | Uncertain | 0.537 | 0.447 | 0.500 | -11.821 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.30 | Likely Benign | 0.1 | 0.42 | Likely Benign | 0.36 | Likely Benign | 0.55 | Ambiguous | 0.359 | Likely Benign | -4.03 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.91 | Pathogenic | 0.13 | Tolerated | 0.3267 | 0.1315 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.999A>T | K333N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K333N has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, and SIFT, whereas a larger group predicts pathogenicity: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). The high‑accuracy methods give mixed results: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, whereas Foldetta (combining FoldX‑MD and Rosetta outputs) reports a benign stability change. premPS is inconclusive and is treated as unavailable. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.311707 | Structured | 0.330781 | Uncertain | 0.537 | 0.447 | 0.500 | -11.821 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.30 | Likely Benign | 0.1 | 0.42 | Likely Benign | 0.36 | Likely Benign | 0.55 | Ambiguous | 0.366 | Likely Benign | -4.03 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.91 | Pathogenic | 0.13 | Tolerated | 0.3267 | 0.1315 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1015A>G | K339E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K339E missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and premPS. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar annotation because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -14.284 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.13 | Likely Benign | 0.1 | -0.03 | Likely Benign | 0.05 | Likely Benign | 0.40 | Likely Benign | 0.482 | Likely Benign | -3.00 | Deleterious | 0.939 | Possibly Damaging | 0.670 | Possibly Damaging | 1.92 | Pathogenic | 0.03 | Affected | 0.3421 | 0.0882 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1016A>T | K339M 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 K339M missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include FoldX and premPS, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools with uncertain or inconclusive results are Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for K339M. This conclusion is not contradicted by ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -13.387 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.23 | Likely Benign | 0.0 | 0.88 | Ambiguous | 0.56 | Ambiguous | -0.37 | Likely Benign | 0.575 | Likely Pathogenic | -4.95 | Deleterious | 0.999 | Probably Damaging | 0.964 | Probably Damaging | 1.92 | Pathogenic | 0.01 | Affected | 0.0967 | 0.3541 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1019C>G | A340G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A340G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive, so they provide no evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of reliable predictors indicate a benign effect, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | -3.763 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.66 | Ambiguous | 0.2 | 1.44 | Ambiguous | 1.05 | Ambiguous | 0.50 | Likely Benign | 0.044 | Likely Benign | -0.34 | Neutral | 0.267 | Benign | 0.127 | Benign | 1.92 | Pathogenic | 0.42 | Tolerated | 0.2355 | 0.4470 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1453C>G | R485G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R485G is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (gnomAD ID: 6‑33438485‑C‑G). Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tools predict a benign outcome. Uncertain or inconclusive predictions come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Uncertain. Overall, the evidence strongly favors a pathogenic classification, and this conclusion does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.377409 | Uncertain | 0.805 | 0.246 | 0.125 | 6-33438485-C-G | 1 | 6.20e-7 | -15.777 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.84 | Ambiguous | 0.1 | 1.60 | Ambiguous | 1.22 | Ambiguous | 0.98 | Ambiguous | 0.631 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.3140 | 0.2678 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1454G>T | R485L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R485L is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include FoldX, Rosetta, Foldetta, and premPS, whereas the remaining tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus—predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta as benign. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which is currently unreported. Thus, the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.377409 | Uncertain | 0.805 | 0.246 | 0.125 | -15.807 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.23 | Likely Benign | 0.2 | 0.14 | Likely Benign | 0.19 | Likely Benign | 0.39 | Likely Benign | 0.631 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.1715 | 0.3784 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.2345A>T | D782V 2D AIThe SynGAP1 missense variant D782V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (which reports “Likely Pathogenic”). The high‑accuracy AlphaMissense‑Optimized tool yields an uncertain result, and the Foldetta stability assessment is unavailable. Overall, the consensus of the available predictions strongly favors a pathogenic effect for D782V. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -8.250 | Likely Pathogenic | 0.931 | Likely Pathogenic | Ambiguous | 0.462 | Likely Benign | -3.59 | Deleterious | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.0803 | 0.6477 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2468G>T | S823I 2D  AIThe SynGAP1 missense variant S823I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only REVEL predicts a benign outcome, while ESM1b remains uncertain. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.685117 | Disordered | 0.627336 | Binding | 0.358 | 0.884 | 0.750 | -7.332 | In-Between | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.287 | Likely Benign | -4.26 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.0964 | 0.5848 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2533G>C | D845H 2D  AIThe SynGAP1 missense variant D845H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL indicates a benign likelihood, whereas the remaining predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. The consensus score from the SGM framework, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic.” High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM consensus also reports a likely pathogenic status. Foldetta, a protein‑folding stability method that combines FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion is consistent with the absence of a ClinVar entry (no contradictory status). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -8.613 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.382 | Likely Benign | -5.08 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.1394 | 0.7515 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.3298G>C | G1100R 2D  AIThe SynGAP1 missense variant G1100R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of available predictions lean toward pathogenicity, and this does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -2.923 | Likely Benign | 0.603 | Likely Pathogenic | Likely Benign | 0.176 | Likely Benign | -2.05 | Neutral | 0.992 | Probably Damaging | 0.906 | Possibly Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.0986 | 0.5217 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.826C>T | P276S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P276S missense change is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign calls from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic calls from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Four tools predict benign while five predict pathogenic, and four additional methods (FoldX, Rosetta, Foldetta, premPS) return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive; Foldetta also reports an uncertain stability change. Taken together, the preponderance of evidence leans toward a pathogenic effect, and this assessment does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.037156 | Structured | 0.338937 | Uncertain | 0.724 | 0.230 | 0.250 | -5.946 | Likely Benign | 0.142 | Likely Benign | Likely Benign | 1.78 | Ambiguous | 0.2 | 0.96 | Ambiguous | 1.37 | Ambiguous | 0.65 | Ambiguous | 0.205 | Likely Benign | -3.25 | Deleterious | 0.835 | Possibly Damaging | 0.468 | Possibly Damaging | 1.92 | Pathogenic | 0.05 | Affected | 0.3191 | 0.3736 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||
| c.835C>G | R279G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R279G is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated tools predict a pathogenic impact: FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote) which labels it “Likely Pathogenic.” AlphaMissense‑Optimized is uncertain, providing no definitive evidence. High‑accuracy assessments further support pathogenicity: the SGM‑Consensus remains “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic effect. Based on the overwhelming majority of predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.309382 | Uncertain | 0.887 | 0.257 | 0.125 | -9.941 | Likely Pathogenic | 0.920 | Likely Pathogenic | Ambiguous | 3.10 | Destabilizing | 0.7 | 2.85 | Destabilizing | 2.98 | Destabilizing | 1.11 | Destabilizing | 0.442 | Likely Benign | -5.37 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.92 | Pathogenic | 0.02 | Affected | 0.2961 | 0.2209 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.847G>A | E283K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E283K is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL and FoldX, whereas the majority of algorithms predict it to be pathogenic: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Uncertain results come from Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.098513 | Structured | 0.358602 | Uncertain | 0.950 | 0.249 | 0.000 | -14.350 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.30 | Likely Benign | 0.1 | 0.82 | Ambiguous | 0.56 | Ambiguous | 0.62 | Ambiguous | 0.443 | Likely Benign | -3.68 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.92 | Pathogenic | 0.01 | Affected | 0.2989 | 0.5714 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.868C>A | L290M 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L290M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, FoldX, and PROVEAN, whereas pathogenic predictions are reported by polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Tools with uncertain outcomes include Foldetta, premPS, AlphaMissense‑Optimized, and Rosetta. High‑accuracy analyses indicate that AlphaMissense‑Optimized is inconclusive, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. Overall, the majority of evidence leans toward a pathogenic effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.127496 | Structured | 0.399723 | Uncertain | 0.904 | 0.255 | 0.000 | -8.393 | Likely Pathogenic | 0.901 | Likely Pathogenic | Ambiguous | 0.48 | Likely Benign | 0.2 | 0.98 | Ambiguous | 0.73 | Ambiguous | 0.59 | Ambiguous | 0.438 | Likely Benign | -1.84 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.92 | Pathogenic | 0.02 | Affected | 0.0823 | 0.4167 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.886T>C | S296P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S296P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, FoldX, Rosetta, and SIFT, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. When predictions are grouped by agreement, four tools predict benign and seven predict pathogenic, with premPS remaining uncertain. High‑accuracy methods give a mixed signal: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus, derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a benign effect. Thus, the majority of evidence leans toward pathogenicity, and this conclusion is consistent with the lack of ClinVar annotation and gnomAD absence, which do not contradict the prediction. Overall, the variant is most likely pathogenic based on the current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.444081 | Structured | 0.282669 | Uncertain | 0.887 | 0.284 | 0.250 | -8.302 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.04 | Likely Benign | 0.4 | 0.18 | Likely Benign | 0.11 | Likely Benign | 0.72 | Ambiguous | 0.439 | Likely Benign | -3.74 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.92 | Pathogenic | 0.12 | Tolerated | 0.2273 | 0.6320 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.893C>A | P298H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 P298H missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Remaining tools (AlphaMissense‑Default, FoldX, Rosetta, Foldetta, premPS) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta is uncertain. Overall, the majority of predictions (five pathogenic vs. three benign) and the SGM Consensus support a pathogenic interpretation. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.328603 | Structured | 0.268765 | Uncertain | 0.860 | 0.283 | 0.500 | -9.777 | Likely Pathogenic | 0.443 | Ambiguous | Likely Benign | 1.57 | Ambiguous | 0.2 | 1.49 | Ambiguous | 1.53 | Ambiguous | 0.83 | Ambiguous | 0.313 | Likely Benign | -2.37 | Neutral | 0.999 | Probably Damaging | 0.964 | Probably Damaging | 1.92 | Pathogenic | 0.04 | Affected | 0.1769 | 0.4943 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||
| c.920T>G | F307C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 F307C missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). In silico predictors overwhelmingly indicate a deleterious effect: all tools that provide a definitive call—REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The only predictions that are inconclusive are FoldX, Rosetta, and Foldetta, which are treated as unavailable. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely pathogenic status; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. **Based on the consensus of the available predictions, the variant is most likely pathogenic, and this assessment is not contradicted by any ClinVar status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.298791 | Structured | 0.327302 | Uncertain | 0.900 | 0.315 | 0.125 | -11.484 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.36 | Ambiguous | 0.1 | 1.44 | Ambiguous | 1.40 | Ambiguous | 1.05 | Destabilizing | 0.754 | Likely Pathogenic | -7.35 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.2729 | 0.1628 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.955G>A | A319T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A319T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen2_HumVar. Those that predict a pathogenic effect are polyPhen2_HumDiv and FATHMM. The remaining tools—FoldX, Rosetta, Foldetta, and ESM1b—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of reliable predictors classify the variant as benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.410405 | Uncertain | 0.879 | 0.254 | 0.125 | -7.841 | In-Between | 0.098 | Likely Benign | Likely Benign | 0.55 | Ambiguous | 0.3 | 0.70 | Ambiguous | 0.63 | Ambiguous | 0.30 | Likely Benign | 0.116 | Likely Benign | -1.35 | Neutral | 0.775 | Possibly Damaging | 0.306 | Benign | 1.92 | Pathogenic | 0.09 | Tolerated | 0.1274 | 0.6083 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||
| c.961C>G | R321G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R321G is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, Rosetta, AlphaMissense‑Default, AlphaMissense‑Optimized, and premPS. Tools that predict a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of conventional predictors lean toward pathogenicity, while the high‑accuracy subset is mixed, with one benign, one pathogenic, and one uncertain result. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.175930 | Structured | 0.423273 | Uncertain | 0.931 | 0.297 | 0.125 | -10.554 | Likely Pathogenic | 0.293 | Likely Benign | Likely Benign | 0.90 | Ambiguous | 0.1 | 0.28 | Likely Benign | 0.59 | Ambiguous | 0.49 | Likely Benign | 0.430 | Likely Benign | -3.70 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.92 | Pathogenic | 0.04 | Affected | 0.3224 | 0.2898 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.962G>A | R321H 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant R321H is listed in ClinVar as Benign (ClinVar ID 1940706) and is present in gnomAD (ID 6-33437867-G-A). Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools predicting a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM; premPS is reported as Uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, Foldetta as Benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence supports a benign effect, aligning with the ClinVar classification and indicating no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.175930 | Structured | 0.423273 | Uncertain | 0.931 | 0.297 | 0.125 | Benign | 1 | 6-33437867-G-A | 8 | 4.96e-6 | -8.751 | Likely Pathogenic | 0.136 | Likely Benign | Likely Benign | 0.48 | Likely Benign | 0.1 | -0.36 | Likely Benign | 0.06 | Likely Benign | 0.59 | Ambiguous | 0.323 | Likely Benign | -1.43 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.92 | Pathogenic | 0.25 | Tolerated | 3.38 | 23 | 0.2930 | 0.0936 | 2 | 0 | 1.3 | -19.05 | 218.5 | 86.9 | 1.1 | 0.0 | 0.3 | 0.0 | X | Potentially Benign | The guanidinium group of Arg321, located in a β hairpin loop linking two anti-parallel β sheet strands (res. Thr305-Asn315, res. Ala322-Asp330), faces outward without forming any stable interactions in the WT simulations. Similarly, in the variant simulations, the imidazole ring of His321 also points outward without making any stable intra-protein interactions. Thus, the residue swap does not seem to cause adverse effects on the protein structure based on the simulations. However, β hairpins are potential nucleation sites during the initial stages of protein folding, so even minor changes in them could be significant. | ||||||||||||||
| c.962G>C | R321P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R321P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. FoldX and Rosetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools (8/12) predict pathogenicity, whereas 4/12 predict benign, with two high‑accuracy tools supporting benign and one supporting pathogenic. Thus, the variant is most likely pathogenic based on the preponderance of evidence, and this assessment does not contradict ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.175930 | Structured | 0.423273 | Uncertain | 0.931 | 0.297 | 0.125 | -11.576 | Likely Pathogenic | 0.740 | Likely Pathogenic | Likely Benign | 1.22 | Ambiguous | 0.3 | -0.77 | Ambiguous | 0.23 | Likely Benign | 0.41 | Likely Benign | 0.488 | Likely Benign | -3.59 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.92 | Pathogenic | 0.03 | Affected | 0.2186 | 0.3661 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.971G>A | R324Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R324Q is listed in ClinVar with an uncertain significance (ClinVar ID 2572558.0) and is present in gnomAD (ID 6‑33437876‑G‑A). Prediction tools that classify the variant as benign include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates a likely benign outcome. Protein‑stability predictions from FoldX, Rosetta, and the combined Foldetta method are all uncertain. Overall, the consensus of available computational evidence points to a benign effect for R324Q, which is consistent with its ClinVar status of uncertain significance rather than a pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.257454 | Structured | 0.426893 | Uncertain | 0.954 | 0.397 | 0.000 | Uncertain | 3 | 6-33437876-G-A | 3 | 1.86e-6 | -5.001 | Likely Benign | 0.173 | Likely Benign | Likely Benign | 0.56 | Ambiguous | 0.1 | 0.63 | Ambiguous | 0.60 | Ambiguous | 1.02 | Destabilizing | 0.307 | Likely Benign | -1.17 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.92 | Pathogenic | 0.41 | Tolerated | 3.39 | 22 | 0.3804 | 0.3214 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||
| c.976C>T | H326Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H326Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, and premPS, whereas a majority of tools (REVEL, SIFT, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic impact. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the preponderance of evidence points to a pathogenic effect for H326Y, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.342579 | Structured | 0.418150 | Uncertain | 0.944 | 0.455 | 0.000 | -10.896 | Likely Pathogenic | 0.855 | Likely Pathogenic | Ambiguous | 0.03 | Likely Benign | 0.4 | 0.25 | Likely Benign | 0.14 | Likely Benign | 0.07 | Likely Benign | 0.562 | Likely Pathogenic | -5.32 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.92 | Pathogenic | 0.02 | Affected | 0.0796 | 0.4323 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||
| c.977A>C | H326P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H326P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions are absent; pathogenic predictions are reported by REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). The only inconclusive result is premPS, which is listed as Uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts Pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports Pathogenic. Based on the consensus of these predictions, the variant is most likely pathogenic, and this conclusion is consistent with the absence of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.342579 | Structured | 0.418150 | Uncertain | 0.944 | 0.455 | 0.000 | -18.452 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.67 | Destabilizing | 0.3 | 2.59 | Destabilizing | 2.63 | Destabilizing | 0.88 | Ambiguous | 0.711 | Likely Pathogenic | -8.73 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.92 | Pathogenic | 0.03 | Affected | 0.2383 | 0.4492 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||
| c.1018G>A | A340T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A340T is reported in gnomAD (ID 6‑33437923‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus all classify the change as benign or likely benign. Only two tools predict pathogenicity—polyPhen‑2 HumDiv and FATHMM—while stability‑based methods (FoldX, Rosetta, premPS, Foldetta) return uncertain or inconclusive results. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely benign, and Foldetta provides no definitive stability change. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | 6-33437923-G-A | -3.286 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.84 | Ambiguous | 0.2 | 0.96 | Ambiguous | 0.90 | Ambiguous | -0.54 | Ambiguous | 0.105 | Likely Benign | 0.62 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 1.93 | Pathogenic | 0.47 | Tolerated | 3.42 | 13 | 0.1740 | 0.7297 | 0 | 1 | -2.5 | 30.03 | ||||||||||||||||||||||||||
| c.1018G>T | A340S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A340S is reported in gnomAD (variant ID 6‑33437923‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only FATHMM predicts pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a benign majority (3 benign vs. 1 pathogenic). High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign; the SGM‑Consensus is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | 6-33437923-G-T | 1 | 6.20e-7 | -0.705 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.15 | Likely Benign | 0.0 | 0.27 | Likely Benign | 0.21 | Likely Benign | -0.46 | Likely Benign | 0.083 | Likely Benign | 1.62 | Neutral | 0.007 | Benign | 0.008 | Benign | 1.93 | Pathogenic | 0.51 | Tolerated | 3.42 | 13 | 0.2852 | 0.6309 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||
| c.1453C>A | R485S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R485S is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FoldX, which scores the variant as benign. In contrast, the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that return uncertain results are Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for R485S, which is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.377409 | Uncertain | 0.805 | 0.246 | 0.125 | Uncertain | 1 | -15.603 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.40 | Likely Benign | 0.1 | 1.07 | Ambiguous | 0.74 | Ambiguous | 0.82 | Ambiguous | 0.609 | Likely Pathogenic | -5.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.2968 | 0.3266 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||
| c.1454G>A | R485H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R485H missense variant is listed in ClinVar as Benign (ClinVar ID 3707943.0) and is present in the gnomAD database (gnomAD ID 6‑33438486‑G‑A). Functional prediction tools that agree on a benign effect are Rosetta and Foldetta, while the majority of tools (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the preponderance of evidence points to a pathogenic effect, which contradicts the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.377409 | Uncertain | 0.805 | 0.246 | 0.125 | Likely Benign | 1 | 6-33438486-G-A | 13 | 8.05e-6 | -13.628 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 0.77 | Ambiguous | 0.1 | 0.12 | Likely Benign | 0.45 | Likely Benign | 1.13 | Destabilizing | 0.618 | Likely Pathogenic | -4.97 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.2990 | 0.1602 | 0 | 2 | 1.3 | -19.05 | ||||||||||||||||||||||
| c.1627C>A | L543M 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L543M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL and PROVEAN, whereas pathogenic calls are made by polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Rosetta. Predictions that are uncertain (FoldX, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments give AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the majority of available tools predict a deleterious effect, indicating that the variant is most likely pathogenic. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.020918 | Uncertain | 0.963 | 0.314 | 0.000 | -11.452 | Likely Pathogenic | 0.922 | Likely Pathogenic | Ambiguous | 0.68 | Ambiguous | 0.2 | 2.53 | Destabilizing | 1.61 | Ambiguous | 0.99 | Ambiguous | 0.382 | Likely Benign | -1.99 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.93 | Pathogenic | 0.01 | Affected | 0.0788 | 0.2172 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.2344G>C | D782H 2D AIThe SynGAP1 missense variant D782H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). Only REVEL predicts a benign outcome, while AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show the SGM‑Consensus as Likely Pathogenic, whereas AlphaMissense‑Optimized remains inconclusive and Foldetta data are unavailable. Taken together, the majority of evidence supports a pathogenic interpretation, and this is consistent with the absence of a ClinVar assertion. Therefore, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -8.528 | Likely Pathogenic | 0.937 | Likely Pathogenic | Ambiguous | 0.311 | Likely Benign | -2.63 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.1333 | 0.7286 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.2356C>A | L786I 2D AIThe SynGAP1 missense variant L786I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence, especially from the high‑accuracy methods, points to a benign impact. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | -5.464 | Likely Benign | 0.215 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -1.12 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.1056 | 0.4199 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||||||||||
| c.2467A>C | S823R 2D  AIThe SynGAP1 missense variant S823R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all indicate likely pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta results are not available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy prediction tools points to a pathogenic effect for S823R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.685117 | Disordered | 0.627336 | Binding | 0.358 | 0.884 | 0.750 | -6.612 | Likely Benign | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.293 | Likely Benign | -3.47 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.0893 | 0.4046 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2468G>A | S823N 2D  AIThe SynGAP1 missense variant S823N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b. Tools that agree on a pathogenic effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic, two benign votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.685117 | Disordered | 0.627336 | Binding | 0.358 | 0.884 | 0.750 | -6.880 | Likely Benign | 0.974 | Likely Pathogenic | Likely Pathogenic | 0.170 | Likely Benign | -1.76 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.1370 | 0.5172 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2469C>A | S823R 2D AIThe SynGAP1 missense variant S823R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus also indicates Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.685117 | Disordered | 0.627336 | Binding | 0.358 | 0.884 | 0.750 | -6.612 | Likely Benign | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.196 | Likely Benign | -3.47 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.0893 | 0.4046 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2469C>G | S823R 2D AIThe SynGAP1 missense variant S823R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus also indicates Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.685117 | Disordered | 0.627336 | Binding | 0.358 | 0.884 | 0.750 | -6.612 | Likely Benign | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.196 | Likely Benign | -3.47 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.0893 | 0.4046 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2867C>A | S956Y 2D  AIThe SynGAP1 missense variant S956Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Thus, the variant is most likely benign, with no contradiction from ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.957345 | Binding | 0.364 | 0.917 | 0.750 | -4.920 | Likely Benign | 0.233 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -0.90 | Neutral | 0.411 | Benign | 0.097 | Benign | 1.93 | Pathogenic | 0.22 | Tolerated | 0.1449 | 0.4932 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||||
| c.2867C>T | S956F 2D  AIThe SynGAP1 missense variant S956F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.957345 | Binding | 0.364 | 0.917 | 0.750 | -6.654 | Likely Benign | 0.226 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -1.04 | Neutral | 0.832 | Possibly Damaging | 0.398 | Benign | 1.93 | Pathogenic | 0.39 | Tolerated | 0.1315 | 0.4943 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.3299G>A | G1100D 2D AIThe SynGAP1 missense variant G1100D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -5.235 | Likely Benign | 0.577 | Likely Pathogenic | Likely Benign | 0.141 | Likely Benign | -1.67 | Neutral | 0.049 | Benign | 0.030 | Benign | 1.93 | Pathogenic | 0.01 | Affected | 0.1924 | 0.3043 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3299G>T | G1100V 2D  AIThe SynGAP1 missense variant G1100V is reported in gnomAD (variant ID 6‑33443851‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign); pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the balance of evidence favors a benign classification for G1100V, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | 6-33443851-G-T | 1 | 6.51e-7 | -2.362 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.198 | Likely Benign | -2.43 | Neutral | 0.992 | Probably Damaging | 0.906 | Possibly Damaging | 1.93 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1297 | 0.4036 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||
| c.3764A>G | K1255R 2D AIThe SynGAP1 K1255R missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and PROVEAN, whereas a majority of tools predict a pathogenic impact: polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all classify the change as damaging. The high‑accuracy consensus approach (SGM‑Consensus) – a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN – yields a pathogenic verdict (3 pathogenic vs. 1 benign). AlphaMissense‑Optimized is uncertain, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for K1255R. This prediction is consistent with the lack of ClinVar annotation; there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.637480 | Disordered | 0.417615 | Uncertain | 0.880 | 0.563 | 0.625 | -9.687 | Likely Pathogenic | 0.866 | Likely Pathogenic | Ambiguous | 0.171 | Likely Benign | -2.43 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.3727 | 0.0878 | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||||||
| c.800G>T | W267L 2D  AIThe SynGAP1 missense variant W267L is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. No tool in the dataset predicts a benign effect. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS, which are treated as unavailable evidence. High‑accuracy assessments further support a damaging effect: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic,” while Foldetta’s stability prediction is uncertain. Overall, the evidence strongly indicates that W267L is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.298060 | Uncertain | 0.943 | 0.274 | 0.000 | -13.670 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.19 | Ambiguous | 0.7 | 1.31 | Ambiguous | 1.25 | Ambiguous | 0.75 | Ambiguous | 0.628 | Likely Pathogenic | -11.95 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.93 | Pathogenic | 0.01 | Affected | 0.2122 | 0.2843 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||
| c.884C>A | T295N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 T295N missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, ESM1b, and Foldetta, while those that predict a pathogenic outcome are SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, seven tools predict pathogenicity versus four predicting benign, with no ClinVar evidence to contradict this assessment. Thus, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.401658 | Structured | 0.295548 | Uncertain | 0.881 | 0.288 | 0.125 | -6.588 | Likely Benign | 0.793 | Likely Pathogenic | Ambiguous | 0.24 | Likely Benign | 0.1 | 0.66 | Ambiguous | 0.45 | Likely Benign | 0.89 | Ambiguous | 0.438 | Likely Benign | -3.51 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.93 | Pathogenic | 0.03 | Affected | 0.1522 | 0.4457 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.887C>A | S296Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S296Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and premPS, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX and Foldetta give uncertain results. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic, and Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic impact. The variant is most likely pathogenic based on predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.444081 | Structured | 0.282669 | Uncertain | 0.887 | 0.284 | 0.250 | -12.148 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.46 | Ambiguous | 0.2 | 0.48 | Likely Benign | 0.97 | Ambiguous | 0.15 | Likely Benign | 0.486 | Likely Benign | -4.34 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.93 | Pathogenic | 0.05 | Affected | 0.0800 | 0.5610 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||
| c.938A>T | E313V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 E313V missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, and premPS, whereas a majority of tools (REVEL, SIFT, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic impact. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the preponderance of evidence points to a pathogenic effect for E313V. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.170161 | Structured | 0.366526 | Uncertain | 0.898 | 0.304 | 0.125 | -13.169 | Likely Pathogenic | 0.821 | Likely Pathogenic | Ambiguous | 0.34 | Likely Benign | 0.1 | -0.02 | Likely Benign | 0.16 | Likely Benign | 0.21 | Likely Benign | 0.655 | Likely Pathogenic | -5.73 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.93 | Pathogenic | 0.05 | Affected | 0.1132 | 0.7900 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.943A>T | N315Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N315Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. The high‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the balance of evidence slightly favors a pathogenic interpretation, with six pathogenic‑predicted tools versus five benign‑predicted tools, and the high‑accuracy consensus leaning pathogenic. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -8.303 | Likely Pathogenic | 0.339 | Likely Benign | Likely Benign | -1.03 | Ambiguous | 0.5 | -0.85 | Ambiguous | -0.94 | Ambiguous | -0.10 | Likely Benign | 0.420 | Likely Benign | -3.81 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.93 | Pathogenic | 1.00 | Tolerated | 0.0613 | 0.6732 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.959T>A | V320D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V320D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL and SIFT; pathogenic predictions from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Stability‑based methods FoldX and Rosetta returned uncertain results, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also yielded an uncertain prediction. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points toward a pathogenic effect, which is consistent with the lack of ClinVar annotation and gnomAD absence. Thus, the variant is most likely pathogenic, and this prediction does not contradict ClinVar status because ClinVar has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.185198 | Structured | 0.419626 | Uncertain | 0.905 | 0.266 | 0.125 | -11.269 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.99 | Ambiguous | 1.0 | 1.67 | Ambiguous | 1.83 | Ambiguous | 1.50 | Destabilizing | 0.405 | Likely Benign | -5.58 | Deleterious | 0.999 | Probably Damaging | 0.972 | Probably Damaging | 1.93 | Pathogenic | 0.07 | Tolerated | 0.1101 | 0.0482 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.962G>T | R321L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R321L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include SIFT, polyPhen‑2 (HumDiv and HumVar), PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign; FoldX and Rosetta individually are inconclusive. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.175930 | Structured | 0.423273 | Uncertain | 0.931 | 0.297 | 0.125 | -11.709 | Likely Pathogenic | 0.551 | Ambiguous | Likely Benign | 0.54 | Ambiguous | 0.0 | -0.58 | Ambiguous | -0.02 | Likely Benign | 0.18 | Likely Benign | 0.451 | Likely Benign | -4.06 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.93 | Pathogenic | 0.03 | Affected | 0.1745 | 0.4048 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.1016A>C | K339T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K339T missense variant is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools are split: benign calls come from FoldX, Rosetta, Foldetta, premPS, and SIFT, whereas pathogenic calls are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further highlight this discordance: the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect, whereas Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, predicts a benign impact. AlphaMissense‑Optimized returned an uncertain result and is treated as unavailable. Overall, the majority of robust predictors lean toward pathogenicity, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -10.061 | Likely Pathogenic | 0.910 | Likely Pathogenic | Ambiguous | 0.32 | Likely Benign | 0.0 | 0.10 | Likely Benign | 0.21 | Likely Benign | -0.04 | Likely Benign | 0.512 | Likely Pathogenic | -4.73 | Deleterious | 0.991 | Probably Damaging | 0.795 | Possibly Damaging | 1.94 | Pathogenic | 0.10 | Tolerated | 0.1900 | 0.3020 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1016A>G | K339R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K339R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is benign. No predictions or stability results are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -5.773 | Likely Benign | 0.120 | Likely Benign | Likely Benign | -0.15 | Likely Benign | 0.1 | 0.46 | Likely Benign | 0.16 | Likely Benign | 0.08 | Likely Benign | 0.134 | Likely Benign | -2.05 | Neutral | 0.046 | Benign | 0.040 | Benign | 1.94 | Pathogenic | 0.48 | Tolerated | 0.4197 | 0.0976 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.1454G>C | R485P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R485P is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return pathogenic or likely pathogenic scores. No tool in the dataset predicts a benign effect. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a pathogenic impact. Based on the unanimous pathogenic predictions and the absence of any benign calls, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.377409 | Uncertain | 0.805 | 0.246 | 0.125 | -16.356 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 5.26 | Destabilizing | 0.3 | 6.86 | Destabilizing | 6.06 | Destabilizing | 0.56 | Ambiguous | 0.692 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.94 | Pathogenic | 0.00 | Affected | 0.2059 | 0.3941 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.2473T>C | S825P 2D AIThe SynGAP1 missense variant S825P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. Foldetta results are not available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that S825P is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.618614 | Binding | 0.384 | 0.886 | 0.750 | -3.227 | Likely Benign | 0.959 | Likely Pathogenic | Likely Pathogenic | 0.285 | Likely Benign | -3.12 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.94 | Pathogenic | 0.02 | Affected | 0.2060 | 0.5998 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2474C>T | S825L 2D AIThe SynGAP1 missense variant S825L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443026‑C‑T). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain,” and the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a pathogenic effect, which is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.618614 | Binding | 0.384 | 0.886 | 0.750 | Uncertain | 1 | 6-33443026-C-T | 1 | 6.20e-7 | -4.987 | Likely Benign | 0.910 | Likely Pathogenic | Ambiguous | 0.249 | Likely Benign | -4.30 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.94 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1252 | 0.5747 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||
| c.2867C>G | S956C 2D AIThe SynGAP1 missense variant S956C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. four benign) indicate a likely pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant has no existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984871 | Disordered | 0.957345 | Binding | 0.364 | 0.917 | 0.750 | -9.292 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.107 | Likely Benign | -0.34 | Neutral | 0.938 | Possibly Damaging | 0.665 | Possibly Damaging | 1.94 | Pathogenic | 0.03 | Affected | 0.1833 | 0.5470 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3604A>C | I1202L 2D AIThe SynGAP1 I1202L missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, and SIFT, whereas those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) remains Likely Pathogenic; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation (none exists). Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.529623 | Disordered | 0.510422 | Binding | 0.874 | 0.593 | 0.250 | -8.026 | Likely Pathogenic | 0.953 | Likely Pathogenic | Ambiguous | 0.119 | Likely Benign | -1.27 | Neutral | 0.981 | Probably Damaging | 0.970 | Probably Damaging | 1.94 | Pathogenic | 0.59 | Tolerated | 0.0788 | 0.2874 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||||||||||
| c.3784A>C | I1262L 2D AIThe SynGAP1 missense variant I1262L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and PROVEAN, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; ESM1b remains uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (pathogenic), and PROVEAN (benign)—also indicates pathogenic. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic impact for I1262L, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.497853 | Structured | 0.707863 | Binding | 0.886 | 0.576 | 0.125 | -7.334 | In-Between | 0.962 | Likely Pathogenic | Likely Pathogenic | 0.269 | Likely Benign | -1.66 | Neutral | 0.981 | Probably Damaging | 0.970 | Probably Damaging | 1.94 | Pathogenic | 0.00 | Affected | 0.0735 | 0.3246 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3787A>C | I1263L 2D AIThe SynGAP1 missense variant I1263L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign, two pathogenic). Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.425610 | Structured | 0.740957 | Binding | 0.867 | 0.574 | 0.000 | -1.210 | Likely Benign | 0.699 | Likely Pathogenic | Likely Benign | 0.194 | Likely Benign | -1.66 | Neutral | 0.011 | Benign | 0.022 | Benign | 1.94 | Pathogenic | 0.00 | Affected | 0.0717 | 0.2890 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.793A>G | K265E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K265E missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, and Foldetta. Those that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are uncertain or inconclusive are AlphaMissense‑Optimized, FoldX, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evaluated tools (eight pathogenic vs. three benign) indicate a pathogenic effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.209395 | Structured | 0.309758 | Uncertain | 0.936 | 0.275 | 0.000 | -12.163 | Likely Pathogenic | 0.952 | Likely Pathogenic | Ambiguous | 0.63 | Ambiguous | 0.2 | 0.00 | Likely Benign | 0.32 | Likely Benign | 0.95 | Ambiguous | 0.461 | Likely Benign | -2.79 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.94 | Pathogenic | 0.05 | Affected | 0.3801 | 0.0882 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.836G>A | R279Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R279Q is reported in gnomAD (ID 6‑33437741‑G‑A) but has no ClinVar entry. Prediction tools that agree on a benign effect are SIFT and AlphaMissense‑Optimized; those that agree on a pathogenic effect are SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. FoldX, Rosetta, and Foldetta give uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta remains uncertain. Overall, the majority of reliable predictors (nine pathogenic vs two benign) indicate a pathogenic effect. Thus, the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.309382 | Uncertain | 0.887 | 0.257 | 0.125 | 6-33437741-G-A | 6 | 3.72e-6 | -8.730 | Likely Pathogenic | 0.761 | Likely Pathogenic | Likely Benign | 1.37 | Ambiguous | 0.1 | 0.88 | Ambiguous | 1.13 | Ambiguous | 1.35 | Destabilizing | 0.554 | Likely Pathogenic | -2.61 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.94 | Pathogenic | 0.06 | Tolerated | 3.39 | 18 | 0.2680 | 0.1792 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||
| c.838T>C | Y280H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y280H is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Among the available in‑silico predictors, none indicate a benign effect; all 14 tools that produced a classification predict pathogenicity: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are inconclusive or missing. Based on the unanimous pathogenic predictions and the absence of any benign calls, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status (which is currently unreported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.161087 | Structured | 0.321243 | Uncertain | 0.917 | 0.248 | 0.125 | -9.970 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.01 | Destabilizing | 0.3 | 2.58 | Destabilizing | 2.80 | Destabilizing | 1.95 | Destabilizing | 0.588 | Likely Pathogenic | -4.60 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.94 | Pathogenic | 0.01 | Affected | 0.2021 | 0.0212 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||
| c.838T>G | Y280D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y280D is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a pathogenic outcome. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. With all available evidence pointing to a damaging effect and no ClinVar entry to contradict, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.161087 | Structured | 0.321243 | Uncertain | 0.917 | 0.248 | 0.125 | -16.885 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.91 | Destabilizing | 0.2 | 5.89 | Destabilizing | 5.90 | Destabilizing | 2.00 | Destabilizing | 0.580 | Likely Pathogenic | -9.19 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.94 | Pathogenic | 0.02 | Affected | 0.3593 | 0.0212 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.866T>A | M289K 2D  3DClick to see structure in 3D Viewer AISynGAP1 M289K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Because the majority of consensus and individual predictors lean toward pathogenicity, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status since none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.127496 | Structured | 0.403499 | Uncertain | 0.886 | 0.276 | 0.000 | -12.192 | Likely Pathogenic | 0.738 | Likely Pathogenic | Likely Benign | 0.36 | Likely Benign | 0.2 | 0.60 | Ambiguous | 0.48 | Likely Benign | 0.94 | Ambiguous | 0.229 | Likely Benign | -2.52 | Deleterious | 0.891 | Possibly Damaging | 0.492 | Possibly Damaging | 1.94 | Pathogenic | 0.12 | Tolerated | 0.1271 | 0.0879 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||
| c.887C>T | S296F 2D AIThe SynGAP1 missense variant S296F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, and premPS. In contrast, the majority of tools predict a pathogenic impact: SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic; SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive and therefore unavailable as evidence. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.444081 | Structured | 0.282669 | Uncertain | 0.887 | 0.284 | 0.250 | -11.721 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.29 | Ambiguous | 1.6 | 0.24 | Likely Benign | 0.77 | Ambiguous | 0.29 | Likely Benign | 0.494 | Likely Benign | -4.34 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.94 | Pathogenic | 0.04 | Affected | 0.0720 | 0.5905 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||
| c.892C>G | P298A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P298A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No evidence from FoldX or Rosetta alone is conclusive. Based on the preponderance of benign predictions and the lack of pathogenic consensus, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.328603 | Structured | 0.268765 | Uncertain | 0.860 | 0.283 | 0.500 | -6.082 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 1.22 | Ambiguous | 0.1 | 1.17 | Ambiguous | 1.20 | Ambiguous | 0.50 | Likely Benign | 0.191 | Likely Benign | -0.98 | Neutral | 0.885 | Possibly Damaging | 0.589 | Possibly Damaging | 1.94 | Pathogenic | 0.66 | Tolerated | 0.3761 | 0.5787 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.898T>A | S300T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S300T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only FATHMM indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM‑Consensus (3 benign vs. 1 pathogenic) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. No prediction or stability result is inconclusive. Overall, the evidence overwhelmingly supports a benign classification, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.256848 | Uncertain | 0.742 | 0.280 | 0.375 | -4.648 | Likely Benign | 0.069 | Likely Benign | Likely Benign | 0.85 | Ambiguous | 0.1 | -0.30 | Likely Benign | 0.28 | Likely Benign | -0.33 | Likely Benign | 0.056 | Likely Benign | 0.50 | Neutral | 0.003 | Benign | 0.002 | Benign | 1.94 | Pathogenic | 0.85 | Tolerated | 0.1409 | 0.6428 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.931C>G | H311D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H311D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that assess evolutionary conservation and protein function uniformly indicate a deleterious effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as pathogenic. The majority‑vote consensus (SGM‑Consensus) also reports it as likely pathogenic. Tools that evaluate structural stability give inconclusive results: FoldX, Rosetta, Foldetta, and premPS are listed as uncertain. High‑accuracy assessments further support a damaging outcome: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (derived from the same set of predictors) reports likely pathogenic, while Foldetta’s combined FoldX‑MD/Rosetta stability analysis remains uncertain. Overall, the preponderance of evidence from multiple independent predictors points to a pathogenic effect, which is consistent with the absence of a benign ClinVar annotation and the lack of population frequency data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.229226 | Structured | 0.354792 | Uncertain | 0.902 | 0.314 | 0.125 | -13.513 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 1.27 | Ambiguous | 0.0 | 1.83 | Ambiguous | 1.55 | Ambiguous | 0.89 | Ambiguous | 0.633 | Likely Pathogenic | -6.94 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 1.94 | Pathogenic | 0.02 | Affected | 0.2301 | 0.2008 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||
| c.932A>G | H311R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H311R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, SIFT, and AlphaMissense‑Optimized, whereas a majority of tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic outcome. Tools with uncertain or inconclusive results are Rosetta, Foldetta, and premPS. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized indicates benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates pathogenic, and Foldetta remains uncertain. Overall, the balance of evidence favors a pathogenic classification, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.229226 | Structured | 0.354792 | Uncertain | 0.902 | 0.314 | 0.125 | -9.825 | Likely Pathogenic | 0.719 | Likely Pathogenic | Likely Benign | 0.43 | Likely Benign | 0.1 | 0.85 | Ambiguous | 0.64 | Ambiguous | 0.70 | Ambiguous | 0.532 | Likely Pathogenic | -5.72 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 1.94 | Pathogenic | 0.14 | Tolerated | 0.1931 | 0.2490 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||
| c.933C>A | H311Q 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 H311Q missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, and Foldetta. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and premPS. The high‑accuracy consensus methods give mixed signals: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, more tools (seven) predict pathogenicity than benign (four), and the SGM Consensus supports a pathogenic classification, while Foldetta offers a contrary benign prediction. The variant is most likely pathogenic based on the aggregate predictions, and this assessment does not contradict the absence of a ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.229226 | Structured | 0.354792 | Uncertain | 0.902 | 0.314 | 0.125 | -8.656 | Likely Pathogenic | 0.792 | Likely Pathogenic | Ambiguous | 0.15 | Likely Benign | 0.1 | 0.39 | Likely Benign | 0.27 | Likely Benign | 0.92 | Ambiguous | 0.414 | Likely Benign | -5.95 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.94 | Pathogenic | 0.03 | Affected | 0.1490 | 0.3775 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.933C>G | H311Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H311Q missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, and Foldetta. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. With seven pathogenic versus four benign predictions, the overall consensus leans toward pathogenicity. This conclusion does not contradict ClinVar status, as no ClinVar classification is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.229226 | Structured | 0.354792 | Uncertain | 0.902 | 0.314 | 0.125 | -8.656 | Likely Pathogenic | 0.792 | Likely Pathogenic | Ambiguous | 0.15 | Likely Benign | 0.1 | 0.39 | Likely Benign | 0.27 | Likely Benign | 0.92 | Ambiguous | 0.414 | Likely Benign | -5.95 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.94 | Pathogenic | 0.03 | Affected | 0.1490 | 0.3775 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.965C>G | A322G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A322G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score. Only FATHMM predicts a pathogenic outcome. Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain or inconclusive and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Based on the preponderance of benign predictions and the lack of contradictory evidence, the variant is most likely benign; this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.175930 | Structured | 0.425745 | Uncertain | 0.938 | 0.334 | 0.000 | -5.230 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.84 | Ambiguous | 0.1 | 1.43 | Ambiguous | 1.14 | Ambiguous | 0.64 | Ambiguous | 0.054 | Likely Benign | -1.07 | Neutral | 0.139 | Benign | 0.088 | Benign | 1.94 | Pathogenic | 0.19 | Tolerated | 0.2513 | 0.4683 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1066C>G | R356G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R356G missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and SIFT, while the majority of tools predict a pathogenic outcome: SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are uncertain and therefore treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. Overall, the preponderance of pathogenic predictions indicates that the variant is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which contains no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.219301 | Structured | 0.395028 | Uncertain | 0.802 | 0.373 | 0.250 | -12.305 | Likely Pathogenic | 0.883 | Likely Pathogenic | Ambiguous | 1.02 | Ambiguous | 0.0 | 1.80 | Ambiguous | 1.41 | Ambiguous | 1.06 | Destabilizing | 0.271 | Likely Benign | -6.20 | Deleterious | 0.993 | Probably Damaging | 0.982 | Probably Damaging | 1.95 | Pathogenic | 0.08 | Tolerated | 0.3505 | 0.3793 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.2345A>C | D782A 2D  AIThe SynGAP1 missense variant D782A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all classify the variant as damaging. Uncertain predictions come from ESM1b and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, reports the variant as Likely Pathogenic, while AlphaMissense‑Optimized remains uncertain and Foldetta results are unavailable. Taken together, the preponderance of evidence from multiple in silico predictors and the SGM‑Consensus suggests that D782A is most likely pathogenic. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -7.054 | In-Between | 0.892 | Likely Pathogenic | Ambiguous | 0.345 | Likely Benign | -3.33 | Deleterious | 0.990 | Probably Damaging | 0.932 | Probably Damaging | 1.95 | Pathogenic | 0.01 | Affected | 0.3819 | 0.6121 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2345A>G | D782G 2D  AIThe SynGAP1 missense variant D782G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic effect: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Pathogenic” verdict (3 pathogenic vs. 1 benign). AlphaMissense‑Optimized is uncertain, and Foldetta results are unavailable. Based on the overall pattern of predictions, the variant is most likely pathogenic, and this assessment does not contradict any existing ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -6.811 | Likely Benign | 0.858 | Likely Pathogenic | Ambiguous | 0.291 | Likely Benign | -3.27 | Deleterious | 0.995 | Probably Damaging | 0.950 | Probably Damaging | 1.95 | Pathogenic | 0.02 | Affected | 0.3816 | 0.6318 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2534A>C | D845A 2D AIThe SynGAP1 missense variant D845A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also predicts pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that D845A is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -6.482 | Likely Benign | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.376 | Likely Benign | -5.67 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.95 | Pathogenic | 0.00 | Affected | 0.3999 | 0.6731 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2866T>C | S956P 2D AIThe SynGAP1 missense variant S956P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.957345 | Binding | 0.364 | 0.917 | 0.750 | -3.526 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.157 | Likely Benign | -0.55 | Neutral | 0.000 | Benign | 0.001 | Benign | 1.95 | Pathogenic | 0.05 | Affected | 0.2670 | 0.5337 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3755A>C | Q1252P 2D  AIThe SynGAP1 missense variant Q1252P is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.759478 | Disordered | 0.371411 | Uncertain | 0.850 | 0.544 | 0.875 | -14.386 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.352 | Likely Benign | -4.75 | Deleterious | 0.998 | Probably Damaging | 0.995 | Probably Damaging | 1.95 | Pathogenic | 0.00 | Affected | 0.1998 | 0.3742 | 0 | -1 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||||||
| c.3756G>C | Q1252H 2D  AIThe SynGAP1 missense variant Q1252H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all classify the variant as deleterious. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.759478 | Disordered | 0.371411 | Uncertain | 0.850 | 0.544 | 0.875 | -6.891 | Likely Benign | 0.951 | Likely Pathogenic | Ambiguous | 0.175 | Likely Benign | -4.02 | Deleterious | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 1.95 | Pathogenic | 0.00 | Affected | 0.1038 | 0.2149 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||||||
| c.3756G>T | Q1252H 2D AIThe SynGAP1 missense variant Q1252H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all classify the variant as damaging. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, SGM‑Consensus is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the preponderance of evidence from multiple in‑silico predictors indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.759478 | Disordered | 0.371411 | Uncertain | 0.850 | 0.544 | 0.875 | -6.891 | Likely Benign | 0.951 | Likely Pathogenic | Ambiguous | 0.175 | Likely Benign | -4.02 | Deleterious | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 1.95 | Pathogenic | 0.00 | Affected | 0.1038 | 0.2149 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||||||
| c.3773A>C | Q1258P 2D AIThe SynGAP1 missense variant Q1258P is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.486429 | Structured | 0.525814 | Binding | 0.859 | 0.577 | 0.250 | -16.904 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.440 | Likely Benign | -4.79 | Deleterious | 0.998 | Probably Damaging | 0.995 | Probably Damaging | 1.95 | Pathogenic | 0.00 | Affected | 0.1727 | 0.4187 | 0 | -1 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||||||
| c.3774G>C | Q1258H 2D AIThe SynGAP1 missense variant Q1258H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also leans pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.486429 | Structured | 0.525814 | Binding | 0.859 | 0.577 | 0.250 | -5.465 | Likely Benign | 0.960 | Likely Pathogenic | Likely Pathogenic | 0.172 | Likely Benign | -3.99 | Deleterious | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 1.95 | Pathogenic | 0.00 | Affected | 0.0735 | 0.3066 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||||||
| c.3774G>T | Q1258H 2D AIThe SynGAP1 missense variant Q1258H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote) is likely pathogenic; Foldetta results are unavailable. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.486429 | Structured | 0.525814 | Binding | 0.859 | 0.577 | 0.250 | -5.465 | Likely Benign | 0.960 | Likely Pathogenic | Likely Pathogenic | 0.172 | Likely Benign | -3.99 | Deleterious | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 1.95 | Pathogenic | 0.00 | Affected | 0.0735 | 0.3066 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||||||
