
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain and Structure information: based on WT protein | Annotated databases | Deep learning-based pathogenicity predictions | Folding stability-based pathogenicity predictions | Sequence/structure-based pathogenicity predictions | Phase Separation | Evolutionary/physical properties | Molecular Dynamics-based analysis | DOI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | IUPred2 | ANCHOR2 | AlphaFold | MobiDB | PhosphoSitePlus | ClinVar | gnomAD | ESM1b | AlphaMissense | FoldX | Rosetta | Foldetta | PremPS | REVEL | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PSMutPred | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | |||||||||||||||||||||||||||||||||||||||||||||
| Score | Prediction | Score | Prediction | pLDDT | disorder | disorder | LTP | HTP | KL | PTM | Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | IP RF | SP RF | Prediction | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.2055G>T | L685F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L685F is not reported in ClinVar and has no gnomAD entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, and FATHMM, whereas pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the change as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic, while Foldetta’s stability analysis is inconclusive. FoldX and Rosetta predictions are uncertain and are treated as unavailable. Overall, the preponderance of evidence points to a pathogenic impact for the variant, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.162061 | Uncertain | 0.913 | 0.280 | 0.000 | -12.304 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.62 | Ambiguous | 0.2 | 1.80 | Ambiguous | 1.71 | Ambiguous | 0.50 | Likely Benign | 0.300 | Likely Benign | -3.99 | Deleterious | 0.999 | Probably Damaging | 0.895 | Possibly Damaging | 3.33 | Benign | 0.01 | Affected | 0.0724 | 0.2367 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.2056G>A | G686S 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant G686S is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include FoldX, Foldetta, SIFT, and FATHMM. Those that predict pathogenicity are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; the SGM‑Consensus score is also “Likely Pathogenic.” Uncertain predictions from Rosetta and premPS are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta (a combined FoldX‑MD and Rosetta stability method) as benign. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.177104 | Uncertain | 0.919 | 0.268 | 0.000 | -10.884 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.30 | Likely Benign | 0.3 | 0.50 | Ambiguous | 0.40 | Likely Benign | 0.69 | Ambiguous | 0.537 | Likely Pathogenic | -5.29 | Deleterious | 0.998 | Probably Damaging | 0.929 | Probably Damaging | 3.46 | Benign | 0.06 | Tolerated | 0.2558 | 0.4355 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.2056G>C | G686R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G686R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta, Foldetta, and FATHMM, whereas the remaining tools (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized) all predict a pathogenic impact; FoldX is listed as uncertain and is treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.177104 | Uncertain | 0.919 | 0.268 | 0.000 | -14.801 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.79 | Ambiguous | 0.3 | 0.15 | Likely Benign | 0.47 | Likely Benign | 1.10 | Destabilizing | 0.503 | Likely Pathogenic | -7.21 | Deleterious | 0.974 | Probably Damaging | 0.449 | Possibly Damaging | 3.46 | Benign | 0.00 | Affected | 0.0951 | 0.3580 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.2056G>T | G686C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G686C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from FoldX, Rosetta, FATHMM, and the protein‑folding stability method Foldetta; pathogenic predictions arise from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, with the SGM‑Consensus score labeling the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as Pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.177104 | Uncertain | 0.919 | 0.268 | 0.000 | -12.790 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.25 | Likely Benign | 0.2 | 0.41 | Likely Benign | 0.33 | Likely Benign | 0.93 | Ambiguous | 0.553 | Likely Pathogenic | -8.14 | Deleterious | 1.000 | Probably Damaging | 0.988 | Probably Damaging | 3.31 | Benign | 0.02 | Affected | 0.1209 | 0.3246 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.2057G>A | G686D 2D  AIThe SynGAP1 missense variant G686D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.177104 | Uncertain | 0.919 | 0.268 | 0.000 | -14.109 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.00 | Destabilizing | 1.7 | 2.61 | Destabilizing | 2.31 | Destabilizing | 1.05 | Destabilizing | 0.563 | Likely Pathogenic | -6.28 | Deleterious | 1.000 | Probably Damaging | 0.967 | Probably Damaging | 3.40 | Benign | 0.00 | Affected | 0.1720 | 0.2196 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.2057G>C | G686A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G686A is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, Rosetta, SIFT, and FATHMM, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy methods give a pathogenic verdict from AlphaMissense‑Optimized and a Likely Pathogenic consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), while Foldetta predicts benign stability. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.177104 | Uncertain | 0.919 | 0.268 | 0.000 | -9.975 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | -0.39 | Likely Benign | 0.2 | -0.46 | Likely Benign | -0.43 | Likely Benign | 0.58 | Ambiguous | 0.321 | Likely Benign | -5.19 | Deleterious | 0.993 | Probably Damaging | 0.732 | Possibly Damaging | 3.37 | Benign | 0.13 | Tolerated | 0.3744 | 0.4131 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.2057G>T | G686V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G686V has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on benign impact are Rosetta and FATHMM, while the majority of other in silico predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score (Likely Pathogenic) indicate a pathogenic effect. Uncertain results come from FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for G686V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.177104 | Uncertain | 0.919 | 0.268 | 0.000 | -13.751 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.89 | Ambiguous | 0.5 | 0.21 | Likely Benign | 0.55 | Ambiguous | 0.74 | Ambiguous | 0.570 | Likely Pathogenic | -8.08 | Deleterious | 1.000 | Probably Damaging | 0.979 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.1059 | 0.3646 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2059C>G | R687G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 R687G missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the remaining tools (SGM‑Consensus, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) uniformly predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta indicates a destabilizing, pathogenic effect. AlphaMissense‑Optimized is uncertain and therefore treated as unavailable. Overall, the variant is most likely pathogenic based on the consensus of predictive tools, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.191060 | Uncertain | 0.914 | 0.259 | 0.000 | -12.900 | Likely Pathogenic | 0.953 | Likely Pathogenic | Ambiguous | 2.94 | Destabilizing | 0.3 | 2.53 | Destabilizing | 2.74 | Destabilizing | 1.27 | Destabilizing | 0.360 | Likely Benign | -6.26 | Deleterious | 1.000 | Probably Damaging | 0.990 | Probably Damaging | 3.88 | Benign | 0.01 | Affected | 0.2508 | 0.2119 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.205A>C | I69L 2D  AIThe SynGAP1 missense variant I69L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates benign; Foldetta results are not available. Overall, the majority of evidence supports a benign impact for I69L, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | -2.065 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -0.32 | Neutral | 0.267 | Benign | 0.141 | Benign | 4.27 | Benign | 0.00 | Affected | 0.0636 | 0.3447 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.205A>G | I69V 2D  AIThe SynGAP1 missense variant I69V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” status. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates benign; Foldetta results are not available. Overall, the consensus of the available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | -2.809 | Likely Benign | 0.176 | Likely Benign | Likely Benign | 0.080 | Likely Benign | 0.06 | Neutral | 0.267 | Benign | 0.141 | Benign | 4.24 | Benign | 0.00 | Affected | 0.0972 | 0.3142 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.205A>T | I69F 2D  AIThe SynGAP1 missense variant I69F is reported in gnomAD (variant ID 6‑33425813‑A‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is also benign. Foldetta results are unavailable. Overall, the preponderance of evidence indicates that I69F is most likely benign, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | 6-33425813-A-T | 2 | 1.24e-6 | -3.747 | Likely Benign | 0.251 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -0.99 | Neutral | 0.824 | Possibly Damaging | 0.507 | Possibly Damaging | 4.12 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0412 | 0.2743 | 0 | 1 | -1.7 | 34.02 | ||||||||||||||||||||||||||||||||||
| c.2060G>A | R687Q 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant R687Q is annotated in ClinVar as benign (ClinVar ID 2693600.0) and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, Foldetta, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant as benign, SGM‑Consensus indicating pathogenicity, and Foldetta (integrating FoldX‑MD and Rosetta outputs) classifying it as benign. With three high‑accuracy tools giving benign or uncertain results and only one (SGM‑Consensus) suggesting pathogenicity, the overall evidence leans toward a benign effect. This prediction aligns with the ClinVar benign classification, indicating no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.191060 | Uncertain | 0.914 | 0.259 | 0.000 | Likely Benign | 1 | -10.002 | Likely Pathogenic | 0.575 | Likely Pathogenic | Likely Benign | 0.92 | Ambiguous | 0.1 | -0.37 | Likely Benign | 0.28 | Likely Benign | 1.55 | Destabilizing | 0.401 | Likely Benign | -3.37 | Deleterious | 1.000 | Probably Damaging | 0.844 | Possibly Damaging | 3.91 | Benign | 0.03 | Affected | 3.42 | 17 | 0.2143 | 0.1952 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||
| c.2060G>C | R687P 2D 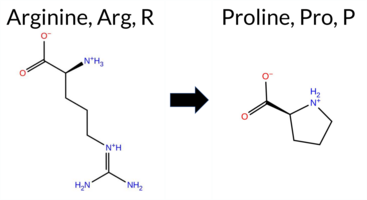 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R687P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, whereas the remaining eleven tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also indicates pathogenicity. With the overwhelming majority of evidence pointing to a damaging effect and no conflicting ClinVar annotation, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.191060 | Uncertain | 0.914 | 0.259 | 0.000 | -15.697 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.31 | Destabilizing | 0.3 | 6.63 | Destabilizing | 4.47 | Destabilizing | 0.89 | Ambiguous | 0.553 | Likely Pathogenic | -6.12 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.87 | Benign | 0.01 | Affected | 0.1811 | 0.3159 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.2060G>T | R687L 2D 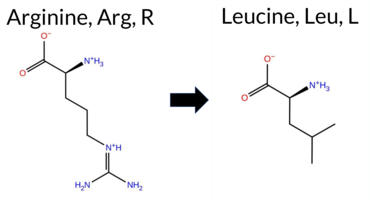 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R687L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on benign include REVEL, SIFT, ESM1b, and FATHMM, while those that agree on pathogenic are AlphaMissense‑Default, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The remaining tools—AlphaMissense‑Optimized, FoldX, Foldetta, and premPS—return uncertain or inconclusive results. High‑accuracy assessments are likewise inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a tie, and Foldetta is uncertain. Consequently, the evidence does not strongly support either benign or pathogenic classification. The variant is therefore most likely inconclusive, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.054297 | Structured | 0.191060 | Uncertain | 0.914 | 0.259 | 0.000 | -6.925 | Likely Benign | 0.901 | Likely Pathogenic | Ambiguous | 1.43 | Ambiguous | 0.3 | 0.05 | Likely Benign | 0.74 | Ambiguous | 0.83 | Ambiguous | 0.448 | Likely Benign | -5.76 | Deleterious | 1.000 | Probably Damaging | 0.987 | Probably Damaging | 3.90 | Benign | 0.10 | Tolerated | 0.1252 | 0.3376 | -3 | -2 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||
| c.2062G>A | E688K 2D 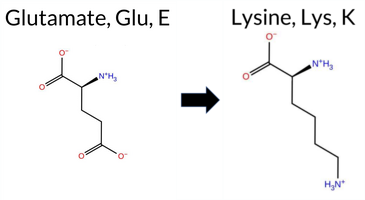 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E688K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, FoldX, FATHMM, and Foldetta, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are reported by Rosetta and premPS. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign stability. Overall, the majority of conventional predictors and the SGM Consensus lean toward pathogenicity, and there is no conflict with ClinVar status because the variant is not yet catalogued. Thus, based on current computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.211124 | Uncertain | 0.947 | 0.223 | 0.000 | -15.177 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.44 | Likely Benign | 0.6 | -0.60 | Ambiguous | -0.08 | Likely Benign | 0.77 | Ambiguous | 0.469 | Likely Benign | -3.49 | Deleterious | 0.998 | Probably Damaging | 0.975 | Probably Damaging | 3.27 | Benign | 0.01 | Affected | 0.2458 | 0.5518 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.2062G>C | E688Q 2D 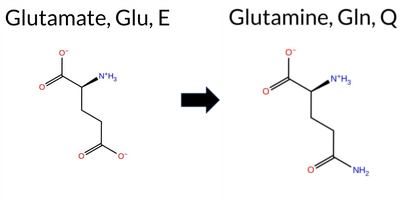 AIThe SynGAP1 missense variant E688Q is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic outcome: SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Two tools (premPS and AlphaMissense‑Optimized) give uncertain results and are treated as unavailable. High‑accuracy assessments further show SGM‑Consensus as Likely Pathogenic, Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign, and AlphaMissense‑Optimized as uncertain. Overall, the balance of evidence leans toward pathogenicity, with no ClinVar annotation to contradict this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.211124 | Uncertain | 0.947 | 0.223 | 0.000 | -9.419 | Likely Pathogenic | 0.943 | Likely Pathogenic | Ambiguous | 0.17 | Likely Benign | 0.9 | 0.15 | Likely Benign | 0.16 | Likely Benign | 0.59 | Ambiguous | 0.302 | Likely Benign | -2.53 | Deleterious | 0.997 | Probably Damaging | 0.973 | Probably Damaging | 3.27 | Benign | 0.15 | Tolerated | 0.1154 | 0.5387 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.2063A>C | E688A 2D 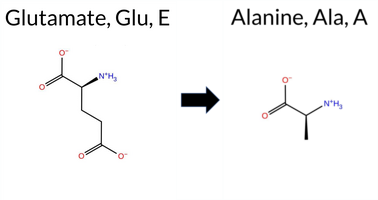 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E688A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Foldetta, and FATHMM, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX, Rosetta, and premPS are inconclusive. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts benign. Overall, the majority of evidence points to a pathogenic effect for E688A. This conclusion is consistent with the absence of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.211124 | Uncertain | 0.947 | 0.223 | 0.000 | -13.556 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.55 | Ambiguous | 0.5 | -0.53 | Ambiguous | 0.01 | Likely Benign | 0.68 | Ambiguous | 0.495 | Likely Benign | -5.55 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.26 | Benign | 0.01 | Affected | 0.3806 | 0.5296 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.2063A>G | E688G 2D 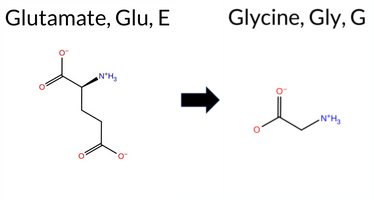 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E688G has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM. Those that predict a pathogenic effect include FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain or inconclusive predictions come from Foldetta, premPS, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain (treated as unavailable). Overall, the majority of reliable tools predict a deleterious effect. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.211124 | Uncertain | 0.947 | 0.223 | 0.000 | -14.338 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 2.17 | Destabilizing | 0.5 | 1.44 | Ambiguous | 1.81 | Ambiguous | 0.86 | Ambiguous | 0.486 | Likely Benign | -6.55 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.27 | Benign | 0.00 | Affected | 0.3059 | 0.4433 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.2063A>T | E688V 2D 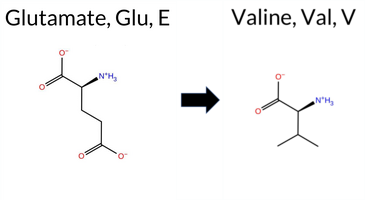 3DClick to see structure in 3D Viewer AIThe SynGAP1 E688V missense change is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from FoldX, Foldetta, premPS, and FATHMM, while pathogenic calls are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; Rosetta is uncertain. High‑accuracy methods give divergent results: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, whereas Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of tools support a pathogenic effect, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.211124 | Uncertain | 0.947 | 0.223 | 0.000 | -14.642 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | -0.02 | Likely Benign | 0.6 | -0.63 | Ambiguous | -0.33 | Likely Benign | 0.37 | Likely Benign | 0.532 | Likely Pathogenic | -6.62 | Deleterious | 0.998 | Probably Damaging | 0.983 | Probably Damaging | 3.19 | Benign | 0.02 | Affected | 0.0681 | 0.5831 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.2064G>C | E688D 2D 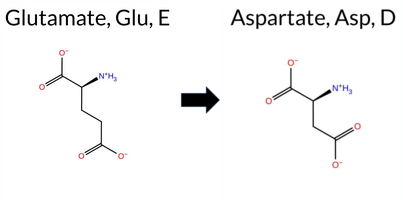 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E688D is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. Tools that predict a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy methods specifically give AlphaMissense‑Optimized a pathogenic prediction, SGM‑Consensus a pathogenic prediction, and Foldetta an uncertain result. Overall, the majority of evidence points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.211124 | Uncertain | 0.947 | 0.223 | 0.000 | -11.890 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 1.79 | Ambiguous | 0.5 | 0.53 | Ambiguous | 1.16 | Ambiguous | 0.91 | Ambiguous | 0.302 | Likely Benign | -2.89 | Deleterious | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.47 | Benign | 0.08 | Tolerated | 0.1722 | 0.3492 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.2064G>T | E688D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E688D is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. Tools that predict a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy methods specifically give: AlphaMissense‑Optimized – pathogenic; SGM‑Consensus – pathogenic; Foldetta – uncertain. Taken together, the majority of evidence points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.211124 | Uncertain | 0.947 | 0.223 | 0.000 | -11.890 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 1.79 | Ambiguous | 0.5 | 0.53 | Ambiguous | 1.16 | Ambiguous | 0.91 | Ambiguous | 0.302 | Likely Benign | -2.89 | Deleterious | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.47 | Benign | 0.08 | Tolerated | 0.1722 | 0.3492 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.2065C>A | L689I 2D 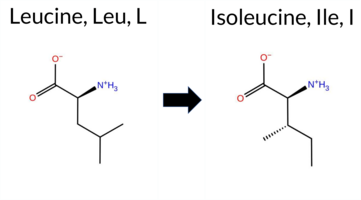 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L689I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar, while those that predict a pathogenic effect are polyPhen2_HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also unavailable. No folding‑stability evidence supports a deleterious change. Overall, the balance of evidence slightly favors a benign interpretation, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.042364 | Structured | 0.227227 | Uncertain | 0.963 | 0.248 | 0.000 | -11.196 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | 1.71 | Ambiguous | 0.1 | 1.12 | Ambiguous | 1.42 | Ambiguous | 0.85 | Ambiguous | 0.180 | Likely Benign | -1.97 | Neutral | 0.822 | Possibly Damaging | 0.381 | Benign | 3.44 | Benign | 0.00 | Affected | 0.0921 | 0.3479 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.2065C>G | L689V 2D 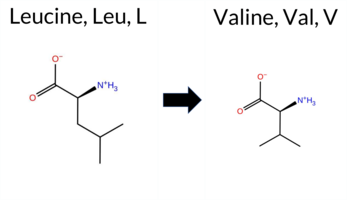 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L689V is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) and the SGM Consensus (Likely Pathogenic) predict a pathogenic impact. The high‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain; SGM Consensus is Likely Pathogenic; Foldetta predicts a pathogenic effect. Taken together, the preponderance of evidence points to a pathogenic effect for L689V. This conclusion is not contradicted by ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.227227 | Uncertain | 0.963 | 0.248 | 0.000 | -11.387 | Likely Pathogenic | 0.862 | Likely Pathogenic | Ambiguous | 2.98 | Destabilizing | 0.1 | 2.25 | Destabilizing | 2.62 | Destabilizing | 1.32 | Destabilizing | 0.234 | Likely Benign | -2.97 | Deleterious | 0.926 | Possibly Damaging | 0.481 | Possibly Damaging | 3.27 | Benign | 0.00 | Affected | 0.1393 | 0.3189 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2065C>T | L689F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L689F is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict a pathogenic impact: FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus. Two tools (Rosetta and premPS) yield uncertain results. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.227227 | Uncertain | 0.963 | 0.248 | 0.000 | -9.817 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 2.45 | Destabilizing | 0.2 | 1.95 | Ambiguous | 2.20 | Destabilizing | 0.67 | Ambiguous | 0.286 | Likely Benign | -3.98 | Deleterious | 0.999 | Probably Damaging | 0.860 | Possibly Damaging | 3.18 | Benign | 0.00 | Affected | 0.0608 | 0.2891 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.2066T>A | L689H 2D 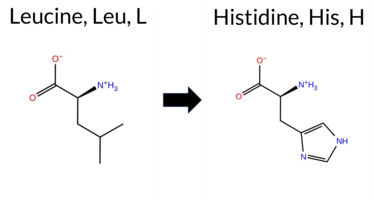 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L689H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods further support this: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.227227 | Uncertain | 0.963 | 0.248 | 0.000 | -14.659 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.40 | Destabilizing | 0.1 | 2.50 | Destabilizing | 2.95 | Destabilizing | 2.21 | Destabilizing | 0.532 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.14 | Benign | 0.00 | Affected | 0.1013 | 0.0456 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2066T>C | L689P 2D 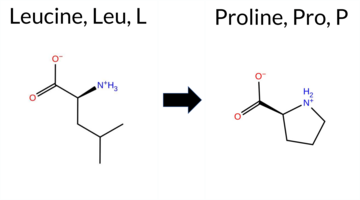 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L689P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.227227 | Uncertain | 0.963 | 0.248 | 0.000 | -17.900 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.76 | Destabilizing | 0.2 | 13.35 | Destabilizing | 10.06 | Destabilizing | 2.29 | Destabilizing | 0.631 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.17 | Benign | 0.00 | Affected | 0.3668 | 0.1353 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2066T>G | L689R 2D 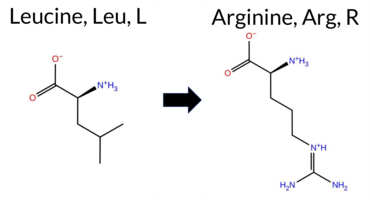 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L689R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (both HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate pathogenicity, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is pathogenic. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.227227 | Uncertain | 0.963 | 0.248 | 0.000 | -17.776 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 5.91 | Destabilizing | 0.6 | 5.61 | Destabilizing | 5.76 | Destabilizing | 2.14 | Destabilizing | 0.609 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 0.932 | Probably Damaging | 3.15 | Benign | 0.00 | Affected | 0.1208 | 0.0530 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2068T>A | S690T 2D 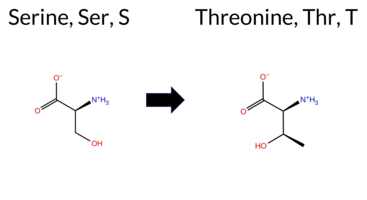 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S690T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, FATHMM, and polyPhen‑2 HumVar, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. Predictions that are inconclusive (FoldX, premPS, AlphaMissense‑Optimized) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the balance of evidence leans toward pathogenicity, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.055536 | Structured | 0.247926 | Uncertain | 0.944 | 0.253 | 0.000 | -11.380 | Likely Pathogenic | 0.845 | Likely Pathogenic | Ambiguous | 0.99 | Ambiguous | 0.2 | -0.21 | Likely Benign | 0.39 | Likely Benign | 0.67 | Ambiguous | 0.311 | Likely Benign | -2.84 | Deleterious | 0.943 | Possibly Damaging | 0.267 | Benign | 3.37 | Benign | 0.01 | Affected | 0.1059 | 0.4674 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.2068T>G | S690A 2D 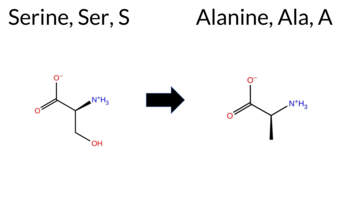 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S690A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta reports an uncertain stability change, so these results are treated as unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.055536 | Structured | 0.247926 | Uncertain | 0.944 | 0.253 | 0.000 | -8.763 | Likely Pathogenic | 0.318 | Likely Benign | Likely Benign | -0.82 | Ambiguous | 0.0 | -2.17 | Stabilizing | -1.50 | Ambiguous | 0.45 | Likely Benign | 0.217 | Likely Benign | -2.55 | Deleterious | 0.887 | Possibly Damaging | 0.738 | Possibly Damaging | 3.45 | Benign | 0.21 | Tolerated | 0.4619 | 0.3131 | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||
| c.2069C>A | S690Y 2D 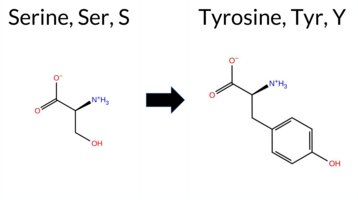 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S690Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM, whereas the majority of other in silico predictors (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) classify the variant as pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus confirms a Likely Pathogenic status, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic effect. Based on the preponderance of pathogenic predictions and the absence of benign consensus, the variant is most likely pathogenic, with no contradiction to ClinVar status (which has no entry for this variant). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.055536 | Structured | 0.247926 | Uncertain | 0.944 | 0.253 | 0.000 | -14.051 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 11.45 | Destabilizing | 3.1 | 3.02 | Destabilizing | 7.24 | Destabilizing | 0.16 | Likely Benign | 0.381 | Likely Benign | -5.76 | Deleterious | 0.999 | Probably Damaging | 0.935 | Probably Damaging | 3.39 | Benign | 0.00 | Affected | 0.0512 | 0.4643 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||
| c.2069C>G | S690C 2D 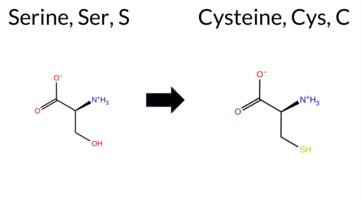 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S690C is not reported in ClinVar and has no gnomAD entry. Consensus predictions from high‑accuracy tools show a split: AlphaMissense‑Optimized rates it benign, whereas the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, also predicts a benign effect. In contrast, the broader set of in silico predictors is divided: benign calls come from REVEL, FoldX, Rosetta, Foldetta, and FATHMM; pathogenic calls arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The premPS score is uncertain. Overall, the majority of tools (seven pathogenic vs. six benign) lean toward a pathogenic interpretation, but the presence of strong benign evidence from several high‑confidence methods tempers this conclusion. Thus, the variant is most likely pathogenic, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.055536 | Structured | 0.247926 | Uncertain | 0.944 | 0.253 | 0.000 | -10.651 | Likely Pathogenic | 0.749 | Likely Pathogenic | Likely Benign | 0.26 | Likely Benign | 0.0 | 0.40 | Likely Benign | 0.33 | Likely Benign | 0.82 | Ambiguous | 0.358 | Likely Benign | -4.69 | Deleterious | 0.999 | Probably Damaging | 0.944 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.0787 | 0.4612 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.2069C>T | S690F 2D 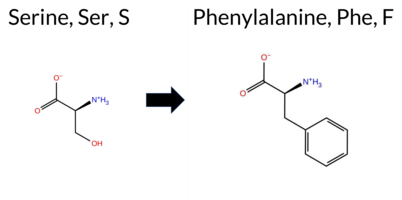 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S690F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of algorithms predict a pathogenic outcome: FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). The high‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are inconclusive or missing. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.055536 | Structured | 0.247926 | Uncertain | 0.944 | 0.253 | 0.000 | -14.325 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 9.85 | Destabilizing | 2.4 | 2.17 | Destabilizing | 6.01 | Destabilizing | 0.51 | Ambiguous | 0.384 | Likely Benign | -5.76 | Deleterious | 0.999 | Probably Damaging | 0.935 | Probably Damaging | 3.39 | Benign | 0.00 | Affected | 0.0498 | 0.4800 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||
| c.206T>A | I69N 2D 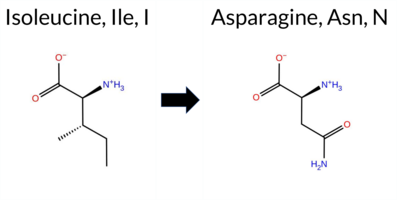 AIThe SynGAP1 missense variant I69N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | -3.220 | Likely Benign | 0.631 | Likely Pathogenic | Likely Benign | 0.110 | Likely Benign | -0.90 | Neutral | 0.943 | Possibly Damaging | 0.781 | Possibly Damaging | 4.10 | Benign | 0.00 | Affected | 0.0859 | 0.0412 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.206T>C | I69T 2D 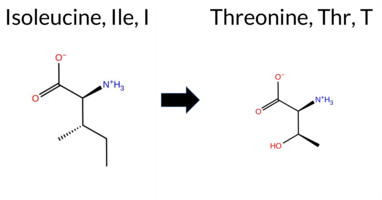 AIThe SynGAP1 missense variant I69T is listed in gnomAD (ID 6‑33425814‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as “Likely Benign” (three benign votes versus one pathogenic). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of evidence points to a benign effect for I69T, and this conclusion does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | 6-33425814-T-C | 1 | 6.20e-7 | -2.978 | Likely Benign | 0.755 | Likely Pathogenic | Likely Benign | 0.116 | Likely Benign | -0.79 | Neutral | 0.824 | Possibly Damaging | 0.507 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1022 | 0.0891 | -1 | 0 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||||
| c.206T>G | I69S 2D 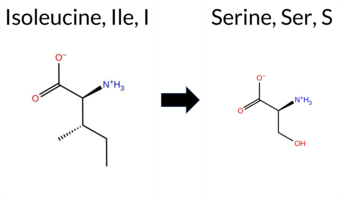 AIThe SynGAP1 I69S missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current predictive data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | -1.880 | Likely Benign | 0.634 | Likely Pathogenic | Likely Benign | 0.152 | Likely Benign | -0.78 | Neutral | 0.824 | Possibly Damaging | 0.507 | Possibly Damaging | 4.14 | Benign | 0.00 | Affected | 0.2672 | 0.0782 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2071A>G | T691A 2D 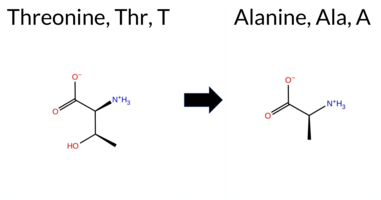 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T691A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign or tolerated. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while premPS and ESM1b are uncertain. The SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, SGM‑Consensus is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No prediction contradicts the ClinVar status, which is currently unreported. Based on the preponderance of evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.271308 | Uncertain | 0.941 | 0.232 | 0.000 | -7.840 | In-Between | 0.086 | Likely Benign | Likely Benign | -0.11 | Likely Benign | 0.0 | 0.22 | Likely Benign | 0.06 | Likely Benign | 0.69 | Ambiguous | 0.063 | Likely Benign | -1.84 | Neutral | 0.751 | Possibly Damaging | 0.131 | Benign | 3.49 | Benign | 0.33 | Tolerated | 0.2819 | 0.3251 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.2071A>T | T691S 2D 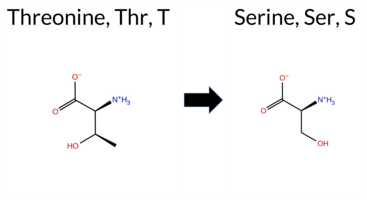 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T691S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall consensus of the available predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.271308 | Uncertain | 0.941 | 0.232 | 0.000 | -9.274 | Likely Pathogenic | 0.123 | Likely Benign | Likely Benign | 0.44 | Likely Benign | 0.1 | 0.23 | Likely Benign | 0.34 | Likely Benign | 0.88 | Ambiguous | 0.041 | Likely Benign | -1.67 | Neutral | 0.860 | Possibly Damaging | 0.584 | Possibly Damaging | 3.49 | Benign | 0.61 | Tolerated | 0.2213 | 0.3343 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.2072C>A | T691K 2D 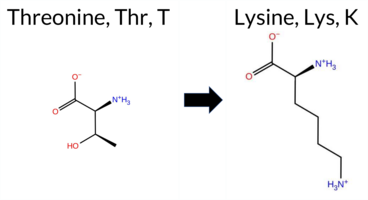 3DClick to see structure in 3D Viewer AIThe SynGAP1 T691K missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, Rosetta, Foldetta, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). FoldX returned an uncertain result and is not counted. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, while the SGM Consensus indicates pathogenic. Overall, the majority of tools and the consensus high‑accuracy prediction lean toward pathogenicity. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.271308 | Uncertain | 0.941 | 0.232 | 0.000 | -12.104 | Likely Pathogenic | 0.715 | Likely Pathogenic | Likely Benign | -0.50 | Ambiguous | 0.1 | -0.31 | Likely Benign | -0.41 | Likely Benign | 1.27 | Destabilizing | 0.147 | Likely Benign | -3.20 | Deleterious | 0.937 | Possibly Damaging | 0.120 | Benign | 3.42 | Benign | 0.04 | Affected | 0.0851 | 0.2628 | 0 | -1 | -3.2 | 27.07 | |||||||||||||||||||||||||||||
| c.2072C>G | T691R 2D 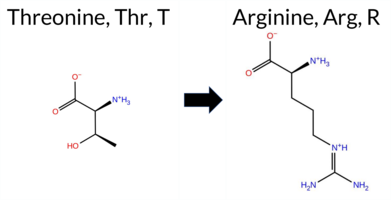 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T691R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as Benign, SGM Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Overall, the preponderance of evidence from standard and high‑accuracy predictors points to a pathogenic effect. Based on the aggregate predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.271308 | Uncertain | 0.941 | 0.232 | 0.000 | -12.228 | Likely Pathogenic | 0.621 | Likely Pathogenic | Likely Benign | -1.27 | Ambiguous | 0.3 | -0.94 | Ambiguous | -1.11 | Ambiguous | 1.35 | Destabilizing | 0.180 | Likely Benign | -3.66 | Deleterious | 0.993 | Probably Damaging | 0.566 | Possibly Damaging | 3.48 | Benign | 0.02 | Affected | 0.0729 | 0.2478 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.2072C>T | T691I 2D 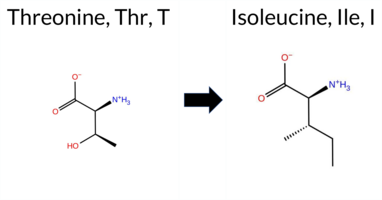 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T691I is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (gnomAD ID: 6‑33441331‑C‑T). Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from FoldX, Foldetta, and premPS. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, while Foldetta remains uncertain. Overall, the evidence overwhelmingly indicates that T691I is most likely benign, and this conclusion does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.271308 | Uncertain | 0.941 | 0.232 | 0.000 | 6-33441331-C-T | 1 | 6.20e-7 | -5.857 | Likely Benign | 0.202 | Likely Benign | Likely Benign | -1.08 | Ambiguous | 0.1 | -2.12 | Stabilizing | -1.60 | Ambiguous | -0.61 | Ambiguous | 0.052 | Likely Benign | -1.19 | Neutral | 0.040 | Benign | 0.003 | Benign | 3.49 | Benign | 0.34 | Tolerated | 3.43 | 14 | 0.0644 | 0.5397 | -1 | 0 | 5.2 | 12.05 | ||||||||||||||||||||||||
| c.2074C>A | L692M 2D 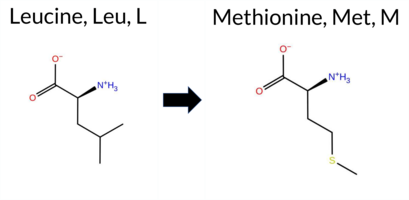 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L692M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2) and Foldetta (combining FoldX‑MD and Rosetta outputs) is also inconclusive. Thus, the overall evidence slightly favors pathogenicity, with a majority of standard tools predicting a deleterious impact. The variant is most likely pathogenic based on current predictions, and this assessment does not contradict the ClinVar status, which has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.064632 | Structured | 0.295225 | Uncertain | 0.966 | 0.243 | 0.000 | -9.659 | Likely Pathogenic | 0.746 | Likely Pathogenic | Likely Benign | 0.49 | Likely Benign | 0.0 | 1.81 | Ambiguous | 1.15 | Ambiguous | 1.01 | Destabilizing | 0.302 | Likely Benign | -1.99 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.07 | Benign | 0.01 | Affected | 0.0754 | 0.2585 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.2074C>G | L692V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L692V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. All other evaluated tools—SGM‑Consensus, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized indicates benign, but the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as pathogenic, and Foldetta also predicts pathogenic. No predictions are missing or inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for the variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.064632 | Structured | 0.295225 | Uncertain | 0.966 | 0.243 | 0.000 | -11.441 | Likely Pathogenic | 0.733 | Likely Pathogenic | Likely Benign | 3.29 | Destabilizing | 0.1 | 2.91 | Destabilizing | 3.10 | Destabilizing | 1.57 | Destabilizing | 0.286 | Likely Benign | -2.99 | Deleterious | 0.978 | Probably Damaging | 0.606 | Possibly Damaging | 3.12 | Benign | 0.01 | Affected | 0.1395 | 0.2460 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2075T>A | L692Q 2D 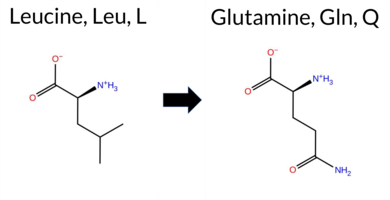 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L692Q is listed in ClinVar as Pathogenic (ClinVar ID 2714634.0) and is not reported in gnomAD. Prediction tools that classify the variant as benign include only FATHMM. All other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic effect. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No prediction or stability result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this conclusion aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.064632 | Structured | 0.295225 | Uncertain | 0.966 | 0.243 | 0.000 | Pathogenic | 1 | -13.873 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.24 | Destabilizing | 0.1 | 3.27 | Destabilizing | 3.26 | Destabilizing | 2.76 | Destabilizing | 0.596 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.06 | Benign | 0.00 | Affected | 3.42 | 17 | 0.1079 | 0.0488 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||
| c.2075T>G | L692R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L692R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.064632 | Structured | 0.295225 | Uncertain | 0.966 | 0.243 | 0.000 | -16.656 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.34 | Destabilizing | 0.0 | 5.51 | Destabilizing | 4.93 | Destabilizing | 1.96 | Destabilizing | 0.611 | Likely Pathogenic | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.895 | Possibly Damaging | 3.07 | Benign | 0.00 | Affected | 0.1204 | 0.0488 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2077C>A | H693N 2D 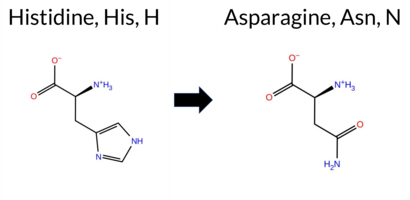 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H693N is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL and FATHMM, whereas the majority of other in silico predictors—premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus—label it pathogenic. FoldX, Rosetta, and Foldetta provide uncertain results. High‑accuracy methods specifically give AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for H693N, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.073402 | Structured | 0.323991 | Uncertain | 0.964 | 0.260 | 0.000 | -12.275 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 1.74 | Ambiguous | 0.1 | 0.80 | Ambiguous | 1.27 | Ambiguous | 1.28 | Destabilizing | 0.436 | Likely Benign | -6.98 | Deleterious | 1.000 | Probably Damaging | 0.987 | Probably Damaging | 3.10 | Benign | 0.01 | Affected | 0.1380 | 0.1849 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||
| c.2077C>G | H693D 2D 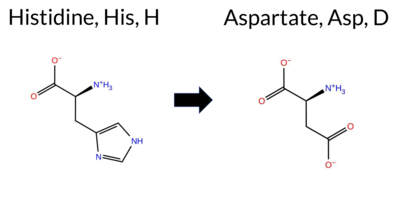 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H693D is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity largely agree: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. Only FATHMM predicts a benign outcome. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No predictions are missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.073402 | Structured | 0.323991 | Uncertain | 0.964 | 0.260 | 0.000 | -15.500 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.60 | Destabilizing | 0.1 | 2.03 | Destabilizing | 2.32 | Destabilizing | 1.62 | Destabilizing | 0.578 | Likely Pathogenic | -8.97 | Deleterious | 1.000 | Probably Damaging | 0.991 | Probably Damaging | 3.09 | Benign | 0.01 | Affected | 0.2166 | 0.1108 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||
| c.2077C>T | H693Y 2D 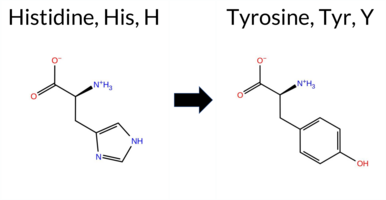 AIThe SynGAP1 H693Y missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, premPS, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain predictions come from Rosetta, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.073402 | Structured | 0.323991 | Uncertain | 0.964 | 0.260 | 0.000 | -7.963 | In-Between | 0.984 | Likely Pathogenic | Likely Pathogenic | -0.16 | Likely Benign | 1.7 | -1.41 | Ambiguous | -0.79 | Ambiguous | 0.10 | Likely Benign | 0.513 | Likely Pathogenic | -5.98 | Deleterious | 0.553 | Possibly Damaging | 0.046 | Benign | 3.13 | Benign | 0.02 | Affected | 0.0819 | 0.3419 | 0 | 2 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||
| c.2078A>C | H693P 2D 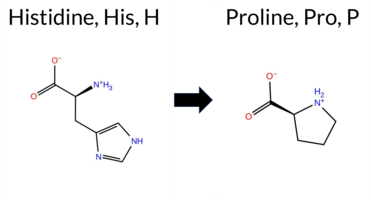 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H693P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are limited to FATHMM, which classifies the variant as benign. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic or likely pathogenic impact. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.073402 | Structured | 0.323991 | Uncertain | 0.964 | 0.260 | 0.000 | -16.281 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 5.54 | Destabilizing | 0.2 | 6.09 | Destabilizing | 5.82 | Destabilizing | 1.06 | Destabilizing | 0.600 | Likely Pathogenic | -9.97 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.09 | Benign | 0.01 | Affected | 0.2080 | 0.3210 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||
| c.2078A>G | H693R 2D 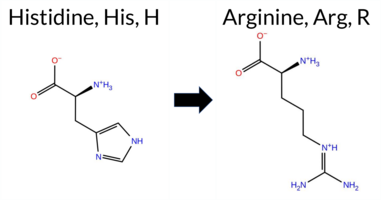 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H693R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the majority of algorithms (SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; FoldX, Rosetta, and Foldetta are inconclusive. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic effect, and Foldetta’s stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the lack of benign evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.073402 | Structured | 0.323991 | Uncertain | 0.964 | 0.260 | 0.000 | -14.326 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.39 | Ambiguous | 0.2 | 1.28 | Ambiguous | 1.34 | Ambiguous | 1.03 | Destabilizing | 0.593 | Likely Pathogenic | -7.97 | Deleterious | 0.998 | Probably Damaging | 0.646 | Possibly Damaging | 3.13 | Benign | 0.01 | Affected | 0.1839 | 0.1670 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||
| c.2078A>T | H693L 2D 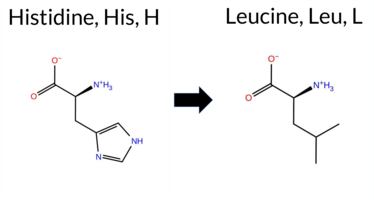 3DClick to see structure in 3D Viewer AISynGAP1 missense variant H693L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from Foldetta, premPS, polyPhen‑2 HumVar, and FATHMM, while pathogenic predictions are made by REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Uncertain results are reported by FoldX and Rosetta. High‑accuracy assessments indicate AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus also leans pathogenic, whereas Foldetta predicts benign stability. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.073402 | Structured | 0.323991 | Uncertain | 0.964 | 0.260 | 0.000 | -14.006 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | -0.53 | Ambiguous | 0.1 | 0.92 | Ambiguous | 0.20 | Likely Benign | -0.29 | Likely Benign | 0.573 | Likely Pathogenic | -10.96 | Deleterious | 0.979 | Probably Damaging | 0.390 | Benign | 3.18 | Benign | 0.01 | Affected | 0.0824 | 0.4675 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.2079T>A | H693Q 2D 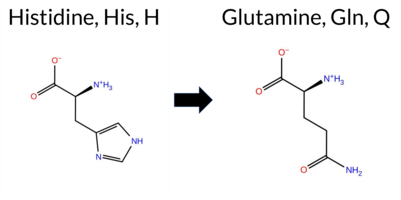 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H693Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL and FATHMM, while the remaining evaluated algorithms (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) all predict pathogenicity. FoldX and Rosetta give uncertain stability changes, and Foldetta likewise reports no definitive effect. High‑accuracy assessments further support a deleterious interpretation: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains inconclusive. Overall, the preponderance of evidence from multiple pathogenic‑oriented predictors and the high‑accuracy consensus indicates that H693Q is most likely pathogenic, a conclusion that aligns with the lack of ClinVar annotation and gnomAD data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.073402 | Structured | 0.323991 | Uncertain | 0.964 | 0.260 | 0.000 | -11.425 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.92 | Ambiguous | 0.1 | 0.78 | Ambiguous | 0.85 | Ambiguous | 1.27 | Destabilizing | 0.386 | Likely Benign | -7.97 | Deleterious | 1.000 | Probably Damaging | 0.921 | Probably Damaging | 3.14 | Benign | 0.01 | Affected | 0.1274 | 0.2955 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.2079T>G | H693Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H693Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL and FATHMM, while the remaining evaluated algorithms (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) all predict pathogenicity. FoldX and Rosetta give uncertain stability changes, and Foldetta likewise reports no definitive effect. High‑accuracy assessments further support a deleterious interpretation: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains inconclusive. Overall, the preponderance of evidence from multiple pathogenic‑oriented predictors and the high‑accuracy consensus indicates that H693Q is most likely pathogenic, a conclusion that aligns with the lack of ClinVar annotation and gnomAD data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.073402 | Structured | 0.323991 | Uncertain | 0.964 | 0.260 | 0.000 | -11.425 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.92 | Ambiguous | 0.1 | 0.78 | Ambiguous | 0.85 | Ambiguous | 1.27 | Destabilizing | 0.386 | Likely Benign | -7.97 | Deleterious | 1.000 | Probably Damaging | 0.921 | Probably Damaging | 3.14 | Benign | 0.01 | Affected | 0.1274 | 0.2955 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.207C>G | I69M 2D  AIThe SynGAP1 missense variant I69M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for I69M, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | -4.071 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.29 | Neutral | 0.943 | Possibly Damaging | 0.703 | Possibly Damaging | 4.17 | Benign | 0.00 | Affected | 0.0579 | 0.2906 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2080G>A | A694T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A694T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Taken together, the overwhelming majority of evidence indicates a benign effect for A694T, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.127496 | Structured | 0.352199 | Uncertain | 0.938 | 0.269 | 0.000 | -3.565 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.55 | Ambiguous | 0.1 | 0.11 | Likely Benign | 0.33 | Likely Benign | -0.26 | Likely Benign | 0.095 | Likely Benign | -0.71 | Neutral | 0.787 | Possibly Damaging | 0.098 | Benign | 3.46 | Benign | 0.17 | Tolerated | 0.1618 | 0.5440 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.2080G>C | A694P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A694P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM, while the majority of other in silico predictors (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a pathogenic impact; premPS is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as Likely Pathogenic, with three of four votes pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. Taken together, the preponderance of evidence points to a pathogenic effect for A694P. This conclusion is consistent with the absence of ClinVar annotation and gnomAD data, and there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.127496 | Structured | 0.352199 | Uncertain | 0.938 | 0.269 | 0.000 | -10.569 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 3.39 | Destabilizing | 0.2 | 5.59 | Destabilizing | 4.49 | Destabilizing | 0.82 | Ambiguous | 0.209 | Likely Benign | -2.77 | Deleterious | 0.988 | Probably Damaging | 0.578 | Possibly Damaging | 3.44 | Benign | 0.03 | Affected | 0.2265 | 0.3829 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.2080G>T | A694S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A694S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree that the substitution is benign: REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The only inconclusive results come from Rosetta and premPS, which are listed as uncertain and do not influence the overall assessment. High‑accuracy predictors reinforce this view: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports benign. Consequently, the variant is most likely benign, and this prediction is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.127496 | Structured | 0.352199 | Uncertain | 0.938 | 0.269 | 0.000 | -0.326 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.1 | 0.59 | Ambiguous | 0.41 | Likely Benign | -0.53 | Ambiguous | 0.092 | Likely Benign | 0.70 | Neutral | 0.013 | Benign | 0.021 | Benign | 3.57 | Benign | 1.00 | Tolerated | 0.2725 | 0.4231 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.2081C>A | A694D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 A694D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, FATHMM, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are premPS, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta is uncertain. Consequently, the variant’s functional impact remains ambiguous. The predictions do not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.127496 | Structured | 0.352199 | Uncertain | 0.938 | 0.269 | 0.000 | -9.542 | Likely Pathogenic | 0.841 | Likely Pathogenic | Ambiguous | 0.35 | Likely Benign | 0.1 | 1.04 | Ambiguous | 0.70 | Ambiguous | 1.01 | Destabilizing | 0.163 | Likely Benign | -2.47 | Neutral | 0.918 | Possibly Damaging | 0.375 | Benign | 3.48 | Benign | 0.05 | Affected | 0.2085 | 0.2216 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||
| c.2081C>G | A694G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A694G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact. The variant’s predicted benign status does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.127496 | Structured | 0.352199 | Uncertain | 0.938 | 0.269 | 0.000 | -3.420 | Likely Benign | 0.144 | Likely Benign | Likely Benign | 0.86 | Ambiguous | 0.0 | 1.16 | Ambiguous | 1.01 | Ambiguous | 0.86 | Ambiguous | 0.093 | Likely Benign | -1.90 | Neutral | 0.866 | Possibly Damaging | 0.171 | Benign | 3.50 | Benign | 0.05 | Affected | 0.2015 | 0.3387 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.2081C>T | A694V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A694V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that predict a pathogenic effect are PROVEAN and polyPhen2_HumDiv, while Rosetta and ESM1b give uncertain results. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign; and Foldetta is benign. No prediction or stability result is missing or inconclusive. Overall, the majority of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.127496 | Structured | 0.352199 | Uncertain | 0.938 | 0.269 | 0.000 | -7.099 | In-Between | 0.221 | Likely Benign | Likely Benign | 0.46 | Likely Benign | 0.1 | -0.76 | Ambiguous | -0.15 | Likely Benign | 0.24 | Likely Benign | 0.149 | Likely Benign | -2.66 | Deleterious | 0.970 | Probably Damaging | 0.207 | Benign | 3.42 | Benign | 0.08 | Tolerated | 0.1238 | 0.4771 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||
| c.2083C>A | L695I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L695I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, and FATHMM, while polyPhen‑2 (HumDiv and HumVar) and ESM1b predict a pathogenic outcome. The remaining tools—AlphaMissense‑Default, Foldetta, premPS, and Rosetta—return uncertain results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, whereas Foldetta remains uncertain. Overall, the majority of reliable predictions support a benign classification, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.118441 | Structured | 0.373419 | Uncertain | 0.942 | 0.258 | 0.000 | -12.096 | Likely Pathogenic | 0.395 | Ambiguous | Likely Benign | 0.47 | Likely Benign | 0.1 | 0.63 | Ambiguous | 0.55 | Ambiguous | 0.93 | Ambiguous | 0.251 | Likely Benign | -2.00 | Neutral | 0.996 | Probably Damaging | 0.905 | Possibly Damaging | 3.24 | Benign | 0.08 | Tolerated | 0.0815 | 0.2759 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.2083C>G | L695V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L695V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. FoldX, Rosetta, and Foldetta give uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs. 2 pathogenic), and Foldetta is also unavailable. Overall, the majority of available predictions (six pathogenic vs. four benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.118441 | Structured | 0.373419 | Uncertain | 0.942 | 0.258 | 0.000 | -10.605 | Likely Pathogenic | 0.317 | Likely Benign | Likely Benign | 1.61 | Ambiguous | 0.0 | 1.57 | Ambiguous | 1.59 | Ambiguous | 1.19 | Destabilizing | 0.274 | Likely Benign | -2.99 | Deleterious | 0.993 | Probably Damaging | 0.694 | Possibly Damaging | 3.20 | Benign | 0.02 | Affected | 0.1158 | 0.2828 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||
| c.2084T>A | L695Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L695Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic impact comprise REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). FoldX and Foldetta provide uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus, derived from a majority of high‑confidence predictors, indicates pathogenicity; Foldetta remains inconclusive. Overall, the majority of reliable tools predict a pathogenic effect, and this is consistent with the lack of ClinVar annotation (no contradiction). Thus, the variant is most likely pathogenic based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.373419 | Uncertain | 0.942 | 0.258 | 0.000 | -12.192 | Likely Pathogenic | 0.706 | Likely Pathogenic | Likely Benign | 1.88 | Ambiguous | 0.1 | 2.03 | Destabilizing | 1.96 | Ambiguous | 1.71 | Destabilizing | 0.554 | Likely Pathogenic | -5.92 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.17 | Benign | 0.08 | Tolerated | 0.0869 | 0.0688 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.2084T>C | L695P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L695P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.373419 | Uncertain | 0.942 | 0.258 | 0.000 | -17.496 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.63 | Destabilizing | 0.2 | 4.73 | Destabilizing | 4.68 | Destabilizing | 2.11 | Destabilizing | 0.612 | Likely Pathogenic | -6.85 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.16 | Benign | 0.00 | Affected | 0.3044 | 0.0992 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2084T>G | L695R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L695R is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, while the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) all classify the variant as pathogenic. AlphaMissense‑Optimized returns an uncertain result. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also pathogenic. Overall, the evidence overwhelmingly indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.373419 | Uncertain | 0.942 | 0.258 | 0.000 | -15.582 | Likely Pathogenic | 0.873 | Likely Pathogenic | Ambiguous | 2.05 | Destabilizing | 0.1 | 2.66 | Destabilizing | 2.36 | Destabilizing | 1.57 | Destabilizing | 0.605 | Likely Pathogenic | -5.92 | Deleterious | 0.993 | Probably Damaging | 0.588 | Possibly Damaging | 3.17 | Benign | 0.00 | Affected | 0.1220 | 0.0530 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2086C>A | L696I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L696I missense change is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. FoldX, Rosetta, Foldetta, and premPS give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta is unavailable. Overall, the majority of conventional tools lean toward pathogenicity, and the high‑accuracy prediction that is available (AlphaMissense‑Optimized) indicates benign, leaving the evidence mixed. Thus, the variant is most likely pathogenic based on the preponderance of predictions, and this does not contradict the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.200174 | Structured | 0.390093 | Uncertain | 0.962 | 0.267 | 0.000 | -10.652 | Likely Pathogenic | 0.698 | Likely Pathogenic | Likely Benign | 1.01 | Ambiguous | 0.1 | 0.58 | Ambiguous | 0.80 | Ambiguous | 0.85 | Ambiguous | 0.250 | Likely Benign | -1.86 | Neutral | 0.996 | Probably Damaging | 0.989 | Probably Damaging | 3.15 | Benign | 0.00 | Affected | 0.0921 | 0.2542 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.2086C>G | L696V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L696V variant is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas the majority of other in silico predictors (FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) report a pathogenic outcome; Rosetta remains inconclusive. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts benign, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Overall, the preponderance of evidence points to a pathogenic effect for the variant, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.390093 | Uncertain | 0.962 | 0.267 | 0.000 | Uncertain | 1 | -11.909 | Likely Pathogenic | 0.745 | Likely Pathogenic | Likely Benign | 2.35 | Destabilizing | 0.1 | 1.85 | Ambiguous | 2.10 | Destabilizing | 1.46 | Destabilizing | 0.351 | Likely Benign | -2.79 | Deleterious | 0.992 | Probably Damaging | 0.970 | Probably Damaging | 3.16 | Benign | 0.00 | Affected | 3.46 | 13 | 0.1307 | 0.2830 | 1 | 2 | 0.4 | -14.03 | |||||||||||||||||||||||||
| c.2086C>T | L696F 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L696F has no ClinVar entry and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not contradict any ClinVar annotation because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.390093 | Uncertain | 0.962 | 0.267 | 0.000 | -9.651 | Likely Pathogenic | 0.897 | Likely Pathogenic | Ambiguous | 0.14 | Likely Benign | 0.1 | 0.74 | Ambiguous | 0.44 | Likely Benign | 0.55 | Ambiguous | 0.422 | Likely Benign | -3.79 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | 3.05 | Benign | 0.00 | Affected | 0.0667 | 0.2008 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.2087T>A | L696H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L696H is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: all available predictors except FATHMM classify the variant as pathogenic (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized). Only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Taken together, the overwhelming majority of computational evidence supports a pathogenic classification, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.390093 | Uncertain | 0.962 | 0.267 | 0.000 | -17.042 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.56 | Destabilizing | 0.0 | 2.55 | Destabilizing | 2.56 | Destabilizing | 2.07 | Destabilizing | 0.569 | Likely Pathogenic | -6.58 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.00 | Benign | 0.00 | Affected | 0.1009 | 0.0488 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2087T>G | L696R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L696R is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: all available predictors except FATHMM (which flags it as benign) report pathogenicity. The benign group contains only FATHMM; the pathogenic group includes SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.390093 | Uncertain | 0.962 | 0.267 | 0.000 | -19.609 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.67 | Destabilizing | 0.0 | 5.36 | Destabilizing | 4.52 | Destabilizing | 2.44 | Destabilizing | 0.624 | Likely Pathogenic | -5.68 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.01 | Benign | 0.00 | Affected | 0.1200 | 0.0688 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2089T>G | W697G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 W697G missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. In contrast, a majority of tools predict a pathogenic outcome: FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar). AlphaMissense‑Default and ESM1b are uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it receives one benign, one pathogenic, and two uncertain calls. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus remains unavailable. Overall, the preponderance of evidence from multiple independent pathogenicity predictors and the protein‑stability assessment by Foldetta indicates that W697G is most likely pathogenic. This conclusion is not contradicted by ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.400169 | Uncertain | 0.945 | 0.297 | 0.000 | -7.429 | In-Between | 0.480 | Ambiguous | Likely Benign | 2.57 | Destabilizing | 0.1 | 2.38 | Destabilizing | 2.48 | Destabilizing | 1.21 | Destabilizing | 0.485 | Likely Benign | -8.93 | Deleterious | 1.000 | Probably Damaging | 0.987 | Probably Damaging | 3.44 | Benign | 0.06 | Tolerated | 0.3965 | 0.1482 | -7 | -2 | 0.5 | -129.16 | ||||||||||||||||||||||||||||||
| c.208C>G | R70G 2D AIThe SynGAP1 missense variant R70G is listed in gnomAD (ID 6‑33425816‑C‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar) and SIFT. AlphaMissense‑Default is uncertain, and Foldetta (FoldX‑MD/Rosetta stability assessment) has no result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also indicates a likely benign outcome; Foldetta data are unavailable. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.458981 | Uncertain | 0.392 | 0.793 | 0.375 | 6-33425816-C-G | 1 | 6.20e-7 | -3.555 | Likely Benign | 0.430 | Ambiguous | Likely Benign | 0.095 | Likely Benign | -0.61 | Neutral | 0.962 | Probably Damaging | 0.726 | Possibly Damaging | 4.11 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3078 | 0.3194 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||
| c.208C>T | R70W 2D  AIThe SynGAP1 missense variant R70W is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.458981 | Uncertain | 0.392 | 0.793 | 0.375 | -3.558 | Likely Benign | 0.588 | Likely Pathogenic | Likely Benign | 0.148 | Likely Benign | -1.83 | Neutral | 0.999 | Probably Damaging | 0.876 | Possibly Damaging | 4.06 | Benign | 0.00 | Affected | 0.1061 | 0.4028 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2090G>C | W697S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W697S is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are premPS, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as unavailable due to uncertainty. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.400169 | Uncertain | 0.945 | 0.297 | 0.000 | -4.900 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 1.90 | Ambiguous | 0.1 | 1.58 | Ambiguous | 1.74 | Ambiguous | 1.13 | Destabilizing | 0.322 | Likely Benign | -8.89 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 3.53 | Benign | 0.13 | Tolerated | 0.3738 | 0.1089 | -2 | -3 | 0.1 | -99.14 | ||||||||||||||||||||||||||||||
| c.2090G>T | W697L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W697L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. No predictions are missing or inconclusive. Overall, the majority of tools (seven pathogenic vs. six benign) lean toward a pathogenic interpretation, and this does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.400169 | Uncertain | 0.945 | 0.297 | 0.000 | -9.057 | Likely Pathogenic | 0.676 | Likely Pathogenic | Likely Benign | 0.20 | Likely Benign | 0.1 | -0.17 | Likely Benign | 0.02 | Likely Benign | 0.90 | Ambiguous | 0.280 | Likely Benign | -8.48 | Deleterious | 1.000 | Probably Damaging | 0.991 | Probably Damaging | 3.58 | Benign | 0.02 | Affected | 0.2222 | 0.2848 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||
| c.2091G>C | W697C 2D  3DClick to see structure in 3D Viewer AISynGAP1 W697C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, ESM1b, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The remaining tools—FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized—return uncertain or inconclusive results. High‑accuracy assessments are likewise ambiguous: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a 2‑vs‑2 tie, and Foldetta is uncertain. Consequently, the evidence does not favor either outcome; the variant’s impact remains indeterminate. This lack of consensus does not contradict ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.400169 | Uncertain | 0.945 | 0.297 | 0.000 | -5.683 | Likely Benign | 0.811 | Likely Pathogenic | Ambiguous | 1.84 | Ambiguous | 0.1 | 1.01 | Ambiguous | 1.43 | Ambiguous | 0.98 | Ambiguous | 0.318 | Likely Benign | -8.65 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.48 | Benign | 0.10 | Tolerated | 0.3575 | 0.1015 | -8 | -2 | 3.4 | -83.07 | ||||||||||||||||||||||||||||||
| c.2091G>T | W697C 2D 3DClick to see structure in 3D Viewer AISynGAP1 W697C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, ESM1b, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Five tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) yield uncertain or missing results. High‑accuracy assessments are likewise inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is a 2‑vs‑2 tie; and Foldetta is uncertain. Consequently, the evidence does not strongly favor either outcome. The variant is therefore best classified as of uncertain significance; this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.400169 | Uncertain | 0.945 | 0.297 | 0.000 | -5.683 | Likely Benign | 0.811 | Likely Pathogenic | Ambiguous | 1.84 | Ambiguous | 0.1 | 1.01 | Ambiguous | 1.43 | Ambiguous | 0.98 | Ambiguous | 0.318 | Likely Benign | -8.65 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.48 | Benign | 0.10 | Tolerated | 0.3575 | 0.1015 | -8 | -2 | 3.4 | -83.07 | ||||||||||||||||||||||||||||||