| c.818A>G | E273G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 E273G missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, premPS, SIFT, and ESM1b; pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and FATHMM. Four tools (FoldX, Rosetta, AlphaMissense‑Default, Foldetta) returned uncertain results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels it pathogenic; Foldetta remains uncertain. Given the split between benign and pathogenic signals and the lack of a ClinVar classification, the variant is best described as of uncertain significance, with a slight inclination toward benign based on the most reliable single‑tool prediction. This assessment does not contradict any existing ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.071867 | Structured | 0.398918 | Uncertain | 0.863 | 0.196 | 0.125 | -4.784 | Likely Benign | 0.373 | Ambiguous | Likely Benign | -0.65 | Ambiguous | 0.2 | -0.93 | Ambiguous | -0.79 | Ambiguous | -0.46 | Likely Benign | 0.225 | Likely Benign | -2.71 | Deleterious | 0.896 | Possibly Damaging | 0.519 | Possibly Damaging | 1.95 | Pathogenic | 0.26 | Tolerated | 0.2763 | 0.3341 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||
| c.838T>A | Y280N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y280N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only SIFT, whereas all other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Based on the overwhelming consensus of pathogenic predictions and the absence of any benign consensus, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for Y280N. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.161087 | Structured | 0.321243 | Uncertain | 0.917 | 0.248 | 0.125 | -13.090 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 4.26 | Destabilizing | 0.2 | 4.18 | Destabilizing | 4.22 | Destabilizing | 1.82 | Destabilizing | 0.589 | Likely Pathogenic | -8.27 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.95 | Pathogenic | 0.06 | Tolerated | 0.1902 | 0.0212 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.944A>C | N315T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N315T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), and FATHMM. Two tools, premPS and ESM1b, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of individual predictors (six benign vs. four pathogenic) and the Foldetta result support a benign classification, while the SGM Consensus suggests pathogenicity. Thus, the variant is most likely benign based on the preponderance of evidence, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -7.071 | In-Between | 0.211 | Likely Benign | Likely Benign | 0.31 | Likely Benign | 0.1 | -0.36 | Likely Benign | -0.03 | Likely Benign | 0.62 | Ambiguous | 0.333 | Likely Benign | -2.91 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.95 | Pathogenic | 0.53 | Tolerated | 0.1471 | 0.8068 | 0 | 0 | 2.8 | -13.00 | ||||||||||||||||||||||||||||||
| c.964G>A | A322T 2D AIThe SynGAP1 missense variant A322T is not reported in ClinVar and is absent from gnomAD. Consensus from most in silico predictors classifies the change as benign: SGM‑Consensus (Likely Benign), REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a neutral effect. Only FATHMM indicates a pathogenic signal, while FoldX and premPS are inconclusive. High‑accuracy assessments reinforce the benign interpretation: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a benign impact on protein stability. Overall, the majority of evidence supports a benign effect for A322T, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.175930 | Structured | 0.425745 | Uncertain | 0.938 | 0.334 | 0.000 | -2.252 | Likely Benign | 0.064 | Likely Benign | Likely Benign | 0.58 | Ambiguous | 0.9 | 0.22 | Likely Benign | 0.40 | Likely Benign | -0.52 | Ambiguous | 0.112 | Likely Benign | 0.20 | Neutral | 0.010 | Benign | 0.005 | Benign | 1.95 | Pathogenic | 0.73 | Tolerated | 0.1367 | 0.6432 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.976C>A | H326N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H326N has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are REVEL and SIFT, whereas a majority (seven) predict a pathogenic outcome: SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the balance of evidence points to a pathogenic impact for H326N, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.342579 | Structured | 0.418150 | Uncertain | 0.944 | 0.455 | 0.000 | -8.914 | Likely Pathogenic | 0.925 | Likely Pathogenic | Ambiguous | 0.81 | Ambiguous | 0.2 | 1.48 | Ambiguous | 1.15 | Ambiguous | 0.97 | Ambiguous | 0.409 | Likely Benign | -5.63 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.95 | Pathogenic | 0.11 | Tolerated | 0.1929 | 0.2552 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||
| c.977A>T | H326L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H326L missense variant is not reported in ClinVar (status: None) and has no entry in gnomAD. Prediction tools that agree on a benign effect include SIFT, premPS, Rosetta, and Foldetta. Tools that agree on a pathogenic effect include REVEL, SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic; and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) predicts benign. No evidence is available from FoldX or AlphaMissense‑Optimized to support either outcome. Overall, the majority of predictions (nine pathogenic vs. four benign) indicate that H326L is most likely pathogenic, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.342579 | Structured | 0.418150 | Uncertain | 0.944 | 0.455 | 0.000 | -10.421 | Likely Pathogenic | 0.888 | Likely Pathogenic | Ambiguous | -0.85 | Ambiguous | 0.2 | 0.36 | Likely Benign | -0.25 | Likely Benign | 0.44 | Likely Benign | 0.627 | Likely Pathogenic | -9.64 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.95 | Pathogenic | 0.08 | Tolerated | 0.0992 | 0.5799 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.2467A>G | S823G 2D  AIThe SynGAP1 missense variant S823G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the Foldetta stability analysis is unavailable. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.685117 | Disordered | 0.627336 | Binding | 0.358 | 0.884 | 0.750 | -6.034 | Likely Benign | 0.822 | Likely Pathogenic | Ambiguous | 0.202 | Likely Benign | -2.59 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 1.96 | Pathogenic | 0.00 | Affected | 0.2855 | 0.5287 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2533G>A | D845N 2D  AIThe SynGAP1 missense variant D845N is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. When the high‑accuracy consensus is considered, AlphaMissense‑Optimized remains pathogenic, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates pathogenicity. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -6.586 | Likely Benign | 0.961 | Likely Pathogenic | Likely Pathogenic | 0.264 | Likely Benign | -3.42 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 1.96 | Pathogenic | 0.00 | Affected | 0.1216 | 0.7112 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.799T>G | W267G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W267G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify it as pathogenic: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy methods corroborate this: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Thus, the variant is most likely pathogenic, with no contradiction to its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.298060 | Uncertain | 0.943 | 0.274 | 0.000 | -15.020 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 4.40 | Destabilizing | 0.2 | 3.99 | Destabilizing | 4.20 | Destabilizing | 1.20 | Destabilizing | 0.678 | Likely Pathogenic | -11.95 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.96 | Pathogenic | 0.03 | Affected | 0.4049 | 0.1481 | -7 | -2 | 0.5 | -129.16 | |||||||||||||||||||||||||||||
| c.839A>G | Y280C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y280C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions are absent; all evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) classify the change as pathogenic or likely pathogenic. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a likely pathogenic verdict, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a pathogenic effect on protein stability. Consequently, the variant is most likely pathogenic, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.161087 | Structured | 0.321243 | Uncertain | 0.917 | 0.248 | 0.125 | -10.645 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 3.85 | Destabilizing | 0.2 | 4.45 | Destabilizing | 4.15 | Destabilizing | 1.68 | Destabilizing | 0.629 | Likely Pathogenic | -8.24 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.96 | Pathogenic | 0.01 | Affected | 0.2877 | 0.1296 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||
| c.883A>G | T295A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 T295A missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into benign (REVEL, FoldX, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized) and pathogenic (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM). Four tools are uncertain (Rosetta, Foldetta, premPS, ESM1b). High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as inconclusive. Overall, the majority of conventional predictors lean toward a benign effect, whereas the SGM Consensus indicates a pathogenic signal. Thus, the variant is most likely benign based on the bulk of evidence, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.401658 | Structured | 0.295548 | Uncertain | 0.881 | 0.288 | 0.125 | -7.276 | In-Between | 0.336 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.1 | 1.17 | Ambiguous | 0.70 | Ambiguous | 0.56 | Ambiguous | 0.340 | Likely Benign | -3.51 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.96 | Pathogenic | 0.11 | Tolerated | 0.4623 | 0.4190 | 1 | 0 | 2.5 | -30.03 | ||||||||||||||||||||||||||||||
| c.892C>A | P298T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P298T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar) and FATHMM. The remaining tools—FoldX, Rosetta, Foldetta, and ESM1b—return uncertain results and are treated as unavailable for pathogenicity inference. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, while Foldetta remains uncertain. Overall, the preponderance of evidence points to a benign effect for P298T, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.328603 | Structured | 0.268765 | Uncertain | 0.860 | 0.283 | 0.500 | -7.366 | In-Between | 0.104 | Likely Benign | Likely Benign | 1.44 | Ambiguous | 0.2 | 1.03 | Ambiguous | 1.24 | Ambiguous | 0.04 | Likely Benign | 0.209 | Likely Benign | -0.13 | Neutral | 0.939 | Possibly Damaging | 0.739 | Possibly Damaging | 1.96 | Pathogenic | 1.00 | Tolerated | 0.1595 | 0.6349 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||
| c.920T>A | F307Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F307Y is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, and premPS, whereas the majority of tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the balance of evidence favors a pathogenic classification, and this conclusion does not contradict the ClinVar status, which currently contains no assertion for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.298791 | Structured | 0.327302 | Uncertain | 0.900 | 0.315 | 0.125 | -9.870 | Likely Pathogenic | 0.889 | Likely Pathogenic | Ambiguous | 0.36 | Likely Benign | 0.1 | -0.21 | Likely Benign | 0.08 | Likely Benign | 0.11 | Likely Benign | 0.596 | Likely Pathogenic | -2.76 | Deleterious | 0.997 | Probably Damaging | 0.987 | Probably Damaging | 1.96 | Pathogenic | 0.05 | Affected | 0.1570 | 0.2325 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.965C>A | A322D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 A322D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The remaining tools—FoldX, Rosetta, ESM1b, and AlphaMissense‑Default—return uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta indicates no significant stability change (uncertain). Overall, the preponderance of evidence (seven benign predictions versus one pathogenic) suggests the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.175930 | Structured | 0.425745 | Uncertain | 0.938 | 0.334 | 0.000 | -7.184 | In-Between | 0.411 | Ambiguous | Likely Benign | 0.65 | Ambiguous | 0.1 | 1.35 | Ambiguous | 1.00 | Ambiguous | 0.34 | Likely Benign | 0.246 | Likely Benign | -0.95 | Neutral | 0.270 | Benign | 0.136 | Benign | 1.96 | Pathogenic | 0.32 | Tolerated | 0.1646 | 0.1660 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||
| c.965C>T | A322V 2D AIThe SynGAP1 missense variant A322V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No predictions or stability results are missing or inconclusive. Based on the overwhelming consensus of the available tools, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.175930 | Structured | 0.425745 | Uncertain | 0.938 | 0.334 | 0.000 | -4.905 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.02 | Likely Benign | 1.0 | -0.42 | Likely Benign | -0.20 | Likely Benign | 0.19 | Likely Benign | 0.052 | Likely Benign | -1.63 | Neutral | 0.270 | Benign | 0.136 | Benign | 1.96 | Pathogenic | 0.22 | Tolerated | 0.1279 | 0.6126 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.998A>C | K333T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K333T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include Rosetta, Foldetta, premPS, and SIFT, whereas a larger set—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score—predict a pathogenic impact. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is inconclusive, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) reports a benign effect. Overall, the majority of evidence points to a pathogenic effect for K333T, and this conclusion does not conflict with any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.311707 | Structured | 0.330781 | Uncertain | 0.537 | 0.447 | 0.500 | -11.358 | Likely Pathogenic | 0.949 | Likely Pathogenic | Ambiguous | 0.51 | Ambiguous | 0.0 | -0.15 | Likely Benign | 0.18 | Likely Benign | 0.46 | Likely Benign | 0.506 | Likely Pathogenic | -4.82 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.96 | Pathogenic | 0.08 | Tolerated | 0.1839 | 0.3354 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.2453C>A | P818Q 2D  AIThe SynGAP1 missense variant P818Q is not listed in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, SIFT, and ESM1b, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that P818Q is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.715889 | Binding | 0.371 | 0.893 | 0.625 | -6.855 | Likely Benign | 0.969 | Likely Pathogenic | Likely Pathogenic | 0.303 | Likely Benign | -4.07 | Deleterious | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 1.97 | Pathogenic | 0.27 | Tolerated | 0.1569 | 0.5456 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2453C>G | P818R 2D AIThe SynGAP1 missense variant P818R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated predictors—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. ESM1b is uncertain and does not contribute to a consensus. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Pathogenic” designation. AlphaMissense‑Optimized independently predicts pathogenicity, while Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.715889 | Binding | 0.371 | 0.893 | 0.625 | -7.267 | In-Between | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.374 | Likely Benign | -5.12 | Deleterious | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 1.97 | Pathogenic | 0.03 | Affected | 0.1499 | 0.4547 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2525C>A | S842Y 2D AIThe SynGAP1 missense variant S842Y is listed in ClinVar as Pathogenic (ClinVar ID 624244.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized returns a pathogenic score, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is labeled Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, in agreement with its ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.617281 | Binding | 0.274 | 0.861 | 0.250 | Likely Pathogenic | 1 | -16.124 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.191 | Likely Benign | -4.28 | Deleterious | 0.944 | Possibly Damaging | 0.676 | Possibly Damaging | 1.97 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.0576 | 0.5403 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||
| c.3755A>T | Q1252L 2D  AIThe SynGAP1 missense variant Q1252L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.759478 | Disordered | 0.371411 | Uncertain | 0.850 | 0.544 | 0.875 | -8.110 | Likely Pathogenic | 0.833 | Likely Pathogenic | Ambiguous | 0.295 | Likely Benign | -5.62 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 1.97 | Pathogenic | 0.00 | Affected | 0.0593 | 0.3689 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3773A>T | Q1258L 2D AIThe SynGAP1 missense variant Q1258L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: REVEL scores the variant as benign, whereas PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenicity. Grouping by consensus, the majority of tools (seven) predict pathogenic, while only one tool (REVEL) predicts benign. High‑accuracy assessments further support a deleterious effect: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic; AlphaMissense‑Optimized remains uncertain, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic. This conclusion aligns with the lack of ClinVar annotation and gnomAD absence, indicating no conflicting evidence from population databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.486429 | Structured | 0.525814 | Binding | 0.859 | 0.577 | 0.250 | -10.302 | Likely Pathogenic | 0.895 | Likely Pathogenic | Ambiguous | 0.341 | Likely Benign | -5.55 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 1.97 | Pathogenic | 0.00 | Affected | 0.0448 | 0.4137 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.869T>A | L290Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L290Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only SIFT, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which itself is “Likely Pathogenic”). Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, and Foldetta’s stability prediction is unavailable. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.127496 | Structured | 0.399723 | Uncertain | 0.904 | 0.255 | 0.000 | -12.776 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.12 | Ambiguous | 0.1 | 1.38 | Ambiguous | 1.25 | Ambiguous | 0.68 | Ambiguous | 0.697 | Likely Pathogenic | -5.52 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.97 | Pathogenic | 0.06 | Tolerated | 0.1079 | 0.1014 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.886T>G | S296A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S296A (ClinVar ID 469166.0) is listed as ClinVar status Uncertain and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign or neutral. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict pathogenic. Grouping by consensus, the benign‑predicting tools outnumber the pathogenic ones. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a benign effect. No evidence from the available tools suggests a deleterious impact. Therefore, the variant is most likely benign, and this conclusion is consistent with its ClinVar status of Uncertain rather than pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.282669 | Uncertain | 0.887 | 0.284 | 0.250 | Uncertain | 1 | -6.847 | Likely Benign | 0.247 | Likely Benign | Likely Benign | 0.50 | Ambiguous | 0.3 | -0.26 | Likely Benign | 0.12 | Likely Benign | 0.35 | Likely Benign | 0.209 | Likely Benign | -1.79 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 1.97 | Pathogenic | 0.65 | Tolerated | 3.40 | 16 | 0.4958 | 0.5522 | 1 | 1 | 2.6 | -16.00 | 182.5 | 26.6 | -0.2 | 0.1 | -0.5 | 0.0 | X | Potentially Pathogenic | The hydroxyl group of the Ser296 side chain, located in an anti-parallel β sheet strand (res. Met289-Pro298), stably hydrogen bonds with the carboxylate group of Asp330 in a neighboring β strand (res. Ala322-Asp332). The backbone carbonyl group of Ser296 also hydrogen bonds with the guanidinium group of Arg279 in another nearby β strand (res. Arg279-Cys285). In the variant simulations, the methyl group of the Ala296 side chain cannot hydrogen bond with Asp330, causing the carboxylate group positioning to fluctuate more than in the WT simulations.Although the residue swap does not seem to affect the anti-parallel β sheet assembly during the simulations, it is possible that the Ser296-Asp330 hydrogen bond plays a crucial role in maintaining the C2 domain fold. Notably, because Ser296 is located near the membrane interface, the potential effect of the residue swap on the SynGAP-membrane association cannot be addressed by solvent-only simulations. | ||||||||||||||||
| c.920T>C | F307S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F307S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. In silico predictors that provide a definitive call all classify the variant as pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely pathogenic verdict. Predictions that are inconclusive or uncertain include FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.298791 | Structured | 0.327302 | Uncertain | 0.900 | 0.315 | 0.125 | -12.282 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 1.58 | Ambiguous | 0.2 | 1.22 | Ambiguous | 1.40 | Ambiguous | 0.99 | Ambiguous | 0.766 | Likely Pathogenic | -7.32 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.97 | Pathogenic | 0.01 | Affected | 0.4576 | 0.0758 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.955G>C | A319P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A319P is catalogued in gnomAD (ID 6‑33437860‑G‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions that are inconclusive are Rosetta, ESM1b, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign, while Foldetta remains uncertain. Overall, the majority of evidence points to a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.410405 | Uncertain | 0.879 | 0.254 | 0.125 | 6-33437860-G-C | 3 | 1.86e-6 | -7.213 | In-Between | 0.109 | Likely Benign | Likely Benign | -0.02 | Likely Benign | 0.9 | -1.18 | Ambiguous | -0.60 | Ambiguous | 0.22 | Likely Benign | 0.286 | Likely Benign | 0.11 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 1.97 | Pathogenic | 1.00 | Tolerated | 3.38 | 23 | 0.1839 | 0.5067 | -1 | 1 | -3.4 | 26.04 | |||||||||||||||||||||||||
| c.977A>G | H326R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H326R missense variant is not listed in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include SIFT and Foldetta, whereas the remaining tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict a pathogenic impact. High‑accuracy methods give the following results: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports it as likely pathogenic; Foldetta, a protein‑folding stability approach that integrates FoldX‑MD and Rosetta outputs, predicts a benign effect. Because the majority of evidence points to a deleterious change, the variant is most likely pathogenic, and this assessment is not contradicted by the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.342579 | Structured | 0.418150 | Uncertain | 0.944 | 0.455 | 0.000 | -12.369 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | -0.65 | Ambiguous | 0.1 | 1.40 | Ambiguous | 0.38 | Likely Benign | 0.83 | Ambiguous | 0.601 | Likely Pathogenic | -6.89 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 1.97 | Pathogenic | 0.10 | Tolerated | 0.1832 | 0.2413 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||
| c.1027G>A | V343I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343I is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33437932‑G‑A). Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to “Likely Benign” (3 benign vs. 1 pathogenic). High‑accuracy assessments are consistent: AlphaMissense‑Optimized is benign; the SGM‑Consensus is likely benign; and Foldetta, combining FoldX‑MD and Rosetta outputs, is benign. Overall, the collective evidence strongly supports a benign classification, and this does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | Uncertain | 2 | 6-33437932-G-A | 1 | 6.20e-7 | -6.020 | Likely Benign | 0.117 | Likely Benign | Likely Benign | -0.27 | Likely Benign | 0.0 | -0.04 | Likely Benign | -0.16 | Likely Benign | -0.39 | Likely Benign | 0.020 | Likely Benign | -0.14 | Neutral | 0.159 | Benign | 0.084 | Benign | 1.98 | Pathogenic | 0.27 | Tolerated | 3.37 | 25 | 0.1095 | 0.4536 | 4 | 3 | 0.3 | 14.03 | 240.2 | -26.9 | -0.2 | 0.2 | -0.2 | 0.2 | X | Potentially Benign | The iso-propyl side chain of Val343, located in an anti-parallel β sheet strand (res. Gly341-Pro349), is packing against multiple hydrophobic residues of the C2 domain (e.g., Leu327, Leu274, Val365). In the variant simulations, the sec-butyl side chain of Ile343 is basically able to form the same interactions as valine due to its similar hydrophobic profile. The residue swap also does not seem to cause negative effects on the protein structure based on the simulations. | |||||||||||||
| c.2344G>A | D782N 2D AIThe SynGAP1 missense variant D782N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs four benign) lean toward a pathogenic impact. This conclusion does not contradict ClinVar status, as the variant has no existing ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -6.857 | Likely Benign | 0.766 | Likely Pathogenic | Likely Benign | 0.131 | Likely Benign | -2.26 | Neutral | 0.995 | Probably Damaging | 0.950 | Probably Damaging | 1.98 | Pathogenic | 0.02 | Affected | 0.1167 | 0.6884 | 2 | 1 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2453C>T | P818L 2D AIThe SynGAP1 missense variant P818L is catalogued in gnomAD (ID 6‑33443005‑C‑T) but has no ClinVar entry. Functional prediction tools fall into two consensus groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). AlphaMissense‑Optimized returns an uncertain result. High‑accuracy assessments are limited: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus indicates “Likely Pathogenic,” and Foldetta data are unavailable. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Thus, the variant is most likely pathogenic based on current predictive tools. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.715889 | Binding | 0.371 | 0.893 | 0.625 | 6-33443005-C-T | 1 | 6.20e-7 | -6.064 | Likely Benign | 0.938 | Likely Pathogenic | Ambiguous | 0.285 | Likely Benign | -5.81 | Deleterious | 0.997 | Probably Damaging | 0.954 | Probably Damaging | 1.98 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.2351 | 0.6951 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.2525C>G | S842C 2D AIThe SynGAP1 missense variant S842C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—predict a pathogenic or likely pathogenic outcome. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments show the SGM‑Consensus as Likely Pathogenic, while the Foldetta stability analysis is unavailable. Based on the collective evidence, the variant is most likely pathogenic; this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.617281 | Binding | 0.274 | 0.861 | 0.250 | -12.405 | Likely Pathogenic | 0.863 | Likely Pathogenic | Ambiguous | 0.233 | Likely Benign | -3.93 | Deleterious | 0.999 | Probably Damaging | 0.944 | Probably Damaging | 1.98 | Pathogenic | 0.00 | Affected | 0.0806 | 0.5506 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2525C>T | S842F 2D AIThe SynGAP1 missense variant S842F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls from REVEL, polyPhen‑2 HumDiv and HumVar, whereas pathogenic calls come from PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default and AlphaMissense‑Optimized. The consensus predictor SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus result is consistent with this. Foldetta, a protein‑folding stability method that combines FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.617281 | Binding | 0.274 | 0.861 | 0.250 | -14.590 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.188 | Likely Benign | -4.12 | Deleterious | 0.029 | Benign | 0.043 | Benign | 1.98 | Pathogenic | 0.00 | Affected | 0.0584 | 0.5692 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.826C>G | P276A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P276A is reported in gnomAD (variant ID 6‑33437731‑C‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The high‑accuracy consensus methods give a benign signal: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” while Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar status (none is available). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.037156 | Structured | 0.338937 | Uncertain | 0.724 | 0.230 | 0.250 | 6-33437731-C-G | 5 | 3.10e-6 | -3.414 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 1.42 | Ambiguous | 0.1 | 1.01 | Ambiguous | 1.22 | Ambiguous | 0.50 | Likely Benign | 0.187 | Likely Benign | -2.31 | Neutral | 0.044 | Benign | 0.030 | Benign | 1.98 | Pathogenic | 0.57 | Tolerated | 3.38 | 19 | 0.3149 | 0.3669 | -1 | 1 | 3.4 | -26.04 | ||||||||||||||||||||||||
| c.839A>T | Y280F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y280F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The remaining tools—premPS, ESM1b, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, while the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive and therefore unavailable. Overall, the majority of available predictions favor a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.161087 | Structured | 0.321243 | Uncertain | 0.917 | 0.248 | 0.125 | -7.844 | In-Between | 0.414 | Ambiguous | Likely Benign | 0.49 | Likely Benign | 0.2 | 0.03 | Likely Benign | 0.26 | Likely Benign | 0.87 | Ambiguous | 0.544 | Likely Pathogenic | -3.68 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 1.98 | Pathogenic | 0.02 | Affected | 0.2263 | 0.2398 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||
| c.868C>G | L290V 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant L290V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and Rosetta, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive or unavailable are FoldX, premPS, ESM1b, AlphaMissense‑Optimized, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence from multiple pathogenic‑predicting tools and the SGM Consensus suggests that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.127496 | Structured | 0.399723 | Uncertain | 0.904 | 0.255 | 0.000 | -7.392 | In-Between | 0.834 | Likely Pathogenic | Ambiguous | 1.17 | Ambiguous | 0.1 | 0.35 | Likely Benign | 0.76 | Ambiguous | 0.62 | Ambiguous | 0.366 | Likely Benign | -2.76 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.98 | Pathogenic | 0.05 | Affected | 0.1574 | 0.4177 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.945C>A | N315K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N315K is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and Foldetta. Tools that agree on a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and premPS. High‑accuracy assessments show that the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. No other high‑accuracy tool provides a conclusive result. Overall, the majority of predictions (seven pathogenic vs. five benign, with two uncertain) indicate that the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -10.380 | Likely Pathogenic | 0.872 | Likely Pathogenic | Ambiguous | -0.03 | Likely Benign | 0.1 | 0.08 | Likely Benign | 0.03 | Likely Benign | 0.87 | Ambiguous | 0.340 | Likely Benign | -3.27 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.98 | Pathogenic | 0.54 | Tolerated | 0.2268 | 0.6436 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.945C>G | N315K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N315K is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and Foldetta. Tools that agree on a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and premPS. High‑accuracy assessments show that AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of evidence (seven pathogenic vs. five benign, with two uncertain) points to a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -10.380 | Likely Pathogenic | 0.872 | Likely Pathogenic | Ambiguous | -0.03 | Likely Benign | 0.1 | 0.08 | Likely Benign | 0.03 | Likely Benign | 0.87 | Ambiguous | 0.340 | Likely Benign | -3.27 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.98 | Pathogenic | 0.54 | Tolerated | 0.2268 | 0.6436 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.1627C>G | L543V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L543V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—classify the variant as pathogenic. AlphaMissense‑Optimized is inconclusive (uncertain). High‑accuracy assessments further support pathogenicity: the SGM‑Consensus predicts “Likely Pathogenic,” and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a destabilizing, pathogenic effect. AlphaMissense‑Optimized remains uncertain. Based on the overwhelming majority of predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.020918 | Uncertain | 0.963 | 0.314 | 0.000 | -11.561 | Likely Pathogenic | 0.908 | Likely Pathogenic | Ambiguous | 3.09 | Destabilizing | 0.3 | 2.03 | Destabilizing | 2.56 | Destabilizing | 1.28 | Destabilizing | 0.398 | Likely Benign | -2.99 | Deleterious | 0.998 | Probably Damaging | 0.992 | Probably Damaging | 1.99 | Pathogenic | 0.01 | Affected | 0.1334 | 0.2028 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2524T>C | S842P 2D AIThe SynGAP1 missense variant S842P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess pathogenicity largely agree: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all predict a deleterious effect. Only REVEL classifies the variant as benign. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus indicates “Likely Pathogenic.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the consensus of the majority of tools, including the high‑accuracy predictors, indicates that S842P is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.617281 | Binding | 0.274 | 0.861 | 0.250 | -13.890 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | 0.309 | Likely Benign | -3.43 | Deleterious | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 1.99 | Pathogenic | 0.00 | Affected | 0.1494 | 0.5342 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3784A>G | I1262V 2D AIThe SynGAP1 I1262V missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments are less decisive: AlphaMissense‑Optimized rates the variant as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta folding‑stability data are unavailable. Overall, the majority of standard predictors lean toward pathogenicity, but the most reliable tools provide no clear verdict. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.497853 | Structured | 0.707863 | Binding | 0.886 | 0.576 | 0.125 | -6.773 | Likely Benign | 0.814 | Likely Pathogenic | Ambiguous | 0.237 | Likely Benign | -0.82 | Neutral | 0.958 | Probably Damaging | 0.970 | Probably Damaging | 1.99 | Pathogenic | 0.00 | Affected | 0.1049 | 0.3270 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3787A>G | I1263V 2D AIThe SynGAP1 missense variant I1263V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two benign vs. two pathogenic votes), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.425610 | Structured | 0.740957 | Binding | 0.867 | 0.574 | 0.000 | -4.230 | Likely Benign | 0.729 | Likely Pathogenic | Likely Benign | 0.221 | Likely Benign | -0.83 | Neutral | 0.437 | Benign | 0.170 | Benign | 1.99 | Pathogenic | 0.00 | Affected | 0.1136 | 0.3102 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2356C>G | L786V 2D AIThe SynGAP1 missense variant L786V is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict the ClinVar status, which currently has no classification for L786V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | -4.607 | Likely Benign | 0.310 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -1.66 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.00 | Pathogenic | 0.00 | Affected | 0.1683 | 0.4049 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.2866T>A | S956T 2D AIThe SynGAP1 missense variant S956T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for S956T, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.957345 | Binding | 0.364 | 0.917 | 0.750 | -5.404 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -0.45 | Neutral | 0.369 | Benign | 0.159 | Benign | 2.00 | Pathogenic | 0.08 | Tolerated | 0.2244 | 0.5727 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3194C>A | P1065Q 2D AIThe SynGAP1 missense variant P1065Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -3.928 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -2.44 | Neutral | 0.102 | Benign | 0.057 | Benign | 2.00 | Pathogenic | 0.00 | Affected | 0.1487 | 0.5478 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3194C>G | P1065R 2D AIThe SynGAP1 missense variant P1065R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -3.237 | Likely Benign | 0.228 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -2.46 | Neutral | 0.102 | Benign | 0.052 | Benign | 2.00 | Pathogenic | 0.00 | Affected | 0.1439 | 0.4369 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3604A>G | I1202V 2D AIThe SynGAP1 I1202V missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all predict a pathogenic outcome. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split; Foldetta results are not available. Overall, the majority of evidence (five pathogenic vs. three benign predictions) points to a likely pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.529623 | Disordered | 0.510422 | Binding | 0.874 | 0.593 | 0.250 | -5.494 | Likely Benign | 0.947 | Likely Pathogenic | Ambiguous | 0.093 | Likely Benign | -0.80 | Neutral | 0.958 | Probably Damaging | 0.970 | Probably Damaging | 2.00 | Pathogenic | 0.05 | Affected | 0.1109 | 0.2697 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3755A>G | Q1252R 2D  AIThe SynGAP1 missense variant Q1252R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: REVEL scores the variant as benign, whereas PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenicity. Grouping by consensus, the majority of tools (seven) support a pathogenic effect, while only one tool (REVEL) indicates benign. High‑accuracy assessments further support a deleterious impact: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic; AlphaMissense‑Optimized remains uncertain, and Foldetta data are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus result, the variant is most likely pathogenic. This conclusion aligns with the lack of ClinVar annotation and gnomAD absence, and there is no contradiction with ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.759478 | Disordered | 0.371411 | Uncertain | 0.850 | 0.544 | 0.875 | -11.890 | Likely Pathogenic | 0.913 | Likely Pathogenic | Ambiguous | 0.249 | Likely Benign | -3.26 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.00 | Pathogenic | 0.00 | Affected | 0.1284 | 0.0854 | 1 | 1 | -1.0 | 28.06 | ||||||||||||||||||||||||||||||||||||||
| c.3773A>G | Q1258R 2D AIThe SynGAP1 missense variant Q1258R is listed in ClinVar with an uncertain significance (ClinVar ID 3359527.0) and is not observed in gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default, while only REVEL predicts a benign outcome. The high‑accuracy predictors give the following results: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic classification; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no available output for this variant. Based on the preponderance of pathogenic predictions and the SGM Consensus, the variant is most likely pathogenic, which is consistent with its ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.486429 | Structured | 0.525814 | Binding | 0.859 | 0.577 | 0.250 | Uncertain | 1 | -10.971 | Likely Pathogenic | 0.931 | Likely Pathogenic | Ambiguous | 0.316 | Likely Benign | -3.19 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.00 | Pathogenic | 0.00 | Affected | 0.1027 | 0.0991 | 1 | 1 | -1.0 | 28.06 | ||||||||||||||||||||||||||||||||||||
| c.799T>A | W267R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W267R has no ClinVar entry and is not reported in gnomAD. Prediction tools that assess pathogenicity all agree on a deleterious effect: SGM‑Consensus (Likely Pathogenic), REVEL, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return pathogenic or likely pathogenic scores. Tools that are inconclusive (FoldX and premPS) are noted as uncertain. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No benign predictions are present. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.298060 | Uncertain | 0.943 | 0.274 | 0.000 | -13.868 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.94 | Ambiguous | 0.1 | 2.47 | Destabilizing | 2.21 | Destabilizing | 0.51 | Ambiguous | 0.672 | Likely Pathogenic | -12.87 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.00 | Pathogenic | 0.01 | Affected | 0.3763 | 0.0571 | 2 | -3 | -3.6 | -30.03 | |||||||||||||||||||||||||||||
| c.799T>C | W267R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W267R is not reported in ClinVar (ClinVar status: None) and has no entries in gnomAD (gnomAD ID: None). Prediction tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), REVEL, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tools predict a benign outcome. Uncertain predictions are provided by FoldX and premPS. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts Likely Pathogenic; and Foldetta predicts Pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.298060 | Uncertain | 0.943 | 0.274 | 0.000 | -13.868 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.94 | Ambiguous | 0.1 | 2.47 | Destabilizing | 2.21 | Destabilizing | 0.51 | Ambiguous | 0.672 | Likely Pathogenic | -12.87 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.00 | Pathogenic | 0.01 | Affected | 0.3763 | 0.0571 | 2 | -3 | -3.6 | -30.03 | |||||||||||||||||||||||||||||
| c.819G>C | E273D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E273D missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools largely agree on a benign effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign or likely benign. Only FATHMM predicts a pathogenic outcome, while premPS remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Overall, the majority of evidence supports a benign impact for E273D, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.071867 | Structured | 0.398918 | Uncertain | 0.863 | 0.196 | 0.125 | -1.811 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.26 | Likely Benign | 0.1 | -0.48 | Likely Benign | -0.11 | Likely Benign | -0.63 | Ambiguous | 0.094 | Likely Benign | 1.99 | Neutral | 0.004 | Benign | 0.010 | Benign | 2.00 | Pathogenic | 1.00 | Tolerated | 3.38 | 18 | 0.1711 | 0.1859 | 3 | 2 | 0.0 | -14.03 | 223.1 | 22.1 | 0.2 | 0.0 | 0.0 | 0.1 | X | Potentially Benign | The negatively charged residue Glu273, located in a β hairpin loop (res. Glu273-Lys278) that connects two anti-parallel β sheet strands, is replaced with another negatively charged residue, aspartate. Because the C2 domain loop faces the membrane surface, the potentially crucial role of the carboxylate group of Glu273 or Asp273 on SynGAP-membrane association cannot be fully explored via solvent-only simulations.As a minor note, the neighboring residue Arg272, which stacks with the indole ring of the Trp362 side chain and directly faces RasGTPase, forms a salt bridge more often with Asp273 than with the non-mutated Glu273 in the simulations. Regardless, due to the similar physicochemical properties of the WT and variant residues at the membrane interface, the residue swap is likely to be well tolerated. | ||||||||||||||||||
| c.819G>T | E273D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E273D is listed in ClinVar (ID 1471608) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33437724‑G‑T). Prediction tools that report a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score. Only FATHMM predicts a pathogenic outcome, while premPS is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.071867 | Structured | 0.398918 | Uncertain | 0.863 | 0.196 | 0.125 | Conflicting | 2 | 6-33437724-G-T | 2 | 1.24e-6 | -1.811 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.26 | Likely Benign | 0.1 | -0.48 | Likely Benign | -0.11 | Likely Benign | -0.63 | Ambiguous | 0.092 | Likely Benign | 1.99 | Neutral | 0.004 | Benign | 0.010 | Benign | 2.00 | Pathogenic | 1.00 | Tolerated | 3.38 | 18 | 0.1711 | 0.1859 | 3 | 2 | 0.0 | -14.03 | 223.1 | 22.1 | 0.2 | 0.0 | 0.0 | 0.1 | X | Potentially Benign | The negatively charged residue Glu273, located in a β hairpin loop (res. Glu273-Lys278) that connects two anti-parallel β sheet strands, is replaced with another negatively charged residue, aspartate. Because the C2 domain loop faces the membrane surface, the potentially crucial role of the carboxylate group of Glu273 or Asp273 on SynGAP-membrane association cannot be fully explored via solvent-only simulations.As a minor note, the neighboring residue Arg272, which stacks with the indole ring of the Trp362 side chain and directly faces RasGTPase, forms a salt bridge more often with Asp273 than with the non-mutated Glu273 in the simulations. Regardless, due to the similar physicochemical properties of the WT and variant residues at the membrane interface, the residue swap is likely to be well tolerated. | |||||||||||||
| c.839A>C | Y280S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y280S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions are absent; all evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) classify the substitution as pathogenic. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a likely pathogenic verdict, and Foldetta (integrating FoldX‑MD and Rosetta outputs) also reports pathogenic. Consequently, the variant is most likely pathogenic, and this prediction is consistent with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.161087 | Structured | 0.321243 | Uncertain | 0.917 | 0.248 | 0.125 | -13.491 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 4.63 | Destabilizing | 0.3 | 4.80 | Destabilizing | 4.72 | Destabilizing | 2.03 | Destabilizing | 0.635 | Likely Pathogenic | -8.27 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.00 | Pathogenic | 0.05 | Affected | 0.3993 | 0.1227 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||
| c.947A>T | N316I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N316I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and premPS, whereas the majority of tools predict a pathogenic impact: FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. No prediction is missing or inconclusive beyond the uncertain AlphaMissense‑Optimized result. Based on the preponderance of pathogenic predictions and the high‑accuracy tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.118441 | Structured | 0.385187 | Uncertain | 0.817 | 0.246 | 0.125 | -11.164 | Likely Pathogenic | 0.899 | Likely Pathogenic | Ambiguous | 2.74 | Destabilizing | 0.2 | 4.10 | Destabilizing | 3.42 | Destabilizing | 0.18 | Likely Benign | 0.318 | Likely Benign | -6.37 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.00 | Pathogenic | 0.03 | Affected | 0.0734 | 0.7422 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.961C>A | R321S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R321S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Two tools—FoldX and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of predictions lean toward a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.175930 | Structured | 0.423273 | Uncertain | 0.931 | 0.297 | 0.125 | -10.146 | Likely Pathogenic | 0.497 | Ambiguous | Likely Benign | 0.66 | Ambiguous | 0.1 | -0.30 | Likely Benign | 0.18 | Likely Benign | 0.24 | Likely Benign | 0.379 | Likely Benign | -2.27 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.00 | Pathogenic | 0.10 | Tolerated | 0.2958 | 0.2785 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||
| c.964G>T | A322S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A322S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only FATHMM predicts a pathogenic outcome. Grouping by consensus, the benign‑predicting tools outnumber the single pathogenic prediction. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also indicates a benign effect. No prediction or stability result is missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.175930 | Structured | 0.425745 | Uncertain | 0.938 | 0.334 | 0.000 | -1.424 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.41 | Likely Benign | 0.2 | 0.38 | Likely Benign | 0.40 | Likely Benign | -0.44 | Likely Benign | 0.127 | Likely Benign | 1.09 | Neutral | 0.010 | Benign | 0.010 | Benign | 2.00 | Pathogenic | 0.67 | Tolerated | 0.2696 | 0.5253 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1003C>G | R335G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R335G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, and SIFT, whereas those that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, more tools predict pathogenicity than benignity, and the high‑accuracy consensus also leans pathogenic. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.305330 | Structured | 0.331028 | Uncertain | 0.483 | 0.428 | 0.500 | -11.860 | Likely Pathogenic | 0.880 | Likely Pathogenic | Ambiguous | 1.01 | Ambiguous | 0.1 | 0.44 | Likely Benign | 0.73 | Ambiguous | 0.20 | Likely Benign | 0.194 | Likely Benign | -4.77 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.01 | Pathogenic | 0.10 | Tolerated | 0.3000 | 0.3554 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.2452C>A | P818T 2D AIThe SynGAP1 missense variant P818T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default all classify the variant as damaging. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.715889 | Binding | 0.371 | 0.893 | 0.625 | -6.864 | Likely Benign | 0.936 | Likely Pathogenic | Ambiguous | 0.265 | Likely Benign | -4.58 | Deleterious | 0.994 | Probably Damaging | 0.927 | Probably Damaging | 2.01 | Pathogenic | 0.02 | Affected | 0.1797 | 0.6453 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2644G>T | G882W 2D  AIThe SynGAP1 missense variant G882W has no ClinVar entry and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion does not conflict with the ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.690604 | Disordered | 0.632888 | Binding | 0.306 | 0.878 | 0.250 | -8.637 | Likely Pathogenic | 0.606 | Likely Pathogenic | Likely Benign | 0.166 | Likely Benign | -2.35 | Neutral | 0.983 | Probably Damaging | 0.813 | Possibly Damaging | 2.01 | Pathogenic | 0.00 | Affected | 0.0829 | 0.4616 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.3194C>T | P1065L 2D AIThe SynGAP1 missense variant P1065L is listed in ClinVar as Benign and is present in gnomAD (ID 6‑33443746‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no reported result for this variant. Overall, the balance of evidence (5 benign vs. 4 pathogenic predictions) and the high‑accuracy benign call support a benign classification, aligning with the ClinVar status and indicating no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | Likely Benign | 1 | 6-33443746-C-T | 14 | 8.71e-6 | -5.085 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -2.94 | Deleterious | 0.950 | Possibly Damaging | 0.419 | Benign | 2.01 | Pathogenic | 0.00 | Affected | 4.32 | 2 | 0.2286 | 0.6922 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||
| c.3299G>C | G1100A 2D  AIThe SynGAP1 missense variant G1100A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign based on current computational evidence, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -2.862 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -0.95 | Neutral | 0.943 | Possibly Damaging | 0.667 | Possibly Damaging | 2.01 | Pathogenic | 0.05 | Affected | 0.3442 | 0.5154 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3754C>G | Q1252E 2D  AIThe SynGAP1 missense variant Q1252E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs. three benign) lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.759478 | Disordered | 0.371411 | Uncertain | 0.850 | 0.544 | 0.875 | -10.181 | Likely Pathogenic | 0.485 | Ambiguous | Likely Benign | 0.152 | Likely Benign | -2.32 | Neutral | 0.985 | Probably Damaging | 0.981 | Probably Damaging | 2.01 | Pathogenic | 0.00 | Affected | 0.1130 | 0.1266 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3772C>G | Q1258E 2D AIThe SynGAP1 missense variant Q1258E is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, PROVEAN, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to a likely pathogenic verdict (3/4 pathogenic votes). High‑accuracy assessments further support this: AlphaMissense‑Optimized indicates a benign effect, whereas the SGM‑Consensus remains pathogenic; Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points toward a pathogenic impact, which is consistent with the lack of ClinVar annotation and gnomAD absence. Thus, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.486429 | Structured | 0.525814 | Binding | 0.859 | 0.577 | 0.250 | -9.894 | Likely Pathogenic | 0.666 | Likely Pathogenic | Likely Benign | 0.206 | Likely Benign | -2.39 | Neutral | 0.985 | Probably Damaging | 0.981 | Probably Damaging | 2.01 | Pathogenic | 0.00 | Affected | 0.0958 | 0.1897 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||||||||||
| c.4003G>T | G1335C 2D AIThe SynGAP1 missense variant G1335C is listed in gnomAD (variant ID 6-33451877‑G‑T) but has no ClinVar entry. Functional prediction tools split into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote) also indicates Likely Pathogenic. Foldetta results are not available for this variant. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.967705 | Binding | 0.323 | 0.724 | 0.750 | 6-33451877-G-T | 1 | 7.91e-7 | -6.878 | Likely Benign | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.426 | Likely Benign | -5.51 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.01 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1376 | 0.4513 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||
| c.869T>C | L290P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L290P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity unanimously favor a deleterious effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all predict pathogenicity. No tool in the dataset predicts a benign outcome; the remaining tools (FoldX, Rosetta, Foldetta, premPS) return uncertain results, which are treated as unavailable evidence. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is likely pathogenic, and Foldetta is inconclusive. Based on the collective predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.127496 | Structured | 0.399723 | Uncertain | 0.904 | 0.255 | 0.000 | -11.796 | Likely Pathogenic | 0.973 | Likely Pathogenic | Likely Pathogenic | 0.79 | Ambiguous | 0.1 | 1.10 | Ambiguous | 0.95 | Ambiguous | 0.56 | Ambiguous | 0.727 | Likely Pathogenic | -6.43 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.01 | Pathogenic | 0.03 | Affected | 0.3646 | 0.1703 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.956C>A | A319D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A319D missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, FoldX, premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized; pathogenic predictions from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a likely pathogenic verdict (3/4 pathogenic votes). High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, whereas the SGM‑Consensus remains pathogenic. Foldetta, which integrates FoldX‑MD (benign) and Rosetta (uncertain), is considered unavailable. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.179055 | Structured | 0.410405 | Uncertain | 0.879 | 0.254 | 0.125 | -11.144 | Likely Pathogenic | 0.752 | Likely Pathogenic | Likely Benign | -0.02 | Likely Benign | 0.2 | -0.84 | Ambiguous | -0.43 | Likely Benign | 0.30 | Likely Benign | 0.373 | Likely Benign | -2.38 | Neutral | 0.998 | Probably Damaging | 0.966 | Probably Damaging | 2.01 | Pathogenic | 0.20 | Tolerated | 0.1504 | 0.1360 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.998A>G | K333R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K333R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus “Likely Benign”) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates benign stability. No prediction or folding result is missing or inconclusive. Overall, the majority of evidence points to a benign effect for K333R, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.311707 | Structured | 0.330781 | Uncertain | 0.537 | 0.447 | 0.500 | -4.897 | Likely Benign | 0.120 | Likely Benign | Likely Benign | -0.23 | Likely Benign | 0.0 | 0.15 | Likely Benign | -0.04 | Likely Benign | 0.33 | Likely Benign | 0.232 | Likely Benign | -2.02 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.01 | Pathogenic | 0.14 | Tolerated | 0.4294 | 0.1134 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.2366C>T | P789L 2D AIThe SynGAP1 P789L missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence (five pathogenic vs. three benign predictions, with the SGM Consensus supporting pathogenicity) indicates that P789L is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -4.623 | Likely Benign | 0.457 | Ambiguous | Likely Benign | 0.303 | Likely Benign | -5.91 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1958 | 0.4866 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2535C>A | D845E 2D  AIThe SynGAP1 missense variant D845E is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas seven tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic outcome. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and Foldetta stability analysis is unavailable. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. Overall, the balance of evidence points to a pathogenic effect for D845E, and this assessment does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -6.979 | Likely Benign | 0.914 | Likely Pathogenic | Ambiguous | 0.196 | Likely Benign | -2.67 | Deleterious | 0.992 | Probably Damaging | 0.992 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1404 | 0.6998 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2535C>G | D845E 2D AIThe SynGAP1 missense variant D845E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all indicate pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to a pathogenic effect for D845E, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -6.979 | Likely Benign | 0.914 | Likely Pathogenic | Ambiguous | 0.196 | Likely Benign | -2.67 | Deleterious | 0.992 | Probably Damaging | 0.992 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1404 | 0.6998 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2947A>T | S983C 2D AIThe SynGAP1 missense variant S983C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools cluster into two groups: benign predictions come from REVEL and AlphaMissense‑Optimized, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, whereas the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points toward a pathogenic impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -7.083 | In-Between | 0.741 | Likely Pathogenic | Likely Benign | 0.162 | Likely Benign | -2.64 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1657 | 0.5298 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2948G>T | S983I 2D AIThe SynGAP1 missense variant S983I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). In silico predictors that agree on a benign effect are REVEL and ESM1b, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (3 pathogenic vs. 1 benign) is likely pathogenic. Foldetta results are unavailable. Overall, the preponderance of evidence indicates that S983I is most likely pathogenic, and this conclusion is not contradicted by the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -6.259 | Likely Benign | 0.968 | Likely Pathogenic | Likely Pathogenic | 0.190 | Likely Benign | -2.67 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1380 | 0.4625 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3809A>T | E1270V 2D AIThe SynGAP1 missense variant E1270V is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus agrees. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that E1270V is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.490133 | Structured | 0.771865 | Binding | 0.805 | 0.659 | 0.250 | -13.293 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.408 | Likely Benign | -5.90 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.0570 | 0.6461 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3812A>T | E1271V 2D AISynGAP1 missense variant E1271V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. AlphaMissense‑Optimized yields an uncertain result, and no Foldetta stability assessment is available. Considering the majority of high‑confidence tools and the consensus score, the variant is most likely pathogenic. This assessment does not contradict any ClinVar annotation, as no ClinVar entry exists for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.767529 | Binding | 0.832 | 0.666 | 0.375 | -6.961 | Likely Benign | 0.848 | Likely Pathogenic | Ambiguous | 0.303 | Likely Benign | -5.64 | Deleterious | 0.995 | Probably Damaging | 0.846 | Possibly Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.0620 | 0.6106 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.4003G>C | G1335R 2D AIThe SynGAP1 missense variant G1335R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic. Foldetta results are not available. Overall, the preponderance of evidence supports a pathogenic classification for G1335R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.967705 | Binding | 0.323 | 0.724 | 0.750 | -4.921 | Likely Benign | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.394 | Likely Benign | -5.06 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1071 | 0.5024 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.4004G>A | G1335D 2D AIThe SynGAP1 missense variant G1335D is reported in ClinVar as “not listed” and is present in gnomAD (variant ID 6‑33451878‑G‑A). Functional prediction tools that agree on a benign effect are REVEL and ESM1b, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (majority vote) also indicates Likely Pathogenic. Foldetta results are not available for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and the SGM‑Consensus suggests that G1335D is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.967705 | Binding | 0.323 | 0.724 | 0.750 | 6-33451878-G-A | -5.687 | Likely Benign | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.404 | Likely Benign | -4.42 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2120 | 0.3702 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||
| c.4004G>T | G1335V 2D AIThe SynGAP1 missense variant G1335V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this assessment does not conflict with the ClinVar status, which currently has no entry for G1335V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.967705 | Binding | 0.323 | 0.724 | 0.750 | -4.982 | Likely Benign | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.394 | Likely Benign | -5.65 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1124 | 0.4195 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.830A>C | K277T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K277T missense variant is not reported in ClinVar (status: None) and has no entry in gnomAD. Prediction tools that agree on a benign effect are Rosetta and premPS, while the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict pathogenicity. FoldX and Foldetta return uncertain results. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also pathogenic; Foldetta remains uncertain. Overall, the preponderance of evidence indicates that K277T is most likely pathogenic, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.061840 | Structured | 0.321811 | Uncertain | 0.649 | 0.247 | 0.250 | -11.688 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 1.46 | Ambiguous | 0.1 | 0.14 | Likely Benign | 0.80 | Ambiguous | 0.17 | Likely Benign | 0.573 | Likely Pathogenic | -5.52 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.02 | Pathogenic | 0.01 | Affected | 0.1837 | 0.2220 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.883A>T | T295S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T295S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; premPS is inconclusive. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority (3 benign vs. 1 pathogenic); and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.401658 | Structured | 0.295548 | Uncertain | 0.881 | 0.288 | 0.125 | -4.904 | Likely Benign | 0.335 | Likely Benign | Likely Benign | -0.16 | Likely Benign | 0.2 | 0.28 | Likely Benign | 0.06 | Likely Benign | 0.54 | Ambiguous | 0.218 | Likely Benign | -2.16 | Neutral | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.02 | Pathogenic | 0.14 | Tolerated | 0.3910 | 0.4473 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.884C>G | T295S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T295S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; premPS is inconclusive. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority (3 benign vs. 1 pathogenic); and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.401658 | Structured | 0.295548 | Uncertain | 0.881 | 0.288 | 0.125 | -4.904 | Likely Benign | 0.335 | Likely Benign | Likely Benign | -0.16 | Likely Benign | 0.2 | 0.28 | Likely Benign | 0.06 | Likely Benign | 0.54 | Ambiguous | 0.196 | Likely Benign | -2.16 | Neutral | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.02 | Pathogenic | 0.14 | Tolerated | 0.3910 | 0.4473 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.943A>G | N315D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N315D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar) and FATHMM. The remaining tools (FoldX, premPS, AlphaMissense‑Default) are inconclusive. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority; and Foldetta predicts a benign stability change. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -5.667 | Likely Benign | 0.536 | Ambiguous | Likely Benign | 1.27 | Ambiguous | 0.6 | -0.30 | Likely Benign | 0.49 | Likely Benign | 0.88 | Ambiguous | 0.229 | Likely Benign | -2.41 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.02 | Pathogenic | 0.45 | Tolerated | 0.1972 | 0.4488 | 2 | 1 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.955G>T | A319S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A319S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as benign. No prediction or stability result is missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.410405 | Uncertain | 0.879 | 0.254 | 0.125 | -7.109 | In-Between | 0.093 | Likely Benign | Likely Benign | 0.16 | Likely Benign | 0.2 | 0.02 | Likely Benign | 0.09 | Likely Benign | 0.13 | Likely Benign | 0.165 | Likely Benign | -0.58 | Neutral | 0.978 | Probably Damaging | 0.754 | Possibly Damaging | 2.02 | Pathogenic | 0.12 | Tolerated | 0.2537 | 0.4703 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||
| c.1025A>T | Y342F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y342F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive and therefore treated as unavailable. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.366687 | Structured | 0.408200 | Uncertain | 0.866 | 0.487 | 0.250 | -6.987 | Likely Benign | 0.145 | Likely Benign | Likely Benign | -0.13 | Likely Benign | 0.1 | 0.42 | Likely Benign | 0.15 | Likely Benign | 0.05 | Likely Benign | 0.160 | Likely Benign | -2.67 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.03 | Pathogenic | 0.26 | Tolerated | 0.2626 | 0.3615 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||
| c.2171C>A | A724D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A724D is not reported in ClinVar or gnomAD. Prediction tools that indicate a benign effect include REVEL, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. Four methods (FoldX, Rosetta, Foldetta, premPS) returned uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic; Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for A724D, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -12.233 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 1.06 | Ambiguous | 0.2 | 0.86 | Ambiguous | 0.96 | Ambiguous | 0.60 | Ambiguous | 0.335 | Likely Benign | -4.44 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1596 | 0.1812 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.2366C>A | P789Q 2D AIThe SynGAP1 P789Q missense change is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the ClinVar status, which currently has no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -5.191 | Likely Benign | 0.465 | Ambiguous | Likely Benign | 0.235 | Likely Benign | -4.53 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1284 | 0.3215 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2366C>G | P789R 2D AIThe SynGAP1 missense variant P789R is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. In contrast, the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all indicate pathogenicity, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains Likely Pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -2.503 | Likely Benign | 0.668 | Likely Pathogenic | Likely Benign | 0.354 | Likely Benign | -5.04 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1353 | 0.2129 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||
| c.2947A>C | S983R 2D AIThe SynGAP1 missense variant S983R is reported in ClinVar as “Not submitted” and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and also indicates a likely pathogenic outcome. AlphaMissense‑Optimized independently predicts pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for S983R, and this conclusion does not contradict any ClinVar annotation, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -4.733 | Likely Benign | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.156 | Likely Benign | -2.66 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1163 | 0.3740 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2949C>A | S983R 2D AIThe SynGAP1 missense variant S983R is reported in ClinVar as “Not submitted” and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (3 pathogenic vs. 1 benign) is likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that S983R is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently lacks a classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -4.733 | Likely Benign | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.190 | Likely Benign | -2.66 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1163 | 0.3740 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2949C>G | S983R 2D AIThe SynGAP1 missense variant S983R is reported in ClinVar as “Not submitted” and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (3 pathogenic vs. 1 benign) is likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S983R is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently lacks a pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -4.733 | Likely Benign | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.190 | Likely Benign | -2.66 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1163 | 0.3740 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.3635C>A | S1212Y 2D AIThe SynGAP1 missense variant S1212Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic status. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the convergence of multiple prediction algorithms, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.566480 | Disordered | 0.548409 | Binding | 0.852 | 0.565 | 0.500 | -12.186 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.304 | Likely Benign | -4.55 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.0546 | 0.4291 | -3 | -2 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||||
| c.3635C>T | S1212F 2D AIThe SynGAP1 missense variant S1212F is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) score—predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from multiple independent predictors indicates that the variant is most likely pathogenic, which is consistent with its ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.566480 | Disordered | 0.548409 | Binding | 0.852 | 0.565 | 0.500 | Conflicting | 2 | -14.445 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.271 | Likely Benign | -4.52 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.0503 | 0.4579 | -3 | -2 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||
| c.3754C>A | Q1252K 2D 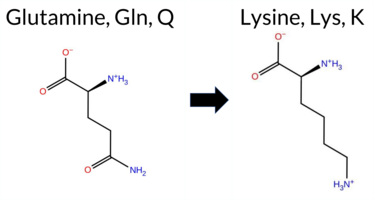 AIThe SynGAP1 missense variant Q1252K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict a pathogenic impact, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the predominance of pathogenic predictions, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.759478 | Disordered | 0.371411 | Uncertain | 0.850 | 0.544 | 0.875 | -13.590 | Likely Pathogenic | 0.878 | Likely Pathogenic | Ambiguous | 0.217 | Likely Benign | -3.22 | Deleterious | 0.985 | Probably Damaging | 0.981 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1460 | 0.2518 | 1 | 1 | -0.4 | 0.04 | ||||||||||||||||||||||||||||||||||||||
| c.3772C>A | Q1258K 2D AIThe SynGAP1 missense variant Q1258K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: REVEL scores the variant as benign, whereas the majority of other in silico predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—classify it as pathogenic. Grouping by consensus, the benign prediction is represented only by REVEL, while the pathogenic predictions are supported by seven distinct algorithms. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized returns an uncertain result, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels the variant as likely pathogenic, and Foldetta data are not available. Taken together, the preponderance of evidence from multiple pathogenic predictors and the SGM‑Consensus suggests that the variant is most likely pathogenic, which is consistent with the absence of a ClinVar entry and gnomAD observation. Thus, the variant is most likely pathogenic, and this prediction does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.486429 | Structured | 0.525814 | Binding | 0.859 | 0.577 | 0.250 | -10.927 | Likely Pathogenic | 0.912 | Likely Pathogenic | Ambiguous | 0.227 | Likely Benign | -3.19 | Deleterious | 0.985 | Probably Damaging | 0.981 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1151 | 0.2761 | 1 | 1 | -0.4 | 0.04 | ||||||||||||||||||||||||||||||||||||||
| c.800G>C | W267S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W267S is not listed in ClinVar and has no allele in gnomAD. Functional prediction tools largely agree on a deleterious effect: SIFT is the sole benign predictor, whereas the remaining 12 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all report pathogenicity; premPS is uncertain. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.298060 | Uncertain | 0.943 | 0.274 | 0.000 | -12.833 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 3.42 | Destabilizing | 0.3 | 3.20 | Destabilizing | 3.31 | Destabilizing | 0.87 | Ambiguous | 0.593 | Likely Pathogenic | -12.87 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.03 | Pathogenic | 0.09 | Tolerated | 0.3809 | 0.1415 | -2 | -3 | 0.1 | -99.14 | |||||||||||||||||||||||||||||
| c.869T>G | L290R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L290R is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions are limited to FoldX, whereas the remaining tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all classify the variant as pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus also indicates likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result. Overall, the consensus of the majority of evidence points to a pathogenic impact. This conclusion is consistent with the absence of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.127496 | Structured | 0.399723 | Uncertain | 0.904 | 0.255 | 0.000 | -13.938 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.48 | Likely Benign | 0.1 | 0.88 | Ambiguous | 0.68 | Ambiguous | 0.89 | Ambiguous | 0.705 | Likely Pathogenic | -5.52 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.03 | Pathogenic | 0.01 | Affected | 0.1215 | 0.1056 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.892C>T | P298S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P298S is listed in ClinVar as Benign (ClinVar ID 2965590.0) and is present in gnomAD (ID 6‑33437797‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. No evidence from FoldX, Rosetta, or premPS is available to support either outcome. Overall, the majority of predictions support a benign impact, aligning with the ClinVar designation. Thus, the variant is most likely benign and does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.328603 | Structured | 0.268765 | Uncertain | 0.860 | 0.283 | 0.500 | Benign | 1 | 6-33437797-C-T | 5 | 3.10e-6 | -6.342 | Likely Benign | 0.144 | Likely Benign | Likely Benign | 1.38 | Ambiguous | 0.2 | 1.41 | Ambiguous | 1.40 | Ambiguous | 0.58 | Ambiguous | 0.189 | Likely Benign | -1.20 | Neutral | 0.991 | Probably Damaging | 0.898 | Possibly Damaging | 2.03 | Pathogenic | 0.85 | Tolerated | 3.39 | 20 | 0.3678 | 0.5855 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||
| c.893C>G | P298R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P298R has no ClinVar entry and is not reported in gnomAD. Computational predictors fall into two consensus groups: benign predictions come from REVEL, FoldX, PROVEAN, SIFT, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results are reported only by Foldetta and premPS. High‑accuracy tools give a mixed picture: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta reports an uncertain stability change. Overall, the majority of evidence points toward a pathogenic effect, and this assessment is not contradicted by ClinVar, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.268765 | Uncertain | 0.860 | 0.283 | 0.500 | -11.427 | Likely Pathogenic | 0.733 | Likely Pathogenic | Likely Benign | 0.45 | Likely Benign | 0.0 | 2.08 | Destabilizing | 1.27 | Ambiguous | 0.61 | Ambiguous | 0.280 | Likely Benign | -1.83 | Neutral | 0.997 | Probably Damaging | 0.952 | Probably Damaging | 2.03 | Pathogenic | 0.09 | Tolerated | 0.1299 | 0.3872 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.944A>G | N315S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N315S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact for the variant, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -4.847 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.85 | Ambiguous | 0.2 | 0.78 | Ambiguous | 0.82 | Ambiguous | 0.68 | Ambiguous | 0.250 | Likely Benign | -1.84 | Neutral | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.03 | Pathogenic | 0.60 | Tolerated | 0.4022 | 0.7395 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||
| c.2170G>C | A724P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A724P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: REVEL predicts a benign effect, whereas the remaining 13 tools (FoldX, Rosetta, Foldetta, premPS [uncertain], PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized returns a pathogenic score; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, classifies the variant as pathogenic. No tool suggests a benign outcome, and the single benign prediction (REVEL) is outweighed by the consensus of pathogenic predictions. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this mutation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -12.817 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.90 | Destabilizing | 0.3 | 6.35 | Destabilizing | 4.63 | Destabilizing | 0.52 | Ambiguous | 0.391 | Likely Benign | -3.88 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.04 | Pathogenic | 0.04 | Affected | 0.1642 | 0.4273 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.2452C>T | P818S 2D AIThe SynGAP1 missense variant P818S is catalogued in gnomAD (ID 6‑33443004‑C‑T) but has no ClinVar entry. Functional prediction tools split in two groups: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The consensus predictor SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments are limited: AlphaMissense‑Optimized yields an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this residue. Taken together, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.715889 | Binding | 0.371 | 0.893 | 0.625 | 6-33443004-C-T | 1 | 6.20e-7 | -5.740 | Likely Benign | 0.932 | Likely Pathogenic | Ambiguous | 0.203 | Likely Benign | -4.38 | Deleterious | 0.989 | Probably Damaging | 0.824 | Possibly Damaging | 2.04 | Pathogenic | 0.04 | Affected | 3.77 | 5 | 0.3581 | 0.6199 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.2473T>G | S825A 2D AIThe SynGAP1 missense variant S825A is not reported in ClinVar (no entry) and is absent from gnomAD. Prediction tools that agree on benign include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on pathogenic are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of standard predictors indicate a pathogenic effect, while the high‑accuracy AlphaMissense‑Optimized suggests benign and the consensus is neutral. Given the preponderance of pathogenic predictions and the lack of conflicting evidence from ClinVar or population databases, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.675549 | Disordered | 0.618614 | Binding | 0.384 | 0.886 | 0.750 | -4.878 | Likely Benign | 0.569 | Likely Pathogenic | Likely Benign | 0.135 | Likely Benign | -2.30 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.04 | Pathogenic | 0.05 | Affected | 0.4794 | 0.5749 | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||||
| c.2644G>A | G882R 2D AIThe SynGAP1 missense variant G882R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive because it yields a 2‑to‑2 split. Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.632888 | Binding | 0.306 | 0.878 | 0.250 | -4.715 | Likely Benign | 0.758 | Likely Pathogenic | Likely Benign | 0.051 | Likely Benign | -1.48 | Neutral | 0.001 | Benign | 0.003 | Benign | 2.04 | Pathogenic | 0.01 | Affected | 0.0905 | 0.4126 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.2644G>C | G882R 2D AIThe SynGAP1 missense variant G882R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs 2 pathogenic). Foldetta results are unavailable. Overall, the majority of predictions (six benign vs three pathogenic) support a benign classification. This consensus does not contradict any ClinVar status, as the variant is not yet catalogued there. Thus, the variant is most likely benign based on current predictive evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.632888 | Binding | 0.306 | 0.878 | 0.250 | -4.715 | Likely Benign | 0.758 | Likely Pathogenic | Likely Benign | 0.051 | Likely Benign | -1.48 | Neutral | 0.001 | Benign | 0.003 | Benign | 2.04 | Pathogenic | 0.01 | Affected | 0.0905 | 0.4126 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.2645G>A | G882E 2D  AIThe SynGAP1 missense variant G882E is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split; Foldetta results are unavailable. Overall, more tools (six) predict benign than pathogenic (three), and no ClinVar evidence contradicts this assessment. Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.632888 | Binding | 0.306 | 0.878 | 0.250 | -4.246 | Likely Benign | 0.613 | Likely Pathogenic | Likely Benign | 0.079 | Likely Benign | -1.16 | Neutral | 0.004 | Benign | 0.005 | Benign | 2.04 | Pathogenic | 0.01 | Affected | 0.1263 | 0.3244 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||||||||
| c.2948G>A | S983N 2D AISynGAP1 missense variant S983N is listed as Benign in ClinVar (ID 469153) and is present in gnomAD (6‑33443500‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive; Foldetta results are not available. Overall, the majority of available predictions (five pathogenic vs. three benign) suggest a pathogenic impact, which contradicts the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | Likely Benign | 1 | 6-33443500-G-A | 6 | 3.72e-6 | -5.604 | Likely Benign | 0.909 | Likely Pathogenic | Ambiguous | 0.136 | Likely Benign | -1.78 | Neutral | 0.991 | Probably Damaging | 0.988 | Probably Damaging | 2.04 | Pathogenic | 0.00 | Affected | 4.32 | 1 | 0.1933 | 0.4069 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||
| c.3193C>A | P1065T 2D AIThe SynGAP1 missense variant P1065T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign classification. There is no ClinVar status to contradict this assessment, so the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -5.392 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -1.23 | Neutral | 0.770 | Possibly Damaging | 0.481 | Possibly Damaging | 2.04 | Pathogenic | 0.00 | Affected | 0.1577 | 0.6788 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3635C>G | S1212C 2D AIThe SynGAP1 missense variant S1212C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, while the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all indicate pathogenicity. The SGM‑Consensus, a majority‑vote method from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. ESM1b is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. High‑accuracy tools therefore give a benign call from AlphaMissense‑Optimized, a pathogenic call from SGM‑Consensus, and no data from Foldetta. Overall, the preponderance of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.566480 | Disordered | 0.548409 | Binding | 0.852 | 0.565 | 0.500 | -7.938 | In-Between | 0.701 | Likely Pathogenic | Likely Benign | 0.245 | Likely Benign | -3.58 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.04 | Pathogenic | 0.00 | Affected | 0.0693 | 0.4631 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||
| c.4003G>A | G1335S 2D AIThe SynGAP1 missense variant G1335S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33451877‑G‑A). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence indicates that G1335S is most likely pathogenic, which is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.967705 | Binding | 0.323 | 0.724 | 0.750 | Conflicting | 2 | 6-33451877-G-A | 3 | 2.37e-6 | -4.495 | Likely Benign | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.362 | Likely Benign | -3.79 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.04 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2452 | 0.5895 | 1 | 0 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||||
| c.919T>C | F307L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F307L is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Rosetta and Foldetta, whereas the majority of tools predict it to be pathogenic: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools (FoldX and premPS) give uncertain results. High‑accuracy methods give mixed evidence: AlphaMissense‑Optimized and the SGM Consensus both indicate a likely pathogenic effect, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a benign outcome. Overall, the bulk of evidence points to a pathogenic impact, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.298791 | Structured | 0.327302 | Uncertain | 0.900 | 0.315 | 0.125 | -10.173 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.54 | Ambiguous | 0.1 | 0.14 | Likely Benign | 0.34 | Likely Benign | 0.57 | Ambiguous | 0.597 | Likely Pathogenic | -5.52 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.04 | Pathogenic | 0.01 | Affected | 0.2800 | 0.4207 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.921C>A | F307L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F307L is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Rosetta and Foldetta, whereas the majority of tools predict it to be pathogenic: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools (FoldX and premPS) give uncertain results. High‑accuracy methods give mixed evidence: AlphaMissense‑Optimized and the SGM Consensus both indicate a likely pathogenic effect, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a benign outcome. Overall, the bulk of evidence points to a pathogenic impact, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.298791 | Structured | 0.327302 | Uncertain | 0.900 | 0.315 | 0.125 | -10.173 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.54 | Ambiguous | 0.1 | 0.14 | Likely Benign | 0.34 | Likely Benign | 0.57 | Ambiguous | 0.529 | Likely Pathogenic | -5.52 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.04 | Pathogenic | 0.01 | Affected | 0.2800 | 0.4207 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.921C>G | F307L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F307L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta and Foldetta, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) classify the change as pathogenic. Uncertain results are reported by FoldX and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as benign. Overall, the consensus of the majority of tools points to a pathogenic effect, and this is consistent with the lack of ClinVar annotation; there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.298791 | Structured | 0.327302 | Uncertain | 0.900 | 0.315 | 0.125 | -10.173 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.54 | Ambiguous | 0.1 | 0.14 | Likely Benign | 0.34 | Likely Benign | 0.57 | Ambiguous | 0.529 | Likely Pathogenic | -5.52 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.04 | Pathogenic | 0.01 | Affected | 0.2800 | 0.4207 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.976C>G | H326D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H326D missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only SIFT, which scores the variant as benign. All other evaluated algorithms—REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic outcome. Tools with inconclusive results are FoldX, Foldetta, and premPS, which are listed as uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a pathogenic verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, reports an uncertain result. Overall, the preponderance of evidence indicates the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.342579 | Structured | 0.418150 | Uncertain | 0.944 | 0.455 | 0.000 | -13.000 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.87 | Ambiguous | 0.6 | 2.00 | Destabilizing | 1.44 | Ambiguous | 0.96 | Ambiguous | 0.561 | Likely Pathogenic | -7.67 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 2.04 | Pathogenic | 0.10 | Tolerated | 0.2612 | 0.1611 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||
| c.2468G>C | S823T 2D AIThe SynGAP1 missense variant S823T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus (SGM Consensus) derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN is inconclusive (two benign, two pathogenic). AlphaMissense‑Optimized predicts benign, while Foldetta (combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the majority of standard predictors indicate a pathogenic effect, and this assessment does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.685117 | Disordered | 0.627336 | Binding | 0.358 | 0.884 | 0.750 | -6.036 | Likely Benign | 0.717 | Likely Pathogenic | Likely Benign | 0.165 | Likely Benign | -2.02 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.05 | Pathogenic | 0.00 | Affected | 0.1453 | 0.6580 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3634T>C | S1212P 2D AIThe SynGAP1 missense variant S1212P is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools that classify the variant as benign include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic effect. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus likewise reports likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of computational evidence points to the variant being most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.566480 | Disordered | 0.548409 | Binding | 0.852 | 0.565 | 0.500 | -13.336 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.239 | Likely Benign | -3.79 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.05 | Pathogenic | 0.00 | Affected | 0.1585 | 0.4370 | 1 | -1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||||||
| c.3809A>G | E1270G 2D AIThe SynGAP1 missense variant E1270G is listed in gnomAD (ID 6‑33447857‑A‑G) and has no ClinVar entry. Prediction tools cluster into two groups: the single benign predictor REVEL, and a consensus of pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic, while Foldetta’s protein‑folding stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the absence of a benign consensus, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.490133 | Structured | 0.771865 | Binding | 0.805 | 0.659 | 0.250 | 6-33447857-A-G | -12.022 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.385 | Likely Benign | -5.90 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.05 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2415 | 0.5452 | -2 | 0 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||
| c.3812A>G | E1271G 2D AIThe SynGAP1 missense variant E1271G is catalogued in gnomAD (ID 6‑33447860‑A‑G) but has no entry in ClinVar. Functional prediction tools show a split consensus: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts a benign effect, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (two pathogenic versus one benign vote). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence, including the SGM Consensus, indicates a pathogenic impact. This prediction is not contradicted by ClinVar, as no ClinVar classification exists for E1271G. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.483068 | Structured | 0.767529 | Binding | 0.832 | 0.666 | 0.375 | 6-33447860-A-G | -4.857 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.288 | Likely Benign | -5.37 | Deleterious | 0.905 | Possibly Damaging | 0.538 | Possibly Damaging | 2.05 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2513 | 0.5485 | -2 | 0 | 3.1 | -72.06 | 10.1016/j.ajhg.2020.11.011 | ||||||||||||||||||||||||||||||||||||
| c.4004G>C | G1335A 2D AIThe SynGAP1 missense variant G1335A is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely pathogenic. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect for G1335A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.967705 | Binding | 0.323 | 0.724 | 0.750 | -4.942 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.357 | Likely Benign | -3.69 | Deleterious | 0.996 | Probably Damaging | 0.992 | Probably Damaging | 2.05 | Pathogenic | 0.00 | Affected | 0.3383 | 0.4953 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.863A>G | D288G 2D 3DClick to see structure in 3D Viewer AISynGAP1 D288G is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, Rosetta, premPS, SIFT, AlphaMissense‑Optimized, and Foldetta. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. No folding‑stability result is available from FoldX. Overall, the balance of evidence slightly favors a pathogenic interpretation, with one high‑accuracy tool supporting benignity. The prediction does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.125101 | Structured | 0.395525 | Uncertain | 0.873 | 0.261 | 0.000 | -8.531 | Likely Pathogenic | 0.739 | Likely Pathogenic | Likely Benign | -0.71 | Ambiguous | 0.4 | 0.21 | Likely Benign | -0.25 | Likely Benign | -0.15 | Likely Benign | 0.436 | Likely Benign | -5.03 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.05 | Pathogenic | 0.13 | Tolerated | 0.4126 | 0.5934 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.2365C>A | P789T 2D AIThe SynGAP1 P789T missense variant has no ClinVar entry and is not present in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further clarify the variant’s likely effect: AlphaMissense‑Optimized predicts a benign outcome, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that P789T is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -5.242 | Likely Benign | 0.356 | Ambiguous | Likely Benign | 0.224 | Likely Benign | -4.77 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.06 | Pathogenic | 0.00 | Affected | 0.1371 | 0.3837 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2534A>G | D845G 2D AIThe SynGAP1 missense variant D845G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL classifies it as benign, whereas the remaining 11 predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a Likely Pathogenic status; Foldetta results are not available. Taken together, the preponderance of evidence indicates that D845G is most likely pathogenic, and this assessment does not conflict with the current ClinVar record, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.599971 | Binding | 0.297 | 0.827 | 0.500 | -8.209 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.399 | Likely Benign | -4.96 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 2.06 | Pathogenic | 0.00 | Affected | 0.3953 | 0.6740 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2645G>T | G882V 2D AIThe SynGAP1 missense variant G882V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation—there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.632888 | Binding | 0.306 | 0.878 | 0.250 | -5.893 | Likely Benign | 0.257 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -2.41 | Neutral | 0.224 | Benign | 0.066 | Benign | 2.06 | Pathogenic | 0.02 | Affected | 0.1178 | 0.4717 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3193C>T | P1065S 2D AIThe SynGAP1 missense variant P1065S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -5.512 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -2.07 | Neutral | 0.770 | Possibly Damaging | 0.255 | Benign | 2.06 | Pathogenic | 0.00 | Affected | 0.3172 | 0.6021 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3808G>C | E1270Q 2D AIThe SynGAP1 missense variant E1270Q is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Prediction tools that agree on a benign effect include only REVEL, which scores the substitution as benign. In contrast, the majority of in silico predictors—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—classify the change as pathogenic. The high‑accuracy AlphaMissense‑Optimized assessment is uncertain, while the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic outcome. Foldetta predictions are unavailable for this variant. Overall, the preponderance of pathogenic predictions, together with the SGM Consensus result, indicates that E1270Q is most likely pathogenic; this conclusion does not contradict ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.490133 | Structured | 0.771865 | Binding | 0.805 | 0.659 | 0.250 | -8.645 | Likely Pathogenic | 0.935 | Likely Pathogenic | Ambiguous | 0.330 | Likely Benign | -2.53 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.06 | Pathogenic | 0.00 | Affected | 0.0919 | 0.5858 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||
| c.3809A>C | E1270A 2D AIThe SynGAP1 missense change E1270A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: REVEL is the only score that flags the variant as benign, whereas PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a pathogenic effect, and the SGM‑Consensus also indicates a likely pathogenic outcome. No Foldetta (FoldX‑MD/Rosetta stability) result is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a pathogenic impact for E1270A, and this conclusion is consistent with the absence of a ClinVar entry (i.e., no contradictory clinical classification). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.490133 | Structured | 0.771865 | Binding | 0.805 | 0.659 | 0.250 | -12.081 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 0.388 | Likely Benign | -5.04 | Deleterious | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.06 | Pathogenic | 0.00 | Affected | 0.3024 | 0.5126 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||
| c.3811G>C | E1271Q 2D AIThe SynGAP1 missense variant E1271Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of predictions (six benign vs. four pathogenic) support a benign classification. This conclusion is not contradicted by ClinVar, which has no entry for this variant. Thus, based on the available computational evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.483068 | Structured | 0.767529 | Binding | 0.832 | 0.666 | 0.375 | -3.085 | Likely Benign | 0.282 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -2.40 | Neutral | 0.951 | Possibly Damaging | 0.617 | Possibly Damaging | 2.06 | Pathogenic | 0.00 | Affected | 0.0955 | 0.5470 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3812A>C | E1271A 2D AIThe SynGAP1 missense variant E1271A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessment shows AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of conventional tools and the high‑accuracy consensus favor a pathogenic interpretation. This prediction does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.483068 | Structured | 0.767529 | Binding | 0.832 | 0.666 | 0.375 | -3.817 | Likely Benign | 0.375 | Ambiguous | Likely Benign | 0.298 | Likely Benign | -4.79 | Deleterious | 0.951 | Possibly Damaging | 0.433 | Benign | 2.06 | Pathogenic | 0.00 | Affected | 0.2985 | 0.4959 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3863A>T | K1288M 2D AIThe SynGAP1 missense variant K1288M is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus indicates a likely pathogenic outcome; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of computational predictions (seven pathogenic vs. three benign) point to a pathogenic impact for K1288M. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.827927 | Disordered | 0.814714 | Binding | 0.538 | 0.784 | 0.625 | -3.355 | Likely Benign | 0.660 | Likely Pathogenic | Likely Benign | 0.246 | Likely Benign | -4.89 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.06 | Pathogenic | 0.00 | Affected | 0.0985 | 0.3140 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||||||||||||
| c.919T>A | F307I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F307I is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign are FoldX, Rosetta, and Foldetta. Tools that predict it as pathogenic include SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy assessments give AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Benign. Overall, the majority of predictions (10/13) indicate pathogenicity, while only three tools suggest benign. Therefore, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.298791 | Structured | 0.327302 | Uncertain | 0.900 | 0.315 | 0.125 | -13.114 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.49 | Likely Benign | 0.2 | 0.19 | Likely Benign | 0.34 | Likely Benign | 0.61 | Ambiguous | 0.632 | Likely Pathogenic | -5.52 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 2.06 | Pathogenic | 0.01 | Affected | 0.2531 | 0.3036 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.2171C>G | A724G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A724G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -8.908 | Likely Pathogenic | 0.580 | Likely Pathogenic | Likely Benign | 1.45 | Ambiguous | 0.1 | 1.73 | Ambiguous | 1.59 | Ambiguous | 0.56 | Ambiguous | 0.286 | Likely Benign | -3.10 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.07 | Pathogenic | 0.08 | Tolerated | 0.2001 | 0.3609 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.2171C>T | A724V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A724V missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign, SGM Consensus predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. With no ClinVar annotation, there is no contradiction between the predictions and existing clinical data. Overall, the evidence is mixed, but the majority of high‑confidence tools lean toward a benign effect, suggesting the variant is most likely benign rather than pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -9.000 | Likely Pathogenic | 0.471 | Ambiguous | Likely Benign | 0.42 | Likely Benign | 0.1 | 0.35 | Likely Benign | 0.39 | Likely Benign | 0.24 | Likely Benign | 0.241 | Likely Benign | -3.28 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | 2.07 | Pathogenic | 0.01 | Affected | 0.0872 | 0.5620 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.2284G>T | D762Y 2D AIThe SynGAP1 D762Y variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas a larger group predicts a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus methods give the following results: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic”; Foldetta (a protein‑folding stability approach combining FoldX‑MD and Rosetta) has no available output for this variant. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -6.959 | Likely Benign | 0.905 | Likely Pathogenic | Ambiguous | 0.219 | Likely Benign | -3.24 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 2.07 | Pathogenic | 0.01 | Affected | 0.0700 | 0.7929 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2473T>A | S825T 2D AIThe SynGAP1 missense variant S825T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus (SGM Consensus) derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN is inconclusive (two benign, two pathogenic). AlphaMissense‑Optimized, a high‑accuracy model, predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of standard tools (5 pathogenic vs. 4 benign) suggest a pathogenic impact, while the single high‑accuracy model indicates benign. Thus, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.675549 | Disordered | 0.618614 | Binding | 0.384 | 0.886 | 0.750 | -4.659 | Likely Benign | 0.615 | Likely Pathogenic | Likely Benign | 0.155 | Likely Benign | -2.26 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.07 | Pathogenic | 0.05 | Affected | 0.1432 | 0.6658 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3620A>T | E1207V 2D AIThe SynGAP1 missense change E1207V is not reported in ClinVar (ClinVar ID = None) and has no entries in gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments show the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, while AlphaMissense‑Optimized remains uncertain and Foldetta data are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for E1207V, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.604312 | Disordered | 0.562696 | Binding | 0.912 | 0.571 | 0.375 | -9.580 | Likely Pathogenic | 0.821 | Likely Pathogenic | Ambiguous | 0.342 | Likely Benign | -5.00 | Deleterious | 0.999 | Probably Damaging | 0.958 | Probably Damaging | 2.07 | Pathogenic | 0.00 | Affected | 0.0511 | 0.4439 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3730A>T | S1244C 2D AIThe SynGAP1 missense variant S1244C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Benign predictions are limited to REVEL and AlphaMissense‑Optimized. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized predicts benign, whereas the SGM‑Consensus (majority vote) predicts likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.648219 | Disordered | 0.411055 | Uncertain | 0.833 | 0.549 | 0.500 | -9.792 | Likely Pathogenic | 0.625 | Likely Pathogenic | Likely Benign | 0.270 | Likely Benign | -3.63 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.07 | Pathogenic | 0.04 | Affected | 0.0875 | 0.4616 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||
| c.3808G>A | E1270K 2D AIThe SynGAP1 missense variant E1270K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: REVEL predicts a benign outcome, whereas all other evaluated algorithms (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence indicates that E1270K is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.490133 | Structured | 0.771865 | Binding | 0.805 | 0.659 | 0.250 | -12.549 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.413 | Likely Benign | -3.37 | Deleterious | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.07 | Pathogenic | 0.00 | Affected | 0.1780 | 0.6276 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3811G>A | E1271K 2D AIThe SynGAP1 missense variant E1271K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further reveal AlphaMissense‑Optimized as benign, whereas Foldetta (combining FoldX‑MD and Rosetta) has no available result. Overall, the preponderance of evidence—both from general predictors and the SGM Consensus—leans toward pathogenicity. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.767529 | Binding | 0.832 | 0.666 | 0.375 | -2.295 | Likely Benign | 0.689 | Likely Pathogenic | Likely Benign | 0.192 | Likely Benign | -3.24 | Deleterious | 0.905 | Possibly Damaging | 0.433 | Benign | 2.07 | Pathogenic | 0.00 | Affected | 0.1780 | 0.5888 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.978T>A | H326Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H326Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, and SIFT, whereas a majority of tools predict a pathogenic outcome: premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). Two tools, Foldetta (combining FoldX‑MD and Rosetta) and Rosetta alone, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for H326Q. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.342579 | Structured | 0.418150 | Uncertain | 0.944 | 0.455 | 0.000 | -8.688 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.46 | Likely Benign | 0.1 | 1.67 | Ambiguous | 1.07 | Ambiguous | 1.00 | Destabilizing | 0.444 | Likely Benign | -6.89 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.07 | Pathogenic | 0.10 | Tolerated | 0.1485 | 0.3500 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.978T>G | H326Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H326Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, and SIFT, whereas a majority of tools predict a pathogenic outcome: premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). Two tools, Foldetta (combining FoldX‑MD and Rosetta) and Rosetta alone, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus also indicates likely pathogenic, while Foldetta remains uncertain. Overall, the preponderance of evidence from multiple independent predictors points to a pathogenic effect for H326Q. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.342579 | Structured | 0.418150 | Uncertain | 0.944 | 0.455 | 0.000 | -8.688 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.46 | Likely Benign | 0.1 | 1.67 | Ambiguous | 1.07 | Ambiguous | 1.00 | Destabilizing | 0.444 | Likely Benign | -6.89 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.07 | Pathogenic | 0.10 | Tolerated | 0.1485 | 0.3500 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.2284G>C | D762H 2D AIThe SynGAP1 D762H missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -4.643 | Likely Benign | 0.909 | Likely Pathogenic | Ambiguous | 0.212 | Likely Benign | -2.73 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 2.08 | Pathogenic | 0.02 | Affected | 0.2007 | 0.9102 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.2285A>T | D762V 2D AIThe SynGAP1 D762V missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b. Tools that agree on a pathogenic effect include polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returned an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of consensus tools predict a pathogenic impact, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -6.122 | Likely Benign | 0.949 | Likely Pathogenic | Ambiguous | 0.229 | Likely Benign | -1.98 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.08 | Pathogenic | 0.01 | Affected | 0.1105 | 0.8785 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.2948G>C | S983T 2D AIThe SynGAP1 missense variant S983T is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign). High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus and Foldetta (which would combine FoldX‑MD and Rosetta outputs) are unavailable. Overall, the balance of evidence leans toward pathogenicity, with five tools supporting a deleterious effect versus four supporting benign. This prediction does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -4.493 | Likely Benign | 0.588 | Likely Pathogenic | Likely Benign | 0.172 | Likely Benign | -1.65 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 2.08 | Pathogenic | 0.00 | Affected | 0.2042 | 0.5517 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3695A>T | K1232I 2D AIThe SynGAP1 K1232I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. The high‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is classified as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.505461 | Disordered | 0.542907 | Binding | 0.894 | 0.535 | 0.125 | -12.225 | Likely Pathogenic | 0.896 | Likely Pathogenic | Ambiguous | 0.197 | Likely Benign | -5.98 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.08 | Pathogenic | 0.00 | Affected | 0.0778 | 0.3321 | -2 | -3 | 8.4 | -15.01 | ||||||||||||||||||||||||||||||||||||||
| c.3731G>T | S1244I 2D AIThe SynGAP1 missense variant S1244I is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as likely pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as likely pathogenic; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.648219 | Disordered | 0.411055 | Uncertain | 0.833 | 0.549 | 0.500 | -13.073 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.284 | Likely Benign | -4.39 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.08 | Pathogenic | 0.02 | Affected | 0.0780 | 0.4684 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||
| c.3805G>A | V1269M 2D  AIThe SynGAP1 missense variant V1269M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (majority vote) is pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect for V1269M, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.787464 | Binding | 0.843 | 0.647 | 0.125 | -3.743 | Likely Benign | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.300 | Likely Benign | -2.53 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.08 | Pathogenic | 0.00 | Affected | 0.0582 | 0.3676 | 2 | 1 | -2.3 | 32.06 | ||||||||||||||||||||||||||||||||||||||
| c.1019C>T | A340V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A340V variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) is uncertain and therefore unavailable for interpretation. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | -6.427 | Likely Benign | 0.174 | Likely Benign | Likely Benign | 0.69 | Ambiguous | 0.3 | 0.32 | Likely Benign | 0.51 | Ambiguous | 0.40 | Likely Benign | 0.102 | Likely Benign | -1.81 | Neutral | 0.801 | Possibly Damaging | 0.315 | Benign | 2.09 | Pathogenic | 0.57 | Tolerated | 0.1197 | 0.5780 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.2170G>A | A724T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A724T missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that indicate a benign effect include REVEL, premPS, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Predictions that are inconclusive are AlphaMissense‑Default, FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evaluated tools (seven pathogenic vs. three benign) suggest a pathogenic impact. This conclusion is consistent with the lack of ClinVar annotation, as there is no existing classification to contradict. Thus, the variant is most likely pathogenic based on current predictive evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -8.047 | Likely Pathogenic | 0.456 | Ambiguous | Likely Benign | 0.95 | Ambiguous | 0.1 | 1.10 | Ambiguous | 1.03 | Ambiguous | 0.40 | Likely Benign | 0.273 | Likely Benign | -2.87 | Deleterious | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 2.09 | Pathogenic | 0.01 | Affected | 0.1149 | 0.5897 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.2524T>G | S842A 2D AIThe SynGAP1 missense variant S842A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Overall, the majority of predictions (seven pathogenic vs. three benign) indicate a pathogenic impact. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.617281 | Binding | 0.274 | 0.861 | 0.250 | -13.601 | Likely Pathogenic | 0.656 | Likely Pathogenic | Likely Benign | 0.180 | Likely Benign | -2.37 | Neutral | 0.889 | Possibly Damaging | 0.614 | Possibly Damaging | 2.09 | Pathogenic | 0.00 | Affected | 0.3971 | 0.4223 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3620A>G | E1207G 2D AIThe SynGAP1 missense variant E1207G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) all predict a pathogenic impact. High‑accuracy assessments further support a deleterious interpretation: AlphaMissense‑Optimized classifies the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Pathogenic, and a Foldetta stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the SGM Consensus result, the variant is most likely pathogenic, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.604312 | Disordered | 0.562696 | Binding | 0.912 | 0.571 | 0.375 | -8.886 | Likely Pathogenic | 0.641 | Likely Pathogenic | Likely Benign | 0.311 | Likely Benign | -4.84 | Deleterious | 0.978 | Probably Damaging | 0.871 | Possibly Damaging | 2.09 | Pathogenic | 0.01 | Affected | 0.2621 | 0.4030 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||||||
| c.3730A>C | S1244R 2D AIThe SynGAP1 missense variant S1244R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (majority vote) confirms a likely pathogenic status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.648219 | Disordered | 0.411055 | Uncertain | 0.833 | 0.549 | 0.500 | -10.986 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.309 | Likely Benign | -3.49 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.09 | Pathogenic | 0.04 | Affected | 0.0778 | 0.3098 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||
| c.3732T>A | S1244R 2D AIThe SynGAP1 missense variant S1244R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.648219 | Disordered | 0.411055 | Uncertain | 0.833 | 0.549 | 0.500 | -10.986 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.274 | Likely Benign | -3.49 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.09 | Pathogenic | 0.04 | Affected | 0.0778 | 0.3098 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||
| c.3732T>G | S1244R 2D AIThe SynGAP1 missense variant S1244R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.648219 | Disordered | 0.411055 | Uncertain | 0.833 | 0.549 | 0.500 | -10.986 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.274 | Likely Benign | -3.49 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.09 | Pathogenic | 0.04 | Affected | 0.0778 | 0.3098 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||
| c.3806T>A | V1269E 2D  AIThe SynGAP1 missense change V1269E is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that flag the variant as benign include only REVEL, whereas the remaining predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classify it as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a “Likely Pathogenic” outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as likely pathogenic; the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a pathogenic effect, which does not contradict the ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.787464 | Binding | 0.843 | 0.647 | 0.125 | Uncertain | 1 | -11.418 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.403 | Likely Benign | -5.05 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.09 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.0899 | 0.1557 | -2 | -2 | -7.7 | 29.98 | ||||||||||||||||||||||||||||||||||
| c.3863A>C | K1288T 2D AIThe SynGAP1 missense variant K1288T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, more tools (five) predict pathogenicity than benign (three), and the high‑accuracy consensus also leans pathogenic. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.814714 | Binding | 0.538 | 0.784 | 0.625 | -2.616 | Likely Benign | 0.477 | Ambiguous | Likely Benign | 0.227 | Likely Benign | -4.84 | Deleterious | 0.991 | Probably Damaging | 0.982 | Probably Damaging | 2.09 | Pathogenic | 0.00 | Affected | 0.2066 | 0.2614 | 0 | -1 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||||||||||||
| c.2285A>G | D762G 2D AIThe SynGAP1 missense variant D762G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, and ESM1b, whereas those that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the majority of predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -1.062 | Likely Benign | 0.812 | Likely Pathogenic | Ambiguous | 0.170 | Likely Benign | -2.55 | Deleterious | 0.998 | Probably Damaging | 0.949 | Probably Damaging | 2.10 | Pathogenic | 0.08 | Tolerated | 0.4710 | 0.7841 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2957A>T | E986V 2D AIThe SynGAP1 E986V missense variant is not reported in ClinVar and has no gnomAD entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b, whereas pathogenic predictions arise from PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (benign), FATHMM (pathogenic), and PROVEAN (pathogenic)—also indicates pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a pathogenic effect for E986V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.511 | Likely Benign | 0.965 | Likely Pathogenic | Likely Pathogenic | 0.220 | Likely Benign | -3.48 | Deleterious | 0.018 | Benign | 0.028 | Benign | 2.10 | Pathogenic | 0.00 | Affected | 0.1146 | 0.7960 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3298G>A | G1100S 2D AIThe SynGAP1 missense variant G1100S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for G1100S, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.972009 | Binding | 0.360 | 0.865 | 0.875 | -2.898 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -0.65 | Neutral | 0.943 | Possibly Damaging | 0.595 | Possibly Damaging | 2.10 | Pathogenic | 0.28 | Tolerated | 0.2501 | 0.5699 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3634T>A | S1212T 2D AIThe SynGAP1 missense variant S1212T is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; SGM Consensus is unavailable; Foldetta, which combines FoldX‑MD and Rosetta stability calculations, has no reported output for this variant. Overall, the balance of evidence slightly favors a pathogenic interpretation, but the single high‑accuracy benign prediction and the lack of a consensus from SGM and Foldetta leave the assessment uncertain. There is no conflict with ClinVar status, as the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.566480 | Disordered | 0.548409 | Binding | 0.852 | 0.565 | 0.500 | -4.972 | Likely Benign | 0.759 | Likely Pathogenic | Likely Benign | 0.147 | Likely Benign | -2.19 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.10 | Pathogenic | 0.00 | Affected | 0.0997 | 0.4618 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3695A>C | K1232T 2D AIThe SynGAP1 missense variant K1232T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.505461 | Disordered | 0.542907 | Binding | 0.894 | 0.535 | 0.125 | -9.276 | Likely Pathogenic | 0.568 | Likely Pathogenic | Likely Benign | 0.189 | Likely Benign | -4.49 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.10 | Pathogenic | 0.00 | Affected | 0.1846 | 0.3196 | 0 | -1 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||||||||||
| c.3696A>C | K1232N 2D AIThe SynGAP1 missense variant K1232N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: REVEL scores the variant as benign, whereas PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict it to be pathogenic. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized returns an uncertain result, and Foldetta data are not available. Based on the overall evidence, the variant is most likely pathogenic, which is consistent with the lack of ClinVar annotation and gnomAD absence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.505461 | Disordered | 0.542907 | Binding | 0.894 | 0.535 | 0.125 | -8.657 | Likely Pathogenic | 0.834 | Likely Pathogenic | Ambiguous | 0.167 | Likely Benign | -3.72 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.10 | Pathogenic | 0.00 | Affected | 0.2913 | 0.1464 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||||||||||
| c.3696A>T | K1232N 2D AIThe SynGAP1 missense variant K1232N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—consistently predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.505461 | Disordered | 0.542907 | Binding | 0.894 | 0.535 | 0.125 | -8.657 | Likely Pathogenic | 0.834 | Likely Pathogenic | Ambiguous | 0.167 | Likely Benign | -3.72 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.10 | Pathogenic | 0.00 | Affected | 0.2913 | 0.1464 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||||||||||
| c.3731G>A | S1244N 2D AIThe SynGAP1 missense variant S1244N is listed in ClinVar with an “Uncertain” status (ClinVar ID 931075.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and AlphaMissense‑Optimized, whereas PolyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default all predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—indicates pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence leans toward a pathogenic effect, which does not contradict the ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.648219 | Disordered | 0.411055 | Uncertain | 0.833 | 0.549 | 0.500 | Uncertain | 1 | -9.008 | Likely Pathogenic | 0.751 | Likely Pathogenic | Likely Benign | 0.154 | Likely Benign | -1.87 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.10 | Pathogenic | 0.15 | Tolerated | 3.77 | 5 | 0.1033 | 0.3464 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||||
| c.3797T>C | L1266P 2D AIThe SynGAP1 missense variant L1266P is reported in gnomAD (6‑33447845‑T‑C) but has no ClinVar entry. Prediction tools cluster into two groups: benign predictions are made only by REVEL, whereas all other evaluated algorithms (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic effect. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized returns a pathogenic score, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Pathogenic.” The Foldetta stability analysis is unavailable for this variant. Overall, the consensus of both general and high‑accuracy predictors points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.802655 | Binding | 0.868 | 0.602 | 0.000 | 6-33447845-T-C | -16.566 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.424 | Likely Benign | -5.83 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.10 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3150 | 0.2283 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.3806T>G | V1269G 2D AIThe SynGAP1 missense variant V1269G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include only REVEL, whereas all other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic outcome. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenicity. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus is pathogenic; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.433034 | Structured | 0.787464 | Binding | 0.843 | 0.647 | 0.125 | -9.927 | Likely Pathogenic | 0.969 | Likely Pathogenic | Likely Pathogenic | 0.420 | Likely Benign | -5.91 | Deleterious | 0.995 | Probably Damaging | 0.999 | Probably Damaging | 2.10 | Pathogenic | 0.00 | Affected | 0.2180 | 0.2557 | -1 | -3 | -4.6 | -42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3864G>C | K1288N 2D AIThe SynGAP1 missense variant K1288N has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no result for this variant. High‑accuracy assessments therefore show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta as unavailable. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.827927 | Disordered | 0.814714 | Binding | 0.538 | 0.784 | 0.625 | -3.670 | Likely Benign | 0.801 | Likely Pathogenic | Ambiguous | 0.128 | Likely Benign | -4.05 | Deleterious | 0.991 | Probably Damaging | 0.987 | Probably Damaging | 2.10 | Pathogenic | 0.00 | Affected | 0.3513 | 0.1101 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3864G>T | K1288N 2D AIThe SynGAP1 missense variant K1288N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and ESM1b, while the majority of tools predict a pathogenic effect: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy assessments therefore show an uncertain AlphaMissense‑Optimized prediction, a Likely Pathogenic SGM‑Consensus, and no Foldetta data. Overall, the preponderance of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.827927 | Disordered | 0.814714 | Binding | 0.538 | 0.784 | 0.625 | -3.670 | Likely Benign | 0.801 | Likely Pathogenic | Ambiguous | 0.128 | Likely Benign | -4.05 | Deleterious | 0.991 | Probably Damaging | 0.987 | Probably Damaging | 2.10 | Pathogenic | 0.00 | Affected | 0.3513 | 0.1101 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.2158G>T | D720Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D720Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and premPS, while pathogenic predictions are made by SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Predictions from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive. The high‑accuracy consensus (SGM Consensus) classifies the variant as Likely Pathogenic, and Foldetta likewise yields an uncertain stability change. AlphaMissense‑Optimized remains inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -14.771 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | -0.74 | Ambiguous | 0.1 | -1.38 | Ambiguous | -1.06 | Ambiguous | 0.20 | Likely Benign | 0.499 | Likely Benign | -7.05 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.11 | Pathogenic | 0.00 | Affected | 0.0626 | 0.5973 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.2365C>G | P789A 2D AIThe SynGAP1 P789A missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a tie (2 benign, 2 pathogenic) and is therefore inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus remains inconclusive and Foldetta data are unavailable. Overall, the majority of tools (five out of nine) predict pathogenicity, but the presence of four benign predictions and the inconclusive high‑accuracy results suggest uncertainty. The variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -4.777 | Likely Benign | 0.235 | Likely Benign | Likely Benign | 0.258 | Likely Benign | -4.92 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.11 | Pathogenic | 0.00 | Affected | 0.3120 | 0.3052 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2417T>G | F806C 2D AIThe SynGAP1 missense variant F806C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict a pathogenic impact, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while SGM‑Consensus remains likely pathogenic; Foldetta results are unavailable. Based on the predominance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | -8.565 | Likely Pathogenic | 0.809 | Likely Pathogenic | Ambiguous | 0.266 | Likely Benign | -4.46 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.11 | Pathogenic | 0.00 | Affected | 0.3071 | 0.1125 | -4 | -2 | -0.3 | -44.04 | ||||||||||||||||||||||||||||||||||||||