| c.2092G>A | E698K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E698K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) indicates a benign impact. Because the high‑accuracy predictions are split, the overall evidence is inconclusive, but the majority of tools lean toward pathogenicity. The variant is therefore most likely pathogenic based on the preponderance of predictions, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.120615 | Structured | 0.417514 | Uncertain | 0.922 | 0.315 | 0.000 | -8.881 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 0.23 | Likely Benign | 0.1 | 0.38 | Likely Benign | 0.31 | Likely Benign | -0.07 | Likely Benign | 0.466 | Likely Benign | -3.79 | Deleterious | 0.991 | Probably Damaging | 0.951 | Probably Damaging | 3.37 | Benign | 0.01 | Affected | 0.2218 | 0.4383 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.2092G>C | E698Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E698Q is not reported in ClinVar and has no entries in gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized. Tools that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. No prediction or folding stability result is missing or inconclusive. Overall, the evidence is split, with an equal number of benign and pathogenic calls; the high‑accuracy tools lean toward benign but are not definitive. Thus, the variant’s pathogenicity remains uncertain and does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.120615 | Structured | 0.417514 | Uncertain | 0.922 | 0.315 | 0.000 | -8.369 | Likely Pathogenic | 0.714 | Likely Pathogenic | Likely Benign | 0.00 | Likely Benign | 0.1 | 0.35 | Likely Benign | 0.18 | Likely Benign | -0.02 | Likely Benign | 0.326 | Likely Benign | -2.86 | Deleterious | 0.987 | Probably Damaging | 0.946 | Probably Damaging | 3.35 | Benign | 0.01 | Affected | 0.1029 | 0.3702 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.2093A>C | E698A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E698A missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM, while those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. No prediction or folding result is missing. Overall, the majority of tools (seven versus six) favor a pathogenic interpretation, and this does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.120615 | Structured | 0.417514 | Uncertain | 0.922 | 0.315 | 0.000 | -10.962 | Likely Pathogenic | 0.822 | Likely Pathogenic | Ambiguous | 0.31 | Likely Benign | 0.0 | 0.26 | Likely Benign | 0.29 | Likely Benign | 0.25 | Likely Benign | 0.476 | Likely Benign | -5.57 | Deleterious | 0.997 | Probably Damaging | 0.991 | Probably Damaging | 3.35 | Benign | 0.01 | Affected | 0.3147 | 0.4160 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.2093A>G | E698G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E698G missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM. In contrast, a majority of tools predict a pathogenic impact: SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all indicate deleteriousness. FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive, providing no definitive evidence. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E698G. This prediction aligns with the absence of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.120615 | Structured | 0.417514 | Uncertain | 0.922 | 0.315 | 0.000 | -10.617 | Likely Pathogenic | 0.786 | Likely Pathogenic | Ambiguous | 0.57 | Ambiguous | 0.0 | 1.25 | Ambiguous | 0.91 | Ambiguous | 0.29 | Likely Benign | 0.439 | Likely Benign | -6.37 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.2529 | 0.3286 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.2093A>T | E698V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E698V missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, and FATHMM, whereas those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. Tools with inconclusive results are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts a likely pathogenic outcome. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports an uncertain result. Overall, the majority of evidence points to a pathogenic impact for E698V, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.120615 | Structured | 0.417514 | Uncertain | 0.922 | 0.315 | 0.000 | -12.797 | Likely Pathogenic | 0.945 | Likely Pathogenic | Ambiguous | 0.76 | Ambiguous | 0.0 | 0.39 | Likely Benign | 0.58 | Ambiguous | 0.26 | Likely Benign | 0.480 | Likely Benign | -6.51 | Deleterious | 0.992 | Probably Damaging | 0.967 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.0577 | 0.4546 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.2094G>C | E698D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E698D is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. No predictions are missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.120615 | Structured | 0.417514 | Uncertain | 0.922 | 0.315 | 0.000 | -6.716 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.1 | 0.35 | Likely Benign | 0.28 | Likely Benign | 0.16 | Likely Benign | 0.155 | Likely Benign | -2.45 | Neutral | 0.977 | Probably Damaging | 0.921 | Probably Damaging | 3.35 | Benign | 0.03 | Affected | 0.1643 | 0.2597 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.2094G>T | E698D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E698D is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. No predictions are missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.120615 | Structured | 0.417514 | Uncertain | 0.922 | 0.315 | 0.000 | -6.716 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.1 | 0.35 | Likely Benign | 0.28 | Likely Benign | 0.16 | Likely Benign | 0.155 | Likely Benign | -2.45 | Neutral | 0.977 | Probably Damaging | 0.921 | Probably Damaging | 3.35 | Benign | 0.03 | Affected | 0.1643 | 0.2597 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.2095G>C | V699L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V699L has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome, while FoldX, premPS, and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority; and Foldetta also predicts benign stability. No prediction or folding result is missing or inconclusive. Overall, the variant is most likely benign based on the preponderance of evidence, and this assessment does not contradict any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.069024 | Structured | 0.432975 | Uncertain | 0.935 | 0.315 | 0.000 | -8.301 | Likely Pathogenic | 0.558 | Ambiguous | Likely Benign | -0.58 | Ambiguous | 0.1 | -0.36 | Likely Benign | -0.47 | Likely Benign | 0.69 | Ambiguous | 0.150 | Likely Benign | -2.14 | Neutral | 0.448 | Benign | 0.153 | Benign | 3.48 | Benign | 0.10 | Tolerated | 0.0900 | 0.3289 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||
| c.2095G>T | V699L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V699L has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome, while FoldX, premPS, and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority; and Foldetta also predicts benign stability. No prediction or folding result is missing or inconclusive. Overall, the variant is most likely benign based on the consensus of the available tools, and this benign prediction does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.069024 | Structured | 0.432975 | Uncertain | 0.935 | 0.315 | 0.000 | -8.301 | Likely Pathogenic | 0.558 | Ambiguous | Likely Benign | -0.58 | Ambiguous | 0.1 | -0.36 | Likely Benign | -0.47 | Likely Benign | 0.69 | Ambiguous | 0.150 | Likely Benign | -2.14 | Neutral | 0.448 | Benign | 0.153 | Benign | 3.48 | Benign | 0.10 | Tolerated | 0.0900 | 0.3289 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||
| c.2096T>A | V699E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 V699E missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL and FATHMM, whereas the majority of other in‑silico predictors (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score) indicate a pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Pathogenic. Based on the preponderance of pathogenic predictions and the high‑accuracy tools’ results, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.069024 | Structured | 0.432975 | Uncertain | 0.935 | 0.315 | 0.000 | -12.647 | Likely Pathogenic | 0.813 | Likely Pathogenic | Ambiguous | 2.41 | Destabilizing | 0.1 | 2.08 | Destabilizing | 2.25 | Destabilizing | 1.82 | Destabilizing | 0.468 | Likely Benign | -4.56 | Deleterious | 1.000 | Probably Damaging | 0.986 | Probably Damaging | 3.42 | Benign | 0.02 | Affected | 0.1092 | 0.1326 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.2096T>C | V699A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V699A has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while premPS, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar predict a pathogenic impact. Tools with inconclusive results are FoldX, Rosetta, Foldetta, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is Benign, and the high‑accuracy AlphaMissense‑Optimized also reports Benign. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is Uncertain. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.069024 | Structured | 0.432975 | Uncertain | 0.935 | 0.315 | 0.000 | -6.017 | Likely Benign | 0.426 | Ambiguous | Likely Benign | 1.37 | Ambiguous | 0.0 | 1.30 | Ambiguous | 1.34 | Ambiguous | 1.21 | Destabilizing | 0.236 | Likely Benign | -2.39 | Neutral | 0.861 | Possibly Damaging | 0.625 | Possibly Damaging | 3.42 | Benign | 0.54 | Tolerated | 0.2450 | 0.2124 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.2096T>G | V699G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V699G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include FATHMM and AlphaMissense‑Optimized, while the remaining tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b) uniformly predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic verdict; and Foldetta also predicts pathogenic. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.069024 | Structured | 0.432975 | Uncertain | 0.935 | 0.315 | 0.000 | -11.912 | Likely Pathogenic | 0.481 | Ambiguous | Likely Benign | 2.22 | Destabilizing | 0.0 | 3.25 | Destabilizing | 2.74 | Destabilizing | 1.77 | Destabilizing | 0.514 | Likely Pathogenic | -5.11 | Deleterious | 0.972 | Probably Damaging | 0.999 | Probably Damaging | 3.37 | Benign | 0.01 | Affected | 0.1830 | 0.2240 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.2098C>A | L700M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L700M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only Rosetta predicts a pathogenic outcome, while Foldetta’s stability assessment is uncertain. The high‑accuracy consensus (SGM Consensus) is derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, all of which are benign, yielding a “Likely Benign” classification. AlphaMissense‑Optimized also predicts benign, whereas Foldetta remains inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.113710 | Structured | 0.416255 | Uncertain | 0.927 | 0.331 | 0.000 | -4.617 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.11 | Likely Benign | 0.0 | 2.79 | Destabilizing | 1.45 | Ambiguous | 0.35 | Likely Benign | 0.039 | Likely Benign | -0.32 | Neutral | 0.211 | Benign | 0.081 | Benign | 3.29 | Benign | 0.08 | Tolerated | 0.0733 | 0.2397 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.2098C>G | L700V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L700V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. In contrast, protein‑stability methods predict a deleterious impact: FoldX and Rosetta both score the variant as pathogenic, and the combined Foldetta analysis (FoldX‑MD + Rosetta) also reports a pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, while Foldetta remains pathogenic. Overall, the majority of predictions support a benign classification, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.113710 | Structured | 0.416255 | Uncertain | 0.927 | 0.331 | 0.000 | -3.703 | Likely Benign | 0.192 | Likely Benign | Likely Benign | 2.52 | Destabilizing | 0.0 | 3.16 | Destabilizing | 2.84 | Destabilizing | 0.13 | Likely Benign | 0.056 | Likely Benign | 0.37 | Neutral | 0.003 | Benign | 0.005 | Benign | 3.46 | Benign | 0.76 | Tolerated | 0.1424 | 0.2460 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2099T>A | L700Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L700Q is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. All other evaluated tools—SGM‑Consensus, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—predict a pathogenic impact. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized indicates a benign change, whereas the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta outputs) both predict pathogenicity. No predictions are missing or inconclusive. Overall, the preponderance of evidence from the majority of tools points to a pathogenic effect, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.416255 | Uncertain | 0.927 | 0.331 | 0.000 | -10.522 | Likely Pathogenic | 0.783 | Likely Pathogenic | Likely Benign | 2.41 | Destabilizing | 0.1 | 4.02 | Destabilizing | 3.22 | Destabilizing | 1.35 | Destabilizing | 0.321 | Likely Benign | -3.79 | Deleterious | 0.994 | Probably Damaging | 0.896 | Possibly Damaging | 3.34 | Benign | 0.01 | Affected | 0.1074 | 0.0488 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.2099T>C | L700P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L700P is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while only FATHMM predicts it benign. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Pathogenic” verdict (3 pathogenic vs. 1 benign). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.416255 | Uncertain | 0.927 | 0.331 | 0.000 | -13.092 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 7.29 | Destabilizing | 0.5 | 12.85 | Destabilizing | 10.07 | Destabilizing | 1.84 | Destabilizing | 0.541 | Likely Pathogenic | -4.31 | Deleterious | 0.999 | Probably Damaging | 0.966 | Probably Damaging | 3.30 | Benign | 0.01 | Affected | 0.3603 | 0.1025 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2099T>G | L700R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L700R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign calls come from REVEL, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic calls are made by Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is inconclusive, SGM Consensus remains Likely Pathogenic, and Foldetta predicts a destabilizing, pathogenic effect. Overall, the preponderance of evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.416255 | Uncertain | 0.927 | 0.331 | 0.000 | -12.389 | Likely Pathogenic | 0.938 | Likely Pathogenic | Ambiguous | 1.82 | Ambiguous | 0.1 | 4.19 | Destabilizing | 3.01 | Destabilizing | 1.89 | Destabilizing | 0.485 | Likely Benign | -4.29 | Deleterious | 0.728 | Possibly Damaging | 0.249 | Benign | 3.35 | Benign | 0.01 | Affected | 0.1210 | 0.0488 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.209G>A | R70Q 2D AIThe SynGAP1 missense variant R70Q is reported in gnomAD (variant ID 6-33425817‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: the majority (REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a benign effect, while a minority (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT) predict pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized classifies the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Benign. No Foldetta stability data are available, so it does not influence the conclusion. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a benign impact. This assessment is not in conflict with ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.458981 | Uncertain | 0.392 | 0.793 | 0.375 | 6-33425817-G-A | 1 | 6.20e-7 | -3.399 | Likely Benign | 0.180 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.04 | Neutral | 0.983 | Probably Damaging | 0.602 | Possibly Damaging | 4.25 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2512 | 0.2506 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||||
| c.209G>C | R70P 2D AIThe SynGAP1 missense variant R70P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.458981 | Uncertain | 0.392 | 0.793 | 0.375 | -2.914 | Likely Benign | 0.633 | Likely Pathogenic | Likely Benign | 0.170 | Likely Benign | -1.33 | Neutral | 0.989 | Probably Damaging | 0.859 | Possibly Damaging | 4.08 | Benign | 0.00 | Affected | 0.1903 | 0.4304 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.209G>T | R70L 2D AIThe SynGAP1 missense variant R70L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.458981 | Uncertain | 0.392 | 0.793 | 0.375 | -3.422 | Likely Benign | 0.636 | Likely Pathogenic | Likely Benign | 0.125 | Likely Benign | -1.39 | Neutral | 0.962 | Probably Damaging | 0.726 | Possibly Damaging | 4.11 | Benign | 0.00 | Affected | 0.1455 | 0.4554 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.20C>A | S7Y 2D AIThe SynGAP1 missense variant S7Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar annotation, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | -5.240 | Likely Benign | 0.230 | Likely Benign | Likely Benign | 0.193 | Likely Benign | -0.36 | Neutral | 0.561 | Possibly Damaging | 0.047 | Benign | 4.06 | Benign | 0.00 | Affected | 0.0708 | 0.5053 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||||
| c.20C>G | S7C 2D AIThe SynGAP1 missense variant S7C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, all of which are benign, and therefore SGM‑Consensus also predicts benign. AlphaMissense‑Optimized independently predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | -5.066 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.111 | Likely Benign | 0.41 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.05 | Benign | 0.00 | Affected | 0.1231 | 0.5672 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.20C>T | S7F 2D AIThe SynGAP1 missense variant S7F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. AlphaMissense‑Optimized independently predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | -4.346 | Likely Benign | 0.250 | Likely Benign | Likely Benign | 0.171 | Likely Benign | -0.24 | Neutral | 0.296 | Benign | 0.032 | Benign | 4.06 | Benign | 0.00 | Affected | 0.0622 | 0.5177 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.2101C>A | P701T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P701T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a tolerated change. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports benign. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive, and FoldX alone is uncertain, so these results are treated as unavailable. Overall, the evidence strongly supports a benign effect for P701T, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.074921 | Structured | 0.404318 | Uncertain | 0.918 | 0.345 | 0.000 | -6.920 | Likely Benign | 0.272 | Likely Benign | Likely Benign | 1.61 | Ambiguous | 0.0 | 0.11 | Likely Benign | 0.86 | Ambiguous | -0.18 | Likely Benign | 0.144 | Likely Benign | -0.43 | Neutral | 0.084 | Benign | 0.050 | Benign | 3.44 | Benign | 0.70 | Tolerated | 0.1398 | 0.4160 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||
| c.2101C>G | P701A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P701A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; all of these classify the substitution as benign. No tool predicts a pathogenic outcome. Predictions that are inconclusive are FoldX (uncertain) and Foldetta (uncertain). High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign, while Foldetta remains uncertain. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.074921 | Structured | 0.404318 | Uncertain | 0.918 | 0.345 | 0.000 | -6.836 | Likely Benign | 0.217 | Likely Benign | Likely Benign | 1.35 | Ambiguous | 0.0 | 0.23 | Likely Benign | 0.79 | Ambiguous | -0.16 | Likely Benign | 0.058 | Likely Benign | -0.55 | Neutral | 0.002 | Benign | 0.011 | Benign | 3.50 | Benign | 0.82 | Tolerated | 0.3143 | 0.3765 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.2101C>T | P701S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P701S (ClinVar ID 2995856.0) is listed as “Uncertain” in ClinVar and is present in gnomAD (ID 6‑33441360‑C‑T). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). No tool in the dataset predicts a pathogenic outcome; all remaining predictions are either benign or uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.074921 | Structured | 0.404318 | Uncertain | 0.918 | 0.345 | 0.000 | Uncertain | 1 | 6-33441360-C-T | 3 | 1.86e-6 | -4.375 | Likely Benign | 0.221 | Likely Benign | Likely Benign | 1.33 | Ambiguous | 0.0 | 0.12 | Likely Benign | 0.73 | Ambiguous | -0.36 | Likely Benign | 0.132 | Likely Benign | 0.78 | Neutral | 0.044 | Benign | 0.025 | Benign | 3.48 | Benign | 1.00 | Tolerated | 3.47 | 10 | 0.3149 | 0.3782 | -1 | 1 | 0.8 | -10.04 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||
| c.2102C>A | P701H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 P701H missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Predictions that are uncertain or inconclusive are FoldX, Foldetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the balance of evidence favors a benign classification, and this conclusion does not contradict the ClinVar status, which contains no pathogenic assertion for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.404318 | Uncertain | 0.918 | 0.345 | 0.000 | -8.786 | Likely Pathogenic | 0.553 | Ambiguous | Likely Benign | 1.40 | Ambiguous | 0.0 | -0.41 | Likely Benign | 0.50 | Ambiguous | 0.62 | Ambiguous | 0.141 | Likely Benign | -2.12 | Neutral | 0.936 | Possibly Damaging | 0.539 | Possibly Damaging | 3.38 | Benign | 0.03 | Affected | 0.1495 | 0.3495 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||
| c.2102C>G | P701R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 P701R missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. Two tools (FoldX and premPS) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta (combining FoldX‑MD and Rosetta outputs) as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.404318 | Uncertain | 0.918 | 0.345 | 0.000 | -11.060 | Likely Pathogenic | 0.779 | Likely Pathogenic | Likely Benign | 0.54 | Ambiguous | 0.0 | -0.19 | Likely Benign | 0.18 | Likely Benign | 0.65 | Ambiguous | 0.088 | Likely Benign | -2.09 | Neutral | 0.784 | Possibly Damaging | 0.278 | Benign | 3.41 | Benign | 0.08 | Tolerated | 0.1365 | 0.2500 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||
| c.2102C>T | P701L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 P701L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and ESM1b. The remaining tools (FoldX, Rosetta, AlphaMissense‑Default) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of predictions (7 benign vs. 3 pathogenic) and the two high‑accuracy benign calls suggest that the variant is most likely benign. This conclusion does not contradict any ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.404318 | Uncertain | 0.918 | 0.345 | 0.000 | -10.185 | Likely Pathogenic | 0.515 | Ambiguous | Likely Benign | 1.15 | Ambiguous | 0.0 | -0.68 | Ambiguous | 0.24 | Likely Benign | 0.12 | Likely Benign | 0.116 | Likely Benign | -3.04 | Deleterious | 0.642 | Possibly Damaging | 0.087 | Benign | 3.50 | Benign | 0.09 | Tolerated | 0.2018 | 0.5546 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||
| c.2104C>A | Q702K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q702K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and Foldetta as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence supports a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.397258 | Uncertain | 0.907 | 0.345 | 0.000 | -8.750 | Likely Pathogenic | 0.338 | Likely Benign | Likely Benign | -0.23 | Likely Benign | 0.0 | 0.26 | Likely Benign | 0.02 | Likely Benign | 0.08 | Likely Benign | 0.224 | Likely Benign | -2.86 | Deleterious | 0.863 | Possibly Damaging | 0.773 | Possibly Damaging | 3.46 | Benign | 0.08 | Tolerated | 0.1346 | 0.2524 | 1 | 1 | -0.4 | 0.04 | ||||||||||||||||||||||||||||||
| c.2104C>G | Q702E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q702E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Rosetta and Foldetta give uncertain results. High‑accuracy methods further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign; Foldetta remains inconclusive. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.074921 | Structured | 0.397258 | Uncertain | 0.907 | 0.345 | 0.000 | -10.974 | Likely Pathogenic | 0.230 | Likely Benign | Likely Benign | 0.48 | Likely Benign | 0.0 | 0.72 | Ambiguous | 0.60 | Ambiguous | 0.27 | Likely Benign | 0.196 | Likely Benign | -2.27 | Neutral | 0.989 | Probably Damaging | 0.930 | Probably Damaging | 3.55 | Benign | 0.15 | Tolerated | 0.1021 | 0.1609 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.2105A>C | Q702P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q702P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b; FoldX is uncertain and therefore not counted. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, more tools predict pathogenicity (7) than benignity (5), and the high‑accuracy pathogenic prediction from Foldetta further supports this. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.397258 | Uncertain | 0.907 | 0.345 | 0.000 | -10.361 | Likely Pathogenic | 0.324 | Likely Benign | Likely Benign | 1.76 | Ambiguous | 0.1 | 7.05 | Destabilizing | 4.41 | Destabilizing | -0.04 | Likely Benign | 0.369 | Likely Benign | -4.71 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.42 | Benign | 0.05 | Affected | 0.1877 | 0.3371 | 0 | -1 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||
| c.2105A>T | Q702L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q702L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the balance of evidence leans toward a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.397258 | Uncertain | 0.907 | 0.345 | 0.000 | -9.954 | Likely Pathogenic | 0.149 | Likely Benign | Likely Benign | -0.13 | Likely Benign | 0.0 | 0.09 | Likely Benign | -0.02 | Likely Benign | 0.16 | Likely Benign | 0.392 | Likely Benign | -5.66 | Deleterious | 0.939 | Possibly Damaging | 0.838 | Possibly Damaging | 3.42 | Benign | 0.00 | Affected | 0.0584 | 0.3628 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||
| c.2106G>C | Q702H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q702H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, Rosetta, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Uncertain results come from FoldX and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign outcome (2 benign vs. 1 pathogenic vote), and Foldetta predicts benign stability. No prediction or folding result is missing or inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.397258 | Uncertain | 0.907 | 0.345 | 0.000 | -7.966 | In-Between | 0.211 | Likely Benign | Likely Benign | 0.55 | Ambiguous | 0.0 | 0.13 | Likely Benign | 0.34 | Likely Benign | 0.12 | Likely Benign | 0.233 | Likely Benign | -4.11 | Deleterious | 0.982 | Probably Damaging | 0.947 | Probably Damaging | 3.45 | Benign | 0.00 | Affected | 0.0811 | 0.2555 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||
| c.2106G>T | Q702H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q702H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, Rosetta, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Uncertain results come from FoldX and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign outcome (2 benign vs. 1 pathogenic vote), and Foldetta predicts benign stability. No prediction or folding result is missing or inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.397258 | Uncertain | 0.907 | 0.345 | 0.000 | -7.966 | In-Between | 0.211 | Likely Benign | Likely Benign | 0.55 | Ambiguous | 0.0 | 0.13 | Likely Benign | 0.34 | Likely Benign | 0.12 | Likely Benign | 0.233 | Likely Benign | -4.11 | Deleterious | 0.982 | Probably Damaging | 0.947 | Probably Damaging | 3.45 | Benign | 0.00 | Affected | 0.0811 | 0.2555 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||
| c.2107C>A | L703I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L703I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, while those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and Rosetta. Predictions that are uncertain or inconclusive (FoldX, Foldetta, premPS, AlphaMissense‑Default) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of high‑confidence tools predict a benign impact, and this conclusion does not contradict the ClinVar status, which has no pathogenic classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.144935 | Structured | 0.388282 | Uncertain | 0.929 | 0.353 | 0.000 | -9.332 | Likely Pathogenic | 0.345 | Ambiguous | Likely Benign | 1.44 | Ambiguous | 0.1 | 2.21 | Destabilizing | 1.83 | Ambiguous | 0.61 | Ambiguous | 0.108 | Likely Benign | -1.50 | Neutral | 0.982 | Probably Damaging | 0.758 | Possibly Damaging | 3.38 | Benign | 0.00 | Affected | 0.0909 | 0.3079 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.2107C>G | L703V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L703V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumDiv, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Because the variant is not present in ClinVar or gnomAD, there is no existing clinical classification to contradict. Overall, the majority of predictions and the two high‑accuracy benign assessments suggest the variant is most likely benign, although the Foldetta result indicates a potential pathogenic effect that warrants further functional investigation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.144935 | Structured | 0.388282 | Uncertain | 0.929 | 0.353 | 0.000 | -10.086 | Likely Pathogenic | 0.301 | Likely Benign | Likely Benign | 2.32 | Destabilizing | 0.1 | 2.61 | Destabilizing | 2.47 | Destabilizing | 1.07 | Destabilizing | 0.080 | Likely Benign | -2.22 | Neutral | 0.789 | Possibly Damaging | 0.352 | Benign | 3.19 | Benign | 0.00 | Affected | 0.1383 | 0.2621 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2107C>T | L703F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L703F is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, premPS, FATHMM, and AlphaMissense‑Optimized, while those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta remains unavailable. Overall, the majority of reliable predictions lean toward pathogenicity, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.144935 | Structured | 0.388282 | Uncertain | 0.929 | 0.353 | 0.000 | -10.929 | Likely Pathogenic | 0.768 | Likely Pathogenic | Likely Benign | 1.03 | Ambiguous | 0.1 | 0.59 | Ambiguous | 0.81 | Ambiguous | 0.50 | Likely Benign | 0.247 | Likely Benign | -3.25 | Deleterious | 0.994 | Probably Damaging | 0.806 | Possibly Damaging | 3.12 | Benign | 0.00 | Affected | 0.0600 | 0.2555 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.2108T>A | L703H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L703H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from REVEL and FATHMM, whereas pathogenic predictions are made by FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts Pathogenic; the SGM‑Consensus itself is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts Pathogenic. Taken together, the overwhelming majority of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.144935 | Structured | 0.388282 | Uncertain | 0.929 | 0.353 | 0.000 | -12.886 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 2.52 | Destabilizing | 0.0 | 2.29 | Destabilizing | 2.41 | Destabilizing | 1.75 | Destabilizing | 0.420 | Likely Benign | -5.71 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.09 | Benign | 0.00 | Affected | 0.1038 | 0.0288 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2108T>C | L703P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L703P is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic, while only FATHMM predicts it benign. The SGM‑Consensus, which is a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Pathogenic” result (3 pathogenic vs. 1 benign). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; SGM‑Consensus is likely pathogenic; and Foldetta, integrating FoldX‑MD and Rosetta outputs, is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.144935 | Structured | 0.388282 | Uncertain | 0.929 | 0.353 | 0.000 | -13.766 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 4.44 | Destabilizing | 0.1 | 9.38 | Destabilizing | 6.91 | Destabilizing | 1.54 | Destabilizing | 0.559 | Likely Pathogenic | -5.70 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.11 | Benign | 0.00 | Affected | 0.3698 | 0.1186 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2108T>G | L703R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L703R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus) all classify the change as pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. The preponderance of pathogenic predictions, together with the high‑accuracy tools’ positive results, suggests that L703R is most likely pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.144935 | Structured | 0.388282 | Uncertain | 0.929 | 0.353 | 0.000 | -13.244 | Likely Pathogenic | 0.935 | Likely Pathogenic | Ambiguous | 2.85 | Destabilizing | 0.0 | 3.74 | Destabilizing | 3.30 | Destabilizing | 1.79 | Destabilizing | 0.459 | Likely Benign | -4.92 | Deleterious | 0.994 | Probably Damaging | 0.806 | Possibly Damaging | 3.13 | Benign | 0.00 | Affected | 0.1223 | 0.0488 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2110A>C | S704R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S704R is not reported in gnomAD and has no ClinVar entry. Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. FoldX, Rosetta, and Foldetta provide uncertain stability results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the evidence points to the variant being most likely pathogenic, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.096677 | Structured | 0.383620 | Uncertain | 0.928 | 0.363 | 0.000 | -9.417 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.81 | Ambiguous | 0.1 | 0.92 | Ambiguous | 0.87 | Ambiguous | 0.31 | Likely Benign | 0.178 | Likely Benign | -2.65 | Deleterious | 0.997 | Probably Damaging | 0.822 | Possibly Damaging | 3.53 | Benign | 0.05 | Affected | 0.0714 | 0.3038 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.2110A>G | S704G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S704G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, premPS, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Taken together, the majority of reliable predictors indicate a benign impact, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.096677 | Structured | 0.383620 | Uncertain | 0.928 | 0.363 | 0.000 | -7.827 | In-Between | 0.169 | Likely Benign | Likely Benign | 1.05 | Ambiguous | 0.1 | 1.33 | Ambiguous | 1.19 | Ambiguous | 0.53 | Ambiguous | 0.091 | Likely Benign | -2.25 | Neutral | 0.981 | Probably Damaging | 0.514 | Possibly Damaging | 3.42 | Benign | 0.07 | Tolerated | 0.2224 | 0.3378 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||
| c.2110A>T | S704C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S704C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Other stability‑based tools such as FoldX, Rosetta, and premPS also return uncertain results. Overall, the majority of individual predictors lean toward pathogenicity, and the high‑accuracy consensus also supports a pathogenic classification. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.096677 | Structured | 0.383620 | Uncertain | 0.928 | 0.363 | 0.000 | -10.438 | Likely Pathogenic | 0.423 | Ambiguous | Likely Benign | 1.56 | Ambiguous | 0.1 | 0.98 | Ambiguous | 1.27 | Ambiguous | 0.52 | Ambiguous | 0.251 | Likely Benign | -3.52 | Deleterious | 0.997 | Probably Damaging | 0.789 | Possibly Damaging | 3.40 | Benign | 0.01 | Affected | 0.0789 | 0.4612 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||
| c.2111G>T | S704I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S704I lies in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus) report a pathogenic or likely pathogenic outcome. FoldX, Rosetta, and Foldetta provide uncertain results. High‑accuracy methods specifically give AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, and Foldetta as uncertain. Based on the overall consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.096677 | Structured | 0.383620 | Uncertain | 0.928 | 0.363 | 0.000 | -14.222 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 1.63 | Ambiguous | 0.1 | 1.32 | Ambiguous | 1.48 | Ambiguous | 0.29 | Likely Benign | 0.232 | Likely Benign | -4.05 | Deleterious | 0.997 | Probably Damaging | 0.758 | Possibly Damaging | 3.49 | Benign | 0.02 | Affected | 0.0727 | 0.4798 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.2112C>A | S704R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S704R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic, the SGM Consensus confirms Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result, providing no additional evidence. Overall, the preponderance of evidence from multiple independent predictors indicates that S704R is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.096677 | Structured | 0.383620 | Uncertain | 0.928 | 0.363 | 0.000 | -9.417 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.81 | Ambiguous | 0.1 | 0.92 | Ambiguous | 0.87 | Ambiguous | 0.31 | Likely Benign | 0.200 | Likely Benign | -2.65 | Deleterious | 0.997 | Probably Damaging | 0.822 | Possibly Damaging | 3.53 | Benign | 0.05 | Affected | 0.0714 | 0.3038 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.2112C>G | S704R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S704R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). FoldX and Rosetta give uncertain results, and Foldetta (a combined FoldX‑MD/Rosetta stability assessment) is also uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic; this conclusion does not contradict any ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.096677 | Structured | 0.383620 | Uncertain | 0.928 | 0.363 | 0.000 | -9.417 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.81 | Ambiguous | 0.1 | 0.92 | Ambiguous | 0.87 | Ambiguous | 0.31 | Likely Benign | 0.200 | Likely Benign | -2.65 | Deleterious | 0.997 | Probably Damaging | 0.822 | Possibly Damaging | 3.53 | Benign | 0.05 | Affected | 0.0714 | 0.3038 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.2113A>C | K705Q 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 K705Q missense variant (ClinVar ID 3699560.0) is listed as “Uncertain” in ClinVar and is present in gnomAD (variant ID 6‑33441372‑A‑C). Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (uncertain), ESM1b (benign), FATHMM (benign), and PROVEAN (benign)—also yields a benign classification; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the preponderance of evidence supports a benign impact for K705Q, and this conclusion does not contradict the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.134866 | Structured | 0.379324 | Uncertain | 0.922 | 0.364 | 0.000 | Uncertain | 1 | 6-33441372-A-C | 1 | 6.20e-7 | -5.787 | Likely Benign | 0.436 | Ambiguous | Likely Benign | -0.10 | Likely Benign | 0.0 | 0.33 | Likely Benign | 0.12 | Likely Benign | -0.02 | Likely Benign | 0.142 | Likely Benign | -0.24 | Neutral | 0.997 | Probably Damaging | 0.969 | Probably Damaging | 3.42 | Benign | 0.78 | Tolerated | 3.47 | 10 | 0.3063 | 0.1014 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||
| c.2113A>G | K705E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 K705E missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, AlphaMissense‑Optimized, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta predicts a benign stability change. Overall, the majority of evidence (9 benign vs. 4 pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.134866 | Structured | 0.379324 | Uncertain | 0.922 | 0.364 | 0.000 | -9.371 | Likely Pathogenic | 0.704 | Likely Pathogenic | Likely Benign | 0.10 | Likely Benign | 0.0 | -0.09 | Likely Benign | 0.01 | Likely Benign | 0.47 | Likely Benign | 0.075 | Likely Benign | -1.53 | Neutral | 0.935 | Possibly Damaging | 0.537 | Possibly Damaging | 3.33 | Benign | 0.14 | Tolerated | 0.2601 | 0.0789 | 0 | 1 | 0.4 | 0.94 | ||||||||||||||||||||||||||||||
| c.2114A>C | K705T 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant K705T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Uncertain calls are made by FoldX and AlphaMissense‑Optimized. High‑accuracy assessments indicate that the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts a pathogenic effect, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a benign outcome. Overall, the balance of evidence leans toward a pathogenic interpretation, and this assessment does not conflict with ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.134866 | Structured | 0.379324 | Uncertain | 0.922 | 0.364 | 0.000 | -8.617 | Likely Pathogenic | 0.826 | Likely Pathogenic | Ambiguous | 0.60 | Ambiguous | 0.0 | 0.04 | Likely Benign | 0.32 | Likely Benign | 0.17 | Likely Benign | 0.272 | Likely Benign | -4.05 | Deleterious | 0.995 | Probably Damaging | 0.991 | Probably Damaging | 3.38 | Benign | 0.02 | Affected | 0.1299 | 0.2612 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.2114A>G | K705R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K705R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate a benign or likely benign outcome. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Thus, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.134866 | Structured | 0.379324 | Uncertain | 0.922 | 0.364 | 0.000 | -4.679 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 0.35 | Likely Benign | 0.23 | Likely Benign | 0.18 | Likely Benign | 0.094 | Likely Benign | -1.66 | Neutral | 0.960 | Probably Damaging | 0.545 | Possibly Damaging | 3.33 | Benign | 0.06 | Tolerated | 0.3456 | 0.0789 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.2114A>T | K705M 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant K705M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, FoldX, Foldetta, premPS, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Uncertain results are reported by Rosetta and AlphaMissense‑Optimized. The high‑accuracy consensus (SGM‑Consensus) aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and yields a pathogenic verdict (3/4 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the majority of evidence points toward a pathogenic impact, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.134866 | Structured | 0.379324 | Uncertain | 0.922 | 0.364 | 0.000 | -9.595 | Likely Pathogenic | 0.939 | Likely Pathogenic | Ambiguous | -0.13 | Likely Benign | 0.0 | 0.53 | Ambiguous | 0.20 | Likely Benign | 0.17 | Likely Benign | 0.306 | Likely Benign | -3.65 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.26 | Benign | 0.00 | Affected | 0.0724 | 0.3057 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.2116G>C | E706Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E706Q missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are AlphaMissense‑Default and ESM1b. Rosetta and Foldetta provide uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta also yields an uncertain outcome. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.200174 | Structured | 0.377033 | Uncertain | 0.929 | 0.363 | 0.000 | -8.171 | Likely Pathogenic | 0.588 | Likely Pathogenic | Likely Benign | 0.44 | Likely Benign | 0.0 | 0.61 | Ambiguous | 0.53 | Ambiguous | -0.14 | Likely Benign | 0.059 | Likely Benign | -1.00 | Neutral | 0.433 | Benign | 0.051 | Benign | 4.13 | Benign | 0.31 | Tolerated | 0.0941 | 0.3831 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.2117A>C | E706A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E706A has no ClinVar entry and is not reported in gnomAD. Prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen2_HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign. Only polyPhen2_HumDiv suggests pathogenicity, while FoldX, Foldetta, and AlphaMissense‑Default are uncertain. High‑accuracy methods reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of ClinVar classification or gnomAD observation. Thus, the variant is most likely benign, and this is consistent with the lack of ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.200174 | Structured | 0.377033 | Uncertain | 0.929 | 0.363 | 0.000 | -4.604 | Likely Benign | 0.443 | Ambiguous | Likely Benign | 0.80 | Ambiguous | 0.0 | 0.48 | Likely Benign | 0.64 | Ambiguous | -0.12 | Likely Benign | 0.117 | Likely Benign | -0.81 | Neutral | 0.613 | Possibly Damaging | 0.180 | Benign | 4.20 | Benign | 0.65 | Tolerated | 0.3413 | 0.4089 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.2117A>G | E706G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E706G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar all classify the substitution as benign or tolerated. Only polyPhen2_HumDiv predicts a pathogenic effect. Tools with uncertain outcomes—AlphaMissense‑Default, FoldX, Rosetta, and Foldetta—do not provide a definitive assessment. High‑accuracy predictors reinforce the benign consensus: AlphaMissense‑Optimized reports a benign change; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Overall, the majority of evidence supports a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this is not contradictory to ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.200174 | Structured | 0.377033 | Uncertain | 0.929 | 0.363 | 0.000 | -5.289 | Likely Benign | 0.535 | Ambiguous | Likely Benign | 1.22 | Ambiguous | 0.0 | 1.32 | Ambiguous | 1.27 | Ambiguous | 0.12 | Likely Benign | 0.071 | Likely Benign | -1.71 | Neutral | 0.931 | Possibly Damaging | 0.138 | Benign | 4.07 | Benign | 0.23 | Tolerated | 0.2781 | 0.3614 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.2117A>T | E706V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E706V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. Two tools, FoldX and Foldetta, give uncertain or inconclusive results. High‑accuracy methods give mixed evidence: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta remains uncertain. Overall, the majority of predictions lean toward a benign impact, and this assessment does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.377033 | Uncertain | 0.929 | 0.363 | 0.000 | -9.306 | Likely Pathogenic | 0.667 | Likely Pathogenic | Likely Benign | 1.05 | Ambiguous | 0.0 | 0.30 | Likely Benign | 0.68 | Ambiguous | 0.05 | Likely Benign | 0.099 | Likely Benign | -2.63 | Deleterious | 0.555 | Possibly Damaging | 0.109 | Benign | 4.07 | Benign | 0.16 | Tolerated | 0.0528 | 0.4275 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.2118A>C | E706D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E706D is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the consensus SGM score (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign effect. No tool predicts pathogenicity. High‑accuracy assessments likewise support a benign outcome: AlphaMissense‑Optimized is benign, the SGM Consensus is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. **Thus, the variant is most likely benign, and this prediction does not contradict any ClinVar status (none is available).** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.200174 | Structured | 0.377033 | Uncertain | 0.929 | 0.363 | 0.000 | -4.216 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.0 | 0.02 | Likely Benign | 0.12 | Likely Benign | -0.16 | Likely Benign | 0.182 | Likely Benign | -0.01 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.17 | Benign | 0.41 | Tolerated | 0.1681 | 0.2726 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.2118A>T | E706D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E706D is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the consensus SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts pathogenicity. High‑accuracy assessments further support a benign effect: AlphaMissense‑Optimized predicts benign, the SGM Consensus indicates “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. With all evidence pointing to a neutral impact and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.200174 | Structured | 0.377033 | Uncertain | 0.929 | 0.363 | 0.000 | -4.216 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.0 | 0.02 | Likely Benign | 0.12 | Likely Benign | -0.16 | Likely Benign | 0.182 | Likely Benign | -0.01 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.17 | Benign | 0.41 | Tolerated | 0.1681 | 0.2726 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.2119G>A | A707T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A707T is reported in gnomAD (ID 6‑33441584‑G‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. The Foldetta stability prediction is inconclusive and therefore not considered evidence. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.203355 | Structured | 0.371229 | Uncertain | 0.927 | 0.365 | 0.000 | 6-33441584-G-A | 1 | 6.20e-7 | -0.836 | Likely Benign | 0.175 | Likely Benign | Likely Benign | 0.50 | Ambiguous | 0.0 | 2.36 | Destabilizing | 1.43 | Ambiguous | 0.10 | Likely Benign | 0.252 | Likely Benign | -0.57 | Neutral | 0.980 | Probably Damaging | 0.947 | Probably Damaging | 3.52 | Benign | 0.44 | Tolerated | 3.50 | 9 | 0.0880 | 0.4330 | 0 | 1 | -2.5 | 30.03 | ||||||||||||||||||||||||
| c.2119G>C | A707P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A707P is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, SIFT, and FATHMM, whereas pathogenic predictions come from Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as Pathogenic. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.203355 | Structured | 0.371229 | Uncertain | 0.927 | 0.365 | 0.000 | -8.082 | Likely Pathogenic | 0.885 | Likely Pathogenic | Ambiguous | 0.28 | Likely Benign | 0.1 | 5.75 | Destabilizing | 3.02 | Destabilizing | 0.76 | Ambiguous | 0.228 | Likely Benign | -2.92 | Deleterious | 0.996 | Probably Damaging | 0.983 | Probably Damaging | 3.41 | Benign | 0.09 | Tolerated | 0.1375 | 0.3095 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.2119G>T | A707S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A707S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign. In contrast, polyPhen‑2 HumDiv and HumVar classify the variant as pathogenic. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence supports a benign impact, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.203355 | Structured | 0.371229 | Uncertain | 0.927 | 0.365 | 0.000 | -4.432 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.70 | Ambiguous | 0.1 | 0.63 | Ambiguous | 0.67 | Ambiguous | 0.28 | Likely Benign | 0.150 | Likely Benign | -1.07 | Neutral | 0.848 | Possibly Damaging | 0.945 | Probably Damaging | 3.43 | Benign | 0.08 | Tolerated | 0.1825 | 0.2924 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.211G>A | D71N 2D  AIThe SynGAP1 D71N missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -4.279 | Likely Benign | 0.370 | Ambiguous | Likely Benign | 0.136 | Likely Benign | -1.25 | Neutral | 0.198 | Benign | 0.021 | Benign | 4.08 | Benign | 0.00 | Affected | 0.1331 | 0.6021 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.211G>C | D71H 2D  AIThe SynGAP1 D71H missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -3.974 | Likely Benign | 0.653 | Likely Pathogenic | Likely Benign | 0.099 | Likely Benign | -1.66 | Neutral | 0.637 | Possibly Damaging | 0.136 | Benign | 4.01 | Benign | 0.00 | Affected | 0.1655 | 0.6446 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.211G>T | D71Y 2D  AIThe SynGAP1 missense variant D71Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -4.090 | Likely Benign | 0.740 | Likely Pathogenic | Likely Benign | 0.188 | Likely Benign | -2.49 | Neutral | 0.842 | Possibly Damaging | 0.189 | Benign | 4.00 | Benign | 0.00 | Affected | 0.0584 | 0.6086 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2120C>A | A707D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A707D is not reported in ClinVar (status: None) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, FATHMM, AlphaMissense‑Optimized, and Foldetta. Tools that predict a pathogenic effect are SGM‑Consensus, AlphaMissense‑Default, ESM1b, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Two tools give uncertain results: premPS and Rosetta. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta predicts benign. Overall, the majority of evidence points to a pathogenic impact, and this assessment does not contradict any ClinVar classification because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.203355 | Structured | 0.371229 | Uncertain | 0.927 | 0.365 | 0.000 | -9.160 | Likely Pathogenic | 0.772 | Likely Pathogenic | Likely Benign | 0.29 | Likely Benign | 0.0 | -0.61 | Ambiguous | -0.16 | Likely Benign | 0.89 | Ambiguous | 0.225 | Likely Benign | -3.16 | Deleterious | 0.996 | Probably Damaging | 0.983 | Probably Damaging | 3.39 | Benign | 0.02 | Affected | 0.1376 | 0.1741 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.2120C>G | A707G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A707G missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. The remaining tools (FoldX, Rosetta, Foldetta, premPS, ESM1b) give uncertain or inconclusive results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign, while Foldetta remains uncertain. Overall, the majority of definitive predictions lean toward a benign effect, and there is no ClinVar status to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.203355 | Structured | 0.371229 | Uncertain | 0.927 | 0.365 | 0.000 | -7.480 | In-Between | 0.224 | Likely Benign | Likely Benign | 1.45 | Ambiguous | 0.0 | 1.52 | Ambiguous | 1.49 | Ambiguous | 0.62 | Ambiguous | 0.198 | Likely Benign | -2.60 | Deleterious | 0.991 | Probably Damaging | 0.960 | Probably Damaging | 3.42 | Benign | 0.01 | Affected | 0.1678 | 0.2388 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||
| c.2120C>T | A707V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A707V variant is not reported in ClinVar (no ClinVar ID) but is present in gnomAD (variant ID 6‑33441585‑C‑T). Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign classification. Only three tools—Rosetta, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar—predict pathogenicity, while Foldetta reports an uncertain stability change. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, the SGM‑Consensus is benign, and Foldetta remains uncertain. Overall, the majority of evidence supports a benign impact for A707V, and this conclusion does not contradict any ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.203355 | Structured | 0.371229 | Uncertain | 0.927 | 0.365 | 0.000 | 6-33441585-C-T | 1 | 6.20e-7 | -6.479 | Likely Benign | 0.277 | Likely Benign | Likely Benign | 0.05 | Likely Benign | 0.0 | 2.17 | Destabilizing | 1.11 | Ambiguous | -0.30 | Likely Benign | 0.212 | Likely Benign | -1.62 | Neutral | 0.991 | Probably Damaging | 0.912 | Probably Damaging | 3.45 | Benign | 1.00 | Tolerated | 3.50 | 9 | 0.0794 | 0.4048 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||
| c.2122C>A | L708I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L708I is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify the substitution as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, Rosetta, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, and FATHMM. No tool predicts pathogenicity. High‑accuracy assessments concur: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | -4.406 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.42 | Likely Benign | 0.0 | -0.21 | Likely Benign | 0.11 | Likely Benign | -0.05 | Likely Benign | 0.108 | Likely Benign | 0.02 | Neutral | 0.022 | Benign | 0.021 | Benign | 3.34 | Benign | 0.20 | Tolerated | 0.0749 | 0.2729 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.2122C>G | L708V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L708V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results are from FoldX (uncertain) and Foldetta (uncertain). High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign, while Foldetta remains uncertain. Overall, the evidence strongly indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | -4.361 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 1.59 | Ambiguous | 0.0 | 0.12 | Likely Benign | 0.86 | Ambiguous | -0.02 | Likely Benign | 0.122 | Likely Benign | -0.23 | Neutral | 0.158 | Benign | 0.035 | Benign | 3.35 | Benign | 0.94 | Tolerated | 0.1094 | 0.2460 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2122C>T | L708F 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L708F is not reported in ClinVar and is present in gnomAD (ID 6‑33441587‑C‑T). Functional prediction tools that reach consensus classify the change as benign: REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Pathogenic predictions are limited to polyPhen‑2 HumDiv and ESM1b. The remaining tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Default) return uncertain or no result. High‑accuracy assessments give AlphaMissense‑Optimized a benign score, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, and Foldetta indicates no significant destabilization (uncertain). Overall, the preponderance of evidence supports a benign effect for L708F, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | 6-33441587-C-T | 2 | 1.24e-6 | -9.154 | Likely Pathogenic | 0.436 | Ambiguous | Likely Benign | 1.48 | Ambiguous | 0.3 | 0.93 | Ambiguous | 1.21 | Ambiguous | 0.37 | Likely Benign | 0.110 | Likely Benign | -2.46 | Neutral | 0.931 | Possibly Damaging | 0.326 | Benign | 3.29 | Benign | 0.07 | Tolerated | 3.50 | 9 | 0.0497 | 0.2366 | 0 | 2 | -1.0 | 34.02 | |||||||||||||||||||||||||
| c.2123T>A | L708H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L708H is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default; the SGM Consensus score is also labeled Likely Pathogenic. Stability‑based methods FoldX and Rosetta return uncertain results, and Foldetta likewise is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the majority of conventional predictors lean toward pathogenicity, whereas a few high‑accuracy tools suggest benign or are inconclusive. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | -8.059 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | 1.84 | Ambiguous | 0.1 | 1.55 | Ambiguous | 1.70 | Ambiguous | 1.44 | Destabilizing | 0.243 | Likely Benign | -4.68 | Deleterious | 1.000 | Probably Damaging | 0.981 | Probably Damaging | 3.25 | Benign | 0.02 | Affected | 0.0824 | 0.0288 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2123T>C | L708P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L708P is not reported in ClinVar and is absent from gnomAD. Consensus among most in‑silico predictors is strongly pathogenic: FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a deleterious effect. Only REVEL and FATHMM predict a benign outcome, while AlphaMissense‑Optimized is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is inconclusive, SGM Consensus reports “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic change. Taken together, the preponderance of evidence points to a pathogenic impact for L708P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | -10.268 | Likely Pathogenic | 0.932 | Likely Pathogenic | Ambiguous | 4.29 | Destabilizing | 0.1 | 6.43 | Destabilizing | 5.36 | Destabilizing | 1.62 | Destabilizing | 0.298 | Likely Benign | -4.47 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 3.39 | Benign | 0.04 | Affected | 0.2800 | 0.0825 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2123T>G | L708R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L708R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports it as Likely Pathogenic. FoldX, Rosetta, and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the majority of tools (six pathogenic vs. four benign) and the SGM Consensus support a pathogenic classification, whereas AlphaMissense‑Optimized suggests benign. The variant is most likely pathogenic based on the collective predictions, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.365875 | Uncertain | 0.931 | 0.378 | 0.000 | -9.154 | Likely Pathogenic | 0.638 | Likely Pathogenic | Likely Benign | 0.87 | Ambiguous | 0.0 | 0.90 | Ambiguous | 0.89 | Ambiguous | 1.24 | Destabilizing | 0.249 | Likely Benign | -4.18 | Deleterious | 0.988 | Probably Damaging | 0.598 | Possibly Damaging | 3.27 | Benign | 0.19 | Tolerated | 0.1091 | 0.0488 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2125C>A | L709M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L709M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign or likely benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. No predictions are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.243554 | Structured | 0.365830 | Uncertain | 0.934 | 0.379 | 0.000 | -5.242 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.18 | Likely Benign | 0.0 | 0.40 | Likely Benign | 0.29 | Likely Benign | -0.10 | Likely Benign | 0.065 | Likely Benign | 0.03 | Neutral | 0.688 | Possibly Damaging | 0.235 | Benign | 3.41 | Benign | 0.17 | Tolerated | 0.0629 | 0.2409 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.2125C>G | L709V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L709V is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, every tool that produced a definitive call classifies the variant as benign: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. The only inconclusive results are FoldX (uncertain) and Foldetta (uncertain), which are treated as unavailable evidence. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also benign; Foldetta remains uncertain. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.243554 | Structured | 0.365830 | Uncertain | 0.934 | 0.379 | 0.000 | -4.406 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.75 | Ambiguous | 0.0 | 0.42 | Likely Benign | 0.59 | Ambiguous | 0.03 | Likely Benign | 0.063 | Likely Benign | -0.36 | Neutral | 0.062 | Benign | 0.016 | Benign | 3.42 | Benign | 0.45 | Tolerated | 0.1162 | 0.2460 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2126T>A | L709Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L709Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate benign or likely benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Thus, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.243554 | Structured | 0.365830 | Uncertain | 0.934 | 0.379 | 0.000 | -5.758 | Likely Benign | 0.179 | Likely Benign | Likely Benign | 0.13 | Likely Benign | 0.0 | 0.40 | Likely Benign | 0.27 | Likely Benign | 0.18 | Likely Benign | 0.033 | Likely Benign | -0.78 | Neutral | 0.998 | Probably Damaging | 0.923 | Probably Damaging | 3.52 | Benign | 0.48 | Tolerated | 0.0838 | 0.0488 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.2126T>C | L709P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L709P is not reported in ClinVar and is absent from gnomAD. Benign predictions come from REVEL, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from FoldX, Rosetta, Foldetta, polyPhen2_HumDiv, polyPhen2_HumVar, and AlphaMissense‑Default. High‑accuracy tools give mixed results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, FATHMM, PROVEAN) also predicts benign; Foldetta predicts pathogenic. Because the majority of standard predictors and the SGM Consensus favor benign, the variant is most likely benign, and this is consistent with the lack of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.243554 | Structured | 0.365830 | Uncertain | 0.934 | 0.379 | 0.000 | -7.517 | In-Between | 0.767 | Likely Pathogenic | Likely Benign | 2.81 | Destabilizing | 0.0 | 5.65 | Destabilizing | 4.23 | Destabilizing | 0.31 | Likely Benign | 0.167 | Likely Benign | -1.93 | Neutral | 1.000 | Probably Damaging | 0.975 | Probably Damaging | 3.47 | Benign | 0.37 | Tolerated | 0.3004 | 0.0825 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||
| c.2126T>G | L709R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L709R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. Tools with uncertain or inconclusive results are Rosetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign consensus; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Overall, the majority of evidence indicates a benign impact for the variant, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.243554 | Structured | 0.365830 | Uncertain | 0.934 | 0.379 | 0.000 | -8.636 | Likely Pathogenic | 0.482 | Ambiguous | Likely Benign | 0.22 | Likely Benign | 0.0 | 0.68 | Ambiguous | 0.45 | Likely Benign | 0.62 | Ambiguous | 0.061 | Likely Benign | -1.56 | Neutral | 0.906 | Possibly Damaging | 0.314 | Benign | 3.50 | Benign | 0.44 | Tolerated | 0.1127 | 0.0488 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||