| c.3619G>C | E1207Q 2D AIThe SynGAP1 missense variant E1207Q is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, while Foldetta results are unavailable. Taken together, the majority of evidence points to a benign effect, and this conclusion does not conflict with any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.604312 | Disordered | 0.562696 | Binding | 0.912 | 0.571 | 0.375 | -6.789 | Likely Benign | 0.538 | Ambiguous | Likely Benign | 0.167 | Likely Benign | -1.95 | Neutral | 0.989 | Probably Damaging | 0.904 | Possibly Damaging | 2.11 | Pathogenic | 0.03 | Affected | 0.0808 | 0.4185 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3620A>C | E1207A 2D AIThe SynGAP1 missense variant E1207A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and AlphaMissense‑Optimized, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized indicates a benign effect, but the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels the variant as Likely Pathogenic. No Foldetta stability analysis is available for this residue. Overall, the preponderance of evidence points to a pathogenic effect for E1207A, and this conclusion is not contradicted by any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.604312 | Disordered | 0.562696 | Binding | 0.912 | 0.571 | 0.375 | -8.277 | Likely Pathogenic | 0.679 | Likely Pathogenic | Likely Benign | 0.295 | Likely Benign | -4.27 | Deleterious | 0.989 | Probably Damaging | 0.829 | Possibly Damaging | 2.11 | Pathogenic | 0.02 | Affected | 0.3019 | 0.4305 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||
| c.3862A>C | K1288Q 2D AIThe SynGAP1 missense variant K1288Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a 2‑to‑2 split and is therefore inconclusive. Foldetta, a protein‑folding stability method that combines FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence is divided, with an equal number of benign and pathogenic calls, leaving the variant’s clinical significance uncertain. This uncertainty does not contradict ClinVar, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.814714 | Binding | 0.538 | 0.784 | 0.625 | -2.369 | Likely Benign | 0.172 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -2.51 | Deleterious | 0.991 | Probably Damaging | 0.987 | Probably Damaging | 2.11 | Pathogenic | 0.00 | Affected | 0.4149 | 0.0657 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||||||||||||
| c.1027G>C | V343L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all predict benign or likely benign. Only FATHMM predicts a pathogenic outcome, while FoldX and Rosetta provide uncertain results and are therefore not considered decisive. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates benign. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of ClinVar reporting. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -6.268 | Likely Benign | 0.310 | Likely Benign | Likely Benign | -0.93 | Ambiguous | 0.2 | 0.66 | Ambiguous | -0.14 | Likely Benign | 0.16 | Likely Benign | 0.033 | Likely Benign | -1.09 | Neutral | 0.005 | Benign | 0.013 | Benign | 2.12 | Pathogenic | 0.64 | Tolerated | 0.1361 | 0.5008 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.2159A>T | D720V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D720V has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, Foldetta, and premPS, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -12.730 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 0.08 | Likely Benign | 0.0 | -0.78 | Ambiguous | -0.35 | Likely Benign | 0.20 | Likely Benign | 0.437 | Likely Benign | -7.18 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 2.12 | Pathogenic | 0.00 | Affected | 0.0822 | 0.5708 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.2179A>T | N727Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727Y has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Two tools remain inconclusive: AlphaMissense‑Default and Rosetta. Separately, the high‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign. Overall, the majority of individual predictors and the SGM Consensus lean toward a pathogenic interpretation, while the high‑accuracy folding‑stability assessment is benign. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -10.106 | Likely Pathogenic | 0.426 | Ambiguous | Likely Benign | -0.12 | Likely Benign | 0.1 | -0.52 | Ambiguous | -0.32 | Likely Benign | 0.35 | Likely Benign | 0.347 | Likely Benign | -5.34 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.12 | Pathogenic | 0.02 | Affected | 0.0563 | 0.6091 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||
| c.2285A>C | D762A 2D AIThe SynGAP1 D762A missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool yields an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward a pathogenic impact. This assessment does not contradict any ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -4.510 | Likely Benign | 0.912 | Likely Pathogenic | Ambiguous | 0.178 | Likely Benign | -2.40 | Neutral | 0.994 | Probably Damaging | 0.900 | Possibly Damaging | 2.12 | Pathogenic | 0.05 | Affected | 0.4632 | 0.8428 | 0 | -2 | 5.3 | -44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3619G>A | E1207K 2D AIThe SynGAP1 missense variant E1207K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for E1207K. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.604312 | Disordered | 0.562696 | Binding | 0.912 | 0.571 | 0.375 | -8.145 | Likely Pathogenic | 0.908 | Likely Pathogenic | Ambiguous | 0.261 | Likely Benign | -2.88 | Deleterious | 0.978 | Probably Damaging | 0.829 | Possibly Damaging | 2.12 | Pathogenic | 0.02 | Affected | 0.1796 | 0.4234 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3730A>G | S1244G 2D AIThe SynGAP1 missense variant S1244G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy evidence therefore comes only from AlphaMissense‑Optimized, which predicts benign, while the other high‑accuracy tools are unavailable. Overall, the balance of evidence (five benign vs four pathogenic predictions, with the most reliable tool indicating benign) suggests that the variant is most likely benign. This conclusion does not contradict ClinVar status, as the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.648219 | Disordered | 0.411055 | Uncertain | 0.833 | 0.549 | 0.500 | -8.304 | Likely Pathogenic | 0.221 | Likely Benign | Likely Benign | 0.130 | Likely Benign | -1.77 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.12 | Pathogenic | 0.65 | Tolerated | 0.2267 | 0.3678 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3796C>A | L1266M 2D AIThe SynGAP1 missense variant L1266M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and PROVEAN, whereas a larger set—polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments are mixed: AlphaMissense‑Optimized yields an uncertain result, SGM‑Consensus remains likely pathogenic, and Foldetta data are unavailable. Overall, the majority of evidence points toward a pathogenic effect for L1266M. This conclusion does not conflict with ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.802655 | Binding | 0.868 | 0.602 | 0.000 | -8.257 | Likely Pathogenic | 0.938 | Likely Pathogenic | Ambiguous | 0.126 | Likely Benign | -1.67 | Neutral | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 2.12 | Pathogenic | 0.00 | Affected | 0.0627 | 0.3113 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||||
| c.3797T>A | L1266Q 2D AIThe SynGAP1 missense variant L1266Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates likely pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence points to a pathogenic effect for L1266Q, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.802655 | Binding | 0.868 | 0.602 | 0.000 | -16.101 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.374 | Likely Benign | -5.02 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.12 | Pathogenic | 0.00 | Affected | 0.0906 | 0.1249 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3818T>C | L1273P 2D AIThe SynGAP1 missense variant L1273P is not reported in ClinVar (ClinVar status: not listed) but is present in the gnomAD database (gnomAD ID: 6‑33447866‑T‑C). Functional prediction tools uniformly indicate a deleterious effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. No tool in the dataset predicts a benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the consensus of predictive algorithms strongly supports a pathogenic classification, and this assessment does not contradict the ClinVar status, which simply lacks an entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.773625 | Binding | 0.747 | 0.677 | 0.500 | 6-33447866-T-C | -9.443 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.523 | Likely Pathogenic | -5.87 | Deleterious | 0.997 | Probably Damaging | 0.950 | Probably Damaging | 2.12 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3836 | 0.1343 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||
| c.3830A>C | H1277P 2D AIThe SynGAP1 missense variant H1277P is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33447878‑A‑C). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, and this is consistent with the lack of a ClinVar pathogenic annotation. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | 6-33447878-A-C | -3.829 | Likely Benign | 0.324 | Likely Benign | Likely Benign | 0.237 | Likely Benign | -7.14 | Deleterious | 0.586 | Possibly Damaging | 0.287 | Benign | 2.12 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2266 | 0.3393 | -2 | 0 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||
| c.2158G>C | D720H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D720H missense variant is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, and premPS. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and is labeled Likely Pathogenic. The high‑accuracy AlphaMissense‑Optimized score is Uncertain, while Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, indicates a Benign effect. Considering the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -12.355 | Likely Pathogenic | 0.947 | Likely Pathogenic | Ambiguous | 0.03 | Likely Benign | 0.0 | -0.87 | Ambiguous | -0.42 | Likely Benign | 0.48 | Likely Benign | 0.444 | Likely Benign | -5.27 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.13 | Pathogenic | 0.01 | Affected | 0.1363 | 0.6198 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.2179A>C | N727H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727H is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Two tools (premPS and ESM1b) return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of predictions (six benign vs. five pathogenic) lean toward a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -7.308 | In-Between | 0.224 | Likely Benign | Likely Benign | 0.13 | Likely Benign | 0.0 | -0.02 | Likely Benign | 0.06 | Likely Benign | 0.51 | Ambiguous | 0.171 | Likely Benign | -3.18 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.13 | Pathogenic | 0.03 | Affected | 0.1320 | 0.7186 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||
| c.2180A>T | N727I 2D 3DClick to see structure in 3D Viewer AISynGAP1 N727I is not reported in ClinVar and is absent from gnomAD. Benign predictions come from REVEL, FoldX, premPS, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen2_HumDiv, polyPhen2_HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Foldetta and Rosetta provide inconclusive results. High‑accuracy tools give a mixed picture: AlphaMissense‑Optimized predicts benign, SGM‑Consensus predicts likely pathogenic, and Foldetta is uncertain. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -10.230 | Likely Pathogenic | 0.577 | Likely Pathogenic | Likely Benign | 0.17 | Likely Benign | 0.1 | 0.90 | Ambiguous | 0.54 | Ambiguous | 0.43 | Likely Benign | 0.319 | Likely Benign | -5.93 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.13 | Pathogenic | 0.03 | Affected | 0.0666 | 0.5917 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||
| c.2284G>A | D762N 2D AIThe SynGAP1 D762N missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign impact for D762N, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -3.323 | Likely Benign | 0.640 | Likely Pathogenic | Likely Benign | 0.110 | Likely Benign | -1.51 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.13 | Pathogenic | 0.11 | Tolerated | 0.1698 | 0.8797 | 2 | 1 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2957A>G | E986G 2D AIThe SynGAP1 missense variant E986G is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas seven tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this assessment does not conflict with the ClinVar status, which currently contains no classification for E986G. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -5.584 | Likely Benign | 0.834 | Likely Pathogenic | Ambiguous | 0.219 | Likely Benign | -3.14 | Deleterious | 0.924 | Possibly Damaging | 0.784 | Possibly Damaging | 2.13 | Pathogenic | 0.00 | Affected | 0.2866 | 0.6225 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3647T>C | L1216P 2D AIThe SynGAP1 missense variant L1216P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool predicts a benign outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as likely pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions strongly suggests that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.580690 | Disordered | 0.504713 | Binding | 0.863 | 0.563 | 0.250 | -16.029 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.524 | Likely Pathogenic | -5.28 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.13 | Pathogenic | 0.00 | Affected | 0.3281 | 0.1048 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||
| c.3694A>G | K1232E 2D AIThe SynGAP1 missense variant K1232E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) all predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as benign, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels it as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.505461 | Disordered | 0.542907 | Binding | 0.894 | 0.535 | 0.125 | -10.690 | Likely Pathogenic | 0.695 | Likely Pathogenic | Likely Benign | 0.165 | Likely Benign | -2.76 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.13 | Pathogenic | 0.00 | Affected | 0.2965 | 0.1039 | 0 | 1 | 0.4 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3797T>G | L1266R 2D AISynGAP1 missense variant L1266R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic, and the SGM‑Consensus confirms a likely pathogenic classification. Foldetta results are unavailable for this variant. Overall, the preponderance of evidence from multiple in silico predictors and high‑accuracy tools points to a pathogenic effect, with no conflict from ClinVar status (which has no entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.802655 | Binding | 0.868 | 0.602 | 0.000 | -16.676 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.391 | Likely Benign | -5.04 | Deleterious | 0.996 | Probably Damaging | 0.951 | Probably Damaging | 2.13 | Pathogenic | 0.00 | Affected | 0.1063 | 0.0891 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||
| c.3818T>A | L1273Q 2D AIThe SynGAP1 missense variant L1273Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic and the SGM‑Consensus is labeled Likely Pathogenic; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.773625 | Binding | 0.747 | 0.677 | 0.500 | -6.813 | Likely Benign | 0.957 | Likely Pathogenic | Likely Pathogenic | 0.423 | Likely Benign | -5.05 | Deleterious | 0.997 | Probably Damaging | 0.950 | Probably Damaging | 2.13 | Pathogenic | 0.00 | Affected | 0.1139 | 0.1119 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3818T>G | L1273R 2D AIThe SynGAP1 missense variant L1273R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all classify the variant as damaging. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic; this assessment is not contradicted by any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.773625 | Binding | 0.747 | 0.677 | 0.500 | -6.252 | Likely Benign | 0.946 | Likely Pathogenic | Ambiguous | 0.431 | Likely Benign | -5.05 | Deleterious | 0.997 | Probably Damaging | 0.934 | Probably Damaging | 2.13 | Pathogenic | 0.00 | Affected | 0.1369 | 0.0761 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3862A>G | K1288E 2D AISynGAP1 missense variant K1288E is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33447910‑A‑G). Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this change. Overall, the preponderance of evidence points to a pathogenic effect, which is consistent with the ClinVar designation of uncertain significance rather than a clear benign classification. Thus, the variant is most likely pathogenic, and this does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.814714 | Binding | 0.538 | 0.784 | 0.625 | Uncertain | 1 | 6-33447910-A-G | 5 | 3.22e-6 | -2.751 | Likely Benign | 0.407 | Ambiguous | Likely Benign | 0.185 | Likely Benign | -3.27 | Deleterious | 0.979 | Probably Damaging | 0.973 | Probably Damaging | 2.13 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3627 | 0.0676 | 1 | 0 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||
| c.886T>A | S296T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 S296T missense variant is not reported in ClinVar and is absent from gnomAD. Consensus from most in silico predictors shows a split: six tools (REVEL, FoldX, Rosetta, premPS, PROVEAN, SIFT) and AlphaMissense‑Optimized predict a benign effect, whereas four tools (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default) predict pathogenicity. ESM1b remains uncertain. High‑accuracy assessments give a more definitive picture: AlphaMissense‑Optimized classifies the variant as benign, the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans toward pathogenicity, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a benign outcome. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.444081 | Structured | 0.282669 | Uncertain | 0.887 | 0.284 | 0.250 | -7.020 | In-Between | 0.645 | Likely Pathogenic | Likely Benign | 0.09 | Likely Benign | 0.4 | 0.38 | Likely Benign | 0.24 | Likely Benign | 0.19 | Likely Benign | 0.201 | Likely Benign | -2.10 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.13 | Pathogenic | 0.13 | Tolerated | 0.1437 | 0.6340 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||
| c.2346C>A | D782E 2D AIThe SynGAP1 missense variant D782E is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -4.447 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.127 | Likely Benign | -1.75 | Neutral | 0.561 | Possibly Damaging | 0.207 | Benign | 2.14 | Pathogenic | 0.01 | Affected | 0.1351 | 0.7036 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2346C>G | D782E 2D AIThe SynGAP1 missense variant D782E is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a majority benign vote (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -4.447 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.127 | Likely Benign | -1.75 | Neutral | 0.561 | Possibly Damaging | 0.207 | Benign | 2.14 | Pathogenic | 0.01 | Affected | 0.1351 | 0.7036 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2538A>C | L846F 2D  AIThe SynGAP1 missense variant L846F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. In contrast, the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all classify the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. AlphaMissense‑Optimized yields an uncertain result, and no Foldetta (FoldX‑MD/Rosetta) stability assessment is available. Overall, the preponderance of evidence from multiple high‑accuracy predictors indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.653063 | Disordered | 0.589606 | Binding | 0.349 | 0.825 | 0.500 | -10.559 | Likely Pathogenic | 0.862 | Likely Pathogenic | Ambiguous | 0.206 | Likely Benign | -2.88 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.14 | Pathogenic | 0.00 | Affected | 0.0665 | 0.2912 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2538A>T | L846F 2D AIThe SynGAP1 missense variant L846F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict a pathogenic or deleterious impact, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Pathogenic. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Overall, the majority of predictions indicate a pathogenic effect, and this conclusion does not contradict any ClinVar status because none is available. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.653063 | Disordered | 0.589606 | Binding | 0.349 | 0.825 | 0.500 | -10.559 | Likely Pathogenic | 0.862 | Likely Pathogenic | Ambiguous | 0.206 | Likely Benign | -2.88 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.14 | Pathogenic | 0.00 | Affected | 0.0665 | 0.2912 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2645G>C | G882A 2D AIThe SynGAP1 missense variant G882A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, whereas only FATHMM predicts pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence indicates the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.632888 | Binding | 0.306 | 0.878 | 0.250 | -3.876 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -1.05 | Neutral | 0.004 | Benign | 0.002 | Benign | 2.14 | Pathogenic | 0.79 | Tolerated | 0.3345 | 0.5268 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2956G>C | E986Q 2D AIThe SynGAP1 missense variant E986Q is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward a pathogenic interpretation. This assessment does not contradict ClinVar status, as the variant is not yet catalogued there. Thus, based on current computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.471 | Likely Benign | 0.839 | Likely Pathogenic | Ambiguous | 0.160 | Likely Benign | -1.66 | Neutral | 0.974 | Probably Damaging | 0.842 | Possibly Damaging | 2.14 | Pathogenic | 0.00 | Affected | 0.1700 | 0.7589 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2957A>C | E986A 2D AIThe SynGAP1 missense variant E986A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, and ESM1b, whereas tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split; Foldetta stability analysis is unavailable. Overall, the evidence is evenly divided, with no clear majority. Based on the current predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.653 | Likely Benign | 0.895 | Likely Pathogenic | Ambiguous | 0.160 | Likely Benign | -2.32 | Neutral | 0.552 | Possibly Damaging | 0.388 | Benign | 2.14 | Pathogenic | 0.00 | Affected | 0.4207 | 0.7733 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3694A>C | K1232Q 2D AIThe SynGAP1 missense variant K1232Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of individual predictors (five pathogenic vs four benign) lean toward a pathogenic interpretation. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict the ClinVar status, which has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.505461 | Disordered | 0.542907 | Binding | 0.894 | 0.535 | 0.125 | -6.905 | Likely Benign | 0.247 | Likely Benign | Likely Benign | 0.102 | Likely Benign | -2.86 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.14 | Pathogenic | 0.00 | Affected | 0.3457 | 0.1419 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3805G>C | V1269L 2D AIThe SynGAP1 missense variant V1269L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.787464 | Binding | 0.843 | 0.647 | 0.125 | -3.572 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.299 | Likely Benign | -2.53 | Deleterious | 0.992 | Probably Damaging | 0.983 | Probably Damaging | 2.14 | Pathogenic | 0.00 | Affected | 0.0683 | 0.4082 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3805G>T | V1269L 2D AIThe SynGAP1 missense variant V1269L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of predictions points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.787464 | Binding | 0.843 | 0.647 | 0.125 | -3.572 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.299 | Likely Benign | -2.53 | Deleterious | 0.992 | Probably Damaging | 0.983 | Probably Damaging | 2.14 | Pathogenic | 0.00 | Affected | 0.0683 | 0.4082 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3817C>A | L1273M 2D AIThe SynGAP1 missense variant L1273M is catalogued in gnomAD (ID 6‑33447865‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, and the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect for the variant. This conclusion is not contradicted by ClinVar, as no ClinVar classification exists for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.773625 | Binding | 0.747 | 0.677 | 0.500 | 6-33447865-C-A | -5.375 | Likely Benign | 0.503 | Ambiguous | Likely Benign | 0.161 | Likely Benign | -1.68 | Neutral | 0.983 | Probably Damaging | 0.874 | Possibly Damaging | 2.14 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.0824 | 0.3932 | 2 | 4 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||
| c.3829C>A | H1277N 2D AIThe SynGAP1 missense variant H1277N is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33447877‑C‑A). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | 6-33447877-C-A | -3.347 | Likely Benign | 0.193 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -4.96 | Deleterious | 0.224 | Benign | 0.120 | Benign | 2.14 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1562 | 0.1250 | 1 | 2 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||
| c.3829C>G | H1277D 2D AIThe SynGAP1 missense variant H1277D is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33447877‑C‑G). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), and ESM1b, while those that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. AlphaMissense‑Default is uncertain, whereas AlphaMissense‑Optimized predicts benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a pathogenic outcome (two pathogenic, one benign, one uncertain). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑accuracy and consensus predictions indicate a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | 6-33447877-C-G | -4.632 | Likely Benign | 0.537 | Ambiguous | Likely Benign | 0.172 | Likely Benign | -6.38 | Deleterious | 0.411 | Benign | 0.091 | Benign | 2.14 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2389 | 0.1266 | -1 | 1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||||||||||
| c.3831C>A | H1277Q 2D AIThe SynGAP1 missense variant H1277Q is reported in gnomAD (ID 6‑33447879‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b; pathogenic predictions come from PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta data are unavailable. Overall, the majority of evidence points toward a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | 6-33447879-C-A | -3.323 | Likely Benign | 0.325 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -5.34 | Deleterious | 0.004 | Benign | 0.010 | Benign | 2.14 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1311 | 0.2351 | 0 | 3 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||
| c.3831C>G | H1277Q 2D AIThe SynGAP1 missense variant H1277Q is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b; pathogenic predictions come from PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs. 2 pathogenic). Foldetta results are not available. Overall, the majority of evidence points toward a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | -3.323 | Likely Benign | 0.325 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -5.34 | Deleterious | 0.004 | Benign | 0.010 | Benign | 2.14 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1311 | 0.2351 | 0 | 3 | -0.3 | -9.01 | ||||||||||||||||||||||||||||||||||||||
| c.3863A>G | K1288R 2D AIThe SynGAP1 missense variant K1288R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.814714 | Binding | 0.538 | 0.784 | 0.625 | -2.367 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 0.141 | Likely Benign | -2.22 | Neutral | 0.979 | Probably Damaging | 0.973 | Probably Damaging | 2.14 | Pathogenic | 0.00 | Affected | 0.4189 | 0.0782 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||||||||
| c.3880G>C | A1294P 2D AIThe SynGAP1 missense variant A1294P is catalogued in gnomAD (6‑33447928‑G‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a 2‑to‑2 split and is therefore inconclusive; Foldetta results are not available. Overall, the balance of evidence leans toward a pathogenic effect, though the single high‑accuracy benign call and the inconclusive consensus temper certainty. This assessment does not conflict with ClinVar, which currently contains no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | 6-33447928-G-C | 1 | 6.45e-7 | -2.688 | Likely Benign | 0.220 | Likely Benign | Likely Benign | 0.367 | Likely Benign | -3.79 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.14 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1682 | 0.3207 | -1 | 1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||
| c.3881C>A | A1294D 2D AIThe SynGAP1 missense variant A1294D is listed in gnomAD (ID 6‑33447929‑C‑A) but has no ClinVar entry. Functional prediction tools show mixed results: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessment further clarifies the picture: AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans toward pathogenicity. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a pathogenic effect, which is consistent with the lack of a benign ClinVar classification and the presence of the variant in a general population database. Thus, the variant is most likely pathogenic, and this prediction does not contradict any ClinVar status because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | 6-33447929-C-A | -4.179 | Likely Benign | 0.369 | Ambiguous | Likely Benign | 0.224 | Likely Benign | -4.17 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.14 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1893 | 0.2262 | -2 | 0 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||
| c.2159A>G | D720G 2D AISynGAP1 missense variant D720G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Foldetta, and premPS, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools remain inconclusive: AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta outputs) as benign. Overall, the balance of evidence favors a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -11.130 | Likely Pathogenic | 0.911 | Likely Pathogenic | Ambiguous | 0.45 | Likely Benign | 0.4 | 0.53 | Ambiguous | 0.49 | Likely Benign | 0.46 | Likely Benign | 0.427 | Likely Benign | -5.67 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.15 | Pathogenic | 0.01 | Affected | 0.4027 | 0.5011 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.2416T>A | F806I 2D AIThe SynGAP1 missense variant F806I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect; Foldetta results are unavailable. Overall, the balance of evidence from the consensus of prediction algorithms points to a pathogenic impact for F806I. This conclusion is not contradicted by ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | -5.040 | Likely Benign | 0.744 | Likely Pathogenic | Likely Benign | 0.182 | Likely Benign | -3.19 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 2.15 | Pathogenic | 0.03 | Affected | 0.2583 | 0.1357 | 1 | 0 | 1.7 | -34.02 | ||||||||||||||||||||||||||||||||||||||
| c.2452C>G | P818A 2D AIThe SynGAP1 missense variant P818A is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split opinion: benign predictions come from REVEL, polyPhen‑2 HumVar, SIFT, and ESM1b, while pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, FATHMM, and AlphaMissense‑Default. When the predictions are grouped by consensus, four tools favor benign and four favor pathogenic. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized returns an uncertain result, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Taken together, the preponderance of evidence from both consensus and high‑accuracy tools indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.715889 | Binding | 0.371 | 0.893 | 0.625 | -6.084 | Likely Benign | 0.820 | Likely Pathogenic | Ambiguous | 0.157 | Likely Benign | -4.37 | Deleterious | 0.543 | Possibly Damaging | 0.306 | Benign | 2.15 | Pathogenic | 0.06 | Tolerated | 0.3574 | 0.5724 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2524T>A | S842T 2D AIThe SynGAP1 missense variant S842T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, while the SGM‑Consensus (majority of the four high‑accuracy predictors) is Pathogenic; Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact for S842T, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.617281 | Binding | 0.274 | 0.861 | 0.250 | -13.443 | Likely Pathogenic | 0.725 | Likely Pathogenic | Likely Benign | 0.163 | Likely Benign | -2.23 | Neutral | 0.983 | Probably Damaging | 0.702 | Possibly Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.0974 | 0.5391 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2656G>C | A886P 2D AIThe SynGAP1 missense variant A886P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) support a benign classification. This consensus does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.619166 | Binding | 0.359 | 0.922 | 0.500 | -1.931 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.073 | Likely Benign | -1.20 | Neutral | 0.812 | Possibly Damaging | 0.575 | Possibly Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.1985 | 0.4773 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2956G>A | E986K 2D AIThe SynGAP1 missense variant E986K is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are REVEL and PROVEAN, while those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The remaining tools, ESM1b and AlphaMissense‑Optimized, return uncertain results and are not considered evidence for either side. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a pathogenic verdict (2 pathogenic vs. 1 benign, with one uncertain). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑accuracy predictors classify the variant as pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -7.174 | In-Between | 0.950 | Likely Pathogenic | Ambiguous | 0.164 | Likely Benign | -2.19 | Neutral | 0.924 | Possibly Damaging | 0.722 | Possibly Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.2760 | 0.7973 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.3655T>A | Y1219N 2D AIThe SynGAP1 missense variant Y1219N is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess functional impact largely agree on a deleterious effect: SIFT, polyPhen‑2 (HumDiv and HumVar), PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while the only benign prediction comes from REVEL. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Pathogenic.” High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic status. Foldetta results are not available, so they do not influence the overall assessment. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.613573 | Disordered | 0.474748 | Uncertain | 0.855 | 0.557 | 0.375 | -11.679 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.366 | Likely Benign | -6.41 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.2478 | 0.0573 | -2 | -2 | -2.2 | -49.07 | ||||||||||||||||||||||||||||||||||||||
| c.3655T>C | Y1219H 2D AIThe SynGAP1 missense variant Y1219H is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic)—all predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely pathogenic, which does not contradict its current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.613573 | Disordered | 0.474748 | Uncertain | 0.855 | 0.557 | 0.375 | Uncertain | 1 | -9.511 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.363 | Likely Benign | -3.62 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.15 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2234 | 0.0573 | 0 | 2 | -1.9 | -26.03 | ||||||||||||||||||||||||||||||||||
| c.3655T>G | Y1219D 2D AIThe SynGAP1 missense variant Y1219D is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta results are not available for this variant. Based on the preponderance of pathogenic predictions, Y1219D is most likely pathogenic, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.613573 | Disordered | 0.474748 | Uncertain | 0.855 | 0.557 | 0.375 | -15.860 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.443 | Likely Benign | -7.11 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.4549 | 0.0573 | -4 | -3 | -2.2 | -48.09 | ||||||||||||||||||||||||||||||||||||||
| c.3806T>C | V1269A 2D  AIThe SynGAP1 missense variant V1269A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas a larger group predicts pathogenicity: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). AlphaMissense‑Optimized is uncertain, and Foldetta results are unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and no Foldetta data. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.787464 | Binding | 0.843 | 0.647 | 0.125 | -6.115 | Likely Benign | 0.954 | Likely Pathogenic | Ambiguous | 0.291 | Likely Benign | -3.38 | Deleterious | 0.992 | Probably Damaging | 0.983 | Probably Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.2699 | 0.2312 | 0 | 0 | -2.4 | -28.05 | ||||||||||||||||||||||||||||||||||||||
| c.3830A>G | H1277R 2D AIThe SynGAP1 missense variant H1277R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs. two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | -2.940 | Likely Benign | 0.306 | Likely Benign | Likely Benign | 0.164 | Likely Benign | -5.60 | Deleterious | 0.259 | Benign | 0.066 | Benign | 2.15 | Pathogenic | 0.00 | Affected | 0.1710 | 0.1562 | 2 | 0 | -1.3 | 19.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3877G>T | D1293Y 2D AIThe SynGAP1 missense variant D1293Y is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. three benign) and the SGM Consensus support a pathogenic classification, with no ClinVar entry to contradict this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | -5.452 | Likely Benign | 0.392 | Ambiguous | Likely Benign | 0.310 | Likely Benign | -6.15 | Deleterious | 0.995 | Probably Damaging | 0.897 | Possibly Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.0627 | 0.3577 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||||
| c.3881C>G | A1294G 2D AIThe SynGAP1 missense variant A1294G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑accuracy predictors (AlphaMissense‑Optimized) indicate a benign outcome, while the consensus of other tools is split. Thus, the variant is most likely benign based on current predictions, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | -3.242 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.201 | Likely Benign | -3.06 | Deleterious | 0.992 | Probably Damaging | 0.983 | Probably Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.1980 | 0.2695 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.802A>G | I268V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I268V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign, while Foldetta’s stability analysis is uncertain. Overall, the majority of reliable predictors indicate a benign impact, and this is consistent with the lack of ClinVar evidence; there is no contradiction with ClinVar status. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.216401 | Structured | 0.314336 | Uncertain | 0.951 | 0.264 | 0.000 | -4.553 | Likely Benign | 0.147 | Likely Benign | Likely Benign | 1.46 | Ambiguous | 0.0 | 0.95 | Ambiguous | 1.21 | Ambiguous | 0.71 | Ambiguous | 0.139 | Likely Benign | -0.56 | Neutral | 0.958 | Probably Damaging | 0.970 | Probably Damaging | 2.15 | Pathogenic | 0.71 | Tolerated | 0.0845 | 0.2489 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||
| c.2159A>C | D720A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D720A missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and SIFT, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show that the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome, whereas Foldetta (combining FoldX‑MD and Rosetta) predicts a benign impact, and AlphaMissense‑Optimized remains uncertain. Overall, the predictions are split, with a slight tilt toward pathogenicity from the consensus and high‑accuracy methods. Thus, the variant is most likely pathogenic based on the available predictions, and this does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -10.999 | Likely Pathogenic | 0.871 | Likely Pathogenic | Ambiguous | -0.14 | Likely Benign | 0.0 | -0.35 | Likely Benign | -0.25 | Likely Benign | 0.40 | Likely Benign | 0.424 | Likely Benign | -6.20 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.16 | Pathogenic | 0.11 | Tolerated | 0.3726 | 0.5551 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.2416T>G | F806V 2D  AIThe SynGAP1 missense variant F806V is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; a Foldetta stability analysis is unavailable. Overall, the balance of evidence from the majority of prediction algorithms points to a pathogenic impact, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | -4.548 | Likely Benign | 0.676 | Likely Pathogenic | Likely Benign | 0.192 | Likely Benign | -3.79 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.16 | Pathogenic | 0.02 | Affected | 0.2464 | 0.1325 | -1 | -1 | 1.4 | -48.04 | ||||||||||||||||||||||||||||||||||||||
| c.3328A>T | S1110C 2D AIThe SynGAP1 missense variant S1110C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also resolves to benign (2 benign vs. 1 pathogenic vote). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy consensus—points to a benign impact. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -7.250 | In-Between | 0.096 | Likely Benign | Likely Benign | 0.027 | Likely Benign | -2.12 | Neutral | 0.898 | Possibly Damaging | 0.477 | Possibly Damaging | 2.16 | Pathogenic | 0.01 | Affected | 0.1214 | 0.5954 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.3646C>A | L1216M 2D AIThe SynGAP1 missense variant L1216M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs 2 benign), and Foldetta results are unavailable. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward a pathogenic interpretation. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.580690 | Disordered | 0.504713 | Binding | 0.863 | 0.563 | 0.250 | -6.590 | Likely Benign | 0.806 | Likely Pathogenic | Ambiguous | 0.142 | Likely Benign | -1.40 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.16 | Pathogenic | 0.00 | Affected | 0.0612 | 0.2092 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3647T>G | L1216R 2D AIThe SynGAP1 missense variant L1216R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus agrees. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.580690 | Disordered | 0.504713 | Binding | 0.863 | 0.563 | 0.250 | -9.700 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.387 | Likely Benign | -4.39 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.16 | Pathogenic | 0.00 | Affected | 0.1071 | 0.0488 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||
| c.3796C>G | L1266V 2D AIThe SynGAP1 missense variant L1266V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: REVEL predicts a benign outcome, whereas all other evaluated algorithms (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.802655 | Binding | 0.868 | 0.602 | 0.000 | -10.024 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.125 | Likely Benign | -2.53 | Deleterious | 0.995 | Probably Damaging | 0.890 | Possibly Damaging | 2.16 | Pathogenic | 0.00 | Affected | 0.1301 | 0.3118 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3878A>T | D1293V 2D AIThe SynGAP1 missense variant D1293V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of available predictions (five pathogenic vs. four benign) lean toward a pathogenic impact. This assessment does not contradict ClinVar status, as the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | -3.817 | Likely Benign | 0.328 | Likely Benign | Likely Benign | 0.343 | Likely Benign | -6.03 | Deleterious | 0.960 | Probably Damaging | 0.679 | Possibly Damaging | 2.16 | Pathogenic | 0.00 | Affected | 0.0896 | 0.3603 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.2455G>C | A819P 2D AIThe SynGAP1 missense variant A819P is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to a pathogenic impact for A819P, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.694846 | Disordered | 0.707644 | Binding | 0.317 | 0.892 | 0.625 | -5.542 | Likely Benign | 0.789 | Likely Pathogenic | Ambiguous | 0.342 | Likely Benign | -3.18 | Deleterious | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 2.17 | Pathogenic | 0.01 | Affected | 0.1920 | 0.5753 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2657C>A | A886E 2D AIThe SynGAP1 missense variant A886E is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors a benign outcome. Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.716283 | Disordered | 0.619166 | Binding | 0.359 | 0.922 | 0.500 | -3.747 | Likely Benign | 0.422 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -1.31 | Neutral | 0.423 | Benign | 0.230 | Benign | 2.17 | Pathogenic | 0.00 | Affected | 0.1483 | 0.2008 | 0 | -1 | -5.3 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2657C>T | A886V 2D AIThe SynGAP1 missense variant A886V is listed in ClinVar with an “Uncertain” status and is present in the gnomAD database (gnomAD ID 6‑33443209‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus call (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.619166 | Binding | 0.359 | 0.922 | 0.500 | Uncertain | 1 | 6-33443209-C-T | 18 | 1.12e-5 | -4.478 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.061 | Likely Benign | -0.20 | Neutral | 0.888 | Possibly Damaging | 0.314 | Benign | 2.17 | Pathogenic | 0.00 | Affected | 4.32 | 4 | 0.1131 | 0.5471 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||
| c.2947A>G | S983G 2D AIThe SynGAP1 missense variant S983G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 vs 2). High‑accuracy methods show AlphaMissense‑Optimized as benign; the SGM Consensus is unavailable, and Foldetta results are not provided, so no folding‑stability evidence is available. Overall, the balance of evidence (five pathogenic vs four benign predictions) suggests the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -5.206 | Likely Benign | 0.638 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | -2.22 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 2.17 | Pathogenic | 0.00 | Affected | 0.2521 | 0.4480 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3329G>T | S1110I 2D AIThe SynGAP1 missense variant S1110I is catalogued in gnomAD (ID 6‑33443881‑G‑T) but has no ClinVar submission. Functional prediction tools show a split: six algorithms (REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a benign effect, whereas three (PROVEAN, SIFT, FATHMM) predict pathogenicity. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized classifies the variant as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic); Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no available result for this residue. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | 6-33443881-G-T | -6.124 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -2.99 | Deleterious | 0.007 | Benign | 0.003 | Benign | 2.17 | Pathogenic | 0.01 | Affected | 4.32 | 2 | 0.1203 | 0.4971 | -2 | -1 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||
| c.3725A>T | E1242V 2D AIThe E1242V missense change occurs in the coiled‑coil domain of SynGAP1. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect are REVEL and AlphaMissense‑Optimized. Those that predict a pathogenic outcome include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (which is “Likely Pathogenic”). ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which simply lacks an entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.557691 | Disordered | 0.456349 | Uncertain | 0.870 | 0.549 | 0.500 | -7.456 | In-Between | 0.691 | Likely Pathogenic | Likely Benign | 0.267 | Likely Benign | -5.46 | Deleterious | 0.991 | Probably Damaging | 0.898 | Possibly Damaging | 2.17 | Pathogenic | 0.00 | Affected | 0.0447 | 0.4320 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3830A>T | H1277L 2D AIThe SynGAP1 missense variant H1277L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs. two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | -2.949 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.215 | Likely Benign | -7.93 | Deleterious | 0.224 | Benign | 0.091 | Benign | 2.17 | Pathogenic | 0.00 | Affected | 0.0822 | 0.4158 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3877G>C | D1293H 2D AIThe SynGAP1 missense variant D1293H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. three benign) and the SGM Consensus support a pathogenic classification, whereas AlphaMissense‑Optimized suggests benign. The variant is most likely pathogenic based on the collective evidence, and this conclusion does not contradict any ClinVar status because the variant is not yet reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | -4.422 | Likely Benign | 0.391 | Ambiguous | Likely Benign | 0.278 | Likely Benign | -4.59 | Deleterious | 0.984 | Probably Damaging | 0.888 | Possibly Damaging | 2.17 | Pathogenic | 0.00 | Affected | 0.1609 | 0.3731 | 1 | -1 | 0.3 | 22.05 | ||||||||||||||||||||||||||||||||||||||||
| c.2158G>A | D720N 2D 3DClick to see structure in 3D Viewer AISynGAP1 D720N is listed in ClinVar as benign (ClinVar ID 2837618.0) and is present in gnomAD (ID 6‑33441623‑G‑A). Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus as pathogenic. With seven pathogenic versus six benign predictions overall, the variant is most likely pathogenic according to in‑silico evidence, which contradicts the benign classification in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | Likely Benign | 1 | 6-33441623-G-A | 5 | 3.10e-6 | -9.135 | Likely Pathogenic | 0.654 | Likely Pathogenic | Likely Benign | 0.01 | Likely Benign | 0.0 | -0.20 | Likely Benign | -0.10 | Likely Benign | 0.46 | Likely Benign | 0.289 | Likely Benign | -3.74 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.18 | Pathogenic | 0.01 | Affected | 3.50 | 9 | 0.1216 | 0.5513 | 1 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||
| c.2179A>G | N727D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N727D has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default, while the SGM‑Consensus score is labeled Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions lean toward a benign effect, and this does not contradict any ClinVar annotation, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -5.640 | Likely Benign | 0.601 | Likely Pathogenic | Likely Benign | 0.22 | Likely Benign | 0.0 | 0.35 | Likely Benign | 0.29 | Likely Benign | 0.36 | Likely Benign | 0.142 | Likely Benign | -2.93 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.18 | Pathogenic | 0.08 | Tolerated | 0.1899 | 0.4309 | 2 | 1 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.2181C>A | N727K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N727K is catalogued in gnomAD (ID 6‑33441646‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, SIFT, and the protein‑folding stability method Foldetta; pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the consensus score SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, SGM Consensus indicates likely pathogenic, and Foldetta reports benign stability. Overall, the majority of evidence points to a pathogenic effect, and this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | 6-33441646-C-A | 1 | 6.19e-7 | -10.601 | Likely Pathogenic | 0.884 | Likely Pathogenic | Ambiguous | -0.12 | Likely Benign | 0.2 | -0.44 | Likely Benign | -0.28 | Likely Benign | 0.86 | Ambiguous | 0.148 | Likely Benign | -3.82 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 2.18 | Pathogenic | 0.12 | Tolerated | 3.59 | 7 | 0.2002 | 0.5590 | 0 | 1 | -0.4 | 14.07 | |||||||||||||||||||||||||
| c.2181C>G | N727K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727K is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and Foldetta. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and premPS. High‑accuracy assessments show that AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of evidence (seven pathogenic vs. five benign, with two uncertain) points to a pathogenic impact. This conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -10.601 | Likely Pathogenic | 0.884 | Likely Pathogenic | Ambiguous | -0.12 | Likely Benign | 0.2 | -0.44 | Likely Benign | -0.28 | Likely Benign | 0.86 | Ambiguous | 0.148 | Likely Benign | -3.82 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 2.18 | Pathogenic | 0.12 | Tolerated | 3.59 | 7 | 0.2002 | 0.5590 | 0 | 1 | -0.4 | 14.07 | ||||||||||||||||||||||||||||
| c.2286C>A | D762E 2D AIThe SynGAP1 D762E missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence predictions lean toward a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -4.332 | Likely Benign | 0.459 | Ambiguous | Likely Benign | 0.144 | Likely Benign | -1.59 | Neutral | 0.994 | Probably Damaging | 0.891 | Possibly Damaging | 2.18 | Pathogenic | 0.05 | Affected | 0.1911 | 0.8236 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2286C>G | D762E 2D AIThe SynGAP1 D762E missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence predictions lean toward a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -4.332 | Likely Benign | 0.459 | Ambiguous | Likely Benign | 0.144 | Likely Benign | -1.59 | Neutral | 0.994 | Probably Damaging | 0.891 | Possibly Damaging | 2.18 | Pathogenic | 0.05 | Affected | 0.1911 | 0.8236 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2416T>C | F806L 2D AIThe SynGAP1 missense variant F806L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, SIFT, and ESM1b, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the balance of evidence—five pathogenic versus three benign predictions, with a consensus pathogenic signal from SGM‑Consensus—suggests that F806L is most likely pathogenic. This conclusion does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | -3.079 | Likely Benign | 0.941 | Likely Pathogenic | Ambiguous | 0.142 | Likely Benign | -2.96 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.18 | Pathogenic | 0.32 | Tolerated | 0.2618 | 0.2329 | 2 | 0 | 1.0 | -34.02 | ||||||||||||||||||||||||||||||||||||||
| c.2418C>A | F806L 2D AIThe SynGAP1 missense variant F806L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, SIFT, and ESM1b, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the balance of evidence—five pathogenic versus three benign predictions, with a consensus pathogenic signal from SGM‑Consensus—suggests that F806L is most likely pathogenic. This conclusion does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | -3.079 | Likely Benign | 0.941 | Likely Pathogenic | Ambiguous | 0.170 | Likely Benign | -2.96 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.18 | Pathogenic | 0.32 | Tolerated | 0.2618 | 0.2329 | 2 | 0 | 1.0 | -34.02 | ||||||||||||||||||||||||||||||||||||||
| c.2418C>G | F806L 2D AIThe SynGAP1 missense variant F806L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show discordant results: benign predictions come from REVEL, SIFT, and ESM1b, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus method SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a likely pathogenic verdict (3/4 pathogenic votes). AlphaMissense‑Optimized returns an uncertain result, and no Foldetta stability assessment is available. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion aligns with the SGM‑Consensus prediction; it does not contradict any ClinVar status because no ClinVar entry exists. Thus, the variant is most likely pathogenic, and this assessment is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | -3.079 | Likely Benign | 0.941 | Likely Pathogenic | Ambiguous | 0.170 | Likely Benign | -2.96 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.18 | Pathogenic | 0.32 | Tolerated | 0.2618 | 0.2329 | 2 | 0 | 1.0 | -34.02 | ||||||||||||||||||||||||||||||||||||||
| c.2536T>A | L846I 2D AIThe SynGAP1 missense variant L846I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Two tools, ESM1b and AlphaMissense‑Default, return uncertain results. For high‑accuracy assessment, AlphaMissense‑Optimized classifies the variant as benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because the votes are split (one pathogenic, one benign, two uncertain). Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no available output for this variant. Overall, the balance of evidence from the majority of prediction tools suggests the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.653063 | Disordered | 0.589606 | Binding | 0.349 | 0.825 | 0.500 | -7.882 | In-Between | 0.474 | Ambiguous | Likely Benign | 0.151 | Likely Benign | -1.38 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 0.0973 | 0.3475 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||||||||||||
| c.2717T>A | L906H 2D  AIThe SynGAP1 missense variant L906H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs. two pathogenic votes) and Foldetta results are unavailable. Overall, the majority of standard predictors lean toward pathogenicity, but the most reliable high‑accuracy tool suggests a benign outcome and the consensus is unresolved. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.644316 | Binding | 0.315 | 0.920 | 0.250 | -4.367 | Likely Benign | 0.732 | Likely Pathogenic | Likely Benign | 0.124 | Likely Benign | -1.01 | Neutral | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.18 | Pathogenic | 0.02 | Affected | 0.1125 | 0.1073 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2717T>C | L906P 2D AIThe SynGAP1 missense variant L906P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome. Foldetta, which would provide a protein‑folding stability estimate, is unavailable for this variant. Overall, the preponderance of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.644316 | Binding | 0.315 | 0.920 | 0.250 | -3.662 | Likely Benign | 0.478 | Ambiguous | Likely Benign | 0.150 | Likely Benign | 0.08 | Neutral | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 2.18 | Pathogenic | 0.10 | Tolerated | 0.3430 | 0.1458 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2717T>G | L906R 2D AIThe SynGAP1 missense variant L906R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of standard predictors (5 vs 4) lean toward pathogenicity, while the high‑accuracy AlphaMissense‑Optimized predicts benign and the consensus remains unresolved. Thus, the variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.644316 | Binding | 0.315 | 0.920 | 0.250 | -3.440 | Likely Benign | 0.769 | Likely Pathogenic | Likely Benign | 0.160 | Likely Benign | -0.26 | Neutral | 1.000 | Probably Damaging | 0.990 | Probably Damaging | 2.18 | Pathogenic | 0.04 | Affected | 0.1228 | 0.0947 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2816C>A | P939Q 2D AIThe SynGAP1 missense variant P939Q is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus remains inconclusive and Foldetta is unavailable. Overall, five of the nine evaluated tools predict pathogenicity versus four predicting benignity, giving a slight tilt toward a pathogenic interpretation. This prediction does not contradict ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -4.950 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.156 | Likely Benign | -3.45 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 0.1551 | 0.4342 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||||||
| c.2816C>G | P939R 2D AIThe SynGAP1 missense variant P939R is reported in gnomAD (ID 6‑33443368‑C‑G) but has no ClinVar entry. Functional prediction tools show mixed results: benign calls come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Grouping by consensus, the benign‑predicted tools outnumbered the pathogenic ones by one, but the overall tally favors pathogenicity (5 pathogenic vs 4 benign). High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is benign, but the SGM Consensus (a 2‑vs‑2 majority among AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields no clear verdict, and Foldetta data are unavailable. Consequently, the variant is most likely pathogenic according to the available predictions, and this assessment does not contradict any ClinVar status because none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | 6-33443368-C-G | 1 | 6.20e-7 | -4.863 | Likely Benign | 0.260 | Likely Benign | Likely Benign | 0.199 | Likely Benign | -3.79 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1360 | 0.3336 | -2 | 0 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||
| c.2816C>T | P939L 2D AIThe SynGAP1 missense variant P939L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta are unavailable. Overall, five tools predict pathogenicity versus four predicting benign, so the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -4.191 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.159 | Likely Benign | -3.69 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 0.2113 | 0.5142 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2866T>G | S956A 2D AIThe SynGAP1 missense variant S956A is not reported in ClinVar or gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely benign. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta stability analysis is unavailable. Overall, the collective evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.957345 | Binding | 0.364 | 0.917 | 0.750 | -4.468 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -0.47 | Neutral | 0.112 | Benign | 0.039 | Benign | 2.18 | Pathogenic | 0.13 | Tolerated | 0.4239 | 0.4838 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3482T>G | M1161R 2D  AIThe SynGAP1 missense variant M1161R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, SIFT, and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as likely pathogenic; Foldetta results are unavailable. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.580690 | Disordered | 0.864109 | Binding | 0.372 | 0.830 | 0.375 | -2.653 | Likely Benign | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.220 | Likely Benign | -2.97 | Deleterious | 0.968 | Probably Damaging | 0.978 | Probably Damaging | 2.18 | Pathogenic | 0.13 | Tolerated | 0.1689 | 0.0809 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||||||||||||
| c.3656A>C | Y1219S 2D AIThe SynGAP1 missense variant Y1219S is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.613573 | Disordered | 0.474748 | Uncertain | 0.855 | 0.557 | 0.375 | -11.227 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.424 | Likely Benign | -6.05 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 0.5047 | 0.1403 | Weaken | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||||||||||
| c.3880G>A | A1294T 2D AIThe SynGAP1 missense variant A1294T is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33447928‑G‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta remain unavailable. Overall, five tools predict pathogenicity versus four predicting benign, and the lack of ClinVar evidence does not contradict these findings. Thus, the variant is most likely pathogenic based on the current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | 6-33447928-G-A | -3.966 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.290 | Likely Benign | -3.04 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1244 | 0.5479 | 0 | 1 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||