| c.2128A>C | K710Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K710Q missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The high‑accuracy AlphaMissense‑Optimized score classifies the variant as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (uncertain), ESM1b (pathogenic), FATHMM (benign), and PROVEAN (pathogenic)—indicates pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, also predicts a benign effect. Overall, the majority of evidence points to a benign impact, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.321458 | Structured | 0.370438 | Uncertain | 0.949 | 0.368 | 0.000 | -8.920 | Likely Pathogenic | 0.372 | Ambiguous | Likely Benign | 0.12 | Likely Benign | 0.0 | 0.02 | Likely Benign | 0.07 | Likely Benign | 0.49 | Likely Benign | 0.183 | Likely Benign | -3.58 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 3.39 | Benign | 0.01 | Affected | 0.3043 | 0.1214 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||
| c.2128A>G | K710E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K710E missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, and FATHMM, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. Overall, the majority of evidence points toward a pathogenic effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.321458 | Structured | 0.370438 | Uncertain | 0.949 | 0.368 | 0.000 | -11.405 | Likely Pathogenic | 0.885 | Likely Pathogenic | Ambiguous | 0.56 | Ambiguous | 0.0 | 0.94 | Ambiguous | 0.75 | Ambiguous | 0.39 | Likely Benign | 0.178 | Likely Benign | -3.53 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 3.45 | Benign | 0.08 | Tolerated | 0.2655 | 0.0789 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.2129A>C | K710T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K710T is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as uncertain (no definitive stability change). Other stability predictions (FoldX, Rosetta) are also uncertain and thus unavailable for interpretation. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.321458 | Structured | 0.370438 | Uncertain | 0.949 | 0.368 | 0.000 | -10.454 | Likely Pathogenic | 0.759 | Likely Pathogenic | Likely Benign | 1.02 | Ambiguous | 0.0 | 1.57 | Ambiguous | 1.30 | Ambiguous | 0.21 | Likely Benign | 0.305 | Likely Benign | -5.45 | Deleterious | 0.999 | Probably Damaging | 1.000 | Probably Damaging | 3.41 | Benign | 0.06 | Tolerated | 0.1487 | 0.3000 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.2129A>G | K710R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K710R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments—all of which are available—show benign predictions: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.321458 | Structured | 0.370438 | Uncertain | 0.949 | 0.368 | 0.000 | -6.295 | Likely Benign | 0.141 | Likely Benign | Likely Benign | 0.04 | Likely Benign | 0.0 | -0.14 | Likely Benign | -0.05 | Likely Benign | 0.29 | Likely Benign | 0.209 | Likely Benign | -2.78 | Deleterious | 0.983 | Probably Damaging | 0.962 | Probably Damaging | 3.44 | Benign | 0.02 | Affected | 0.3351 | 0.0789 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.2129A>T | K710M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K710M missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM Consensus as Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of individual predictors (seven pathogenic vs. six benign) and the SGM Consensus lean toward a pathogenic interpretation, while the high‑accuracy Foldetta result is contradictory. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.321458 | Structured | 0.370438 | Uncertain | 0.949 | 0.368 | 0.000 | -13.081 | Likely Pathogenic | 0.822 | Likely Pathogenic | Ambiguous | -0.13 | Likely Benign | 0.0 | 0.28 | Likely Benign | 0.08 | Likely Benign | 0.20 | Likely Benign | 0.298 | Likely Benign | -5.61 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.38 | Benign | 0.00 | Affected | 0.0835 | 0.3444 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.212A>C | D71A 2D  AIThe SynGAP1 D71A missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -3.422 | Likely Benign | 0.619 | Likely Pathogenic | Likely Benign | 0.077 | Likely Benign | -1.89 | Neutral | 0.092 | Benign | 0.011 | Benign | 4.06 | Benign | 0.00 | Affected | 0.3876 | 0.6040 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.212A>G | D71G 2D  AIThe SynGAP1 missense variant D71G is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33425820‑A‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | 6-33425820-A-G | 1 | 6.20e-7 | -3.136 | Likely Benign | 0.439 | Ambiguous | Likely Benign | 0.111 | Likely Benign | -1.82 | Neutral | 0.171 | Benign | 0.021 | Benign | 4.03 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3802 | 0.5704 | -1 | 1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||||||
| c.212A>T | D71V 2D  AIThe SynGAP1 D71V missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to a likely benign classification (3 benign vs. 1 pathogenic votes). High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -3.549 | Likely Benign | 0.780 | Likely Pathogenic | Likely Benign | 0.183 | Likely Benign | -2.28 | Neutral | 0.334 | Benign | 0.060 | Benign | 4.01 | Benign | 0.00 | Affected | 0.0771 | 0.6197 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2130G>C | K710N 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant K710N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, premPS, and FATHMM, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). Uncertain or unavailable results are reported for FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is inconclusive, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic impact for K710N, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.321458 | Structured | 0.370438 | Uncertain | 0.949 | 0.368 | 0.000 | -9.330 | Likely Pathogenic | 0.943 | Likely Pathogenic | Ambiguous | 0.85 | Ambiguous | 0.0 | 0.50 | Ambiguous | 0.68 | Ambiguous | 0.33 | Likely Benign | 0.201 | Likely Benign | -4.39 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.42 | Benign | 0.04 | Affected | 0.2485 | 0.1124 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.2130G>T | K710N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K710N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM. In contrast, a majority of algorithms predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the balance of evidence favors a pathogenic interpretation, and this conclusion does not conflict with the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.321458 | Structured | 0.370438 | Uncertain | 0.949 | 0.368 | 0.000 | -9.330 | Likely Pathogenic | 0.943 | Likely Pathogenic | Ambiguous | 0.85 | Ambiguous | 0.0 | 0.50 | Ambiguous | 0.68 | Ambiguous | 0.33 | Likely Benign | 0.201 | Likely Benign | -4.39 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.42 | Benign | 0.04 | Affected | 0.2485 | 0.1124 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.2131C>A | L711M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L711M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta reports an uncertain stability change, which is treated as unavailable evidence. Overall, the majority of individual predictors (six pathogenic vs. five benign) and the available high‑accuracy results lean toward a pathogenic interpretation. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.308712 | Structured | 0.377436 | Uncertain | 0.950 | 0.364 | 0.000 | -9.368 | Likely Pathogenic | 0.679 | Likely Pathogenic | Likely Benign | 0.39 | Likely Benign | 0.0 | 1.11 | Ambiguous | 0.75 | Ambiguous | 1.40 | Destabilizing | 0.216 | Likely Benign | -1.86 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.0755 | 0.2758 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.2131C>G | L711V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L711V is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33441596‑C‑G). Prediction tools that indicate a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. The majority of other in silico predictors—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—classify the change as pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as likely pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the preponderance of evidence points to a pathogenic effect, which does not conflict with the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.308712 | Structured | 0.377436 | Uncertain | 0.950 | 0.364 | 0.000 | Uncertain | 1 | 6-33441596-C-G | 1 | 6.20e-7 | -10.045 | Likely Pathogenic | 0.709 | Likely Pathogenic | Likely Benign | 3.48 | Destabilizing | 0.1 | 2.22 | Destabilizing | 2.85 | Destabilizing | 1.40 | Destabilizing | 0.170 | Likely Benign | -2.59 | Deleterious | 0.992 | Probably Damaging | 0.970 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 3.50 | 9 | 0.1318 | 0.3010 | 1 | 2 | 0.4 | -14.03 | ||||||||||||||||||||||
| c.2132T>A | L711Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L711Q is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are REVEL and FATHMM. Tools that predict a pathogenic effect include FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, SGM Consensus, and Foldetta; Rosetta is uncertain. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized predicts Pathogenic, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Pathogenic. No predictions are missing or inconclusive. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.308712 | Structured | 0.377436 | Uncertain | 0.950 | 0.364 | 0.000 | -11.792 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.93 | Destabilizing | 0.0 | 1.68 | Ambiguous | 2.31 | Destabilizing | 1.63 | Destabilizing | 0.388 | Likely Benign | -5.67 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.31 | Benign | 0.00 | Affected | 0.1041 | 0.0488 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.2132T>C | L711P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L711P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and FATHMM; all other evaluated algorithms—including FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.308712 | Structured | 0.377436 | Uncertain | 0.950 | 0.364 | 0.000 | -12.128 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 5.70 | Destabilizing | 0.1 | 8.15 | Destabilizing | 6.93 | Destabilizing | 2.09 | Destabilizing | 0.375 | Likely Benign | -6.59 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.28 | Benign | 0.00 | Affected | 0.3300 | 0.0986 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2132T>G | L711R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L711R lies in the GAP domain. ClinVar has no entry for this variant, and it is not present in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM; all other evaluated algorithms (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact, and the SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions or stability results are missing or inconclusive. Based on the overwhelming majority of computational evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.308712 | Structured | 0.377436 | Uncertain | 0.950 | 0.364 | 0.000 | -14.009 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.71 | Destabilizing | 0.0 | 3.98 | Destabilizing | 3.85 | Destabilizing | 2.13 | Destabilizing | 0.432 | Likely Benign | -5.71 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.35 | Benign | 0.00 | Affected | 0.1345 | 0.0488 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2134G>A | G712S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G712S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic impact comprise FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. Uncertain results come from premPS, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive and therefore unavailable. Overall, the majority of evidence points to a pathogenic effect for G712S. This conclusion does not contradict ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | -7.942 | In-Between | 0.422 | Ambiguous | Likely Benign | 2.30 | Destabilizing | 0.1 | 4.79 | Destabilizing | 3.55 | Destabilizing | 0.62 | Ambiguous | 0.282 | Likely Benign | -4.84 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.42 | Benign | 0.02 | Affected | 0.2862 | 0.4501 | 1 | 0 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||
| c.2134G>C | G712R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G712R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors—PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, SIFT, ESM1b, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, Foldetta, Rosetta, and the SGM Consensus—classify it as pathogenic. Two tools, FoldX and premPS, return uncertain results. High‑accuracy methods all support pathogenicity: AlphaMissense‑Optimized, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta each predict a deleterious effect. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | -11.776 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 1.75 | Ambiguous | 0.0 | 4.68 | Destabilizing | 3.22 | Destabilizing | 0.82 | Ambiguous | 0.458 | Likely Benign | -6.58 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.35 | Benign | 0.01 | Affected | 0.1229 | 0.4482 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.2134G>T | G712C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G712C is catalogued in gnomAD (6‑33441599‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the majority of algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | 6-33441599-G-T | 1 | 6.20e-7 | -11.376 | Likely Pathogenic | 0.829 | Likely Pathogenic | Ambiguous | 2.54 | Destabilizing | 0.0 | 5.72 | Destabilizing | 4.13 | Destabilizing | 0.56 | Ambiguous | 0.516 | Likely Pathogenic | -7.75 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.31 | Benign | 0.00 | Affected | 3.50 | 9 | 0.1513 | 0.3947 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||
| c.2135G>A | G712D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G712D is catalogued in gnomAD (ID 6‑33441600‑G‑A) but has no ClinVar entry. In silico predictors show mixed results: benign calls come from REVEL, SIFT, and FATHMM, while pathogenic calls are made by FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Uncertain predictions are reported by premPS and AlphaMissense‑Optimized. The high‑accuracy consensus tools give a pathogenic verdict: AlphaMissense‑Optimized is inconclusive, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Pathogenic. Overall, the majority of reliable predictors and the consensus methods lean toward a pathogenic effect, and this assessment does not conflict with ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | 6-33441600-G-A | 1 | 6.20e-7 | -8.211 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 2.87 | Destabilizing | 0.1 | 5.37 | Destabilizing | 4.12 | Destabilizing | 0.90 | Ambiguous | 0.320 | Likely Benign | -5.69 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.67 | Benign | 0.12 | Tolerated | 3.50 | 9 | 0.2146 | 0.2533 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||
| c.2135G>C | G712A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G712A missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions arise from FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. Two tools report uncertainty: premPS and AlphaMissense‑Default. High‑accuracy assessments further refine the picture. AlphaMissense‑Optimized classifies the variant as benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign verdict (2 benign vs. 1 pathogenic vote). Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a pathogenic effect. Because the majority of conventional predictors (7/11) indicate pathogenicity, the overall consensus leans toward a pathogenic impact, despite the mixed high‑accuracy results. This conclusion does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | -6.444 | Likely Benign | 0.553 | Ambiguous | Likely Benign | 2.89 | Destabilizing | 0.1 | 4.98 | Destabilizing | 3.94 | Destabilizing | 0.70 | Ambiguous | 0.281 | Likely Benign | -4.99 | Deleterious | 0.999 | Probably Damaging | 0.991 | Probably Damaging | 3.44 | Benign | 0.02 | Affected | 0.3993 | 0.4105 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||
| c.2135G>T | G712V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G712V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, while the majority of tools (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic impact. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | -10.466 | Likely Pathogenic | 0.877 | Likely Pathogenic | Ambiguous | 3.79 | Destabilizing | 0.0 | 6.18 | Destabilizing | 4.99 | Destabilizing | 0.79 | Ambiguous | 0.410 | Likely Benign | -7.55 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.32 | Benign | 0.00 | Affected | 0.1395 | 0.3712 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2137C>A | P713T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P713T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments further clarify the picture: the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic, whereas AlphaMissense‑Optimized and Foldetta provide inconclusive results and are treated as unavailable. Taken together, the preponderance of evidence points to a pathogenic effect for P713T, and this assessment does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.393235 | Uncertain | 0.961 | 0.371 | 0.000 | -9.915 | Likely Pathogenic | 0.819 | Likely Pathogenic | Ambiguous | 1.15 | Ambiguous | 0.0 | 0.46 | Likely Benign | 0.81 | Ambiguous | 0.65 | Ambiguous | 0.225 | Likely Benign | -6.72 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.1417 | 0.3818 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||
| c.2137C>G | P713A 2D 3DClick to see structure in 3D Viewer AISynGAP1 P713A is not reported in ClinVar and is absent from gnomAD. Benign predictions are provided by REVEL, Rosetta, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default; the SGM‑Consensus score is labeled Likely Pathogenic. FoldX, Foldetta, and premPS give uncertain results. High‑accuracy tools: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic; Foldetta remains uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this assessment does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.393235 | Uncertain | 0.961 | 0.371 | 0.000 | -8.535 | Likely Pathogenic | 0.689 | Likely Pathogenic | Likely Benign | 1.04 | Ambiguous | 0.1 | 0.15 | Likely Benign | 0.60 | Ambiguous | 0.75 | Ambiguous | 0.235 | Likely Benign | -6.80 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.3159 | 0.3475 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.2137C>T | P713S 2D AIThe SynGAP1 missense variant P713S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, Rosetta, and FATHMM, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). Uncertain results are provided by FoldX, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments further highlight this ambiguity: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus indicates a likely pathogenic effect, and Foldetta likewise yields an uncertain stability change. Overall, the majority of tools lean toward a pathogenic interpretation, and this aligns with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.393235 | Uncertain | 0.961 | 0.371 | 0.000 | -8.496 | Likely Pathogenic | 0.846 | Likely Pathogenic | Ambiguous | 0.91 | Ambiguous | 0.7 | 0.19 | Likely Benign | 0.55 | Ambiguous | 0.62 | Ambiguous | 0.261 | Likely Benign | -6.60 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 0.3234 | 0.3674 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||
| c.2138C>A | P713Q 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant P713Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, FoldX, Foldetta, and FATHMM, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). Uncertain calls are made by Rosetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments further highlight this discordance: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a benign effect. Overall, the majority of evidence leans toward a benign interpretation, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.393235 | Uncertain | 0.961 | 0.371 | 0.000 | -10.253 | Likely Pathogenic | 0.875 | Likely Pathogenic | Ambiguous | 0.26 | Likely Benign | 0.0 | -0.77 | Ambiguous | -0.26 | Likely Benign | 0.95 | Ambiguous | 0.364 | Likely Benign | -6.38 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.37 | Benign | 0.00 | Affected | 0.1225 | 0.3388 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||
| c.2138C>G | P713R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P713R has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and FATHMM. Those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The remaining tools, premPS and AlphaMissense‑Optimized, are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of predictions lean toward pathogenicity, while the most accurate methods give conflicting results. Thus, the variant is most likely pathogenic based on the current evidence, and this assessment does not contradict ClinVar status, which has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.393235 | Uncertain | 0.961 | 0.371 | 0.000 | -12.101 | Likely Pathogenic | 0.930 | Likely Pathogenic | Ambiguous | 0.29 | Likely Benign | 0.0 | 0.21 | Likely Benign | 0.25 | Likely Benign | 0.86 | Ambiguous | 0.331 | Likely Benign | -7.42 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 0.1366 | 0.2335 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.2138C>T | P713L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P713L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact; AlphaMissense‑Optimized and premPS are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the balance of evidence favors a pathogenic classification, and this conclusion does not contradict the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.393235 | Uncertain | 0.961 | 0.371 | 0.000 | -11.323 | Likely Pathogenic | 0.850 | Likely Pathogenic | Ambiguous | 0.18 | Likely Benign | 0.1 | -0.03 | Likely Benign | 0.08 | Likely Benign | 0.55 | Ambiguous | 0.324 | Likely Benign | -8.60 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.1993 | 0.5261 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.213C>A | D71E 2D  AIThe SynGAP1 missense variant D71E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -3.276 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -0.28 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.39 | Benign | 0.00 | Affected | 0.1490 | 0.5922 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.213C>G | D71E 2D AIThe SynGAP1 missense variant D71E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign impact, and this is consistent with the lack of ClinVar evidence or population data. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | -3.276 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -0.28 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.39 | Benign | 0.00 | Affected | 0.1490 | 0.5922 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2140C>A | L714M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L714M is not reported in ClinVar (no entry) and is absent from gnomAD. Benign predictions are provided by REVEL, FoldX, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (likely benign). Pathogenic predictions come from Rosetta, Foldetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy tools give mixed results: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign; Foldetta (combining FoldX‑MD and Rosetta) predicts pathogenic. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.314870 | Structured | 0.402311 | Uncertain | 0.961 | 0.369 | 0.000 | -0.989 | Likely Benign | 0.827 | Likely Pathogenic | Ambiguous | 0.45 | Likely Benign | 0.0 | 4.49 | Destabilizing | 2.47 | Destabilizing | 1.00 | Destabilizing | 0.249 | Likely Benign | -1.86 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.09 | Benign | 0.00 | Affected | 0.0687 | 0.2626 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.2140C>G | L714V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L714V has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Overall, the majority of predictions (10 pathogenic vs. 4 benign) indicate a likely pathogenic impact. This conclusion is not contradicted by ClinVar status, which has no record for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.314870 | Structured | 0.402311 | Uncertain | 0.961 | 0.369 | 0.000 | -4.966 | Likely Benign | 0.781 | Likely Pathogenic | Likely Benign | 3.91 | Destabilizing | 0.1 | 6.19 | Destabilizing | 5.05 | Destabilizing | 1.21 | Destabilizing | 0.259 | Likely Benign | -2.76 | Deleterious | 0.995 | Probably Damaging | 0.970 | Probably Damaging | 3.16 | Benign | 0.00 | Affected | 0.1421 | 0.2689 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||
| c.2141T>A | L714Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L714Q is not reported in ClinVar (ClinVar: not reported) and is absent from gnomAD (gnomAD: not found). Prediction tools that agree on a benign effect are REVEL and FATHMM. All other evaluated tools—including FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.402311 | Uncertain | 0.961 | 0.369 | 0.000 | -12.145 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.91 | Destabilizing | 0.0 | 5.23 | Destabilizing | 4.07 | Destabilizing | 2.13 | Destabilizing | 0.378 | Likely Benign | -5.56 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.11 | Benign | 0.00 | Affected | 0.1096 | 0.0558 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.2141T>C | L714P 2D AIThe SynGAP1 missense variant L714P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the preponderance of evidence from these tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.402311 | Uncertain | 0.961 | 0.369 | 0.000 | -12.562 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 7.48 | Destabilizing | 1.9 | 13.54 | Destabilizing | 10.51 | Destabilizing | 2.26 | Destabilizing | 0.441 | Likely Benign | -6.44 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.08 | Benign | 0.00 | Affected | 0.3572 | 0.1253 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2141T>G | L714R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L714R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.402311 | Uncertain | 0.961 | 0.369 | 0.000 | -12.763 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.38 | Destabilizing | 0.2 | 7.54 | Destabilizing | 5.96 | Destabilizing | 2.09 | Destabilizing | 0.402 | Likely Benign | -5.62 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.09 | Benign | 0.00 | Affected | 0.1186 | 0.0558 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2143C>A | P715T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P715T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, Rosetta, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus. Two tools, Foldetta and premPS, returned uncertain results. High‑accuracy analyses further clarify the picture: AlphaMissense‑Optimized predicts a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates pathogenicity; Foldetta remains inconclusive. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.243554 | Structured | 0.409757 | Uncertain | 0.956 | 0.362 | 0.000 | -10.501 | Likely Pathogenic | 0.744 | Likely Pathogenic | Likely Benign | 2.55 | Destabilizing | 0.1 | 0.29 | Likely Benign | 1.42 | Ambiguous | 0.72 | Ambiguous | 0.368 | Likely Benign | -7.23 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.37 | Benign | 0.01 | Affected | 0.1357 | 0.4072 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||
| c.2143C>G | P715A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant P715A has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions are reported by FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments further indicate AlphaMissense‑Optimized predicts Benign, whereas the SGM‑Consensus remains Likely Pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, is Uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.243554 | Structured | 0.409757 | Uncertain | 0.956 | 0.362 | 0.000 | -9.261 | Likely Pathogenic | 0.597 | Likely Pathogenic | Likely Benign | 2.69 | Destabilizing | 0.0 | 0.28 | Likely Benign | 1.49 | Ambiguous | 0.77 | Ambiguous | 0.229 | Likely Benign | -7.13 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.45 | Benign | 0.02 | Affected | 0.2993 | 0.3560 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.2144C>A | P715H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P715H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM, whereas a majority of predictors (FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus) indicate a pathogenic impact. Tools with inconclusive results (Foldetta, premPS, AlphaMissense‑Optimized) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the consensus of the majority of evidence points to a pathogenic effect for P715H. Based on the aggregate predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.243554 | Structured | 0.409757 | Uncertain | 0.956 | 0.362 | 0.000 | -10.523 | Likely Pathogenic | 0.906 | Likely Pathogenic | Ambiguous | 2.80 | Destabilizing | 0.0 | 0.28 | Likely Benign | 1.54 | Ambiguous | 0.56 | Ambiguous | 0.271 | Likely Benign | -7.73 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.37 | Benign | 0.00 | Affected | 0.1413 | 0.3684 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||
| c.2144C>G | P715R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P715R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors (SGM‑Consensus, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) classify the change as pathogenic. High‑accuracy assessments show that the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts a likely pathogenic outcome, AlphaMissense‑Optimized is inconclusive, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also inconclusive. Because the pathogenic predictions outnumber the benign ones and the high‑accuracy consensus supports a deleterious effect, the variant is most likely pathogenic. This assessment does not contradict ClinVar, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.243554 | Structured | 0.409757 | Uncertain | 0.956 | 0.362 | 0.000 | -12.191 | Likely Pathogenic | 0.940 | Likely Pathogenic | Ambiguous | 2.19 | Destabilizing | 0.1 | 0.53 | Ambiguous | 1.36 | Ambiguous | 0.87 | Ambiguous | 0.324 | Likely Benign | -8.09 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.53 | Benign | 0.01 | Affected | 0.1389 | 0.2620 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.2144C>T | P715L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P715L is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL, Rosetta, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX and premPS give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. Because the majority of consensus and individual predictors indicate pathogenicity, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for P715L. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.243554 | Structured | 0.409757 | Uncertain | 0.956 | 0.362 | 0.000 | -12.207 | Likely Pathogenic | 0.764 | Likely Pathogenic | Likely Benign | 1.43 | Ambiguous | 0.1 | 0.06 | Likely Benign | 0.75 | Ambiguous | 0.58 | Ambiguous | 0.318 | Likely Benign | -9.10 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.39 | Benign | 0.01 | Affected | 0.1944 | 0.5105 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.2146C>G | R716G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R716G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Other stability predictors (FoldX, Rosetta, premPS) are also Uncertain. Overall, the balance of evidence from the majority of tools and the SGM‑Consensus indicates a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.247041 | Structured | 0.419135 | Uncertain | 0.962 | 0.379 | 0.000 | -9.927 | Likely Pathogenic | 0.728 | Likely Pathogenic | Likely Benign | 1.32 | Ambiguous | 0.1 | 1.63 | Ambiguous | 1.48 | Ambiguous | 0.72 | Ambiguous | 0.359 | Likely Benign | -5.70 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.36 | Benign | 0.01 | Affected | 0.3437 | 0.2466 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.2146C>T | R716W 2D 3DClick to see structure in 3D Viewer AIClinVar has no entry for this SynGAP1 R716W variant, and it is present in the gnomAD database (ID 6‑33441611‑C‑T). Prediction tools that agree on a benign effect include REVEL, FoldX, FATHMM, AlphaMissense‑Optimized, premPS, and Foldetta, while those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic. With two high‑accuracy tools supporting benign and one supporting pathogenic, the overall prediction leans toward a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.247041 | Structured | 0.419135 | Uncertain | 0.962 | 0.379 | 0.000 | 6-33441611-C-T | 5 | 3.10e-6 | -11.543 | Likely Pathogenic | 0.766 | Likely Pathogenic | Likely Benign | -0.11 | Likely Benign | 0.0 | 0.87 | Ambiguous | 0.38 | Likely Benign | 0.31 | Likely Benign | 0.339 | Likely Benign | -6.72 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 3.32 | Benign | 0.00 | Affected | 3.50 | 9 | 0.1303 | 0.3252 | -3 | 2 | 3.6 | 30.03 | ||||||||||||||||||||||||
| c.2147G>C | R716P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R716P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, premPS, and FATHMM; pathogenic predictions come from AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, Rosetta, Foldetta, and the SGM‑Consensus score. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.247041 | Structured | 0.419135 | Uncertain | 0.962 | 0.379 | 0.000 | -10.744 | Likely Pathogenic | 0.956 | Likely Pathogenic | Likely Pathogenic | 0.18 | Likely Benign | 0.1 | 5.67 | Destabilizing | 2.93 | Destabilizing | 0.49 | Likely Benign | 0.320 | Likely Benign | -5.75 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.33 | Benign | 0.01 | Affected | 0.2140 | 0.3576 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.2147G>T | R716L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R716L is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, FATHMM, premPS, AlphaMissense‑Optimized, and Foldetta. Tools that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as benign. Overall, the majority of predictions (7/13) lean toward pathogenicity, with a near‑even split and a slight edge for pathogenic. The variant is therefore most likely pathogenic based on the current computational evidence, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists for R716L. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.247041 | Structured | 0.419135 | Uncertain | 0.962 | 0.379 | 0.000 | -10.690 | Likely Pathogenic | 0.713 | Likely Pathogenic | Likely Benign | 0.31 | Likely Benign | 0.0 | 0.51 | Ambiguous | 0.41 | Likely Benign | 0.37 | Likely Benign | 0.289 | Likely Benign | -5.70 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.46 | Benign | 0.01 | Affected | 0.1701 | 0.3775 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.2149C>A | L717I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L717I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only two tools—polyPhen‑2 HumDiv and HumVar—predict a pathogenic outcome, while premPS is inconclusive. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts Benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) reports Benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.239899 | Structured | 0.429342 | Uncertain | 0.969 | 0.397 | 0.000 | -4.836 | Likely Benign | 0.168 | Likely Benign | Likely Benign | 0.27 | Likely Benign | 0.0 | 0.33 | Likely Benign | 0.30 | Likely Benign | -0.73 | Ambiguous | 0.182 | Likely Benign | 0.60 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 3.43 | Benign | 1.00 | Tolerated | 0.0712 | 0.2570 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.2149C>G | L717V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L717V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Two tools predict a damaging effect: polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Stability‑based methods (FoldX, Rosetta, Foldetta) return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as inconclusive. Taken together, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar assertion, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.239899 | Structured | 0.429342 | Uncertain | 0.969 | 0.397 | 0.000 | -6.164 | Likely Benign | 0.308 | Likely Benign | Likely Benign | 1.53 | Ambiguous | 0.0 | 1.25 | Ambiguous | 1.39 | Ambiguous | 0.09 | Likely Benign | 0.119 | Likely Benign | -0.25 | Neutral | 0.995 | Probably Damaging | 0.970 | Probably Damaging | 3.38 | Benign | 0.51 | Tolerated | 0.1226 | 0.2489 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2149C>T | L717F 2D AIThe SynGAP1 missense variant L717F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL and FATHMM, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also uncertain. No folding‑stability metrics (FoldX, Rosetta, premPS) provide decisive evidence. Overall, the majority of predictions lean toward pathogenicity, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.429342 | Uncertain | 0.969 | 0.397 | 0.000 | -10.917 | Likely Pathogenic | 0.903 | Likely Pathogenic | Ambiguous | 0.74 | Ambiguous | 0.6 | 0.59 | Ambiguous | 0.67 | Ambiguous | 0.63 | Ambiguous | 0.157 | Likely Benign | -2.56 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | 3.36 | Benign | 0.02 | Affected | 0.0444 | 0.2207 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.214C>G | R72G 2D AIThe SynGAP1 missense variant R72G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar classification; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.455349 | Uncertain | 0.355 | 0.819 | 0.375 | -3.580 | Likely Benign | 0.344 | Ambiguous | Likely Benign | 0.121 | Likely Benign | -0.93 | Neutral | 0.686 | Possibly Damaging | 0.250 | Benign | 4.11 | Benign | 0.00 | Affected | 0.3900 | 0.2847 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.214C>T | R72W 2D AIThe SynGAP1 missense variant R72W is catalogued in gnomAD (ID 6‑33425822‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions are reported by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus also indicates a likely benign outcome. The Foldetta stability analysis is unavailable, providing no additional evidence. Overall, the preponderance of computational evidence points to a benign effect for R72W, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.455349 | Uncertain | 0.355 | 0.819 | 0.375 | 6-33425822-C-T | 1 | 6.20e-7 | -5.546 | Likely Benign | 0.430 | Ambiguous | Likely Benign | 0.145 | Likely Benign | -1.82 | Neutral | 0.994 | Probably Damaging | 0.689 | Possibly Damaging | 4.07 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1601 | 0.3652 | -3 | 2 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||
| c.2150T>A | L717H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L717H occurs in the GAP domain. ClinVar has no entry for this variant, and it is not present in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM; all other evaluated algorithms (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact, and the SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions or stability results are missing or inconclusive. Based on the overwhelming majority of computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.429342 | Uncertain | 0.969 | 0.397 | 0.000 | -11.107 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 2.27 | Destabilizing | 0.1 | 2.04 | Destabilizing | 2.16 | Destabilizing | 1.05 | Destabilizing | 0.317 | Likely Benign | -5.23 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 0.0918 | 0.0558 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2150T>C | L717P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L717P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from REVEL and FATHMM, while the remaining 13 tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus) all classify the variant as pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts a pathogenic effect. No predictions are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.429342 | Uncertain | 0.969 | 0.397 | 0.000 | -10.214 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.96 | Destabilizing | 0.2 | 6.56 | Destabilizing | 5.26 | Destabilizing | 1.66 | Destabilizing | 0.447 | Likely Benign | -5.32 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.28 | Benign | 0.00 | Affected | 0.3065 | 0.1053 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2150T>G | L717R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L717R is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL and FATHMM, while the remaining 13 tools (SGM‑Consensus, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the majority‑vote SGM Consensus) predict pathogenicity; FoldX is uncertain. High‑accuracy methods reinforce a pathogenic verdict: AlphaMissense‑Optimized scores it as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta also predicts pathogenic. No prediction is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.429342 | Uncertain | 0.969 | 0.397 | 0.000 | -11.352 | Likely Pathogenic | 0.973 | Likely Pathogenic | Likely Pathogenic | 1.66 | Ambiguous | 0.1 | 3.78 | Destabilizing | 2.72 | Destabilizing | 1.57 | Destabilizing | 0.353 | Likely Benign | -4.98 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.28 | Benign | 0.00 | Affected | 0.1169 | 0.0558 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2152C>A | L718I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L718I is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Three tools (Foldetta, premPS, Rosetta) give uncertain results and are not considered evidence. High‑accuracy methods specifically show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Because the majority of reliable predictors (eight out of eleven) indicate pathogenicity, the variant is most likely pathogenic. This assessment does not contradict ClinVar, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.438417 | Uncertain | 0.966 | 0.385 | 0.000 | -10.560 | Likely Pathogenic | 0.615 | Likely Pathogenic | Likely Benign | 2.21 | Destabilizing | 0.2 | 1.37 | Ambiguous | 1.79 | Ambiguous | 0.89 | Ambiguous | 0.296 | Likely Benign | -1.90 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.37 | Pathogenic | 0.00 | Affected | 0.0876 | 0.3206 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.2152C>G | L718V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L718V is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) all predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized reports benign, the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.438417 | Uncertain | 0.966 | 0.385 | 0.000 | -11.585 | Likely Pathogenic | 0.693 | Likely Pathogenic | Likely Benign | 3.15 | Destabilizing | 0.1 | 2.11 | Destabilizing | 2.63 | Destabilizing | 1.14 | Destabilizing | 0.237 | Likely Benign | -2.83 | Deleterious | 0.998 | Probably Damaging | 0.992 | Probably Damaging | 1.36 | Pathogenic | 0.00 | Affected | 0.1485 | 0.2656 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2152C>T | L718F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L718F lies in the GAP domain. ClinVar has no entry for this variant, and it is not present in gnomAD. Prediction tools cluster into two groups: benign predictions are made only by REVEL, whereas the remaining tools (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity; premPS is uncertain and is not counted as evidence. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the overwhelming consensus of these predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.438417 | Uncertain | 0.966 | 0.385 | 0.000 | -11.302 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 3.87 | Destabilizing | 0.2 | 2.64 | Destabilizing | 3.26 | Destabilizing | 0.78 | Ambiguous | 0.347 | Likely Benign | -3.73 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 1.32 | Pathogenic | 0.00 | Affected | 0.0590 | 0.2979 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.2153T>A | L718H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L718H is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools cluster into two groups: the single benign predictor REVEL, and a consensus of pathogenic predictions from FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are inconclusive or missing. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.438417 | Uncertain | 0.966 | 0.385 | 0.000 | -14.923 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.84 | Destabilizing | 0.1 | 2.68 | Destabilizing | 3.26 | Destabilizing | 2.69 | Destabilizing | 0.460 | Likely Benign | -6.64 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.1020 | 0.0526 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.2153T>C | L718P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L718P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity are unanimous: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool in the dataset predicts a benign effect, so the benign‑prediction group is empty. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Consequently, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.438417 | Uncertain | 0.966 | 0.385 | 0.000 | -15.643 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.56 | Destabilizing | 0.1 | 9.69 | Destabilizing | 8.13 | Destabilizing | 2.35 | Destabilizing | 0.660 | Likely Pathogenic | -6.64 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.3854 | 0.1221 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2153T>G | L718R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L718R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.438417 | Uncertain | 0.966 | 0.385 | 0.000 | -16.119 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 5.90 | Destabilizing | 0.2 | 5.05 | Destabilizing | 5.48 | Destabilizing | 2.57 | Destabilizing | 0.484 | Likely Benign | -5.69 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.1254 | 0.0600 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2155A>C | N719H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only two tools—polyPhen‑2 HumDiv and polyPhen‑2 HumVar—predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized reports Benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates Benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -6.310 | Likely Benign | 0.130 | Likely Benign | Likely Benign | -0.04 | Likely Benign | 0.0 | -0.36 | Likely Benign | -0.20 | Likely Benign | 0.12 | Likely Benign | 0.095 | Likely Benign | -2.22 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.71 | Benign | 0.19 | Tolerated | 0.0782 | 0.4209 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.2155A>G | N719D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, FoldX, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and Foldetta. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Two tools give uncertain results: Rosetta and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of individual predictors (seven benign vs. four pathogenic) support a benign effect, and this conclusion does not contradict the lack of ClinVar annotation. Thus, based on the available predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -9.016 | Likely Pathogenic | 0.416 | Ambiguous | Likely Benign | -0.03 | Likely Benign | 0.0 | 0.64 | Ambiguous | 0.31 | Likely Benign | 0.46 | Likely Benign | 0.106 | Likely Benign | -2.56 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.75 | Benign | 0.84 | Tolerated | 0.1581 | 0.2345 | 2 | 1 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.2155A>T | N719Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Rosetta and Foldetta provide uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta is also uncertain. Overall, the majority of evaluated tools (7 benign vs 4 pathogenic) support a benign classification. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -11.005 | Likely Pathogenic | 0.302 | Likely Benign | Likely Benign | -0.38 | Likely Benign | 0.0 | -0.62 | Ambiguous | -0.50 | Ambiguous | 0.33 | Likely Benign | 0.187 | Likely Benign | -4.15 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.70 | Benign | 0.09 | Tolerated | 0.0412 | 0.3951 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||
| c.2156A>C | N719T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Foldetta, premPS, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign) all predict a neutral impact. In contrast, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar predict a pathogenic effect, while Rosetta remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -6.251 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.39 | Likely Benign | 0.0 | -0.59 | Ambiguous | -0.10 | Likely Benign | 0.44 | Likely Benign | 0.078 | Likely Benign | -2.60 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.76 | Benign | 0.38 | Tolerated | 0.0851 | 0.4620 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.2156A>G | N719S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719S is reported in gnomAD (variant ID 6‑33441621‑A‑G) but has no ClinVar entry. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign status: AlphaMissense‑Optimized predicts Benign; the SGM‑Consensus itself is Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts Benign. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | 6-33441621-A-G | 2 | 1.24e-6 | -5.190 | Likely Benign | 0.072 | Likely Benign | Likely Benign | -0.06 | Likely Benign | 0.0 | -0.29 | Likely Benign | -0.18 | Likely Benign | 0.46 | Likely Benign | 0.087 | Likely Benign | -1.83 | Neutral | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.85 | Benign | 0.40 | Tolerated | 3.50 | 9 | 0.2611 | 0.4653 | 1 | 1 | 2.7 | -27.03 | ||||||||||||||||||||||||
| c.2156A>T | N719I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Two tools report uncertainty: Rosetta and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of predictions lean toward a benign impact, with no conflict with ClinVar status. Thus, the variant is most likely benign based on the current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -10.794 | Likely Pathogenic | 0.399 | Ambiguous | Likely Benign | -0.19 | Likely Benign | 0.0 | -0.74 | Ambiguous | -0.47 | Likely Benign | 0.40 | Likely Benign | 0.146 | Likely Benign | -4.88 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.71 | Benign | 0.10 | Tolerated | 0.0408 | 0.4493 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||
| c.2157C>A | N719K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The remaining tools (FoldX, Rosetta, ESM1b, and Foldetta) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of reliable predictions indicate a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -7.454 | In-Between | 0.651 | Likely Pathogenic | Likely Benign | -0.64 | Ambiguous | 0.0 | -0.62 | Ambiguous | -0.63 | Ambiguous | 0.42 | Likely Benign | 0.250 | Likely Benign | -1.84 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.80 | Benign | 0.55 | Tolerated | 0.1424 | 0.3112 | 1 | 0 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||
| c.2157C>G | N719K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N719K is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors shows a predominance of benign calls: REVEL, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized all predict a neutral effect, whereas polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default predict pathogenicity. FoldX, Rosetta, ESM1b, and Foldetta are inconclusive. High‑accuracy assessment further supports a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign outcome; Foldetta remains uncertain. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.384043 | Structured | 0.445381 | Uncertain | 0.961 | 0.386 | 0.000 | -7.454 | In-Between | 0.651 | Likely Pathogenic | Likely Benign | -0.64 | Ambiguous | 0.0 | -0.62 | Ambiguous | -0.63 | Ambiguous | 0.42 | Likely Benign | 0.244 | Likely Benign | -1.84 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.80 | Benign | 0.55 | Tolerated | 0.1424 | 0.3112 | 1 | 0 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||
| c.2158G>A | D720N 2D 3DClick to see structure in 3D Viewer AISynGAP1 D720N is listed in ClinVar as benign (ClinVar ID 2837618.0) and is present in gnomAD (ID 6‑33441623‑G‑A). Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus as pathogenic. With seven pathogenic versus six benign predictions overall, the variant is most likely pathogenic according to in‑silico evidence, which contradicts the benign classification in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | Likely Benign | 1 | 6-33441623-G-A | 5 | 3.10e-6 | -9.135 | Likely Pathogenic | 0.654 | Likely Pathogenic | Likely Benign | 0.01 | Likely Benign | 0.0 | -0.20 | Likely Benign | -0.10 | Likely Benign | 0.46 | Likely Benign | 0.289 | Likely Benign | -3.74 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.18 | Pathogenic | 0.01 | Affected | 3.50 | 9 | 0.1216 | 0.5513 | 1 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||
| c.2158G>C | D720H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D720H missense variant is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, and premPS. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and is labeled Likely Pathogenic. The high‑accuracy AlphaMissense‑Optimized score is Uncertain, while Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, indicates a Benign effect. Considering the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -12.355 | Likely Pathogenic | 0.947 | Likely Pathogenic | Ambiguous | 0.03 | Likely Benign | 0.0 | -0.87 | Ambiguous | -0.42 | Likely Benign | 0.48 | Likely Benign | 0.444 | Likely Benign | -5.27 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.13 | Pathogenic | 0.01 | Affected | 0.1363 | 0.6198 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.2158G>T | D720Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D720Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and premPS, while pathogenic predictions are made by SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Predictions from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive. The high‑accuracy consensus (SGM Consensus) classifies the variant as Likely Pathogenic, and Foldetta likewise yields an uncertain stability change. AlphaMissense‑Optimized remains inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -14.771 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | -0.74 | Ambiguous | 0.1 | -1.38 | Ambiguous | -1.06 | Ambiguous | 0.20 | Likely Benign | 0.499 | Likely Benign | -7.05 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.11 | Pathogenic | 0.00 | Affected | 0.0626 | 0.5973 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.2159A>C | D720A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D720A missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and SIFT, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show that the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome, whereas Foldetta (combining FoldX‑MD and Rosetta) predicts a benign impact, and AlphaMissense‑Optimized remains uncertain. Overall, the predictions are split, with a slight tilt toward pathogenicity from the consensus and high‑accuracy methods. Thus, the variant is most likely pathogenic based on the available predictions, and this does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -10.999 | Likely Pathogenic | 0.871 | Likely Pathogenic | Ambiguous | -0.14 | Likely Benign | 0.0 | -0.35 | Likely Benign | -0.25 | Likely Benign | 0.40 | Likely Benign | 0.424 | Likely Benign | -6.20 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.16 | Pathogenic | 0.11 | Tolerated | 0.3726 | 0.5551 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.2159A>G | D720G 2D AISynGAP1 missense variant D720G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Foldetta, and premPS, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools remain inconclusive: AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta outputs) as benign. Overall, the balance of evidence favors a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -11.130 | Likely Pathogenic | 0.911 | Likely Pathogenic | Ambiguous | 0.45 | Likely Benign | 0.4 | 0.53 | Ambiguous | 0.49 | Likely Benign | 0.46 | Likely Benign | 0.427 | Likely Benign | -5.67 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.15 | Pathogenic | 0.01 | Affected | 0.4027 | 0.5011 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.2159A>T | D720V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D720V has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, Foldetta, and premPS, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -12.730 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 0.08 | Likely Benign | 0.0 | -0.78 | Ambiguous | -0.35 | Likely Benign | 0.20 | Likely Benign | 0.437 | Likely Benign | -7.18 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 2.12 | Pathogenic | 0.00 | Affected | 0.0822 | 0.5708 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.215G>A | R72Q 2D AIThe SynGAP1 missense variant R72Q is reported in gnomAD (variant ID 6-33425823‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for R72Q, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.455349 | Uncertain | 0.355 | 0.819 | 0.375 | 6-33425823-G-A | -4.413 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -0.13 | Neutral | 0.829 | Possibly Damaging | 0.238 | Benign | 4.20 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3570 | 0.2380 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||||||
| c.215G>C | R72P 2D AIThe SynGAP1 missense variant R72P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R72P, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.455349 | Uncertain | 0.355 | 0.819 | 0.375 | -3.394 | Likely Benign | 0.510 | Ambiguous | Likely Benign | 0.149 | Likely Benign | -0.93 | Neutral | 0.841 | Possibly Damaging | 0.453 | Possibly Damaging | 4.10 | Benign | 0.00 | Affected | 0.2506 | 0.4157 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.215G>T | R72L 2D AIThe SynGAP1 missense variant R72L is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Pathogenicity is suggested only by polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for R72L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.455349 | Uncertain | 0.355 | 0.819 | 0.375 | -3.102 | Likely Benign | 0.476 | Ambiguous | Likely Benign | 0.108 | Likely Benign | -1.49 | Neutral | 0.686 | Possibly Damaging | 0.250 | Benign | 4.12 | Benign | 0.00 | Affected | 0.2104 | 0.4352 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2160C>A | D720E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D720E missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority, and Foldetta also predicts benign stability. No prediction or folding result is missing or inconclusive. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -5.527 | Likely Benign | 0.391 | Ambiguous | Likely Benign | -0.31 | Likely Benign | 0.1 | -0.41 | Likely Benign | -0.36 | Likely Benign | 0.23 | Likely Benign | 0.131 | Likely Benign | -2.46 | Neutral | 0.975 | Probably Damaging | 0.969 | Probably Damaging | 2.46 | Pathogenic | 0.15 | Tolerated | 0.1310 | 0.5275 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||
| c.2160C>G | D720E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D720E missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority, and Foldetta also predicts benign stability. No prediction or folding result is missing or inconclusive. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.374039 | Structured | 0.450695 | Uncertain | 0.955 | 0.417 | 0.125 | -5.527 | Likely Benign | 0.391 | Ambiguous | Likely Benign | -0.31 | Likely Benign | 0.1 | -0.41 | Likely Benign | -0.36 | Likely Benign | 0.23 | Likely Benign | 0.131 | Likely Benign | -2.46 | Neutral | 0.975 | Probably Damaging | 0.969 | Probably Damaging | 2.46 | Pathogenic | 0.15 | Tolerated | 0.1310 | 0.5275 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||
| c.2161A>C | I721L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I721L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. Two tools (premPS and ESM1b) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the predictions are split, but the majority of high‑confidence tools lean toward a benign interpretation. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -7.843 | In-Between | 0.757 | Likely Pathogenic | Likely Benign | 0.35 | Likely Benign | 0.0 | 0.34 | Likely Benign | 0.35 | Likely Benign | 0.65 | Ambiguous | 0.188 | Likely Benign | -1.83 | Neutral | 0.955 | Possibly Damaging | 0.985 | Probably Damaging | 2.37 | Pathogenic | 0.02 | Affected | 0.0627 | 0.3446 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.2161A>G | I721V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Predictions that are uncertain or inconclusive are FoldX, Rosetta, Foldetta, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) remains uncertain. Overall, the majority of reliable predictors classify the variant as benign, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -6.730 | Likely Benign | 0.380 | Ambiguous | Likely Benign | 1.22 | Ambiguous | 0.0 | 1.11 | Ambiguous | 1.17 | Ambiguous | 0.48 | Likely Benign | 0.098 | Likely Benign | -0.40 | Neutral | 0.969 | Probably Damaging | 0.960 | Probably Damaging | 2.60 | Benign | 0.45 | Tolerated | 0.0976 | 0.3070 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||
| c.2161A>T | I721F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic or likely pathogenic impact, while premPS remains uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Based on the overwhelming agreement among these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -12.559 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 4.61 | Destabilizing | 0.1 | 2.74 | Destabilizing | 3.68 | Destabilizing | 0.66 | Ambiguous | 0.295 | Likely Benign | -3.74 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.0420 | 0.2931 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.2162T>A | I721N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -14.905 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.82 | Destabilizing | 0.0 | 3.21 | Destabilizing | 3.02 | Destabilizing | 2.10 | Destabilizing | 0.425 | Likely Benign | -6.30 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.19 | Pathogenic | 0.00 | Affected | 0.0737 | 0.0340 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.2162T>C | I721T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721T is not reported in ClinVar (ClinVar status: None) and has no entries in gnomAD (gnomAD ID: None). Prediction tools that indicate a benign effect include only REVEL. All other evaluated tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote) yields Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts Pathogenic. No prediction or folding result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | -10.374 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 2.73 | Destabilizing | 0.0 | 2.56 | Destabilizing | 2.65 | Destabilizing | 2.11 | Destabilizing | 0.417 | Likely Benign | -4.18 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.0918 | 0.1019 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.2163C>G | I721M 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant I721M is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, Rosetta, and PROVEAN, whereas pathogenic predictions are made by SGM‑Consensus, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Tools with inconclusive results are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of reliable predictors indicate a pathogenic effect, which aligns with the ClinVar designation of uncertain significance rather than contradicting it. Therefore, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | Uncertain | 1 | -9.767 | Likely Pathogenic | 0.872 | Likely Pathogenic | Ambiguous | 0.71 | Ambiguous | 0.0 | 0.45 | Likely Benign | 0.58 | Ambiguous | 1.00 | Destabilizing | 0.225 | Likely Benign | -2.40 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.0576 | 0.2726 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||
| c.2164A>C | S722R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S722R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, SIFT, and FATHMM, whereas those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools and the high‑accuracy methods lean toward a pathogenic effect. Thus, the variant is most likely pathogenic based on current predictions, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.468512 | Structured | 0.457186 | Uncertain | 0.950 | 0.431 | 0.375 | -10.731 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.05 | Likely Benign | 0.1 | -0.03 | Likely Benign | 0.01 | Likely Benign | 0.72 | Ambiguous | 0.306 | Likely Benign | -2.66 | Deleterious | 0.999 | Probably Damaging | 0.948 | Probably Damaging | 2.52 | Benign | 0.09 | Tolerated | 0.0886 | 0.2797 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.2164A>G | S722G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S722G is not reported in ClinVar and is present in gnomAD (ID 6‑33441629‑A‑G). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and the protein‑folding stability method Foldetta. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. The consensus predictor SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta as benign. No evidence from the high‑accuracy tools contradicts the benign predictions, but the consensus and several individual pathogenic predictors suggest a potential deleterious impact. Based on the overall pattern of predictions, the variant is most likely pathogenic, which is consistent with the lack of ClinVar annotation and the presence of multiple pathogenic signals. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.468512 | Structured | 0.457186 | Uncertain | 0.950 | 0.431 | 0.375 | 6-33441629-A-G | 2 | 1.24e-6 | -9.141 | Likely Pathogenic | 0.214 | Likely Benign | Likely Benign | 0.24 | Likely Benign | 0.1 | 0.67 | Ambiguous | 0.46 | Likely Benign | 0.50 | Likely Benign | 0.270 | Likely Benign | -2.72 | Deleterious | 0.998 | Probably Damaging | 0.863 | Possibly Damaging | 2.49 | Pathogenic | 0.14 | Tolerated | 3.50 | 8 | 0.2202 | 0.3400 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||