| c.850C>G | L284V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L284V missense variant has no ClinVar entry and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are Rosetta, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Predictions that are uncertain or inconclusive are FoldX, AlphaMissense‑Default, and Foldetta. High‑accuracy methods give a benign consensus: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, while Foldetta remains uncertain. Overall, the balance of evidence leans toward a pathogenic effect, and this assessment does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.094817 | Structured | 0.371601 | Uncertain | 0.950 | 0.255 | 0.000 | -6.726 | Likely Benign | 0.347 | Ambiguous | Likely Benign | 1.65 | Ambiguous | 0.2 | 2.08 | Destabilizing | 1.87 | Ambiguous | 1.38 | Destabilizing | 0.248 | Likely Benign | -2.32 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 2.18 | Pathogenic | 0.03 | Affected | 0.1259 | 0.2456 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||
| c.2162T>A | I721N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -14.905 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.82 | Destabilizing | 0.0 | 3.21 | Destabilizing | 3.02 | Destabilizing | 2.10 | Destabilizing | 0.425 | Likely Benign | -6.30 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.19 | Pathogenic | 0.00 | Affected | 0.0737 | 0.0340 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.2180A>G | N727S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727S is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of evidence supports a benign effect. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -6.195 | Likely Benign | 0.184 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.1 | 0.28 | Likely Benign | 0.30 | Likely Benign | 0.18 | Likely Benign | 0.118 | Likely Benign | -2.67 | Deleterious | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 2.19 | Pathogenic | 0.23 | Tolerated | 0.3833 | 0.6680 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||
| c.2365C>T | P789S 2D AIThe SynGAP1 P789S variant has no ClinVar entry (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that classify it as benign include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence leans toward pathogenicity, with five tools supporting a deleterious effect versus three supporting benignity. This prediction does not contradict ClinVar status, which currently has no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -4.793 | Likely Benign | 0.409 | Ambiguous | Likely Benign | 0.221 | Likely Benign | -4.64 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.19 | Pathogenic | 0.00 | Affected | 0.3100 | 0.3436 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2456C>A | A819E 2D AIThe SynGAP1 missense variant A819E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. In contrast, the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. ESM1b is uncertain, and no Foldetta stability assessment is available. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic, and Foldetta data are unavailable. Based on the preponderance of pathogenic predictions and the lack of any benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.694846 | Disordered | 0.707644 | Binding | 0.317 | 0.892 | 0.625 | -7.333 | In-Between | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.292 | Likely Benign | -2.85 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.19 | Pathogenic | 0.00 | Affected | 0.1189 | 0.2037 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2456C>T | A819V 2D AIThe SynGAP1 missense variant A819V is catalogued in gnomAD (ID 6‑33443008‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas a majority of tools predict a pathogenic outcome: polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs. two pathogenic), and Foldetta stability analysis is unavailable. Overall, the balance of evidence favors a pathogenic classification for A819V, and this assessment does not contradict any ClinVar status because the variant is not yet reported in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.694846 | Disordered | 0.707644 | Binding | 0.317 | 0.892 | 0.625 | 6-33443008-C-T | 1 | 6.19e-7 | -4.944 | Likely Benign | 0.797 | Likely Pathogenic | Ambiguous | 0.246 | Likely Benign | -2.40 | Neutral | 0.998 | Probably Damaging | 0.963 | Probably Damaging | 2.19 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1098 | 0.6664 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||
| c.3193C>G | P1065A 2D AIThe SynGAP1 missense variant P1065A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for P1065A, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.959518 | Binding | 0.424 | 0.917 | 0.875 | -4.043 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.050 | Likely Benign | -1.85 | Neutral | 0.580 | Possibly Damaging | 0.184 | Benign | 2.19 | Pathogenic | 0.00 | Affected | 0.3140 | 0.5790 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3328A>G | S1110G 2D AIThe SynGAP1 missense variant S1110G is listed in ClinVar (ID 1722210.0) as benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion aligns with the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | Likely Benign | 1 | -4.674 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -2.26 | Neutral | 0.036 | Benign | 0.026 | Benign | 2.19 | Pathogenic | 0.08 | Tolerated | 4.32 | 2 | 0.2559 | 0.4806 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||
| c.3482T>A | M1161K 2D AIThe SynGAP1 missense variant M1161K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, SIFT, and ESM1b, whereas tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as pathogenic and the SGM‑Consensus labeling it as Likely Pathogenic; Foldetta results are unavailable. Overall, the majority of computational predictions support a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Thus, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.580690 | Disordered | 0.864109 | Binding | 0.372 | 0.830 | 0.375 | -3.005 | Likely Benign | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.236 | Likely Benign | -2.91 | Deleterious | 0.968 | Probably Damaging | 0.969 | Probably Damaging | 2.19 | Pathogenic | 0.17 | Tolerated | 0.1629 | 0.0828 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.3482T>C | M1161T 2D  AIThe SynGAP1 missense variant M1161T is listed in gnomAD (ID 6‑33444517‑T‑C) but has no ClinVar entry. Functional prediction tools fall into two groups: benign predictions come from REVEL, SIFT, and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.580690 | Disordered | 0.864109 | Binding | 0.372 | 0.830 | 0.375 | 6-33444517-T-C | 1 | 6.20e-7 | -2.620 | Likely Benign | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.148 | Likely Benign | -2.75 | Deleterious | 0.968 | Probably Damaging | 0.954 | Probably Damaging | 2.19 | Pathogenic | 0.27 | Tolerated | 3.77 | 5 | 0.2218 | 0.1786 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.3725A>G | E1242G 2D AIThe SynGAP1 missense variant E1242G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and ESM1b, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Optimized also predicts a benign outcome, whereas AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple high‑accuracy predictors points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.557691 | Disordered | 0.456349 | Uncertain | 0.870 | 0.549 | 0.500 | -6.674 | Likely Benign | 0.528 | Ambiguous | Likely Benign | 0.214 | Likely Benign | -5.33 | Deleterious | 0.939 | Possibly Damaging | 0.670 | Possibly Damaging | 2.19 | Pathogenic | 0.00 | Affected | 0.2431 | 0.4295 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3878A>G | D1293G 2D AIThe SynGAP1 missense variant D1293G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the balance of evidence (5 benign vs 4 pathogenic predictions, with the most accurate tool indicating benign) suggests the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | -3.060 | Likely Benign | 0.261 | Likely Benign | Likely Benign | 0.332 | Likely Benign | -4.91 | Deleterious | 0.872 | Possibly Damaging | 0.399 | Benign | 2.19 | Pathogenic | 0.00 | Affected | 0.3022 | 0.3944 | 1 | -1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3880G>T | A1294S 2D AIThe SynGAP1 missense variant A1294S is reported in ClinVar as not yet classified (no ClinVar ID) and is present in gnomAD (variant ID 6-33447928‑G‑T). Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. No Foldetta stability analysis is available. Overall, the majority of evidence points to a benign effect for A1294S, and this conclusion does not contradict the current ClinVar status, which remains unclassified. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | 6-33447928-G-T | -3.229 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.276 | Likely Benign | -2.21 | Neutral | 0.997 | Probably Damaging | 0.991 | Probably Damaging | 2.19 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2302 | 0.3990 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||||||||
| c.2170G>T | A724S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A724S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain. Overall, the majority of evidence points to a benign impact for A724S, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -6.893 | Likely Benign | 0.204 | Likely Benign | Likely Benign | 0.93 | Ambiguous | 0.1 | 0.88 | Ambiguous | 0.91 | Ambiguous | 0.57 | Ambiguous | 0.265 | Likely Benign | -1.68 | Neutral | 0.983 | Probably Damaging | 0.993 | Probably Damaging | 2.20 | Pathogenic | 0.25 | Tolerated | 0.2308 | 0.4518 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.2536T>G | L846V 2D AIThe SynGAP1 missense variant L846V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Pathogenic, while AlphaMissense‑Optimized remains benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this assessment does not conflict with any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.653063 | Disordered | 0.589606 | Binding | 0.349 | 0.825 | 0.500 | -8.326 | Likely Pathogenic | 0.569 | Likely Pathogenic | Likely Benign | 0.165 | Likely Benign | -2.07 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.20 | Pathogenic | 0.00 | Affected | 0.1540 | 0.3126 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3329G>A | S1110N 2D AIThe SynGAP1 missense variant S1110N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S1110N, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -5.028 | Likely Benign | 0.167 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -1.99 | Neutral | 0.144 | Benign | 0.078 | Benign | 2.20 | Pathogenic | 0.01 | Affected | 0.1542 | 0.4716 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3646C>G | L1216V 2D AIThe SynGAP1 missense variant L1216V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two agreement groups: benign predictions come from REVEL and PROVEAN, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. Two tools—ESM1b and AlphaMissense‑Optimized—return uncertain results. High‑accuracy assessment further shows that AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.580690 | Disordered | 0.504713 | Binding | 0.863 | 0.563 | 0.250 | -7.842 | In-Between | 0.861 | Likely Pathogenic | Ambiguous | 0.126 | Likely Benign | -2.26 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 2.20 | Pathogenic | 0.00 | Affected | 0.1294 | 0.1883 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3781A>T | S1261C 2D AIThe SynGAP1 missense variant S1261C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the variant as damaging. The SGM‑Consensus, which aggregates these predictions, reports the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized remains benign, but the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple prediction algorithms indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.501700 | Disordered | 0.671500 | Binding | 0.889 | 0.574 | 0.250 | -8.275 | Likely Pathogenic | 0.322 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -3.51 | Deleterious | 0.999 | Probably Damaging | 0.947 | Probably Damaging | 2.20 | Pathogenic | 0.04 | Affected | 0.0760 | 0.4580 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||
| c.3817C>G | L1273V 2D AIThe SynGAP1 missense variant L1273V is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (two pathogenic versus one benign vote). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence, particularly the SGM Consensus, indicates that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.773625 | Binding | 0.747 | 0.677 | 0.500 | -6.014 | Likely Benign | 0.539 | Ambiguous | Likely Benign | 0.111 | Likely Benign | -2.50 | Deleterious | 0.773 | Possibly Damaging | 0.287 | Benign | 2.20 | Pathogenic | 0.00 | Affected | 0.1556 | 0.3178 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3878A>C | D1293A 2D AIThe SynGAP1 missense variant D1293A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the balance of evidence (5 benign vs 4 pathogenic predictions) leans toward a benign classification. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | -2.251 | Likely Benign | 0.197 | Likely Benign | Likely Benign | 0.302 | Likely Benign | -5.19 | Deleterious | 0.770 | Possibly Damaging | 0.255 | Benign | 2.20 | Pathogenic | 0.00 | Affected | 0.3267 | 0.3447 | 0 | -2 | 5.3 | -44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.2162T>G | I721S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721S is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that assess the variant’s effect fall into two groups: the single benign prediction comes from REVEL, while all other evaluated algorithms (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic outcome. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, which does not contradict its current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | Uncertain | 1 | -14.032 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 3.91 | Destabilizing | 0.1 | 3.96 | Destabilizing | 3.94 | Destabilizing | 2.28 | Destabilizing | 0.466 | Likely Benign | -5.26 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.21 | Pathogenic | 0.00 | Affected | 3.50 | 9 | 0.2606 | 0.1110 | -1 | -2 | -5.3 | -26.08 | 203.3 | 49.3 | -0.1 | 0.0 | -1.1 | 0.0 | X | Uncertain | The sec-butyl side chain of Ile721, located on an α-helix (res. Leu714-Arg726), engages in hydrophobic packing with other residues in the hydrophobic inter-helix space, such as Phe420, Tyr417, His693, and Leu717. In the variant simulations, the hydroxyl side chain of Ser721 forms hydrogen bonds with nearby residues, such as Leu717 and His693. Although no major structural changes are observed during the variant simulations, the hydrophilic residue Ser721 could disrupt the hydrophobic packing during folding. However, because the model ends abruptly at the C-terminus, no definite conclusions can be drawn based on the simulations. | ||||||||||||||||
| c.2417T>C | F806S 2D AIThe SynGAP1 missense variant F806S is catalogued in gnomAD (ID 6‑33442969‑T‑C) but has no ClinVar entry. Functional prediction tools split into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, whereas the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because none is reported. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | 6-33442969-T-C | 1 | 6.20e-7 | -6.959 | Likely Benign | 0.911 | Likely Pathogenic | Ambiguous | 0.269 | Likely Benign | -4.13 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.21 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.5067 | 0.0391 | Weaken | -2 | -3 | -3.6 | -60.10 | ||||||||||||||||||||||||||||||||
| c.2486A>T | E829V 2D AIThe SynGAP1 missense variant E829V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority of the four high‑accuracy inputs) remains pathogenic; Foldetta results are unavailable. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.562014 | Disordered | 0.626045 | Binding | 0.326 | 0.882 | 0.375 | -5.142 | Likely Benign | 0.719 | Likely Pathogenic | Likely Benign | 0.296 | Likely Benign | -4.86 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.21 | Pathogenic | 0.00 | Affected | 0.0835 | 0.7984 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2656G>A | A886T 2D AIThe SynGAP1 missense variant A886T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only SIFT and FATHMM predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.619166 | Binding | 0.359 | 0.922 | 0.500 | -4.319 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.78 | Neutral | 0.369 | Benign | 0.237 | Benign | 2.21 | Pathogenic | 0.00 | Affected | 0.1581 | 0.6737 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2845G>C | G949R 2D AIThe SynGAP1 missense variant G949R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Consensus from standard in silico predictors shows a split: benign calls come from REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls come from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessment further supports a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign outcome, and Foldetta data are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not conflict with any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | -7.650 | In-Between | 0.301 | Likely Benign | Likely Benign | 0.317 | Likely Benign | -0.14 | Neutral | 0.997 | Probably Damaging | 0.934 | Probably Damaging | 2.21 | Pathogenic | 0.01 | Affected | 0.1095 | 0.4569 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.2845G>T | G949C 2D AIThe SynGAP1 missense variant G949C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. four benign) indicate that the variant is most likely pathogenic. This assessment does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | -10.580 | Likely Pathogenic | 0.091 | Likely Benign | Likely Benign | 0.345 | Likely Benign | -1.02 | Neutral | 1.000 | Probably Damaging | 0.975 | Probably Damaging | 2.21 | Pathogenic | 0.01 | Affected | 0.1480 | 0.4097 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2846G>A | G949D 2D AIThe SynGAP1 missense variant G949D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑confidence predictors (5 pathogenic vs. 4 benign) indicate a pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | -8.692 | Likely Pathogenic | 0.239 | Likely Benign | Likely Benign | 0.316 | Likely Benign | 0.25 | Neutral | 0.945 | Possibly Damaging | 0.753 | Possibly Damaging | 2.21 | Pathogenic | 0.02 | Affected | 0.1927 | 0.2826 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2846G>T | G949V 2D AIThe SynGAP1 missense variant G949V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | -7.364 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.328 | Likely Benign | -0.55 | Neutral | 0.992 | Probably Damaging | 0.834 | Possibly Damaging | 2.21 | Pathogenic | 0.01 | Affected | 0.1493 | 0.3165 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3329G>C | S1110T 2D AIThe SynGAP1 missense variant S1110T is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the preponderance of benign predictions and the consensus from high‑accuracy tools, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -3.989 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.032 | Likely Benign | -1.76 | Neutral | 0.001 | Benign | 0.003 | Benign | 2.21 | Pathogenic | 0.04 | Affected | 0.1581 | 0.6224 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3653A>T | E1218V 2D AISynGAP1 missense variant E1218V is listed in ClinVar with an uncertain significance (ClinVar ID 1015602.0) and is not reported in gnomAD. Functional prediction tools show a split opinion: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. When the high‑accuracy consensus is considered, AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this change. Overall, the majority of evidence points toward a pathogenic effect, which is consistent with the ClinVar designation of uncertain significance rather than a benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.595080 | Disordered | 0.483050 | Uncertain | 0.898 | 0.565 | 0.375 | Uncertain | 2 | -5.647 | Likely Benign | 0.936 | Likely Pathogenic | Ambiguous | 0.418 | Likely Benign | -5.68 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.21 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.0491 | 0.4120 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||
| c.3724G>C | E1242Q 2D AIThe SynGAP1 missense variant E1242Q is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta results are unavailable. Consequently, the aggregate evidence favors a benign effect for E1242Q, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.557691 | Disordered | 0.456349 | Uncertain | 0.870 | 0.549 | 0.500 | -7.036 | In-Between | 0.291 | Likely Benign | Likely Benign | 0.167 | Likely Benign | -2.33 | Neutral | 0.969 | Probably Damaging | 0.843 | Possibly Damaging | 2.21 | Pathogenic | 0.00 | Affected | 0.0763 | 0.3666 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3725A>C | E1242A 2D AIThe SynGAP1 missense variant E1242A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated tools (five pathogenic vs. three benign) predict a pathogenic impact. This prediction does not contradict ClinVar status, as the variant is not yet catalogued there. Thus, based on current computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.557691 | Disordered | 0.456349 | Uncertain | 0.870 | 0.549 | 0.500 | -5.654 | Likely Benign | 0.437 | Ambiguous | Likely Benign | 0.187 | Likely Benign | -4.66 | Deleterious | 0.939 | Possibly Damaging | 0.735 | Possibly Damaging | 2.21 | Pathogenic | 0.00 | Affected | 0.2738 | 0.4169 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.3734A>T | E1245V 2D AIThe SynGAP1 missense change E1245V is not reported in ClinVar and is absent from gnomAD. Prediction tools that flag the variant as benign include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic effect. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus likewise reports a likely pathogenic outcome. Foldetta results are not available for this variant. Overall, the preponderance of computational evidence points to a pathogenic effect for E1245V, and this conclusion does not conflict with the current ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.712013 | Disordered | 0.387847 | Uncertain | 0.869 | 0.554 | 0.625 | -12.988 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.319 | Likely Benign | -5.65 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.21 | Pathogenic | 0.00 | Affected | 0.0518 | 0.6856 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3781A>C | S1261R 2D AIThe SynGAP1 missense variant S1261R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple independent predictors points to a pathogenic effect for S1261R, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.501700 | Disordered | 0.671500 | Binding | 0.889 | 0.574 | 0.250 | -4.363 | Likely Benign | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.152 | Likely Benign | -2.97 | Deleterious | 0.996 | Probably Damaging | 0.898 | Possibly Damaging | 2.21 | Pathogenic | 0.04 | Affected | 0.0661 | 0.2642 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||
| c.3782G>T | S1261I 2D AIThe SynGAP1 missense variant S1261I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and AlphaMissense‑Optimized, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score indicates a benign effect, whereas the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. No Foldetta stability assessment is available for this residue. Overall, the preponderance of evidence from standard and high‑accuracy predictors points to a pathogenic impact for S1261I. This conclusion is not contradicted by ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.501700 | Disordered | 0.671500 | Binding | 0.889 | 0.574 | 0.250 | -8.835 | Likely Pathogenic | 0.761 | Likely Pathogenic | Likely Benign | 0.244 | Likely Benign | -4.20 | Deleterious | 0.996 | Probably Damaging | 0.898 | Possibly Damaging | 2.21 | Pathogenic | 0.02 | Affected | 0.0631 | 0.4410 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||
| c.3783C>A | S1261R 2D AIThe SynGAP1 missense variant S1261R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from multiple in silico tools points to a pathogenic classification, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.501700 | Disordered | 0.671500 | Binding | 0.889 | 0.574 | 0.250 | -4.363 | Likely Benign | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.190 | Likely Benign | -2.97 | Deleterious | 0.996 | Probably Damaging | 0.898 | Possibly Damaging | 2.21 | Pathogenic | 0.04 | Affected | 0.0661 | 0.2642 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||
| c.3783C>G | S1261R 2D AIThe SynGAP1 missense variant S1261R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence indicates that S1261R is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.501700 | Disordered | 0.671500 | Binding | 0.889 | 0.574 | 0.250 | -4.363 | Likely Benign | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.190 | Likely Benign | -2.97 | Deleterious | 0.996 | Probably Damaging | 0.898 | Possibly Damaging | 2.21 | Pathogenic | 0.04 | Affected | 0.0661 | 0.2642 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||
| c.2161A>T | I721F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic or likely pathogenic impact, while premPS remains uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Based on the overwhelming agreement among these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -12.559 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 4.61 | Destabilizing | 0.1 | 2.74 | Destabilizing | 3.68 | Destabilizing | 0.66 | Ambiguous | 0.295 | Likely Benign | -3.74 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.0420 | 0.2931 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.2162T>C | I721T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721T is not reported in ClinVar (ClinVar status: None) and has no entries in gnomAD (gnomAD ID: None). Prediction tools that indicate a benign effect include only REVEL. All other evaluated tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote) yields Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts Pathogenic. No prediction or folding result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -10.374 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 2.73 | Destabilizing | 0.0 | 2.56 | Destabilizing | 2.65 | Destabilizing | 2.11 | Destabilizing | 0.417 | Likely Benign | -4.18 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.0918 | 0.1019 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.2353C>T | R785C 2D AIThe SynGAP1 R785C missense variant is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33442905‑C‑T). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—indicates a likely pathogenic outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points toward a pathogenic impact, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | Uncertain | 1 | 6-33442905-C-T | 29 | 1.80e-5 | -5.887 | Likely Benign | 0.662 | Likely Pathogenic | Likely Benign | 0.126 | Likely Benign | -5.06 | Deleterious | 0.144 | Benign | 0.046 | Benign | 2.22 | Pathogenic | 0.00 | Affected | 3.64 | 6 | 0.3775 | 0.3530 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||
| c.2716C>T | L906F 2D AIThe SynGAP1 missense variant L906F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for the variant. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.604312 | Disordered | 0.644316 | Binding | 0.315 | 0.920 | 0.250 | -4.700 | Likely Benign | 0.304 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -1.79 | Neutral | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 2.22 | Pathogenic | 0.18 | Tolerated | 0.0753 | 0.3342 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2815C>A | P939T 2D AIThe SynGAP1 missense variant P939T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions show AlphaMissense‑Optimized as benign, while the SGM Consensus remains inconclusive and Foldetta is unavailable. Overall, more tools (five) predict pathogenicity than benign (four), suggesting the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -5.739 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -3.34 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.1788 | 0.5310 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||||||
| c.3653A>G | E1218G 2D AIThe SynGAP1 missense variant E1218G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all indicate pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as likely pathogenic. AlphaMissense‑Optimized returns an uncertain result, and no Foldetta (FoldX‑MD/Rosetta) stability assessment is available. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.595080 | Disordered | 0.483050 | Uncertain | 0.898 | 0.565 | 0.375 | -5.595 | Likely Benign | 0.864 | Likely Pathogenic | Ambiguous | 0.413 | Likely Benign | -5.61 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.2496 | 0.4095 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||||||
| c.3724G>A | E1242K 2D AIThe SynGAP1 missense variant E1242K is catalogued in gnomAD (6‑33446716‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: the benign‑predicting REVEL score contrasts with a pathogenic consensus from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic,” and the protein‑folding stability method Foldetta is not available for this variant. Taken together, the majority of evidence points to a pathogenic impact, and this conclusion does not conflict with ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.557691 | Disordered | 0.456349 | Uncertain | 0.870 | 0.549 | 0.500 | 6-33446716-G-A | 1 | 6.20e-7 | -10.075 | Likely Pathogenic | 0.798 | Likely Pathogenic | Ambiguous | 0.179 | Likely Benign | -3.13 | Deleterious | 0.939 | Possibly Damaging | 0.670 | Possibly Damaging | 2.22 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1520 | 0.4024 | 1 | 0 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||
| c.3734A>G | E1245G 2D AIThe SynGAP1 missense variant E1245G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. The high‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is classified as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.712013 | Disordered | 0.387847 | Uncertain | 0.869 | 0.554 | 0.625 | -12.113 | Likely Pathogenic | 0.901 | Likely Pathogenic | Ambiguous | 0.299 | Likely Benign | -5.65 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.2145 | 0.5447 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||||||
| c.3815A>G | E1272G 2D AIThe SynGAP1 missense variant E1272G is catalogued in gnomAD (ID 6‑33447863‑A‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score, which is labeled Likely Pathogenic. The high‑accuracy AlphaMissense‑Optimized assessment is uncertain, and the Foldetta protein‑folding stability analysis is not available for this residue. Overall, the majority of evidence points toward a deleterious effect, so the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, as no ClinVar classification exists for E1272G. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.766082 | Binding | 0.799 | 0.677 | 0.500 | 6-33447863-A-G | -1.919 | Likely Benign | 0.863 | Likely Pathogenic | Ambiguous | 0.287 | Likely Benign | -5.89 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2804 | 0.5072 | -2 | 0 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||||
| c.3815A>T | E1272V 2D AIThe SynGAP1 missense variant E1272V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all indicate pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. AlphaMissense‑Optimized returns an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.766082 | Binding | 0.799 | 0.677 | 0.500 | -3.628 | Likely Benign | 0.934 | Likely Pathogenic | Ambiguous | 0.278 | Likely Benign | -5.90 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.0424 | 0.5894 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3881C>T | A1294V 2D AIThe SynGAP1 missense variant A1294V is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33447929‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑benign/2‑pathogenic split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools specifically show AlphaMissense‑Optimized as benign, SGM Consensus as inconclusive, and Foldetta as unavailable. Overall, the predictions are mixed, with one more pathogenic than benign calls. Thus, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | 6-33447929-C-T | 1 | 6.45e-7 | -3.842 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.164 | Likely Benign | -3.16 | Deleterious | 0.997 | Probably Damaging | 0.983 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1033 | 0.4413 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||
| c.2845G>A | G949S 2D AIThe SynGAP1 missense variant G949S is listed in ClinVar as a benign alteration (ClinVar ID 212352.0) and is present in the gnomAD database (6‑33443397‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, aligning with the ClinVar classification and indicating no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | Benign/Likely benign | 4 | 6-33443397-G-A | 122 | 7.56e-5 | -5.693 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.321 | Likely Benign | 0.30 | Neutral | 0.611 | Possibly Damaging | 0.102 | Benign | 2.23 | Pathogenic | 0.00 | Affected | 4.32 | 4 | 0.2528 | 0.5159 | 1 | 0 | -0.4 | 30.03 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||
| c.3647T>A | L1216Q 2D AIThe SynGAP1 missense variant L1216Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as pathogenic; Foldetta results are not available. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.580690 | Disordered | 0.504713 | Binding | 0.863 | 0.563 | 0.250 | -8.731 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.404 | Likely Benign | -4.12 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.23 | Pathogenic | 0.00 | Affected | 0.0939 | 0.0488 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3656A>G | Y1219C 2D AIThe SynGAP1 missense variant Y1219C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. ESM1b is uncertain and does not contribute to a consensus. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic; Foldetta results are unavailable. Overall, the evidence strongly favors a pathogenic classification for Y1219C, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.613573 | Disordered | 0.474748 | Uncertain | 0.855 | 0.557 | 0.375 | -7.776 | In-Between | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.310 | Likely Benign | -5.41 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.23 | Pathogenic | 0.00 | Affected | 0.3446 | 0.1672 | 0 | -2 | 3.8 | -60.04 | ||||||||||||||||||||||||||||||||||||||
| c.3656A>T | Y1219F 2D AIThe SynGAP1 missense variant Y1219F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments show that the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome, while AlphaMissense‑Optimized remains uncertain. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence points to a pathogenic effect for Y1219F, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.613573 | Disordered | 0.474748 | Uncertain | 0.855 | 0.557 | 0.375 | -7.202 | In-Between | 0.837 | Likely Pathogenic | Ambiguous | 0.272 | Likely Benign | -2.79 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.23 | Pathogenic | 0.00 | Affected | 0.1998 | 0.2583 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.3782G>A | S1261N 2D AIThe SynGAP1 missense variant S1261N is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions, including the high‑accuracy tools, point to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.501700 | Disordered | 0.671500 | Binding | 0.889 | 0.574 | 0.250 | Uncertain | 1 | -4.850 | Likely Benign | 0.294 | Likely Benign | Likely Benign | 0.123 | Likely Benign | -0.92 | Neutral | 0.959 | Probably Damaging | 0.551 | Possibly Damaging | 2.23 | Pathogenic | 0.12 | Tolerated | 0.0917 | 0.2820 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||||||
| c.3793A>T | R1265W 2D AIThe SynGAP1 missense variant R1265W is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly classify the variant as deleterious: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool in the dataset predicts a benign outcome, so the benign group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates a pathogenic change, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. Foldetta results are not available, so they do not influence the conclusion. Overall, the variant is most likely pathogenic based on the consensus of predictive tools, and this assessment is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.414856 | Structured | 0.782497 | Binding | 0.887 | 0.592 | 0.000 | -14.584 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.505 | Likely Pathogenic | -6.54 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.23 | Pathogenic | 0.00 | Affected | 0.1166 | 0.3868 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||
| c.3884A>C | Q1295P 2D AIThe SynGAP1 missense variant Q1295P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of predictions (5/9) indicate pathogenicity, while 4/9 suggest benignity. Thus, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.892719 | Binding | 0.499 | 0.801 | 0.625 | -2.511 | Likely Benign | 0.168 | Likely Benign | Likely Benign | 0.372 | Likely Benign | -4.95 | Deleterious | 0.968 | Probably Damaging | 0.954 | Probably Damaging | 2.23 | Pathogenic | 0.00 | Affected | 0.2299 | 0.4738 | 0 | -1 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||||||||
| c.2354G>C | R785P 2D AIThe SynGAP1 missense variant R785P is catalogued in gnomAD (6‑33442906‑G‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as Benign, while the SGM‑Consensus remains Likely Pathogenic; Foldetta results are unavailable. Overall, the balance of evidence, with seven pathogenic versus three benign predictions and a pathogenic consensus from SGM, indicates that R785P is most likely pathogenic. This conclusion does not contradict ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | 6-33442906-G-C | -3.603 | Likely Benign | 0.721 | Likely Pathogenic | Likely Benign | 0.203 | Likely Benign | -4.12 | Deleterious | 0.998 | Probably Damaging | 0.958 | Probably Damaging | 2.24 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.2370 | 0.4485 | -2 | 0 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||
| c.2486A>G | E829G 2D AIThe SynGAP1 missense variant E829G is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the remaining tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and the Foldetta stability analysis is unavailable. Overall, the majority of predictions lean toward pathogenicity, with one high‑accuracy tool suggesting benign. No ClinVar annotation exists, so there is no contradiction with clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.562014 | Disordered | 0.626045 | Binding | 0.326 | 0.882 | 0.375 | -4.152 | Likely Benign | 0.593 | Likely Pathogenic | Likely Benign | 0.316 | Likely Benign | -4.52 | Deleterious | 0.994 | Probably Damaging | 0.927 | Probably Damaging | 2.24 | Pathogenic | 0.00 | Affected | 0.3517 | 0.6557 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.2656G>T | A886S 2D AIThe SynGAP1 missense variant A886S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.619166 | Binding | 0.359 | 0.922 | 0.500 | -2.366 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.50 | Neutral | 0.224 | Benign | 0.185 | Benign | 2.24 | Pathogenic | 0.00 | Affected | 0.2755 | 0.5639 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2815C>T | P939S 2D AIThe SynGAP1 missense variant P939S is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools specifically show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta remain unavailable. Overall, the majority of predictions (5 pathogenic vs. 4 benign) indicate that P939S is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -4.331 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -3.44 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.24 | Pathogenic | 0.00 | Affected | 0.3372 | 0.4517 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3052A>C | T1018P 2D AIThe SynGAP1 missense variant T1018P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.959985 | Binding | 0.348 | 0.801 | 0.500 | -2.046 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.218 | Likely Benign | -1.39 | Neutral | 0.586 | Possibly Damaging | 0.302 | Benign | 2.24 | Pathogenic | 0.02 | Affected | 0.2155 | 0.3960 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3053C>T | T1018I 2D AIThe SynGAP1 missense variant T1018I is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443605‑C‑T). Prediction tools that agree on benign impact include REVEL, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized, while those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta results are unavailable. Overall, the predictions are split, with no clear majority leaning toward either benign or pathogenic. Thus, the variant’s impact remains inconclusive, and this uncertainty aligns with ClinVar’s current “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.849326 | Disordered | 0.959985 | Binding | 0.348 | 0.801 | 0.500 | Uncertain | 1 | 6-33443605-C-T | 4 | 2.48e-6 | -3.264 | Likely Benign | 0.524 | Ambiguous | Likely Benign | 0.076 | Likely Benign | -2.55 | Deleterious | 0.586 | Possibly Damaging | 0.304 | Benign | 2.24 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1022 | 0.4776 | -1 | 0 | 5.2 | 12.05 | |||||||||||||||||||||||||||||||||
| c.3731G>C | S1244T 2D AISynGAP1 missense variant S1244T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on benign effect include REVEL, PROVEAN, SIFT, and AlphaMissense‑Optimized. Tools that agree on pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments give AlphaMissense‑Optimized a benign score, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—predicts pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence leans toward pathogenicity, with four high‑confidence pathogenic predictions versus four benign predictions, and the SGM Consensus tipping the scale. This conclusion does not contradict ClinVar status, as the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.648219 | Disordered | 0.411055 | Uncertain | 0.833 | 0.549 | 0.500 | -9.020 | Likely Pathogenic | 0.405 | Ambiguous | Likely Benign | 0.149 | Likely Benign | -1.82 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.24 | Pathogenic | 0.11 | Tolerated | 0.1129 | 0.4603 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3885G>C | Q1295H 2D AIThe SynGAP1 missense variant Q1295H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show discordant results: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further highlight this split: AlphaMissense‑Optimized predicts a benign effect, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome. No Foldetta stability analysis is available for this residue. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not contradict any existing ClinVar annotation, as none is present. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.852992 | Disordered | 0.892719 | Binding | 0.499 | 0.801 | 0.625 | -4.851 | Likely Benign | 0.588 | Likely Pathogenic | Likely Benign | 0.349 | Likely Benign | -4.08 | Deleterious | 0.991 | Probably Damaging | 0.986 | Probably Damaging | 2.24 | Pathogenic | 0.00 | Affected | 0.1345 | 0.3203 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3885G>T | Q1295H 2D AIThe SynGAP1 missense variant Q1295H is catalogued in gnomAD (ID 6‑33447933‑G‑T) but has no ClinVar submission. Functional prediction tools show a split: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. When predictions are grouped, 3 tools predict benign and 6 predict pathogenic. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized remains benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this substitution. Overall, the preponderance of evidence points to a pathogenic effect, and this assessment does not conflict with ClinVar, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.852992 | Disordered | 0.892719 | Binding | 0.499 | 0.801 | 0.625 | 6-33447933-G-T | -4.851 | Likely Benign | 0.588 | Likely Pathogenic | Likely Benign | 0.349 | Likely Benign | -4.08 | Deleterious | 0.991 | Probably Damaging | 0.986 | Probably Damaging | 2.24 | Pathogenic | 0.00 | Affected | 0.1345 | 0.3203 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||||||
| c.2180A>C | N727T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727T is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that classify the variant as benign include REVEL, Rosetta, premPS, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. FoldX gives an uncertain result and is therefore treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta (combining FoldX‑MD and Rosetta outputs) as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -6.900 | Likely Benign | 0.335 | Likely Benign | Likely Benign | 0.52 | Ambiguous | 0.1 | -0.18 | Likely Benign | 0.17 | Likely Benign | 0.04 | Likely Benign | 0.125 | Likely Benign | -3.08 | Deleterious | 0.987 | Probably Damaging | 0.980 | Probably Damaging | 2.25 | Pathogenic | 0.74 | Tolerated | 0.1315 | 0.7181 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||
| c.2353C>G | R785G 2D AIThe SynGAP1 missense variant R785G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome. High‑accuracy assessments further reveal that AlphaMissense‑Optimized predicts a benign effect, while the SGM‑Consensus again suggests pathogenicity; Foldetta stability analysis is not available for this variant. Overall, the majority of predictions lean toward pathogenicity, and this is consistent with the SGM‑Consensus result. Because the variant is not present in ClinVar, there is no existing clinical classification to contradict; thus, based on the available computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | -3.684 | Likely Benign | 0.697 | Likely Pathogenic | Likely Benign | 0.190 | Likely Benign | -3.44 | Deleterious | 0.980 | Probably Damaging | 0.818 | Possibly Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.3695 | 0.3686 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2354G>A | R785H 2D AIThe SynGAP1 R785H missense variant (ClinVar ID 2321588.0) is listed as “Uncertain” in ClinVar and is present in gnomAD (ID 6‑33442906‑G‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, whereas the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a pathogenic outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, does not provide a result for this variant. Overall, the majority of computational predictions (five pathogenic versus three benign) lean toward a pathogenic interpretation. Thus, the variant is most likely pathogenic based on current predictions, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | Uncertain | 2 | 6-33442906-G-A | 4 | 2.50e-6 | -4.782 | Likely Benign | 0.388 | Ambiguous | Likely Benign | 0.129 | Likely Benign | -2.61 | Deleterious | 0.999 | Probably Damaging | 0.947 | Probably Damaging | 2.25 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.3260 | 0.1589 | 2 | 0 | 1.3 | -19.05 | ||||||||||||||||||||||||||||||||
| c.2368A>C | T790P 2D AIThe SynGAP1 missense variant T790P has no ClinVar entry (ClinVar status: not reported) but is present in gnomAD (ID 6‑33442920‑A‑C). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of conventional tools (5 pathogenic vs 4 benign) lean toward a pathogenic interpretation, while the single high‑accuracy tool suggests benign. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | 6-33442920-A-C | -3.564 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 0.250 | Likely Benign | -3.55 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.25 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.2147 | 0.4748 | -1 | 0 | -0.9 | -3.99 | ||||||||||||||||||||||||||||||||||||
| c.2537T>C | L846S 2D  AIThe SynGAP1 missense variant L846S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL classifies it as benign, whereas the remaining 11 predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus result is consistent with a likely pathogenic classification. Foldetta predictions are unavailable. Taken together, the preponderance of evidence indicates that the variant is most likely pathogenic, and this assessment does not conflict with the current ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.653063 | Disordered | 0.589606 | Binding | 0.349 | 0.825 | 0.500 | -10.944 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.366 | Likely Benign | -3.44 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.3128 | 0.1070 | -3 | -2 | -4.6 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3053C>A | T1018N 2D AIThe SynGAP1 missense variant T1018N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar status, as no ClinVar claim exists for this variant. Thus, based on current computational predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.959985 | Binding | 0.348 | 0.801 | 0.500 | -3.888 | Likely Benign | 0.165 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -1.74 | Neutral | 0.411 | Benign | 0.139 | Benign | 2.25 | Pathogenic | 0.01 | Affected | 0.1537 | 0.3979 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||||||||||||
| c.3499G>T | D1167Y 2D AIThe SynGAP1 missense variant D1167Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a 2‑to‑2 split and is therefore inconclusive. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑confidence predictors (six pathogenic vs. three benign) indicate that D1167Y is most likely pathogenic. This assessment does not contradict any ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.783999 | Binding | 0.336 | 0.798 | 0.500 | -4.050 | Likely Benign | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.281 | Likely Benign | -2.46 | Neutral | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.0640 | 0.7307 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||||
| c.3653A>C | E1218A 2D AIThe SynGAP1 missense variant E1218A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and ESM1b, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic outcome. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the overall consensus of the available predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.595080 | Disordered | 0.483050 | Uncertain | 0.898 | 0.565 | 0.375 | -3.698 | Likely Benign | 0.819 | Likely Pathogenic | Ambiguous | 0.370 | Likely Benign | -4.75 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.3005 | 0.3969 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||
| c.3680A>T | E1227V 2D AIThe SynGAP1 missense variant E1227V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess the variant’s effect fall into two groups: the single benign prediction comes from REVEL, whereas all other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—classify it as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus also indicates likely pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from multiple independent prediction tools and high‑accuracy methods indicates that E1227V is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.513880 | Disordered | 0.433399 | Uncertain | 0.860 | 0.544 | 0.500 | -12.852 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.355 | Likely Benign | -5.49 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.0405 | 0.6559 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3734A>C | E1245A 2D AIThe SynGAP1 missense variant E1245A is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess the variant’s effect largely agree on a deleterious outcome: REVEL is the sole tool that predicts a benign effect, whereas PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus likewise indicates a likely pathogenic effect. Foldetta results are not available for this variant. Overall, the consensus of the available predictions indicates that E1245A is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.712013 | Disordered | 0.387847 | Uncertain | 0.869 | 0.554 | 0.625 | -11.433 | Likely Pathogenic | 0.960 | Likely Pathogenic | Likely Pathogenic | 0.311 | Likely Benign | -4.85 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.2555 | 0.5721 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||
| c.3794G>T | R1265M 2D  AIThe SynGAP1 missense variant R1265M is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic status; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that R1265M is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.414856 | Structured | 0.782497 | Binding | 0.887 | 0.592 | 0.000 | -13.657 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.495 | Likely Benign | -4.97 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.1442 | 0.4082 | 0 | -1 | 6.4 | -24.99 | ||||||||||||||||||||||||||||||||||||||
| c.3814G>C | E1272Q 2D AIThe SynGAP1 E1272Q missense variant is catalogued in gnomAD (variant ID 6‑33447862‑G‑C) but has no ClinVar entry. Prediction tools cluster into two groups: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, a majority‑vote aggregator of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic, and Foldetta (a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the preponderance of evidence (seven pathogenic versus three benign predictions) indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.766082 | Binding | 0.799 | 0.677 | 0.500 | 6-33447862-G-C | 1 | 6.45e-7 | -3.000 | Likely Benign | 0.651 | Likely Pathogenic | Likely Benign | 0.207 | Likely Benign | -2.53 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.0746 | 0.5258 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||
| c.3884A>T | Q1295L 2D AIThe SynGAP1 missense variant Q1295L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact for Q1295L, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.892719 | Binding | 0.499 | 0.801 | 0.625 | -2.862 | Likely Benign | 0.348 | Ambiguous | Likely Benign | 0.379 | Likely Benign | -5.63 | Deleterious | 0.925 | Possibly Damaging | 0.932 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.0810 | 0.5672 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.2354G>T | R785L 2D AIThe SynGAP1 missense variant R785L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; Foldetta results are unavailable. Overall, the balance of evidence leans toward a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | -4.457 | Likely Benign | 0.699 | Likely Pathogenic | Likely Benign | 0.158 | Likely Benign | -4.43 | Deleterious | 0.960 | Probably Damaging | 0.627 | Possibly Damaging | 2.26 | Pathogenic | 0.01 | Affected | 0.1993 | 0.4539 | -3 | -2 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||||||
| c.2485G>C | E829Q 2D AIThe SynGAP1 missense variant E829Q is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of reliable tools predict a benign impact, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.562014 | Disordered | 0.626045 | Binding | 0.326 | 0.882 | 0.375 | -4.985 | Likely Benign | 0.531 | Ambiguous | Likely Benign | 0.225 | Likely Benign | -1.83 | Neutral | 0.994 | Probably Damaging | 0.946 | Probably Damaging | 2.26 | Pathogenic | 0.00 | Affected | 0.1321 | 0.7248 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2486A>C | E829A 2D AIThe SynGAP1 missense variant E829A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) results are unavailable. Overall, the majority of evidence—including the SGM‑Consensus—points to a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.562014 | Disordered | 0.626045 | Binding | 0.326 | 0.882 | 0.375 | -4.096 | Likely Benign | 0.574 | Likely Pathogenic | Likely Benign | 0.265 | Likely Benign | -3.75 | Deleterious | 0.994 | Probably Damaging | 0.926 | Probably Damaging | 2.26 | Pathogenic | 0.00 | Affected | 0.4707 | 0.7255 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2540A>C | Q847P 2D AIThe SynGAP1 missense variant Q847P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑vs‑2 split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑accuracy predictors (AlphaMissense‑Optimized and AlphaMissense‑Default) indicate a benign outcome, while the consensus of other tools is mixed. Thus, the variant is most likely benign based on current predictions, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.653063 | Disordered | 0.577677 | Binding | 0.282 | 0.818 | 0.500 | -6.742 | Likely Benign | 0.320 | Likely Benign | Likely Benign | 0.331 | Likely Benign | -3.83 | Deleterious | 0.990 | Probably Damaging | 0.892 | Possibly Damaging | 2.26 | Pathogenic | 0.00 | Affected | 0.2290 | 0.4831 | 0 | -1 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||||||||
| c.2848G>C | G950R 2D AIThe SynGAP1 missense variant G950R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for G950R, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -6.929 | Likely Benign | 0.318 | Likely Benign | Likely Benign | 0.388 | Likely Benign | -1.10 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.26 | Pathogenic | 0.01 | Affected | 0.1009 | 0.4733 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2848G>T | G950C 2D AIThe SynGAP1 missense variant G950C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑confidence predictors (five pathogenic vs. four benign) indicate a pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -9.589 | Likely Pathogenic | 0.092 | Likely Benign | Likely Benign | 0.395 | Likely Benign | -0.35 | Neutral | 0.983 | Probably Damaging | 0.750 | Possibly Damaging | 2.26 | Pathogenic | 0.01 | Affected | 0.1406 | 0.4439 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2849G>A | G950D 2D AIThe SynGAP1 missense variant G950D is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -7.515 | In-Between | 0.245 | Likely Benign | Likely Benign | 0.402 | Likely Benign | -0.61 | Neutral | 0.411 | Benign | 0.131 | Benign | 2.26 | Pathogenic | 0.02 | Affected | 0.1874 | 0.2826 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2849G>T | G950V 2D AIThe SynGAP1 missense variant G950V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that G950V is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -7.796 | In-Between | 0.093 | Likely Benign | Likely Benign | 0.428 | Likely Benign | -0.91 | Neutral | 0.411 | Benign | 0.239 | Benign | 2.26 | Pathogenic | 0.01 | Affected | 0.1392 | 0.3507 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3304G>C | A1102P 2D AIThe SynGAP1 missense variant A1102P is listed in ClinVar (ID 2789225.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | Uncertain | 1 | -5.120 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -0.97 | Neutral | 0.000 | Benign | 0.002 | Benign | 2.26 | Pathogenic | 0.13 | Tolerated | 3.77 | 5 | 0.1978 | 0.5919 | -1 | 1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||
| c.3500A>T | D1167V 2D AIThe SynGAP1 missense variant D1167V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Based on the preponderance of pathogenic predictions and the high‑accuracy tool results, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.783999 | Binding | 0.336 | 0.798 | 0.500 | -2.750 | Likely Benign | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.288 | Likely Benign | -3.34 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.26 | Pathogenic | 0.00 | Affected | 0.0946 | 0.7515 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3621G>C | E1207D 2D AIThe SynGAP1 missense variant E1207D is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign) all classify the change as tolerated or benign. Only FATHMM predicts a pathogenic outcome. High‑accuracy tools that were available give a benign verdict: AlphaMissense‑Optimized is benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign. Foldetta results are not reported, so they do not influence the assessment. Overall, the majority of evidence supports a benign interpretation, and this is consistent with the lack of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.604312 | Disordered | 0.562696 | Binding | 0.912 | 0.571 | 0.375 | -3.924 | Likely Benign | 0.249 | Likely Benign | Likely Benign | 0.010 | Likely Benign | -1.16 | Neutral | 0.121 | Benign | 0.069 | Benign | 2.26 | Pathogenic | 0.51 | Tolerated | 0.1485 | 0.2810 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3621G>T | E1207D 2D AIThe SynGAP1 missense variant E1207D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only FATHMM predicts it as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of tools, including the high‑accuracy predictors, indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.604312 | Disordered | 0.562696 | Binding | 0.912 | 0.571 | 0.375 | -3.924 | Likely Benign | 0.249 | Likely Benign | Likely Benign | 0.010 | Likely Benign | -1.16 | Neutral | 0.121 | Benign | 0.069 | Benign | 2.26 | Pathogenic | 0.51 | Tolerated | 0.1485 | 0.2810 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3810G>C | E1270D 2D AIThe SynGAP1 missense variant E1270D is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.490133 | Structured | 0.771865 | Binding | 0.805 | 0.659 | 0.250 | -9.379 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.220 | Likely Benign | -2.53 | Deleterious | 0.992 | Probably Damaging | 0.983 | Probably Damaging | 2.26 | Pathogenic | 0.00 | Affected | 0.1655 | 0.3271 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3810G>T | E1270D 2D AIThe SynGAP1 missense variant E1270D is catalogued in gnomAD (6-33447858‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: the single benign prediction comes from REVEL, while all other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Pathogenic.” The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence indicates that E1270D is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.490133 | Structured | 0.771865 | Binding | 0.805 | 0.659 | 0.250 | 6-33447858-G-T | -9.379 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.219 | Likely Benign | -2.53 | Deleterious | 0.992 | Probably Damaging | 0.983 | Probably Damaging | 2.26 | Pathogenic | 0.00 | Affected | 0.1655 | 0.3271 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.3813G>C | E1271D 2D AIThe SynGAP1 missense variant E1271D is not reported in ClinVar and has no entry in gnomAD, indicating it is not catalogued in these databases. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign; the Foldetta stability prediction is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.483068 | Structured | 0.767529 | Binding | 0.832 | 0.666 | 0.375 | -4.816 | Likely Benign | 0.179 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -1.59 | Neutral | 0.004 | Benign | 0.008 | Benign | 2.26 | Pathogenic | 0.00 | Affected | 0.1729 | 0.3471 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3813G>T | E1271D 2D AIThe SynGAP1 missense variant E1271D is not reported in ClinVar and has no entry in gnomAD, indicating it is not catalogued in these databases. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign; the Foldetta stability prediction is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.483068 | Structured | 0.767529 | Binding | 0.832 | 0.666 | 0.375 | -4.816 | Likely Benign | 0.179 | Likely Benign | Likely Benign | 0.121 | Likely Benign | -1.59 | Neutral | 0.004 | Benign | 0.008 | Benign | 2.26 | Pathogenic | 0.00 | Affected | 0.1729 | 0.3471 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3815A>C | E1272A 2D AIThe SynGAP1 missense variant E1272A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus indicates a likely pathogenic outcome; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points toward a pathogenic impact for E1272A. This prediction is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.766082 | Binding | 0.799 | 0.677 | 0.500 | -0.783 | Likely Benign | 0.779 | Likely Pathogenic | Likely Benign | 0.261 | Likely Benign | -5.05 | Deleterious | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.26 | Pathogenic | 0.00 | Affected | 0.3243 | 0.5547 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2369C>A | T790N 2D AIThe SynGAP1 missense variant T790N is listed in ClinVar with an “Uncertain” status and is present in the gnomAD database (ID 6‑33442921‑C‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive and therefore unavailable, and Foldetta results are not reported. Overall, the majority of conventional tools (5 pathogenic vs. 4 benign) lean toward a pathogenic interpretation, while the single high‑accuracy tool suggests benign. The variant’s ClinVar status remains uncertain, so there is no contradiction with the current clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | Conflicting | 3 | 6-33442921-C-A | 69 | 4.28e-5 | -5.243 | Likely Benign | 0.276 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -2.54 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.27 | Pathogenic | 0.02 | Affected | 3.64 | 6 | 0.1446 | 0.4653 | 0 | 0 | -2.8 | 13.00 | ||||||||||||||||||||||||||||||||