| c.2164A>T | S722C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S722C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as benign. Overall, the majority of individual predictors and the high‑accuracy methods lean toward a benign impact, with only the SGM Consensus suggesting pathogenicity. Thus, the variant is most likely benign based on the available predictions, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.468512 | Structured | 0.457186 | Uncertain | 0.950 | 0.431 | 0.375 | -8.060 | Likely Pathogenic | 0.273 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.0 | -0.23 | Likely Benign | -0.01 | Likely Benign | 0.28 | Likely Benign | 0.362 | Likely Benign | -3.44 | Deleterious | 1.000 | Probably Damaging | 0.968 | Probably Damaging | 2.46 | Pathogenic | 0.05 | Affected | 0.1132 | 0.4306 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.2165G>A | S722N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S722N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach consensus classify the change as benign: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict a neutral effect. No tool predicts pathogenicity; the only inconclusive results come from premPS and AlphaMissense‑Default, which are treated as unavailable evidence. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign outcome, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts a benign impact. Taken together, the evidence overwhelmingly supports a benign classification, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.468512 | Structured | 0.457186 | Uncertain | 0.950 | 0.431 | 0.375 | -4.399 | Likely Benign | 0.379 | Ambiguous | Likely Benign | 0.18 | Likely Benign | 0.1 | 0.15 | Likely Benign | 0.17 | Likely Benign | 0.79 | Ambiguous | 0.051 | Likely Benign | -0.91 | Neutral | 0.072 | Benign | 0.028 | Benign | 2.58 | Benign | 0.27 | Tolerated | 0.1258 | 0.3452 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||
| c.2165G>C | S722T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S722T is reported in gnomAD (6‑33441630‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts it as pathogenic, while FoldX is uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. No prediction or stability result is missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.468512 | Structured | 0.457186 | Uncertain | 0.950 | 0.431 | 0.375 | 6-33441630-G-C | 1 | 6.20e-7 | -5.734 | Likely Benign | 0.202 | Likely Benign | Likely Benign | 0.53 | Ambiguous | 0.0 | -0.32 | Likely Benign | 0.11 | Likely Benign | -0.12 | Likely Benign | 0.118 | Likely Benign | -0.57 | Neutral | 0.921 | Possibly Damaging | 0.414 | Benign | 2.57 | Benign | 1.00 | Tolerated | 3.50 | 8 | 0.1357 | 0.4363 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||
| c.2165G>T | S722I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S722I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, premPS, SIFT, and the folding‑stability method Foldetta, whereas pathogenic predictions are reported by SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results are provided by AlphaMissense‑Optimized, FoldX, and Rosetta. High‑accuracy analyses further clarify the picture: AlphaMissense‑Optimized remains inconclusive; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect; Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a benign impact on protein stability. Overall, the majority of evidence leans toward a pathogenic interpretation, and this conclusion does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.468512 | Structured | 0.457186 | Uncertain | 0.950 | 0.431 | 0.375 | -11.165 | Likely Pathogenic | 0.867 | Likely Pathogenic | Ambiguous | 0.69 | Ambiguous | 0.1 | -0.65 | Ambiguous | 0.02 | Likely Benign | 0.18 | Likely Benign | 0.232 | Likely Benign | -3.88 | Deleterious | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.48 | Pathogenic | 0.07 | Tolerated | 0.0776 | 0.4187 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.2166C>A | S722R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S722R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, SIFT, and FATHMM, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus (SGM Consensus) derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN is pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the majority of evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.468512 | Structured | 0.457186 | Uncertain | 0.950 | 0.431 | 0.375 | -10.731 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.05 | Likely Benign | 0.1 | -0.03 | Likely Benign | 0.01 | Likely Benign | 0.72 | Ambiguous | 0.221 | Likely Benign | -2.66 | Deleterious | 0.999 | Probably Damaging | 0.948 | Probably Damaging | 2.52 | Benign | 0.09 | Tolerated | 0.0886 | 0.2797 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.2166C>G | S722R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S722R is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, SIFT, and FATHMM, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a pathogenic bias: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign stability. Overall, the balance of evidence leans toward pathogenicity, and this conclusion does not contradict any ClinVar annotation (none is available). Thus, based on the current computational predictions, the S722R variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.468512 | Structured | 0.457186 | Uncertain | 0.950 | 0.431 | 0.375 | -10.731 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.05 | Likely Benign | 0.1 | -0.03 | Likely Benign | 0.01 | Likely Benign | 0.72 | Ambiguous | 0.220 | Likely Benign | -2.66 | Deleterious | 0.999 | Probably Damaging | 0.948 | Probably Damaging | 2.52 | Benign | 0.09 | Tolerated | 0.0886 | 0.2797 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.2167A>C | T723P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T723P is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, FATHMM, and AlphaMissense‑Optimized. Tools that predict pathogenicity are SGM Consensus, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic; Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. premPS is uncertain and does not influence the overall assessment. Overall, the majority of tools and the high‑accuracy methods support a pathogenic effect. Thus, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458243 | Uncertain | 0.945 | 0.447 | 0.375 | -9.231 | Likely Pathogenic | 0.741 | Likely Pathogenic | Likely Benign | 3.98 | Destabilizing | 0.1 | 6.10 | Destabilizing | 5.04 | Destabilizing | 0.54 | Ambiguous | 0.085 | Likely Benign | -2.51 | Deleterious | 0.995 | Probably Damaging | 0.929 | Probably Damaging | 3.49 | Benign | 0.04 | Affected | 0.1826 | 0.4406 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.2167A>G | T723A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T723A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess sequence conservation, structural impact, or functional effect all converge on a benign outcome: SIFT, PolyPhen‑2 (HumDiv and HumVar), PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, REVEL, and the protein‑stability predictors FoldX, Rosetta, premPS, and Foldetta all indicate a benign effect. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Based on the uniform predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.476583 | Structured | 0.458243 | Uncertain | 0.945 | 0.447 | 0.375 | -4.572 | Likely Benign | 0.064 | Likely Benign | Likely Benign | -0.47 | Likely Benign | 0.0 | 0.16 | Likely Benign | -0.16 | Likely Benign | 0.25 | Likely Benign | 0.053 | Likely Benign | -0.91 | Neutral | 0.161 | Benign | 0.045 | Benign | 3.51 | Benign | 0.06 | Tolerated | 0.3902 | 0.3639 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.2167A>T | T723S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T723S is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign or tolerated. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The high‑accuracy consensus methods confirm the benign assessment: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta) predicts benign stability. Overall, the majority of evidence supports a benign impact for T723S, and this conclusion is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.476583 | Structured | 0.458243 | Uncertain | 0.945 | 0.447 | 0.375 | -4.792 | Likely Benign | 0.091 | Likely Benign | Likely Benign | -0.26 | Likely Benign | 0.0 | 0.28 | Likely Benign | 0.01 | Likely Benign | 0.23 | Likely Benign | 0.037 | Likely Benign | -0.89 | Neutral | 0.673 | Possibly Damaging | 0.678 | Possibly Damaging | 3.55 | Benign | 0.06 | Tolerated | 0.3171 | 0.3731 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.2168C>A | T723K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T723K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, while FoldX and AlphaMissense‑Default are uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, the SGM Consensus is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the preponderance of evidence supports a benign classification for T723K, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.476583 | Structured | 0.458243 | Uncertain | 0.945 | 0.447 | 0.375 | -4.624 | Likely Benign | 0.482 | Ambiguous | Likely Benign | -0.77 | Ambiguous | 0.1 | -0.04 | Likely Benign | -0.41 | Likely Benign | 0.25 | Likely Benign | 0.121 | Likely Benign | -1.40 | Neutral | 0.674 | Possibly Damaging | 0.118 | Benign | 3.46 | Benign | 0.51 | Tolerated | 0.0924 | 0.2798 | 0 | -1 | -3.2 | 27.07 | |||||||||||||||||||||||||||||
| c.2168C>G | T723R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T723R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify it as benign. Two tools (polyPhen‑2 HumDiv and HumVar) predict it to be pathogenic, while FoldX, Foldetta, and AlphaMissense‑Default are uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM Consensus as likely benign, and Foldetta as uncertain. Overall, the preponderance of evidence points to a benign impact for T723R, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.476583 | Structured | 0.458243 | Uncertain | 0.945 | 0.447 | 0.375 | -3.654 | Likely Benign | 0.408 | Ambiguous | Likely Benign | -0.90 | Ambiguous | 0.1 | -0.13 | Likely Benign | -0.52 | Ambiguous | 0.44 | Likely Benign | 0.146 | Likely Benign | -1.50 | Neutral | 0.931 | Possibly Damaging | 0.458 | Possibly Damaging | 3.51 | Benign | 0.29 | Tolerated | 0.0794 | 0.2437 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.2170G>A | A724T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A724T missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that indicate a benign effect include REVEL, premPS, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Predictions that are inconclusive are AlphaMissense‑Default, FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evaluated tools (seven pathogenic vs. three benign) suggest a pathogenic impact. This conclusion is consistent with the lack of ClinVar annotation, as there is no existing classification to contradict. Thus, the variant is most likely pathogenic based on current predictive evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -8.047 | Likely Pathogenic | 0.456 | Ambiguous | Likely Benign | 0.95 | Ambiguous | 0.1 | 1.10 | Ambiguous | 1.03 | Ambiguous | 0.40 | Likely Benign | 0.273 | Likely Benign | -2.87 | Deleterious | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 2.09 | Pathogenic | 0.01 | Affected | 0.1149 | 0.5897 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.2170G>C | A724P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A724P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: REVEL predicts a benign effect, whereas the remaining 13 tools (FoldX, Rosetta, Foldetta, premPS [uncertain], PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized returns a pathogenic score; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, classifies the variant as pathogenic. No tool suggests a benign outcome, and the single benign prediction (REVEL) is outweighed by the consensus of pathogenic predictions. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this mutation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -12.817 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.90 | Destabilizing | 0.3 | 6.35 | Destabilizing | 4.63 | Destabilizing | 0.52 | Ambiguous | 0.391 | Likely Benign | -3.88 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.04 | Pathogenic | 0.04 | Affected | 0.1642 | 0.4273 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.2170G>T | A724S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A724S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain. Overall, the majority of evidence points to a benign impact for A724S, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -6.893 | Likely Benign | 0.204 | Likely Benign | Likely Benign | 0.93 | Ambiguous | 0.1 | 0.88 | Ambiguous | 0.91 | Ambiguous | 0.57 | Ambiguous | 0.265 | Likely Benign | -1.68 | Neutral | 0.983 | Probably Damaging | 0.993 | Probably Damaging | 2.20 | Pathogenic | 0.25 | Tolerated | 0.2308 | 0.4518 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.2171C>A | A724D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A724D is not reported in ClinVar or gnomAD. Prediction tools that indicate a benign effect include REVEL, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. Four methods (FoldX, Rosetta, Foldetta, premPS) returned uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic; Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for A724D, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -12.233 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 1.06 | Ambiguous | 0.2 | 0.86 | Ambiguous | 0.96 | Ambiguous | 0.60 | Ambiguous | 0.335 | Likely Benign | -4.44 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1596 | 0.1812 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.2171C>G | A724G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A724G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -8.908 | Likely Pathogenic | 0.580 | Likely Pathogenic | Likely Benign | 1.45 | Ambiguous | 0.1 | 1.73 | Ambiguous | 1.59 | Ambiguous | 0.56 | Ambiguous | 0.286 | Likely Benign | -3.10 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.07 | Pathogenic | 0.08 | Tolerated | 0.2001 | 0.3609 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.2171C>T | A724V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A724V missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign, SGM Consensus predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. With no ClinVar annotation, there is no contradiction between the predictions and existing clinical data. Overall, the evidence is mixed, but the majority of high‑confidence tools lean toward a benign effect, suggesting the variant is most likely benign rather than pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.476583 | Structured | 0.458050 | Uncertain | 0.923 | 0.483 | 0.250 | -9.000 | Likely Pathogenic | 0.471 | Ambiguous | Likely Benign | 0.42 | Likely Benign | 0.1 | 0.35 | Likely Benign | 0.39 | Likely Benign | 0.24 | Likely Benign | 0.241 | Likely Benign | -3.28 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | 2.07 | Pathogenic | 0.01 | Affected | 0.0872 | 0.5620 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.2173C>A | L725M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L725M is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Uncertain or inconclusive predictions come from Foldetta, premPS, AlphaMissense‑Default, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leans pathogenic (2 pathogenic vs. 1 benign). Foldetta remains uncertain and is not used as evidence. Overall, the majority of tools, including the high‑accuracy consensus, predict a pathogenic impact. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.557691 | Disordered | 0.455613 | Uncertain | 0.911 | 0.491 | 0.625 | -9.531 | Likely Pathogenic | 0.436 | Ambiguous | Likely Benign | 0.42 | Likely Benign | 0.1 | 1.05 | Ambiguous | 0.74 | Ambiguous | 0.80 | Ambiguous | 0.172 | Likely Benign | -1.81 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.32 | Pathogenic | 0.04 | Affected | 0.0854 | 0.4156 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||
| c.2173C>G | L725V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L725V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The remaining methods (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) are inconclusive. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome, and Foldetta provides no definitive stability change. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.455613 | Uncertain | 0.911 | 0.491 | 0.625 | -8.291 | Likely Pathogenic | 0.461 | Ambiguous | Likely Benign | 1.76 | Ambiguous | 0.1 | 1.87 | Ambiguous | 1.82 | Ambiguous | 0.77 | Ambiguous | 0.183 | Likely Benign | -2.69 | Deleterious | 0.993 | Probably Damaging | 0.992 | Probably Damaging | 1.36 | Pathogenic | 0.01 | Affected | 0.1739 | 0.3977 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||
| c.2174T>A | L725Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L725Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to REVEL, which scores the variant as benign. The majority of tools predict a pathogenic impact: premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, Rosetta, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Uncertain or inconclusive results come from FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Taken together, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.455613 | Uncertain | 0.911 | 0.491 | 0.625 | -13.952 | Likely Pathogenic | 0.888 | Likely Pathogenic | Ambiguous | 1.55 | Ambiguous | 0.1 | 2.09 | Destabilizing | 1.82 | Ambiguous | 1.88 | Destabilizing | 0.319 | Likely Benign | -5.43 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.1198 | 0.1203 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||
| c.2174T>C | L725P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L725P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. **Based on the collective predictions, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.455613 | Uncertain | 0.911 | 0.491 | 0.625 | -15.390 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 4.91 | Destabilizing | 0.1 | 9.02 | Destabilizing | 6.97 | Destabilizing | 1.82 | Destabilizing | 0.396 | Likely Benign | -6.08 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.3796 | 0.1664 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||
| c.2174T>G | L725R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L725R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, Rosetta, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—classify the variant as pathogenic. FoldX and Foldetta report uncertain results and are therefore not considered evidence for either side. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus indicates likely pathogenic, while Foldetta remains uncertain. Based on the overwhelming majority of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, which is consistent with the absence of a ClinVar entry and gnomAD observation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.455613 | Uncertain | 0.911 | 0.491 | 0.625 | -15.383 | Likely Pathogenic | 0.961 | Likely Pathogenic | Likely Pathogenic | 0.69 | Ambiguous | 0.3 | 2.16 | Destabilizing | 1.43 | Ambiguous | 1.49 | Destabilizing | 0.345 | Likely Benign | -5.46 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.1374 | 0.0846 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||
| c.2176A>G | R726G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R726G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Predictions that are uncertain or inconclusive are AlphaMissense‑Default, FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain (treated as unavailable). Overall, the majority of evidence points to a benign impact for R726G, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.449098 | Uncertain | 0.888 | 0.513 | 0.625 | -5.879 | Likely Benign | 0.528 | Ambiguous | Likely Benign | 0.80 | Ambiguous | 0.1 | 0.66 | Ambiguous | 0.73 | Ambiguous | 0.39 | Likely Benign | 0.159 | Likely Benign | -1.59 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.61 | Benign | 0.08 | Tolerated | 0.3343 | 0.3575 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||
| c.2176A>T | R726W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R726W has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a split: benign calls from REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further reveal AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. No prediction or stability result is missing or inconclusive. Overall, the majority of tools (seven) predict a benign effect, but the SGM‑Consensus and several high‑accuracy methods indicate pathogenicity, leaving the variant’s clinical significance uncertain. The predictions do not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.521092 | Disordered | 0.449098 | Uncertain | 0.888 | 0.513 | 0.625 | -10.091 | Likely Pathogenic | 0.580 | Likely Pathogenic | Likely Benign | 0.46 | Likely Benign | 0.1 | 0.46 | Likely Benign | 0.46 | Likely Benign | 0.15 | Likely Benign | 0.217 | Likely Benign | -3.72 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.57 | Benign | 0.01 | Affected | 0.1161 | 0.4252 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||
| c.2177G>A | R726K 2D 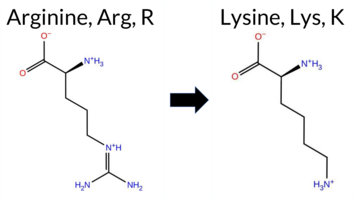 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R726K is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only two tools—polyPhen‑2 HumDiv and polyPhen‑2 HumVar—predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign”; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.449098 | Uncertain | 0.888 | 0.513 | 0.625 | -5.344 | Likely Benign | 0.231 | Likely Benign | Likely Benign | 0.28 | Likely Benign | 0.0 | -0.02 | Likely Benign | 0.13 | Likely Benign | 0.20 | Likely Benign | 0.154 | Likely Benign | -0.63 | Neutral | 0.990 | Probably Damaging | 0.998 | Probably Damaging | 2.67 | Benign | 0.16 | Tolerated | 0.4920 | 0.4042 | 3 | 2 | 0.6 | -28.01 | ||||||||||||||||||||||||||||||
| c.2177G>C | R726T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R726T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default; FoldX is uncertain. High‑accuracy methods give a consistent benign signal: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the aggregate predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.449098 | Uncertain | 0.888 | 0.513 | 0.625 | -5.249 | Likely Benign | 0.661 | Likely Pathogenic | Likely Benign | 0.82 | Ambiguous | 0.0 | -0.11 | Likely Benign | 0.36 | Likely Benign | -0.01 | Likely Benign | 0.200 | Likely Benign | -0.90 | Neutral | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.74 | Benign | 1.00 | Tolerated | 0.1685 | 0.4569 | -1 | -1 | 3.8 | -55.08 | ||||||||||||||||||||||||||||||
| c.2177G>T | R726M 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R726M has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, Foldetta, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX is uncertain and is not counted as evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of tools and the two high‑accuracy methods predict a benign effect. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.521092 | Disordered | 0.449098 | Uncertain | 0.888 | 0.513 | 0.625 | -8.611 | Likely Pathogenic | 0.750 | Likely Pathogenic | Likely Benign | 0.53 | Ambiguous | 0.1 | 0.31 | Likely Benign | 0.42 | Likely Benign | 0.20 | Likely Benign | 0.199 | Likely Benign | -2.02 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.59 | Benign | 0.03 | Affected | 0.1489 | 0.4051 | 0 | -1 | 6.4 | -24.99 | |||||||||||||||||||||||||||||||
| c.2178G>C | R726S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R726S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign; the SGM‑Consensus (majority vote) is Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts Benign. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.449098 | Uncertain | 0.888 | 0.513 | 0.625 | -4.780 | Likely Benign | 0.753 | Likely Pathogenic | Likely Benign | 0.40 | Likely Benign | 0.1 | -0.07 | Likely Benign | 0.17 | Likely Benign | -0.16 | Likely Benign | 0.232 | Likely Benign | -0.41 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.66 | Benign | 0.92 | Tolerated | 0.2993 | 0.3956 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||
| c.2178G>T | R726S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R726S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign; the SGM‑Consensus (majority vote) is Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts Benign. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.449098 | Uncertain | 0.888 | 0.513 | 0.625 | -4.780 | Likely Benign | 0.753 | Likely Pathogenic | Likely Benign | 0.40 | Likely Benign | 0.1 | -0.07 | Likely Benign | 0.17 | Likely Benign | -0.16 | Likely Benign | 0.232 | Likely Benign | -0.41 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.66 | Benign | 0.92 | Tolerated | 0.2993 | 0.3956 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||
| c.2179A>C | N727H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727H is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Two tools (premPS and ESM1b) return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of predictions (six benign vs. five pathogenic) lean toward a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -7.308 | In-Between | 0.224 | Likely Benign | Likely Benign | 0.13 | Likely Benign | 0.0 | -0.02 | Likely Benign | 0.06 | Likely Benign | 0.51 | Ambiguous | 0.171 | Likely Benign | -3.18 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.13 | Pathogenic | 0.03 | Affected | 0.1320 | 0.7186 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||
| c.2179A>G | N727D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N727D has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default, while the SGM‑Consensus score is labeled Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions lean toward a benign effect, and this does not contradict any ClinVar annotation, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -5.640 | Likely Benign | 0.601 | Likely Pathogenic | Likely Benign | 0.22 | Likely Benign | 0.0 | 0.35 | Likely Benign | 0.29 | Likely Benign | 0.36 | Likely Benign | 0.142 | Likely Benign | -2.93 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.18 | Pathogenic | 0.08 | Tolerated | 0.1899 | 0.4309 | 2 | 1 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.2179A>T | N727Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727Y has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Two tools remain inconclusive: AlphaMissense‑Default and Rosetta. Separately, the high‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign. Overall, the majority of individual predictors and the SGM Consensus lean toward a pathogenic interpretation, while the high‑accuracy folding‑stability assessment is benign. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -10.106 | Likely Pathogenic | 0.426 | Ambiguous | Likely Benign | -0.12 | Likely Benign | 0.1 | -0.52 | Ambiguous | -0.32 | Likely Benign | 0.35 | Likely Benign | 0.347 | Likely Benign | -5.34 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.12 | Pathogenic | 0.02 | Affected | 0.0563 | 0.6091 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||
| c.217A>G | R73G 2D AIThe SynGAP1 missense variant R73G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.453164 | Uncertain | 0.332 | 0.826 | 0.375 | -3.556 | Likely Benign | 0.241 | Likely Benign | Likely Benign | 0.133 | Likely Benign | -1.48 | Neutral | 0.028 | Benign | 0.004 | Benign | 4.03 | Benign | 0.00 | Affected | 0.3465 | 0.3608 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.217A>T | R73W 2D AIThe SynGAP1 missense variant R73W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.453164 | Uncertain | 0.332 | 0.826 | 0.375 | -5.874 | Likely Benign | 0.318 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -1.96 | Neutral | 0.962 | Probably Damaging | 0.274 | Benign | 3.98 | Benign | 0.00 | Affected | 0.1505 | 0.4035 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2180A>C | N727T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727T is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that classify the variant as benign include REVEL, Rosetta, premPS, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. FoldX gives an uncertain result and is therefore treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta (combining FoldX‑MD and Rosetta outputs) as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -6.900 | Likely Benign | 0.335 | Likely Benign | Likely Benign | 0.52 | Ambiguous | 0.1 | -0.18 | Likely Benign | 0.17 | Likely Benign | 0.04 | Likely Benign | 0.125 | Likely Benign | -3.08 | Deleterious | 0.987 | Probably Damaging | 0.980 | Probably Damaging | 2.25 | Pathogenic | 0.74 | Tolerated | 0.1315 | 0.7181 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||
| c.2180A>G | N727S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727S is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of evidence supports a benign effect. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -6.195 | Likely Benign | 0.184 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.1 | 0.28 | Likely Benign | 0.30 | Likely Benign | 0.18 | Likely Benign | 0.118 | Likely Benign | -2.67 | Deleterious | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 2.19 | Pathogenic | 0.23 | Tolerated | 0.3833 | 0.6680 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||
| c.2180A>T | N727I 2D 3DClick to see structure in 3D Viewer AISynGAP1 N727I is not reported in ClinVar and is absent from gnomAD. Benign predictions come from REVEL, FoldX, premPS, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen2_HumDiv, polyPhen2_HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Foldetta and Rosetta provide inconclusive results. High‑accuracy tools give a mixed picture: AlphaMissense‑Optimized predicts benign, SGM‑Consensus predicts likely pathogenic, and Foldetta is uncertain. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -10.230 | Likely Pathogenic | 0.577 | Likely Pathogenic | Likely Benign | 0.17 | Likely Benign | 0.1 | 0.90 | Ambiguous | 0.54 | Ambiguous | 0.43 | Likely Benign | 0.319 | Likely Benign | -5.93 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.13 | Pathogenic | 0.03 | Affected | 0.0666 | 0.5917 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||
| c.2181C>A | N727K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N727K is catalogued in gnomAD (ID 6‑33441646‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, SIFT, and the protein‑folding stability method Foldetta; pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the consensus score SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, SGM Consensus indicates likely pathogenic, and Foldetta reports benign stability. Overall, the majority of evidence points to a pathogenic effect, and this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | 6-33441646-C-A | 1 | 6.19e-7 | -10.601 | Likely Pathogenic | 0.884 | Likely Pathogenic | Ambiguous | -0.12 | Likely Benign | 0.2 | -0.44 | Likely Benign | -0.28 | Likely Benign | 0.86 | Ambiguous | 0.148 | Likely Benign | -3.82 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 2.18 | Pathogenic | 0.12 | Tolerated | 3.59 | 7 | 0.2002 | 0.5590 | 0 | 1 | -0.4 | 14.07 | |||||||||||||||||||||||||
| c.2181C>G | N727K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N727K is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and Foldetta. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and premPS. High‑accuracy assessments show that AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of evidence (seven pathogenic vs. five benign, with two uncertain) points to a pathogenic impact. This conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.442107 | Uncertain | 0.843 | 0.542 | 0.625 | -10.601 | Likely Pathogenic | 0.884 | Likely Pathogenic | Ambiguous | -0.12 | Likely Benign | 0.2 | -0.44 | Likely Benign | -0.28 | Likely Benign | 0.86 | Ambiguous | 0.148 | Likely Benign | -3.82 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 2.18 | Pathogenic | 0.12 | Tolerated | 3.59 | 7 | 0.2002 | 0.5590 | 0 | 1 | -0.4 | 14.07 | ||||||||||||||||||||||||||||
| c.2182C>A | P728T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P728T has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only REVEL, while the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. Predictions that are inconclusive or uncertain are FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for P728T, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.632174 | Disordered | 0.434760 | Uncertain | 0.725 | 0.567 | 0.625 | -9.605 | Likely Pathogenic | 0.863 | Likely Pathogenic | Ambiguous | 1.06 | Ambiguous | 0.0 | 1.27 | Ambiguous | 1.17 | Ambiguous | 0.62 | Ambiguous | 0.298 | Likely Benign | -6.21 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 0.67 | Pathogenic | 0.00 | Affected | 0.1843 | 0.3917 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||
| c.2182C>G | P728A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P728A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, whereas the majority of tools predict a pathogenic effect: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show that AlphaMissense‑Optimized is uncertain, the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta is uncertain. Overall, the preponderance of evidence from multiple in silico tools indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.632174 | Disordered | 0.434760 | Uncertain | 0.725 | 0.567 | 0.625 | -9.350 | Likely Pathogenic | 0.800 | Likely Pathogenic | Ambiguous | 0.78 | Ambiguous | 0.1 | 0.79 | Ambiguous | 0.79 | Ambiguous | 0.69 | Ambiguous | 0.277 | Likely Benign | -6.59 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 0.68 | Pathogenic | 0.00 | Affected | 0.3568 | 0.3148 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||
| c.2182C>T | P728S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant P728S is not reported in ClinVar and is present in gnomAD (ID 6‑33441647‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, whereas the majority of tools predict pathogenicity: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results from FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized are treated as unavailable. High‑accuracy consensus methods give a Likely Pathogenic verdict from the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and an Uncertain outcome from AlphaMissense‑Optimized; Foldetta also reports Uncertain. Overall, the preponderance of evidence points to a pathogenic effect for P728S, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.632174 | Disordered | 0.434760 | Uncertain | 0.725 | 0.567 | 0.625 | 6-33441647-C-T | 1 | 6.20e-7 | -9.047 | Likely Pathogenic | 0.897 | Likely Pathogenic | Ambiguous | 0.89 | Ambiguous | 0.0 | 0.98 | Ambiguous | 0.94 | Ambiguous | 0.54 | Ambiguous | 0.280 | Likely Benign | -6.38 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 0.68 | Pathogenic | 0.00 | Affected | 3.59 | 7 | 0.3571 | 0.3571 | -1 | 1 | 0.8 | -10.04 | |||||||||||||||||||||||||
| c.2183C>A | P728H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P728H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default, all of which predict a deleterious impact. Predictions that are inconclusive or uncertain are FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show that AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Overall, the majority of evidence points to a pathogenic effect for P728H, and this conclusion does not contradict any ClinVar status because the variant is not yet classified in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.632174 | Disordered | 0.434760 | Uncertain | 0.725 | 0.567 | 0.625 | -8.897 | Likely Pathogenic | 0.945 | Likely Pathogenic | Ambiguous | 0.94 | Ambiguous | 0.0 | 0.86 | Ambiguous | 0.90 | Ambiguous | 0.64 | Ambiguous | 0.402 | Likely Benign | -7.23 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 0.65 | Pathogenic | 0.00 | Affected | 0.1993 | 0.3016 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||
| c.2183C>G | P728R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P728R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and FoldX, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain or inconclusive results are reported by Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments further indicate a likely pathogenic status from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) and an uncertain outcome from Foldetta (combining FoldX‑MD and Rosetta). AlphaMissense‑Optimized also remains uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.632174 | Disordered | 0.434760 | Uncertain | 0.725 | 0.567 | 0.625 | -10.309 | Likely Pathogenic | 0.938 | Likely Pathogenic | Ambiguous | 0.45 | Likely Benign | 0.1 | 0.59 | Ambiguous | 0.52 | Ambiguous | 0.70 | Ambiguous | 0.418 | Likely Benign | -7.46 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 0.66 | Pathogenic | 0.00 | Affected | 0.1728 | 0.2865 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||
| c.2183C>T | P728L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P728L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, and premPS, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact; FoldX and AlphaMissense‑Optimized are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the preponderance of evidence from standard and high‑accuracy predictors points to a pathogenic effect for P728L. This conclusion is not contradicted by any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.632174 | Disordered | 0.434760 | Uncertain | 0.725 | 0.567 | 0.625 | -11.125 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 0.79 | Ambiguous | 0.0 | 0.15 | Likely Benign | 0.47 | Likely Benign | 0.20 | Likely Benign | 0.402 | Likely Benign | -8.27 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 0.66 | Pathogenic | 0.00 | Affected | 0.2321 | 0.4713 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||
| c.2185A>C | N729H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N729H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from Rosetta (uncertain) and Foldetta (uncertain). High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign, while Foldetta remains uncertain. Overall, the evidence overwhelmingly supports a benign classification, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.426547 | Uncertain | 0.651 | 0.583 | 0.625 | -0.670 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.27 | Likely Benign | 0.0 | 0.84 | Ambiguous | 0.56 | Ambiguous | 0.00 | Likely Benign | 0.080 | Likely Benign | -0.92 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.28 | Benign | 0.17 | Tolerated | 0.1197 | 0.4602 | 2 | 1 | 0.3 | 23.04 | ||||||||||||||||||||||||||||||
| c.2185A>G | N729D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N729D is predicted to be benign by all available in‑silico tools. Consensus predictors (REVEL, SIFT, polyPhen‑2 HumDiv/HumVar, PROVEAN, premPS, FoldX, Rosetta, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly report a benign effect, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a benign impact on protein stability. ClinVar contains no entry for this variant, and it is not listed in gnomAD. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.426547 | Uncertain | 0.651 | 0.583 | 0.625 | -5.117 | Likely Benign | 0.270 | Likely Benign | Likely Benign | 0.03 | Likely Benign | 0.2 | 0.10 | Likely Benign | 0.07 | Likely Benign | 0.14 | Likely Benign | 0.054 | Likely Benign | -1.22 | Neutral | 0.390 | Benign | 0.144 | Benign | 3.41 | Benign | 0.55 | Tolerated | 0.1931 | 0.2250 | 2 | 1 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.2185A>T | N729Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N729Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while Rosetta remains uncertain. The high‑accuracy consensus methods give a consistent benign signal: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Taken together, the overwhelming majority of evidence points to a benign effect. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.426547 | Uncertain | 0.651 | 0.583 | 0.625 | -2.284 | Likely Benign | 0.216 | Likely Benign | Likely Benign | 0.00 | Likely Benign | 0.1 | 0.82 | Ambiguous | 0.41 | Likely Benign | 0.08 | Likely Benign | 0.060 | Likely Benign | -2.35 | Neutral | 0.575 | Possibly Damaging | 0.053 | Benign | 3.27 | Benign | 0.14 | Tolerated | 0.0570 | 0.4073 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||
| c.2186A>C | N729T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N729T is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (gnomAD ID 6‑33441651‑A‑C). Consensus among the majority of in‑silico predictors is benign: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Benign”; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, yields an uncertain result. Overall, the evidence strongly supports a benign effect, and this conclusion does not contradict ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.426547 | Uncertain | 0.651 | 0.583 | 0.625 | 6-33441651-A-C | 1 | 6.20e-7 | -1.952 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.52 | Ambiguous | 0.3 | 1.73 | Ambiguous | 1.13 | Ambiguous | -0.34 | Likely Benign | 0.052 | Likely Benign | -0.52 | Neutral | 0.123 | Benign | 0.042 | Benign | 3.33 | Benign | 1.00 | Tolerated | 3.59 | 7 | 0.1201 | 0.4805 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||
| c.2186A>G | N729S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N729S is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Across the available in‑silico predictors, the majority (REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) classify the change as benign, while Rosetta and Foldetta return uncertain results. No tool predicts pathogenicity. High‑accuracy assessments reinforce this benign trend: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta’s stability prediction is inconclusive. Overall, the computational evidence strongly supports a benign effect, and this does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.426547 | Uncertain | 0.651 | 0.583 | 0.625 | Conflicting | 2 | -1.578 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 0.14 | Likely Benign | 0.1 | 1.34 | Ambiguous | 0.74 | Ambiguous | -0.36 | Likely Benign | 0.063 | Likely Benign | -0.42 | Neutral | 0.221 | Benign | 0.027 | Benign | 3.38 | Benign | 0.93 | Tolerated | 3.59 | 7 | 0.3411 | 0.4854 | 1 | 1 | 2.7 | -27.03 | ||||||||||||||||||||||||||
| c.2186A>T | N729I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N729I is listed in gnomAD (ID 6‑33441651‑A‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, premPS, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicting benign, while Foldetta’s stability analysis is inconclusive. Overall, the majority of evidence points to a benign impact. There is no ClinVar status to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.426547 | Uncertain | 0.651 | 0.583 | 0.625 | 6-33441651-A-T | 1 | 6.20e-7 | -3.308 | Likely Benign | 0.234 | Likely Benign | Likely Benign | 0.54 | Ambiguous | 0.6 | 0.79 | Ambiguous | 0.67 | Ambiguous | 0.29 | Likely Benign | 0.043 | Likely Benign | -2.96 | Deleterious | 0.506 | Possibly Damaging | 0.243 | Benign | 3.26 | Benign | 0.13 | Tolerated | 3.59 | 7 | 0.0625 | 0.4698 | -3 | -2 | 8.0 | -0.94 | |||||||||||||||||||||||||
| c.2187C>A | N729K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N729K has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus call (Likely Benign). Only AlphaMissense‑Default predicts a pathogenic outcome. Tools with uncertain or mixed results are Foldetta (protein‑folding stability) and Rosetta. High‑accuracy assessments: AlphaMissense‑Optimized reports a benign effect; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely benign; Foldetta’s stability prediction is inconclusive. Overall, the majority of evidence points to a benign impact for the variant, and this conclusion does not contradict the current ClinVar status, which contains no report for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.426547 | Uncertain | 0.651 | 0.583 | 0.625 | -5.101 | Likely Benign | 0.648 | Likely Pathogenic | Likely Benign | -0.03 | Likely Benign | 0.1 | 1.92 | Ambiguous | 0.95 | Ambiguous | 0.12 | Likely Benign | 0.036 | Likely Benign | -1.39 | Neutral | 0.109 | Benign | 0.033 | Benign | 3.51 | Benign | 0.47 | Tolerated | 0.1948 | 0.3612 | 1 | 0 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||
| c.2187C>G | N729K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N729K has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus call (Likely Benign). Only AlphaMissense‑Default predicts a pathogenic outcome. Tools with uncertain or mixed results are Foldetta (protein‑folding stability) and Rosetta. High‑accuracy assessments: AlphaMissense‑Optimized reports a benign effect; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely benign; Foldetta’s stability prediction is inconclusive. Overall, the majority of evidence points to a benign impact for the variant, and this conclusion does not contradict the current ClinVar status, which contains no report for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.426547 | Uncertain | 0.651 | 0.583 | 0.625 | -5.101 | Likely Benign | 0.648 | Likely Pathogenic | Likely Benign | -0.03 | Likely Benign | 0.1 | 1.92 | Ambiguous | 0.95 | Ambiguous | 0.12 | Likely Benign | 0.036 | Likely Benign | -1.39 | Neutral | 0.109 | Benign | 0.033 | Benign | 3.51 | Benign | 0.47 | Tolerated | 0.1948 | 0.3612 | 1 | 0 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||
| c.2188A>C | I730L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I730L is reported as “Likely Benign” in ClinVar and is not present in gnomAD. All available in‑silico predictors classify the change as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments concur: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a benign effect. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -1.681 | Likely Benign | 0.069 | Likely Benign | Likely Benign | -0.05 | Likely Benign | 0.0 | -0.31 | Likely Benign | -0.18 | Likely Benign | -0.17 | Likely Benign | 0.028 | Likely Benign | -0.63 | Neutral | 0.000 | Benign | 0.005 | Benign | 3.53 | Benign | 0.47 | Tolerated | 0.0739 | 0.2869 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.2188A>G | I730V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I730V is reported as “Likely Benign” in ClinVar and is not present in gnomAD. All available in‑silico predictors classify the change as benign: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -3.960 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.25 | Likely Benign | 0.1 | 0.04 | Likely Benign | 0.15 | Likely Benign | 0.03 | Likely Benign | 0.028 | Likely Benign | -0.14 | Neutral | 0.112 | Benign | 0.033 | Benign | 3.48 | Benign | 0.39 | Tolerated | 0.1010 | 0.2303 | 4 | 3 | -0.3 | -14.03 | ||||||||||||||||||||||||||||||
| c.2188A>T | I730F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I730F has no ClinVar entry and is not reported in gnomAD. Prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen2_HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign or tolerated. Only polyPhen2_HumDiv classifies the change as pathogenic. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores the variant as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, reports a benign effect. No prediction tool or stability analysis is inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -4.953 | Likely Benign | 0.173 | Likely Benign | Likely Benign | -0.54 | Ambiguous | 0.1 | -0.01 | Likely Benign | -0.28 | Likely Benign | -0.01 | Likely Benign | 0.055 | Likely Benign | -1.86 | Neutral | 0.699 | Possibly Damaging | 0.152 | Benign | 3.45 | Benign | 0.08 | Tolerated | 0.0509 | 0.2082 | 1 | 0 | -1.7 | 34.02 | ||||||||||||||||||||||||||||||
| c.2189T>A | I730N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I730N is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar) and SIFT. The remaining tools—premPS, ESM1b, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -7.987 | In-Between | 0.369 | Ambiguous | Likely Benign | 0.09 | Likely Benign | 0.2 | 0.04 | Likely Benign | 0.07 | Likely Benign | 0.70 | Ambiguous | 0.080 | Likely Benign | -1.60 | Neutral | 1.000 | Probably Damaging | 0.986 | Probably Damaging | 3.46 | Benign | 0.02 | Affected | 0.0969 | 0.0533 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||
| c.2189T>C | I730T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant I730T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Predictions that are inconclusive are premPS and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign classification, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a benign effect. Overall, the consensus of both general and high‑accuracy predictors points to a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -5.641 | Likely Benign | 0.404 | Ambiguous | Likely Benign | 0.18 | Likely Benign | 0.2 | 0.12 | Likely Benign | 0.15 | Likely Benign | 0.54 | Ambiguous | 0.103 | Likely Benign | -0.83 | Neutral | 0.995 | Probably Damaging | 0.934 | Probably Damaging | 3.49 | Benign | 0.08 | Tolerated | 0.1074 | 0.0885 | 0 | -1 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||
| c.2189T>G | I730S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I730S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar; AlphaMissense‑Default remains uncertain. High‑accuracy assessments—AlphaMissense‑Optimized, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta outputs)—all uniformly predict a benign impact. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -6.220 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.25 | Likely Benign | 0.2 | -0.12 | Likely Benign | 0.07 | Likely Benign | 0.47 | Likely Benign | 0.123 | Likely Benign | -0.51 | Neutral | 1.000 | Probably Damaging | 0.967 | Probably Damaging | 3.60 | Benign | 0.34 | Tolerated | 0.2646 | 0.0903 | -1 | -2 | -5.3 | -26.08 | ||||||||||||||||||||||||||||||
| c.218G>A | R73K 2D AIThe SynGAP1 missense variant R73K is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33425826‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized returns a benign prediction, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.453164 | Uncertain | 0.332 | 0.826 | 0.375 | Uncertain | 1 | 6-33425826-G-A | 2 | 1.24e-6 | -4.033 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.46 | Neutral | 0.053 | Benign | 0.007 | Benign | 4.14 | Benign | 0.00 | Affected | 4.32 | 1 | 0.5194 | 0.4428 | Weaken | 2 | 3 | 0.6 | -28.01 | |||||||||||||||||||||||||||||||
| c.218G>C | R73T 2D AIThe SynGAP1 missense variant R73T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable and therefore not considered. Overall, the preponderance of evidence from multiple prediction algorithms and consensus methods indicates that R73T is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.453164 | Uncertain | 0.332 | 0.826 | 0.375 | -4.061 | Likely Benign | 0.343 | Ambiguous | Likely Benign | 0.070 | Likely Benign | -1.05 | Neutral | 0.115 | Benign | 0.012 | Benign | 4.05 | Benign | 0.00 | Affected | 0.1980 | 0.4439 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||||||||||||
| c.218G>T | R73M 2D AIThe SynGAP1 R73M missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus methods give a benign signal: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.453164 | Uncertain | 0.332 | 0.826 | 0.375 | -5.343 | Likely Benign | 0.495 | Ambiguous | Likely Benign | 0.135 | Likely Benign | -1.10 | Neutral | 0.872 | Possibly Damaging | 0.113 | Benign | 4.00 | Benign | 0.00 | Affected | 0.1910 | 0.4238 | 0 | -1 | 6.4 | -24.99 | |||||||||||||||||||||||||||||||||||||||
| c.2190C>G | I730M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I730M is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments—AlphaMissense‑Optimized, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta outputs)—all uniformly indicate a benign impact. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -3.149 | Likely Benign | 0.110 | Likely Benign | Likely Benign | -0.15 | Likely Benign | 0.1 | 0.31 | Likely Benign | 0.08 | Likely Benign | -0.15 | Likely Benign | 0.042 | Likely Benign | -0.77 | Neutral | 0.993 | Probably Damaging | 0.914 | Probably Damaging | 3.43 | Benign | 0.09 | Tolerated | 0.0698 | 0.2159 | 2 | 1 | -2.6 | 18.03 | ||||||||||||||||||||||||||||||
| c.2191C>A | Q731K 2D AIThe SynGAP1 missense variant Q731K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.415202 | Uncertain | 0.507 | 0.654 | 0.750 | -6.686 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.066 | Likely Benign | -1.58 | Neutral | 0.490 | Possibly Damaging | 0.149 | Benign | 2.67 | Benign | 0.20 | Tolerated | 0.1998 | 0.3932 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.2191C>G | Q731E 2D AIThe SynGAP1 missense variant Q731E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while ESM1b is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.415202 | Uncertain | 0.507 | 0.654 | 0.750 | -7.371 | In-Between | 0.161 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -1.21 | Neutral | 0.935 | Possibly Damaging | 0.405 | Benign | 2.66 | Benign | 0.17 | Tolerated | 0.1426 | 0.2479 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2192A>C | Q731P 2D AIThe SynGAP1 missense variant Q731P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic effect. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.415202 | Uncertain | 0.507 | 0.654 | 0.750 | -4.103 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -2.08 | Neutral | 1.000 | Probably Damaging | 0.987 | Probably Damaging | 2.64 | Benign | 0.26 | Tolerated | 0.2285 | 0.5385 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2192A>G | Q731R 2D  AIThe SynGAP1 missense variant Q731R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.415202 | Uncertain | 0.507 | 0.654 | 0.750 | -5.873 | Likely Benign | 0.267 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -1.63 | Neutral | 0.604 | Possibly Damaging | 0.293 | Benign | 2.66 | Benign | 0.14 | Tolerated | 0.1571 | 0.1775 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.2192A>T | Q731L 2D AIThe SynGAP1 missense variant Q731L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign impact for Q731L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.415202 | Uncertain | 0.507 | 0.654 | 0.750 | -4.251 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.161 | Likely Benign | -1.27 | Neutral | 0.825 | Possibly Damaging | 0.270 | Benign | 2.75 | Benign | 0.12 | Tolerated | 0.0879 | 0.5694 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2193A>C | Q731H 2D AIThe SynGAP1 missense variant Q731H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta results are not available for this variant. Overall, the majority of evidence points to a benign effect. The prediction is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.415202 | Uncertain | 0.507 | 0.654 | 0.750 | -5.268 | Likely Benign | 0.171 | Likely Benign | Likely Benign | 0.034 | Likely Benign | -1.46 | Neutral | 0.003 | Benign | 0.004 | Benign | 2.64 | Benign | 0.05 | Affected | 0.1520 | 0.3830 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2193A>T | Q731H 2D AIThe SynGAP1 missense variant Q731H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta results are not available for this variant. Overall, the majority of evidence points to a benign effect. The prediction is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.415202 | Uncertain | 0.507 | 0.654 | 0.750 | -5.268 | Likely Benign | 0.171 | Likely Benign | Likely Benign | 0.034 | Likely Benign | -1.46 | Neutral | 0.003 | Benign | 0.004 | Benign | 2.64 | Benign | 0.05 | Affected | 0.1520 | 0.3830 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2194A>G | R732G 2D AIThe SynGAP1 R732G missense variant is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (ID 6‑33441659‑A‑G). Prediction tools that agree on a benign effect include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs. 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of standard predictors lean toward pathogenicity, while the most accurate single predictor (AlphaMissense‑Optimized) suggests a benign outcome. Given the lack of ClinVar evidence, there is no contradiction; the variant is most likely pathogenic based on the collective predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | 6-33441659-A-G | 1 | 6.20e-7 | -9.348 | Likely Pathogenic | 0.295 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -2.98 | Deleterious | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.56 | Benign | 0.03 | Affected | 3.59 | 7 | 0.3080 | 0.2942 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||
| c.2194A>T | R732W 2D AIThe SynGAP1 missense variant R732W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, five tools predict pathogenicity versus four predicting benign, with no ClinVar evidence to contradict these computational findings. Thus, the variant is most likely pathogenic based on the current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | -11.976 | Likely Pathogenic | 0.252 | Likely Benign | Likely Benign | 0.161 | Likely Benign | -3.57 | Deleterious | 1.000 | Probably Damaging | 0.973 | Probably Damaging | 2.53 | Benign | 0.01 | Affected | 0.1332 | 0.2748 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2195G>A | R732K 2D AIThe SynGAP1 missense variant R732K is listed in ClinVar (ID 537019.0) with an “Uncertain” clinical significance and is present in gnomAD (6‑33441660‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this consensus does not conflict with the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | Conflicting | 2 | 6-33441660-G-A | 4 | 2.48e-6 | -5.278 | Likely Benign | 0.240 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.82 | Neutral | 0.973 | Probably Damaging | 0.943 | Probably Damaging | 2.69 | Benign | 0.21 | Tolerated | 3.59 | 7 | 0.4194 | 0.3923 | 3 | 2 | 0.6 | -28.01 | ||||||||||||||||||||||||||||||||
| c.2195G>C | R732T 2D AISynGAP1 missense variant R732T is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign (REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Optimized) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b). AlphaMissense‑Default remains uncertain. The high‑accuracy AlphaMissense‑Optimized predicts a benign effect, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also favors a benign outcome. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a benign impact, which does not contradict the current ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | Uncertain | 1 | -8.545 | Likely Pathogenic | 0.434 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -1.96 | Neutral | 0.999 | Probably Damaging | 0.892 | Possibly Damaging | 2.59 | Benign | 0.12 | Tolerated | 3.59 | 7 | 0.1915 | 0.3153 | -1 | -1 | 3.8 | -55.08 | ||||||||||||||||||||||||||||||||||||
| c.2195G>T | R732M 2D AIThe SynGAP1 missense variant R732M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and ESM1b. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors a benign outcome. Foldetta results are unavailable. Overall, the balance of evidence (5 benign vs 3 pathogenic predictions) indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | -9.956 | Likely Pathogenic | 0.414 | Ambiguous | Likely Benign | 0.098 | Likely Benign | -1.42 | Neutral | 0.840 | Possibly Damaging | 0.357 | Benign | 2.55 | Benign | 0.01 | Affected | 0.1432 | 0.2980 | 0 | -1 | 6.4 | -24.99 | ||||||||||||||||||||||||||||||||||||||||
| c.2196G>C | R732S 2D AIThe SynGAP1 missense variant R732S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the balance of evidence (five benign versus four pathogenic predictions) favors a benign classification. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | -8.019 | Likely Pathogenic | 0.599 | Likely Pathogenic | Likely Benign | 0.115 | Likely Benign | -1.81 | Neutral | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.61 | Benign | 0.14 | Tolerated | 0.2850 | 0.3129 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.2196G>T | R732S 2D AIThe SynGAP1 missense variant R732S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign impact for R732S, and this conclusion does not contradict any ClinVar annotation because the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | -8.019 | Likely Pathogenic | 0.599 | Likely Pathogenic | Likely Benign | 0.115 | Likely Benign | -1.81 | Neutral | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.61 | Benign | 0.14 | Tolerated | 0.2850 | 0.3129 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.2197C>A | Q733K 2D AIThe SynGAP1 missense variant Q733K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for Q733K, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.410831 | Uncertain | 0.331 | 0.686 | 0.875 | -6.779 | Likely Benign | 0.274 | Likely Benign | Likely Benign | 0.112 | Likely Benign | -1.66 | Neutral | 0.797 | Possibly Damaging | 0.312 | Benign | 2.61 | Benign | 0.05 | Affected | 0.1572 | 0.2790 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.2197C>G | Q733E 2D AIThe SynGAP1 missense variant Q733E is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign predictions from REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions from polyPhen‑2 (HumDiv and HumVar) and SIFT; ESM1b is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus itself is Benign. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of ClinVar pathogenic reports. Thus, the variant is most likely benign, and this is not contradictory to ClinVar, which has no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.410831 | Uncertain | 0.331 | 0.686 | 0.875 | -7.651 | In-Between | 0.152 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -1.38 | Neutral | 0.983 | Probably Damaging | 0.637 | Possibly Damaging | 2.60 | Benign | 0.03 | Affected | 0.1363 | 0.1130 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2198A>C | Q733P 2D AIThe SynGAP1 missense variant Q733P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.410831 | Uncertain | 0.331 | 0.686 | 0.875 | -4.249 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.172 | Likely Benign | -1.91 | Neutral | 0.220 | Benign | 0.308 | Benign | 2.52 | Benign | 0.04 | Affected | 0.2182 | 0.3894 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2198A>G | Q733R 2D AIThe SynGAP1 missense variant Q733R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q733R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.410831 | Uncertain | 0.331 | 0.686 | 0.875 | -5.986 | Likely Benign | 0.291 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -1.96 | Neutral | 0.950 | Possibly Damaging | 0.612 | Possibly Damaging | 2.57 | Benign | 0.04 | Affected | 0.1419 | 0.1279 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.2198A>T | Q733L 2D AIThe SynGAP1 missense variant Q733L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.410831 | Uncertain | 0.331 | 0.686 | 0.875 | -3.465 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -2.04 | Neutral | 0.905 | Possibly Damaging | 0.408 | Benign | 2.55 | Benign | 1.00 | Tolerated | 0.0736 | 0.4291 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2199G>C | Q733H 2D AIThe SynGAP1 missense variant Q733H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; the Foldetta stability analysis is not available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.410831 | Uncertain | 0.331 | 0.686 | 0.875 | -5.741 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -2.47 | Neutral | 0.990 | Probably Damaging | 0.780 | Possibly Damaging | 2.52 | Benign | 0.02 | Affected | 0.1341 | 0.2263 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2199G>T | Q733H 2D AIThe SynGAP1 missense variant Q733H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q733H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.410831 | Uncertain | 0.331 | 0.686 | 0.875 | -5.741 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -2.47 | Neutral | 0.990 | Probably Damaging | 0.780 | Possibly Damaging | 2.52 | Benign | 0.02 | Affected | 0.1341 | 0.2263 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.219G>C | R73S 2D AIThe SynGAP1 missense variant R73S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM‑Consensus likewise indicates Likely Benign, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.453164 | Uncertain | 0.332 | 0.826 | 0.375 | -2.919 | Likely Benign | 0.476 | Ambiguous | Likely Benign | 0.107 | Likely Benign | -0.89 | Neutral | 0.028 | Benign | 0.004 | Benign | 4.07 | Benign | 0.00 | Affected | 0.3336 | 0.4026 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.219G>T | R73S 2D AIThe SynGAP1 missense variant R73S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM‑Consensus (majority vote) also indicates Likely Benign, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.453164 | Uncertain | 0.332 | 0.826 | 0.375 | -2.919 | Likely Benign | 0.476 | Ambiguous | Likely Benign | 0.107 | Likely Benign | -0.89 | Neutral | 0.028 | Benign | 0.004 | Benign | 4.07 | Benign | 0.00 | Affected | 0.3336 | 0.4026 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2200C>A | P734T 2D AIThe SynGAP1 missense variant P734T is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus (SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as “Likely Benign,” and AlphaMissense‑Optimized also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no result available for this variant. Based on the unanimous benign predictions and the lack of any pathogenic evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.411273 | Uncertain | 0.368 | 0.721 | 0.875 | -4.469 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -2.08 | Neutral | 0.040 | Benign | 0.013 | Benign | 2.78 | Benign | 0.09 | Tolerated | 0.1764 | 0.4076 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2200C>G | P734A 2D AIThe SynGAP1 missense variant P734A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect. Consensus predictors such as SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and high‑accuracy methods including AlphaMissense‑Optimized all classify the variant as benign. Additional in silico assessments—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—also predict a benign outcome. No tool in the dataset suggests pathogenicity. Protein‑stability analysis via Foldetta is unavailable for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.411273 | Uncertain | 0.368 | 0.721 | 0.875 | -3.907 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.031 | Likely Benign | -2.19 | Neutral | 0.022 | Benign | 0.074 | Benign | 2.74 | Benign | 0.24 | Tolerated | 0.3801 | 0.3306 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2200C>T | P734S 2D AIThe SynGAP1 missense variant P734S is listed in ClinVar with an uncertain significance (ClinVar ID 2283225.0) and is present in the gnomAD database (gnomAD ID 6‑33441665‑C‑T). Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign effects. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this benign assessment: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the computational evidence strongly supports a benign classification, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.411273 | Uncertain | 0.368 | 0.721 | 0.875 | Uncertain | 2 | 6-33441665-C-T | 2 | 1.24e-6 | -4.291 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -2.44 | Neutral | 0.344 | Benign | 0.048 | Benign | 2.77 | Benign | 0.11 | Tolerated | 3.64 | 6 | 0.3775 | 0.3650 | 1 | -1 | 0.8 | -10.04 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||
| c.2201C>A | P734Q 2D AIThe SynGAP1 missense variant P734Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.411273 | Uncertain | 0.368 | 0.721 | 0.875 | -4.392 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.080 | Likely Benign | -1.92 | Neutral | 0.959 | Probably Damaging | 0.569 | Possibly Damaging | 2.87 | Benign | 0.06 | Tolerated | 0.1608 | 0.3364 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2201C>G | P734R 2D AIThe SynGAP1 missense variant P734R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.411273 | Uncertain | 0.368 | 0.721 | 0.875 | -6.099 | Likely Benign | 0.288 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -3.00 | Deleterious | 0.984 | Probably Damaging | 0.682 | Possibly Damaging | 2.71 | Benign | 0.07 | Tolerated | 0.1664 | 0.2464 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2201C>T | P734L 2D AIThe SynGAP1 missense variant P734L is reported in gnomAD (variant ID 6‑33441666‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, creating a single discordant call. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.411273 | Uncertain | 0.368 | 0.721 | 0.875 | 6-33441666-C-T | 3 | 1.86e-6 | -3.472 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -2.11 | Neutral | 0.897 | Possibly Damaging | 0.330 | Benign | 2.69 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.2390 | 0.5059 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.2203A>C | S735R 2D AIThe SynGAP1 missense variant S735R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, while the SGM‑Consensus (majority vote) remains Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -6.318 | Likely Benign | 0.784 | Likely Pathogenic | Ambiguous | 0.126 | Likely Benign | -1.25 | Neutral | 0.997 | Probably Damaging | 0.933 | Probably Damaging | 2.66 | Benign | 0.72 | Tolerated | 0.0913 | 0.2861 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2203A>G | S735G 2D AIThe SynGAP1 missense variant S735G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the majority of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -5.986 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.55 | Neutral | 0.953 | Possibly Damaging | 0.744 | Possibly Damaging | 2.68 | Benign | 0.23 | Tolerated | 0.2777 | 0.4089 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2203A>T | S735C 2D AIThe SynGAP1 missense variant S735C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S735C, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -7.291 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.174 | Likely Benign | -2.22 | Neutral | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.60 | Benign | 0.05 | Affected | 0.1136 | 0.5464 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2204G>A | S735N 2D AIThe SynGAP1 missense variant S735N is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -6.697 | Likely Benign | 0.142 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.68 | Neutral | 0.400 | Benign | 0.138 | Benign | 2.65 | Benign | 0.18 | Tolerated | 0.1375 | 0.3827 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.2204G>C | S735T 2D AIThe SynGAP1 missense variant S735T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -5.340 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -1.03 | Neutral | 0.980 | Probably Damaging | 0.799 | Possibly Damaging | 2.67 | Benign | 0.46 | Tolerated | 0.1464 | 0.5321 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2204G>T | S735I 2D AIThe SynGAP1 missense variant S735I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S735I, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -5.669 | Likely Benign | 0.167 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -1.71 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.64 | Benign | 0.09 | Tolerated | 0.0933 | 0.5069 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2205C>A | S735R 2D AIThe SynGAP1 missense variant S735R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, whereas the SGM‑Consensus (majority vote) supports a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -6.318 | Likely Benign | 0.784 | Likely Pathogenic | Ambiguous | 0.108 | Likely Benign | -1.25 | Neutral | 0.997 | Probably Damaging | 0.933 | Probably Damaging | 2.66 | Benign | 0.72 | Tolerated | 0.0913 | 0.2861 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2205C>G | S735R 2D AIThe SynGAP1 missense variant S735R has no ClinVar record and is not listed in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, whereas the SGM‑Consensus (majority vote) remains Benign; Foldetta results are unavailable. Overall, the balance of evidence—five benign versus three pathogenic predictions, a benign SGM‑Consensus, and no contradictory ClinVar annotation—indicates that the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -6.318 | Likely Benign | 0.784 | Likely Pathogenic | Ambiguous | 0.108 | Likely Benign | -1.25 | Neutral | 0.997 | Probably Damaging | 0.933 | Probably Damaging | 2.66 | Benign | 0.72 | Tolerated | 0.0913 | 0.2861 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2206C>A | R736S 2D AIThe SynGAP1 missense variant R736S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools (polyPhen‑2 HumDiv and SIFT) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | -3.864 | Likely Benign | 0.223 | Likely Benign | Likely Benign | 0.073 | Likely Benign | -1.17 | Neutral | 0.653 | Possibly Damaging | 0.361 | Benign | 2.63 | Benign | 0.00 | Affected | 0.3422 | 0.2163 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2206C>G | R736G 2D AIThe SynGAP1 missense variant R736G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv and SIFT predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | -4.100 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -2.05 | Neutral | 0.653 | Possibly Damaging | 0.361 | Benign | 2.51 | Benign | 0.00 | Affected | 0.3708 | 0.2554 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2206C>T | R736C 2D  AISynGAP1 missense variant R736C is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33441671‑C‑T). Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized scores benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also returns benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence indicates a benign effect, which does not conflict with the ClinVar uncertain designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | Conflicting | 3 | 6-33441671-C-T | 8 | 4.96e-6 | -7.113 | In-Between | 0.120 | Likely Benign | Likely Benign | 0.190 | Likely Benign | -2.06 | Neutral | 0.999 | Probably Damaging | 0.825 | Possibly Damaging | 2.48 | Pathogenic | 0.00 | Affected | 4.07 | 3 | 0.3740 | 0.1691 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||
| c.2207G>A | R736H 2D  AIThe SynGAP1 missense variant R736H is listed in ClinVar (ID 1351080.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33441672‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign. Foldetta results are not available. Overall, the majority of computational evidence indicates a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | Uncertain | 1 | 6-33441672-G-A | 6 | 3.72e-6 | -5.409 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -0.12 | Neutral | 0.004 | Benign | 0.001 | Benign | 2.50 | Benign | 0.00 | Affected | 4.07 | 3 | 0.2846 | 0.0921 | 2 | 0 | 1.3 | -19.05 | ||||||||||||||||||||||||||||||||
| c.2207G>C | R736P 2D AIThe SynGAP1 missense variant R736P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the overall assessment. Overall, the majority of evidence points to a benign effect for R736P, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | -5.246 | Likely Benign | 0.152 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -1.83 | Neutral | 0.966 | Probably Damaging | 0.638 | Possibly Damaging | 2.50 | Benign | 0.00 | Affected | 0.2396 | 0.3237 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2207G>T | R736L 2D AIThe SynGAP1 missense variant R736L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools (polyPhen‑2 HumDiv and SIFT) predict pathogenicity, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign classification, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.415259 | Uncertain | 0.305 | 0.771 | 0.875 | -4.173 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 0.061 | Likely Benign | -1.27 | Neutral | 0.653 | Possibly Damaging | 0.361 | Benign | 2.60 | Benign | 0.00 | Affected | 0.1856 | 0.3180 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2209C>A | Q737K 2D AIThe SynGAP1 missense variant Q737K is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Taken together, the majority of evidence points to a benign effect for Q737K, and this conclusion is not in conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | -5.841 | Likely Benign | 0.190 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -1.16 | Neutral | 0.906 | Possibly Damaging | 0.551 | Possibly Damaging | 2.77 | Benign | 0.07 | Tolerated | 0.2081 | 0.4009 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.2209C>G | Q737E 2D AIThe SynGAP1 missense variant Q737E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | -5.288 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -1.05 | Neutral | 0.906 | Possibly Damaging | 0.629 | Possibly Damaging | 2.76 | Benign | 0.04 | Affected | 0.1635 | 0.2355 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.220A>C | S74R 2D AIThe SynGAP1 missense variant S74R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority of the high‑accuracy tools) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | -3.271 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.34 | Neutral | 0.361 | Benign | 0.019 | Benign | 4.08 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0943 | 0.3562 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.220A>G | S74G 2D AIThe SynGAP1 missense variant S74G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | -3.540 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.028 | Likely Benign | -1.30 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.08 | Benign | 0.00 | Affected | 0.2347 | 0.3749 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.220A>T | S74C 2D AIThe SynGAP1 missense variant S74C has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | -5.213 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -1.29 | Neutral | 0.704 | Possibly Damaging | 0.089 | Benign | 4.04 | Benign | 0.00 | Affected | 0.1224 | 0.4659 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2210A>C | Q737P 2D AIThe SynGAP1 missense variant Q737P is listed in ClinVar (ID 2580571.0) with an uncertain significance designation and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only SIFT indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta stability analysis is unavailable. Taken together, the preponderance of evidence supports a benign classification for Q737P, which is consistent with its ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | Uncertain | 1 | -2.407 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.154 | Likely Benign | -1.22 | Neutral | 0.005 | Benign | 0.013 | Benign | 2.78 | Benign | 0.04 | Affected | 4.07 | 3 | 0.2366 | 0.4981 | -1 | 0 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||
| c.2210A>G | Q737R 2D AIThe SynGAP1 missense variant Q737R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign impact for Q737R, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | -4.524 | Likely Benign | 0.159 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -1.02 | Neutral | 0.986 | Probably Damaging | 0.793 | Possibly Damaging | 2.74 | Benign | 0.06 | Tolerated | 0.1688 | 0.1852 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.2210A>T | Q737L 2D AIThe SynGAP1 missense variant Q737L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also likely benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | -2.789 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -2.44 | Neutral | 0.959 | Probably Damaging | 0.721 | Possibly Damaging | 2.80 | Benign | 1.00 | Tolerated | 0.0958 | 0.5494 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2211G>C | Q737H 2D AIThe SynGAP1 missense variant Q737H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of computational evidence points to a benign effect for Q737H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | -4.517 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -1.55 | Neutral | 0.996 | Probably Damaging | 0.930 | Probably Damaging | 2.72 | Benign | 0.04 | Affected | 0.1683 | 0.3707 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2211G>T | Q737H 2D AIThe SynGAP1 missense variant Q737H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q737H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | -4.517 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.132 | Likely Benign | -1.55 | Neutral | 0.996 | Probably Damaging | 0.930 | Probably Damaging | 2.72 | Benign | 0.04 | Affected | 0.1683 | 0.3707 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2212A>C | S738R 2D AIThe SynGAP1 missense variant S738R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S738R, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | -4.241 | Likely Benign | 0.570 | Likely Pathogenic | Likely Benign | 0.066 | Likely Benign | -1.55 | Neutral | 0.473 | Possibly Damaging | 0.193 | Benign | 2.69 | Benign | 0.01 | Affected | 4.32 | 2 | 0.0887 | 0.2891 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2212A>G | S738G 2D AIThe SynGAP1 missense variant S738G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | -3.863 | Likely Benign | 0.053 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -1.53 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.66 | Benign | 0.01 | Affected | 0.2032 | 0.3385 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2212A>T | S738C 2D AIThe SynGAP1 missense variant S738C is reported in ClinVar as not yet classified and is present in gnomAD (variant ID 6‑33441677‑A‑T). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict pathogenic. High‑accuracy methods give a benign result from AlphaMissense‑Optimized and a likely benign consensus from SGM‑Consensus; Foldetta predictions are unavailable. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | 6-33441677-A-T | 4 | 2.48e-6 | -6.373 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -2.09 | Neutral | 0.975 | Probably Damaging | 0.815 | Possibly Damaging | 2.63 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1112 | 0.3982 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||
| c.2213G>A | S738N 2D AIThe SynGAP1 missense variant S738N is reported in ClinVar as having no entry and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a benign majority vote, matching the SGM‑Consensus label of “Likely Benign.” Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | -5.005 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -0.53 | Neutral | 0.425 | Benign | 0.233 | Benign | 2.69 | Benign | 0.02 | Affected | 0.1284 | 0.3251 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.2213G>C | S738T 2D AIThe SynGAP1 missense variant S738T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the variant as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | -3.926 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -0.68 | Neutral | 0.010 | Benign | 0.010 | Benign | 2.73 | Benign | 0.51 | Tolerated | 0.1336 | 0.4227 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2213G>T | S738I 2D AIThe SynGAP1 missense variant S738I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | -4.312 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -1.78 | Neutral | 0.642 | Possibly Damaging | 0.393 | Benign | 2.66 | Benign | 0.01 | Affected | 0.0847 | 0.3636 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2214T>A | S738R 2D AIThe SynGAP1 missense variant S738R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus (majority vote) as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | -4.241 | Likely Benign | 0.570 | Likely Pathogenic | Likely Benign | 0.068 | Likely Benign | -1.55 | Neutral | 0.473 | Possibly Damaging | 0.193 | Benign | 2.69 | Benign | 0.01 | Affected | 4.32 | 2 | 0.0887 | 0.2891 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2214T>G | S738R 2D AIThe SynGAP1 missense variant S738R is listed in ClinVar (ID 1592652.0) as Benign and is present in gnomAD (variant ID 6‑33441679‑T‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Benign; Foldetta’s protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, aligning with the ClinVar designation and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | Benign | 1 | 6-33441679-T-G | 1 | 6.20e-7 | -4.241 | Likely Benign | 0.570 | Likely Pathogenic | Likely Benign | 0.068 | Likely Benign | -1.55 | Neutral | 0.473 | Possibly Damaging | 0.193 | Benign | 2.69 | Benign | 0.01 | Affected | 4.32 | 2 | 0.0887 | 0.2891 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||
| c.2215G>A | E739K 2D AIThe SynGAP1 missense variant E739K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.456400 | Uncertain | 0.313 | 0.834 | 0.875 | -5.420 | Likely Benign | 0.343 | Ambiguous | Likely Benign | 0.107 | Likely Benign | -1.49 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 2.55 | Benign | 0.00 | Affected | 0.2697 | 0.7044 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2215G>C | E739Q 2D AIThe SynGAP1 missense variant E739Q is listed in ClinVar (ID 2429558.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.456400 | Uncertain | 0.313 | 0.834 | 0.875 | Uncertain | 1 | -2.846 | Likely Benign | 0.161 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -1.06 | Neutral | 0.801 | Possibly Damaging | 0.339 | Benign | 2.57 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1425 | 0.7060 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||
| c.2216A>C | E739A 2D AIThe SynGAP1 missense variant E739A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools (polyPhen‑2 HumDiv and SIFT) predict pathogenicity, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of evidence points to a benign impact. The variant’s predicted benign nature does not contradict any ClinVar annotation, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.456400 | Uncertain | 0.313 | 0.834 | 0.875 | -2.337 | Likely Benign | 0.184 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -1.85 | Neutral | 0.625 | Possibly Damaging | 0.252 | Benign | 2.52 | Benign | 0.00 | Affected | 0.4643 | 0.7148 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2216A>G | E739G 2D AIThe SynGAP1 missense variant E739G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of predictions (5 benign vs 4 pathogenic) lean toward a benign impact, and this is consistent with the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational evidence, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.456400 | Uncertain | 0.313 | 0.834 | 0.875 | -3.104 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -2.53 | Deleterious | 0.625 | Possibly Damaging | 0.252 | Benign | 2.49 | Pathogenic | 0.00 | Affected | 0.3335 | 0.5983 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||||||||
| c.2216A>T | E739V 2D AIThe SynGAP1 missense variant E739V is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM all predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a benign effect, and this conclusion does not contradict the current ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.456400 | Uncertain | 0.313 | 0.834 | 0.875 | Uncertain | 1 | -3.136 | Likely Benign | 0.274 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -1.86 | Neutral | 0.891 | Possibly Damaging | 0.575 | Possibly Damaging | 2.47 | Pathogenic | 0.00 | Affected | 4.32 | 2 | 0.0953 | 0.7431 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||
| c.2217G>C | E739D 2D AIThe SynGAP1 missense variant E739D is listed in ClinVar (ID 3661302.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this is not in conflict with the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.456400 | Uncertain | 0.313 | 0.834 | 0.875 | Uncertain | 1 | -3.369 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.49 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.59 | Benign | 0.00 | Affected | 0.2145 | 0.4732 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.2217G>T | E739D 2D AIIn silico analysis of the SynGAP1 E739D variant shows a consensus toward benign impact. Benign predictions come from SGM‑Consensus, REVEL, PROVEAN, polyPhen2_HumDiv, polyPhen2_HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas only SIFT predicts pathogenicity. High‑accuracy tools AlphaMissense‑Optimized and the SGM Consensus both classify the variant as benign; Foldetta results are unavailable. ClinVar has no entry for this variant and it is not present in gnomAD, so there is no conflicting evidence. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.456400 | Uncertain | 0.313 | 0.834 | 0.875 | -3.369 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.49 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.59 | Benign | 0.00 | Affected | 0.2145 | 0.4732 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2218C>G | R740G 2D AIThe SynGAP1 missense variant R740G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.475392 | Uncertain | 0.269 | 0.849 | 0.875 | -4.556 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.138 | Likely Benign | -2.55 | Deleterious | 0.993 | Probably Damaging | 0.887 | Possibly Damaging | 2.56 | Benign | 0.03 | Affected | 0.3739 | 0.3149 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2218C>T | R740W 2D AIThe SynGAP1 missense variant R740W is listed in ClinVar with an uncertain significance and is present in the gnomAD database (ID 6‑33441683‑C‑T). Prediction tools that classify the variant as benign include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while those that predict pathogenicity are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized predicting a benign effect; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two benign vs. two pathogenic calls) and is treated as unavailable, and no Foldetta stability data are reported. Overall, the majority of conventional tools (five pathogenic vs. four benign) suggest a pathogenic impact, whereas the single high‑accuracy tool indicates benign. Thus, the variant is most likely pathogenic based on the aggregate predictions, and this assessment does not contradict the ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.771762 | Disordered | 0.475392 | Uncertain | 0.269 | 0.849 | 0.875 | Uncertain | 2 | 6-33441683-C-T | 6 | 3.72e-6 | -8.561 | Likely Pathogenic | 0.168 | Likely Benign | Likely Benign | 0.180 | Likely Benign | -3.09 | Deleterious | 1.000 | Probably Damaging | 0.938 | Probably Damaging | 2.52 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1566 | 0.3375 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||
| c.2219G>A | R740Q 2D AIThe SynGAP1 missense variant R740Q is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33441684‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant, so it does not influence the assessment. Overall, the majority of predictions indicate that R740Q is most likely benign, which is consistent with the ClinVar “Uncertain” classification and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.475392 | Uncertain | 0.269 | 0.849 | 0.875 | Uncertain | 1 | 6-33441684-G-A | 4 | 2.48e-6 | -5.195 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.102 | Likely Benign | -0.67 | Neutral | 0.999 | Probably Damaging | 0.881 | Possibly Damaging | 2.60 | Benign | 0.08 | Tolerated | 4.32 | 2 | 0.3454 | 0.2203 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||
| c.2219G>C | R740P 2D AIThe SynGAP1 missense variant R740P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for R740P, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.475392 | Uncertain | 0.269 | 0.849 | 0.875 | -4.163 | Likely Benign | 0.155 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -1.99 | Neutral | 0.998 | Probably Damaging | 0.951 | Probably Damaging | 2.55 | Benign | 0.02 | Affected | 0.2426 | 0.4338 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2219G>T | R740L 2D AIThe SynGAP1 missense variant R740L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.475392 | Uncertain | 0.269 | 0.849 | 0.875 | -4.958 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -2.30 | Neutral | 0.064 | Benign | 0.040 | Benign | 2.57 | Benign | 0.03 | Affected | 0.2341 | 0.4243 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.221G>A | S74N 2D AIThe SynGAP1 missense variant S74N is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33425829‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” No Foldetta stability result is available. Overall, the majority of computational evidence indicates that the variant is most likely benign, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | Uncertain | 1 | 6-33425829-G-A | 5 | 3.10e-6 | -5.156 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.031 | Likely Benign | -0.89 | Neutral | 0.043 | Benign | 0.007 | Benign | 4.09 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1434 | 0.4193 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||
| c.221G>C | S74T 2D AIThe SynGAP1 missense variant S74T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | -3.874 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.53 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.22 | Benign | 0.00 | Affected | 0.1573 | 0.4704 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.221G>T | S74I 2D AIThe SynGAP1 missense variant S74I is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as likely benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is benign; Foldetta results are not available. Overall, the consensus of available predictions indicates that S74I is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | -4.668 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.036 | Likely Benign | -1.78 | Neutral | 0.099 | Benign | 0.007 | Benign | 4.06 | Benign | 0.00 | Affected | 0.0886 | 0.4680 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2221C>A | P741T 2D AIThe SynGAP1 missense variant P741T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.493550 | Uncertain | 0.354 | 0.859 | 0.875 | -4.626 | Likely Benign | 0.061 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.54 | Neutral | 0.010 | Benign | 0.022 | Benign | 2.86 | Benign | 0.05 | Affected | 0.1209 | 0.5080 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2221C>G | P741A 2D AIThe SynGAP1 missense variant P741A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” while Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.493550 | Uncertain | 0.354 | 0.859 | 0.875 | -3.995 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -0.33 | Neutral | 0.425 | Benign | 0.136 | Benign | 3.01 | Benign | 0.98 | Tolerated | 0.2942 | 0.3883 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2221C>T | P741S 2D AIThe SynGAP1 missense variant P741S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33441686‑C‑T). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. Grouping by consensus, the benign‑predicting tools outnumber the pathogenic one. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” No Foldetta stability data are available, so it does not influence the conclusion. Overall, the computational evidence indicates that the variant is most likely benign, and this assessment does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.493550 | Uncertain | 0.354 | 0.859 | 0.875 | Uncertain | 2 | 6-33441686-C-T | 3 | 1.86e-6 | -3.700 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -0.27 | Neutral | 0.270 | Benign | 0.136 | Benign | 2.92 | Benign | 0.00 | Affected | 4.32 | 2 | 0.2888 | 0.4278 | 1 | -1 | 0.8 | -10.04 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||
| c.2222C>A | P741H 2D AIThe SynGAP1 missense variant P741H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The prediction is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.493550 | Uncertain | 0.354 | 0.859 | 0.875 | -5.592 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -0.99 | Neutral | 0.006 | Benign | 0.007 | Benign | 2.81 | Benign | 0.01 | Affected | 0.1372 | 0.3831 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2222C>G | P741R 2D AIThe SynGAP1 missense variant P741R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.493550 | Uncertain | 0.354 | 0.859 | 0.875 | -4.434 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.070 | Likely Benign | -1.19 | Neutral | 0.642 | Possibly Damaging | 0.393 | Benign | 2.85 | Benign | 0.02 | Affected | 0.1303 | 0.2712 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2222C>T | P741L 2D AIThe SynGAP1 missense variant P741L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.493550 | Uncertain | 0.354 | 0.859 | 0.875 | -4.850 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.63 | Neutral | 0.001 | Benign | 0.003 | Benign | 2.84 | Benign | 0.03 | Affected | 0.1978 | 0.5780 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2224C>G | R742G 2D AIThe SynGAP1 missense variant R742G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.509587 | Binding | 0.309 | 0.856 | 0.875 | -4.065 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -1.25 | Neutral | 0.524 | Possibly Damaging | 0.259 | Benign | 2.70 | Benign | 0.02 | Affected | 0.3974 | 0.2805 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2224C>T | R742W 2D AIThe SynGAP1 missense variant R742W is listed in ClinVar (ID 2581888.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33441689‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign impact, which is consistent with the ClinVar “Uncertain” classification and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.509587 | Binding | 0.309 | 0.856 | 0.875 | Uncertain | 1 | 6-33441689-C-T | 6 | 3.72e-6 | -7.725 | In-Between | 0.133 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -1.71 | Neutral | 0.992 | Probably Damaging | 0.684 | Possibly Damaging | 2.66 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1638 | 0.2839 | -3 | 2 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||
| c.2225G>A | R742Q 2D AIThe SynGAP1 missense variant R742Q is listed in ClinVar (ID 928481.0) with an uncertain significance annotation and is observed in gnomAD (variant ID 6‑33441690‑G‑A). Consensus from multiple in‑silico predictors—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—uniformly classify the change as benign. No tool in the dataset reports a pathogenic prediction. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. A protein‑folding stability analysis via Foldetta is not available for this variant. Overall, the computational evidence strongly favors a benign interpretation, which is consistent with the ClinVar uncertain status rather than contradicting it. The variant is most likely benign, and this assessment does not contradict its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.509587 | Binding | 0.309 | 0.856 | 0.875 | Uncertain | 2 | 6-33441690-G-A | 24 | 1.49e-5 | -4.090 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -0.19 | Neutral | 0.032 | Benign | 0.007 | Benign | 2.73 | Benign | 0.07 | Tolerated | 4.32 | 2 | 0.3660 | 0.1530 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||
| c.2225G>C | R742P 2D AIThe SynGAP1 missense variant R742P is catalogued in gnomAD (6-33441690‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign or likely benign outcome. Only SIFT classifies the change as pathogenic, representing the sole discordant prediction. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise reports likely benign. The Foldetta protein‑folding stability analysis is not available for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.509587 | Binding | 0.309 | 0.856 | 0.875 | 6-33441690-G-C | 1 | 6.20e-7 | -2.920 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -0.51 | Neutral | 0.001 | Benign | 0.004 | Benign | 2.84 | Benign | 0.03 | Affected | 4.32 | 2 | 0.2598 | 0.3637 | -2 | 0 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||||
| c.2225G>T | R742L 2D AIThe SynGAP1 missense variant R742L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.509587 | Binding | 0.309 | 0.856 | 0.875 | -3.778 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -0.77 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.71 | Benign | 0.16 | Tolerated | 0.2342 | 0.3831 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2227C>A | P743T 2D AIThe SynGAP1 missense variant P743T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, creating a single discordant prediction. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence supports a benign classification, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.526809 | Binding | 0.317 | 0.862 | 0.875 | -4.892 | Likely Benign | 0.061 | Likely Benign | Likely Benign | 0.050 | Likely Benign | -1.11 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 2.75 | Benign | 0.07 | Tolerated | 0.1461 | 0.4750 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2227C>G | P743A 2D AIThe SynGAP1 missense variant P743A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.526809 | Binding | 0.317 | 0.862 | 0.875 | -4.253 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -1.10 | Neutral | 0.005 | Benign | 0.008 | Benign | 2.78 | Benign | 0.12 | Tolerated | 0.3158 | 0.3604 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2227C>T | P743S 2D AIThe SynGAP1 missense variant P743S is listed in gnomAD (ID 6‑33441692‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the change as benign. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is “Likely Benign.” No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that P743S is most likely benign, and this conclusion is not contradicted by any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.526809 | Binding | 0.317 | 0.862 | 0.875 | 6-33441692-C-T | 1 | 6.19e-7 | -4.286 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -0.33 | Neutral | 0.021 | Benign | 0.015 | Benign | 2.93 | Benign | 0.00 | Affected | 4.32 | 2 | 0.3087 | 0.3948 | -1 | 1 | 0.8 | -10.04 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||||
| c.2228C>A | P743H 2D AIThe SynGAP1 missense variant P743H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the overall assessment. Overall, the majority of computational evidence points to a benign effect for P743H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.526809 | Binding | 0.317 | 0.862 | 0.875 | -5.649 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.121 | Likely Benign | -1.99 | Neutral | 0.989 | Probably Damaging | 0.870 | Possibly Damaging | 2.70 | Benign | 0.01 | Affected | 0.1653 | 0.3762 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2228C>G | P743R 2D AIThe SynGAP1 missense variant P743R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.526809 | Binding | 0.317 | 0.862 | 0.875 | -4.295 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -1.96 | Neutral | 0.966 | Probably Damaging | 0.494 | Possibly Damaging | 2.73 | Benign | 0.02 | Affected | 0.1488 | 0.2819 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2228C>T | P743L 2D AIThe SynGAP1 missense variant P743L is listed in gnomAD (ID 6‑33441693‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.526809 | Binding | 0.317 | 0.862 | 0.875 | 6-33441693-C-T | 1 | 6.19e-7 | -4.838 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.112 | Likely Benign | -2.21 | Neutral | 0.801 | Possibly Damaging | 0.192 | Benign | 2.73 | Benign | 0.00 | Affected | 4.32 | 2 | 0.2166 | 0.5533 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||
| c.222C>A | S74R 2D AIThe SynGAP1 missense variant S74R is catalogued in gnomAD (ID 6‑33425830‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all report benign or likely benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments confirm the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus, derived from the majority of the high‑confidence predictors, is benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S74R is most likely benign, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | 6-33425830-C-A | 1 | 6.20e-7 | -3.271 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.070 | Likely Benign | -1.34 | Neutral | 0.361 | Benign | 0.019 | Benign | 4.08 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0943 | 0.3562 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||
| c.222C>G | S74R 2D AIThe SynGAP1 missense variant S74R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority of the high‑accuracy tools) is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | -3.271 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.070 | Likely Benign | -1.34 | Neutral | 0.361 | Benign | 0.019 | Benign | 4.08 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0943 | 0.3562 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2230C>A | Q744K 2D AIThe SynGAP1 missense variant Q744K is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -3.929 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.22 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.79 | Benign | 0.07 | Tolerated | 0.1784 | 0.3757 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.2230C>G | Q744E 2D AIThe SynGAP1 missense variant Q744E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” while Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -4.053 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.57 | Neutral | 0.065 | Benign | 0.038 | Benign | 2.77 | Benign | 0.08 | Tolerated | 0.1453 | 0.1892 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2231A>C | Q744P 2D AIThe SynGAP1 missense variant Q744P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign impact for Q744P, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -2.062 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.080 | Likely Benign | -0.46 | Neutral | 0.784 | Possibly Damaging | 0.206 | Benign | 2.82 | Benign | 0.12 | Tolerated | 0.2584 | 0.4397 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2231A>G | Q744R 2D AIThe SynGAP1 missense variant Q744R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -2.207 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -0.43 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.80 | Benign | 0.05 | Affected | 0.1437 | 0.1588 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.2231A>T | Q744L 2D AIThe SynGAP1 missense variant Q744L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta results are not available for this variant. Overall, the preponderance of evidence points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -3.724 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.052 | Likely Benign | -1.96 | Neutral | 0.425 | Benign | 0.158 | Benign | 2.70 | Benign | 0.02 | Affected | 0.0813 | 0.4731 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2232G>C | Q744H 2D AIThe SynGAP1 missense variant Q744H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q744H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -4.359 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.83 | Neutral | 0.975 | Probably Damaging | 0.574 | Possibly Damaging | 2.68 | Benign | 0.01 | Affected | 0.1452 | 0.3443 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2232G>T | Q744H 2D AIThe SynGAP1 missense variant Q744H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q744H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.540428 | Binding | 0.316 | 0.866 | 0.875 | -4.359 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.83 | Neutral | 0.975 | Probably Damaging | 0.574 | Possibly Damaging | 2.68 | Benign | 0.01 | Affected | 0.1452 | 0.3443 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2233C>A | P745T 2D AIThe SynGAP1 missense variant P745T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | -5.119 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.203 | Likely Benign | -2.78 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.52 | Benign | 0.02 | Affected | 0.1657 | 0.4593 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2233C>G | P745A 2D AIThe SynGAP1 missense variant P745A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for P745A, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | -4.599 | Likely Benign | 0.057 | Likely Benign | Likely Benign | 0.171 | Likely Benign | -2.88 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.55 | Benign | 0.17 | Tolerated | 0.3424 | 0.3458 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2233C>T | P745S 2D AIThe SynGAP1 missense variant P745S is reported in gnomAD (variant ID 6-33441698-C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence from multiple prediction algorithms and consensus methods points to a benign effect for P745S, and this conclusion is not contradicted by any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | 6-33441698-C-T | 1 | 6.19e-7 | -4.300 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.160 | Likely Benign | -2.32 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.54 | Benign | 0.05 | Affected | 4.32 | 2 | 0.3397 | 0.3882 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.2234C>A | P745H 2D AIThe SynGAP1 missense variant P745H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | -5.569 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.200 | Likely Benign | -2.84 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.53 | Benign | 0.00 | Affected | 0.1912 | 0.3403 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2234C>G | P745R 2D AIThe SynGAP1 missense variant P745R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the majority of computational evidence points to a benign effect for P745R, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | -4.564 | Likely Benign | 0.187 | Likely Benign | Likely Benign | 0.245 | Likely Benign | -2.90 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.51 | Benign | 0.01 | Affected | 0.1571 | 0.2852 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2234C>T | P745L 2D AIThe SynGAP1 missense variant P745L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus (majority vote) as Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | -6.303 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.211 | Likely Benign | -3.79 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.53 | Benign | 0.01 | Affected | 0.2205 | 0.5188 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2236G>A | V746M 2D  AIThe SynGAP1 missense variant V746M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the consensus of available predictions strongly suggests that V746M is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.808535 | Disordered | 0.576597 | Binding | 0.336 | 0.867 | 0.875 | -4.194 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.010 | Likely Benign | -0.58 | Neutral | 0.065 | Benign | 0.037 | Benign | 2.79 | Benign | 0.60 | Tolerated | 0.0756 | 0.4576 | 2 | 1 | -2.3 | 32.06 | |||||||||||||||||||||||||||||||||||||||
| c.2236G>C | V746L 2D AIThe SynGAP1 missense variant V746L is catalogued in gnomAD (ID 6‑33441701‑G‑C) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool reports a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the set predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available for this variant. Taken together, the computational evidence overwhelmingly supports a benign effect, and this conclusion does not conflict with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.808535 | Disordered | 0.576597 | Binding | 0.336 | 0.867 | 0.875 | 6-33441701-G-C | 1 | 6.19e-7 | -3.260 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.015 | Likely Benign | -0.68 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.82 | Benign | 0.08 | Tolerated | 4.32 | 2 | 0.0896 | 0.5065 | 1 | 2 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||||||
| c.2236G>T | V746L 2D AIThe SynGAP1 missense variant V746L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that V746L is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.808535 | Disordered | 0.576597 | Binding | 0.336 | 0.867 | 0.875 | -3.260 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.015 | Likely Benign | -0.68 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.82 | Benign | 0.08 | Tolerated | 4.32 | 2 | 0.0896 | 0.5065 | 1 | 2 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.2237T>A | V746E 2D AIThe SynGAP1 missense variant V746E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts benign. No Foldetta stability data are available. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.808535 | Disordered | 0.576597 | Binding | 0.336 | 0.867 | 0.875 | -4.136 | Likely Benign | 0.315 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -0.94 | Neutral | 0.642 | Possibly Damaging | 0.316 | Benign | 2.85 | Benign | 0.05 | Affected | 0.0928 | 0.1813 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2237T>C | V746A 2D AIThe SynGAP1 missense variant V746A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.808535 | Disordered | 0.576597 | Binding | 0.336 | 0.867 | 0.875 | -2.875 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.29 | Neutral | 0.010 | Benign | 0.005 | Benign | 2.80 | Benign | 0.21 | Tolerated | 0.2566 | 0.2531 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2237T>G | V746G 2D AIThe SynGAP1 missense variant V746G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized scores benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, V746G is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.808535 | Disordered | 0.576597 | Binding | 0.336 | 0.867 | 0.875 | -2.971 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -0.89 | Neutral | 0.136 | Benign | 0.270 | Benign | 2.74 | Benign | 0.02 | Affected | 0.1897 | 0.2550 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2239G>A | V747I 2D  AIThe SynGAP1 missense variant V747I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | -3.981 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -0.08 | Neutral | 0.002 | Benign | 0.008 | Benign | 2.70 | Benign | 0.00 | Affected | 0.0560 | 0.3018 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2239G>C | V747L 2D AIThe SynGAP1 missense variant V747L (ClinVar ID 1985039.0) is listed as ClinVar status Uncertain and is present in gnomAD (6‑33441704‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of computational evidence supports a benign classification, which is consistent with the ClinVar Uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | Uncertain | 1 | 6-33441704-G-C | 2 | 1.24e-6 | -2.790 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.52 | Neutral | 0.065 | Benign | 0.033 | Benign | 2.67 | Benign | 0.00 | Affected | 4.32 | 2 | 0.0699 | 0.3674 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||||
| c.2239G>T | V747L 2D AIThe SynGAP1 missense variant V747L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | -2.790 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.52 | Neutral | 0.065 | Benign | 0.033 | Benign | 2.67 | Benign | 0.00 | Affected | 4.32 | 2 | 0.0699 | 0.3674 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.223G>A | E75K 2D AIThe SynGAP1 missense variant E75K is listed in ClinVar as Benign (ClinVar ID 3360083.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, aligning with the ClinVar classification and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.443881 | Uncertain | 0.303 | 0.822 | 0.500 | Benign/Likely benign | 2 | -4.020 | Likely Benign | 0.358 | Ambiguous | Likely Benign | 0.134 | Likely Benign | -1.12 | Neutral | 0.748 | Possibly Damaging | 0.017 | Benign | 4.07 | Benign | 0.00 | Affected | 0.2565 | 0.6908 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||
| c.223G>C | E75Q 2D AIThe SynGAP1 missense variant E75Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.443881 | Uncertain | 0.303 | 0.822 | 0.500 | -3.772 | Likely Benign | 0.194 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -0.76 | Neutral | 0.731 | Possibly Damaging | 0.058 | Benign | 4.04 | Benign | 0.00 | Affected | 0.1461 | 0.6634 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2240T>A | V747E 2D AIThe SynGAP1 missense variant V747E is evaluated by multiple in silico tools. ClinVar has no entry for this change, and it is not reported in gnomAD. Consensus from the SGM‑Consensus algorithm classifies the variant as Likely Benign. When grouping predictions by agreement, benign calls come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign, and no Foldetta stability data are available. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Based on the aggregate predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | -4.124 | Likely Benign | 0.464 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -1.31 | Neutral | 0.917 | Possibly Damaging | 0.666 | Possibly Damaging | 2.56 | Benign | 0.00 | Affected | 0.0965 | 0.1559 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2240T>C | V747A 2D AIThe SynGAP1 missense variant V747A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of computational evidence supports a benign classification for V747A, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | -3.118 | Likely Benign | 0.171 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -0.69 | Neutral | 0.425 | Benign | 0.252 | Benign | 2.73 | Benign | 0.00 | Affected | 0.2585 | 0.1714 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2240T>G | V747G 2D AIThe SynGAP1 missense variant V747G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as benign, and Foldetta results are unavailable. Overall, the consensus of the available predictions indicates that V747G is most likely benign, and this conclusion does not contradict any ClinVar status because the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | -3.883 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -1.22 | Neutral | 0.589 | Possibly Damaging | 0.917 | Probably Damaging | 2.56 | Benign | 0.00 | Affected | 0.1960 | 0.2374 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2242C>A | L748M 2D AIThe SynGAP1 missense variant L748M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for the variant, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.611637 | Binding | 0.339 | 0.863 | 0.750 | -3.935 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.12 | Neutral | 0.991 | Probably Damaging | 0.852 | Possibly Damaging | 2.70 | Benign | 0.05 | Affected | 0.0909 | 0.3863 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2242C>G | L748V 2D AIThe SynGAP1 missense variant L748V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.611637 | Binding | 0.339 | 0.863 | 0.750 | -3.454 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.42 | Neutral | 0.679 | Possibly Damaging | 0.216 | Benign | 2.74 | Benign | 0.05 | Affected | 0.1680 | 0.3485 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2243T>A | L748Q 2D AIThe SynGAP1 missense variant L748Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for L748Q, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.611637 | Binding | 0.339 | 0.863 | 0.750 | -3.177 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.46 | Neutral | 0.912 | Possibly Damaging | 0.611 | Possibly Damaging | 2.74 | Benign | 0.01 | Affected | 0.1235 | 0.1246 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2243T>C | L748P 2D AIThe SynGAP1 missense variant L748P is reported in gnomAD (ID 6‑33441708‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions are made by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments reinforce the benign view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.611637 | Binding | 0.339 | 0.863 | 0.750 | 6-33441708-T-C | 1 | 6.20e-7 | -2.732 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.69 | Neutral | 0.912 | Possibly Damaging | 0.611 | Possibly Damaging | 2.69 | Benign | 0.02 | Affected | 4.32 | 2 | 0.3503 | 0.1399 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||
| c.2243T>G | L748R 2D AIThe SynGAP1 missense variant L748R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33441708‑T‑G). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the current ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.611637 | Binding | 0.339 | 0.863 | 0.750 | Conflicting | 2 | 6-33441708-T-G | 3 | 1.86e-6 | -3.331 | Likely Benign | 0.245 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.67 | Neutral | 0.912 | Possibly Damaging | 0.448 | Possibly Damaging | 2.73 | Benign | 0.02 | Affected | 4.32 | 2 | 0.1342 | 0.0888 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||
| c.2245C>G | R749G 2D AIThe SynGAP1 missense variant R749G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for R749G, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.626050 | Binding | 0.337 | 0.860 | 0.625 | -3.045 | Likely Benign | 0.285 | Likely Benign | Likely Benign | 0.161 | Likely Benign | -1.03 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.68 | Benign | 0.02 | Affected | 0.3690 | 0.4196 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2245C>T | R749W 2D AIThe SynGAP1 missense variant R749W is listed in ClinVar as benign and is observed in gnomAD (ID 6‑33441710‑C‑T). Prediction tools that classify the variant as benign include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also returns benign, and Foldetta stability analysis is unavailable. Overall, the majority of evidence, especially from high‑confidence methods, supports a benign effect. This consensus aligns with the ClinVar designation, so there is no contradiction between the predictions and the reported clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.675549 | Disordered | 0.626050 | Binding | 0.337 | 0.860 | 0.625 | Likely Benign | 1 | 6-33441710-C-T | 3 | 1.86e-6 | -7.647 | In-Between | 0.338 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -2.62 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.59 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1215 | 0.4088 | 2 | -3 | 3.6 | 30.03 | |||||||||||||||||||||||||||||||||
| c.2246G>A | R749Q 2D AIThe SynGAP1 missense variant R749Q is listed in ClinVar (ID 793884.0) as Benign and is present in gnomAD (6‑33441711‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a Likely Benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence—including high‑accuracy predictions—supports a benign classification, which is consistent with the ClinVar status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.626050 | Binding | 0.337 | 0.860 | 0.625 | Likely Benign | 1 | 6-33441711-G-A | 4 | 2.48e-6 | -3.069 | Likely Benign | 0.212 | Likely Benign | Likely Benign | 0.152 | Likely Benign | -1.00 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 2.64 | Benign | 0.03 | Affected | 4.32 | 2 | 0.3467 | 0.2529 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||
| c.2246G>C | R749P 2D AIThe SynGAP1 missense variant R749P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for R749P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.626050 | Binding | 0.337 | 0.860 | 0.625 | -2.467 | Likely Benign | 0.311 | Likely Benign | Likely Benign | 0.170 | Likely Benign | -1.54 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.61 | Benign | 0.02 | Affected | 0.2195 | 0.4878 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2246G>T | R749L 2D AIThe SynGAP1 missense variant R749L is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. The AlphaMissense‑Default score is uncertain, and no Foldetta stability assessment is available. High‑accuracy predictors that are available—AlphaMissense‑Optimized and the SGM‑Consensus—both support a benign classification. Consequently, the overall evidence points to the variant being most likely benign, and this assessment does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.626050 | Binding | 0.337 | 0.860 | 0.625 | -3.926 | Likely Benign | 0.413 | Ambiguous | Likely Benign | 0.168 | Likely Benign | -2.15 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.65 | Benign | 0.01 | Affected | 0.2106 | 0.5154 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2248G>A | G750R 2D AIThe SynGAP1 missense variant G750R is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33441713‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs four benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | 6-33441713-G-A | 17 | 1.05e-5 | -3.861 | Likely Benign | 0.619 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -2.09 | Neutral | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.45 | Pathogenic | 0.00 | Affected | 3.99 | 5 | 0.0960 | 0.4015 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.2248G>C | G750R 2D AIThe SynGAP1 missense variant G750R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments are mixed: AlphaMissense‑Optimized predicts benign, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2), and Foldetta data are unavailable. Overall, the majority of standard predictors (5 pathogenic vs 4 benign) lean toward pathogenicity, and no ClinVar annotation contradicts this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -3.861 | Likely Benign | 0.619 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -2.09 | Neutral | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.45 | Pathogenic | 0.00 | Affected | 3.99 | 5 | 0.0960 | 0.4015 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2248G>T | G750W 2D  AIThe SynGAP1 missense variant G750W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, while the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the variant as damaging. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the preponderance of evidence from multiple in silico predictors indicates that G750W is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -8.433 | Likely Pathogenic | 0.427 | Ambiguous | Likely Benign | 0.135 | Likely Benign | -3.30 | Deleterious | 1.000 | Probably Damaging | 0.994 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 0.0738 | 0.4009 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.2249G>A | G750E 2D  AISynGAP1 missense variant G750E is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | Uncertain | 1 | -2.618 | Likely Benign | 0.413 | Ambiguous | Likely Benign | 0.146 | Likely Benign | -2.27 | Neutral | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.49 | Pathogenic | 0.01 | Affected | 3.99 | 5 | 0.1326 | 0.3768 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||||
| c.2249G>C | G750A 2D AIThe SynGAP1 missense variant G750A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -3.263 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -1.65 | Neutral | 0.996 | Probably Damaging | 0.887 | Possibly Damaging | 2.52 | Benign | 0.04 | Affected | 0.3683 | 0.4792 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2249G>T | G750V 2D AIThe SynGAP1 missense variant G750V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions therefore show AlphaMissense‑Optimized as benign, while the remaining high‑confidence tools (PROVEAN, polyPhen‑2, SIFT, FATHMM) all indicate pathogenicity. Overall, the majority of evidence (five pathogenic vs four benign) points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -4.512 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -2.54 | Deleterious | 0.984 | Probably Damaging | 0.850 | Possibly Damaging | 2.45 | Pathogenic | 0.01 | Affected | 0.1270 | 0.4212 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.224A>C | E75A 2D AIThe SynGAP1 missense variant E75A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions point to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.443881 | Uncertain | 0.303 | 0.822 | 0.500 | -3.111 | Likely Benign | 0.194 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -1.19 | Neutral | 0.345 | Benign | 0.021 | Benign | 4.05 | Benign | 0.00 | Affected | 0.4248 | 0.6413 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.224A>G | E75G 2D AIThe SynGAP1 missense variant E75G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels it “Likely Benign.” No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that E75G is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.443881 | Uncertain | 0.303 | 0.822 | 0.500 | -2.991 | Likely Benign | 0.175 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -1.74 | Neutral | 0.345 | Benign | 0.023 | Benign | 4.01 | Benign | 0.00 | Affected | 0.3078 | 0.5738 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.224A>T | E75V 2D AIThe SynGAP1 missense variant E75V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.443881 | Uncertain | 0.303 | 0.822 | 0.500 | -3.426 | Likely Benign | 0.305 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -1.72 | Neutral | 0.789 | Possibly Damaging | 0.095 | Benign | 4.02 | Benign | 0.00 | Affected | 0.0878 | 0.7398 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2251C>A | P751T 2D AIThe SynGAP1 missense variant P751T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.667683 | Binding | 0.386 | 0.866 | 0.625 | -5.111 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -1.65 | Neutral | 0.679 | Possibly Damaging | 0.348 | Benign | 2.71 | Benign | 1.00 | Tolerated | 0.1613 | 0.5704 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2251C>G | P751A 2D AIThe SynGAP1 missense variant P751A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.667683 | Binding | 0.386 | 0.866 | 0.625 | -4.612 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -1.42 | Neutral | 0.028 | Benign | 0.009 | Benign | 2.73 | Benign | 0.26 | Tolerated | 0.3592 | 0.5487 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2251C>T | P751S 2D AIThe SynGAP1 missense variant P751S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions indicate that P751S is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.667683 | Binding | 0.386 | 0.866 | 0.625 | -4.157 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.80 | Neutral | 0.514 | Possibly Damaging | 0.216 | Benign | 2.70 | Benign | 0.33 | Tolerated | 0.3446 | 0.5837 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2252C>A | P751Q 2D AIThe SynGAP1 missense variant P751Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.667683 | Binding | 0.386 | 0.866 | 0.625 | -5.273 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -2.07 | Neutral | 0.837 | Possibly Damaging | 0.531 | Possibly Damaging | 2.68 | Benign | 0.05 | Affected | 0.1488 | 0.5073 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2252C>G | P751R 2D AIThe SynGAP1 missense variant P751R is catalogued in gnomAD (ID 6‑33441717‑C‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are PROVEAN and polyPhen‑2 HumDiv. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.667683 | Binding | 0.386 | 0.866 | 0.625 | 6-33441717-C-G | 1 | 6.20e-7 | -5.646 | Likely Benign | 0.296 | Likely Benign | Likely Benign | 0.157 | Likely Benign | -2.61 | Deleterious | 0.719 | Possibly Damaging | 0.295 | Benign | 2.68 | Benign | 0.06 | Tolerated | 3.99 | 5 | 0.1390 | 0.3644 | -2 | 0 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||