| c.2423T>G | V808G 2D AIThe SynGAP1 V808G missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of individual predictors (five benign vs. three pathogenic) support a benign classification, and this is not contradicted by any ClinVar annotation. Thus, based on the available predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.699094 | Disordered | 0.856438 | Binding | 0.289 | 0.903 | 0.500 | -6.635 | Likely Benign | 0.460 | Ambiguous | Likely Benign | 0.268 | Likely Benign | -2.94 | Deleterious | 0.069 | Benign | 0.135 | Benign | 2.27 | Pathogenic | 0.00 | Affected | 0.1925 | 0.3336 | -1 | -3 | -4.6 | -42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2485G>A | E829K 2D AIThe SynGAP1 missense variant E829K is listed in ClinVar as Pathogenic (ClinVar ID 1721258.0) and is not reported in gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only REVEL predicts a benign outcome, while ESM1b and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments show the SGM‑Consensus as Likely Pathogenic, AlphaMissense‑Optimized as uncertain, and Foldetta results are unavailable. Overall, the preponderance of evidence indicates that E829K is most likely pathogenic, and this conclusion aligns with the ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.562014 | Disordered | 0.626045 | Binding | 0.326 | 0.882 | 0.375 | Pathogenic | 1 | -7.527 | In-Between | 0.807 | Likely Pathogenic | Ambiguous | 0.194 | Likely Benign | -2.65 | Deleterious | 0.994 | Probably Damaging | 0.900 | Possibly Damaging | 2.27 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2400 | 0.7372 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||
| c.2541G>C | Q847H 2D AIThe SynGAP1 missense variant Q847H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default—predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and the Foldetta stability analysis is unavailable. Overall, the majority of predictions (seven pathogenic vs. three benign) and the SGM‑Consensus result point to a pathogenic effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.653063 | Disordered | 0.577677 | Binding | 0.282 | 0.818 | 0.500 | -4.208 | Likely Benign | 0.720 | Likely Pathogenic | Likely Benign | 0.204 | Likely Benign | -2.82 | Deleterious | 0.990 | Probably Damaging | 0.925 | Probably Damaging | 2.27 | Pathogenic | 0.00 | Affected | 0.1429 | 0.3634 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2541G>T | Q847H 2D AIThe SynGAP1 missense variant Q847H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default—predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and the Foldetta stability analysis is unavailable. Overall, the majority of predictions (seven pathogenic vs. three benign) and the SGM‑Consensus support a pathogenic interpretation. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.653063 | Disordered | 0.577677 | Binding | 0.282 | 0.818 | 0.500 | -4.208 | Likely Benign | 0.720 | Likely Pathogenic | Likely Benign | 0.204 | Likely Benign | -2.82 | Deleterious | 0.990 | Probably Damaging | 0.925 | Probably Damaging | 2.27 | Pathogenic | 0.00 | Affected | 0.1429 | 0.3634 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3305C>A | A1102D 2D AIThe SynGAP1 missense variant A1102D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | -4.647 | Likely Benign | 0.388 | Ambiguous | Likely Benign | 0.081 | Likely Benign | -1.38 | Neutral | 0.033 | Benign | 0.028 | Benign | 2.27 | Pathogenic | 0.12 | Tolerated | 0.2045 | 0.2984 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3481A>G | M1161V 2D  AISynGAP1 missense variant M1161V is not reported in ClinVar and is present in gnomAD (ID 6‑33444516‑A‑G). Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, SIFT, and ESM1b; pathogenic predictions from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized remains uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta data are unavailable. Consequently, the evidence does not favor either outcome. The variant is most likely neither clearly benign nor pathogenic based on current predictions, and this lack of consensus does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.580690 | Disordered | 0.864109 | Binding | 0.372 | 0.830 | 0.375 | 6-33444516-A-G | 2 | 1.24e-6 | -3.060 | Likely Benign | 0.915 | Likely Pathogenic | Ambiguous | 0.150 | Likely Benign | -1.85 | Neutral | 0.843 | Possibly Damaging | 0.926 | Probably Damaging | 2.27 | Pathogenic | 0.22 | Tolerated | 3.77 | 5 | 0.3422 | 0.3292 | 1 | 2 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||
| c.3483G>A | M1161I 2D  AIThe SynGAP1 missense variant M1161I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the majority of evidence (five pathogenic vs. four benign predictions) indicates that the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.580690 | Disordered | 0.864109 | Binding | 0.372 | 0.830 | 0.375 | -3.124 | Likely Benign | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.155 | Likely Benign | -1.92 | Neutral | 0.925 | Possibly Damaging | 0.954 | Probably Damaging | 2.27 | Pathogenic | 0.25 | Tolerated | 3.77 | 5 | 0.1437 | 0.3190 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||||||
| c.3483G>C | M1161I 2D AIThe SynGAP1 missense variant M1161I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs. four benign) indicate that the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.580690 | Disordered | 0.864109 | Binding | 0.372 | 0.830 | 0.375 | -3.124 | Likely Benign | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.155 | Likely Benign | -1.92 | Neutral | 0.925 | Possibly Damaging | 0.954 | Probably Damaging | 2.27 | Pathogenic | 0.25 | Tolerated | 3.77 | 5 | 0.1437 | 0.3190 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||||||
| c.3483G>T | M1161I 2D AIThe SynGAP1 missense variant M1161I is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33444518‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b, whereas polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, there are five pathogenic predictions versus four benign ones, tipping the balance toward a likely pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.580690 | Disordered | 0.864109 | Binding | 0.372 | 0.830 | 0.375 | 6-33444518-G-T | 1 | 6.20e-7 | -3.124 | Likely Benign | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.155 | Likely Benign | -1.92 | Neutral | 0.925 | Possibly Damaging | 0.954 | Probably Damaging | 2.27 | Pathogenic | 0.25 | Tolerated | 3.77 | 5 | 0.1437 | 0.3190 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||
| c.3499G>C | D1167H 2D AIThe SynGAP1 missense variant D1167H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b. Tools that agree on a pathogenic effect include polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic, two benign). Foldetta predictions are unavailable. Overall, the majority of evaluated tools (six pathogenic vs. three benign) indicate a pathogenic effect. This conclusion is consistent with the lack of ClinVar reporting; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.783999 | Binding | 0.336 | 0.798 | 0.500 | -3.774 | Likely Benign | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.257 | Likely Benign | -2.36 | Neutral | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.27 | Pathogenic | 0.00 | Affected | 0.1514 | 0.8433 | 1 | -1 | 0.3 | 22.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3634T>G | S1212A 2D AIThe SynGAP1 missense variant S1212A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta results are unavailable. Consequently, the collective evidence points to a benign classification for S1212A, and this conclusion does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.566480 | Disordered | 0.548409 | Binding | 0.852 | 0.565 | 0.500 | -4.705 | Likely Benign | 0.403 | Ambiguous | Likely Benign | 0.109 | Likely Benign | -1.66 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.27 | Pathogenic | 0.00 | Affected | 0.3901 | 0.3823 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3782G>C | S1261T 2D AIThe SynGAP1 missense variant S1261T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate a benign or likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports likely benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for S1261T, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.501700 | Disordered | 0.671500 | Binding | 0.889 | 0.574 | 0.250 | -4.800 | Likely Benign | 0.203 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -2.01 | Neutral | 0.906 | Possibly Damaging | 0.629 | Possibly Damaging | 2.27 | Pathogenic | 0.11 | Tolerated | 0.1007 | 0.4249 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3793A>G | R1265G 2D AIThe SynGAP1 missense variant R1265G is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all label the change as pathogenic. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, while a Foldetta stability analysis is unavailable. Based on the unanimous pathogenic predictions and the lack of any benign calls, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status (which has no entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.414856 | Structured | 0.782497 | Binding | 0.887 | 0.592 | 0.000 | -12.732 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.523 | Likely Pathogenic | -5.80 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 2.27 | Pathogenic | 0.00 | Affected | 0.3520 | 0.3759 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3816G>C | E1272D 2D AIThe SynGAP1 missense variant E1272D has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the remaining tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) all predict a pathogenic impact. High‑accuracy assessments further show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely pathogenic; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a pathogenic effect. There is no ClinVar classification to contradict this conclusion, so the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.766082 | Binding | 0.799 | 0.677 | 0.500 | -4.781 | Likely Benign | 0.751 | Likely Pathogenic | Likely Benign | 0.189 | Likely Benign | -2.53 | Deleterious | 0.992 | Probably Damaging | 0.983 | Probably Damaging | 2.27 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1450 | 0.3471 | 2 | 3 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.3816G>T | E1272D 2D AIThe SynGAP1 missense variant E1272D is listed in ClinVar with no submitted interpretation and is present in gnomAD (variant ID 6‑33447864‑G‑T). Functional prediction tools show mixed results: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. The high‑accuracy AlphaMissense‑Optimized tool classifies the change as benign, while the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect. No Foldetta stability assessment is available for this variant. Overall, the majority of conventional predictors and the consensus score lean toward pathogenicity, which is consistent with the SGM‑Consensus designation but contradicts the benign calls from AlphaMissense‑Optimized and a few other tools. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion aligns with the SGM‑Consensus prediction rather than the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.766082 | Binding | 0.799 | 0.677 | 0.500 | 6-33447864-G-T | -4.781 | Likely Benign | 0.751 | Likely Pathogenic | Likely Benign | 0.188 | Likely Benign | -2.53 | Deleterious | 0.992 | Probably Damaging | 0.983 | Probably Damaging | 2.27 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1450 | 0.3471 | 2 | 3 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||
| c.2335A>T | S779C 2D AIThe SynGAP1 missense variant S779C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for this variant. This conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -6.375 | Likely Benign | 0.196 | Likely Benign | Likely Benign | 0.230 | Likely Benign | -1.88 | Neutral | 0.992 | Probably Damaging | 0.905 | Possibly Damaging | 2.28 | Pathogenic | 0.05 | Affected | 0.1177 | 0.6350 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2336G>T | S779I 2D AIThe SynGAP1 missense variant S779I is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy methods give a benign call from AlphaMissense‑Optimized; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of tools (five pathogenic vs. four benign) suggest a pathogenic impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -6.261 | Likely Benign | 0.578 | Likely Pathogenic | Likely Benign | 0.198 | Likely Benign | -2.10 | Neutral | 0.918 | Possibly Damaging | 0.827 | Possibly Damaging | 2.28 | Pathogenic | 0.05 | Affected | 0.1046 | 0.6248 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2369C>T | T790I 2D AIThe SynGAP1 missense variant T790I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic. Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict the ClinVar status, which currently has no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | -3.556 | Likely Benign | 0.482 | Ambiguous | Likely Benign | 0.190 | Likely Benign | -3.08 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.28 | Pathogenic | 0.01 | Affected | 0.0987 | 0.5431 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||||||||||||
| c.2423T>A | V808E 2D AIThe SynGAP1 missense variant V808E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: the single benign prediction comes from REVEL, while the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—indicate pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized returns an uncertain result, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels the variant as Likely Pathogenic, and Foldetta data are not available. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status, as none exists for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.699094 | Disordered | 0.856438 | Binding | 0.289 | 0.903 | 0.500 | -9.078 | Likely Pathogenic | 0.888 | Likely Pathogenic | Ambiguous | 0.307 | Likely Benign | -2.84 | Deleterious | 0.999 | Probably Damaging | 0.958 | Probably Damaging | 2.28 | Pathogenic | 0.00 | Affected | 0.1129 | 0.2787 | -2 | -2 | -7.7 | 29.98 | ||||||||||||||||||||||||||||||||||||||
| c.2657C>G | A886G 2D AIThe SynGAP1 missense variant A886G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for A886G, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.619166 | Binding | 0.359 | 0.922 | 0.500 | -1.993 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -0.70 | Neutral | 0.597 | Possibly Damaging | 0.366 | Benign | 2.28 | Pathogenic | 0.00 | Affected | 0.2173 | 0.3913 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2846G>C | G949A 2D AIThe SynGAP1 missense variant G949A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign” (3 benign vs. 1 pathogenic votes). High‑accuracy methods confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | -6.838 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.274 | Likely Benign | -0.08 | Neutral | 0.779 | Possibly Damaging | 0.425 | Benign | 2.28 | Pathogenic | 0.08 | Tolerated | 0.3385 | 0.4414 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2849G>C | G950A 2D AIThe SynGAP1 missense variant G950A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only FATHMM predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict, reflecting the majority of benign calls. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -6.557 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.339 | Likely Benign | -0.13 | Neutral | 0.059 | Benign | 0.061 | Benign | 2.28 | Pathogenic | 0.08 | Tolerated | 0.3343 | 0.4957 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3652G>A | E1218K 2D AIThe SynGAP1 missense variant E1218K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—all predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus indicates it is likely pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from both general and high‑accuracy prediction tools indicates that E1218K is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.595080 | Disordered | 0.483050 | Uncertain | 0.898 | 0.565 | 0.375 | -8.932 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.336 | Likely Benign | -3.12 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.28 | Pathogenic | 0.00 | Affected | 0.1672 | 0.4024 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3680A>G | E1227G 2D AIThe SynGAP1 missense variant E1227G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. In contrast, the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all classify the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. AlphaMissense‑Optimized yields an uncertain result, and Foldetta (FoldX‑MD/Rosetta stability analysis) is not available for this variant. Overall, the preponderance of evidence from high‑accuracy predictors and consensus methods indicates that E1227G is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.513880 | Disordered | 0.433399 | Uncertain | 0.860 | 0.544 | 0.500 | -9.328 | Likely Pathogenic | 0.903 | Likely Pathogenic | Ambiguous | 0.336 | Likely Benign | -5.26 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.28 | Pathogenic | 0.00 | Affected | 0.2725 | 0.5550 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||||||
| c.3733G>A | E1245K 2D AIThe SynGAP1 missense variant E1245K is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this view: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that E1245K is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.712013 | Disordered | 0.387847 | Uncertain | 0.869 | 0.554 | 0.625 | -11.911 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 0.276 | Likely Benign | -3.22 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.28 | Pathogenic | 0.00 | Affected | 0.1627 | 0.6574 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3814G>A | E1272K 2D AIThe SynGAP1 E1272K missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions are made by REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; the SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic, and Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.766082 | Binding | 0.799 | 0.677 | 0.500 | -4.227 | Likely Benign | 0.956 | Likely Pathogenic | Likely Pathogenic | 0.312 | Likely Benign | -3.37 | Deleterious | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.28 | Pathogenic | 0.00 | Affected | 0.1544 | 0.5488 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.1394T>A | L465H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L465H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic. Based on the consensus of all available predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.346032 | Structured | 0.319240 | Uncertain | 0.956 | 0.202 | 0.000 | -16.751 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.15 | Destabilizing | 0.4 | 2.29 | Destabilizing | 2.72 | Destabilizing | 2.54 | Destabilizing | 0.764 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.1289 | 0.1089 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1394T>C | L465P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L465P is listed in ClinVar as Pathogenic (ClinVar ID 1067821.0) and is not reported in gnomAD. Prediction tools that assess functional impact uniformly classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this conclusion aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.346032 | Structured | 0.319240 | Uncertain | 0.956 | 0.202 | 0.000 | Likely Pathogenic | 1 | -14.824 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 7.18 | Destabilizing | 0.3 | 10.85 | Destabilizing | 9.02 | Destabilizing | 2.73 | Destabilizing | 0.778 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 3.37 | 34 | 0.3387 | 0.1582 | -3 | -3 | -5.4 | -16.04 | 211.1 | 65.9 | 0.1 | 0.0 | -0.2 | 0.0 | X | Potentially Pathogenic | The iso-butyl side chain of Leu465, located in the middle of an α helix (res. Ala461–Phe476), packs with hydrophobic residues (e.g., Phe464, Met468, Tyr497, Ile494) in an inter-helix space formed with two other α helices (res. Ala461–Phe476 and res. Thr488-Gly502). In the variant simulations, the cyclic five-membered pyrrolidine ring of Pro465 is not as optimal as the side chain of Leu465 for filling the three α helix hydrophobic niche. Although the residue swap does not cause a large-scale conformational shift during the simulations, the H-bond between the backbone amide group of Leu465 and the backbone carbonyl group of Ala461 is lost. This, in turn, breaks the continuity of the α helix secondary structure element. | ||||||||||||||||
| c.1394T>G | L465R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L465R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. No inconclusive or missing results are present. Based on the consensus of all available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.346032 | Structured | 0.319240 | Uncertain | 0.956 | 0.202 | 0.000 | -17.976 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.52 | Destabilizing | 0.7 | 4.44 | Destabilizing | 4.48 | Destabilizing | 2.41 | Destabilizing | 0.761 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.1597 | 0.0963 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1415A>T | E472V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E472V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Foldetta and premPS, whereas the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as benign. No prediction or folding‑stability result is missing or inconclusive; uncertain outputs from FoldX and Rosetta are treated as unavailable. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -14.957 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 1.05 | Ambiguous | 0.3 | -0.64 | Ambiguous | 0.21 | Likely Benign | 0.37 | Likely Benign | 0.733 | Likely Pathogenic | -6.90 | Deleterious | 0.996 | Probably Damaging | 0.991 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.0881 | 0.6347 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.2335A>C | S779R 2D AIThe SynGAP1 missense variant S779R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain, and both the SGM Consensus and Foldetta (which would combine FoldX‑MD and Rosetta outputs) are unavailable. Consequently, the overall prediction is ambiguous. The variant is most likely benign based on the balance of evidence, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.044 | Likely Benign | 0.950 | Likely Pathogenic | Ambiguous | 0.103 | Likely Benign | -0.74 | Neutral | 0.846 | Possibly Damaging | 0.627 | Possibly Damaging | 2.29 | Pathogenic | 0.12 | Tolerated | 0.0961 | 0.4046 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.2337C>A | S779R 2D AIThe SynGAP1 missense variant S779R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain, and both the SGM Consensus and Foldetta (which would combine FoldX‑MD and Rosetta outputs) are unavailable. Consequently, the overall prediction is ambiguous. The variant is most likely benign based on the balance of evidence, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.044 | Likely Benign | 0.950 | Likely Pathogenic | Ambiguous | 0.119 | Likely Benign | -0.74 | Neutral | 0.846 | Possibly Damaging | 0.627 | Possibly Damaging | 2.29 | Pathogenic | 0.12 | Tolerated | 0.0961 | 0.4046 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.2337C>G | S779R 2D AIThe SynGAP1 missense variant S779R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain, and both the SGM Consensus and Foldetta (which would combine FoldX‑MD and Rosetta outputs) are unavailable. Consequently, the overall prediction is ambiguous. The variant is most likely benign based on the balance of evidence, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.044 | Likely Benign | 0.950 | Likely Pathogenic | Ambiguous | 0.124 | Likely Benign | -0.74 | Neutral | 0.846 | Possibly Damaging | 0.627 | Possibly Damaging | 2.29 | Pathogenic | 0.12 | Tolerated | 0.0961 | 0.4046 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.2455G>A | A819T 2D AIThe SynGAP1 missense variant A819T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs 2 benign) and Foldetta data are unavailable. Overall, the majority of conventional predictors lean toward pathogenicity, but the single high‑accuracy tool predicts benign and the consensus is unresolved. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.694846 | Disordered | 0.707644 | Binding | 0.317 | 0.892 | 0.625 | -3.421 | Likely Benign | 0.675 | Likely Pathogenic | Likely Benign | 0.287 | Likely Benign | -2.23 | Neutral | 0.998 | Probably Damaging | 0.963 | Probably Damaging | 2.29 | Pathogenic | 0.01 | Affected | 0.1443 | 0.7583 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2456C>G | A819G 2D AIThe SynGAP1 missense variant A819G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic). Foldetta results are unavailable. Overall, the majority of predictions (six benign vs three pathogenic) support a benign classification. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.694846 | Disordered | 0.707644 | Binding | 0.317 | 0.892 | 0.625 | -5.730 | Likely Benign | 0.714 | Likely Pathogenic | Likely Benign | 0.116 | Likely Benign | -2.17 | Neutral | 0.104 | Benign | 0.121 | Benign | 2.29 | Pathogenic | 0.03 | Affected | 0.2363 | 0.5396 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2803G>C | A935P 2D AIThe SynGAP1 missense variant A935P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, and Foldetta results are unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no conflict with ClinVar status. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.736850 | Disordered | 0.980490 | Binding | 0.286 | 0.865 | 0.625 | -3.239 | Likely Benign | 0.371 | Ambiguous | Likely Benign | 0.269 | Likely Benign | -2.08 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.2062 | 0.5466 | 1 | -1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3305C>T | A1102V 2D AIThe SynGAP1 missense variant A1102V is listed in ClinVar (ID 2846719.0) as Benign and is present in gnomAD (variant ID 6‑33443857‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion aligns with the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | Benign | 1 | 6-33443857-C-T | -2.440 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -1.27 | Neutral | 0.017 | Benign | 0.028 | Benign | 2.29 | Pathogenic | 0.12 | Tolerated | 3.77 | 5 | 0.1333 | 0.6264 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||
| c.3473T>A | V1158D 2D AIThe SynGAP1 missense variant V1158D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence indicates that V1158D is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.877504 | Binding | 0.369 | 0.847 | 0.250 | -4.076 | Likely Benign | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.486 | Likely Benign | -4.56 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.1502 | 0.1820 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3500A>G | D1167G 2D AIThe SynGAP1 missense variant D1167G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that D1167G is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.783999 | Binding | 0.336 | 0.798 | 0.500 | -2.967 | Likely Benign | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.246 | Likely Benign | -2.57 | Deleterious | 0.995 | Probably Damaging | 0.949 | Probably Damaging | 2.29 | Pathogenic | 0.01 | Affected | 0.4172 | 0.7305 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.3680A>C | E1227A 2D AIThe SynGAP1 missense variant E1227A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled “Likely Pathogenic.” The Foldetta protein‑folding stability analysis is unavailable for this variant. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.513880 | Disordered | 0.433399 | Uncertain | 0.860 | 0.544 | 0.500 | -9.111 | Likely Pathogenic | 0.961 | Likely Pathogenic | Likely Pathogenic | 0.341 | Likely Benign | -4.63 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.3142 | 0.6025 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||
| c.3779A>T | K1260M 2D AIThe SynGAP1 missense variant K1260M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the majority of algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—classify the substitution as pathogenic. High‑accuracy assessments further support a deleterious impact: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports “Likely Pathogenic”; AlphaMissense‑Optimized yields an uncertain result, and Foldetta’s stability prediction is unavailable. Taken together, the evidence overwhelmingly points to a pathogenic effect for K1260M. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.509769 | Disordered | 0.625808 | Binding | 0.890 | 0.575 | 0.250 | -10.938 | Likely Pathogenic | 0.887 | Likely Pathogenic | Ambiguous | 0.400 | Likely Benign | -4.72 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.0746 | 0.3366 | 0 | -1 | 5.8 | 3.02 | ||||||||||||||||||||||||||||||||||||||
| c.3794G>C | R1265T 2D  AIThe SynGAP1 missense variant R1265T is reported in ClinVar as Pathogenic (ClinVar ID 522047.0) and is not found in gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. No tool in the dataset predicts a benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenicity, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a Likely Pathogenic verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions supports a pathogenic classification, which is consistent with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.414856 | Structured | 0.782497 | Binding | 0.887 | 0.592 | 0.000 | Likely Pathogenic | 1 | -10.129 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.529 | Likely Pathogenic | -4.97 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1947 | 0.4618 | -1 | -1 | 3.8 | -55.08 | ||||||||||||||||||||||||||||||||||
| c.3883C>G | Q1295E 2D AIThe SynGAP1 missense variant Q1295E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.892719 | Binding | 0.499 | 0.801 | 0.625 | -3.192 | Likely Benign | 0.177 | Likely Benign | Likely Benign | 0.264 | Likely Benign | -2.46 | Neutral | 0.843 | Possibly Damaging | 0.848 | Possibly Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.1428 | 0.1492 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.919T>G | F307V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F307V is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect include Rosetta and premPS, whereas the majority of other in silico predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) and the consensus score SGM‑Consensus all classify the change as pathogenic or likely pathogenic. High‑accuracy methods give a pathogenic call from AlphaMissense‑Optimized, a likely pathogenic result from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and an inconclusive result from Foldetta (combining FoldX‑MD and Rosetta). No prediction is missing or inconclusive enough to alter the overall assessment. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.298791 | Structured | 0.327302 | Uncertain | 0.900 | 0.315 | 0.125 | -12.262 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.91 | Ambiguous | 0.1 | 0.10 | Likely Benign | 0.51 | Ambiguous | 0.36 | Likely Benign | 0.605 | Likely Pathogenic | -6.43 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.29 | Pathogenic | 0.05 | Affected | 0.2523 | 0.3393 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1415A>G | E472G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E472G is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that assess the variant’s effect largely agree on a deleterious outcome: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic or likely pathogenic. Only premPS predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -15.239 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 3.06 | Destabilizing | 0.2 | 2.86 | Destabilizing | 2.96 | Destabilizing | 0.24 | Likely Benign | 0.744 | Likely Pathogenic | -6.83 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.3343 | 0.4950 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.2163C>G | I721M 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant I721M is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, Rosetta, and PROVEAN, whereas pathogenic predictions are made by SGM‑Consensus, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Tools with inconclusive results are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of reliable predictors indicate a pathogenic effect, which aligns with the ClinVar designation of uncertain significance rather than contradicting it. Therefore, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | Uncertain | 1 | -9.767 | Likely Pathogenic | 0.872 | Likely Pathogenic | Ambiguous | 0.71 | Ambiguous | 0.0 | 0.45 | Likely Benign | 0.58 | Ambiguous | 1.00 | Destabilizing | 0.225 | Likely Benign | -2.40 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.0576 | 0.2726 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||
| c.2336G>A | S779N 2D AIThe SynGAP1 missense variant S779N is listed in gnomAD (ID 6‑33442494‑G‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. A high‑accuracy consensus (SGM Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a benign majority. The AlphaMissense‑Optimized score is benign, and no Foldetta stability assessment is available. Taken together, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | 6-33442494-G-A | -4.880 | Likely Benign | 0.384 | Ambiguous | Likely Benign | 0.087 | Likely Benign | -0.75 | Neutral | 0.021 | Benign | 0.026 | Benign | 2.30 | Pathogenic | 0.23 | Tolerated | 3.64 | 6 | 0.1327 | 0.4984 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||
| c.2540A>G | Q847R 2D AIThe SynGAP1 missense variant Q847R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Optimized; pathogenic calls come from PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further reveal AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Thus, the overall prediction leans toward benign based on the majority of tools, but the high‑accuracy SGM‑Consensus contradicts this by indicating likely pathogenic. No ClinVar annotation exists, so there is no conflict with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.653063 | Disordered | 0.577677 | Binding | 0.282 | 0.818 | 0.500 | -3.232 | Likely Benign | 0.662 | Likely Pathogenic | Likely Benign | 0.256 | Likely Benign | -2.63 | Deleterious | 0.014 | Benign | 0.026 | Benign | 2.30 | Pathogenic | 0.00 | Affected | 0.1404 | 0.1979 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.2795T>G | F932C 2D AIThe SynGAP1 missense variant F932C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts Pathogenic, and the SGM‑Consensus also indicates Likely Pathogenic. Foldetta results are unavailable. Overall, the preponderance of evidence from multiple in silico predictors and high‑accuracy tools indicates that the F932C variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.989197 | Binding | 0.293 | 0.858 | 0.500 | -8.601 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.309 | Likely Benign | -4.62 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.2446 | 0.1506 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||||||||||||
| c.2804C>T | A935V 2D AIThe SynGAP1 missense variant A935V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also leans benign (2 benign vs. 1 pathogenic votes). Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for A935V, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.736850 | Disordered | 0.980490 | Binding | 0.286 | 0.865 | 0.625 | -4.750 | Likely Benign | 0.397 | Ambiguous | Likely Benign | 0.194 | Likely Benign | -2.06 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.1340 | 0.5941 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3305C>G | A1102G 2D AIThe SynGAP1 A1102G missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The high‑accuracy consensus (SGM‑Consensus) classifies the variant as “Likely Benign,” and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | -3.070 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.060 | Likely Benign | 0.39 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.30 | Pathogenic | 0.14 | Tolerated | 0.1947 | 0.4958 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3449C>A | A1150D 2D AIThe SynGAP1 missense variant A1150D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated tools (five pathogenic vs. three benign) predict a pathogenic impact. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.762850 | Disordered | 0.795712 | Binding | 0.371 | 0.831 | 0.625 | -3.923 | Likely Benign | 0.859 | Likely Pathogenic | Ambiguous | 0.156 | Likely Benign | -2.30 | Neutral | 0.995 | Probably Damaging | 0.940 | Probably Damaging | 2.30 | Pathogenic | 0.01 | Affected | 0.1754 | 0.2143 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3473T>G | V1158G 2D AIThe SynGAP1 missense variant V1158G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic outcome. High‑accuracy assessments reinforce this pattern: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a pathogenic verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that V1158G is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.877504 | Binding | 0.369 | 0.847 | 0.250 | -3.058 | Likely Benign | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.433 | Likely Benign | -4.62 | Deleterious | 0.997 | Probably Damaging | 0.999 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.1870 | 0.3289 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3500A>C | D1167A 2D AIThe SynGAP1 missense variant D1167A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all indicate likely pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the preponderance of pathogenic predictions and the high‑accuracy tool outputs, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.783999 | Binding | 0.336 | 0.798 | 0.500 | -1.281 | Likely Benign | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.244 | Likely Benign | -3.11 | Deleterious | 0.986 | Probably Damaging | 0.926 | Probably Damaging | 2.30 | Pathogenic | 0.01 | Affected | 0.4156 | 0.7359 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.3654G>C | E1218D 2D AIThe SynGAP1 missense variant E1218D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, and no Foldetta stability data are available. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.595080 | Disordered | 0.483050 | Uncertain | 0.898 | 0.565 | 0.375 | -3.574 | Likely Benign | 0.517 | Ambiguous | Likely Benign | 0.138 | Likely Benign | -2.42 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.1485 | 0.2475 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3654G>T | E1218D 2D AIThe SynGAP1 missense variant E1218D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, and no Foldetta stability data are available. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.595080 | Disordered | 0.483050 | Uncertain | 0.898 | 0.565 | 0.375 | -3.574 | Likely Benign | 0.517 | Ambiguous | Likely Benign | 0.138 | Likely Benign | -2.42 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.1485 | 0.2475 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3679G>A | E1227K 2D AIThe SynGAP1 missense variant E1227K is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.513880 | Disordered | 0.433399 | Uncertain | 0.860 | 0.544 | 0.500 | -11.825 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.280 | Likely Benign | -2.94 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.1661 | 0.6348 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3679G>C | E1227Q 2D AIThe SynGAP1 missense variant E1227Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and PROVEAN, whereas a majority of tools predict a pathogenic impact: polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, while the SGM‑Consensus remains pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple in silico predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.513880 | Disordered | 0.433399 | Uncertain | 0.860 | 0.544 | 0.500 | -9.212 | Likely Pathogenic | 0.860 | Likely Pathogenic | Ambiguous | 0.277 | Likely Benign | -2.28 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.0761 | 0.6204 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||
| c.3713A>C | Q1238P 2D AIThe SynGAP1 missense variant Q1238P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic impact. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as pathogenic; Foldetta results are not available. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.562014 | Disordered | 0.548882 | Binding | 0.855 | 0.545 | 0.250 | -13.929 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.442 | Likely Benign | -4.06 | Deleterious | 0.998 | Probably Damaging | 0.995 | Probably Damaging | 2.30 | Pathogenic | 0.01 | Affected | 0.1549 | 0.3619 | 0 | -1 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||||||
| c.3735G>C | E1245D 2D AIThe SynGAP1 missense variant E1245D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas a majority of tools predict a pathogenic impact: polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and no consensus could be drawn from the SGM Consensus (a tie between pathogenic and benign votes from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the balance of evidence from the available predictors leans toward a pathogenic effect. This conclusion is consistent with the lack of ClinVar annotation, as there is no reported ClinVar status to contradict the prediction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.712013 | Disordered | 0.387847 | Uncertain | 0.869 | 0.554 | 0.625 | -6.075 | Likely Benign | 0.928 | Likely Pathogenic | Ambiguous | 0.157 | Likely Benign | -2.46 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.1585 | 0.3097 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3735G>T | E1245D 2D AIThe SynGAP1 missense variant E1245D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas a majority of tools predict a pathogenic impact: polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and no consensus could be drawn from the SGM Consensus (a tie between pathogenic and benign votes from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the balance of evidence from the available predictors leans toward a pathogenic effect. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.712013 | Disordered | 0.387847 | Uncertain | 0.869 | 0.554 | 0.625 | -6.075 | Likely Benign | 0.928 | Likely Pathogenic | Ambiguous | 0.157 | Likely Benign | -2.46 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.1585 | 0.3097 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3800T>G | M1267R 2D AIThe SynGAP1 missense variant M1267R is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. Grouping by consensus, two tools predict benign and six predict pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic impact, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.429200 | Structured | 0.812047 | Binding | 0.847 | 0.614 | 0.000 | -4.990 | Likely Benign | 0.899 | Likely Pathogenic | Ambiguous | 0.366 | Likely Benign | -5.03 | Deleterious | 0.982 | Probably Damaging | 0.757 | Possibly Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.1555 | 0.0837 | 0 | -1 | -6.4 | 24.99 | ||||||||||||||||||||||||||||||||||||||
| c.2408A>T | K803I 2D AIThe SynGAP1 K803I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. The high‑accuracy AlphaMissense‑Optimized assessment is uncertain, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently contains no classification for K803I. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.827927 | Disordered | 0.733908 | Binding | 0.349 | 0.900 | 0.625 | -5.207 | Likely Benign | 0.894 | Likely Pathogenic | Ambiguous | 0.196 | Likely Benign | -4.06 | Deleterious | 0.995 | Probably Damaging | 0.913 | Probably Damaging | 2.31 | Pathogenic | 0.00 | Affected | 0.1425 | 0.3889 | -2 | -3 | 8.4 | -15.01 | ||||||||||||||||||||||||||||||||||||||
| c.2423T>C | V808A 2D AIThe SynGAP1 missense variant V808A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign verdict, while Foldetta results are unavailable. Taken together, the majority of evidence points to a benign impact, and this conclusion does not contradict the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.699094 | Disordered | 0.856438 | Binding | 0.289 | 0.903 | 0.500 | -5.091 | Likely Benign | 0.541 | Ambiguous | Likely Benign | 0.177 | Likely Benign | -1.96 | Neutral | 0.953 | Possibly Damaging | 0.621 | Possibly Damaging | 2.31 | Pathogenic | 0.00 | Affected | 0.2748 | 0.2809 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2539C>G | Q847E 2D AISynGAP1 missense variant Q847E is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The remaining tools, ESM1b and AlphaMissense‑Default, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is unavailable due to no majority, and Foldetta stability analysis is also unavailable. Overall, the predictions are split, providing no clear bias toward benign or pathogenic. Thus the variant is most likely of uncertain significance, which does not contradict its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.653063 | Disordered | 0.577677 | Binding | 0.282 | 0.818 | 0.500 | Uncertain | 1 | -7.864 | In-Between | 0.377 | Ambiguous | Likely Benign | 0.140 | Likely Benign | -2.12 | Neutral | 0.649 | Possibly Damaging | 0.535 | Possibly Damaging | 2.31 | Pathogenic | 0.00 | Affected | 0.1376 | 0.2083 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||||||||||
| c.2795T>C | F932S 2D AIThe SynGAP1 missense variant F932S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. In contrast, the majority of tools predict a pathogenic effect: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. ESM1b is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus also indicates Likely Pathogenic. Foldetta results are unavailable. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools indicates that F932S is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.989197 | Binding | 0.293 | 0.858 | 0.500 | -7.196 | In-Between | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.260 | Likely Benign | -4.45 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.31 | Pathogenic | 0.00 | Affected | 0.4257 | 0.0584 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||||||||||||
| c.2804C>A | A935D 2D AIThe SynGAP1 missense variant A935D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to a pathogenic impact for A935D, and this conclusion does not conflict with the current ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.736850 | Disordered | 0.980490 | Binding | 0.286 | 0.865 | 0.625 | -4.089 | Likely Benign | 0.817 | Likely Pathogenic | Ambiguous | 0.247 | Likely Benign | -2.69 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.31 | Pathogenic | 0.00 | Affected | 0.1832 | 0.1860 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.3328A>C | S1110R 2D AIThe SynGAP1 missense variant S1110R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the balance of evidence (six benign versus three pathogenic predictions) indicates that the variant is most likely benign. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -5.075 | Likely Benign | 0.773 | Likely Pathogenic | Likely Benign | 0.065 | Likely Benign | -2.46 | Neutral | 0.144 | Benign | 0.042 | Benign | 2.31 | Pathogenic | 0.01 | Affected | 0.1057 | 0.3571 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3330C>A | S1110R 2D AIThe SynGAP1 missense variant S1110R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs. three pathogenic predictions) points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -5.075 | Likely Benign | 0.773 | Likely Pathogenic | Likely Benign | 0.043 | Likely Benign | -2.46 | Neutral | 0.144 | Benign | 0.042 | Benign | 2.31 | Pathogenic | 0.01 | Affected | 0.1057 | 0.3571 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3330C>G | S1110R 2D AIThe SynGAP1 missense variant S1110R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs. three pathogenic predictions) points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | -5.075 | Likely Benign | 0.773 | Likely Pathogenic | Likely Benign | 0.043 | Likely Benign | -2.46 | Neutral | 0.144 | Benign | 0.042 | Benign | 2.31 | Pathogenic | 0.01 | Affected | 0.1057 | 0.3571 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.3713A>T | Q1238L 2D AIThe SynGAP1 missense variant Q1238L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—classify the variant as pathogenic. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments show the SGM‑Consensus as “Likely Pathogenic,” while AlphaMissense‑Optimized remains uncertain and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.562014 | Disordered | 0.548882 | Binding | 0.855 | 0.545 | 0.250 | -14.299 | Likely Pathogenic | 0.876 | Likely Pathogenic | Ambiguous | 0.353 | Likely Benign | -4.89 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.31 | Pathogenic | 0.01 | Affected | 0.0543 | 0.3744 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3714G>C | Q1238H 2D AIThe SynGAP1 missense variant Q1238H is reported in gnomAD (variant ID 6‑33446706‑G‑C) but has no ClinVar entry. Functional prediction tools split into two groups: benign predictions come from REVEL and AlphaMissense‑Optimized, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar classification (none exists). Thus, based on current predictions, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.562014 | Disordered | 0.548882 | Binding | 0.855 | 0.545 | 0.250 | 6-33446706-G-C | 1 | 6.20e-7 | -8.647 | Likely Pathogenic | 0.757 | Likely Pathogenic | Likely Benign | 0.202 | Likely Benign | -3.49 | Deleterious | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 2.31 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1007 | 0.3004 | 0 | 3 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||
| c.3714G>T | Q1238H 2D AIThe SynGAP1 missense variant Q1238H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) all indicate a pathogenic impact. High‑accuracy assessments further support this dichotomy: AlphaMissense‑Optimized classifies the variant as benign, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it as Likely Pathogenic. No Foldetta stability prediction is available. Overall, the preponderance of evidence from multiple independent tools points to a pathogenic effect for Q1238H. This conclusion is not contradicted by ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.562014 | Disordered | 0.548882 | Binding | 0.855 | 0.545 | 0.250 | -8.647 | Likely Pathogenic | 0.757 | Likely Pathogenic | Likely Benign | 0.202 | Likely Benign | -3.49 | Deleterious | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 2.31 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1007 | 0.3004 | 0 | 3 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||||||||
| c.3761A>T | E1254V 2D AIThe SynGAP1 missense variant E1254V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: REVEL scores the variant as benign, whereas the remaining seven tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict it to be pathogenic. Grouping by consensus, the benign prediction is represented only by REVEL, while the pathogenic predictions are supported by the majority of in silico methods. High‑accuracy assessments further reinforce a pathogenic interpretation: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic, which is consistent with the lack of ClinVar annotation and gnomAD absence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.657645 | Disordered | 0.403242 | Uncertain | 0.886 | 0.555 | 0.625 | -9.913 | Likely Pathogenic | 0.814 | Likely Pathogenic | Ambiguous | 0.350 | Likely Benign | -5.15 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.31 | Pathogenic | 0.00 | Affected | 0.0481 | 0.5894 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3800T>A | M1267K 2D AIThe SynGAP1 missense variant M1267K is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a pathogenic outcome (3/4 pathogenic). AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. Overall, the majority of evidence points to a deleterious effect, classifying the variant as most likely pathogenic. This assessment does not conflict with ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.429200 | Structured | 0.812047 | Binding | 0.847 | 0.614 | 0.000 | -6.415 | Likely Benign | 0.917 | Likely Pathogenic | Ambiguous | 0.366 | Likely Benign | -5.01 | Deleterious | 0.884 | Possibly Damaging | 0.581 | Possibly Damaging | 2.31 | Pathogenic | 0.00 | Affected | 0.1373 | 0.0488 | 0 | -1 | -5.8 | -3.02 | ||||||||||||||||||||||||||||||||||||||
| c.3922C>T | R1308C 2D AIThe SynGAP1 missense variant R1308C is listed in ClinVar with an “Uncertain” significance and is present in the gnomAD database (ID 6‑33451796‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. The high‑accuracy consensus (SGM Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a pathogenic verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this assessment does not contradict the current ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.741537 | Disordered | 0.930652 | Binding | 0.378 | 0.904 | 0.750 | Conflicting | 2 | 6-33451796-C-T | 4 | 2.48e-6 | -4.994 | Likely Benign | 0.421 | Ambiguous | Likely Benign | 0.352 | Likely Benign | -4.89 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 2.31 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3139 | 0.4274 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.1415A>C | E472A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E472A is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; all these methods uniformly classify the change as deleterious. Tools that are inconclusive or uncertain for this variant are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts a pathogenic outcome, the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates pathogenicity, while Foldetta’s stability analysis remains uncertain. Taken together, the overwhelming majority of evidence points to a pathogenic effect for E472A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -15.356 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 1.81 | Ambiguous | 0.3 | 0.67 | Ambiguous | 1.24 | Ambiguous | 0.69 | Ambiguous | 0.732 | Likely Pathogenic | -5.90 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.32 | Pathogenic | 0.01 | Affected | 0.4639 | 0.6315 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.2417T>A | F806Y 2D AIThe SynGAP1 missense variant F806Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also classifies it as benign, and Foldetta results are unavailable. Taken together, the majority of evidence points to a benign effect, and this conclusion does not contradict the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | -5.229 | Likely Benign | 0.404 | Ambiguous | Likely Benign | 0.162 | Likely Benign | -1.51 | Neutral | 0.997 | Probably Damaging | 0.987 | Probably Damaging | 2.32 | Pathogenic | 0.03 | Affected | 0.1746 | 0.1437 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2428C>T | R810C 2D AIThe SynGAP1 missense variant R810C is listed in gnomAD (6‑33442980‑C‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.486429 | Structured | 0.851848 | Binding | 0.263 | 0.907 | 0.375 | 6-33442980-C-T | 2 | 1.24e-6 | -8.925 | Likely Pathogenic | 0.839 | Likely Pathogenic | Ambiguous | 0.245 | Likely Benign | -4.91 | Deleterious | 1.000 | Probably Damaging | 0.991 | Probably Damaging | 2.32 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3517 | 0.4263 | -3 | -4 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.2492A>T | E831V 2D AIThe SynGAP1 missense variant E831V is not reported in ClinVar and has no gnomAD allele. Prediction tools show a split: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts a benign effect, SGM‑Consensus indicates a likely pathogenic outcome, and Foldetta data are unavailable. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.626927 | Disordered | 0.617732 | Binding | 0.319 | 0.874 | 0.375 | -6.327 | Likely Benign | 0.629 | Likely Pathogenic | Likely Benign | 0.204 | Likely Benign | -3.43 | Deleterious | 0.891 | Possibly Damaging | 0.492 | Possibly Damaging | 2.32 | Pathogenic | 0.02 | Affected | 0.0604 | 0.7407 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2539C>A | Q847K 2D AIThe SynGAP1 missense variant Q847K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign calls come from REVEL, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic calls come from PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a Likely Pathogenic verdict. High‑accuracy assessments further indicate that AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus remains Likely Pathogenic; no Foldetta stability data are available. Overall, the majority of predictions lean toward pathogenicity, and this is consistent with the SGM Consensus result. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar annotation exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.653063 | Disordered | 0.577677 | Binding | 0.282 | 0.818 | 0.500 | -5.507 | Likely Benign | 0.736 | Likely Pathogenic | Likely Benign | 0.214 | Likely Benign | -2.82 | Deleterious | 0.481 | Possibly Damaging | 0.373 | Benign | 2.32 | Pathogenic | 0.00 | Affected | 0.1694 | 0.4129 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.2707G>T | G903C 2D AIThe SynGAP1 missense variant G903C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain and does not influence the consensus. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, which would provide a protein‑folding stability estimate, is unavailable for this variant. Overall, the majority of evidence (five pathogenic versus three benign predictions) points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.680603 | Disordered | 0.549818 | Binding | 0.291 | 0.917 | 0.375 | -6.593 | Likely Benign | 0.471 | Ambiguous | Likely Benign | 0.151 | Likely Benign | -3.28 | Deleterious | 1.000 | Probably Damaging | 0.990 | Probably Damaging | 2.32 | Pathogenic | 0.02 | Affected | 0.1158 | 0.4420 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2794T>A | F932I 2D AIThe SynGAP1 missense variant F932I is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as Likely Pathogenic because three of the four constituent tools predict pathogenicity. High‑accuracy assessments further support this: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (majority vote) is also pathogenic; Foldetta results are unavailable. Consequently, the collective evidence points to a pathogenic effect for F932I. This conclusion is consistent with the absence of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.989197 | Binding | 0.293 | 0.858 | 0.500 | -4.963 | Likely Benign | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.348 | Likely Benign | -3.45 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 2.32 | Pathogenic | 0.01 | Affected | 0.2202 | 0.2914 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2794T>G | F932V 2D AIThe SynGAP1 missense variant F932V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic outcome. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely pathogenic status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.989197 | Binding | 0.293 | 0.858 | 0.500 | -5.728 | Likely Benign | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.392 | Likely Benign | -3.68 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.32 | Pathogenic | 0.01 | Affected | 0.2137 | 0.3082 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2804C>G | A935G 2D AIThe SynGAP1 missense variant A935G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.980490 | Binding | 0.286 | 0.865 | 0.625 | -2.868 | Likely Benign | 0.179 | Likely Benign | Likely Benign | 0.152 | Likely Benign | -1.97 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.2318 | 0.4679 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2848G>A | G950S 2D AIThe SynGAP1 missense variant G950S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -5.286 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.288 | Likely Benign | 0.46 | Neutral | 0.004 | Benign | 0.008 | Benign | 2.32 | Pathogenic | 0.52 | Tolerated | 0.2459 | 0.5502 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2864C>A | S955Y 2D AIThe SynGAP1 missense variant S955Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) support a benign classification. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.945325 | Binding | 0.350 | 0.924 | 0.750 | -6.212 | Likely Benign | 0.216 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -1.62 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.1321 | 0.4932 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||||
| c.2864C>G | S955C 2D AIThe SynGAP1 missense variant S955C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (5 pathogenic vs. 4 benign) indicate a likely pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant has no existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984871 | Disordered | 0.945325 | Binding | 0.350 | 0.924 | 0.750 | -8.675 | Likely Pathogenic | 0.117 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -1.48 | Neutral | 0.977 | Probably Damaging | 0.796 | Possibly Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.1741 | 0.5470 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.2864C>T | S955F 2D AISynGAP1 missense variant S955F is listed in ClinVar as uncertain and is present in gnomAD (ID 6‑33443416‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM; ESM1b is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also returns benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence predictions favor a benign impact, and this does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984871 | Disordered | 0.945325 | Binding | 0.350 | 0.924 | 0.750 | Conflicting | 4 | 6-33443416-C-T | 95 | 5.89e-5 | -7.374 | In-Between | 0.176 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -1.73 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 2.32 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1246 | 0.4943 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||
| c.2958G>C | E986D 2D AIThe SynGAP1 missense variant E986D is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs two benign votes) and Foldetta results are unavailable. Overall, the majority of conventional tools lean toward pathogenicity, but the single high‑accuracy benign prediction and the inconclusive consensus leave the variant’s impact uncertain. There is no contradiction with ClinVar status, as the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.001 | Likely Benign | 0.628 | Likely Pathogenic | Likely Benign | 0.129 | Likely Benign | -1.22 | Neutral | 0.974 | Probably Damaging | 0.715 | Possibly Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.2081 | 0.5364 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2958G>T | E986D 2D AIThe SynGAP1 missense variant E986D is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs two benign votes) and Foldetta results are unavailable. Overall, the majority of conventional tools lean toward pathogenicity, but the single high‑accuracy benign prediction and the inconclusive consensus leave the variant’s impact uncertain. There is no contradiction with ClinVar status, as the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.001 | Likely Benign | 0.628 | Likely Pathogenic | Likely Benign | 0.129 | Likely Benign | -1.22 | Neutral | 0.974 | Probably Damaging | 0.715 | Possibly Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.2081 | 0.5364 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3304G>A | A1102T 2D AIThe SynGAP1 missense variant A1102T is listed in ClinVar (ID 1422927) as Benign and is present in gnomAD (6‑33443856‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence supports a benign classification, which is consistent with the ClinVar status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.915074 | Disordered | 0.962659 | Binding | 0.388 | 0.859 | 0.875 | Benign | 1 | 6-33443856-G-A | 11 | 7.17e-6 | -3.540 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.30 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.32 | Pathogenic | 0.95 | Tolerated | 3.77 | 5 | 0.1755 | 0.7699 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||
| c.3449C>G | A1150G 2D AIThe SynGAP1 A1150G missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.795712 | Binding | 0.371 | 0.831 | 0.625 | -3.089 | Likely Benign | 0.253 | Likely Benign | Likely Benign | 0.158 | Likely Benign | -2.37 | Neutral | 0.983 | Probably Damaging | 0.818 | Possibly Damaging | 2.32 | Pathogenic | 0.09 | Tolerated | 0.2263 | 0.4741 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3449C>T | A1150V 2D AIThe SynGAP1 missense variant A1150V is listed in ClinVar (ID 589625.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33444484‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are SIFT and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also benign. Foldetta, a protein‑folding stability method, did not provide a result for this variant. Overall, the majority of computational evidence indicates that A1150V is most likely benign, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.795712 | Binding | 0.371 | 0.831 | 0.625 | Uncertain | 1 | 6-33444484-C-T | 3 | 1.86e-6 | -3.648 | Likely Benign | 0.192 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -2.22 | Neutral | 0.114 | Benign | 0.055 | Benign | 2.32 | Pathogenic | 0.04 | Affected | 3.77 | 5 | 0.1242 | 0.6335 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||