| c.2252C>T | P751L 2D AIThe SynGAP1 missense variant P751L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.667683 | Binding | 0.386 | 0.866 | 0.625 | -3.558 | Likely Benign | 0.143 | Likely Benign | Likely Benign | 0.207 | Likely Benign | -1.62 | Neutral | 0.316 | Benign | 0.062 | Benign | 2.94 | Benign | 0.24 | Tolerated | 0.2318 | 0.6451 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2254T>A | S752T 2D AIThe SynGAP1 missense variant S752T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.604312 | Disordered | 0.690594 | Binding | 0.365 | 0.877 | 0.625 | -4.040 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -1.33 | Neutral | 0.248 | Benign | 0.137 | Benign | 1.55 | Pathogenic | 0.07 | Tolerated | 0.1633 | 0.6342 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2254T>C | S752P 2D  AIThe SynGAP1 missense variant S752P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for S752P, and this conclusion does not contradict the ClinVar status, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.604312 | Disordered | 0.690594 | Binding | 0.365 | 0.877 | 0.625 | -3.491 | Likely Benign | 0.158 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -1.09 | Neutral | 0.998 | Probably Damaging | 0.912 | Probably Damaging | 1.51 | Pathogenic | 0.02 | Affected | 0.2288 | 0.5882 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2254T>G | S752A 2D AIThe SynGAP1 missense variant S752A has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.604312 | Disordered | 0.690594 | Binding | 0.365 | 0.877 | 0.625 | -3.258 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -1.42 | Neutral | 0.910 | Possibly Damaging | 0.524 | Possibly Damaging | 1.59 | Pathogenic | 0.04 | Affected | 0.5092 | 0.5634 | Strenghten | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.2255C>G | S752W 2D  AIThe SynGAP1 missense variant S752W is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas seven tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic, and the Foldetta stability analysis is unavailable. Overall, the majority of predictions lean toward pathogenicity, and this conclusion is not contradicted by ClinVar status (which is absent). Thus, the variant is most likely pathogenic based on the collective evidence, despite the single benign prediction from AlphaMissense‑Optimized. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.690594 | Binding | 0.365 | 0.877 | 0.625 | -6.771 | Likely Benign | 0.565 | Likely Pathogenic | Likely Benign | 0.341 | Likely Benign | -3.54 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 1.49 | Pathogenic | 0.00 | Affected | 0.0837 | 0.6241 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2255C>T | S752L 2D  AIThe SynGAP1 missense variant S752L is listed in ClinVar with an “Uncertain” status (ClinVar ID 2143952.0) and is present in gnomAD (ID 6‑33441720‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.604312 | Disordered | 0.690594 | Binding | 0.365 | 0.877 | 0.625 | Uncertain | 2 | 6-33441720-C-T | 6 | 3.72e-6 | -3.386 | Likely Benign | 0.182 | Likely Benign | Likely Benign | 0.195 | Likely Benign | -2.09 | Neutral | 0.993 | Probably Damaging | 0.641 | Possibly Damaging | 1.51 | Pathogenic | 0.01 | Affected | 3.99 | 5 | 0.1308 | 0.5909 | -3 | -2 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||
| c.2257G>A | A753T 2D AIThe SynGAP1 missense variant A753T is catalogued in gnomAD (ID 6‑33441722‑G‑A) but has no ClinVar entry. Across the spectrum of in‑silico predictors, every tool listed—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classifies the change as benign. No pathogenic predictions are reported. Grouping by agreement, all available tools fall into the benign category, with no tools indicating pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status remains unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.722781 | Binding | 0.381 | 0.873 | 0.625 | 6-33441722-G-A | 2 | 1.24e-6 | -4.298 | Likely Benign | 0.069 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -0.42 | Neutral | 0.022 | Benign | 0.018 | Benign | 2.70 | Benign | 0.28 | Tolerated | 3.99 | 5 | 0.1672 | 0.6997 | 0 | 1 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||||
| c.2257G>C | A753P 2D AIThe SynGAP1 missense variant A753P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation—there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.722781 | Binding | 0.381 | 0.873 | 0.625 | -2.486 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.05 | Neutral | 0.966 | Probably Damaging | 0.575 | Possibly Damaging | 2.59 | Benign | 0.30 | Tolerated | 0.2143 | 0.5442 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2257G>T | A753S 2D AIThe SynGAP1 missense variant A753S is reported as “Likely Benign” in ClinVar and is not present in gnomAD. Prediction tools that assess functional impact all converge on a benign outcome: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool in the dataset predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. Foldetta results are unavailable. Overall, the variant is most likely benign, and this assessment does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.722781 | Binding | 0.381 | 0.873 | 0.625 | -3.656 | Likely Benign | 0.069 | Likely Benign | Likely Benign | 0.105 | Likely Benign | 0.25 | Neutral | 0.062 | Benign | 0.015 | Benign | 3.03 | Benign | 0.59 | Tolerated | 0.2905 | 0.5899 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2258C>A | A753D 2D AISynGAP1 missense variant A753D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all score benign, while only polyPhen‑2 HumDiv predicts pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy tools corroborate this view: AlphaMissense‑Optimized reports a benign outcome, SGM‑Consensus likewise indicates Likely Benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign classification, and this conclusion does not contradict the ClinVar status. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.722781 | Binding | 0.381 | 0.873 | 0.625 | -5.836 | Likely Benign | 0.408 | Ambiguous | Likely Benign | 0.113 | Likely Benign | -1.66 | Neutral | 0.669 | Possibly Damaging | 0.265 | Benign | 2.60 | Benign | 0.60 | Tolerated | 0.2151 | 0.2200 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2258C>G | A753G 2D AIThe SynGAP1 missense variant A753G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta results are not available, so they do not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.722781 | Binding | 0.381 | 0.873 | 0.625 | -4.257 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -1.10 | Neutral | 0.625 | Possibly Damaging | 0.192 | Benign | 2.62 | Benign | 0.65 | Tolerated | 0.2447 | 0.5089 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2258C>T | A753V 2D AIThe SynGAP1 missense variant A753V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of computational evidence indicates that A753V is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.722781 | Binding | 0.381 | 0.873 | 0.625 | -3.759 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -1.55 | Neutral | 0.669 | Possibly Damaging | 0.192 | Benign | 2.71 | Benign | 0.18 | Tolerated | 0.1344 | 0.5953 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.225G>C | E75D 2D AIThe SynGAP1 missense variant E75D is reported in gnomAD (6‑33425833‑G‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is “Likely Benign.” No Foldetta stability prediction is available. Overall, the preponderance of evidence indicates that E75D is most likely benign, and this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.443881 | Uncertain | 0.303 | 0.822 | 0.500 | 6-33425833-G-C | 1 | 6.20e-7 | -3.710 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.27 | Neutral | 0.001 | Benign | 0.000 | Benign | 4.25 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2026 | 0.3833 | 2 | 3 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.225G>T | E75D 2D AIThe SynGAP1 missense variant E75D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.595080 | Disordered | 0.443881 | Uncertain | 0.303 | 0.822 | 0.500 | -3.710 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.27 | Neutral | 0.001 | Benign | 0.000 | Benign | 4.25 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2026 | 0.3833 | 2 | 3 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.2260G>A | E754K 2D AIThe SynGAP1 missense variant E754K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Consensus among in‑silico predictors shows a predominance of benign calls: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized all predict a benign effect, whereas polyPhen‑2 HumDiv and AlphaMissense‑Default predict pathogenicity; ESM1b remains uncertain. High‑accuracy assessment further supports a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, and Foldetta data are unavailable. Consequently, the variant is most likely benign according to the aggregate predictions, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.505461 | Disordered | 0.750531 | Binding | 0.357 | 0.872 | 0.500 | -7.620 | In-Between | 0.610 | Likely Pathogenic | Likely Benign | 0.138 | Likely Benign | -1.33 | Neutral | 0.801 | Possibly Damaging | 0.412 | Benign | 2.50 | Benign | 0.26 | Tolerated | 0.2159 | 0.7136 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.2260G>C | E754Q 2D AIThe SynGAP1 missense variant E754Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a neutral impact. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign classification, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.750531 | Binding | 0.357 | 0.872 | 0.500 | -5.324 | Likely Benign | 0.314 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -0.76 | Neutral | 0.891 | Possibly Damaging | 0.596 | Possibly Damaging | 2.48 | Pathogenic | 0.27 | Tolerated | 0.1112 | 0.6714 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2261A>C | E754A 2D AIThe SynGAP1 missense variant E754A is not reported in ClinVar (ClinVar status: not reported) and is absent from gnomAD (gnomAD: not present). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction (2 benign vs. 1 pathogenic votes). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.505461 | Disordered | 0.750531 | Binding | 0.357 | 0.872 | 0.500 | -4.688 | Likely Benign | 0.381 | Ambiguous | Likely Benign | 0.049 | Likely Benign | -1.56 | Neutral | 0.801 | Possibly Damaging | 0.412 | Benign | 2.49 | Pathogenic | 0.31 | Tolerated | 0.3549 | 0.6283 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2261A>G | E754G 2D AIThe SynGAP1 missense variant E754G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.750531 | Binding | 0.357 | 0.872 | 0.500 | -5.029 | Likely Benign | 0.313 | Likely Benign | Likely Benign | 0.080 | Likely Benign | -0.52 | Neutral | 0.801 | Possibly Damaging | 0.339 | Benign | 2.94 | Benign | 0.13 | Tolerated | 0.2631 | 0.5820 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.2261A>T | E754V 2D AIThe SynGAP1 missense variant E754V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence (five benign versus four pathogenic predictions) and the high‑accuracy benign call suggest that the variant is most likely benign. This conclusion does not contradict ClinVar status, as the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.505461 | Disordered | 0.750531 | Binding | 0.357 | 0.872 | 0.500 | -6.147 | Likely Benign | 0.601 | Likely Pathogenic | Likely Benign | 0.157 | Likely Benign | -1.86 | Neutral | 0.966 | Probably Damaging | 0.773 | Possibly Damaging | 2.45 | Pathogenic | 0.28 | Tolerated | 0.0730 | 0.7417 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2262G>C | E754D 2D AIThe SynGAP1 missense variant E754D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.750531 | Binding | 0.357 | 0.872 | 0.500 | -2.360 | Likely Benign | 0.065 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -0.27 | Neutral | 0.002 | Benign | 0.007 | Benign | 2.53 | Benign | 0.45 | Tolerated | 0.1725 | 0.3983 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2262G>T | E754D 2D AIThe SynGAP1 missense variant E754D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.750531 | Binding | 0.357 | 0.872 | 0.500 | -2.360 | Likely Benign | 0.065 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -0.27 | Neutral | 0.002 | Benign | 0.007 | Benign | 2.53 | Benign | 0.45 | Tolerated | 0.1725 | 0.3983 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2263A>C | M755L 2D  AIThe SynGAP1 missense variant M755L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -1.298 | Likely Benign | 0.084 | Likely Benign | Likely Benign | 0.067 | Likely Benign | 0.03 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.39 | Benign | 0.22 | Tolerated | 0.1167 | 0.3311 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2263A>G | M755V 2D  AIThe SynGAP1 missense variant M755V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -3.804 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.80 | Neutral | 0.002 | Benign | 0.003 | Benign | 2.73 | Benign | 0.27 | Tolerated | 0.2809 | 0.2873 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||||||
| c.2263A>T | M755L 2D AIThe SynGAP1 missense variant M755L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a neutral effect. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign effect, and the SGM‑Consensus (majority vote) yields a likely benign classification. Foldetta results are not available, so they do not influence the assessment. Overall, the computational evidence strongly supports a benign interpretation of M755L, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -1.298 | Likely Benign | 0.084 | Likely Benign | Likely Benign | 0.067 | Likely Benign | 0.03 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.39 | Benign | 0.22 | Tolerated | 0.1167 | 0.3311 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2264T>A | M755K 2D  AIThe SynGAP1 missense variant M755K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus indicates Likely Benign, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which contains no conflicting report. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -5.642 | Likely Benign | 0.531 | Ambiguous | Likely Benign | 0.101 | Likely Benign | -1.88 | Neutral | 0.468 | Possibly Damaging | 0.206 | Benign | 2.63 | Benign | 0.28 | Tolerated | 0.1183 | 0.0688 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.2264T>C | M755T 2D  AIThe SynGAP1 missense variant M755T is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the change as benign, and AlphaMissense‑Optimized also predicts benign. No tool predicts pathogenicity; AlphaMissense‑Default is uncertain. The SGM‑Consensus, which aggregates majority votes from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta data are not available. Overall, the computational evidence overwhelmingly suggests the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -4.116 | Likely Benign | 0.358 | Ambiguous | Likely Benign | 0.040 | Likely Benign | -1.23 | Neutral | 0.159 | Benign | 0.053 | Benign | 2.65 | Benign | 0.19 | Tolerated | 0.1944 | 0.1767 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2264T>G | M755R 2D  AIThe SynGAP1 missense variant M755R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools indicates that the M755R variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -4.247 | Likely Benign | 0.453 | Ambiguous | Likely Benign | 0.104 | Likely Benign | -2.06 | Neutral | 0.468 | Possibly Damaging | 0.206 | Benign | 2.63 | Benign | 0.18 | Tolerated | 0.1407 | 0.0837 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||||||||||||
| c.2265G>A | M755I 2D  AIThe SynGAP1 missense variant M755I is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -3.856 | Likely Benign | 0.240 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -0.55 | Neutral | 0.039 | Benign | 0.014 | Benign | 2.81 | Benign | 0.14 | Tolerated | 0.1059 | 0.2403 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2265G>C | M755I 2D AIThe SynGAP1 missense variant M755I is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -3.856 | Likely Benign | 0.240 | Likely Benign | Likely Benign | 0.031 | Likely Benign | -0.55 | Neutral | 0.039 | Benign | 0.014 | Benign | 2.81 | Benign | 0.14 | Tolerated | 0.1059 | 0.2403 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2265G>T | M755I 2D AIThe SynGAP1 missense variant M755I is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -3.856 | Likely Benign | 0.240 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -0.55 | Neutral | 0.039 | Benign | 0.014 | Benign | 2.81 | Benign | 0.14 | Tolerated | 0.1059 | 0.2403 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2266C>A | Q756K 2D AIThe SynGAP1 missense variant Q756K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for Q756K, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.806299 | Binding | 0.340 | 0.866 | 0.250 | -6.059 | Likely Benign | 0.340 | Likely Benign | Likely Benign | 0.199 | Likely Benign | -1.47 | Neutral | 0.985 | Probably Damaging | 0.981 | Probably Damaging | 1.60 | Pathogenic | 0.21 | Tolerated | 0.1816 | 0.4797 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.2266C>G | Q756E 2D AIThe SynGAP1 missense variant Q756E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for Q756E, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.806299 | Binding | 0.340 | 0.866 | 0.250 | -4.149 | Likely Benign | 0.172 | Likely Benign | Likely Benign | 0.174 | Likely Benign | -1.19 | Neutral | 0.985 | Probably Damaging | 0.981 | Probably Damaging | 1.58 | Pathogenic | 0.23 | Tolerated | 0.1465 | 0.2853 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2267A>C | Q756P 2D AIThe SynGAP1 missense variant Q756P has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.806299 | Binding | 0.340 | 0.866 | 0.250 | -4.226 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 0.315 | Likely Benign | -0.93 | Neutral | 0.998 | Probably Damaging | 0.995 | Probably Damaging | 1.54 | Pathogenic | 0.23 | Tolerated | 0.2590 | 0.5186 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2267A>G | Q756R 2D AIThe SynGAP1 missense variant Q756R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for Q756R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.806299 | Binding | 0.340 | 0.866 | 0.250 | -5.044 | Likely Benign | 0.307 | Likely Benign | Likely Benign | 0.245 | Likely Benign | -1.77 | Neutral | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 1.58 | Pathogenic | 0.14 | Tolerated | 0.1500 | 0.2440 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.2267A>T | Q756L 2D AIThe SynGAP1 missense variant Q756L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is labeled “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for Q756L, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.806299 | Binding | 0.340 | 0.866 | 0.250 | -3.697 | Likely Benign | 0.339 | Likely Benign | Likely Benign | 0.274 | Likely Benign | -1.95 | Neutral | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 1.56 | Pathogenic | 0.08 | Tolerated | 0.0797 | 0.5700 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2268G>C | Q756H 2D AIThe SynGAP1 missense variant Q756H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign based on current computational evidence, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.806299 | Binding | 0.340 | 0.866 | 0.250 | -4.625 | Likely Benign | 0.276 | Likely Benign | Likely Benign | 0.197 | Likely Benign | -2.17 | Neutral | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 1.54 | Pathogenic | 0.05 | Affected | 0.1545 | 0.4404 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2268G>T | Q756H 2D AIThe SynGAP1 missense variant Q756H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign based on current computational evidence, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.806299 | Binding | 0.340 | 0.866 | 0.250 | -4.625 | Likely Benign | 0.276 | Likely Benign | Likely Benign | 0.197 | Likely Benign | -2.17 | Neutral | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 1.54 | Pathogenic | 0.05 | Affected | 0.1545 | 0.4404 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2269G>A | G757S 2D AIThe SynGAP1 missense variant G757S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess pathogenicity uniformly predict a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. No tool in the dataset predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.830995 | Binding | 0.310 | 0.869 | 0.375 | -1.492 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.058 | Likely Benign | 0.47 | Neutral | 0.007 | Benign | 0.008 | Benign | 2.73 | Benign | 0.29 | Tolerated | 0.2595 | 0.3877 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2269G>C | G757R 2D AIThe SynGAP1 missense variant G757R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic outcome. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports likely benign; Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.830995 | Binding | 0.310 | 0.869 | 0.375 | -2.254 | Likely Benign | 0.534 | Ambiguous | Likely Benign | 0.158 | Likely Benign | -0.04 | Neutral | 0.801 | Possibly Damaging | 0.494 | Possibly Damaging | 2.79 | Benign | 0.05 | Affected | 0.0903 | 0.3481 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2269G>T | G757C 2D AIThe SynGAP1 missense variant G757C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.830995 | Binding | 0.310 | 0.869 | 0.375 | -6.652 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.179 | Likely Benign | -1.74 | Neutral | 0.997 | Probably Damaging | 0.870 | Possibly Damaging | 2.65 | Benign | 0.04 | Affected | 0.1302 | 0.3630 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.226T>A | S76T 2D AIThe SynGAP1 missense variant S76T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.444487 | Uncertain | 0.279 | 0.826 | 0.500 | -4.000 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -0.73 | Neutral | 0.805 | Possibly Damaging | 0.483 | Possibly Damaging | 3.78 | Benign | 0.00 | Affected | 0.1117 | 0.4774 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.226T>C | S76P 2D AIThe SynGAP1 missense variant S76P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect for S76P, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.444487 | Uncertain | 0.279 | 0.826 | 0.500 | -3.833 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -1.64 | Neutral | 0.909 | Possibly Damaging | 0.665 | Possibly Damaging | 3.77 | Benign | 0.00 | Affected | 0.1790 | 0.4138 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.226T>G | S76A 2D AIThe SynGAP1 missense variant S76A is reported in gnomAD (ID 6‑33425834‑T‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.444487 | Uncertain | 0.279 | 0.826 | 0.500 | 6-33425834-T-G | 1 | 6.20e-7 | -3.230 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -1.10 | Neutral | 0.643 | Possibly Damaging | 0.277 | Benign | 3.86 | Benign | 0.00 | Affected | 4.32 | 1 | 0.4543 | 0.3619 | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||
| c.2270G>A | G757D 2D AIThe SynGAP1 missense variant G757D is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Only polyPhen‑2 HumDiv flags it as pathogenic, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Taken together, the preponderance of evidence supports a benign interpretation, and this assessment does not contradict any ClinVar annotation (none is present). Therefore, the variant is most likely benign, with no conflict with ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.830995 | Binding | 0.310 | 0.869 | 0.375 | -4.613 | Likely Benign | 0.387 | Ambiguous | Likely Benign | 0.150 | Likely Benign | -0.90 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 2.71 | Benign | 0.11 | Tolerated | 0.1826 | 0.1611 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2270G>C | G757A 2D AIThe SynGAP1 missense change G757A is catalogued in ClinVar (ID 3635272.0) with an uncertain significance designation and is not reported in gnomAD. Functional prediction algorithms uniformly classify the variant as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool in the dataset predicts pathogenicity. High‑accuracy consensus methods corroborate this view: the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect, and AlphaMissense‑Optimized also predicts benign. The Foldetta stability assessment is unavailable for this variant. Taken together, the evidence overwhelmingly supports a benign interpretation, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.830995 | Binding | 0.310 | 0.869 | 0.375 | Uncertain | 1 | -2.626 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.45 | Neutral | 0.267 | Benign | 0.127 | Benign | 2.73 | Benign | 0.35 | Tolerated | 0.3690 | 0.3842 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.2270G>T | G757V 2D AIThe SynGAP1 missense variant G757V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.830995 | Binding | 0.310 | 0.869 | 0.375 | -4.840 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -1.47 | Neutral | 0.801 | Possibly Damaging | 0.494 | Possibly Damaging | 2.68 | Benign | 0.07 | Tolerated | 0.1089 | 0.3512 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2272T>A | Y758N 2D 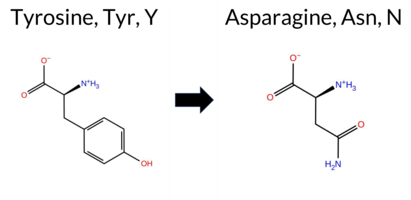 AIThe SynGAP1 missense variant Y758N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.856063 | Binding | 0.289 | 0.871 | 0.375 | -3.316 | Likely Benign | 0.234 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -1.32 | Neutral | 0.837 | Possibly Damaging | 0.631 | Possibly Damaging | 2.72 | Benign | 0.02 | Affected | 0.2467 | 0.0331 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.2272T>C | Y758H 2D 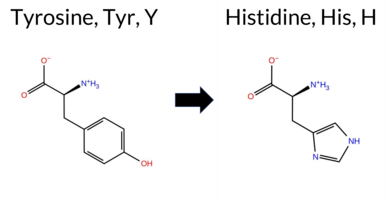 AIThe SynGAP1 missense variant Y758H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.856063 | Binding | 0.289 | 0.871 | 0.375 | -3.194 | Likely Benign | 0.299 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.92 | Neutral | 0.064 | Benign | 0.031 | Benign | 2.71 | Benign | 0.02 | Affected | 0.2519 | 0.0331 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||||||||||||
| c.2272T>G | Y758D 2D 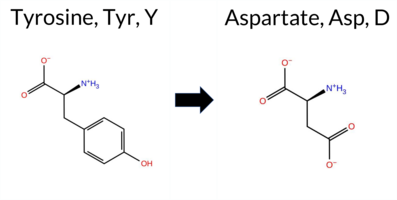 AIThe SynGAP1 missense variant Y758D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized independently predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.856063 | Binding | 0.289 | 0.871 | 0.375 | -3.688 | Likely Benign | 0.481 | Ambiguous | Likely Benign | 0.160 | Likely Benign | -1.89 | Neutral | 0.973 | Probably Damaging | 0.796 | Possibly Damaging | 2.71 | Benign | 0.02 | Affected | 0.4647 | 0.0331 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2273A>C | Y758S 2D 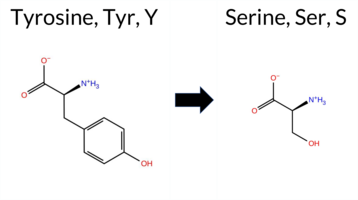 AIThe SynGAP1 missense variant Y758S is reported in gnomAD (variant ID 6‑33441738‑A‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment does not contradict any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.856063 | Binding | 0.289 | 0.871 | 0.375 | 6-33441738-A-C | 1 | 6.20e-7 | -1.912 | Likely Benign | 0.215 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -0.54 | Neutral | 0.912 | Possibly Damaging | 0.629 | Possibly Damaging | 2.75 | Benign | 0.06 | Tolerated | 3.99 | 5 | 0.5377 | 0.1663 | Weaken | -2 | -3 | 0.5 | -76.10 | |||||||||||||||||||||||||||||||||
| c.2273A>G | Y758C 2D  AIThe SynGAP1 missense variant Y758C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.856063 | Binding | 0.289 | 0.871 | 0.375 | -5.256 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -1.91 | Neutral | 0.998 | Probably Damaging | 0.921 | Probably Damaging | 2.71 | Benign | 0.03 | Affected | 0.3375 | 0.1922 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||||||||||||
| c.2273A>T | Y758F 2D  AIThe SynGAP1 missense variant Y758F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.856063 | Binding | 0.289 | 0.871 | 0.375 | -1.431 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.112 | Likely Benign | -0.79 | Neutral | 0.679 | Possibly Damaging | 0.371 | Benign | 2.75 | Benign | 1.00 | Tolerated | 0.2441 | 0.2835 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2275A>C | M759L 2D AIThe SynGAP1 missense variant M759L is listed in ClinVar with an uncertain significance (ClinVar ID 942432.0) and is present in gnomAD (gnomAD ID 6‑33441740‑A‑C). All evaluated in‑silico predictors agree on a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign classifications. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly supports a benign impact, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | Uncertain | 1 | 6-33441740-A-C | 2 | 1.24e-6 | -2.431 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.53 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.84 | Benign | 1.00 | Tolerated | 3.99 | 5 | 0.1308 | 0.4005 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||||
| c.2275A>G | M759V 2D AIThe SynGAP1 missense variant M759V is catalogued in gnomAD (ID 6‑33441740‑A‑G) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool reports a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the set predicts pathogenicity, so the pathogenic group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available for this variant. Taken together, the computational evidence overwhelmingly supports a benign effect, and this conclusion does not conflict with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | 6-33441740-A-G | 20 | 1.24e-5 | -3.985 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -1.00 | Neutral | 0.267 | Benign | 0.127 | Benign | 2.67 | Benign | 0.23 | Tolerated | 3.99 | 5 | 0.2926 | 0.3041 | 1 | 2 | 2.3 | -32.06 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||||
| c.2275A>T | M759L 2D AIThe SynGAP1 missense variant M759L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | -2.431 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.53 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.84 | Benign | 1.00 | Tolerated | 3.99 | 5 | 0.1308 | 0.4005 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||
| c.2276T>A | M759K 2D AIThe SynGAP1 missense variant M759K (ClinVar ID 4178662) is listed as ClinVar status Uncertain and is not present in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, which does not contradict the ClinVar Uncertain designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | Uncertain | 1 | -5.670 | Likely Benign | 0.616 | Likely Pathogenic | Likely Benign | 0.288 | Likely Benign | -1.86 | Neutral | 0.891 | Possibly Damaging | 0.492 | Possibly Damaging | 2.55 | Benign | 0.06 | Tolerated | 0.1338 | 0.0688 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||
| c.2276T>C | M759T 2D AIThe SynGAP1 missense variant M759T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence—including the SGM consensus and AlphaMissense‑Optimized—supports a benign classification, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | -4.202 | Likely Benign | 0.380 | Ambiguous | Likely Benign | 0.197 | Likely Benign | -1.90 | Neutral | 0.891 | Possibly Damaging | 0.315 | Benign | 2.58 | Benign | 0.08 | Tolerated | 0.2063 | 0.1534 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2276T>G | M759R 2D AIThe SynGAP1 missense variant M759R is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign, while the majority of high‑accuracy predictors—AlphaMissense‑Optimized and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—also support a benign classification. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact, but these are the only tools in disagreement. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for M759R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | -4.507 | Likely Benign | 0.531 | Ambiguous | Likely Benign | 0.244 | Likely Benign | -1.82 | Neutral | 0.891 | Possibly Damaging | 0.575 | Possibly Damaging | 2.55 | Benign | 0.06 | Tolerated | 0.1509 | 0.0637 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||||||||||||
| c.2277G>A | M759I 2D AIThe SynGAP1 missense variant M759I is listed in ClinVar (ID 3686687.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33441742‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | Uncertain | 1 | 6-33441742-G-A | 1 | 6.20e-7 | -4.058 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -0.88 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 2.83 | Benign | 0.34 | Tolerated | 3.99 | 5 | 0.1235 | 0.3129 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||
| c.2277G>C | M759I 2D AIThe SynGAP1 missense variant M759I is catalogued in gnomAD (6‑33441742‑G‑C) and has no ClinVar entry. Consensus from multiple in‑silico predictors points to a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all score the variant as benign, while only polyPhen‑2 HumDiv flags it as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy tools reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are not available, so they do not influence the assessment. Overall, the computational evidence overwhelmingly supports a benign classification, and this is consistent with the absence of a pathogenic ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | 6-33441742-G-C | -4.058 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -0.88 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 2.83 | Benign | 0.34 | Tolerated | 3.99 | 5 | 0.1235 | 0.3129 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||||
| c.2277G>T | M759I 2D AIThe SynGAP1 missense variant M759I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the preponderance of evidence from multiple prediction algorithms and high‑accuracy tools points to a benign impact for M759I, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | -4.058 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -0.88 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 2.83 | Benign | 0.34 | Tolerated | 3.99 | 5 | 0.1235 | 0.3129 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||
| c.2278A>C | M760L 2D AIThe SynGAP1 missense variant M760L is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts a pathogenic outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -2.260 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -0.68 | Neutral | 0.065 | Benign | 0.033 | Benign | 2.69 | Benign | 0.15 | Tolerated | 0.1802 | 0.4805 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2278A>G | M760V 2D AIThe SynGAP1 missense variant M760V is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the computational evidence strongly supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -2.803 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -1.20 | Neutral | 0.001 | Benign | 0.008 | Benign | 2.69 | Benign | 0.22 | Tolerated | 0.3974 | 0.4381 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||||||
| c.2278A>T | M760L 2D AIThe SynGAP1 missense variant M760L is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts a pathogenic outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -2.260 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -0.68 | Neutral | 0.065 | Benign | 0.033 | Benign | 2.69 | Benign | 0.15 | Tolerated | 0.1802 | 0.4805 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2279T>A | M760K 2D AIThe SynGAP1 missense variant M760K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -4.069 | Likely Benign | 0.654 | Likely Pathogenic | Likely Benign | 0.108 | Likely Benign | -1.18 | Neutral | 0.784 | Possibly Damaging | 0.492 | Possibly Damaging | 2.75 | Benign | 0.12 | Tolerated | 0.1751 | 0.1071 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.2279T>C | M760T 2D AIThe SynGAP1 missense variant M760T is reported in gnomAD (ID 6‑33441744‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | 6-33441744-T-C | 1 | 6.20e-7 | -2.365 | Likely Benign | 0.519 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -1.56 | Neutral | 0.425 | Benign | 0.252 | Benign | 2.71 | Benign | 0.41 | Tolerated | 3.99 | 5 | 0.2515 | 0.2875 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.2279T>G | M760R 2D AIThe SynGAP1 missense variant M760R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates Likely Benign. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -2.794 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 0.125 | Likely Benign | -1.61 | Neutral | 0.975 | Probably Damaging | 0.690 | Possibly Damaging | 2.71 | Benign | 0.09 | Tolerated | 0.1804 | 0.1318 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||||||||||||
| c.227C>A | S76Y 2D AIThe SynGAP1 missense variant S76Y is listed in ClinVar (ID 4801334) with an uncertain significance status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence points to a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.444487 | Uncertain | 0.279 | 0.826 | 0.500 | Uncertain | 1 | -4.243 | Likely Benign | 0.209 | Likely Benign | Likely Benign | 0.119 | Likely Benign | -2.35 | Neutral | 0.972 | Probably Damaging | 0.831 | Possibly Damaging | 3.71 | Benign | 0.00 | Affected | 0.0540 | 0.4753 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||
| c.227C>G | S76C 2D AIThe SynGAP1 missense variant S76C is listed in ClinVar with an “Uncertain” status (ClinVar ID 1951273.0) and is present in the gnomAD database (gnomAD ID 6‑33425835‑C‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as benign; Foldetta results are not available for this variant. Overall, the majority of computational evidence points to a benign impact, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.444487 | Uncertain | 0.279 | 0.826 | 0.500 | Uncertain | 1 | 6-33425835-C-G | 2 | 1.24e-6 | -5.408 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -1.78 | Neutral | 0.992 | Probably Damaging | 0.869 | Possibly Damaging | 3.71 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0807 | 0.4787 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||
| c.227C>T | S76F 2D AIThe SynGAP1 missense variant S76F is reported in ClinVar as “Not submitted” and is present in gnomAD (variant ID 6-33425835-C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.444487 | Uncertain | 0.279 | 0.826 | 0.500 | 6-33425835-C-T | 4 | 2.48e-6 | -4.557 | Likely Benign | 0.197 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -2.40 | Neutral | 0.972 | Probably Damaging | 0.831 | Possibly Damaging | 3.71 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0501 | 0.4887 | -2 | -3 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||
| c.2280G>A | M760I 2D AIThe SynGAP1 missense variant M760I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective evidence strongly suggests that M760I is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -3.696 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.03 | Neutral | 0.029 | Benign | 0.033 | Benign | 2.67 | Benign | 0.09 | Tolerated | 0.1595 | 0.4054 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2280G>C | M760I 2D AIThe SynGAP1 missense variant M760I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective evidence strongly suggests that M760I is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -3.696 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.03 | Neutral | 0.029 | Benign | 0.033 | Benign | 2.67 | Benign | 0.09 | Tolerated | 0.1595 | 0.4054 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2280G>T | M760I 2D AIThe SynGAP1 missense variant M760I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective evidence strongly suggests that M760I is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | -3.696 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.03 | Neutral | 0.029 | Benign | 0.033 | Benign | 2.67 | Benign | 0.09 | Tolerated | 0.1595 | 0.4054 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2281C>G | R761G 2D AIThe SynGAP1 missense variant R761G is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors indicates a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all score it as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also classifies it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The AlphaMissense‑Default tool remains uncertain, and no Foldetta stability assessment is available. High‑accuracy methods reinforce the benign prediction: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign, with no contradictory Foldetta data. Overall, the majority of evidence supports a benign classification, and this is consistent with the lack of ClinVar annotation; there is no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.538167 | Disordered | 0.900613 | Binding | 0.353 | 0.865 | 0.250 | -4.453 | Likely Benign | 0.427 | Ambiguous | Likely Benign | 0.220 | Likely Benign | -2.07 | Neutral | 0.992 | Probably Damaging | 0.900 | Possibly Damaging | 2.70 | Benign | 0.83 | Tolerated | 0.3333 | 0.3217 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2281C>T | R761W 2D AIThe SynGAP1 missense variant R761W is listed in gnomAD (ID 6‑33441746‑C‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains Likely Pathogenic; Foldetta results are unavailable. Overall, the majority of tools (six pathogenic vs. four benign) suggest a pathogenic effect, and this conclusion does not contradict ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.538167 | Disordered | 0.900613 | Binding | 0.353 | 0.865 | 0.250 | 6-33441746-C-T | 1 | 6.20e-7 | -9.248 | Likely Pathogenic | 0.665 | Likely Pathogenic | Likely Benign | 0.193 | Likely Benign | -3.52 | Deleterious | 1.000 | Probably Damaging | 0.987 | Probably Damaging | 2.66 | Benign | 0.06 | Tolerated | 3.99 | 5 | 0.1018 | 0.3668 | -3 | 2 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||
| c.2282G>A | R761Q 2D AIThe SynGAP1 missense variant R761Q is listed in ClinVar (ID 2882770.0) with an “Uncertain” status and is present in gnomAD (6‑33441747‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.538167 | Disordered | 0.900613 | Binding | 0.353 | 0.865 | 0.250 | Uncertain | 1 | 6-33441747-G-A | 11 | 6.81e-6 | -4.187 | Likely Benign | 0.202 | Likely Benign | Likely Benign | 0.191 | Likely Benign | -0.63 | Neutral | 0.996 | Probably Damaging | 0.878 | Possibly Damaging | 2.75 | Benign | 0.40 | Tolerated | 3.99 | 5 | 0.2640 | 0.2329 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||||||
| c.2282G>C | R761P 2D AIThe SynGAP1 missense variant R761P is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33441747‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions point to a benign impact, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.538167 | Disordered | 0.900613 | Binding | 0.353 | 0.865 | 0.250 | Uncertain | 3 | 6-33441747-G-C | 1 | 6.20e-7 | -5.091 | Likely Benign | 0.640 | Likely Pathogenic | Likely Benign | 0.201 | Likely Benign | -1.89 | Neutral | 0.999 | Probably Damaging | 0.968 | Probably Damaging | 2.69 | Benign | 0.38 | Tolerated | 3.99 | 5 | 0.1998 | 0.4449 | 0 | -2 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||
| c.2282G>T | R761L 2D AIThe SynGAP1 missense variant R761L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact. This consensus does not contradict any ClinVar annotation, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.538167 | Disordered | 0.900613 | Binding | 0.353 | 0.865 | 0.250 | -5.653 | Likely Benign | 0.718 | Likely Pathogenic | Likely Benign | 0.171 | Likely Benign | -2.51 | Deleterious | 0.992 | Probably Damaging | 0.900 | Possibly Damaging | 2.70 | Benign | 0.24 | Tolerated | 0.1786 | 0.4326 | -3 | -2 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2284G>A | D762N 2D AIThe SynGAP1 D762N missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign impact for D762N, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -3.323 | Likely Benign | 0.640 | Likely Pathogenic | Likely Benign | 0.110 | Likely Benign | -1.51 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.13 | Pathogenic | 0.11 | Tolerated | 0.1698 | 0.8797 | 2 | 1 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2284G>C | D762H 2D AIThe SynGAP1 D762H missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence points to a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -4.643 | Likely Benign | 0.909 | Likely Pathogenic | Ambiguous | 0.212 | Likely Benign | -2.73 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 2.08 | Pathogenic | 0.02 | Affected | 0.2007 | 0.9102 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.2284G>T | D762Y 2D AIThe SynGAP1 D762Y variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas a larger group predicts a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus methods give the following results: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic”; Foldetta (a protein‑folding stability approach combining FoldX‑MD and Rosetta) has no available output for this variant. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -6.959 | Likely Benign | 0.905 | Likely Pathogenic | Ambiguous | 0.219 | Likely Benign | -3.24 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 2.07 | Pathogenic | 0.01 | Affected | 0.0700 | 0.7929 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2285A>C | D762A 2D AIThe SynGAP1 D762A missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool yields an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward a pathogenic impact. This assessment does not contradict any ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -4.510 | Likely Benign | 0.912 | Likely Pathogenic | Ambiguous | 0.178 | Likely Benign | -2.40 | Neutral | 0.994 | Probably Damaging | 0.900 | Possibly Damaging | 2.12 | Pathogenic | 0.05 | Affected | 0.4632 | 0.8428 | 0 | -2 | 5.3 | -44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.2285A>G | D762G 2D AIThe SynGAP1 missense variant D762G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, and ESM1b, whereas those that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the majority of predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -1.062 | Likely Benign | 0.812 | Likely Pathogenic | Ambiguous | 0.170 | Likely Benign | -2.55 | Deleterious | 0.998 | Probably Damaging | 0.949 | Probably Damaging | 2.10 | Pathogenic | 0.08 | Tolerated | 0.4710 | 0.7841 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2285A>T | D762V 2D AIThe SynGAP1 D762V missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b. Tools that agree on a pathogenic effect include polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returned an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of consensus tools predict a pathogenic impact, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -6.122 | Likely Benign | 0.949 | Likely Pathogenic | Ambiguous | 0.229 | Likely Benign | -1.98 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.08 | Pathogenic | 0.01 | Affected | 0.1105 | 0.8785 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.2286C>A | D762E 2D AIThe SynGAP1 D762E missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence predictions lean toward a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -4.332 | Likely Benign | 0.459 | Ambiguous | Likely Benign | 0.144 | Likely Benign | -1.59 | Neutral | 0.994 | Probably Damaging | 0.891 | Possibly Damaging | 2.18 | Pathogenic | 0.05 | Affected | 0.1911 | 0.8236 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2286C>G | D762E 2D AIThe SynGAP1 D762E missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence predictions lean toward a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.910475 | Binding | 0.308 | 0.859 | 0.125 | -4.332 | Likely Benign | 0.459 | Ambiguous | Likely Benign | 0.144 | Likely Benign | -1.59 | Neutral | 0.994 | Probably Damaging | 0.891 | Possibly Damaging | 2.18 | Pathogenic | 0.05 | Affected | 0.1911 | 0.8236 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2287C>A | L763I 2D AIThe SynGAP1 missense variant L763I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for this variant, and there is no conflict with ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -4.803 | Likely Benign | 0.150 | Likely Benign | Likely Benign | 0.052 | Likely Benign | -0.55 | Neutral | 0.877 | Possibly Damaging | 0.675 | Possibly Damaging | 2.48 | Pathogenic | 0.31 | Tolerated | 0.0870 | 0.3367 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.2287C>G | L763V 2D AIThe SynGAP1 missense variant L763V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, creating a single discordant signal. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence supports a benign classification, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -5.138 | Likely Benign | 0.164 | Likely Benign | Likely Benign | 0.027 | Likely Benign | -0.94 | Neutral | 0.573 | Possibly Damaging | 0.230 | Benign | 2.57 | Benign | 0.25 | Tolerated | 0.1548 | 0.2817 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2287C>T | L763F 2D AIThe SynGAP1 missense variant L763F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for this variant. There is no ClinVar entry to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -4.127 | Likely Benign | 0.255 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -0.71 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.39 | Pathogenic | 0.19 | Tolerated | 0.0584 | 0.3140 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2288T>A | L763H 2D AIThe SynGAP1 missense variant L763H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on benign include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the predictions are mixed, with a slight tilt toward pathogenicity (5 pathogenic vs. 4 benign). Thus, the variant is most likely pathogenic according to the aggregate predictions, and this assessment does not contradict ClinVar, which currently has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -6.681 | Likely Benign | 0.640 | Likely Pathogenic | Likely Benign | 0.180 | Likely Benign | -0.79 | Neutral | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 2.36 | Pathogenic | 0.03 | Affected | 0.1021 | 0.0887 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2288T>C | L763P 2D AIThe SynGAP1 missense variant L763P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default is uncertain. For high‑accuracy assessment, AlphaMissense‑Optimized predicts benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic, with one uncertain). Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -5.802 | Likely Benign | 0.550 | Ambiguous | Likely Benign | 0.158 | Likely Benign | -0.89 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.36 | Pathogenic | 0.12 | Tolerated | 0.3920 | 0.1182 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2288T>G | L763R 2D AIThe SynGAP1 missense variant L763R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.918636 | Binding | 0.351 | 0.865 | 0.125 | -5.516 | Likely Benign | 0.643 | Likely Pathogenic | Likely Benign | 0.163 | Likely Benign | -1.66 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.38 | Pathogenic | 0.07 | Tolerated | 0.1199 | 0.0761 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2290A>C | N764H 2D AIThe SynGAP1 missense variant N764H is reported in gnomAD (ID 6‑33441755‑A‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions are made by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.919527 | Binding | 0.305 | 0.861 | 0.250 | 6-33441755-A-C | -4.954 | Likely Benign | 0.320 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -2.09 | Neutral | 0.998 | Probably Damaging | 0.985 | Probably Damaging | 2.59 | Benign | 0.02 | Affected | 3.64 | 6 | 0.1236 | 0.5056 | 1 | 2 | 0.3 | 23.04 | ||||||||||||||||||||||||||||||||||||
| c.2290A>G | N764D 2D AIThe SynGAP1 missense variant N764D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, SGM‑Consensus also predicts benign, and Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.919527 | Binding | 0.305 | 0.861 | 0.250 | -6.012 | Likely Benign | 0.572 | Likely Pathogenic | Likely Benign | 0.057 | Likely Benign | -1.00 | Neutral | 0.992 | Probably Damaging | 0.893 | Possibly Damaging | 2.85 | Benign | 0.07 | Tolerated | 0.1769 | 0.2921 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2290A>T | N764Y 2D AIThe SynGAP1 missense variant N764Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy methods give mixed results: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta data are unavailable. Overall, the balance of evidence favors a pathogenic interpretation, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.919527 | Binding | 0.305 | 0.861 | 0.250 | -5.914 | Likely Benign | 0.680 | Likely Pathogenic | Likely Benign | 0.158 | Likely Benign | -2.85 | Deleterious | 0.998 | Probably Damaging | 0.967 | Probably Damaging | 2.58 | Benign | 0.01 | Affected | 0.0552 | 0.4175 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||||||||||||
| c.2291A>C | N764T 2D AIThe SynGAP1 missense variant N764T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of computational evidence points to a benign impact, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.919527 | Binding | 0.305 | 0.861 | 0.250 | -4.214 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.071 | Likely Benign | -1.57 | Neutral | 0.975 | Probably Damaging | 0.850 | Possibly Damaging | 2.63 | Benign | 0.05 | Affected | 0.1272 | 0.5168 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||||||||||
| c.2291A>G | N764S 2D AIThe SynGAP1 missense variant N764S is listed in ClinVar as Benign (ClinVar ID 1948460.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, consistent with the ClinVar classification, and there is no contradiction between the predictions and the reported ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.919527 | Binding | 0.305 | 0.861 | 0.250 | Benign | 1 | -3.149 | Likely Benign | 0.159 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.84 | Neutral | 0.992 | Probably Damaging | 0.846 | Possibly Damaging | 2.65 | Benign | 0.61 | Tolerated | 3.64 | 6 | 0.3762 | 0.5062 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||||||
| c.2291A>T | N764I 2D AIThe SynGAP1 missense variant N764I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, more tools predict pathogenicity (five) than benignity (three), and no ClinVar evidence contradicts this assessment. Thus, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.380708 | Structured | 0.919527 | Binding | 0.305 | 0.861 | 0.250 | -6.879 | Likely Benign | 0.883 | Likely Pathogenic | Ambiguous | 0.115 | Likely Benign | -2.58 | Deleterious | 0.906 | Possibly Damaging | 0.679 | Possibly Damaging | 2.58 | Benign | 0.00 | Affected | 0.0581 | 0.4483 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.2292C>A | N764K 2D AIThe SynGAP1 missense variant N764K is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign classification, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.919527 | Binding | 0.305 | 0.861 | 0.250 | -5.867 | Likely Benign | 0.892 | Likely Pathogenic | Ambiguous | 0.073 | Likely Benign | -1.36 | Neutral | 0.992 | Probably Damaging | 0.921 | Probably Damaging | 2.66 | Benign | 0.02 | Affected | 0.2005 | 0.3539 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||||
| c.2292C>G | N764K 2D AIThe SynGAP1 missense variant N764K is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign classification, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.380708 | Structured | 0.919527 | Binding | 0.305 | 0.861 | 0.250 | -5.867 | Likely Benign | 0.892 | Likely Pathogenic | Ambiguous | 0.073 | Likely Benign | -1.36 | Neutral | 0.992 | Probably Damaging | 0.921 | Probably Damaging | 2.66 | Benign | 0.02 | Affected | 0.2005 | 0.3539 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||||
| c.2293A>C | S765R 2D AIThe SynGAP1 missense variant S765R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus (majority vote) as Benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this conclusion, so the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | -5.422 | Likely Benign | 0.791 | Likely Pathogenic | Ambiguous | 0.157 | Likely Benign | -1.57 | Neutral | 0.996 | Probably Damaging | 0.985 | Probably Damaging | 4.16 | Benign | 0.07 | Tolerated | 3.64 | 6 | 0.0741 | 0.3859 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2293A>G | S765G 2D AIThe SynGAP1 missense variant S765G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to the variant being most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | -4.658 | Likely Benign | 0.152 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.98 | Neutral | 0.963 | Probably Damaging | 0.950 | Probably Damaging | 4.09 | Benign | 0.05 | Affected | 0.2894 | 0.4946 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2293A>T | S765C 2D AIThe SynGAP1 missense variant S765C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence supports a benign classification for S765C, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | -6.875 | Likely Benign | 0.256 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -2.12 | Neutral | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 4.05 | Benign | 0.07 | Tolerated | 0.0893 | 0.6309 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2294G>A | S765N 2D AIThe SynGAP1 missense variant S765N (ClinVar ID 2979632.0) is listed as “Uncertain” in ClinVar and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). In contrast, PolyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, which is consistent with the ClinVar “Uncertain” classification and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | Uncertain | 1 | -5.098 | Likely Benign | 0.378 | Ambiguous | Likely Benign | 0.094 | Likely Benign | -0.94 | Neutral | 0.985 | Probably Damaging | 0.950 | Probably Damaging | 4.11 | Benign | 0.06 | Tolerated | 3.64 | 6 | 0.1141 | 0.4658 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||
| c.2294G>C | S765T 2D AIThe SynGAP1 missense variant S765T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict ClinVar status, as ClinVar contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | -4.233 | Likely Benign | 0.215 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -1.12 | Neutral | 0.963 | Probably Damaging | 0.950 | Probably Damaging | 4.13 | Benign | 0.37 | Tolerated | 0.1312 | 0.6673 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2294G>T | S765I 2D AIThe SynGAP1 missense variant S765I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also resolves to benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | -6.891 | Likely Benign | 0.699 | Likely Pathogenic | Likely Benign | 0.187 | Likely Benign | -1.24 | Neutral | 0.996 | Probably Damaging | 0.985 | Probably Damaging | 4.09 | Benign | 0.69 | Tolerated | 0.0768 | 0.5577 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2295C>A | S765R 2D AIThe SynGAP1 missense variant S765R is reported in gnomAD (ID 6‑33442453‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM. Those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Taken together, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status because none is assigned. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | 6-33442453-C-A | -5.422 | Likely Benign | 0.791 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.57 | Neutral | 0.996 | Probably Damaging | 0.985 | Probably Damaging | 4.16 | Benign | 0.07 | Tolerated | 3.64 | 6 | 0.0741 | 0.3859 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||
| c.2295C>G | S765R 2D AIThe SynGAP1 missense variant S765R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the balance of evidence points to a benign impact for S765R, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | -5.422 | Likely Benign | 0.791 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.57 | Neutral | 0.996 | Probably Damaging | 0.985 | Probably Damaging | 4.16 | Benign | 0.07 | Tolerated | 3.64 | 6 | 0.0741 | 0.3859 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2296T>A | S766T 2D AIThe SynGAP1 missense variant S766T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.405110 | Structured | 0.923125 | Binding | 0.338 | 0.874 | 0.250 | -4.923 | Likely Benign | 0.190 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -1.25 | Neutral | 0.790 | Possibly Damaging | 0.433 | Benign | 4.15 | Benign | 0.02 | Affected | 0.1461 | 0.6511 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2296T>C | S766P 2D AIThe SynGAP1 missense variant S766P is reported in gnomAD (ID 6‑33442454‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while SIFT uniquely predicts it as pathogenic. The remaining tools, ESM1b and AlphaMissense‑Default, are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.923125 | Binding | 0.338 | 0.874 | 0.250 | 6-33442454-T-C | 1 | 1.28e-6 | -7.343 | In-Between | 0.374 | Ambiguous | Likely Benign | 0.193 | Likely Benign | -0.91 | Neutral | 0.006 | Benign | 0.013 | Benign | 4.07 | Benign | 0.01 | Affected | 3.64 | 6 | 0.2216 | 0.5862 | -1 | 1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||
| c.2296T>G | S766A 2D AIThe SynGAP1 missense variant S766A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.405110 | Structured | 0.923125 | Binding | 0.338 | 0.874 | 0.250 | -6.115 | Likely Benign | 0.186 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.98 | Neutral | 0.447 | Benign | 0.198 | Benign | 4.17 | Benign | 0.02 | Affected | 0.5059 | 0.5705 | Strenghten | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.2297C>A | S766Y 2D AIThe SynGAP1 missense variant S766Y is reported in gnomAD (ID 6‑33442455‑C‑A) but has no ClinVar entry. Functional prediction tools show mixed results: benign calls come from REVEL, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts benign, but the SGM‑Consensus (majority vote) predicts pathogenic, and no Foldetta stability data are available. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.923125 | Binding | 0.338 | 0.874 | 0.250 | 6-33442455-C-A | -8.636 | Likely Pathogenic | 0.641 | Likely Pathogenic | Likely Benign | 0.222 | Likely Benign | -2.67 | Deleterious | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 3.64 | 6 | 0.0794 | 0.5609 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||
| c.2297C>G | S766C 2D AIThe SynGAP1 missense variant S766C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S766C, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.405110 | Structured | 0.923125 | Binding | 0.338 | 0.874 | 0.250 | -7.681 | In-Between | 0.326 | Likely Benign | Likely Benign | 0.192 | Likely Benign | -2.02 | Neutral | 0.997 | Probably Damaging | 0.889 | Possibly Damaging | 4.07 | Benign | 0.00 | Affected | 0.1049 | 0.6110 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2297C>T | S766F 2D AIThe SynGAP1 missense variant S766F is listed in gnomAD (ID 6‑33442455‑C‑T) but has no ClinVar entry. Functional prediction tools show a split verdict: benign calls come from REVEL, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments further reveal AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; Foldetta results are unavailable. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.405110 | Structured | 0.923125 | Binding | 0.338 | 0.874 | 0.250 | 6-33442455-C-T | -8.944 | Likely Pathogenic | 0.709 | Likely Pathogenic | Likely Benign | 0.233 | Likely Benign | -2.87 | Deleterious | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 4.08 | Benign | 0.00 | Affected | 3.64 | 6 | 0.0770 | 0.5887 | -2 | -3 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||
| c.2299A>C | I767L 2D AIThe SynGAP1 missense variant I767L is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Based on the collective predictions, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | -1.881 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.159 | Likely Benign | -0.73 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.13 | Benign | 0.34 | Tolerated | 0.1020 | 0.4317 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.2299A>G | I767V 2D AIThe SynGAP1 missense variant I767V is listed in ClinVar (ID 1402700.0) with an “Uncertain” clinical significance and is not reported in gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a “Likely Benign” outcome. No tool predicts pathogenicity. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, while Foldetta (combining FoldX‑MD and Rosetta stability predictions) has no available result for this variant. Overall, the computational evidence strongly supports a benign effect, and this conclusion does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | Uncertain | 1 | -2.791 | Likely Benign | 0.064 | Likely Benign | Likely Benign | 0.096 | Likely Benign | 0.10 | Neutral | 0.072 | Benign | 0.029 | Benign | 4.21 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1418 | 0.4227 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||
| c.2299A>T | I767F 2D AIThe SynGAP1 missense variant I767F is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | -3.618 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.213 | Likely Benign | -1.37 | Neutral | 0.003 | Benign | 0.002 | Benign | 4.04 | Benign | 0.06 | Tolerated | 0.0643 | 0.3567 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.229A>C | S77R 2D AIThe SynGAP1 missense variant S77R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority of the high‑accuracy tools) also indicates Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective evidence points to a benign effect for S77R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | -2.823 | Likely Benign | 0.482 | Ambiguous | Likely Benign | 0.011 | Likely Benign | -1.22 | Neutral | 0.198 | Benign | 0.015 | Benign | 4.09 | Benign | 0.00 | Affected | 0.0752 | 0.3247 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.229A>G | S77G 2D AIThe SynGAP1 missense variant S77G is reported in gnomAD (variant ID 6‑33425837‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact for S77G. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | 6-33425837-A-G | 2 | 1.24e-6 | -3.571 | Likely Benign | 0.064 | Likely Benign | Likely Benign | 0.014 | Likely Benign | -1.23 | Neutral | 0.022 | Benign | 0.003 | Benign | 4.09 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2204 | 0.3866 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||