| c.3652G>C | E1218Q 2D AIThe SynGAP1 E1218Q missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, and ESM1b, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments are less decisive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie and thus inconclusive; Foldetta results are not available. Consequently, the majority of conventional predictors lean toward pathogenicity, but the most reliable tools do not provide a clear verdict. The variant is therefore most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.595080 | Disordered | 0.483050 | Uncertain | 0.898 | 0.565 | 0.375 | -6.490 | Likely Benign | 0.857 | Likely Pathogenic | Ambiguous | 0.226 | Likely Benign | -2.09 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.0784 | 0.3666 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3695A>G | K1232R 2D AIThe SynGAP1 missense variant K1232R is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign impact. This conclusion is consistent with the absence of ClinVar annotation and gnomAD data, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.505461 | Disordered | 0.542907 | Binding | 0.894 | 0.535 | 0.125 | -3.146 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -1.83 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.3515 | 0.0945 | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||||||
| c.3715G>C | A1239P 2D AIThe SynGAP1 missense variant A1239P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL and polyPhen‑2 HumVar, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that A1239P is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.541878 | Disordered | 0.534779 | Binding | 0.887 | 0.542 | 0.250 | -11.055 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.085 | Likely Benign | -2.62 | Deleterious | 0.784 | Possibly Damaging | 0.390 | Benign | 2.32 | Pathogenic | 0.00 | Affected | 0.1438 | 0.3450 | 1 | -1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||||||||||
| c.3733G>C | E1245Q 2D AIThe SynGAP1 missense change E1245Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and PROVEAN, whereas pathogenic predictions are made by polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to a likely pathogenic verdict (3 pathogenic vs. 1 benign). High‑accuracy assessments further support this: AlphaMissense‑Optimized is uncertain, SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is not available. Consequently, the majority of evidence points to a pathogenic effect. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.712013 | Disordered | 0.387847 | Uncertain | 0.869 | 0.554 | 0.625 | -11.323 | Likely Pathogenic | 0.804 | Likely Pathogenic | Ambiguous | 0.210 | Likely Benign | -2.29 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.0819 | 0.5952 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||
| c.3781A>G | S1261G 2D AIThe SynGAP1 missense variant S1261G is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign; Foldetta results are not available. Overall, the majority of evidence supports a benign impact for S1261G, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.501700 | Disordered | 0.671500 | Binding | 0.889 | 0.574 | 0.250 | -5.042 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.46 | Neutral | 0.005 | Benign | 0.013 | Benign | 2.32 | Pathogenic | 0.65 | Tolerated | 0.2063 | 0.3477 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||||||
| c.3800T>C | M1267T 2D AIThe SynGAP1 missense variant M1267T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as likely pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (majority vote) likewise indicates likely pathogenicity. No Foldetta stability prediction is available for this variant. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.429200 | Structured | 0.812047 | Binding | 0.847 | 0.614 | 0.000 | -5.315 | Likely Benign | 0.958 | Likely Pathogenic | Likely Pathogenic | 0.270 | Likely Benign | -5.00 | Deleterious | 0.982 | Probably Damaging | 0.672 | Possibly Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.2036 | 0.1934 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||||||
| c.3923G>C | R1308P 2D AIThe SynGAP1 missense variant R1308P is not reported in ClinVar but is present in gnomAD (ID 6‑33451797‑G‑C). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is a 2‑vs‑2 split and therefore inconclusive. Foldetta, a protein‑folding stability method that combines FoldX‑MD and Rosetta outputs, has no available result for this variant. Because the available predictions are evenly divided between benign and pathogenic, the overall computational assessment is inconclusive; the variant is neither clearly benign nor clearly pathogenic. This lack of consensus does not contradict ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.741537 | Disordered | 0.930652 | Binding | 0.378 | 0.904 | 0.750 | 6-33451797-G-C | -1.320 | Likely Benign | 0.216 | Likely Benign | Likely Benign | 0.158 | Likely Benign | -4.62 | Deleterious | 0.995 | Probably Damaging | 0.992 | Probably Damaging | 2.32 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2070 | 0.5020 | -2 | 0 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||
| c.1414G>A | E472K 2D AIThe SynGAP1 missense variant E472K is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect; the only inconclusive result is from premPS, which is listed as uncertain. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a destabilizing, pathogenic effect. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -15.214 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.01 | Destabilizing | 1.2 | 3.23 | Destabilizing | 2.62 | Destabilizing | 0.78 | Ambiguous | 0.670 | Likely Pathogenic | -3.95 | Deleterious | 0.996 | Probably Damaging | 0.987 | Probably Damaging | 2.33 | Pathogenic | 0.03 | Affected | 0.3077 | 0.5651 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.2422G>A | V808I 2D AIThe SynGAP1 missense variant V808I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenic predictions come from polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign and the SGM‑Consensus (majority vote) is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.699094 | Disordered | 0.856438 | Binding | 0.289 | 0.903 | 0.500 | -4.190 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -0.67 | Neutral | 0.659 | Possibly Damaging | 0.191 | Benign | 2.33 | Pathogenic | 0.00 | Affected | 0.0935 | 0.4282 | 4 | 3 | 0.3 | 14.03 | ||||||||||||||||||||||||||||||||||||||
| c.2422G>C | V808L 2D AIThe SynGAP1 missense variant V808L is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact for V808L, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.699094 | Disordered | 0.856438 | Binding | 0.289 | 0.903 | 0.500 | -4.723 | Likely Benign | 0.306 | Likely Benign | Likely Benign | 0.188 | Likely Benign | -1.82 | Neutral | 0.911 | Possibly Damaging | 0.621 | Possibly Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.1166 | 0.4745 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||||||||||
| c.2422G>T | V808L 2D AIThe SynGAP1 missense variant V808L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.699094 | Disordered | 0.856438 | Binding | 0.289 | 0.903 | 0.500 | -4.723 | Likely Benign | 0.306 | Likely Benign | Likely Benign | 0.188 | Likely Benign | -1.82 | Neutral | 0.911 | Possibly Damaging | 0.621 | Possibly Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.1166 | 0.4745 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||||||||||
| c.2429G>C | R810P 2D AIThe SynGAP1 missense variant R810P is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a pathogenic bias: AlphaMissense‑Optimized indicates a benign effect, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic impact for R810P. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.486429 | Structured | 0.851848 | Binding | 0.263 | 0.907 | 0.375 | -4.120 | Likely Benign | 0.779 | Likely Pathogenic | Likely Benign | 0.316 | Likely Benign | -4.08 | Deleterious | 1.000 | Probably Damaging | 0.977 | Probably Damaging | 2.33 | Pathogenic | 0.01 | Affected | 0.2307 | 0.5478 | 0 | -2 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||||||||
| c.2455G>T | A819S 2D AIThe SynGAP1 missense variant A819S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for A819S, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.694846 | Disordered | 0.707644 | Binding | 0.317 | 0.892 | 0.625 | -3.420 | Likely Benign | 0.331 | Likely Benign | Likely Benign | 0.199 | Likely Benign | -1.49 | Neutral | 0.994 | Probably Damaging | 0.900 | Possibly Damaging | 2.33 | Pathogenic | 0.27 | Tolerated | 0.2729 | 0.6404 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2540A>T | Q847L 2D AIThe SynGAP1 missense variant Q847L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default—predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Pathogenic, and the Foldetta stability analysis is unavailable. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict any existing ClinVar annotation, as none is available. Thus, the variant is most likely pathogenic based on the current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.653063 | Disordered | 0.577677 | Binding | 0.282 | 0.818 | 0.500 | -6.966 | Likely Benign | 0.625 | Likely Pathogenic | Likely Benign | 0.326 | Likely Benign | -4.52 | Deleterious | 0.818 | Possibly Damaging | 0.637 | Possibly Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.0701 | 0.5132 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2707G>C | G903R 2D AIThe SynGAP1 missense variant G903R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of standard predictors lean toward a benign impact, and no ClinVar annotation contradicts this assessment. Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.680603 | Disordered | 0.549818 | Binding | 0.291 | 0.917 | 0.375 | -3.503 | Likely Benign | 0.865 | Likely Pathogenic | Ambiguous | 0.119 | Likely Benign | -2.02 | Neutral | 0.241 | Benign | 0.244 | Benign | 2.33 | Pathogenic | 0.02 | Affected | 0.0872 | 0.4064 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.2708G>A | G903D 2D AIThe SynGAP1 missense variant G903D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward a pathogenic interpretation. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.680603 | Disordered | 0.549818 | Binding | 0.291 | 0.917 | 0.375 | -4.481 | Likely Benign | 0.849 | Likely Pathogenic | Ambiguous | 0.137 | Likely Benign | -1.85 | Neutral | 0.997 | Probably Damaging | 0.939 | Probably Damaging | 2.33 | Pathogenic | 0.01 | Affected | 0.1535 | 0.1321 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2803G>A | A935T 2D AIThe SynGAP1 missense variant A935T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.980490 | Binding | 0.286 | 0.865 | 0.625 | -4.247 | Likely Benign | 0.228 | Likely Benign | Likely Benign | 0.204 | Likely Benign | -1.27 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.1670 | 0.6599 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2863T>C | S955P 2D AIThe SynGAP1 missense variant S955P is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443415‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are SIFT and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.945325 | Binding | 0.350 | 0.924 | 0.750 | Uncertain | 1 | 6-33443415-T-C | 3 | 1.86e-6 | -2.584 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.098 | Likely Benign | -0.75 | Neutral | 0.001 | Benign | 0.004 | Benign | 2.33 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2629 | 0.5537 | 1 | -1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||
| c.2959G>T | D987Y 2D AIThe SynGAP1 missense variant D987Y is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as likely pathogenic (3 pathogenic vs 1 benign). AlphaMissense‑Optimized returns an uncertain result, and Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -6.208 | Likely Benign | 0.831 | Likely Pathogenic | Ambiguous | 0.274 | Likely Benign | -4.41 | Deleterious | 0.999 | Probably Damaging | 0.966 | Probably Damaging | 2.33 | Pathogenic | 0.01 | Affected | 0.0632 | 0.6818 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.3137C>A | P1046H 2D AIThe SynGAP1 missense variant P1046H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for P1046H. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | -5.715 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -1.64 | Neutral | 0.832 | Possibly Damaging | 0.670 | Possibly Damaging | 2.33 | Pathogenic | 0.07 | Tolerated | 0.1585 | 0.5250 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3455A>T | E1152V 2D AIThe SynGAP1 missense variant E1152V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.741537 | Disordered | 0.811118 | Binding | 0.395 | 0.846 | 0.500 | -3.304 | Likely Benign | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.408 | Likely Benign | -4.65 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.1247 | 0.6384 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3472G>T | V1158F 2D  AIThe SynGAP1 missense variant V1158F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, while the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is classified as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. Foldetta results are unavailable. Overall, the majority of evidence indicates that V1158F is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar annotation exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.877504 | Binding | 0.369 | 0.847 | 0.250 | -3.888 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.264 | Likely Benign | -2.66 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.33 | Pathogenic | 0.02 | Affected | 0.0792 | 0.3577 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.3499G>A | D1167N 2D AIThe SynGAP1 missense variant D1167N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated predictors (five pathogenic vs. three benign) lean toward a pathogenic interpretation. This prediction does not contradict any ClinVar status, as the variant is not yet catalogued in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.783999 | Binding | 0.336 | 0.798 | 0.500 | -3.671 | Likely Benign | 0.924 | Likely Pathogenic | Ambiguous | 0.180 | Likely Benign | -1.72 | Neutral | 0.995 | Probably Damaging | 0.963 | Probably Damaging | 2.33 | Pathogenic | 0.01 | Affected | 0.1315 | 0.7940 | 2 | 1 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3650A>T | E1217V 2D AIThe SynGAP1 missense variant E1217V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: REVEL scores the variant as benign, whereas PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenicity. Grouping by consensus, the majority of tools (seven) support a pathogenic effect, while only one tool (REVEL) indicates benign. High‑accuracy assessments further reinforce a deleterious interpretation: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic, and AlphaMissense‑Optimized remains uncertain. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the computational evidence overwhelmingly suggests that E1217V is pathogenic, a finding that aligns with its lack of ClinVar annotation and gnomAD presence. Thus, the variant is most likely pathogenic, and this prediction is consistent with its absence from ClinVar and gnomAD. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.599170 | Disordered | 0.493043 | Uncertain | 0.877 | 0.563 | 0.250 | -12.098 | Likely Pathogenic | 0.843 | Likely Pathogenic | Ambiguous | 0.351 | Likely Benign | -5.48 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.0579 | 0.5348 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3779A>C | K1260T 2D AIThe SynGAP1 missense variant K1260T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (which is “Likely Pathogenic” based on a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Benign predictions are limited to REVEL and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic. No Foldetta stability analysis is available for this variant. Overall, the preponderance of evidence from multiple in‑silico tools indicates that K1260T is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.509769 | Disordered | 0.625808 | Binding | 0.890 | 0.575 | 0.250 | -10.099 | Likely Pathogenic | 0.772 | Likely Pathogenic | Likely Benign | 0.387 | Likely Benign | -4.59 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.1859 | 0.3028 | 0 | -1 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||||||||||
| c.3801G>A | M1267I 2D AIThe SynGAP1 missense variant M1267I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for M1267I. This conclusion is not contradicted by ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.429200 | Structured | 0.812047 | Binding | 0.847 | 0.614 | 0.000 | -1.432 | Likely Benign | 0.936 | Likely Pathogenic | Ambiguous | 0.205 | Likely Benign | -3.33 | Deleterious | 0.959 | Probably Damaging | 0.672 | Possibly Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.1254 | 0.2741 | 2 | 1 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||||||
| c.3801G>C | M1267I 2D AIThe SynGAP1 missense variant M1267I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) remains Likely Pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for M1267I. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.429200 | Structured | 0.812047 | Binding | 0.847 | 0.614 | 0.000 | -1.432 | Likely Benign | 0.936 | Likely Pathogenic | Ambiguous | 0.205 | Likely Benign | -3.33 | Deleterious | 0.959 | Probably Damaging | 0.672 | Possibly Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.1254 | 0.2741 | 2 | 1 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||||||
| c.3801G>T | M1267I 2D AIThe SynGAP1 missense variant M1267I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) remains Likely Pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for M1267I. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.429200 | Structured | 0.812047 | Binding | 0.847 | 0.614 | 0.000 | -1.432 | Likely Benign | 0.936 | Likely Pathogenic | Ambiguous | 0.205 | Likely Benign | -3.33 | Deleterious | 0.959 | Probably Damaging | 0.672 | Possibly Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.1254 | 0.2741 | 2 | 1 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||||||
| c.3877G>A | D1293N 2D AIThe SynGAP1 missense variant D1293N is reported in gnomAD (6‑33447925‑G‑A) but has no ClinVar entry. Functional prediction tools split in a 5‑to‑4 ratio: benign calls come from REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic calls come from PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive and Foldetta results are unavailable. Overall, the majority of evidence points toward a benign effect, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | 6-33447925-G-A | 5 | 3.22e-6 | -3.983 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -3.30 | Deleterious | 0.950 | Possibly Damaging | 0.414 | Benign | 2.33 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1264 | 0.3421 | 1 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||
| c.3922C>G | R1308G 2D AIThe SynGAP1 missense variant R1308G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic interpretation. This assessment does not contradict any ClinVar status, as the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.741537 | Disordered | 0.930652 | Binding | 0.378 | 0.904 | 0.750 | -2.503 | Likely Benign | 0.180 | Likely Benign | Likely Benign | 0.300 | Likely Benign | -4.34 | Deleterious | 0.982 | Probably Damaging | 0.982 | Probably Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.3528 | 0.3830 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.3923G>A | R1308H 2D AISynGAP1 missense variant R1308H is listed in ClinVar as benign and is present in gnomAD (ID 6‑33451797‑G‑A). Functional prediction tools show mixed results: benign calls come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments give a benign result from AlphaMissense‑Optimized; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic), and Foldetta stability analysis is unavailable. Overall, the majority of standard predictors favor a pathogenic effect, whereas the most accurate single‑tool prediction is benign. Thus, the variant is most likely pathogenic according to the aggregate predictions, which contradicts the benign classification in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.741537 | Disordered | 0.930652 | Binding | 0.378 | 0.904 | 0.750 | Likely Benign | 1 | 6-33451797-G-A | 3 | 1.86e-6 | -3.586 | Likely Benign | 0.201 | Likely Benign | Likely Benign | 0.319 | Likely Benign | -3.12 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 2.33 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2955 | 0.2515 | 2 | 0 | 1.3 | -19.05 | |||||||||||||||||||||||||||||||||
| c.1883A>T | K628M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K628M missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX and premPS, whereas the majority of tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) yields an inconclusive result, which is treated as unavailable evidence. Overall, the preponderance of predictions points to a pathogenic effect for K628M, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | -14.949 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | -0.41 | Likely Benign | 0.2 | -0.76 | Ambiguous | -0.59 | Ambiguous | 0.37 | Likely Benign | 0.669 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.0802 | 0.3875 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.2336G>C | S779T 2D AIThe SynGAP1 missense variant S779T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as tolerated or benign. Only two tools—polyPhen‑2 HumDiv and FATHMM—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta results are not available for this variant. Overall, the consensus of available predictions points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.458 | Likely Benign | 0.144 | Likely Benign | Likely Benign | 0.132 | Likely Benign | -0.71 | Neutral | 0.611 | Possibly Damaging | 0.396 | Benign | 2.34 | Pathogenic | 0.79 | Tolerated | 0.1491 | 0.6392 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2353C>A | R785S 2D AIThe SynGAP1 missense variant R785S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. The high‑accuracy consensus from SGM (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors pathogenicity, while the lack of a Foldetta result leaves that evidence inconclusive. Overall, the preponderance of pathogenic predictions indicates that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | -2.926 | Likely Benign | 0.886 | Likely Pathogenic | Ambiguous | 0.157 | Likely Benign | -2.93 | Deleterious | 0.980 | Probably Damaging | 0.765 | Possibly Damaging | 2.34 | Pathogenic | 0.01 | Affected | 0.3523 | 0.3800 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||||||||
| c.2409A>C | K803N 2D AIThe SynGAP1 K803N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool rates the variant as uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, more tools predict pathogenicity (five) than benignity (three), and the high‑accuracy assessments do not overturn this trend. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.827927 | Disordered | 0.733908 | Binding | 0.349 | 0.900 | 0.625 | -5.039 | Likely Benign | 0.807 | Likely Pathogenic | Ambiguous | 0.045 | Likely Benign | -2.13 | Neutral | 0.896 | Possibly Damaging | 0.673 | Possibly Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.3805 | 0.2035 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.2409A>T | K803N 2D AIThe SynGAP1 K803N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, more tools (five) predict pathogenicity than benign (three), and the high‑accuracy assessments do not overturn this trend. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.827927 | Disordered | 0.733908 | Binding | 0.349 | 0.900 | 0.625 | -5.039 | Likely Benign | 0.807 | Likely Pathogenic | Ambiguous | 0.045 | Likely Benign | -2.13 | Neutral | 0.896 | Possibly Damaging | 0.673 | Possibly Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.3805 | 0.2035 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.2960A>T | D987V 2D AIThe SynGAP1 missense variant D987V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that D987V is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -4.647 | Likely Benign | 0.857 | Likely Pathogenic | Ambiguous | 0.389 | Likely Benign | -4.01 | Deleterious | 0.992 | Probably Damaging | 0.913 | Probably Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.0907 | 0.7202 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3026A>T | E1009V 2D AIThe SynGAP1 missense variant E1009V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments give an uncertain result from AlphaMissense‑Optimized, a Likely Pathogenic verdict from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and no available data from Foldetta. Overall, the majority of evidence points to a deleterious effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | -3.660 | Likely Benign | 0.815 | Likely Pathogenic | Ambiguous | 0.156 | Likely Benign | -3.81 | Deleterious | 0.998 | Probably Damaging | 0.924 | Probably Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.1111 | 0.7580 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3052A>G | T1018A 2D AIThe SynGAP1 missense variant T1018A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.959985 | Binding | 0.348 | 0.801 | 0.500 | -3.289 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.55 | Neutral | 0.001 | Benign | 0.004 | Benign | 2.34 | Pathogenic | 0.53 | Tolerated | 0.3859 | 0.3362 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3448G>A | A1150T 2D AIThe SynGAP1 missense variant A1150T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.795712 | Binding | 0.371 | 0.831 | 0.625 | -3.467 | Likely Benign | 0.176 | Likely Benign | Likely Benign | 0.105 | Likely Benign | -2.06 | Neutral | 0.905 | Possibly Damaging | 0.687 | Possibly Damaging | 2.34 | Pathogenic | 0.03 | Affected | 0.1540 | 0.7182 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3473T>C | V1158A 2D AIThe SynGAP1 missense variant V1158A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b. Tools that agree on a pathogenic effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence (six pathogenic vs. three benign predictions) indicates the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.877504 | Binding | 0.369 | 0.847 | 0.250 | -2.726 | Likely Benign | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.232 | Likely Benign | -2.46 | Neutral | 0.992 | Probably Damaging | 0.989 | Probably Damaging | 2.34 | Pathogenic | 0.02 | Affected | 0.2789 | 0.2906 | 0 | 0 | -2.4 | -28.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3761A>G | E1254G 2D AIThe SynGAP1 missense variant E1254G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized indicates a benign change, but the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and the SGM Consensus suggests that the variant is most likely pathogenic, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.657645 | Disordered | 0.403242 | Uncertain | 0.886 | 0.555 | 0.625 | -10.156 | Likely Pathogenic | 0.706 | Likely Pathogenic | Likely Benign | 0.315 | Likely Benign | -4.52 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.34 | Pathogenic | 0.01 | Affected | 0.2636 | 0.5072 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||||||
| c.3839T>C | M1280T 2D AIThe SynGAP1 missense variant M1280T is catalogued in gnomAD (6‑33447887‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign or likely benign outcome. In contrast, SIFT and FATHMM predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic designation. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | 6-33447887-T-C | 3 | 1.93e-6 | -2.305 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.158 | Likely Benign | -2.26 | Neutral | 0.369 | Benign | 0.120 | Benign | 2.34 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1814 | 0.2031 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.914C>A | T305N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T305N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign, and the SGM‑Consensus score is “Likely Benign.” Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, and the combined Foldetta) return uncertain or inconclusive results. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign, and Foldetta remains uncertain. Overall, the majority of evidence supports a benign impact for T305N, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.359901 | Structured | 0.299706 | Uncertain | 0.872 | 0.274 | 0.125 | -3.261 | Likely Benign | 0.158 | Likely Benign | Likely Benign | 1.18 | Ambiguous | 0.2 | 1.65 | Ambiguous | 1.42 | Ambiguous | 0.09 | Likely Benign | 0.098 | Likely Benign | 0.71 | Neutral | 0.046 | Benign | 0.040 | Benign | 2.34 | Pathogenic | 0.67 | Tolerated | 0.1327 | 0.4352 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.2368A>G | T790A 2D AIThe SynGAP1 T790A missense change is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant as benign, while the SGM Consensus remains inconclusive and Foldetta data are unavailable. Overall, the majority of standard predictors indicate pathogenicity, but the single high‑accuracy tool that is available suggests a benign effect, and no high‑accuracy tool provides a definitive pathogenic verdict. Consequently, the variant is most likely pathogenic according to the bulk of predictions, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | -4.337 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.167 | Likely Benign | -2.64 | Deleterious | 0.992 | Probably Damaging | 0.989 | Probably Damaging | 2.35 | Pathogenic | 0.02 | Affected | 0.4157 | 0.4207 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2428C>G | R810G 2D AISynGAP1 missense variant R810G is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: a single benign call from REVEL, and six pathogenic calls from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. Two tools (ESM1b and AlphaMissense‑Optimized) give uncertain results. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized remains uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic, and Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a pathogenic impact for R810G, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.486429 | Structured | 0.851848 | Binding | 0.263 | 0.907 | 0.375 | -7.190 | In-Between | 0.828 | Likely Pathogenic | Ambiguous | 0.202 | Likely Benign | -3.57 | Deleterious | 0.996 | Probably Damaging | 0.925 | Probably Damaging | 2.35 | Pathogenic | 0.01 | Affected | 0.3686 | 0.4207 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2429G>A | R810H 2D AIThe SynGAP1 R810H missense variant is listed in gnomAD (ID 6‑33442981‑G‑A) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas a pathogenic consensus is reached by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as pathogenic, and Foldetta (FoldX‑MD/Rosetta stability analysis) is unavailable. Overall, the majority of tools predict pathogenicity, and the high‑accuracy subset is split, but the pathogenic signal dominates. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.486429 | Structured | 0.851848 | Binding | 0.263 | 0.907 | 0.375 | 6-33442981-G-A | 3 | 1.86e-6 | -6.554 | Likely Benign | 0.550 | Ambiguous | Likely Benign | 0.239 | Likely Benign | -2.80 | Deleterious | 1.000 | Probably Damaging | 0.980 | Probably Damaging | 2.35 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.3149 | 0.2631 | 0 | 2 | 1.3 | -19.05 | ||||||||||||||||||||||||||||||||||
| c.2429G>T | R810L 2D AIThe SynGAP1 R810L missense change is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to REVEL, while the majority of algorithms (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic outcome. Uncertain calls come from ESM1b and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as pathogenic, and Foldetta results are unavailable. Overall, the preponderance of evidence from multiple in‑silico predictors points to a pathogenic impact for R810L. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.486429 | Structured | 0.851848 | Binding | 0.263 | 0.907 | 0.375 | -7.172 | In-Between | 0.834 | Likely Pathogenic | Ambiguous | 0.338 | Likely Benign | -4.57 | Deleterious | 0.996 | Probably Damaging | 0.925 | Probably Damaging | 2.35 | Pathogenic | 0.01 | Affected | 0.2019 | 0.5271 | -3 | -2 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||||||
| c.2959G>C | D987H 2D AIThe SynGAP1 missense variant D987H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and the Foldetta stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus result, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -5.580 | Likely Benign | 0.925 | Likely Pathogenic | Ambiguous | 0.249 | Likely Benign | -3.16 | Deleterious | 0.998 | Probably Damaging | 0.951 | Probably Damaging | 2.35 | Pathogenic | 0.02 | Affected | 0.1553 | 0.7629 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.3137C>T | P1046L 2D AIThe SynGAP1 missense variant P1046L is reported in gnomAD (ID 6‑33443689‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, SIFT and FATHMM predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors and the consensus analysis points to a benign classification. This conclusion is consistent with the absence of a ClinVar pathogenic report, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | 6-33443689-C-T | 1 | 6.20e-7 | -5.022 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -2.11 | Neutral | 0.001 | Benign | 0.005 | Benign | 2.35 | Pathogenic | 0.05 | Affected | 3.77 | 5 | 0.2036 | 0.6442 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.3644A>T | K1215M 2D AIThe SynGAP1 missense variant K1215M is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools largely agree on a deleterious effect: REVEL predicts a benign outcome, whereas all other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely pathogenic status. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the preponderance of computational evidence indicates that K1215M is most likely pathogenic, and this conclusion is consistent with the absence of any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.497853 | Structured | 0.503613 | Binding | 0.888 | 0.568 | 0.375 | -11.140 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.202 | Likely Benign | -4.06 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.35 | Pathogenic | 0.00 | Affected | 0.0922 | 0.3085 | 0 | -1 | 5.8 | 3.02 | ||||||||||||||||||||||||||||||||||||||
| c.3650A>G | E1217G 2D AIThe SynGAP1 missense variant E1217G is reported in gnomAD (ID 6‑33446642‑A‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) all predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for E1217G. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.599170 | Disordered | 0.493043 | Uncertain | 0.877 | 0.563 | 0.250 | 6-33446642-A-G | -10.803 | Likely Pathogenic | 0.620 | Likely Pathogenic | Likely Benign | 0.331 | Likely Benign | -5.38 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.35 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2205 | 0.4927 | -2 | 0 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||
| c.3716C>G | A1239G 2D AIThe SynGAP1 missense variant lies within a coiled‑coil domain. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and the Foldetta stability analysis is unavailable. ClinVar contains no entry for this variant, and it is not present in gnomAD. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.534779 | Binding | 0.887 | 0.542 | 0.250 | -4.188 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -2.09 | Neutral | 0.139 | Benign | 0.088 | Benign | 2.35 | Pathogenic | 0.00 | Affected | 0.1707 | 0.3142 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3716C>T | A1239V 2D AIThe A1239V missense change occurs in a coiled‑coil region of SynGAP1. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. ClinVar has no entry for this variant, and it is not present in gnomAD. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.534779 | Binding | 0.887 | 0.542 | 0.250 | -5.914 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -2.28 | Neutral | 0.425 | Benign | 0.233 | Benign | 2.35 | Pathogenic | 0.00 | Affected | 0.0771 | 0.4518 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||||||
| c.3760G>C | E1254Q 2D AIThe SynGAP1 missense variant E1254Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessment shows AlphaMissense‑Optimized predicts benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—favors pathogenic (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.657645 | Disordered | 0.403242 | Uncertain | 0.886 | 0.555 | 0.625 | -9.041 | Likely Pathogenic | 0.511 | Ambiguous | Likely Benign | 0.207 | Likely Benign | -2.18 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.35 | Pathogenic | 0.03 | Affected | 0.0768 | 0.5258 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3761A>C | E1254A 2D AIThe SynGAP1 missense variant E1254A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining ten tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) all predict a pathogenic impact. High‑accuracy assessments further support this view: AlphaMissense‑Optimized classifies the variant as benign, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates it is likely pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.657645 | Disordered | 0.403242 | Uncertain | 0.886 | 0.555 | 0.625 | -8.943 | Likely Pathogenic | 0.658 | Likely Pathogenic | Likely Benign | 0.270 | Likely Benign | -4.35 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.35 | Pathogenic | 0.02 | Affected | 0.3029 | 0.5547 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||
| c.3799A>G | M1267V 2D AIThe SynGAP1 missense variant M1267V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated tools (five pathogenic vs. three benign) indicate a pathogenic effect. This prediction is not contradicted by ClinVar status, as the variant is not yet catalogued there. Thus, based on current computational evidence, the M1267V variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.429200 | Structured | 0.812047 | Binding | 0.847 | 0.614 | 0.000 | -2.249 | Likely Benign | 0.537 | Ambiguous | Likely Benign | 0.254 | Likely Benign | -3.34 | Deleterious | 0.959 | Probably Damaging | 0.672 | Possibly Damaging | 2.35 | Pathogenic | 0.00 | Affected | 0.3057 | 0.3241 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||||||
| c.3840G>A | M1280I 2D AIThe SynGAP1 missense variant M1280I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | -3.723 | Likely Benign | 0.251 | Likely Benign | Likely Benign | 0.166 | Likely Benign | -2.59 | Deleterious | 0.059 | Benign | 0.041 | Benign | 2.35 | Pathogenic | 0.00 | Affected | 0.0995 | 0.2468 | 2 | 1 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3840G>C | M1280I 2D AIThe SynGAP1 missense variant M1280I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | -3.723 | Likely Benign | 0.251 | Likely Benign | Likely Benign | 0.166 | Likely Benign | -2.59 | Deleterious | 0.059 | Benign | 0.041 | Benign | 2.35 | Pathogenic | 0.00 | Affected | 0.0995 | 0.2468 | 2 | 1 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3840G>T | M1280I 2D AIThe SynGAP1 missense variant M1280I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | -3.723 | Likely Benign | 0.251 | Likely Benign | Likely Benign | 0.166 | Likely Benign | -2.59 | Deleterious | 0.059 | Benign | 0.041 | Benign | 2.35 | Pathogenic | 0.00 | Affected | 0.0995 | 0.2468 | 2 | 1 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||||||||
| c.1883A>C | K628T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K628T is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely converge on a deleterious effect: the majority—including REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus—label the change as pathogenic or likely pathogenic. Only Rosetta predicts a benign outcome, while FoldX, Foldetta, and premPS are inconclusive. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized scores the variant as pathogenic, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic, and Foldetta remains uncertain. Taken together, the preponderance of evidence indicates that K628T is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | -14.675 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.08 | Ambiguous | 0.1 | 0.07 | Likely Benign | 0.58 | Ambiguous | 0.61 | Ambiguous | 0.661 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.36 | Pathogenic | 0.00 | Affected | 0.1622 | 0.3541 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.2288T>A | L763H 2D AIThe SynGAP1 missense variant L763H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on benign include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the predictions are mixed, with a slight tilt toward pathogenicity (5 pathogenic vs. 4 benign). Thus, the variant is most likely pathogenic according to the aggregate predictions, and this assessment does not contradict ClinVar, which currently has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -6.681 | Likely Benign | 0.640 | Likely Pathogenic | Likely Benign | 0.180 | Likely Benign | -0.79 | Neutral | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 2.36 | Pathogenic | 0.03 | Affected | 0.1021 | 0.0887 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2288T>C | L763P 2D AIThe SynGAP1 missense variant L763P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain. For high‑accuracy assessment, AlphaMissense‑Optimized predicts benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic, with one uncertain). Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -5.802 | Likely Benign | 0.550 | Ambiguous | Likely Benign | 0.158 | Likely Benign | -0.89 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.36 | Pathogenic | 0.12 | Tolerated | 0.3920 | 0.1182 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2407A>C | K803Q 2D AIThe SynGAP1 missense variant K803Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of high‑confidence predictions lean toward a benign impact, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.827927 | Disordered | 0.733908 | Binding | 0.349 | 0.900 | 0.625 | -2.792 | Likely Benign | 0.305 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -2.01 | Neutral | 0.984 | Probably Damaging | 0.773 | Possibly Damaging | 2.36 | Pathogenic | 0.00 | Affected | 0.4810 | 0.1398 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||||||||||
| c.2414T>C | L805P 2D AIThe SynGAP1 missense variant L805P is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments give a benign result from AlphaMissense‑Optimized, a pathogenic outcome from the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and no Foldetta data are available. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.775545 | Disordered | 0.827669 | Binding | 0.341 | 0.903 | 0.625 | Uncertain | 1 | -4.661 | Likely Benign | 0.444 | Ambiguous | Likely Benign | 0.272 | Likely Benign | -3.40 | Deleterious | 0.975 | Probably Damaging | 0.767 | Possibly Damaging | 2.36 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3128 | 0.1650 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.2491G>C | E831Q 2D AIThe SynGAP1 missense variant E831Q is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a benign outcome (2 benign vs. 1 pathogenic). High‑accuracy methods give a benign prediction from AlphaMissense‑Optimized and a benign consensus from SGM; Foldetta results are unavailable. Overall, the available computational evidence points to a benign impact for E831Q, and this assessment does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.626927 | Disordered | 0.617732 | Binding | 0.319 | 0.874 | 0.375 | -5.604 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.147 | Likely Benign | -1.23 | Neutral | 0.891 | Possibly Damaging | 0.492 | Possibly Damaging | 2.36 | Pathogenic | 0.09 | Tolerated | 0.1022 | 0.7052 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2492A>C | E831A 2D AIThe SynGAP1 missense variant E831A is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, SIFT, polyPhen‑2 HumVar, and ESM1b, while pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and FATHMM. AlphaMissense‑Default is uncertain, whereas AlphaMissense‑Optimized predicts benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a pathogenic verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools therefore give conflicting signals: AlphaMissense‑Optimized benign versus SGM Consensus pathogenic, with no Foldetta data. Overall, the bulk of predictions lean toward a benign effect, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.626927 | Disordered | 0.617732 | Binding | 0.319 | 0.874 | 0.375 | -4.780 | Likely Benign | 0.429 | Ambiguous | Likely Benign | 0.115 | Likely Benign | -2.56 | Deleterious | 0.625 | Possibly Damaging | 0.315 | Benign | 2.36 | Pathogenic | 0.07 | Tolerated | 0.3845 | 0.6868 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2794T>C | F932L 2D AIThe SynGAP1 missense variant F932L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic. Foldetta results are unavailable. Overall, the consensus of the available predictions points to a pathogenic effect for F932L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.989197 | Binding | 0.293 | 0.858 | 0.500 | -3.390 | Likely Benign | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.309 | Likely Benign | -3.50 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.36 | Pathogenic | 0.03 | Affected | 0.2373 | 0.3895 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2796C>A | F932L 2D AIThe SynGAP1 missense variant F932L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic. Foldetta results are unavailable. Overall, the consensus of the available predictions points to a pathogenic effect for F932L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.989197 | Binding | 0.293 | 0.858 | 0.500 | -3.390 | Likely Benign | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.170 | Likely Benign | -3.50 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.36 | Pathogenic | 0.03 | Affected | 0.2373 | 0.3895 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2796C>G | F932L 2D AIThe SynGAP1 missense variant F932L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic. Foldetta results are unavailable. Overall, the consensus of the available predictions points to a pathogenic effect for F932L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.989197 | Binding | 0.293 | 0.858 | 0.500 | -3.390 | Likely Benign | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.170 | Likely Benign | -3.50 | Deleterious | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.36 | Pathogenic | 0.03 | Affected | 0.2373 | 0.3895 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2797C>T | H933Y 2D AIThe SynGAP1 H933Y variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic prediction. Foldetta results are unavailable. Overall, the majority of evidence (five pathogenic vs. three benign predictions) points to a pathogenic impact for H933Y. This conclusion does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.987531 | Binding | 0.305 | 0.862 | 0.625 | -3.952 | Likely Benign | 0.525 | Ambiguous | Likely Benign | 0.314 | Likely Benign | -3.49 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.36 | Pathogenic | 0.01 | Affected | 0.0968 | 0.4815 | 0 | 2 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2798A>C | H933P 2D AIThe SynGAP1 missense variant H933P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs 2 pathogenic). Foldetta results are unavailable. Overall, six tools predict pathogenicity versus three predicting benign, and the high‑accuracy benign prediction is outweighed by the majority of pathogenic calls. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.987531 | Binding | 0.305 | 0.862 | 0.625 | -3.890 | Likely Benign | 0.332 | Likely Benign | Likely Benign | 0.508 | Likely Pathogenic | -5.39 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.36 | Pathogenic | 0.02 | Affected | 0.1821 | 0.4400 | 0 | -2 | 1.6 | -40.02 | ||||||||||||||||||||||||||||||||||||||||
| c.2825C>A | P942Q 2D AIThe SynGAP1 missense variant P942Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for P942Q, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.878102 | Binding | 0.365 | 0.915 | 0.625 | -6.094 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.028 | Likely Benign | -1.12 | Neutral | 0.026 | Benign | 0.015 | Benign | 2.36 | Pathogenic | 0.00 | Affected | 0.1508 | 0.4703 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2825C>G | P942R 2D AIThe SynGAP1 missense variant P942R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.878102 | Binding | 0.365 | 0.915 | 0.625 | -5.405 | Likely Benign | 0.159 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.78 | Neutral | 0.411 | Benign | 0.139 | Benign | 2.36 | Pathogenic | 0.00 | Affected | 0.1307 | 0.3693 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3026A>G | E1009G 2D AIThe SynGAP1 missense variant E1009G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. In contrast, the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM‑Consensus indicates a likely pathogenic outcome; Foldetta results are unavailable. Overall, the preponderance of evidence from standard and high‑accuracy predictors points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | -2.758 | Likely Benign | 0.610 | Likely Pathogenic | Likely Benign | 0.123 | Likely Benign | -3.06 | Deleterious | 0.961 | Probably Damaging | 0.721 | Possibly Damaging | 2.36 | Pathogenic | 0.01 | Affected | 0.2800 | 0.6003 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3055C>T | R1019C 2D AIThe SynGAP1 missense variant R1019C is listed in ClinVar with an “Uncertain” status (ClinVar ID 1676922.0) and is present in gnomAD (ID 6‑33443607‑C‑T). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact; ESM1b remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote) remains pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.856457 | Disordered | 0.966400 | Binding | 0.315 | 0.794 | 0.500 | Conflicting | 2 | 6-33443607-C-T | 10 | 6.19e-6 | -7.386 | In-Between | 0.646 | Likely Pathogenic | Likely Benign | 0.168 | Likely Benign | -4.00 | Deleterious | 0.999 | Probably Damaging | 0.880 | Possibly Damaging | 2.36 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3016 | 0.3664 | -4 | -3 | 7.0 | -53.05 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||
| c.3455A>G | E1152G 2D AIThe SynGAP1 missense variant E1152G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Overall, the balance of evidence points to a pathogenic effect for E1152G. This conclusion is not contradicted by any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.741537 | Disordered | 0.811118 | Binding | 0.395 | 0.846 | 0.500 | -2.663 | Likely Benign | 0.918 | Likely Pathogenic | Ambiguous | 0.373 | Likely Benign | -3.85 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.36 | Pathogenic | 0.01 | Affected | 0.3242 | 0.5249 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3671T>C | L1224P 2D AIThe SynGAP1 missense variant L1224P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). In silico predictors that agree on a benign effect include REVEL and SIFT, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of predictions indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.525368 | Disordered | 0.441554 | Uncertain | 0.871 | 0.543 | 0.500 | -11.727 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.149 | Likely Benign | -3.18 | Deleterious | 0.998 | Probably Damaging | 0.948 | Probably Damaging | 2.36 | Pathogenic | 0.07 | Tolerated | 0.3618 | 0.1053 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||
| c.3716C>A | A1239D 2D AIThe SynGAP1 missense variant A1239D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs. two pathogenic votes). Foldetta results are unavailable. Overall, the majority of evidence (six benign vs. three pathogenic predictions) supports a benign classification. This conclusion does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.541878 | Disordered | 0.534779 | Binding | 0.887 | 0.542 | 0.250 | -6.177 | Likely Benign | 0.326 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -2.60 | Deleterious | 0.270 | Benign | 0.136 | Benign | 2.36 | Pathogenic | 0.00 | Affected | 0.1521 | 0.2062 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.3760G>A | E1254K 2D AIThe SynGAP1 missense variant E1254K is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for E1254K. This prediction is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.657645 | Disordered | 0.403242 | Uncertain | 0.886 | 0.555 | 0.625 | -11.288 | Likely Pathogenic | 0.872 | Likely Pathogenic | Ambiguous | 0.290 | Likely Benign | -2.97 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.36 | Pathogenic | 0.02 | Affected | 0.1653 | 0.5488 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3778A>C | K1260Q 2D AIThe SynGAP1 missense variant K1260Q is listed in ClinVar with no submitted interpretation and is present in gnomAD (ID 6‑33446770‑A‑C). Functional prediction tools cluster into two groups: benign predictions come from REVEL, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. ESM1b is uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts benign, but the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic verdict, and Foldetta results are unavailable. Overall, the majority of evidence points to pathogenicity, and this conclusion does not contradict the ClinVar status, which remains unclassified. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.509769 | Disordered | 0.625808 | Binding | 0.890 | 0.575 | 0.250 | 6-33446770-A-C | -7.830 | In-Between | 0.325 | Likely Benign | Likely Benign | 0.367 | Likely Benign | -3.06 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.36 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3597 | 0.1102 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||||||||
| c.3799A>C | M1267L 2D  AIThe SynGAP1 missense variant M1267L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence (five pathogenic‑predicting tools versus three benign) indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.429200 | Structured | 0.812047 | Binding | 0.847 | 0.614 | 0.000 | -1.766 | Likely Benign | 0.399 | Ambiguous | Likely Benign | 0.293 | Likely Benign | -2.52 | Deleterious | 0.813 | Possibly Damaging | 0.456 | Possibly Damaging | 2.36 | Pathogenic | 0.00 | Affected | 0.1392 | 0.3626 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3799A>T | M1267L 2D AIThe SynGAP1 missense variant M1267L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence (five pathogenic‑predicting tools versus three benign‑predicting tools) indicates that M1267L is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.429200 | Structured | 0.812047 | Binding | 0.847 | 0.614 | 0.000 | -1.766 | Likely Benign | 0.399 | Ambiguous | Likely Benign | 0.293 | Likely Benign | -2.52 | Deleterious | 0.813 | Possibly Damaging | 0.456 | Possibly Damaging | 2.36 | Pathogenic | 0.00 | Affected | 0.1392 | 0.3626 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3829C>T | H1277Y 2D AIThe SynGAP1 missense variant H1277Y is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the balance of evidence (5 benign vs 4 pathogenic predictions) leans toward a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | -4.288 | Likely Benign | 0.232 | Likely Benign | Likely Benign | 0.136 | Likely Benign | -4.32 | Deleterious | 0.812 | Possibly Damaging | 0.298 | Benign | 2.36 | Pathogenic | 0.00 | Affected | 0.0742 | 0.3034 | 0 | 2 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3838A>G | M1280V 2D AIThe SynGAP1 missense variant M1280V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for M1280V, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | -2.414 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.240 | Likely Benign | -2.24 | Neutral | 0.004 | Benign | 0.008 | Benign | 2.36 | Pathogenic | 0.00 | Affected | 0.2568 | 0.3137 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||||||
| c.3922C>A | R1308S 2D AIThe SynGAP1 missense variant R1308S is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The high‑accuracy AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated tools (five pathogenic vs. three benign) indicate a pathogenic effect. This prediction is not contradicted by ClinVar status, which has no entry for the variant. Thus, based on current computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.741537 | Disordered | 0.930652 | Binding | 0.378 | 0.904 | 0.750 | -2.180 | Likely Benign | 0.419 | Ambiguous | Likely Benign | 0.307 | Likely Benign | -3.76 | Deleterious | 0.982 | Probably Damaging | 0.982 | Probably Damaging | 2.36 | Pathogenic | 0.00 | Affected | 0.3070 | 0.4744 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.1048G>C | V350L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V350L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only FATHMM predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign status. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as uncertain. No other tools provide conflicting evidence. Based on the preponderance of benign predictions and the lack of pathogenic support, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -5.941 | Likely Benign | 0.238 | Likely Benign | Likely Benign | -1.31 | Ambiguous | 0.1 | -0.34 | Likely Benign | -0.83 | Ambiguous | 0.36 | Likely Benign | 0.042 | Likely Benign | -1.63 | Neutral | 0.068 | Benign | 0.022 | Benign | 2.37 | Pathogenic | 0.27 | Tolerated | 0.1173 | 0.4958 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1048G>T | V350L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V350L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only FATHMM predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. No other tools provide conflicting evidence. Based on the preponderance of predictions, V350L is most likely benign, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -5.941 | Likely Benign | 0.238 | Likely Benign | Likely Benign | -1.31 | Ambiguous | 0.1 | -0.34 | Likely Benign | -0.83 | Ambiguous | 0.36 | Likely Benign | 0.042 | Likely Benign | -1.63 | Neutral | 0.068 | Benign | 0.022 | Benign | 2.37 | Pathogenic | 0.27 | Tolerated | 0.1173 | 0.4958 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1882A>G | K628E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K628E missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that are inconclusive or uncertain are FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Uncertain. Overall, the majority of evidence points to a deleterious effect. The variant is most likely pathogenic based on these predictions, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | -14.658 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.04 | Ambiguous | 0.2 | 0.52 | Ambiguous | 0.78 | Ambiguous | 1.06 | Destabilizing | 0.652 | Likely Pathogenic | -3.98 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.37 | Pathogenic | 0.00 | Affected | 0.2909 | 0.0810 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1884G>C | K628N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K628N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—classify the variant as pathogenic or likely pathogenic. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability predictions and are therefore treated as unavailable evidence. High‑accuracy assessments specifically show AlphaMissense‑Optimized predicting pathogenicity, the SGM‑Consensus indicating a likely pathogenic status, and Foldetta yielding an uncertain result. Based on the overwhelming agreement among the majority of prediction tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | -12.284 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.78 | Ambiguous | 0.1 | 0.59 | Ambiguous | 0.69 | Ambiguous | 1.11 | Destabilizing | 0.403 | Likely Benign | -4.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.37 | Pathogenic | 0.00 | Affected | 3.37 | 34 | 0.2740 | 0.1254 | 0 | 1 | 0.4 | -14.07 | |||||||||||||||||||||||||||
| c.1884G>T | K628N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K628N is catalogued in gnomAD (6‑33440936‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: a single benign prediction from REVEL, and a consensus of nine pathogenic predictions from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also labels the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus confirms a likely pathogenic status, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result and therefore does not alter the overall interpretation. Consequently, the variant is most likely pathogenic according to the available computational evidence, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | 6-33440936-G-T | 1 | 6.20e-7 | -12.284 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.78 | Ambiguous | 0.1 | 0.59 | Ambiguous | 0.69 | Ambiguous | 1.11 | Destabilizing | 0.403 | Likely Benign | -4.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.37 | Pathogenic | 0.00 | Affected | 3.37 | 34 | 0.2740 | 0.1254 | 0 | 1 | 0.4 | -14.07 | ||||||||||||||||||||||||
| c.2161A>C | I721L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I721L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. Two tools (premPS and ESM1b) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the predictions are split, but the majority of high‑confidence tools lean toward a benign interpretation. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -7.843 | In-Between | 0.757 | Likely Pathogenic | Likely Benign | 0.35 | Likely Benign | 0.0 | 0.34 | Likely Benign | 0.35 | Likely Benign | 0.65 | Ambiguous | 0.188 | Likely Benign | -1.83 | Neutral | 0.955 | Possibly Damaging | 0.985 | Probably Damaging | 2.37 | Pathogenic | 0.02 | Affected | 0.0627 | 0.3446 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.2414T>A | L805Q 2D AIThe SynGAP1 missense variant L805Q is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts a benign effect, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (2 pathogenic vs. 1 benign, 1 uncertain). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence (five pathogenic vs. three benign predictions, with a pathogenic SGM Consensus) indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.775545 | Disordered | 0.827669 | Binding | 0.341 | 0.903 | 0.625 | -6.244 | Likely Benign | 0.427 | Ambiguous | Likely Benign | 0.152 | Likely Benign | -2.66 | Deleterious | 0.927 | Possibly Damaging | 0.690 | Possibly Damaging | 2.37 | Pathogenic | 0.00 | Affected | 0.1215 | 0.1265 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2414T>G | L805R 2D AIThe SynGAP1 missense variant L805R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. In contrast, the majority of tools predict a pathogenic impact: SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all indicate pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it as likely pathogenic; Foldetta results are unavailable. Overall, the balance of evidence from the broader set of predictors leans toward pathogenicity, and this conclusion does not contradict any existing ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.775545 | Disordered | 0.827669 | Binding | 0.341 | 0.903 | 0.625 | -6.640 | Likely Benign | 0.569 | Likely Pathogenic | Likely Benign | 0.196 | Likely Benign | -3.00 | Deleterious | 0.927 | Possibly Damaging | 0.617 | Possibly Damaging | 2.37 | Pathogenic | 0.00 | Affected | 0.1372 | 0.0908 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||
| c.2491G>A | E831K 2D AIThe SynGAP1 missense variant E831K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, FATHMM, and AlphaMissense‑Default; ESM1b is uncertain. High‑accuracy assessment shows AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of conventional tools lean benign, but the high‑accuracy consensus and several individual pathogenic predictions suggest a pathogenic likelihood. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.626927 | Disordered | 0.617732 | Binding | 0.319 | 0.874 | 0.375 | -7.447 | In-Between | 0.636 | Likely Pathogenic | Likely Benign | 0.167 | Likely Benign | -1.43 | Neutral | 0.625 | Possibly Damaging | 0.252 | Benign | 2.37 | Pathogenic | 0.07 | Tolerated | 0.1989 | 0.6995 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.2801T>G | M934R 2D AISynGAP1 missense variant M934R is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, SIFT, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Overall, the balance of evidence leans toward pathogenicity, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.762850 | Disordered | 0.984677 | Binding | 0.290 | 0.867 | 0.625 | -1.854 | Likely Benign | 0.771 | Likely Pathogenic | Likely Benign | 0.322 | Likely Benign | -3.54 | Deleterious | 0.969 | Probably Damaging | 0.624 | Possibly Damaging | 2.37 | Pathogenic | 0.11 | Tolerated | 0.1902 | 0.1243 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||||||||||||