| c.229A>T | S77C 2D AIThe SynGAP1 missense variant S77C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools (polyPhen‑2 HumDiv and SIFT) predict pathogenicity, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | -5.549 | Likely Benign | 0.061 | Likely Benign | Likely Benign | 0.022 | Likely Benign | -0.94 | Neutral | 0.953 | Possibly Damaging | 0.129 | Benign | 4.04 | Benign | 0.00 | Affected | 0.0954 | 0.5144 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.22A>C | I8L 2D AIThe SynGAP1 missense variant I8L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote from the four high‑accuracy tools) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the I8L variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.543080 | Binding | 0.341 | 0.916 | 0.625 | -1.752 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.124 | Likely Benign | 0.08 | Neutral | 0.002 | Benign | 0.000 | Benign | 4.20 | Benign | 0.00 | Affected | 0.0707 | 0.3776 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.22A>G | I8V 2D AIThe SynGAP1 missense variant I8V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.543080 | Binding | 0.341 | 0.916 | 0.625 | -2.723 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -0.01 | Neutral | 0.005 | Benign | 0.001 | Benign | 4.22 | Benign | 0.00 | Affected | 0.1031 | 0.3470 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.22A>T | I8F 2D AIThe SynGAP1 missense variant I8F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.543080 | Binding | 0.341 | 0.916 | 0.625 | -3.000 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.29 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.02 | Benign | 0.00 | Affected | 0.0483 | 0.2871 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2300T>A | I767N 2D AIThe SynGAP1 missense variant I767N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | -5.117 | Likely Benign | 0.541 | Ambiguous | Likely Benign | 0.122 | Likely Benign | -0.16 | Neutral | 0.977 | Probably Damaging | 0.632 | Possibly Damaging | 4.04 | Benign | 0.06 | Tolerated | 0.1039 | 0.1012 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2300T>C | I767T 2D AIThe SynGAP1 missense variant I767T is listed in ClinVar (ID 1044161.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, creating a single discordant prediction. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification, and AlphaMissense‑Optimized also reports benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | Uncertain | 1 | -3.749 | Likely Benign | 0.252 | Likely Benign | Likely Benign | 0.138 | Likely Benign | -0.78 | Neutral | 0.625 | Possibly Damaging | 0.249 | Benign | 4.12 | Benign | 0.46 | Tolerated | 3.64 | 6 | 0.1274 | 0.1889 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||||||||
| c.2300T>G | I767S 2D AIThe SynGAP1 missense variant I767S is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Only polyPhen‑2 HumDiv flags it as pathogenic, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Taken together, the preponderance of evidence points to a benign classification for I767S, and this assessment does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | -3.030 | Likely Benign | 0.388 | Ambiguous | Likely Benign | 0.126 | Likely Benign | -0.64 | Neutral | 0.925 | Possibly Damaging | 0.329 | Benign | 4.13 | Benign | 0.25 | Tolerated | 0.3242 | 0.1782 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2301C>G | I767M 2D AIThe SynGAP1 missense variant I767M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence supports a benign classification for I767M, and this conclusion does not contradict any ClinVar annotation, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | -2.384 | Likely Benign | 0.084 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -0.60 | Neutral | 0.835 | Possibly Damaging | 0.486 | Possibly Damaging | 4.05 | Benign | 0.11 | Tolerated | 0.0788 | 0.3377 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2302G>A | D768N 2D AIThe SynGAP1 missense variant D768N is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33442460‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus is “Likely Benign,” and Foldetta data are unavailable. Overall, the consensus of available predictions indicates that the variant is most likely benign, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | Uncertain | 1 | 6-33442460-G-A | 2 | 2.57e-6 | -6.892 | Likely Benign | 0.453 | Ambiguous | Likely Benign | 0.048 | Likely Benign | -0.77 | Neutral | 0.106 | Benign | 0.009 | Benign | 4.07 | Benign | 0.96 | Tolerated | 3.64 | 6 | 0.1178 | 0.7843 | 1 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||
| c.2302G>C | D768H 2D AIThe SynGAP1 D768H missense variant is not reported in ClinVar (ClinVar status: not reported) and is absent from gnomAD (gnomAD: not present). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of predictions (5 benign vs. 4 pathogenic) and the high‑accuracy benign call suggest the variant is most likely benign, with no ClinVar status to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | -8.673 | Likely Pathogenic | 0.783 | Likely Pathogenic | Likely Benign | 0.160 | Likely Benign | -1.85 | Neutral | 0.966 | Probably Damaging | 0.737 | Possibly Damaging | 4.03 | Benign | 0.12 | Tolerated | 0.1450 | 0.8136 | 1 | -1 | 0.3 | 22.05 | ||||||||||||||||||||||||||||||||||||||||
| c.2302G>T | D768Y 2D AIThe SynGAP1 missense variant D768Y is listed in ClinVar with status “Uncertain” (ClinVar ID 1061652.0) and is present in gnomAD (variant ID 6‑33442460‑G‑T). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain,” SGM‑Consensus as “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic impact, which does not contradict the ClinVar designation of uncertainty. Thus, based on current predictions, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | Uncertain | 1 | 6-33442460-G-T | -9.866 | Likely Pathogenic | 0.824 | Likely Pathogenic | Ambiguous | 0.234 | Likely Benign | -2.86 | Deleterious | 0.989 | Probably Damaging | 0.806 | Possibly Damaging | 4.01 | Benign | 0.07 | Tolerated | 3.64 | 6 | 0.0581 | 0.7525 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||
| c.2303A>C | D768A 2D AIThe SynGAP1 D768A variant is listed in gnomAD (ID 6‑33442461‑A‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM) and pathogenic predictions (ESM1b, AlphaMissense‑Default). AlphaMissense‑Optimized returns an uncertain result. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is a 2‑vs‑2 tie and thus unavailable; Foldetta stability analysis is not reported. Overall, the preponderance of evidence (six benign vs two pathogenic) points to a benign effect. This conclusion does not contradict ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | 6-33442461-A-C | -8.153 | Likely Pathogenic | 0.786 | Likely Pathogenic | Ambiguous | 0.174 | Likely Benign | -1.84 | Neutral | 0.245 | Benign | 0.096 | Benign | 4.09 | Benign | 0.14 | Tolerated | 3.64 | 6 | 0.3924 | 0.7662 | -2 | 0 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||
| c.2303A>G | D768G 2D AIThe SynGAP1 D768G missense variant is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors shows a predominance of benign calls: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized all predict benign, whereas AlphaMissense‑Default predicts pathogenic. The high‑accuracy AlphaMissense‑Optimized result is benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign classification (2 benign vs. 1 pathogenic, with one uncertain). Foldetta, which would provide a protein‑folding stability assessment, has no available output for this variant. Overall, the preponderance of evidence points to a benign effect, and this assessment does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | -7.150 | In-Between | 0.697 | Likely Pathogenic | Likely Benign | 0.184 | Likely Benign | 0.03 | Neutral | 0.393 | Benign | 0.131 | Benign | 4.09 | Benign | 0.72 | Tolerated | 0.4130 | 0.7476 | 1 | -1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2303A>T | D768V 2D AIThe SynGAP1 D768V variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Based on the preponderance of pathogenic predictions and the SGM‑Consensus result, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | -9.528 | Likely Pathogenic | 0.880 | Likely Pathogenic | Ambiguous | 0.164 | Likely Benign | -2.62 | Deleterious | 0.611 | Possibly Damaging | 0.140 | Benign | 4.04 | Benign | 0.02 | Affected | 0.0802 | 0.8019 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2304C>A | D768E 2D AIThe SynGAP1 missense variant D768E is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the change as benign, and AlphaMissense‑Optimized also predicts a benign outcome. No tool predicts pathogenicity; AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the computational evidence strongly supports a benign effect for D768E, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | -4.611 | Likely Benign | 0.380 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.23 | Neutral | 0.393 | Benign | 0.171 | Benign | 4.16 | Benign | 0.17 | Tolerated | 0.1368 | 0.7729 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2304C>G | D768E 2D AIThe SynGAP1 missense variant D768E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the computational evidence strongly suggests that D768E is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.332115 | Structured | 0.928237 | Binding | 0.314 | 0.877 | 0.250 | -4.611 | Likely Benign | 0.380 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.23 | Neutral | 0.393 | Benign | 0.171 | Benign | 4.16 | Benign | 0.17 | Tolerated | 0.1368 | 0.7729 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2305C>A | L769I 2D AIThe SynGAP1 missense variant L769I is listed in gnomAD (ID 6‑33442463‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | 6-33442463-C-A | -3.993 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -0.15 | Neutral | 0.836 | Possibly Damaging | 0.329 | Benign | 4.09 | Benign | 0.04 | Affected | 3.64 | 6 | 0.0800 | 0.3108 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||||||||
| c.2305C>G | L769V 2D AIThe SynGAP1 missense variant L769V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags it as pathogenic, but this is the sole discordant call. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | -4.585 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.41 | Neutral | 0.625 | Possibly Damaging | 0.249 | Benign | 4.04 | Benign | 0.25 | Tolerated | 0.1364 | 0.2558 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2305C>T | L769F 2D AIThe SynGAP1 missense variant L769F is listed in ClinVar (ID 3617309.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority of the high‑accuracy tools) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions—including the high‑accuracy tools—suggest the variant is most likely benign, which is consistent with its ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | Uncertain | 1 | -5.044 | Likely Benign | 0.146 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.89 | Neutral | 0.925 | Possibly Damaging | 0.510 | Possibly Damaging | 3.94 | Benign | 0.02 | Affected | 0.0554 | 0.2881 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||
| c.2306T>A | L769H 2D AIThe SynGAP1 missense variant L769H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, all of which classify the variant as benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, all of which report the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | -6.038 | Likely Benign | 0.422 | Ambiguous | Likely Benign | 0.235 | Likely Benign | -1.96 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 0.1016 | 0.0828 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2306T>C | L769P 2D AIThe SynGAP1 missense variant L769P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | -6.261 | Likely Benign | 0.338 | Likely Benign | Likely Benign | 0.200 | Likely Benign | -1.75 | Neutral | 0.925 | Possibly Damaging | 0.427 | Benign | 3.90 | Benign | 0.00 | Affected | 0.3573 | 0.1323 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2306T>G | L769R 2D AIThe SynGAP1 missense variant L769R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.928432 | Binding | 0.367 | 0.883 | 0.250 | -6.245 | Likely Benign | 0.493 | Ambiguous | Likely Benign | 0.206 | Likely Benign | -1.66 | Neutral | 0.003 | Benign | 0.006 | Benign | 3.91 | Benign | 0.00 | Affected | 0.1286 | 0.0702 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2308C>A | Q770K 2D AIThe SynGAP1 missense variant Q770K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.923732 | Binding | 0.328 | 0.887 | 0.250 | -4.768 | Likely Benign | 0.367 | Ambiguous | Likely Benign | 0.106 | Likely Benign | -0.72 | Neutral | 0.002 | Benign | 0.003 | Benign | 4.20 | Benign | 0.14 | Tolerated | 0.1984 | 0.4737 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.2308C>G | Q770E 2D AIThe SynGAP1 missense variant Q770E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.923732 | Binding | 0.328 | 0.887 | 0.250 | -4.782 | Likely Benign | 0.201 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.82 | Neutral | 0.002 | Benign | 0.003 | Benign | 4.19 | Benign | 0.04 | Affected | 0.1465 | 0.2833 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2309A>C | Q770P 2D AIThe SynGAP1 missense variant Q770P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for Q770P, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.923732 | Binding | 0.328 | 0.887 | 0.250 | -3.948 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.178 | Likely Benign | -1.00 | Neutral | 0.748 | Possibly Damaging | 0.170 | Benign | 4.10 | Benign | 0.02 | Affected | 0.2435 | 0.5717 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2309A>G | Q770R 2D AIThe SynGAP1 missense variant Q770R is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable. Overall, the collective evidence strongly suggests that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.923732 | Binding | 0.328 | 0.887 | 0.250 | -3.873 | Likely Benign | 0.344 | Ambiguous | Likely Benign | 0.175 | Likely Benign | -1.38 | Neutral | 0.194 | Benign | 0.071 | Benign | 4.14 | Benign | 0.07 | Tolerated | 0.1610 | 0.2580 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.2309A>T | Q770L 2D AIThe SynGAP1 missense variant Q770L is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q770L, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.923732 | Binding | 0.328 | 0.887 | 0.250 | -5.524 | Likely Benign | 0.521 | Ambiguous | Likely Benign | 0.197 | Likely Benign | -2.17 | Neutral | 0.095 | Benign | 0.030 | Benign | 4.14 | Benign | 0.01 | Affected | 0.0776 | 0.6230 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.230G>A | S77N 2D AIThe SynGAP1 missense variant S77N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | -4.597 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.78 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.09 | Benign | 0.00 | Affected | 0.1088 | 0.3774 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||||
| c.230G>C | S77T 2D AIThe SynGAP1 missense variant S77T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of computational evidence points to a benign effect, and this is consistent with the lack of any ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | -4.204 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.03 | Neutral | 0.092 | Benign | 0.007 | Benign | 4.19 | Benign | 0.00 | Affected | 0.1180 | 0.5003 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.230G>T | S77I 2D AIThe SynGAP1 missense variant S77I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | -3.918 | Likely Benign | 0.140 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -1.41 | Neutral | 0.604 | Possibly Damaging | 0.029 | Benign | 4.07 | Benign | 0.00 | Affected | 0.0758 | 0.5165 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2310G>C | Q770H 2D AIThe SynGAP1 missense variant Q770H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for Q770H. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.923732 | Binding | 0.328 | 0.887 | 0.250 | -4.241 | Likely Benign | 0.270 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -1.27 | Neutral | 0.962 | Probably Damaging | 0.515 | Possibly Damaging | 4.11 | Benign | 0.01 | Affected | 0.1579 | 0.4344 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2310G>T | Q770H 2D AIThe SynGAP1 missense variant Q770H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.923732 | Binding | 0.328 | 0.887 | 0.250 | -4.241 | Likely Benign | 0.270 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -1.27 | Neutral | 0.962 | Probably Damaging | 0.515 | Possibly Damaging | 4.11 | Benign | 0.01 | Affected | 0.1579 | 0.4344 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2311T>A | S771T 2D AIThe SynGAP1 missense variant S771T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions point to a benign impact. This conclusion is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.922503 | Binding | 0.306 | 0.883 | 0.250 | -4.765 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -1.38 | Neutral | 0.649 | Possibly Damaging | 0.433 | Benign | 4.07 | Benign | 0.23 | Tolerated | 0.1497 | 0.6310 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2311T>C | S771P 2D AIThe SynGAP1 missense variant S771P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.922503 | Binding | 0.306 | 0.883 | 0.250 | -5.045 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.180 | Likely Benign | -1.32 | Neutral | 0.901 | Possibly Damaging | 0.692 | Possibly Damaging | 4.03 | Benign | 0.19 | Tolerated | 0.2194 | 0.5515 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2311T>G | S771A 2D AIThe SynGAP1 missense variant S771A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification—there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.922503 | Binding | 0.306 | 0.883 | 0.250 | -4.337 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -1.09 | Neutral | 0.025 | Benign | 0.014 | Benign | 4.09 | Benign | 0.62 | Tolerated | 0.5201 | 0.4745 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.2312C>A | S771Y 2D AIThe SynGAP1 missense variant S771Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Two tools, ESM1b and AlphaMissense‑Default, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it contains two benign and two uncertain calls, and Foldetta data are unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.436924 | Structured | 0.922503 | Binding | 0.306 | 0.883 | 0.250 | -7.041 | In-Between | 0.397 | Ambiguous | Likely Benign | 0.176 | Likely Benign | -2.13 | Neutral | 0.990 | Probably Damaging | 0.892 | Possibly Damaging | 4.02 | Benign | 0.05 | Affected | 0.0707 | 0.5155 | -3 | -2 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||||||
| c.2312C>G | S771C 2D AIThe SynGAP1 missense variant S771C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.922503 | Binding | 0.306 | 0.883 | 0.250 | -8.014 | Likely Pathogenic | 0.167 | Likely Benign | Likely Benign | 0.177 | Likely Benign | -1.99 | Neutral | 0.990 | Probably Damaging | 0.917 | Probably Damaging | 4.01 | Benign | 0.07 | Tolerated | 0.1022 | 0.5899 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2312C>T | S771F 2D AIThe SynGAP1 missense variant S771F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Two tools—ESM1b and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is uncertain, and Foldetta stability analysis is unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not conflict with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.436924 | Structured | 0.922503 | Binding | 0.306 | 0.883 | 0.250 | -7.988 | In-Between | 0.525 | Ambiguous | Likely Benign | 0.189 | Likely Benign | -2.29 | Neutral | 0.990 | Probably Damaging | 0.892 | Possibly Damaging | 4.02 | Benign | 0.04 | Affected | 0.0624 | 0.5428 | -3 | -2 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||||||
| c.2314T>A | F772I 2D  AIThe SynGAP1 missense variant F772I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | -2.925 | Likely Benign | 0.255 | Likely Benign | Likely Benign | 0.142 | Likely Benign | -0.38 | Neutral | 0.845 | Possibly Damaging | 0.899 | Possibly Damaging | 4.24 | Benign | 0.41 | Tolerated | 0.1544 | 0.2051 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2314T>C | F772L 2D  AIThe SynGAP1 missense variant F772L is listed in gnomAD (6‑33442472‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | 6-33442472-T-C | 1 | 1.28e-6 | -1.751 | Likely Benign | 0.762 | Likely Pathogenic | Likely Benign | 0.161 | Likely Benign | -0.47 | Neutral | 0.508 | Possibly Damaging | 0.786 | Possibly Damaging | 4.31 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1661 | 0.3033 | 0 | 2 | 1.0 | -34.02 | ||||||||||||||||||||||||||||||||||
| c.2314T>G | F772V 2D  AIThe SynGAP1 missense variant F772V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are not available for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | -2.886 | Likely Benign | 0.191 | Likely Benign | Likely Benign | 0.161 | Likely Benign | -0.43 | Neutral | 0.845 | Possibly Damaging | 0.899 | Possibly Damaging | 4.27 | Benign | 0.37 | Tolerated | 0.1687 | 0.2419 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2315T>A | F772Y 2D  AIThe SynGAP1 missense variant F772Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | -2.657 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -0.54 | Neutral | 0.705 | Possibly Damaging | 0.786 | Possibly Damaging | 4.17 | Benign | 0.35 | Tolerated | 0.1058 | 0.1892 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2315T>C | F772S 2D  AIThe SynGAP1 missense variant F772S is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign, while the majority of other predictors (polyPhen‑2 HumDiv and HumVar) suggest pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus likewise indicates Likely Benign; Foldetta data are not available. Overall, the preponderance of evidence points to a benign effect for F772S, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | -2.722 | Likely Benign | 0.442 | Ambiguous | Likely Benign | 0.138 | Likely Benign | -0.26 | Neutral | 0.845 | Possibly Damaging | 0.899 | Possibly Damaging | 4.21 | Benign | 0.56 | Tolerated | 0.4134 | 0.0558 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||||||||||||
| c.2315T>G | F772C 2D  AIThe SynGAP1 missense variant F772C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict any ClinVar status, as no ClinVar claim exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | -4.498 | Likely Benign | 0.248 | Likely Benign | Likely Benign | 0.156 | Likely Benign | -1.46 | Neutral | 0.979 | Probably Damaging | 0.985 | Probably Damaging | 4.14 | Benign | 0.10 | Tolerated | 0.2450 | 0.1419 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||||||||||||
| c.2316C>A | F772L 2D AIThe SynGAP1 missense variant F772L is catalogued in gnomAD (ID 6‑33442474‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is Likely Benign, while Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the preponderance of evidence indicates that F772L is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | 6-33442474-C-A | -1.751 | Likely Benign | 0.762 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | -0.47 | Neutral | 0.508 | Possibly Damaging | 0.786 | Possibly Damaging | 4.31 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1661 | 0.3033 | 0 | 2 | 1.0 | -34.02 | ||||||||||||||||||||||||||||||||||||
| c.2316C>G | F772L 2D AIThe SynGAP1 missense variant F772L has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability data are available, so it is treated as unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | -1.751 | Likely Benign | 0.762 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | -0.47 | Neutral | 0.508 | Possibly Damaging | 0.786 | Possibly Damaging | 4.31 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1661 | 0.3033 | 0 | 2 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||||
| c.2317A>C | M773L 2D AIThe SynGAP1 missense variant M773L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -3.458 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.211 | Likely Benign | -0.75 | Neutral | 0.038 | Benign | 0.137 | Benign | 4.29 | Benign | 0.81 | Tolerated | 0.1517 | 0.3529 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2317A>G | M773V 2D AIThe SynGAP1 missense variant M773V has no ClinVar entry and is not reported in gnomAD. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the computational evidence indicates that M773V is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -4.353 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.234 | Likely Benign | -0.70 | Neutral | 0.038 | Benign | 0.284 | Benign | 4.30 | Benign | 0.87 | Tolerated | 0.3145 | 0.3081 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||||||
| c.2317A>T | M773L 2D AIThe SynGAP1 missense variant M773L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess pathogenicity uniformly predict a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the consensus of all available predictions points to a benign impact, and this is consistent with the lack of a ClinVar classification—there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -3.458 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.211 | Likely Benign | -0.75 | Neutral | 0.038 | Benign | 0.137 | Benign | 4.29 | Benign | 0.81 | Tolerated | 0.1517 | 0.3529 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2318T>A | M773K 2D AIThe SynGAP1 missense variant M773K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -5.117 | Likely Benign | 0.641 | Likely Pathogenic | Likely Benign | 0.178 | Likely Benign | -1.80 | Neutral | 0.106 | Benign | 0.471 | Possibly Damaging | 4.19 | Benign | 0.02 | Affected | 0.1646 | 0.0851 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.2318T>C | M773T 2D AIThe SynGAP1 missense variant M773T is listed in gnomAD (ID 6‑33442476‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only polyPhen‑2 HumVar predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | 6-33442476-T-C | 1 | 1.28e-6 | -4.225 | Likely Benign | 0.377 | Ambiguous | Likely Benign | 0.112 | Likely Benign | -1.63 | Neutral | 0.106 | Benign | 0.471 | Possibly Damaging | 4.22 | Benign | 0.06 | Tolerated | 3.64 | 6 | 0.2196 | 0.1586 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.2318T>G | M773R 2D AIThe SynGAP1 missense variant M773R is listed in gnomAD (ID 6‑33442476‑T‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates benign. No Foldetta (FoldX‑MD/Rosetta stability) result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | 6-33442476-T-G | 1 | 1.28e-6 | -4.340 | Likely Benign | 0.597 | Likely Pathogenic | Likely Benign | 0.183 | Likely Benign | -1.87 | Neutral | 0.220 | Benign | 0.471 | Possibly Damaging | 4.18 | Benign | 0.02 | Affected | 3.64 | 6 | 0.1778 | 0.0800 | -1 | 0 | -6.4 | 24.99 | ||||||||||||||||||||||||||||||||||
| c.2319G>A | M773I 2D AIThe SynGAP1 missense variant M773I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -4.799 | Likely Benign | 0.567 | Likely Pathogenic | Likely Benign | 0.099 | Likely Benign | -0.83 | Neutral | 0.038 | Benign | 0.284 | Benign | 4.31 | Benign | 0.22 | Tolerated | 0.1406 | 0.3011 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2319G>C | M773I 2D AIThe SynGAP1 missense variant M773I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -4.799 | Likely Benign | 0.567 | Likely Pathogenic | Likely Benign | 0.099 | Likely Benign | -0.83 | Neutral | 0.038 | Benign | 0.284 | Benign | 4.31 | Benign | 0.22 | Tolerated | 0.1406 | 0.3011 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2319G>T | M773I 2D AIThe SynGAP1 missense variant M773I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | -4.799 | Likely Benign | 0.567 | Likely Pathogenic | Likely Benign | 0.099 | Likely Benign | -0.83 | Neutral | 0.038 | Benign | 0.284 | Benign | 4.31 | Benign | 0.22 | Tolerated | 0.1406 | 0.3011 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.231T>A | S77R 2D AIThe SynGAP1 missense variant S77R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority of the high‑accuracy tools) is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | -2.823 | Likely Benign | 0.482 | Ambiguous | Likely Benign | 0.026 | Likely Benign | -1.22 | Neutral | 0.198 | Benign | 0.015 | Benign | 4.09 | Benign | 0.00 | Affected | 0.0752 | 0.3247 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.231T>G | S77R 2D AIThe SynGAP1 missense variant S77R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority of the high‑accuracy tools) is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | -2.823 | Likely Benign | 0.482 | Ambiguous | Likely Benign | 0.026 | Likely Benign | -1.22 | Neutral | 0.198 | Benign | 0.015 | Benign | 4.09 | Benign | 0.00 | Affected | 0.0752 | 0.3247 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2320G>A | A774T 2D AIThe SynGAP1 missense variant A774T is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.905168 | Binding | 0.336 | 0.897 | 0.250 | -3.238 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -0.46 | Neutral | 0.037 | Benign | 0.063 | Benign | 4.20 | Benign | 0.39 | Tolerated | 0.1436 | 0.7578 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2320G>C | A774P 2D AIThe SynGAP1 missense variant A774P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.905168 | Binding | 0.336 | 0.897 | 0.250 | -3.869 | Likely Benign | 0.192 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.94 | Neutral | 0.801 | Possibly Damaging | 0.481 | Possibly Damaging | 4.15 | Benign | 0.18 | Tolerated | 0.1902 | 0.5749 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2320G>T | A774S 2D AIThe SynGAP1 missense variant A774S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.905168 | Binding | 0.336 | 0.897 | 0.250 | -2.780 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.090 | Likely Benign | -0.09 | Neutral | 0.071 | Benign | 0.115 | Benign | 4.27 | Benign | 0.43 | Tolerated | 0.2711 | 0.6399 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2321C>A | A774D 2D AIThe SynGAP1 missense variant A774D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.905168 | Binding | 0.336 | 0.897 | 0.250 | -4.455 | Likely Benign | 0.603 | Likely Pathogenic | Likely Benign | 0.123 | Likely Benign | -0.46 | Neutral | 0.570 | Possibly Damaging | 0.386 | Benign | 4.21 | Benign | 0.06 | Tolerated | 0.1645 | 0.1793 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2321C>G | A774G 2D AIThe SynGAP1 missense variant A774G is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.905168 | Binding | 0.336 | 0.897 | 0.250 | -3.129 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.68 | Neutral | 0.135 | Benign | 0.152 | Benign | 4.16 | Benign | 0.22 | Tolerated | 0.2392 | 0.5436 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2321C>T | A774V 2D AIThe SynGAP1 missense variant A774V is catalogued in gnomAD (ID 6‑33442479‑C‑T) but has no ClinVar entry. Across the spectrum of in‑silico predictors, every tool listed—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently scores the variant as benign. No pathogenic predictions are reported. Grouping by agreement, the benign‑predicting tools comprise the entire set, while the pathogenic group is empty. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.905168 | Binding | 0.336 | 0.897 | 0.250 | 6-33442479-C-T | 1 | 1.28e-6 | -3.075 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.73 | Neutral | 0.000 | Benign | 0.003 | Benign | 4.20 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1070 | 0.6659 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||
| c.2323C>G | R775G 2D AIThe SynGAP1 missense variant R775G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R775G, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.895337 | Binding | 0.320 | 0.896 | 0.250 | -4.186 | Likely Benign | 0.359 | Ambiguous | Likely Benign | 0.118 | Likely Benign | -1.23 | Neutral | 0.933 | Possibly Damaging | 0.871 | Possibly Damaging | 4.12 | Benign | 0.07 | Tolerated | 0.3194 | 0.3761 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2324G>A | R775Q 2D AIThe SynGAP1 missense variant R775Q is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33442482‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from the same set of high‑confidence predictors) is “Likely Benign.” No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.895337 | Binding | 0.320 | 0.896 | 0.250 | Conflicting | 3 | 6-33442482-G-A | 11 | 1.41e-5 | -4.476 | Likely Benign | 0.229 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -0.63 | Neutral | 0.969 | Probably Damaging | 0.863 | Possibly Damaging | 4.17 | Benign | 0.16 | Tolerated | 3.64 | 6 | 0.2844 | 0.2863 | 1 | 1 | 1.0 | -28.06 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||
| c.2324G>C | R775P 2D AIThe SynGAP1 missense variant R775P is listed in ClinVar (ID 2959355) as Benign and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Two tools (polyPhen‑2 HumDiv and HumVar) predict a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, the SGM‑Consensus also indicating a likely benign status, and Foldetta’s protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, which is consistent with the ClinVar classification and does not contradict the reported status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.895337 | Binding | 0.320 | 0.896 | 0.250 | Benign | 2 | -5.072 | Likely Benign | 0.452 | Ambiguous | Likely Benign | 0.168 | Likely Benign | -0.79 | Neutral | 0.971 | Probably Damaging | 0.944 | Probably Damaging | 4.13 | Benign | 0.07 | Tolerated | 3.64 | 6 | 0.1891 | 0.4834 | -2 | 0 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||
| c.2324G>T | R775L 2D AIThe SynGAP1 missense variant R775L (ClinVar ID 4327035) is present in gnomAD (ID 6‑33442482‑G‑T). Prediction tools that agree on benign include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those predicting pathogenic are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also indicates likely benign; Foldetta results are unavailable. Overall, the consensus of computational evidence points to a benign effect, consistent with the ClinVar annotation and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.895337 | Binding | 0.320 | 0.896 | 0.250 | 1 | 6-33442482-G-T | -5.951 | Likely Benign | 0.598 | Likely Pathogenic | Likely Benign | 0.124 | Likely Benign | -1.86 | Neutral | 0.933 | Possibly Damaging | 0.871 | Possibly Damaging | 4.13 | Benign | 0.06 | Tolerated | 3.64 | 6 | 0.1665 | 0.5089 | -2 | -3 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||
| c.2326G>A | G776S 2D AIThe SynGAP1 missense variant G776S is reported in ClinVar as “Not submitted” and is present in gnomAD (variant ID 6-33442484-G-A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus likewise indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of predictive evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which currently contains no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.377384 | Structured | 0.886983 | Binding | 0.296 | 0.888 | 0.250 | 6-33442484-G-A | 1 | 1.28e-6 | -3.334 | Likely Benign | 0.147 | Likely Benign | Likely Benign | 0.163 | Likely Benign | -1.21 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 4.28 | Benign | 0.13 | Tolerated | 3.64 | 6 | 0.2539 | 0.5785 | 0 | 1 | -0.4 | 30.03 | ||||||||||||||||||||||||||||||||||
| c.2326G>C | G776R 2D AIThe SynGAP1 missense variant G776R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which collectively suggest a likely benign outcome. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the Foldetta protein‑folding stability assessment is unavailable for this variant. Overall, the balance of evidence leans toward a benign interpretation, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.377384 | Structured | 0.886983 | Binding | 0.296 | 0.888 | 0.250 | -6.209 | Likely Benign | 0.886 | Likely Pathogenic | Ambiguous | 0.181 | Likely Benign | -2.28 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 4.22 | Benign | 0.01 | Affected | 0.0932 | 0.5115 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2326G>T | G776C 2D AIThe SynGAP1 missense variant G776C is not reported in ClinVar but is present in gnomAD (ID 6‑33442484‑G‑T). Prediction tools cluster into benign (REVEL, FATHMM, AlphaMissense‑Optimized) and pathogenic (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT). Two tools (ESM1b, AlphaMissense‑Default) return uncertain results. High‑accuracy assessments are limited: AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta data are unavailable. Overall, the majority of conventional predictors indicate pathogenicity, whereas the single high‑accuracy tool suggests benign. Given the preponderance of pathogenic predictions and the absence of a ClinVar entry, the variant is most likely pathogenic and does not contradict any existing ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.377384 | Structured | 0.886983 | Binding | 0.296 | 0.888 | 0.250 | 6-33442484-G-T | -7.974 | In-Between | 0.380 | Ambiguous | Likely Benign | 0.181 | Likely Benign | -2.59 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 4.15 | Benign | 0.01 | Affected | 3.64 | 6 | 0.1235 | 0.4403 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||
| c.2327G>A | G776D 2D AIThe SynGAP1 missense variant G776D is catalogued in gnomAD (ID 6‑33442485‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions come from AlphaMissense‑Default, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (majority of the four high‑accuracy tools) also indicates benign. Foldetta stability analysis is unavailable, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for G776D, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.377384 | Structured | 0.886983 | Binding | 0.296 | 0.888 | 0.250 | 6-33442485-G-A | 0.388 | Likely Benign | 0.566 | Likely Pathogenic | Likely Benign | 0.188 | Likely Benign | 0.16 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 4.30 | Benign | 0.10 | Tolerated | 3.64 | 6 | 0.1835 | 0.2971 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||
| c.2327G>C | G776A 2D AIThe SynGAP1 missense variant G776A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for G776A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.377384 | Structured | 0.886983 | Binding | 0.296 | 0.888 | 0.250 | -5.275 | Likely Benign | 0.296 | Likely Benign | Likely Benign | 0.173 | Likely Benign | -1.82 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 4.26 | Benign | 0.03 | Affected | 0.3648 | 0.4971 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2327G>T | G776V 2D AIThe SynGAP1 missense variant G776V is listed in gnomAD (ID 6‑33442485‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar) and SIFT. AlphaMissense‑Default is uncertain, and Foldetta (FoldX‑MD/Rosetta stability assessment) has no result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also indicates a likely benign outcome; Foldetta data are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status because none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.377384 | Structured | 0.886983 | Binding | 0.296 | 0.888 | 0.250 | 6-33442485-G-T | -6.541 | Likely Benign | 0.515 | Ambiguous | Likely Benign | 0.180 | Likely Benign | -2.25 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 4.19 | Benign | 0.01 | Affected | 3.64 | 6 | 0.0993 | 0.4223 | -3 | -1 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||
| c.2329C>A | L777I 2D AIThe SynGAP1 missense variant L777I is listed in gnomAD (ID 6‑33442487‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which reports “Likely Benign.” Pathogenic predictions are made by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for the variant, and this conclusion does not contradict any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | 6-33442487-C-A | -5.346 | Likely Benign | 0.128 | Likely Benign | Likely Benign | 0.096 | Likely Benign | -0.85 | Neutral | 0.843 | Possibly Damaging | 0.920 | Probably Damaging | 4.05 | Benign | 0.02 | Affected | 3.64 | 6 | 0.1041 | 0.4327 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||||||||
| c.2329C>G | L777V 2D AIThe SynGAP1 missense variant L777V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for the variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | -5.693 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.084 | Likely Benign | -1.05 | Neutral | 0.843 | Possibly Damaging | 0.920 | Probably Damaging | 4.07 | Benign | 0.05 | Affected | 0.1651 | 0.3977 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2329C>T | L777F 2D AIThe SynGAP1 missense variant L777F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | -4.499 | Likely Benign | 0.175 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -1.91 | Neutral | 0.968 | Probably Damaging | 0.966 | Probably Damaging | 3.98 | Benign | 0.01 | Affected | 0.0697 | 0.3576 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.232C>A | R78S 2D AIThe SynGAP1 missense variant R78S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized returns an uncertain result. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments are: AlphaMissense‑Optimized (Uncertain), SGM‑Consensus (Likely Benign), and Foldetta (no data available). Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which contains no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.448183 | Uncertain | 0.304 | 0.866 | 0.500 | -3.680 | Likely Benign | 0.792 | Likely Pathogenic | Ambiguous | 0.063 | Likely Benign | -1.11 | Neutral | 0.385 | Benign | 0.015 | Benign | 3.86 | Benign | 0.00 | Affected | 0.3081 | 0.3274 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.232C>G | R78G 2D AIThe SynGAP1 R78G missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign status: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Taken together, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.448183 | Uncertain | 0.304 | 0.866 | 0.500 | -3.972 | Likely Benign | 0.480 | Ambiguous | Likely Benign | 0.072 | Likely Benign | -1.94 | Neutral | 0.385 | Benign | 0.028 | Benign | 3.83 | Benign | 0.00 | Affected | 0.3285 | 0.2986 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.232C>T | R78C 2D AIThe SynGAP1 missense variant R78C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R78C, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.448183 | Uncertain | 0.304 | 0.866 | 0.500 | -6.079 | Likely Benign | 0.467 | Ambiguous | Likely Benign | 0.114 | Likely Benign | -2.13 | Neutral | 0.991 | Probably Damaging | 0.194 | Benign | 3.80 | Benign | 0.00 | Affected | 0.3255 | 0.2804 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||||||||||
| c.2330T>A | L777H 2D AIThe SynGAP1 missense variant L777H has no ClinVar record and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors a benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence—including the consensus of high‑accuracy tools—suggests that the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | -6.719 | Likely Benign | 0.492 | Ambiguous | Likely Benign | 0.230 | Likely Benign | -2.51 | Deleterious | 0.997 | Probably Damaging | 0.993 | Probably Damaging | 3.95 | Benign | 0.00 | Affected | 0.1052 | 0.0972 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2330T>C | L777P 2D AIThe SynGAP1 missense variant L777P is catalogued in gnomAD (6‑33442488‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions are reported by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus also indicates a likely benign outcome. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for the variant, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | 6-33442488-T-C | -5.807 | Likely Benign | 0.361 | Ambiguous | Likely Benign | 0.255 | Likely Benign | -2.13 | Neutral | 0.991 | Probably Damaging | 0.985 | Probably Damaging | 3.94 | Benign | 0.00 | Affected | 3.64 | 6 | 0.3732 | 0.1664 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||
| c.2330T>G | L777R 2D AIThe SynGAP1 missense variant L777R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.876129 | Binding | 0.336 | 0.882 | 0.250 | -6.084 | Likely Benign | 0.650 | Likely Pathogenic | Likely Benign | 0.227 | Likely Benign | -2.20 | Neutral | 0.991 | Probably Damaging | 0.985 | Probably Damaging | 3.97 | Benign | 0.00 | Affected | 0.1249 | 0.0846 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2332A>C | N778H 2D AIThe SynGAP1 missense variant N778H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for this variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.494003 | Structured | 0.853922 | Binding | 0.288 | 0.887 | 0.500 | -4.761 | Likely Benign | 0.172 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -1.62 | Neutral | 0.991 | Probably Damaging | 0.980 | Probably Damaging | 4.17 | Benign | 0.04 | Affected | 0.1400 | 0.7220 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||||||||||
| c.2332A>G | N778D 2D AIThe SynGAP1 missense variant N778D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.494003 | Structured | 0.853922 | Binding | 0.288 | 0.887 | 0.500 | -4.838 | Likely Benign | 0.446 | Ambiguous | Likely Benign | 0.093 | Likely Benign | -0.86 | Neutral | 0.843 | Possibly Damaging | 0.893 | Possibly Damaging | 4.21 | Benign | 0.15 | Tolerated | 0.1896 | 0.4528 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2332A>T | N778Y 2D AIThe SynGAP1 missense variant N778Y is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT). AlphaMissense‑Default is uncertain, while the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Taken together, the preponderance of evidence from both general and high‑accuracy predictors points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.494003 | Structured | 0.853922 | Binding | 0.288 | 0.887 | 0.500 | -5.723 | Likely Benign | 0.421 | Ambiguous | Likely Benign | 0.175 | Likely Benign | -2.48 | Neutral | 0.991 | Probably Damaging | 0.980 | Probably Damaging | 4.16 | Benign | 0.02 | Affected | 0.0589 | 0.6139 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.2333A>C | N778T 2D AIThe SynGAP1 missense variant N778T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.494003 | Structured | 0.853922 | Binding | 0.288 | 0.887 | 0.500 | -3.820 | Likely Benign | 0.222 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -1.52 | Neutral | 0.925 | Possibly Damaging | 0.932 | Probably Damaging | 4.23 | Benign | 0.09 | Tolerated | 0.1441 | 0.7395 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||||||||||
| c.2333A>G | N778S 2D AIThe SynGAP1 missense variant N778S is reported in gnomAD (ID 6‑33442491‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and HumVar—predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.494003 | Structured | 0.853922 | Binding | 0.288 | 0.887 | 0.500 | 6-33442491-A-G | 1 | 1.28e-6 | -2.711 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.137 | Likely Benign | -0.61 | Neutral | 0.843 | Possibly Damaging | 0.893 | Possibly Damaging | 4.32 | Benign | 0.86 | Tolerated | 3.64 | 6 | 0.4285 | 0.6890 | 1 | 1 | 2.7 | -27.03 | ||||||||||||||||||||||||||||||||||
| c.2333A>T | N778I 2D AIThe SynGAP1 missense variant N778I is reported in gnomAD (ID 6‑33442491‑A‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority of the four high‑accuracy predictors) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.494003 | Structured | 0.853922 | Binding | 0.288 | 0.887 | 0.500 | 6-33442491-A-T | -6.659 | Likely Benign | 0.622 | Likely Pathogenic | Likely Benign | 0.150 | Likely Benign | -2.48 | Neutral | 0.991 | Probably Damaging | 0.980 | Probably Damaging | 4.17 | Benign | 0.02 | Affected | 3.64 | 6 | 0.0628 | 0.6128 | -3 | -2 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||
| c.2334C>A | N778K 2D AIThe SynGAP1 missense variant N778K is catalogued in gnomAD (ID 6‑33442492‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, SIFT, ESM1b, and FATHMM; pathogenic predictions from PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome, reflecting the majority of benign calls. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, and Foldetta data are not available. Overall, the majority of evidence points toward a benign effect, and this is consistent with the lack of a ClinVar pathogenic classification. Therefore, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.494003 | Structured | 0.853922 | Binding | 0.288 | 0.887 | 0.500 | 6-33442492-C-A | -6.768 | Likely Benign | 0.798 | Likely Pathogenic | Ambiguous | 0.113 | Likely Benign | -1.57 | Neutral | 0.925 | Possibly Damaging | 0.932 | Probably Damaging | 4.27 | Benign | 0.18 | Tolerated | 3.64 | 6 | 0.2037 | 0.5883 | 0 | 1 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||||||||
| c.2334C>G | N778K 2D AIThe SynGAP1 missense variant N778K has no ClinVar record and is not listed in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yielding a “Likely Benign” classification. AlphaMissense‑Optimized returns an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available output for this variant. Overall, the preponderance of evidence points to a benign effect. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.494003 | Structured | 0.853922 | Binding | 0.288 | 0.887 | 0.500 | -6.768 | Likely Benign | 0.798 | Likely Pathogenic | Ambiguous | 0.114 | Likely Benign | -1.57 | Neutral | 0.925 | Possibly Damaging | 0.932 | Probably Damaging | 4.27 | Benign | 0.18 | Tolerated | 3.64 | 6 | 0.2037 | 0.5883 | 0 | 1 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||
| c.2335A>C | S779R 2D AIThe SynGAP1 missense variant S779R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain, and both the SGM Consensus and Foldetta (which would combine FoldX‑MD and Rosetta outputs) are unavailable. Consequently, the overall prediction is ambiguous. The variant is most likely benign based on the balance of evidence, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.044 | Likely Benign | 0.950 | Likely Pathogenic | Ambiguous | 0.103 | Likely Benign | -0.74 | Neutral | 0.846 | Possibly Damaging | 0.627 | Possibly Damaging | 2.29 | Pathogenic | 0.12 | Tolerated | 0.0961 | 0.4046 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.2335A>G | S779G 2D AIThe SynGAP1 missense variant S779G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.304 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.103 | Likely Benign | 0.38 | Neutral | 0.393 | Benign | 0.324 | Benign | 2.65 | Benign | 0.53 | Tolerated | 0.2894 | 0.5087 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2335A>T | S779C 2D AIThe SynGAP1 missense variant S779C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for this variant. This conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -6.375 | Likely Benign | 0.196 | Likely Benign | Likely Benign | 0.230 | Likely Benign | -1.88 | Neutral | 0.992 | Probably Damaging | 0.905 | Possibly Damaging | 2.28 | Pathogenic | 0.05 | Affected | 0.1177 | 0.6350 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2336G>A | S779N 2D AIThe SynGAP1 missense variant S779N is listed in gnomAD (ID 6‑33442494‑G‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. A high‑accuracy consensus (SGM Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a benign majority. The AlphaMissense‑Optimized score is benign, and no Foldetta stability assessment is available. Taken together, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | 6-33442494-G-A | -4.880 | Likely Benign | 0.384 | Ambiguous | Likely Benign | 0.087 | Likely Benign | -0.75 | Neutral | 0.021 | Benign | 0.026 | Benign | 2.30 | Pathogenic | 0.23 | Tolerated | 3.64 | 6 | 0.1327 | 0.4984 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||||
| c.2336G>C | S779T 2D AIThe SynGAP1 missense variant S779T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as tolerated or benign. Only two tools—polyPhen‑2 HumDiv and FATHMM—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta results are not available for this variant. Overall, the consensus of available predictions points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.458 | Likely Benign | 0.144 | Likely Benign | Likely Benign | 0.132 | Likely Benign | -0.71 | Neutral | 0.611 | Possibly Damaging | 0.396 | Benign | 2.34 | Pathogenic | 0.79 | Tolerated | 0.1491 | 0.6392 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2336G>T | S779I 2D AIThe SynGAP1 missense variant S779I is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy methods give a benign call from AlphaMissense‑Optimized; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of tools (five pathogenic vs. four benign) suggest a pathogenic impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -6.261 | Likely Benign | 0.578 | Likely Pathogenic | Likely Benign | 0.198 | Likely Benign | -2.10 | Neutral | 0.918 | Possibly Damaging | 0.827 | Possibly Damaging | 2.28 | Pathogenic | 0.05 | Affected | 0.1046 | 0.6248 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2337C>A | S779R 2D AIThe SynGAP1 missense variant S779R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain, and both the SGM Consensus and Foldetta (which would combine FoldX‑MD and Rosetta outputs) are unavailable. Consequently, the overall prediction is ambiguous. The variant is most likely benign based on the balance of evidence, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.044 | Likely Benign | 0.950 | Likely Pathogenic | Ambiguous | 0.119 | Likely Benign | -0.74 | Neutral | 0.846 | Possibly Damaging | 0.627 | Possibly Damaging | 2.29 | Pathogenic | 0.12 | Tolerated | 0.0961 | 0.4046 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.2337C>G | S779R 2D AIThe SynGAP1 missense variant S779R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and ESM1b, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑to‑2 split. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain, and both the SGM Consensus and Foldetta (which would combine FoldX‑MD and Rosetta outputs) are unavailable. Consequently, the overall prediction is ambiguous. The variant is most likely benign based on the balance of evidence, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -4.044 | Likely Benign | 0.950 | Likely Pathogenic | Ambiguous | 0.124 | Likely Benign | -0.74 | Neutral | 0.846 | Possibly Damaging | 0.627 | Possibly Damaging | 2.29 | Pathogenic | 0.12 | Tolerated | 0.0961 | 0.4046 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||||
| c.2338T>A | S780T 2D AIThe SynGAP1 missense variant S780T is reported in ClinVar as “Not submitted” and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, while Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.812415 | Binding | 0.283 | 0.883 | 0.500 | -4.911 | Likely Benign | 0.157 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.63 | Neutral | 0.951 | Possibly Damaging | 0.614 | Possibly Damaging | 2.66 | Benign | 0.84 | Tolerated | 0.1445 | 0.6631 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2338T>C | S780P 2D AIThe SynGAP1 missense variant S780P is reported in gnomAD (ID 6‑33442890‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.812415 | Binding | 0.283 | 0.883 | 0.500 | 6-33442890-T-C | 1 | 6.22e-7 | -6.055 | Likely Benign | 0.234 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -1.11 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 2.64 | Benign | 0.30 | Tolerated | 3.64 | 6 | 0.2086 | 0.6171 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||
| c.2338T>G | S780A 2D AIThe SynGAP1 missense variant S780A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S780A is most likely benign, and this conclusion is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.812415 | Binding | 0.283 | 0.883 | 0.500 | -5.627 | Likely Benign | 0.164 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -0.40 | Neutral | 0.798 | Possibly Damaging | 0.340 | Benign | 2.69 | Benign | 0.74 | Tolerated | 0.4936 | 0.5522 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2339C>A | S780Y 2D AIThe SynGAP1 missense variant S780Y is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default; ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.812415 | Binding | 0.283 | 0.883 | 0.500 | -7.682 | In-Between | 0.656 | Likely Pathogenic | Likely Benign | 0.091 | Likely Benign | -1.71 | Neutral | 0.995 | Probably Damaging | 0.925 | Probably Damaging | 2.61 | Benign | 0.11 | Tolerated | 0.0810 | 0.6428 | -3 | -2 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||||||
| c.2339C>G | S780C 2D AIThe SynGAP1 missense variant S780C is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33442891‑C‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). No tool in the dataset predicts a pathogenic outcome; ESM1b is inconclusive and therefore treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while Foldetta results are not reported and thus unavailable. Based on the collective predictions, the variant is most likely benign, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.812415 | Binding | 0.283 | 0.883 | 0.500 | Uncertain | 4 | 6-33442891-C-G | 16 | 9.94e-6 | -7.603 | In-Between | 0.278 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.41 | Neutral | 0.065 | Benign | 0.043 | Benign | 2.59 | Benign | 0.10 | Tolerated | 3.64 | 6 | 0.1063 | 0.6581 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||
| c.2339C>T | S780F 2D AIThe SynGAP1 missense variant S780F is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic) and therefore unavailable as evidence. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is also unavailable for this variant. Overall, the majority of available predictions (five benign vs. four pathogenic) lean toward a benign impact. There is no ClinVar annotation to contradict this assessment, so the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.812415 | Binding | 0.283 | 0.883 | 0.500 | -8.055 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | 0.104 | Likely Benign | -1.42 | Neutral | 0.995 | Probably Damaging | 0.925 | Probably Damaging | 2.61 | Benign | 0.10 | Tolerated | 0.0768 | 0.6706 | -3 | -2 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||||||
| c.233G>A | R78H 2D AIThe SynGAP1 R78H missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.448183 | Uncertain | 0.304 | 0.866 | 0.500 | -4.851 | Likely Benign | 0.250 | Likely Benign | Likely Benign | 0.070 | Likely Benign | -1.38 | Neutral | 0.908 | Possibly Damaging | 0.108 | Benign | 3.83 | Benign | 0.00 | Affected | 0.2453 | 0.1601 | 2 | 0 | 1.3 | -19.05 | |||||||||||||||||||||||||||||||||||||||
| c.233G>C | R78P 2D AIThe SynGAP1 missense variant R78P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R78P, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.448183 | Uncertain | 0.304 | 0.866 | 0.500 | -4.049 | Likely Benign | 0.611 | Likely Pathogenic | Likely Benign | 0.115 | Likely Benign | -1.72 | Neutral | 0.817 | Possibly Damaging | 0.123 | Benign | 3.82 | Benign | 0.00 | Affected | 0.2083 | 0.3750 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.233G>T | R78L 2D AIThe SynGAP1 missense variant R78L is listed in ClinVar (ID 3390541.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.448183 | Uncertain | 0.304 | 0.866 | 0.500 | Uncertain | 1 | -3.389 | Likely Benign | 0.635 | Likely Pathogenic | Likely Benign | 0.062 | Likely Benign | -1.59 | Neutral | 0.385 | Benign | 0.021 | Benign | 3.84 | Benign | 0.00 | Affected | 0.1445 | 0.4276 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||
| c.2341A>C | M781L 2D AIThe SynGAP1 missense variant M781L is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | -2.334 | Likely Benign | 0.140 | Likely Benign | Likely Benign | 0.143 | Likely Benign | -0.41 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.83 | Benign | 0.88 | Tolerated | 0.1530 | 0.3669 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2341A>G | M781V 2D AIThe SynGAP1 missense variant M781V is listed in ClinVar (ID 4746135) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33442893‑A‑G). All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic prediction. High‑accuracy assessments corroborate this benign profile: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the computational evidence strongly supports a benign effect, which is consistent with the ClinVar “Uncertain” status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | Uncertain | 1 | 6-33442893-A-G | 4 | 2.48e-6 | -2.624 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.174 | Likely Benign | 0.00 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.03 | Benign | 0.90 | Tolerated | 3.64 | 6 | 0.3081 | 0.2705 | 1 | 2 | 2.3 | -32.06 | ||||||||||||||||||||||||||||||||
| c.2341A>T | M781L 2D AIThe SynGAP1 missense variant M781L is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this benign classification: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign status. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this prediction is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | -2.334 | Likely Benign | 0.140 | Likely Benign | Likely Benign | 0.143 | Likely Benign | -0.41 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.83 | Benign | 0.88 | Tolerated | 0.1530 | 0.3669 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2342T>A | M781K 2D AIThe SynGAP1 missense variant M781K is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | -6.321 | Likely Benign | 0.734 | Likely Pathogenic | Likely Benign | 0.258 | Likely Benign | -1.47 | Neutral | 0.138 | Benign | 0.150 | Benign | 2.72 | Benign | 0.20 | Tolerated | 0.1550 | 0.0688 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.2342T>C | M781T 2D AIThe SynGAP1 missense variant M781T is catalogued in gnomAD (ID 6‑33442894‑T‑C) and has no ClinVar entry. Across the evaluated in‑silico tools, every predictor classifies the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool reports a pathogenic or likely pathogenic outcome. The high‑accuracy consensus methods reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability predictor, has no available result for this variant. Based on the unanimous benign predictions and the lack of any ClinVar pathogenic annotation, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | 6-33442894-T-C | 1 | 6.21e-7 | -3.774 | Likely Benign | 0.310 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -0.86 | Neutral | 0.015 | Benign | 0.037 | Benign | 2.75 | Benign | 0.59 | Tolerated | 3.64 | 6 | 0.2267 | 0.1599 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.2342T>G | M781R 2D AIThe SynGAP1 missense variant M781R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | -4.990 | Likely Benign | 0.697 | Likely Pathogenic | Likely Benign | 0.265 | Likely Benign | -1.72 | Neutral | 0.327 | Benign | 0.206 | Benign | 2.71 | Benign | 0.14 | Tolerated | 0.1685 | 0.0837 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||||||||||||
| c.2343G>A | M781I 2D AIThe SynGAP1 missense variant M781I is listed in ClinVar (ID 2802065.0) as Benign and is not reported in gnomAD. All available in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the predictions strongly support a benign effect, consistent with the ClinVar designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | Benign | 1 | -2.484 | Likely Benign | 0.323 | Likely Benign | Likely Benign | 0.101 | Likely Benign | 0.05 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.89 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1405 | 0.2793 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||
| c.2343G>C | M781I 2D AIThe SynGAP1 missense variant M781I is catalogued in gnomAD (ID 6‑33442895‑G‑C) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant, so its status is unavailable. Overall, the consensus of all available predictions is benign, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | 6-33442895-G-C | 1 | 6.21e-7 | -2.484 | Likely Benign | 0.323 | Likely Benign | Likely Benign | 0.101 | Likely Benign | 0.05 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.89 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1405 | 0.2793 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||
| c.2343G>T | M781I 2D AIThe SynGAP1 missense variant M781I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the evidence strongly suggests that M781I is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | -2.484 | Likely Benign | 0.323 | Likely Benign | Likely Benign | 0.101 | Likely Benign | 0.05 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.89 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1405 | 0.2793 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||