| c.2825C>T | P942L 2D AIThe SynGAP1 missense variant P942L is listed in ClinVar (ID 2851884.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443377‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.878102 | Binding | 0.365 | 0.915 | 0.625 | Uncertain | 1 | 6-33443377-C-T | 4 | 2.48e-6 | -5.063 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -2.00 | Neutral | 0.411 | Benign | 0.239 | Benign | 2.37 | Pathogenic | 0.00 | Affected | 4.32 | 4 | 0.2094 | 0.5507 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||
| c.3136C>A | P1046T 2D AIThe SynGAP1 missense variant P1046T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | -5.249 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.052 | Likely Benign | -1.18 | Neutral | 0.411 | Benign | 0.131 | Benign | 2.37 | Pathogenic | 0.21 | Tolerated | 0.1430 | 0.5934 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3265G>T | G1089W 2D AIThe SynGAP1 missense variant G1089W is not reported in ClinVar and has no allele in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the consensus SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments give an uncertain result from AlphaMissense‑Optimized, a Likely Pathogenic verdict from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and no available Foldetta stability data. Overall, the majority of evidence points to a deleterious effect, so the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -6.561 | Likely Benign | 0.863 | Likely Pathogenic | Ambiguous | 0.236 | Likely Benign | -3.45 | Deleterious | 1.000 | Probably Damaging | 0.988 | Probably Damaging | 2.37 | Pathogenic | 0.00 | Affected | 0.0840 | 0.4610 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.3448G>T | A1150S 2D AIThe SynGAP1 missense variant A1150S is reported in gnomAD (ID 6‑33444483‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.762850 | Disordered | 0.795712 | Binding | 0.371 | 0.831 | 0.625 | 6-33444483-G-T | 7 | 4.34e-6 | -2.656 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -1.49 | Neutral | 0.951 | Possibly Damaging | 0.752 | Possibly Damaging | 2.37 | Pathogenic | 0.10 | Tolerated | 3.77 | 5 | 0.2656 | 0.5694 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||||||
| c.3454G>C | E1152Q 2D AIThe SynGAP1 missense variant E1152Q is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward a pathogenic interpretation. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.741537 | Disordered | 0.811118 | Binding | 0.395 | 0.846 | 0.500 | -2.798 | Likely Benign | 0.830 | Likely Pathogenic | Ambiguous | 0.291 | Likely Benign | -1.98 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.37 | Pathogenic | 0.03 | Affected | 0.1747 | 0.6395 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3455A>C | E1152A 2D AIThe SynGAP1 missense variant E1152A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Overall, the balance of evidence points to a pathogenic effect for E1152A. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.741537 | Disordered | 0.811118 | Binding | 0.395 | 0.846 | 0.500 | -2.482 | Likely Benign | 0.927 | Likely Pathogenic | Ambiguous | 0.349 | Likely Benign | -3.82 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.37 | Pathogenic | 0.02 | Affected | 0.4434 | 0.6557 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.3617A>T | K1206I 2D AIThe SynGAP1 missense variant K1206I is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are not available. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for K1206I. This conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.585406 | Disordered | 0.555819 | Binding | 0.893 | 0.569 | 0.375 | -13.526 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.302 | Likely Benign | -5.65 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.37 | Pathogenic | 0.01 | Affected | 0.0816 | 0.3521 | -2 | -3 | 8.4 | -15.01 | ||||||||||||||||||||||||||||||||||||||
| c.3712C>G | Q1238E 2D AIThe SynGAP1 missense variant Q1238E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools specifically show AlphaMissense‑Optimized predicting benign, while SGM Consensus and Foldetta are unavailable. Overall, the majority of standard predictors (5 pathogenic vs 4 benign) lean toward a pathogenic interpretation, and this assessment does not contradict ClinVar status because no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.562014 | Disordered | 0.548882 | Binding | 0.855 | 0.545 | 0.250 | -10.421 | Likely Pathogenic | 0.312 | Likely Benign | Likely Benign | 0.201 | Likely Benign | -1.90 | Neutral | 0.985 | Probably Damaging | 0.981 | Probably Damaging | 2.37 | Pathogenic | 0.02 | Affected | 0.1078 | 0.1921 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3715G>A | A1239T 2D AIThe SynGAP1 missense variant A1239T is not reported in ClinVar and has no gnomAD entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the consensus score from SGM‑Consensus is “Likely Benign.” In contrast, two sequence‑based predictors flag it as pathogenic: SIFT and FATHMM. When considering the high‑accuracy subset, AlphaMissense‑Optimized remains benign and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a benign outcome; Foldetta data are unavailable. Overall, the majority of evidence points to a benign impact, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.534779 | Binding | 0.887 | 0.542 | 0.250 | -3.570 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -1.62 | Neutral | 0.270 | Benign | 0.136 | Benign | 2.37 | Pathogenic | 0.00 | Affected | 0.0957 | 0.5384 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||||||||
| c.3726G>C | E1242D 2D AIThe SynGAP1 missense variant E1242D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation—there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.557691 | Disordered | 0.456349 | Uncertain | 0.870 | 0.549 | 0.500 | -2.582 | Likely Benign | 0.158 | Likely Benign | Likely Benign | 0.031 | Likely Benign | -1.96 | Neutral | 0.020 | Benign | 0.018 | Benign | 2.37 | Pathogenic | 0.00 | Affected | 0.1594 | 0.2675 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3726G>T | E1242D 2D AIThe SynGAP1 missense variant E1242D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.557691 | Disordered | 0.456349 | Uncertain | 0.870 | 0.549 | 0.500 | -2.582 | Likely Benign | 0.158 | Likely Benign | Likely Benign | 0.032 | Likely Benign | -1.96 | Neutral | 0.020 | Benign | 0.018 | Benign | 2.37 | Pathogenic | 0.00 | Affected | 0.1594 | 0.2675 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.3839T>A | M1280K 2D AIThe SynGAP1 missense variant M1280K is not reported in ClinVar and has no entry in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and consensus methods points to a benign impact for M1280K, and this conclusion does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | -2.963 | Likely Benign | 0.223 | Likely Benign | Likely Benign | 0.213 | Likely Benign | -1.42 | Neutral | 0.126 | Benign | 0.041 | Benign | 2.37 | Pathogenic | 0.00 | Affected | 0.1096 | 0.0488 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.796C>G | L266V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L266V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are premPS, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. FoldX, Rosetta, and Foldetta give uncertain results and are therefore considered unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta is also inconclusive. Consequently, the variant is most likely benign based on the current predictions, and this assessment does not contradict ClinVar, which has no reported status for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.232838 | Structured | 0.297157 | Uncertain | 0.948 | 0.264 | 0.000 | -8.586 | Likely Pathogenic | 0.153 | Likely Benign | Likely Benign | 1.71 | Ambiguous | 0.1 | 0.97 | Ambiguous | 1.34 | Ambiguous | 1.32 | Destabilizing | 0.193 | Likely Benign | -2.20 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 2.37 | Pathogenic | 0.11 | Tolerated | 0.1152 | 0.2456 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||
| c.2288T>G | L763R 2D AIThe SynGAP1 missense variant L763R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -5.516 | Likely Benign | 0.643 | Likely Pathogenic | Likely Benign | 0.163 | Likely Benign | -1.66 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.38 | Pathogenic | 0.07 | Tolerated | 0.1199 | 0.0761 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2407A>G | K803E 2D AIThe SynGAP1 missense variant K803E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs 2 benign) and Foldetta results are unavailable. Overall, the majority of tools (five pathogenic vs. four benign) predict a pathogenic impact. Thus, the variant is most likely pathogenic, and this prediction does not contradict any ClinVar status because the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.827927 | Disordered | 0.733908 | Binding | 0.349 | 0.900 | 0.625 | -3.889 | Likely Benign | 0.650 | Likely Pathogenic | Likely Benign | 0.172 | Likely Benign | -2.06 | Neutral | 0.896 | Possibly Damaging | 0.596 | Possibly Damaging | 2.38 | Pathogenic | 0.00 | Affected | 0.4043 | 0.1357 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2408A>G | K803R 2D AIThe SynGAP1 missense variant K803R is listed in ClinVar (ID 834618.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—SIFT and FATHMM—predict pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely benign, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.827927 | Disordered | 0.733908 | Binding | 0.349 | 0.900 | 0.625 | Uncertain | 1 | -2.281 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.018 | Likely Benign | -1.52 | Neutral | 0.103 | Benign | 0.038 | Benign | 2.38 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.4857 | 0.1715 | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||
| c.2665G>A | G889R 2D AIThe SynGAP1 missense variant G889R is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (ID 6‑33443217‑G‑A). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized; pathogenic predictions come from SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs three pathogenic) supports a benign impact. This conclusion does not contradict ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | 6-33443217-G-A | 1 | 6.20e-7 | -3.357 | Likely Benign | 0.685 | Likely Pathogenic | Likely Benign | 0.070 | Likely Benign | -1.96 | Neutral | 0.027 | Benign | 0.009 | Benign | 2.38 | Pathogenic | 0.02 | Affected | 4.32 | 4 | 0.0925 | 0.4165 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.2665G>C | G889R 2D AIThe SynGAP1 missense variant G889R is not reported in ClinVar (ClinVar status: not present) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | -3.357 | Likely Benign | 0.685 | Likely Pathogenic | Likely Benign | 0.072 | Likely Benign | -1.96 | Neutral | 0.027 | Benign | 0.009 | Benign | 2.38 | Pathogenic | 0.02 | Affected | 4.32 | 4 | 0.0925 | 0.4165 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2756A>C | Q919P 2D AIThe SynGAP1 missense variant Q919P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign based on current computational evidence, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.911223 | Binding | 0.299 | 0.841 | 0.250 | -2.358 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -1.15 | Neutral | 0.989 | Probably Damaging | 0.834 | Possibly Damaging | 2.38 | Pathogenic | 0.03 | Affected | 0.2364 | 0.6108 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2792T>A | L931H 2D AIThe SynGAP1 missense variant L931H has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. ESM1b and AlphaMissense‑Optimized are uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.549308 | Disordered | 0.989212 | Binding | 0.335 | 0.856 | 0.375 | -7.495 | In-Between | 0.954 | Likely Pathogenic | Ambiguous | 0.301 | Likely Benign | -3.76 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.38 | Pathogenic | 0.00 | Affected | 0.1198 | 0.1205 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2792T>C | L931P 2D AIThe SynGAP1 missense variant L931P is not reported in ClinVar and is absent from gnomAD. Prediction tools show a strong bias toward pathogenicity: SIFT, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, PROVEAN, and AlphaMissense‑Default all predict a deleterious effect, whereas only REVEL indicates a benign outcome. High‑accuracy assessments reinforce this trend: the SGM consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic, and AlphaMissense‑Optimized returns an uncertain result. Foldetta predictions are unavailable. Overall, the preponderance of evidence points to a pathogenic impact for L931P, and this conclusion is consistent with the absence of any ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.549308 | Disordered | 0.989212 | Binding | 0.335 | 0.856 | 0.375 | -8.624 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 0.376 | Likely Benign | -3.43 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.3203 | 0.1698 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2798A>T | H933L 2D AIThe SynGAP1 H933L missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.987531 | Binding | 0.305 | 0.862 | 0.625 | -0.858 | Likely Benign | 0.470 | Ambiguous | Likely Benign | 0.388 | Likely Benign | -6.26 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.38 | Pathogenic | 0.02 | Affected | 0.1003 | 0.5749 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2801T>A | M934K 2D AIThe SynGAP1 missense variant M934K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, and ESM1b, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the balance of evidence from the majority of tools points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.762850 | Disordered | 0.984677 | Binding | 0.290 | 0.867 | 0.625 | -2.457 | Likely Benign | 0.816 | Likely Pathogenic | Ambiguous | 0.333 | Likely Benign | -3.66 | Deleterious | 0.929 | Possibly Damaging | 0.521 | Possibly Damaging | 2.38 | Pathogenic | 0.13 | Tolerated | 0.1828 | 0.1262 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.2863T>A | S955T 2D AIThe SynGAP1 missense variant S955T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S955T, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.945325 | Binding | 0.350 | 0.924 | 0.750 | -4.717 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.070 | Likely Benign | -0.96 | Neutral | 0.451 | Benign | 0.265 | Benign | 2.38 | Pathogenic | 0.00 | Affected | 0.2184 | 0.5927 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3056G>C | R1019P 2D AIThe SynGAP1 missense variant R1019P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on benign impact include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, whereas those that predict pathogenicity are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of conventional tools predict a pathogenic effect, but the most accurate single‑tool prediction is benign and the consensus and folding‑stability analyses are inconclusive. Thus, the variant is most likely pathogenic based on the aggregate predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.856457 | Disordered | 0.966400 | Binding | 0.315 | 0.794 | 0.500 | -3.737 | Likely Benign | 0.697 | Likely Pathogenic | Likely Benign | 0.143 | Likely Benign | -2.48 | Neutral | 0.966 | Probably Damaging | 0.811 | Possibly Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.1899 | 0.4521 | 0 | -2 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||||||||||
| c.3071T>A | L1024H 2D AIThe SynGAP1 missense variant L1024H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized returns an Uncertain result, SGM‑Consensus indicates Likely Pathogenic, and Foldetta data are unavailable. Overall, the majority of evidence points toward a deleterious effect, suggesting the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -3.271 | Likely Benign | 0.868 | Likely Pathogenic | Ambiguous | 0.123 | Likely Benign | -3.09 | Deleterious | 1.000 | Probably Damaging | 0.981 | Probably Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.1198 | 0.1483 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3101C>A | P1034H 2D AIThe SynGAP1 missense variant P1034H is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. three benign) lean toward pathogenicity, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -4.634 | Likely Benign | 0.540 | Ambiguous | Likely Benign | 0.083 | Likely Benign | -3.17 | Deleterious | 0.938 | Possibly Damaging | 0.750 | Possibly Damaging | 2.38 | Pathogenic | 0.02 | Affected | 0.1776 | 0.5451 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3137C>G | P1046R 2D AIThe SynGAP1 missense variant P1046R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all classify the change as benign or likely benign. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM‑Consensus also indicates a likely benign status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | -4.929 | Likely Benign | 0.222 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -1.79 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.38 | Pathogenic | 0.07 | Tolerated | 0.1267 | 0.3841 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3227T>G | L1076W 2D  AIThe SynGAP1 missense variant L1076W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated predictors (five pathogenic vs. three benign) lean toward a pathogenic interpretation. This prediction is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.989617 | Binding | 0.301 | 0.892 | 0.750 | -6.377 | Likely Benign | 0.822 | Likely Pathogenic | Ambiguous | 0.183 | Likely Benign | -1.40 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.38 | Pathogenic | 0.02 | Affected | 0.0749 | 0.3740 | -2 | -2 | -4.7 | 73.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3454G>A | E1152K 2D AIThe SynGAP1 missense variant E1152K is reported in gnomAD (ID 6‑33444489‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic.” High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts a deleterious effect, and the SGM‑Consensus confirms a likely pathogenic outcome. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective evidence points to a pathogenic effect, and this conclusion is not contradicted by ClinVar, which contains no classification for E1152K. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.741537 | Disordered | 0.811118 | Binding | 0.395 | 0.846 | 0.500 | 6-33444489-G-A | 1 | 6.20e-7 | -3.612 | Likely Benign | 0.966 | Likely Pathogenic | Likely Pathogenic | 0.300 | Likely Benign | -2.64 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.38 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.2942 | 0.6397 | 1 | 0 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||
| c.3481A>C | M1161L 2D AIThe SynGAP1 missense variant M1161L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool rates the variant as uncertain, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Consequently, the available computational evidence is conflicting, with an equal number of benign and pathogenic predictions and no definitive high‑accuracy support. The variant is most likely of uncertain significance; this lack of consensus is consistent with its absence from ClinVar and gnomAD. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.580690 | Disordered | 0.864109 | Binding | 0.372 | 0.830 | 0.375 | -2.129 | Likely Benign | 0.862 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.59 | Neutral | 0.699 | Possibly Damaging | 0.833 | Possibly Damaging | 2.38 | Pathogenic | 0.48 | Tolerated | 0.1614 | 0.4244 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3481A>T | M1161L 2D AISynGAP1 missense variant M1161L is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b, while those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain, and both the SGM Consensus and Foldetta (which would combine FoldX‑MD and Rosetta outputs) are unavailable. Consequently, the overall computational evidence does not strongly favor either outcome. The variant is most likely inconclusive in pathogenicity prediction, and this does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.580690 | Disordered | 0.864109 | Binding | 0.372 | 0.830 | 0.375 | -2.129 | Likely Benign | 0.862 | Likely Pathogenic | Ambiguous | 0.156 | Likely Benign | -1.59 | Neutral | 0.699 | Possibly Damaging | 0.833 | Possibly Damaging | 2.38 | Pathogenic | 0.48 | Tolerated | 0.1614 | 0.4244 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3644A>C | K1215T 2D AIThe SynGAP1 missense variant K1215T is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is “Likely Pathogenic.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that K1215T is most likely pathogenic, which does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.497853 | Structured | 0.503613 | Binding | 0.888 | 0.568 | 0.375 | Uncertain | 1 | -12.008 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.204 | Likely Benign | -4.09 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.1972 | 0.2214 | 0 | -1 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||||||||
| c.3645G>C | K1215N 2D AIThe SynGAP1 missense variant K1215N is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that K1215N is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.497853 | Structured | 0.503613 | Binding | 0.888 | 0.568 | 0.375 | -12.569 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.099 | Likely Benign | -3.23 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.3380 | 0.0901 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||||||||||
| c.3645G>T | K1215N 2D AIThe SynGAP1 missense variant K1215N is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that K1215N is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.497853 | Structured | 0.503613 | Binding | 0.888 | 0.568 | 0.375 | -12.569 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.099 | Likely Benign | -3.23 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.3380 | 0.0901 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||||||||||
| c.3649G>C | E1217Q 2D AIThe SynGAP1 missense variant E1217Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of tools (five pathogenic vs. three benign) and the SGM Consensus support a pathogenic classification, which does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.599170 | Disordered | 0.493043 | Uncertain | 0.877 | 0.563 | 0.250 | -8.887 | Likely Pathogenic | 0.398 | Ambiguous | Likely Benign | 0.219 | Likely Benign | -2.16 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.38 | Pathogenic | 0.00 | Affected | 0.0949 | 0.4745 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3650A>C | E1217A 2D AIThe SynGAP1 missense variant E1217A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show a split: AlphaMissense‑Optimized reports benign, while the SGM‑Consensus (majority vote) reports likely pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors points to a pathogenic effect for E1217A, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.599170 | Disordered | 0.493043 | Uncertain | 0.877 | 0.563 | 0.250 | -10.446 | Likely Pathogenic | 0.605 | Likely Pathogenic | Likely Benign | 0.261 | Likely Benign | -4.52 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.38 | Pathogenic | 0.00 | Affected | 0.2585 | 0.4401 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||
| c.3670C>A | L1224M 2D AIThe SynGAP1 missense variant L1224M is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign) all indicate a tolerated change. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact. When focusing on high‑accuracy predictors, AlphaMissense‑Optimized remains benign and the SGM‑Consensus also supports a benign outcome; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.525368 | Disordered | 0.441554 | Uncertain | 0.871 | 0.543 | 0.500 | -5.614 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -0.55 | Neutral | 0.994 | Probably Damaging | 0.925 | Probably Damaging | 2.38 | Pathogenic | 0.18 | Tolerated | 0.0660 | 0.2486 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||||
| c.3778A>G | K1260E 2D AIThe SynGAP1 missense variant K1260E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict a pathogenic impact, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus remains pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions point to a pathogenic effect, and there is no ClinVar annotation to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.509769 | Disordered | 0.625808 | Binding | 0.890 | 0.575 | 0.250 | -10.913 | Likely Pathogenic | 0.846 | Likely Pathogenic | Ambiguous | 0.462 | Likely Benign | -3.06 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.38 | Pathogenic | 0.00 | Affected | 0.3129 | 0.0877 | 0 | 1 | 0.4 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3794G>A | R1265K 2D  AIThe SynGAP1 missense variant R1265K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.414856 | Structured | 0.782497 | Binding | 0.887 | 0.592 | 0.000 | -5.223 | Likely Benign | 0.951 | Likely Pathogenic | Ambiguous | 0.344 | Likely Benign | -2.49 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 2.38 | Pathogenic | 0.00 | Affected | 0.4829 | 0.3632 | 3 | 2 | 0.6 | -28.01 | |||||||||||||||||||||||||||||||||||||||
| c.3883C>A | Q1295K 2D AIThe SynGAP1 missense variant Q1295K is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (gnomAD ID: 6‑33447931‑C‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. The high‑accuracy consensus (SGM Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a pathogenic prediction (2 pathogenic vs. 1 benign vs. 1 uncertain). AlphaMissense‑Optimized remains benign, while Foldetta (combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. three benign) point to a pathogenic effect. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.892719 | Binding | 0.499 | 0.801 | 0.625 | 6-33447931-C-A | -3.419 | Likely Benign | 0.343 | Ambiguous | Likely Benign | 0.313 | Likely Benign | -3.30 | Deleterious | 0.843 | Possibly Damaging | 0.893 | Possibly Damaging | 2.38 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1782 | 0.3517 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||
| c.1414G>C | E472Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E472Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only Foldetta, which classifies the variant as benign. Tools that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Uncertain or inconclusive predictions come from FoldX, Rosetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic impact for E472Q, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -13.760 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 1.34 | Ambiguous | 0.3 | -0.67 | Ambiguous | 0.34 | Likely Benign | 0.89 | Ambiguous | 0.555 | Likely Pathogenic | -2.95 | Deleterious | 0.994 | Probably Damaging | 0.986 | Probably Damaging | 2.39 | Pathogenic | 0.01 | Affected | 0.1388 | 0.5726 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.2287C>T | L763F 2D AIThe SynGAP1 missense variant L763F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for this variant. There is no ClinVar entry to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -4.127 | Likely Benign | 0.255 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -0.71 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.39 | Pathogenic | 0.19 | Tolerated | 0.0584 | 0.3140 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2368A>T | T790S 2D AIThe SynGAP1 missense variant T790S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for T790S, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | -3.914 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.83 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.39 | Pathogenic | 0.33 | Tolerated | 3.64 | 6 | 0.3416 | 0.4449 | 1 | 1 | -0.1 | -14.03 | ||||||||||||||||||||||||||||||||||||
| c.2369C>G | T790S 2D AIThe SynGAP1 missense variant T790S (ClinVar ID 1020340) is listed as Uncertain in ClinVar and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus, which is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN. Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | Uncertain | 2 | -3.914 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -1.83 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.39 | Pathogenic | 0.33 | Tolerated | 3.64 | 6 | 0.3416 | 0.4449 | 1 | 1 | -0.1 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.2413C>A | L805M 2D AIThe SynGAP1 missense variant L805M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and no Foldetta stability data are available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.775545 | Disordered | 0.827669 | Binding | 0.341 | 0.903 | 0.625 | -4.229 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.034 | Likely Benign | -0.67 | Neutral | 0.029 | Benign | 0.033 | Benign | 2.39 | Pathogenic | 0.00 | Affected | 0.0911 | 0.3867 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||||
| c.2449T>C | S817P 2D AIThe SynGAP1 missense variant S817P is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Overall, the majority of tools and the consensus score indicate a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.490133 | Structured | 0.727082 | Binding | 0.314 | 0.901 | 0.625 | -4.633 | Likely Benign | 0.724 | Likely Pathogenic | Likely Benign | 0.201 | Likely Benign | -3.26 | Deleterious | 0.999 | Probably Damaging | 0.966 | Probably Damaging | 2.39 | Pathogenic | 0.00 | Affected | 0.2272 | 0.6023 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2666G>T | G889V 2D AIThe SynGAP1 missense variant G889V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for G889V, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | -5.781 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -2.11 | Neutral | 0.611 | Possibly Damaging | 0.243 | Benign | 2.39 | Pathogenic | 0.03 | Affected | 0.1198 | 0.4523 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2757G>C | Q919H 2D AIThe SynGAP1 missense variant Q919H has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.911223 | Binding | 0.299 | 0.841 | 0.250 | -3.867 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.153 | Likely Benign | -1.83 | Neutral | 0.989 | Probably Damaging | 0.883 | Possibly Damaging | 2.39 | Pathogenic | 0.03 | Affected | 0.1463 | 0.4203 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2757G>T | Q919H 2D AIThe SynGAP1 missense variant Q919H has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.911223 | Binding | 0.299 | 0.841 | 0.250 | -3.867 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.152 | Likely Benign | -1.83 | Neutral | 0.989 | Probably Damaging | 0.883 | Possibly Damaging | 2.39 | Pathogenic | 0.03 | Affected | 0.1463 | 0.4203 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2762T>C | L921P 2D AIThe SynGAP1 missense variant L921P is recorded in gnomAD (ID 6‑33443314‑T‑C) but has no ClinVar submission. Functional prediction tools cluster into two groups: benign calls from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized; pathogenic calls from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because the votes are split (two benign, one pathogenic, one uncertain). Foldetta, a protein‑folding stability predictor that combines FoldX‑MD and Rosetta outputs, has no reported result for this variant. Consequently, the evidence is evenly divided, with high‑accuracy tools not providing a decisive verdict. The variant is therefore inconclusive; it does not contradict ClinVar status, which currently has no entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.608892 | Disordered | 0.943282 | Binding | 0.311 | 0.845 | 0.375 | 6-33443314-T-C | 1 | 6.20e-7 | -2.087 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.215 | Likely Benign | -1.83 | Neutral | 0.998 | Probably Damaging | 0.958 | Probably Damaging | 2.39 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3581 | 0.1343 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.2792T>G | L931R 2D AIThe SynGAP1 missense variant L931R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus remains pathogenic; Foldetta results are not available. Overall, the preponderance of evidence from multiple in‑silico predictors indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.549308 | Disordered | 0.989212 | Binding | 0.335 | 0.856 | 0.375 | -8.606 | Likely Pathogenic | 0.933 | Likely Pathogenic | Ambiguous | 0.344 | Likely Benign | -3.48 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.39 | Pathogenic | 0.01 | Affected | 0.1241 | 0.1205 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2797C>A | H933N 2D AIThe SynGAP1 missense variant H933N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta are unavailable. Overall, five tools predict pathogenicity versus four predicting benignity, suggesting the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.987531 | Binding | 0.305 | 0.862 | 0.625 | -4.333 | Likely Benign | 0.226 | Likely Benign | Likely Benign | 0.261 | Likely Benign | -3.65 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.39 | Pathogenic | 0.04 | Affected | 0.1897 | 0.3439 | 2 | 1 | -0.3 | -23.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2801T>C | M934T 2D AIThe SynGAP1 missense variant M934T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, and ESM1b, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and the Foldetta stability analysis is unavailable. Based on the majority of predictions and the consensus call, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.762850 | Disordered | 0.984677 | Binding | 0.290 | 0.867 | 0.625 | -2.579 | Likely Benign | 0.823 | Likely Pathogenic | Ambiguous | 0.270 | Likely Benign | -3.33 | Deleterious | 0.811 | Possibly Damaging | 0.424 | Benign | 2.39 | Pathogenic | 0.19 | Tolerated | 0.2331 | 0.2267 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2960A>C | D987A 2D AIThe SynGAP1 D987A missense variant is not reported in ClinVar and has no gnomAD entry. Consensus prediction tools that classify the change as benign include REVEL and ESM1b, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) and the SGM‑Consensus score (Likely Pathogenic) indicate a pathogenic effect. Grouping by agreement, benign predictions are limited to two tools, while pathogenic predictions are supported by seven distinct algorithms. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized returns an uncertain result, SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a Likely Pathogenic classification, and the protein‑folding stability method Foldetta is unavailable for this variant. Overall, the preponderance of evidence points to a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -4.880 | Likely Benign | 0.853 | Likely Pathogenic | Ambiguous | 0.261 | Likely Benign | -3.72 | Deleterious | 0.943 | Possibly Damaging | 0.686 | Possibly Damaging | 2.39 | Pathogenic | 0.02 | Affected | 0.3930 | 0.6846 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.3025G>C | E1009Q 2D AIThe SynGAP1 missense variant E1009Q is not reported in ClinVar (ClinVar status: None) but is present in gnomAD (ID 6‑33443577‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs 2 benign votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, five tools predict pathogenicity while four predict benignity, giving a slight tilt toward pathogenicity. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | 6-33443577-G-C | 1 | 6.20e-7 | -3.423 | Likely Benign | 0.615 | Likely Pathogenic | Likely Benign | 0.057 | Likely Benign | -1.65 | Neutral | 0.980 | Probably Damaging | 0.782 | Possibly Damaging | 2.39 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.1577 | 0.7242 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||
| c.3026A>C | E1009A 2D AIThe SynGAP1 missense variant E1009A is listed in ClinVar (ID 2238288.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) results are unavailable. Overall, the majority of predictions (six pathogenic vs. three benign) lean toward a pathogenic impact, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | Uncertain | 1 | -3.118 | Likely Benign | 0.679 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | -3.06 | Deleterious | 0.980 | Probably Damaging | 0.630 | Possibly Damaging | 2.39 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.3959 | 0.7153 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||
| c.3055C>G | R1019G 2D AIThe SynGAP1 missense variant R1019G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas tools predicting a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and the Foldetta stability analysis is unavailable. Overall, the majority of predictions support a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation (none is available). Thus, the variant is most likely pathogenic based on the current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.856457 | Disordered | 0.966400 | Binding | 0.315 | 0.794 | 0.500 | -4.325 | Likely Benign | 0.614 | Likely Pathogenic | Likely Benign | 0.115 | Likely Benign | -3.34 | Deleterious | 0.800 | Possibly Damaging | 0.496 | Possibly Damaging | 2.39 | Pathogenic | 0.00 | Affected | 0.2946 | 0.3489 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3056G>A | R1019H 2D AIThe SynGAP1 missense variant R1019H is listed in ClinVar (ID 1195115.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443608‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) also benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign impact for R1019H, and this conclusion does not contradict the current ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.966400 | Binding | 0.315 | 0.794 | 0.500 | Conflicting | 2 | 6-33443608-G-A | 67 | 4.15e-5 | -4.610 | Likely Benign | 0.258 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -1.95 | Neutral | 0.995 | Probably Damaging | 0.845 | Possibly Damaging | 2.39 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.2400 | 0.1903 | 2 | 0 | 1.3 | -19.05 | ||||||||||||||||||||||||||||||||
| c.3136C>G | P1046A 2D AIThe SynGAP1 missense variant P1046A is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443688‑C‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | Uncertain | 1 | 6-33443688-C-G | 1 | 6.20e-7 | -3.246 | Likely Benign | 0.048 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.67 | Neutral | 0.001 | Benign | 0.008 | Benign | 2.39 | Pathogenic | 0.29 | Tolerated | 3.77 | 5 | 0.2976 | 0.5358 | -1 | 1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||
| c.3136C>T | P1046S 2D AIThe SynGAP1 missense variant P1046S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence indicates a benign effect, and this consensus does not contradict any ClinVar status (none is available). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.970265 | Disordered | 0.942366 | Binding | 0.364 | 0.898 | 0.750 | -3.909 | Likely Benign | 0.059 | Likely Benign | Likely Benign | 0.096 | Likely Benign | -0.72 | Neutral | 0.126 | Benign | 0.096 | Benign | 2.39 | Pathogenic | 0.70 | Tolerated | 0.2929 | 0.5744 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3715G>T | A1239S 2D AIThe SynGAP1 missense variant A1239S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while SIFT and FATHMM predict it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and no Foldetta stability data are available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.534779 | Binding | 0.887 | 0.542 | 0.250 | -2.847 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.019 | Likely Benign | -0.95 | Neutral | 0.003 | Benign | 0.005 | Benign | 2.39 | Pathogenic | 0.00 | Affected | 0.1974 | 0.4095 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||||||||||
| c.3879C>A | D1293E 2D AIThe SynGAP1 missense variant D1293E is reported in gnomAD (variant ID 6‑33447927‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, SIFT and FATHMM predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | 6-33447927-C-A | -2.627 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -1.86 | Neutral | 0.011 | Benign | 0.008 | Benign | 2.39 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1512 | 0.3425 | 2 | 3 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||
| c.3879C>G | D1293E 2D AIThe SynGAP1 missense variant D1293E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for D1293E, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | -2.627 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.86 | Neutral | 0.011 | Benign | 0.008 | Benign | 2.39 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1512 | 0.3425 | 2 | 3 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.3923G>T | R1308L 2D AIThe SynGAP1 missense variant R1308L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic impact. This assessment does not contradict ClinVar status, as the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.741537 | Disordered | 0.930652 | Binding | 0.378 | 0.904 | 0.750 | -1.838 | Likely Benign | 0.257 | Likely Benign | Likely Benign | 0.360 | Likely Benign | -4.04 | Deleterious | 0.982 | Probably Damaging | 0.982 | Probably Damaging | 2.39 | Pathogenic | 0.00 | Affected | 0.1751 | 0.4930 | -3 | -2 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2408A>C | K803T 2D AIThe SynGAP1 missense variant K803T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. In contrast, the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicate it is likely pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized classifies the variant as benign, whereas the SGM‑Consensus indicates it is likely pathogenic; a Foldetta stability analysis is not available. Overall, the balance of evidence points to a pathogenic effect for K803T, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.827927 | Disordered | 0.733908 | Binding | 0.349 | 0.900 | 0.625 | -3.571 | Likely Benign | 0.584 | Likely Pathogenic | Likely Benign | 0.121 | Likely Benign | -2.90 | Deleterious | 0.946 | Possibly Damaging | 0.741 | Possibly Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.2417 | 0.4395 | 0 | -1 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||||||||||
| c.2487G>C | E829D 2D AIThe SynGAP1 missense variant E829D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors indicates that E829D is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.562014 | Disordered | 0.626045 | Binding | 0.326 | 0.882 | 0.375 | -4.015 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -1.89 | Neutral | 0.217 | Benign | 0.121 | Benign | 2.40 | Pathogenic | 0.00 | Affected | 0.1939 | 0.4964 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2487G>T | E829D 2D AIThe SynGAP1 missense variant E829D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors indicates that E829D is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.562014 | Disordered | 0.626045 | Binding | 0.326 | 0.882 | 0.375 | -4.015 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -1.89 | Neutral | 0.217 | Benign | 0.121 | Benign | 2.40 | Pathogenic | 0.00 | Affected | 0.1939 | 0.4964 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2516A>T | K839M 2D AIThe SynGAP1 missense variant K839M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL indicates a benign likelihood, whereas the remaining predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. The consensus score from the SGM framework, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic.” High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM consensus also reports a likely pathogenic outcome. Foldetta, a protein‑folding stability method that combines FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion is consistent with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.642678 | Disordered | 0.611185 | Binding | 0.282 | 0.865 | 0.375 | -13.688 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.241 | Likely Benign | -3.54 | Deleterious | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.1253 | 0.4481 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||||||||||||
| c.2707G>A | G903S 2D AIThe SynGAP1 missense variant G903S is reported in gnomAD (variant ID 6-33443259‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which labels the variant as “Likely Benign.” Pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for G903S, and this conclusion is not contradicted by ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.680603 | Disordered | 0.549818 | Binding | 0.291 | 0.917 | 0.375 | 6-33443259-G-A | 1 | 6.20e-7 | -3.451 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -1.37 | Neutral | 0.989 | Probably Damaging | 0.871 | Possibly Damaging | 2.40 | Pathogenic | 0.04 | Affected | 3.77 | 5 | 0.2447 | 0.4774 | 0 | 1 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||||||
| c.2756A>T | Q919L 2D AIThe SynGAP1 missense variant Q919L is reported in gnomAD (ID 6‑33443308‑A‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.911223 | Binding | 0.299 | 0.841 | 0.250 | 6-33443308-A-T | 1 | 6.20e-7 | -4.492 | Likely Benign | 0.252 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -2.13 | Neutral | 0.891 | Possibly Damaging | 0.596 | Possibly Damaging | 2.40 | Pathogenic | 0.04 | Affected | 4.32 | 4 | 0.0771 | 0.6454 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||
| c.2761C>A | L921M 2D AIThe SynGAP1 missense variant L921M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for L921M, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.943282 | Binding | 0.311 | 0.845 | 0.375 | -4.485 | Likely Benign | 0.142 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.38 | Neutral | 0.489 | Possibly Damaging | 0.137 | Benign | 2.40 | Pathogenic | 0.02 | Affected | 0.0762 | 0.3732 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2797C>G | H933D 2D AIThe SynGAP1 missense variant H933D is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as Likely Pathogenic, matching the majority of individual scores. AlphaMissense‑Optimized returns an Uncertain result, and no Foldetta stability assessment is available. Overall, the preponderance of evidence points to a pathogenic effect for H933D. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with existing database annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.666105 | Disordered | 0.987531 | Binding | 0.305 | 0.862 | 0.625 | -2.888 | Likely Benign | 0.798 | Likely Pathogenic | Ambiguous | 0.320 | Likely Benign | -4.70 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 2.40 | Pathogenic | 0.04 | Affected | 0.2481 | 0.2717 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||||||||||||
| c.2815C>G | P939A 2D AIThe SynGAP1 missense variant P939A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign versus two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools therefore provide a benign prediction from AlphaMissense‑Optimized, an inconclusive SGM Consensus, and no Foldetta data. Overall, the majority of individual predictors (five pathogenic versus four benign) lean toward a pathogenic interpretation, and this does not contradict the lack of ClinVar annotation. **Thus, the variant is most likely pathogenic based on the current computational predictions.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | -4.614 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -3.57 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.3565 | 0.4336 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3056G>T | R1019L 2D AIThe SynGAP1 missense variant R1019L is listed in ClinVar with an “Uncertain” status (ClinVar ID 3364537.0) and is present in gnomAD (gnomAD ID 6‑33443608‑G‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote) remains pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a pathogenic impact, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.856457 | Disordered | 0.966400 | Binding | 0.315 | 0.794 | 0.500 | Uncertain | 1 | 6-33443608-G-T | 2 | 1.24e-6 | -5.194 | Likely Benign | 0.752 | Likely Pathogenic | Likely Benign | 0.110 | Likely Benign | -3.57 | Deleterious | 0.800 | Possibly Damaging | 0.573 | Possibly Damaging | 2.40 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1850 | 0.4886 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||
| c.3070C>T | L1024F 2D AIThe SynGAP1 missense variant L1024F is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it contains both benign and pathogenic calls. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence favors a benign interpretation, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -5.133 | Likely Benign | 0.544 | Ambiguous | Likely Benign | 0.059 | Likely Benign | -2.08 | Neutral | 0.994 | Probably Damaging | 0.924 | Probably Damaging | 2.40 | Pathogenic | 0.07 | Tolerated | 0.0706 | 0.3708 | 2 | 0 | -1.0 | 34.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3071T>G | L1024R 2D AIThe SynGAP1 missense variant L1024R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (5 pathogenic vs. 3 benign) lean toward a pathogenic interpretation. This assessment does not contradict ClinVar status, as the variant is not yet catalogued there. Thus, based on current computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -3.434 | Likely Benign | 0.841 | Likely Pathogenic | Ambiguous | 0.148 | Likely Benign | -2.41 | Neutral | 0.997 | Probably Damaging | 0.962 | Probably Damaging | 2.40 | Pathogenic | 0.02 | Affected | 0.1335 | 0.1557 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3101C>G | P1034R 2D AIThe SynGAP1 P1034R variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic; Foldetta stability analysis is unavailable. Overall, the predictions are mixed, with a slight tilt toward pathogenicity due to the SGM Consensus result and the number of pathogenic calls. The variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -3.666 | Likely Benign | 0.676 | Likely Pathogenic | Likely Benign | 0.073 | Likely Benign | -3.04 | Deleterious | 0.002 | Benign | 0.005 | Benign | 2.40 | Pathogenic | 0.02 | Affected | 0.1366 | 0.4182 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3266G>T | G1089V 2D AIThe SynGAP1 missense variant G1089V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact for G1089V. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -4.809 | Likely Benign | 0.527 | Ambiguous | Likely Benign | 0.182 | Likely Benign | -2.81 | Deleterious | 0.984 | Probably Damaging | 0.722 | Possibly Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.1326 | 0.4413 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.3617A>C | K1206T 2D AIThe SynGAP1 missense variant K1206T is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are not available. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for K1206T. This conclusion is not contradicted by ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.585406 | Disordered | 0.555819 | Binding | 0.893 | 0.569 | 0.375 | -10.161 | Likely Pathogenic | 0.969 | Likely Pathogenic | Likely Pathogenic | 0.290 | Likely Benign | -3.92 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.40 | Pathogenic | 0.01 | Affected | 0.1913 | 0.3354 | 0 | -1 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||||||||||
| c.3643A>G | K1215E 2D AISynGAP1 missense variant K1215E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: the single benign prediction from REVEL versus pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. Foldetta stability analysis is unavailable. Overall, the consensus of available tools indicates that K1215E is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.497853 | Structured | 0.503613 | Binding | 0.888 | 0.568 | 0.375 | -14.365 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.156 | Likely Benign | -2.65 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.40 | Pathogenic | 0.01 | Affected | 0.3403 | 0.0676 | 0 | 1 | 0.4 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3649G>A | E1217K 2D AIThe SynGAP1 missense variant E1217K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. The high‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is classified as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.599170 | Disordered | 0.493043 | Uncertain | 0.877 | 0.563 | 0.250 | -12.869 | Likely Pathogenic | 0.862 | Likely Pathogenic | Ambiguous | 0.306 | Likely Benign | -3.09 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.1826 | 0.5272 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3671T>A | L1224Q 2D AIThe SynGAP1 missense variant L1224Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate a benign or likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports likely benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for L1224Q, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this is not contradictory to ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.525368 | Disordered | 0.441554 | Uncertain | 0.871 | 0.543 | 0.500 | -6.254 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.87 | Neutral | 0.994 | Probably Damaging | 0.900 | Possibly Damaging | 2.40 | Pathogenic | 0.13 | Tolerated | 0.1018 | 0.0558 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3713A>G | Q1238R 2D AIThe SynGAP1 missense variant Q1238R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Consensus from standard predictors shows a split: benign calls come from REVEL, PROVEAN, and AlphaMissense‑Optimized, whereas pathogenic calls arise from polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessment gives AlphaMissense‑Optimized benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence favors a pathogenic interpretation, and this conclusion does not conflict with the ClinVar status, which currently contains no assertion for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.562014 | Disordered | 0.548882 | Binding | 0.855 | 0.545 | 0.250 | -10.216 | Likely Pathogenic | 0.477 | Ambiguous | Likely Benign | 0.229 | Likely Benign | -2.29 | Neutral | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.40 | Pathogenic | 0.04 | Affected | 0.1165 | 0.2019 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3779A>G | K1260R 2D AIThe SynGAP1 missense variant K1260R is catalogued in gnomAD (ID 6‑33446771‑A‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; those that predict pathogenicity are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective predictions point to a benign impact, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.509769 | Disordered | 0.625808 | Binding | 0.890 | 0.575 | 0.250 | 6-33446771-A-G | 1 | 6.20e-7 | -6.033 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.254 | Likely Benign | -2.29 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.40 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3655 | 0.0878 | 2 | 3 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||
| c.3795G>C | R1265S 2D AIThe SynGAP1 missense variant R1265S is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.414856 | Structured | 0.782497 | Binding | 0.887 | 0.592 | 0.000 | -9.874 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.289 | Likely Benign | -4.96 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.3525 | 0.3993 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||||||||
| c.3795G>T | R1265S 2D AIThe SynGAP1 missense variant R1265S is not reported in ClinVar and has no entry in gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.414856 | Structured | 0.782497 | Binding | 0.887 | 0.592 | 0.000 | -9.874 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.289 | Likely Benign | -4.96 | Deleterious | 0.997 | Probably Damaging | 0.994 | Probably Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.3525 | 0.3993 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||||||||
| c.3838A>C | M1280L 2D AIThe SynGAP1 missense variant M1280L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for M1280L, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | -1.391 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.204 | Likely Benign | -2.23 | Neutral | 0.052 | Benign | 0.017 | Benign | 2.40 | Pathogenic | 0.00 | Affected | 0.1113 | 0.3186 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3838A>T | M1280L 2D AIThe SynGAP1 missense variant M1280L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar status, as no ClinVar claim exists for this variant. Thus, based on current computational predictions, the M1280L variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.822030 | Binding | 0.510 | 0.726 | 0.875 | -1.391 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.204 | Likely Benign | -2.23 | Neutral | 0.052 | Benign | 0.017 | Benign | 2.40 | Pathogenic | 0.00 | Affected | 0.1113 | 0.3186 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3926T>A | V1309E 2D AIThe SynGAP1 missense variant V1309E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.948596 | Binding | 0.402 | 0.907 | 0.750 | -3.393 | Likely Benign | 0.333 | Likely Benign | Likely Benign | 0.294 | Likely Benign | -2.40 | Neutral | 0.767 | Possibly Damaging | 0.473 | Possibly Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.1098 | 0.1727 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3926T>G | V1309G 2D AIThe SynGAP1 missense variant V1309G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs. 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence points toward a benign impact, and this conclusion does not contradict any ClinVar annotation because the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.712013 | Disordered | 0.948596 | Binding | 0.402 | 0.907 | 0.750 | -2.618 | Likely Benign | 0.160 | Likely Benign | Likely Benign | 0.326 | Likely Benign | -3.40 | Deleterious | 0.391 | Benign | 0.767 | Possibly Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.1901 | 0.2144 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.1786C>G | R596G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R596G is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity largely agree: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a deleterious effect, while premPS remains uncertain. No tools predict a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Based on the consensus of these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.017797 | Structured | 0.135423 | Uncertain | 0.918 | 0.134 | 0.000 | -13.525 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 4.02 | Destabilizing | 0.1 | 2.36 | Destabilizing | 3.19 | Destabilizing | 0.81 | Ambiguous | 0.629 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 0.3214 | 0.2316 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1786C>T | R596C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R596C is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33440838‑C‑T). Prediction tools that indicate a benign effect include only premPS. All other evaluated algorithms—REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—classify the variant as pathogenic or likely pathogenic, while Rosetta remains inconclusive. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. **Thus, the variant is most likely pathogenic based on the collective predictions, which does not contradict the ClinVar uncertain status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.017797 | Structured | 0.135423 | Uncertain | 0.918 | 0.134 | 0.000 | Conflicting | 2 | 6-33440838-C-T | 6 | 3.72e-6 | -10.805 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | 2.94 | Destabilizing | 0.0 | 1.49 | Ambiguous | 2.22 | Destabilizing | -0.03 | Likely Benign | 0.633 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.3429 | 0.2211 | -4 | -3 | 7.0 | -53.05 | 230.7 | 97.9 | -0.1 | 0.0 | -0.3 | 0.4 | X | X | Potentially Pathogenic | The guanidinium group of Arg596, located in an α helix (res. Glu582-Met603), forms a salt bridge with the carboxylate group of Glu495 from another α helix (res. Leu489-Glu519). In the WT simulations, the side chain of Arg596 hydrogen bonds with the backbone carbonyl groups of Asn487, Glu486, Arg485, and Phe484. Additionally, Arg596 can hydrogen bond with the carboxamide group of the Asn487 side chain on an opposing loop that links two α helices (res. Ala461-Arg475, res. Leu489-Glu519).In the variant simulations, the thiol group of the Cys596 side chain is unable to form salt bridges or any of the hydrogen bonds that the Arg596 side chain can. Thus, the residue swap could affect the tertiary structure assembly more profoundly than observed in the simulations. Notably, Arg596 plays a key role in positioning the aforementioned loop, which is crucial for the placement of the “arginine finger” or the Arg485 side chain during RasGTPase activation. | ||||||||||||
| c.2248G>T | G750W 2D AIThe SynGAP1 missense variant G750W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, while the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the variant as damaging. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the preponderance of evidence from multiple in silico predictors indicates that G750W is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -8.433 | Likely Pathogenic | 0.427 | Ambiguous | Likely Benign | 0.135 | Likely Benign | -3.30 | Deleterious | 1.000 | Probably Damaging | 0.994 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 0.0738 | 0.4009 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.2450C>T | S817L 2D AIThe SynGAP1 missense variant S817L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.490133 | Structured | 0.727082 | Binding | 0.314 | 0.901 | 0.625 | -7.612 | In-Between | 0.659 | Likely Pathogenic | Likely Benign | 0.226 | Likely Benign | -3.90 | Deleterious | 0.997 | Probably Damaging | 0.945 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 0.1226 | 0.5662 | -3 | -2 | 4.6 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2492A>G | E831G 2D AIThe SynGAP1 missense variant E831G is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and pathogenic (polyPhen‑2 HumDiv, SIFT, FATHMM). The high‑accuracy AlphaMissense‑Optimized model predicts a benign effect, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. No Foldetta stability assessment is available. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not contradict any existing ClinVar annotation, as none is present. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.626927 | Disordered | 0.617732 | Binding | 0.319 | 0.874 | 0.375 | -4.769 | Likely Benign | 0.305 | Likely Benign | Likely Benign | 0.119 | Likely Benign | -2.27 | Neutral | 0.625 | Possibly Damaging | 0.252 | Benign | 2.41 | Pathogenic | 0.04 | Affected | 0.2924 | 0.6205 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.2666G>A | G889E 2D AIThe SynGAP1 missense variant G889E is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions lean toward a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | -5.352 | Likely Benign | 0.572 | Likely Pathogenic | Likely Benign | 0.083 | Likely Benign | -1.96 | Neutral | 0.611 | Possibly Damaging | 0.187 | Benign | 2.41 | Pathogenic | 0.02 | Affected | 0.1442 | 0.3719 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||||||||
| c.2762T>A | L921Q 2D AIThe SynGAP1 missense variant L921Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.943282 | Binding | 0.311 | 0.845 | 0.375 | -2.389 | Likely Benign | 0.311 | Likely Benign | Likely Benign | 0.132 | Likely Benign | -0.62 | Neutral | 0.994 | Probably Damaging | 0.940 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 0.1097 | 0.1119 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2762T>G | L921R 2D AIThe SynGAP1 missense variant L921R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no conflict with ClinVar status. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.608892 | Disordered | 0.943282 | Binding | 0.311 | 0.845 | 0.375 | -2.205 | Likely Benign | 0.563 | Ambiguous | Likely Benign | 0.217 | Likely Benign | -1.19 | Neutral | 0.994 | Probably Damaging | 0.912 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 0.1306 | 0.0761 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2791C>T | L931F 2D AIThe SynGAP1 missense variant L931F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, and ESM1b, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. A high‑accuracy consensus (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a 2‑to‑2 split, leaving the consensus inconclusive. Foldetta, which assesses protein‑folding stability via FoldX‑MD and Rosetta, has no available result for this variant. Consequently, the overall evidence leans toward pathogenicity, driven by the majority of standard predictors, and does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.549308 | Disordered | 0.989212 | Binding | 0.335 | 0.856 | 0.375 | -5.101 | Likely Benign | 0.790 | Likely Pathogenic | Ambiguous | 0.167 | Likely Benign | -2.00 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.41 | Pathogenic | 0.04 | Affected | 0.0718 | 0.3208 | 2 | 0 | -1.0 | 34.02 | ||||||||||||||||||||||||||||||||||||||||
| c.2863T>G | S955A 2D AIThe SynGAP1 missense variant S955A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.984871 | Disordered | 0.945325 | Binding | 0.350 | 0.924 | 0.750 | -4.258 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -0.71 | Neutral | 0.004 | Benign | 0.006 | Benign | 2.41 | Pathogenic | 0.00 | Affected | 0.4262 | 0.5038 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
Found 8751 rows. Show 800 rows per page. Page 3/11 « Previous | Next »