| c.2344G>A | D782N 2D AIThe SynGAP1 missense variant D782N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs four benign) lean toward a pathogenic impact. This conclusion does not contradict ClinVar status, as the variant has no existing ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -6.857 | Likely Benign | 0.766 | Likely Pathogenic | Likely Benign | 0.131 | Likely Benign | -2.26 | Neutral | 0.995 | Probably Damaging | 0.950 | Probably Damaging | 1.98 | Pathogenic | 0.02 | Affected | 0.1167 | 0.6884 | 2 | 1 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2344G>C | D782H 2D AIThe SynGAP1 missense variant D782H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). Only REVEL predicts a benign outcome, while AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show the SGM‑Consensus as Likely Pathogenic, whereas AlphaMissense‑Optimized remains inconclusive and Foldetta data are unavailable. Taken together, the majority of evidence supports a pathogenic interpretation, and this is consistent with the absence of a ClinVar assertion. Therefore, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -8.528 | Likely Pathogenic | 0.937 | Likely Pathogenic | Ambiguous | 0.311 | Likely Benign | -2.63 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.1333 | 0.7286 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.2344G>T | D782Y 2D AIThe SynGAP1 missense variant D782Y is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools largely converge on a deleterious effect: pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default, while only REVEL predicts a benign outcome. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is classified as Likely Pathogenic. Foldetta, a protein‑folding stability predictor that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -8.785 | Likely Pathogenic | 0.919 | Likely Pathogenic | Ambiguous | 0.382 | Likely Benign | -3.75 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 1.91 | Pathogenic | 0.00 | Affected | 0.0559 | 0.6202 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2345A>C | D782A 2D AIThe SynGAP1 missense variant D782A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all classify the variant as damaging. Uncertain predictions come from ESM1b and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, reports the variant as Likely Pathogenic, while AlphaMissense‑Optimized remains uncertain and Foldetta results are unavailable. Taken together, the preponderance of evidence from multiple in silico predictors and the SGM‑Consensus suggests that D782A is most likely pathogenic. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -7.054 | In-Between | 0.892 | Likely Pathogenic | Ambiguous | 0.345 | Likely Benign | -3.33 | Deleterious | 0.990 | Probably Damaging | 0.932 | Probably Damaging | 1.95 | Pathogenic | 0.01 | Affected | 0.3819 | 0.6121 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2345A>G | D782G 2D AIThe SynGAP1 missense variant D782G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic effect: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Pathogenic” verdict (3 pathogenic vs. 1 benign). AlphaMissense‑Optimized is uncertain, and Foldetta results are unavailable. Based on the overall pattern of predictions, the variant is most likely pathogenic, and this assessment does not contradict any existing ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -6.811 | Likely Benign | 0.858 | Likely Pathogenic | Ambiguous | 0.291 | Likely Benign | -3.27 | Deleterious | 0.995 | Probably Damaging | 0.950 | Probably Damaging | 1.95 | Pathogenic | 0.02 | Affected | 0.3816 | 0.6318 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2345A>T | D782V 2D AIThe SynGAP1 missense variant D782V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (which reports “Likely Pathogenic”). The high‑accuracy AlphaMissense‑Optimized tool yields an uncertain result, and the Foldetta stability assessment is unavailable. Overall, the consensus of the available predictions strongly favors a pathogenic effect for D782V. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -8.250 | Likely Pathogenic | 0.931 | Likely Pathogenic | Ambiguous | 0.462 | Likely Benign | -3.59 | Deleterious | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.0803 | 0.6477 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2346C>A | D782E 2D AIThe SynGAP1 missense variant D782E is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -4.447 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.127 | Likely Benign | -1.75 | Neutral | 0.561 | Possibly Damaging | 0.207 | Benign | 2.14 | Pathogenic | 0.01 | Affected | 0.1351 | 0.7036 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2346C>G | D782E 2D AIThe SynGAP1 missense variant D782E is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a majority benign vote (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.768342 | Binding | 0.285 | 0.883 | 0.625 | -4.447 | Likely Benign | 0.486 | Ambiguous | Likely Benign | 0.127 | Likely Benign | -1.75 | Neutral | 0.561 | Possibly Damaging | 0.207 | Benign | 2.14 | Pathogenic | 0.01 | Affected | 0.1351 | 0.7036 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2347A>C | M783L 2D AIThe SynGAP1 missense variant M783L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess sequence conservation and structural impact uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the collective evidence strongly supports a benign classification, and this conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -1.915 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -0.48 | Neutral | 0.004 | Benign | 0.006 | Benign | 2.99 | Benign | 1.00 | Tolerated | 0.1727 | 0.4461 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2347A>G | M783V 2D AIThe SynGAP1 missense variant M783V is catalogued in gnomAD (ID 6‑33442899‑A‑G) and has no ClinVar entry. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta results are unavailable. Consequently, the variant is most likely benign, and this prediction does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | 6-33442899-A-G | 1 | 6.21e-7 | -3.453 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -0.96 | Neutral | 0.072 | Benign | 0.026 | Benign | 2.85 | Benign | 0.12 | Tolerated | 3.64 | 6 | 0.3444 | 0.3710 | 1 | 2 | 2.3 | -32.06 | ||||||||||||||||||||||||||||||||||
| c.2347A>T | M783L 2D AIThe SynGAP1 missense variant M783L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess sequence conservation and structural impact (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify the change as benign. No tool in the dataset predicts pathogenicity. High‑accuracy consensus methods reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability approach, has no available result for this variant. Overall, the collective evidence strongly supports a benign interpretation, and this conclusion is consistent with the lack of a ClinVar classification—there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -1.915 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -0.48 | Neutral | 0.004 | Benign | 0.006 | Benign | 2.99 | Benign | 1.00 | Tolerated | 0.1727 | 0.4461 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.2348T>A | M783K 2D AIThe SynGAP1 missense variant M783K is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence points toward a benign impact. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -4.923 | Likely Benign | 0.693 | Likely Pathogenic | Likely Benign | 0.206 | Likely Benign | -2.70 | Deleterious | 0.925 | Possibly Damaging | 0.424 | Benign | 2.66 | Benign | 0.01 | Affected | 0.1730 | 0.0912 | 0 | -1 | -5.8 | -3.02 | ||||||||||||||||||||||||||||||||||||||||
| c.2348T>C | M783T 2D AIThe SynGAP1 missense variant M783T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as tolerated or benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a damaging or pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta results are not available for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and the high‑accuracy consensus points to a benign effect, with no conflict with ClinVar reporting. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -4.064 | Likely Benign | 0.299 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -2.08 | Neutral | 0.625 | Possibly Damaging | 0.265 | Benign | 2.69 | Benign | 0.03 | Affected | 0.2217 | 0.2308 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2348T>G | M783R 2D AIThe SynGAP1 missense variant M783R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 benign vs 2 pathogenic). Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs four benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -3.849 | Likely Benign | 0.655 | Likely Pathogenic | Likely Benign | 0.208 | Likely Benign | -2.69 | Deleterious | 0.925 | Possibly Damaging | 0.529 | Possibly Damaging | 2.66 | Benign | 0.01 | Affected | 0.1815 | 0.0893 | 0 | -1 | -6.4 | 24.99 | ||||||||||||||||||||||||||||||||||||||||
| c.2349G>A | M783I 2D AIThe SynGAP1 missense variant M783I is listed in ClinVar as a benign alteration (ClinVar ID 3618151.0) and is present in the gnomAD database (gnomAD ID 6‑33442901‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a likely benign effect. The Foldetta protein‑folding stability analysis is not available for this variant. Overall, the computational evidence strongly suggests that the variant is most likely benign, in agreement with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | Benign | 1 | 6-33442901-G-A | 6 | 3.72e-6 | -3.560 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.042 | Likely Benign | -0.54 | Neutral | 0.004 | Benign | 0.006 | Benign | 2.87 | Benign | 0.22 | Tolerated | 3.64 | 6 | 0.1540 | 0.3710 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||
| c.2349G>C | M783I 2D AIThe SynGAP1 missense variant M783I is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Consensus from multiple in‑silico predictors classifies the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all return benign scores, and AlphaMissense‑Optimized also predicts a benign effect. No tool in the set indicates pathogenicity. The high‑accuracy assessments corroborate this view: AlphaMissense‑Optimized reports a benign outcome, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a Likely Benign classification, and Foldetta data are unavailable. Consequently, the aggregate evidence strongly supports a benign interpretation for M783I, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -3.560 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.042 | Likely Benign | -0.54 | Neutral | 0.004 | Benign | 0.006 | Benign | 2.87 | Benign | 0.22 | Tolerated | 3.64 | 6 | 0.1540 | 0.3710 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||
| c.2349G>T | M783I 2D AIThe SynGAP1 missense variant M783I is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools uniformly indicate a benign effect. Benign calls are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts pathogenicity; AlphaMissense‑Default remains uncertain. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports a likely benign outcome. Foldetta stability analysis is unavailable, so it does not influence the assessment. Overall, the computational evidence strongly supports a benign classification for M783I, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -3.560 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.042 | Likely Benign | -0.54 | Neutral | 0.004 | Benign | 0.006 | Benign | 2.87 | Benign | 0.22 | Tolerated | 3.64 | 6 | 0.1540 | 0.3710 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||
| c.2350G>A | A784T 2D AIThe SynGAP1 missense variant A784T is listed in ClinVar (ID 962668.0) as Benign and is not reported in gnomAD. Across the available in‑silico predictors, every tool examined—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classifies the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. A Foldetta stability analysis is unavailable, so it does not influence the overall interpretation. Based on the unanimous benign predictions and the ClinVar designation, the variant is most likely benign, with no contradiction to the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.708872 | Binding | 0.314 | 0.896 | 0.625 | Benign | 1 | -3.579 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.046 | Likely Benign | 1.23 | Neutral | 0.001 | Benign | 0.006 | Benign | 2.92 | Benign | 1.00 | Tolerated | 3.64 | 6 | 0.1493 | 0.6659 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||
| c.2350G>C | A784P 2D AIThe SynGAP1 missense variant A784P is reported in gnomAD (variant ID 6-33442902‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact for A784P. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.708872 | Binding | 0.314 | 0.896 | 0.625 | 6-33442902-G-C | 4 | 2.48e-6 | -3.777 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 0.189 | Likely Benign | -0.73 | Neutral | 0.586 | Possibly Damaging | 0.396 | Benign | 2.66 | Benign | 0.19 | Tolerated | 3.64 | 6 | 0.1846 | 0.4771 | -1 | 1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||||||
| c.2350G>T | A784S 2D AIThe SynGAP1 missense variant A784S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess sequence conservation and structural impact uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the collective evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.708872 | Binding | 0.314 | 0.896 | 0.625 | -2.233 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.027 | Likely Benign | 0.41 | Neutral | 0.004 | Benign | 0.010 | Benign | 2.72 | Benign | 0.67 | Tolerated | 0.2624 | 0.5171 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2351C>A | A784D 2D AIThe SynGAP1 missense variant A784D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.708872 | Binding | 0.314 | 0.896 | 0.625 | -3.784 | Likely Benign | 0.766 | Likely Pathogenic | Likely Benign | 0.206 | Likely Benign | -1.21 | Neutral | 0.411 | Benign | 0.237 | Benign | 2.68 | Benign | 0.16 | Tolerated | 0.1824 | 0.2193 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2351C>G | A784G 2D AIThe SynGAP1 missense variant A784G is reported in gnomAD (ID 6‑33442903‑C‑G) and has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Overall, the evidence strongly suggests the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.708872 | Binding | 0.314 | 0.896 | 0.625 | 6-33442903-C-G | 1 | 6.20e-7 | -3.370 | Likely Benign | 0.158 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -1.24 | Neutral | 0.224 | Benign | 0.138 | Benign | 2.67 | Benign | 0.26 | Tolerated | 3.64 | 6 | 0.2303 | 0.4418 | 0 | 1 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.2351C>T | A784V 2D AIThe SynGAP1 missense variant A784V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess sequence conservation and structural impact uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the collective evidence strongly supports a benign classification, and this conclusion is consistent with the lack of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.708872 | Binding | 0.314 | 0.896 | 0.625 | -3.901 | Likely Benign | 0.205 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.97 | Neutral | 0.126 | Benign | 0.138 | Benign | 2.72 | Benign | 0.26 | Tolerated | 0.1150 | 0.5803 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2353C>A | R785S 2D AIThe SynGAP1 missense variant R785S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. The high‑accuracy consensus from SGM (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors pathogenicity, while the lack of a Foldetta result leaves that evidence inconclusive. Overall, the preponderance of pathogenic predictions indicates that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | -2.926 | Likely Benign | 0.886 | Likely Pathogenic | Ambiguous | 0.157 | Likely Benign | -2.93 | Deleterious | 0.980 | Probably Damaging | 0.765 | Possibly Damaging | 2.34 | Pathogenic | 0.01 | Affected | 0.3523 | 0.3800 | 0 | -1 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||||||||
| c.2353C>G | R785G 2D AIThe SynGAP1 missense variant R785G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome. High‑accuracy assessments further reveal that AlphaMissense‑Optimized predicts a benign effect, while the SGM‑Consensus again suggests pathogenicity; Foldetta stability analysis is not available for this variant. Overall, the majority of predictions lean toward pathogenicity, and this is consistent with the SGM‑Consensus result. Because the variant is not present in ClinVar, there is no existing clinical classification to contradict; thus, based on the available computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | -3.684 | Likely Benign | 0.697 | Likely Pathogenic | Likely Benign | 0.190 | Likely Benign | -3.44 | Deleterious | 0.980 | Probably Damaging | 0.818 | Possibly Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.3695 | 0.3686 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2353C>T | R785C 2D AIThe SynGAP1 R785C missense variant is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33442905‑C‑T). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—indicates a likely pathogenic outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points toward a pathogenic impact, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | Uncertain | 1 | 6-33442905-C-T | 29 | 1.80e-5 | -5.887 | Likely Benign | 0.662 | Likely Pathogenic | Likely Benign | 0.126 | Likely Benign | -5.06 | Deleterious | 0.144 | Benign | 0.046 | Benign | 2.22 | Pathogenic | 0.00 | Affected | 3.64 | 6 | 0.3775 | 0.3530 | -4 | -3 | 7.0 | -53.05 | |||||||||||||||||||||||||||||||
| c.2354G>A | R785H 2D AIThe SynGAP1 R785H missense variant (ClinVar ID 2321588.0) is listed as “Uncertain” in ClinVar and is present in gnomAD (ID 6‑33442906‑G‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, whereas the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a pathogenic outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, does not provide a result for this variant. Overall, the majority of computational predictions (five pathogenic versus three benign) lean toward a pathogenic interpretation. Thus, the variant is most likely pathogenic based on current predictions, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | Uncertain | 2 | 6-33442906-G-A | 4 | 2.50e-6 | -4.782 | Likely Benign | 0.388 | Ambiguous | Likely Benign | 0.129 | Likely Benign | -2.61 | Deleterious | 0.999 | Probably Damaging | 0.947 | Probably Damaging | 2.25 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.3260 | 0.1589 | 2 | 0 | 1.3 | -19.05 | ||||||||||||||||||||||||||||||||
| c.2354G>C | R785P 2D AIThe SynGAP1 missense variant R785P is catalogued in gnomAD (6‑33442906‑G‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as Benign, while the SGM‑Consensus remains Likely Pathogenic; Foldetta results are unavailable. Overall, the balance of evidence, with seven pathogenic versus three benign predictions and a pathogenic consensus from SGM, indicates that R785P is most likely pathogenic. This conclusion does not contradict ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | 6-33442906-G-C | -3.603 | Likely Benign | 0.721 | Likely Pathogenic | Likely Benign | 0.203 | Likely Benign | -4.12 | Deleterious | 0.998 | Probably Damaging | 0.958 | Probably Damaging | 2.24 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.2370 | 0.4485 | -2 | 0 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||
| c.2354G>T | R785L 2D AIThe SynGAP1 missense variant R785L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; Foldetta results are unavailable. Overall, the balance of evidence leans toward a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.859585 | Disordered | 0.681730 | Binding | 0.325 | 0.896 | 0.625 | -4.457 | Likely Benign | 0.699 | Likely Pathogenic | Likely Benign | 0.158 | Likely Benign | -4.43 | Deleterious | 0.960 | Probably Damaging | 0.627 | Possibly Damaging | 2.26 | Pathogenic | 0.01 | Affected | 0.1993 | 0.4539 | -3 | -2 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||||||
| c.2356C>A | L786I 2D AIThe SynGAP1 missense variant L786I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence, especially from the high‑accuracy methods, points to a benign impact. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | -5.464 | Likely Benign | 0.215 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -1.12 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 0.1056 | 0.4199 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||||||||||
| c.2356C>G | L786V 2D AIThe SynGAP1 missense variant L786V is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict the ClinVar status, which currently has no classification for L786V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | -4.607 | Likely Benign | 0.310 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -1.66 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.00 | Pathogenic | 0.00 | Affected | 0.1683 | 0.4049 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.2356C>T | L786F 2D AIThe SynGAP1 missense variant L786F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; Foldetta results are unavailable. Overall, the balance of evidence leans toward pathogenicity, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | -4.949 | Likely Benign | 0.578 | Likely Pathogenic | Likely Benign | 0.112 | Likely Benign | -2.53 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.83 | Pathogenic | 0.00 | Affected | 0.0765 | 0.3847 | 2 | 0 | -1.0 | 34.02 | ||||||||||||||||||||||||||||||||||||||
| c.2357T>A | L786H 2D AIThe SynGAP1 missense variant L786H is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. Grouping by agreement, two tools predict benign, seven predict pathogenic, and AlphaMissense‑Optimized remains uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is inconclusive, but the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | -5.892 | Likely Benign | 0.836 | Likely Pathogenic | Ambiguous | 0.168 | Likely Benign | -3.72 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.79 | Pathogenic | 0.00 | Affected | 0.1309 | 0.1214 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||
| c.2357T>C | L786P 2D AISynGAP1 missense variant L786P is reported in gnomAD (ID 6‑33442909‑T‑C) but has no ClinVar entry. Functional prediction tools show discordant results: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further highlight this split: AlphaMissense‑Optimized reports a benign effect, SGM‑Consensus confirms a likely pathogenic outcome, and Foldetta results are unavailable. Overall, the majority of conventional tools and the SGM‑Consensus support a pathogenic interpretation, while one high‑accuracy tool suggests benign. No ClinVar status is present, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | 6-33442909-T-C | -3.217 | Likely Benign | 0.656 | Likely Pathogenic | Likely Benign | 0.219 | Likely Benign | -4.06 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 1.79 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3343 | 0.1814 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.2357T>G | L786R 2D AIThe SynGAP1 missense variant L786R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. The high‑accuracy consensus from SGM (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors pathogenicity, and the lack of a Foldetta result does not alter this conclusion. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant has not yet been catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.882776 | Disordered | 0.655253 | Binding | 0.341 | 0.895 | 0.750 | -4.989 | Likely Benign | 0.842 | Likely Pathogenic | Ambiguous | 0.169 | Likely Benign | -3.07 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.79 | Pathogenic | 0.00 | Affected | 0.1403 | 0.1288 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||||
| c.2359C>A | P787T 2D AISynGAP1 missense variant P787T is listed in ClinVar as benign (ClinVar ID 862728.0) and is present in gnomAD (6‑33442911‑C‑A). Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions are made by AlphaMissense‑Default, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and the SGM‑Consensus score. The high‑accuracy AlphaMissense‑Optimized result is benign, whereas the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic effect, which contradicts the ClinVar benign classification. Thus, the variant is most likely pathogenic, contradicting the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.901269 | Disordered | 0.613211 | Binding | 0.377 | 0.899 | 0.750 | Likely Benign | 1 | 6-33442911-C-A | 17 | 1.05e-5 | -4.813 | Likely Benign | 0.603 | Likely Pathogenic | Likely Benign | 0.258 | Likely Benign | -4.40 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.46 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.1577 | 0.5629 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||
| c.2359C>G | P787A 2D AIThe SynGAP1 P787A missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas a majority (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM) predict a pathogenic impact. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta results are unavailable. Overall, the balance of evidence—five pathogenic versus three benign predictions, with the SGM Consensus supporting pathogenicity—suggests that the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.901269 | Disordered | 0.613211 | Binding | 0.377 | 0.899 | 0.750 | -4.542 | Likely Benign | 0.451 | Ambiguous | Likely Benign | 0.242 | Likely Benign | -4.23 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.48 | Pathogenic | 0.02 | Affected | 0.3425 | 0.4444 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2359C>T | P787S 2D AIThe SynGAP1 P787S variant is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33442911‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. The high‑accuracy consensus (SGM Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a pathogenic majority. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.901269 | Disordered | 0.613211 | Binding | 0.377 | 0.899 | 0.750 | Uncertain | 1 | 6-33442911-C-T | 3 | 1.86e-6 | -4.203 | Likely Benign | 0.564 | Ambiguous | Likely Benign | 0.221 | Likely Benign | -3.81 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.48 | Pathogenic | 0.02 | Affected | 3.64 | 6 | 0.3420 | 0.4675 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||
| c.235A>C | N79H 2D AIThe SynGAP1 missense variant N79H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar classification because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | -2.090 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.72 | Neutral | 0.939 | Possibly Damaging | 0.114 | Benign | 4.16 | Benign | 0.00 | Affected | 0.1247 | 0.4718 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||||||||||
| c.235A>G | N79D 2D AIThe SynGAP1 missense variant N79D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | -3.746 | Likely Benign | 0.281 | Likely Benign | Likely Benign | 0.021 | Likely Benign | -0.92 | Neutral | 0.371 | Benign | 0.018 | Benign | 4.19 | Benign | 0.00 | Affected | 0.1893 | 0.2535 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.235A>T | N79Y 2D AIThe SynGAP1 missense variant N79Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | -2.844 | Likely Benign | 0.250 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -1.35 | Neutral | 0.939 | Possibly Damaging | 0.114 | Benign | 4.13 | Benign | 0.00 | Affected | 0.0510 | 0.4389 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.2360C>A | P787H 2D AIThe SynGAP1 missense variant P787H has no ClinVar entry and is not listed in gnomAD. Prediction tools that classify the variant as benign include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default—predict it to be pathogenic. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic” (3 pathogenic votes versus 1 benign). High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; Foldetta results are unavailable. Overall, the majority of predictions lean toward pathogenicity, but this is contradicted by the benign call from AlphaMissense‑Optimized. Because ClinVar has no reported status, there is no conflict with existing clinical annotations. Thus, the variant is most likely pathogenic based on the prevailing computational evidence, though one high‑accuracy tool suggests a benign effect. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.901269 | Disordered | 0.613211 | Binding | 0.377 | 0.899 | 0.750 | -5.819 | Likely Benign | 0.691 | Likely Pathogenic | Likely Benign | 0.308 | Likely Benign | -4.96 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.44 | Pathogenic | 0.01 | Affected | 0.1790 | 0.4061 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||
| c.2360C>G | P787R 2D AIThe SynGAP1 missense variant P787R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the majority of conventional tools lean toward pathogenicity, whereas the single high‑accuracy tool indicates benign and the consensus is unresolved. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.901269 | Disordered | 0.613211 | Binding | 0.377 | 0.899 | 0.750 | -5.013 | Likely Benign | 0.784 | Likely Pathogenic | Likely Benign | 0.268 | Likely Benign | -4.41 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.50 | Benign | 0.03 | Affected | 0.1373 | 0.2741 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2360C>T | P787L 2D AIThe SynGAP1 missense variant P787L is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that classify the variant as benign include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default—predict it to be pathogenic. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; Foldetta results are unavailable. Overall, the majority of predictions (seven pathogenic vs. three benign) lean toward pathogenicity, and this conclusion is not contradicted by any ClinVar annotation. Thus, the variant is most likely pathogenic based on the current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.901269 | Disordered | 0.613211 | Binding | 0.377 | 0.899 | 0.750 | -3.924 | Likely Benign | 0.747 | Likely Pathogenic | Likely Benign | 0.256 | Likely Benign | -5.89 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.45 | Pathogenic | 0.01 | Affected | 0.2254 | 0.6034 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2362T>A | S788T 2D AIThe SynGAP1 missense variant S788T is listed in ClinVar with an uncertain significance (ClinVar ID 392728.0) and is present in the gnomAD database (gnomAD ID 6‑33442914‑T‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score, which is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN. Tools that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM‑Consensus (majority vote) also favors a benign interpretation. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence points to a benign effect, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.956248 | Disordered | 0.573557 | Binding | 0.349 | 0.895 | 0.750 | Uncertain | 2 | 6-33442914-T-A | 4 | 2.49e-6 | -4.288 | Likely Benign | 0.288 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -2.25 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 1.55 | Pathogenic | 0.02 | Affected | 3.64 | 6 | 0.1794 | 0.6339 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||
| c.2362T>C | S788P 2D AIThe SynGAP1 missense variant S788P is catalogued in gnomAD (ID 6‑33442914‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The AlphaMissense‑Default score is uncertain. A consensus derived from the SGM framework (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is unavailable for this residue. Taken together, the preponderance of evidence from standard predictors and the SGM consensus points to a likely pathogenic effect, which does not contradict any existing ClinVar annotation because none is present. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.956248 | Disordered | 0.573557 | Binding | 0.349 | 0.895 | 0.750 | 6-33442914-T-C | -1.857 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.236 | Likely Benign | -3.91 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 1.51 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.2338 | 0.5537 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||||
| c.2362T>G | S788A 2D AIThe SynGAP1 missense variant S788A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for S788A. This consensus does not contradict ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.956248 | Disordered | 0.573557 | Binding | 0.349 | 0.895 | 0.750 | -5.381 | Likely Benign | 0.255 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -2.24 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 1.59 | Pathogenic | 0.02 | Affected | 0.5079 | 0.5041 | Strenghten | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||
| c.2363C>A | S788Y 2D AIThe SynGAP1 missense variant S788Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments show that the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect, AlphaMissense‑Optimized is uncertain (treated as unavailable), and Foldetta results are not provided. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.956248 | Disordered | 0.573557 | Binding | 0.349 | 0.895 | 0.750 | -8.745 | Likely Pathogenic | 0.795 | Likely Pathogenic | Ambiguous | 0.251 | Likely Benign | -4.56 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 1.50 | Pathogenic | 0.00 | Affected | 0.0844 | 0.5551 | -3 | -2 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||||
| c.2363C>G | S788C 2D AIThe SynGAP1 missense variant S788C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and AlphaMissense‑Optimized, while five tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM) predict a pathogenic outcome. Two tools (ESM1b and AlphaMissense‑Default) return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.956248 | Disordered | 0.573557 | Binding | 0.349 | 0.895 | 0.750 | -7.935 | In-Between | 0.472 | Ambiguous | Likely Benign | 0.269 | Likely Benign | -3.90 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.50 | Pathogenic | 0.00 | Affected | 0.1393 | 0.6073 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2363C>T | S788F 2D AIThe SynGAP1 missense variant S788F is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; Foldetta stability analysis is unavailable. Overall, the preponderance of predictions points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.956248 | Disordered | 0.573557 | Binding | 0.349 | 0.895 | 0.750 | -7.870 | In-Between | 0.749 | Likely Pathogenic | Likely Benign | 0.275 | Likely Benign | -4.73 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 1.50 | Pathogenic | 0.00 | Affected | 0.0781 | 0.5824 | -3 | -2 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||||
| c.2365C>A | P789T 2D AIThe SynGAP1 P789T missense variant has no ClinVar entry and is not present in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further clarify the variant’s likely effect: AlphaMissense‑Optimized predicts a benign outcome, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that P789T is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -5.242 | Likely Benign | 0.356 | Ambiguous | Likely Benign | 0.224 | Likely Benign | -4.77 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.06 | Pathogenic | 0.00 | Affected | 0.1371 | 0.3837 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2365C>G | P789A 2D AIThe SynGAP1 P789A missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a tie (2 benign, 2 pathogenic) and is therefore inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus remains inconclusive and Foldetta data are unavailable. Overall, the majority of tools (five out of nine) predict pathogenicity, but the presence of four benign predictions and the inconclusive high‑accuracy results suggest uncertainty. The variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -4.777 | Likely Benign | 0.235 | Likely Benign | Likely Benign | 0.258 | Likely Benign | -4.92 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.11 | Pathogenic | 0.00 | Affected | 0.3120 | 0.3052 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2365C>T | P789S 2D AIThe SynGAP1 P789S variant has no ClinVar entry (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that classify it as benign include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence leans toward pathogenicity, with five tools supporting a deleterious effect versus three supporting benignity. This prediction does not contradict ClinVar status, which currently has no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -4.793 | Likely Benign | 0.409 | Ambiguous | Likely Benign | 0.221 | Likely Benign | -4.64 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.19 | Pathogenic | 0.00 | Affected | 0.3100 | 0.3436 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2366C>A | P789Q 2D AIThe SynGAP1 P789Q missense change is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the ClinVar status, which currently has no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -5.191 | Likely Benign | 0.465 | Ambiguous | Likely Benign | 0.235 | Likely Benign | -4.53 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1284 | 0.3215 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2366C>G | P789R 2D AIThe SynGAP1 missense variant P789R is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. In contrast, the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all indicate pathogenicity, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains Likely Pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -2.503 | Likely Benign | 0.668 | Likely Pathogenic | Likely Benign | 0.354 | Likely Benign | -5.04 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1353 | 0.2129 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||
| c.2366C>T | P789L 2D AIThe SynGAP1 P789L missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence (five pathogenic vs. three benign predictions, with the SGM Consensus supporting pathogenicity) indicates that P789L is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.963420 | Disordered | 0.541575 | Binding | 0.398 | 0.903 | 0.750 | -4.623 | Likely Benign | 0.457 | Ambiguous | Likely Benign | 0.303 | Likely Benign | -5.91 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1958 | 0.4866 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2368A>C | T790P 2D AIThe SynGAP1 missense variant T790P has no ClinVar entry (ClinVar status: not reported) but is present in gnomAD (ID 6‑33442920‑A‑C). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of conventional tools (5 pathogenic vs 4 benign) lean toward a pathogenic interpretation, while the single high‑accuracy tool suggests benign. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | 6-33442920-A-C | -3.564 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 0.250 | Likely Benign | -3.55 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.25 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.2147 | 0.4748 | -1 | 0 | -0.9 | -3.99 | ||||||||||||||||||||||||||||||||||||
| c.2368A>G | T790A 2D AIThe SynGAP1 T790A missense change is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant as benign, while the SGM Consensus remains inconclusive and Foldetta data are unavailable. Overall, the majority of standard predictors indicate pathogenicity, but the single high‑accuracy tool that is available suggests a benign effect, and no high‑accuracy tool provides a definitive pathogenic verdict. Consequently, the variant is most likely pathogenic according to the bulk of predictions, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | -4.337 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.167 | Likely Benign | -2.64 | Deleterious | 0.992 | Probably Damaging | 0.989 | Probably Damaging | 2.35 | Pathogenic | 0.02 | Affected | 0.4157 | 0.4207 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2368A>T | T790S 2D AIThe SynGAP1 missense variant T790S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for T790S, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | -3.914 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.83 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.39 | Pathogenic | 0.33 | Tolerated | 3.64 | 6 | 0.3416 | 0.4449 | 1 | 1 | -0.1 | -14.03 | ||||||||||||||||||||||||||||||||||||
| c.2369C>A | T790N 2D  AIThe SynGAP1 missense variant T790N is listed in ClinVar with an “Uncertain” status and is present in the gnomAD database (ID 6‑33442921‑C‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive and therefore unavailable, and Foldetta results are not reported. Overall, the majority of conventional tools (5 pathogenic vs. 4 benign) lean toward a pathogenic interpretation, while the single high‑accuracy tool suggests benign. The variant’s ClinVar status remains uncertain, so there is no contradiction with the current clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | Conflicting | 3 | 6-33442921-C-A | 69 | 4.28e-5 | -5.243 | Likely Benign | 0.276 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -2.54 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.27 | Pathogenic | 0.02 | Affected | 3.64 | 6 | 0.1446 | 0.4653 | 0 | 0 | -2.8 | 13.00 | ||||||||||||||||||||||||||||||||
| c.2369C>G | T790S 2D AIThe SynGAP1 missense variant T790S (ClinVar ID 1020340) is listed as Uncertain in ClinVar and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus, which is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN. Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | Uncertain | 2 | -3.914 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -1.83 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.39 | Pathogenic | 0.33 | Tolerated | 3.64 | 6 | 0.3416 | 0.4449 | 1 | 1 | -0.1 | -14.03 | ||||||||||||||||||||||||||||||||||
| c.2369C>T | T790I 2D AIThe SynGAP1 missense variant T790I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic. Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict the ClinVar status, which currently has no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | -3.556 | Likely Benign | 0.482 | Ambiguous | Likely Benign | 0.190 | Likely Benign | -3.08 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.28 | Pathogenic | 0.01 | Affected | 0.0987 | 0.5431 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||||||||||||
| c.236A>C | N79T 2D AIThe SynGAP1 missense variant N79T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | -3.752 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.022 | Likely Benign | -0.61 | Neutral | 0.371 | Benign | 0.018 | Benign | 4.21 | Benign | 0.00 | Affected | 0.1133 | 0.4890 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||||||||||
| c.236A>G | N79S 2D AIThe SynGAP1 missense variant N79S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | -2.311 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.017 | Likely Benign | -0.31 | Neutral | 0.113 | Benign | 0.011 | Benign | 4.29 | Benign | 0.00 | Affected | 0.3155 | 0.4740 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||||||||||
| c.236A>T | N79I 2D AIThe SynGAP1 missense variant N79I is listed in ClinVar (ID 4759645) with an uncertain significance status and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 HumDiv and SIFT predict pathogenicity. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | Uncertain | 1 | -3.958 | Likely Benign | 0.337 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -1.42 | Neutral | 0.939 | Possibly Damaging | 0.080 | Benign | 4.14 | Benign | 0.00 | Affected | 0.0572 | 0.4924 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||
| c.2371A>C | K791Q 2D AIThe SynGAP1 missense variant K791Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that K791Q is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.966441 | Disordered | 0.478670 | Uncertain | 0.356 | 0.896 | 0.875 | -3.418 | Likely Benign | 0.195 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -0.09 | Neutral | 0.802 | Possibly Damaging | 0.335 | Benign | 4.17 | Benign | 0.46 | Tolerated | 0.5322 | 0.1159 | Weaken | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||
| c.2371A>G | K791E 2D AIThe SynGAP1 missense variant K791E is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools uniformly favor a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign; Foldetta results are unavailable. Taken together, the preponderance of evidence supports a benign impact for K791E, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.966441 | Disordered | 0.478670 | Uncertain | 0.356 | 0.896 | 0.875 | -3.823 | Likely Benign | 0.465 | Ambiguous | Likely Benign | 0.053 | Likely Benign | -0.58 | Neutral | 0.451 | Benign | 0.193 | Benign | 4.22 | Benign | 0.65 | Tolerated | 0.4829 | 0.0876 | 0 | 1 | 0.4 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.2372A>C | K791T 2D AIThe SynGAP1 missense variant K791T is listed in gnomAD (ID 6‑33442924‑A‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). No tool predicts pathogenicity. The high‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence overwhelmingly supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.966441 | Disordered | 0.478670 | Uncertain | 0.356 | 0.896 | 0.875 | 6-33442924-A-C | 2 | 1.24e-6 | -1.578 | Likely Benign | 0.415 | Ambiguous | Likely Benign | 0.033 | Likely Benign | -0.92 | Neutral | 0.032 | Benign | 0.017 | Benign | 4.17 | Benign | 0.16 | Tolerated | 3.64 | 6 | 0.2734 | 0.3340 | -1 | 0 | 3.2 | -27.07 | |||||||||||||||||||||||||||||||||
| c.2372A>G | K791R 2D AIThe SynGAP1 missense variant K791R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta results are not available, so they do not influence the assessment. Overall, the preponderance of evidence supports a benign classification for K791R, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.966441 | Disordered | 0.478670 | Uncertain | 0.356 | 0.896 | 0.875 | -2.359 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.028 | Likely Benign | -0.96 | Neutral | 0.802 | Possibly Damaging | 0.249 | Benign | 4.14 | Benign | 0.50 | Tolerated | 0.5520 | 0.1223 | Weaken | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||||||
| c.2372A>T | K791M 2D AIThe SynGAP1 missense variant K791M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence, including the consensus and high‑accuracy predictions, points to a benign impact for K791M. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.966441 | Disordered | 0.478670 | Uncertain | 0.356 | 0.896 | 0.875 | -3.898 | Likely Benign | 0.653 | Likely Pathogenic | Likely Benign | 0.050 | Likely Benign | -1.12 | Neutral | 0.934 | Possibly Damaging | 0.558 | Possibly Damaging | 4.10 | Benign | 0.04 | Affected | 0.1642 | 0.4179 | 0 | -1 | 5.8 | 3.02 | ||||||||||||||||||||||||||||||||||||||
| c.2373G>C | K791N 2D AIThe SynGAP1 missense variant K791N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the majority of high‑accuracy predictions (SGM‑Consensus, AlphaMissense‑Optimized uncertain, Foldetta unavailable) lean toward a benign interpretation, with only two pathogenic calls. Thus, based on the current computational evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.966441 | Disordered | 0.478670 | Uncertain | 0.356 | 0.896 | 0.875 | -4.001 | Likely Benign | 0.794 | Likely Pathogenic | Ambiguous | 0.027 | Likely Benign | -1.26 | Neutral | 0.666 | Possibly Damaging | 0.267 | Benign | 4.14 | Benign | 0.13 | Tolerated | 0.4578 | 0.1354 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||||||||||
| c.2373G>T | K791N 2D AIThe SynGAP1 missense variant K791N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the majority of high‑accuracy predictions (including the SGM‑Consensus) indicate a benign impact, and there is no conflict with ClinVar status. Thus, based on the current computational evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.966441 | Disordered | 0.478670 | Uncertain | 0.356 | 0.896 | 0.875 | -4.001 | Likely Benign | 0.794 | Likely Pathogenic | Ambiguous | 0.027 | Likely Benign | -1.26 | Neutral | 0.666 | Possibly Damaging | 0.267 | Benign | 4.14 | Benign | 0.13 | Tolerated | 0.4578 | 0.1354 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||||||||||
| c.2374G>A | E792K 2D AIThe SynGAP1 missense variant E792K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.974374 | Disordered | 0.452261 | Uncertain | 0.352 | 0.896 | 0.875 | -4.942 | Likely Benign | 0.753 | Likely Pathogenic | Likely Benign | 0.059 | Likely Benign | -2.47 | Neutral | 0.033 | Benign | 0.017 | Benign | 3.90 | Benign | 0.01 | Affected | 0.2646 | 0.7584 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.2374G>C | E792Q 2D AIThe SynGAP1 missense variant E792Q is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” verdict, reflecting the majority of benign calls. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that E792Q is most likely benign, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.974374 | Disordered | 0.452261 | Uncertain | 0.352 | 0.896 | 0.875 | -3.782 | Likely Benign | 0.411 | Ambiguous | Likely Benign | 0.052 | Likely Benign | -1.60 | Neutral | 0.077 | Benign | 0.049 | Benign | 3.89 | Benign | 0.02 | Affected | 0.1469 | 0.7401 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||
| c.2375A>C | E792A 2D AIThe SynGAP1 missense variant E792A is catalogued in gnomAD (ID 6‑33442927‑A‑C) but has no ClinVar submission. Functional prediction tools cluster into two groups: benign (REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized) and pathogenic (PROVEAN, SIFT). The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also favors benign. Foldetta, a protein‑folding stability predictor, has no available result for this residue. Overall, the preponderance of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.974374 | Disordered | 0.452261 | Uncertain | 0.352 | 0.896 | 0.875 | 6-33442927-A-C | 1 | 6.20e-7 | -3.248 | Likely Benign | 0.515 | Ambiguous | Likely Benign | 0.060 | Likely Benign | -3.35 | Deleterious | 0.000 | Benign | 0.001 | Benign | 3.92 | Benign | 0.01 | Affected | 3.64 | 6 | 0.4586 | 0.7544 | -1 | 0 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||
| c.2375A>G | E792G 2D AIThe SynGAP1 E792G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT. AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a benign outcome (2 benign vs. 1 pathogenic vote). High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as benign, and Foldetta results are unavailable. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.974374 | Disordered | 0.452261 | Uncertain | 0.352 | 0.896 | 0.875 | -3.925 | Likely Benign | 0.353 | Ambiguous | Likely Benign | 0.037 | Likely Benign | -3.77 | Deleterious | 0.000 | Benign | 0.000 | Benign | 3.88 | Benign | 0.01 | Affected | 0.3145 | 0.6036 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.2375A>T | E792V 2D AIThe SynGAP1 E792V missense change is not listed in ClinVar and has no allele record in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM, while pathogenic predictions arise from PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence leans toward a benign effect. This conclusion is consistent with the absence of a ClinVar assertion, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.974374 | Disordered | 0.452261 | Uncertain | 0.352 | 0.896 | 0.875 | -4.643 | Likely Benign | 0.640 | Likely Pathogenic | Likely Benign | 0.072 | Likely Benign | -3.85 | Deleterious | 0.000 | Benign | 0.001 | Benign | 3.83 | Benign | 0.00 | Affected | 0.0987 | 0.7772 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2376A>C | E792D 2D AIThe SynGAP1 missense variant E792D is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. No pathogenic predictions are present. High‑accuracy tools reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise predicts likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.974374 | Disordered | 0.452261 | Uncertain | 0.352 | 0.896 | 0.875 | -3.746 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -1.06 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.97 | Benign | 0.26 | Tolerated | 0.2096 | 0.5376 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.2376A>T | E792D 2D AIThe SynGAP1 missense variant E792D is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. No pathogenic predictions are present. High‑accuracy tools reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise predicts likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.974374 | Disordered | 0.452261 | Uncertain | 0.352 | 0.896 | 0.875 | -3.746 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -1.06 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.97 | Benign | 0.26 | Tolerated | 0.2096 | 0.5376 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||
| c.2377A>C | K793Q 2D AIThe SynGAP1 missense variant K793Q is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign status. Foldetta results are unavailable, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.971072 | Disordered | 0.426071 | Uncertain | 0.344 | 0.901 | 0.875 | -2.838 | Likely Benign | 0.256 | Likely Benign | Likely Benign | 0.032 | Likely Benign | -0.83 | Neutral | 0.174 | Benign | 0.099 | Benign | 4.13 | Benign | 0.06 | Tolerated | 0.5276 | 0.1540 | Weaken | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||
| c.2377A>G | K793E 2D AIThe SynGAP1 missense variant K793E is listed in gnomAD (ID 6‑33442929‑A‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.971072 | Disordered | 0.426071 | Uncertain | 0.344 | 0.901 | 0.875 | 6-33442929-A-G | -4.233 | Likely Benign | 0.555 | Ambiguous | Likely Benign | 0.035 | Likely Benign | -1.05 | Neutral | 0.001 | Benign | 0.006 | Benign | 4.17 | Benign | 0.05 | Affected | 4.07 | 3 | 0.4670 | 0.1499 | 1 | 0 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||||
| c.2378A>C | K793T 2D AIThe SynGAP1 missense variant K793T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Only SIFT predicts a pathogenic outcome, and AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for K793T, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.971072 | Disordered | 0.426071 | Uncertain | 0.344 | 0.901 | 0.875 | -3.861 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.066 | Likely Benign | -1.47 | Neutral | 0.174 | Benign | 0.123 | Benign | 4.12 | Benign | 0.03 | Affected | 0.2756 | 0.3473 | 0 | -1 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||||||||||
| c.2378A>G | K793R 2D AIThe SynGAP1 missense variant K793R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess sequence conservation and functional impact all converge on a benign outcome: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. No tool in the dataset indicates pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign,” while Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.971072 | Disordered | 0.426071 | Uncertain | 0.344 | 0.901 | 0.875 | -2.789 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -0.29 | Neutral | 0.001 | Benign | 0.003 | Benign | 4.18 | Benign | 0.22 | Tolerated | 0.5270 | 0.1657 | Weaken | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||||||||||
| c.2378A>T | K793M 2D AIThe SynGAP1 K793M missense change is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is also benign; Foldetta results are not available. Overall, the balance of evidence, including the two high‑accuracy tools, points to a benign effect for K793M. This conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.971072 | Disordered | 0.426071 | Uncertain | 0.344 | 0.901 | 0.875 | -4.762 | Likely Benign | 0.570 | Likely Pathogenic | Likely Benign | 0.073 | Likely Benign | -1.49 | Neutral | 0.820 | Possibly Damaging | 0.601 | Possibly Damaging | 4.06 | Benign | 0.01 | Affected | 0.1674 | 0.4020 | 0 | -1 | 5.8 | 3.02 | ||||||||||||||||||||||||||||||||||||||
| c.2379G>C | K793N 2D AIThe SynGAP1 missense variant K793N is catalogued in gnomAD (6‑33442931‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify it as benign, and the SGM‑Consensus score (Likely Benign) supports this view. Only AlphaMissense‑Default predicts pathogenicity. High‑accuracy assessments further reinforce the benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that K793N is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.971072 | Disordered | 0.426071 | Uncertain | 0.344 | 0.901 | 0.875 | 6-33442931-G-C | 4 | 2.48e-6 | -5.162 | Likely Benign | 0.632 | Likely Pathogenic | Likely Benign | 0.040 | Likely Benign | 1.18 | Neutral | 0.174 | Benign | 0.135 | Benign | 4.13 | Benign | 0.47 | Tolerated | 4.07 | 3 | 0.4429 | 0.1976 | 0 | 1 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||
| c.2379G>T | K793N 2D AIThe SynGAP1 missense variant K793N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. Only AlphaMissense‑Default predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is also benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.971072 | Disordered | 0.426071 | Uncertain | 0.344 | 0.901 | 0.875 | -5.162 | Likely Benign | 0.632 | Likely Pathogenic | Likely Benign | 0.040 | Likely Benign | 1.18 | Neutral | 0.174 | Benign | 0.135 | Benign | 4.13 | Benign | 0.47 | Tolerated | 4.07 | 3 | 0.4429 | 0.1976 | 0 | 1 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||||||||
| c.237C>A | N79K 2D AIThe SynGAP1 missense variant N79K is listed in gnomAD (ID 6‑33425845‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores benign and the SGM‑Consensus indicates “Likely Benign.” No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for the variant, and this conclusion is not contradicted by any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | 6-33425845-C-A | 1 | 6.20e-7 | -2.811 | Likely Benign | 0.422 | Ambiguous | Likely Benign | 0.030 | Likely Benign | -0.91 | Neutral | 0.001 | Benign | 0.000 | Benign | 4.22 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1883 | 0.3929 | 0 | 1 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||||||
| c.237C>G | N79K 2D AIThe SynGAP1 missense variant N79K is listed in gnomAD (ID 6‑33425845‑C‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar classification to contradict this conclusion. Thus, based on current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.457064 | Uncertain | 0.290 | 0.876 | 0.375 | 6-33425845-C-G | 1 | 6.20e-7 | -2.811 | Likely Benign | 0.422 | Ambiguous | Likely Benign | 0.030 | Likely Benign | -0.91 | Neutral | 0.001 | Benign | 0.000 | Benign | 4.22 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1883 | 0.3929 | 0 | 1 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||||||
| c.2380C>A | P794T 2D AIThe SynGAP1 missense variant P794T is catalogued in gnomAD (ID 6‑33442932‑C‑A) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction. The Foldetta stability analysis is not available for this variant. Overall, the consensus of all available predictions is benign, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.979741 | Disordered | 0.408951 | Uncertain | 0.550 | 0.898 | 0.875 | 6-33442932-C-A | 1 | 6.20e-7 | -4.838 | Likely Benign | 0.060 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.34 | Neutral | 0.245 | Benign | 0.138 | Benign | 4.25 | Benign | 0.66 | Tolerated | 4.07 | 3 | 0.1739 | 0.6207 | -1 | 0 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||
| c.2380C>G | P794A 2D AIThe SynGAP1 missense variant P794A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). All evaluated in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta results are unavailable, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.979741 | Disordered | 0.408951 | Uncertain | 0.550 | 0.898 | 0.875 | -3.766 | Likely Benign | 0.056 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -0.63 | Neutral | 0.124 | Benign | 0.089 | Benign | 4.27 | Benign | 0.09 | Tolerated | 0.3692 | 0.5209 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||||||
| c.2380C>T | P794S 2D AIThe SynGAP1 missense variant P794S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are not available, so they do not influence the assessment. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.979741 | Disordered | 0.408951 | Uncertain | 0.550 | 0.898 | 0.875 | -5.086 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.21 | Neutral | 0.025 | Benign | 0.010 | Benign | 4.29 | Benign | 0.13 | Tolerated | 0.3666 | 0.5604 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||||||
| c.2381C>A | P794Q 2D AIThe SynGAP1 missense variant P794Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for P794Q, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.979741 | Disordered | 0.408951 | Uncertain | 0.550 | 0.898 | 0.875 | -4.641 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.31 | Neutral | 0.918 | Possibly Damaging | 0.601 | Possibly Damaging | 4.24 | Benign | 0.02 | Affected | 0.1581 | 0.4971 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||||
| c.2381C>G | P794R 2D AIThe SynGAP1 missense variant P794R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect for P794R, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.979741 | Disordered | 0.408951 | Uncertain | 0.550 | 0.898 | 0.875 | -5.136 | Likely Benign | 0.260 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -0.73 | Neutral | 0.918 | Possibly Damaging | 0.522 | Possibly Damaging | 4.25 | Benign | 0.02 | Affected | 0.1467 | 0.3662 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||
| c.2381C>T | P794L 2D AIThe SynGAP1 missense variant P794L is listed in ClinVar as Benign (ClinVar ID 859213.0) and is present in the gnomAD database (gnomAD ID 6‑33442933‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as benign, while Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the consensus of available predictions indicates that P794L is most likely benign, and this conclusion is consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.979741 | Disordered | 0.408951 | Uncertain | 0.550 | 0.898 | 0.875 | Benign/Likely benign | 2 | 6-33442933-C-T | 73 | 4.52e-5 | -3.808 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -0.80 | Neutral | 0.761 | Possibly Damaging | 0.321 | Benign | 4.24 | Benign | 0.03 | Affected | 4.07 | 3 | 0.2417 | 0.6733 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||
| c.2383C>A | P795T 2D AIThe SynGAP1 missense variant P795T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity, and the only uncertain result comes from ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.972450 | Disordered | 0.410339 | Uncertain | 0.457 | 0.903 | 0.875 | -7.030 | In-Between | 0.060 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.63 | Neutral | 0.036 | Benign | 0.026 | Benign | 4.26 | Benign | 0.09 | Tolerated | 0.1713 | 0.6381 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||||
| c.2383C>G | P795A 2D AIThe SynGAP1 missense variant P795A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly suggests that P795A is most likely benign, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.972450 | Disordered | 0.410339 | Uncertain | 0.457 | 0.903 | 0.875 | -5.348 | Likely Benign | 0.049 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -0.52 | Neutral | 0.016 | Benign | 0.011 | Benign | 4.32 | Benign | 0.16 | Tolerated | 0.3672 | 0.5371 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||||||
| c.2383C>T | P795S 2D AIThe SynGAP1 missense variant P795S is catalogued in gnomAD (6‑33442935‑C‑T) but has no entry in ClinVar. Across the spectrum of in‑silico predictors, every tool listed—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently scores the variant as benign. No pathogenic predictions are reported. Grouping by agreement, all available tools fall into the benign category, with no tools indicating pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. Foldetta results are not provided, so they are treated as unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.972450 | Disordered | 0.410339 | Uncertain | 0.457 | 0.903 | 0.875 | 6-33442935-C-T | 1 | 6.20e-7 | -5.846 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.23 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.30 | Benign | 0.24 | Tolerated | 4.32 | 2 | 0.3517 | 0.5766 | -1 | 1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||
| c.2384C>A | P795H 2D AIThe SynGAP1 missense variant P795H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic outcome. ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.972450 | Disordered | 0.410339 | Uncertain | 0.457 | 0.903 | 0.875 | -7.717 | In-Between | 0.121 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -0.61 | Neutral | 0.898 | Possibly Damaging | 0.477 | Possibly Damaging | 4.24 | Benign | 0.05 | Affected | 0.1947 | 0.5184 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||
| c.2384C>G | P795R 2D AIThe SynGAP1 missense variant P795R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT classifies the change as pathogenic, but this is the sole discordant call. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that P795R is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.972450 | Disordered | 0.410339 | Uncertain | 0.457 | 0.903 | 0.875 | -6.536 | Likely Benign | 0.221 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -1.02 | Neutral | 0.290 | Benign | 0.114 | Benign | 4.25 | Benign | 0.05 | Affected | 0.1430 | 0.4065 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||
| c.2384C>T | P795L 2D AIThe SynGAP1 missense variant P795L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a “Likely Benign” status. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) confirms a benign outcome. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that P795L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.972450 | Disordered | 0.410339 | Uncertain | 0.457 | 0.903 | 0.875 | -2.677 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.45 | Neutral | 0.016 | Benign | 0.010 | Benign | 4.27 | Benign | 0.04 | Affected | 0.2372 | 0.6499 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2386C>A | P796T 2D AIThe SynGAP1 P796T missense variant is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 HumDiv and SIFT predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.936162 | Disordered | 0.426363 | Uncertain | 0.427 | 0.900 | 0.875 | -5.399 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.01 | Neutral | 0.494 | Possibly Damaging | 0.236 | Benign | 4.26 | Benign | 0.04 | Affected | 0.1705 | 0.4961 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||||
| c.2386C>G | P796A 2D AIThe SynGAP1 missense variant P796A is not reported in ClinVar (ClinVar ID: None) but is present in gnomAD (gnomAD ID: 6‑33442938‑C‑G). All evaluated in silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized return benign or benign‑like scores. No tool predicts pathogenicity. Grouping by agreement, the benign‑predicting tools comprise the entire set, while no pathogenic predictions are available. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method, did not provide a result for this variant. Overall, the computational evidence strongly suggests that P796A is most likely benign, and this conclusion does not contradict the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.936162 | Disordered | 0.426363 | Uncertain | 0.427 | 0.900 | 0.875 | 6-33442938-C-G | -6.077 | Likely Benign | 0.050 | Likely Benign | Likely Benign | 0.023 | Likely Benign | -0.31 | Neutral | 0.174 | Benign | 0.063 | Benign | 4.28 | Benign | 0.08 | Tolerated | 4.32 | 2 | 0.3598 | 0.3776 | -1 | 1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||
| c.2386C>T | P796S 2D AIThe SynGAP1 missense variant P796S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) reports likely benign. Foldetta results are not available for this variant. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.936162 | Disordered | 0.426363 | Uncertain | 0.427 | 0.900 | 0.875 | -5.382 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.055 | Likely Benign | 0.05 | Neutral | 0.174 | Benign | 0.091 | Benign | 4.28 | Benign | 0.48 | Tolerated | 0.3560 | 0.4171 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||||||
| c.2387C>A | P796Q 2D AIThe SynGAP1 missense variant P796Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign, while the only tool predicting a deleterious outcome is SIFT, which labels it pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign; no Foldetta stability data are available. Overall, the consensus of the majority of predictors and the high‑accuracy tools points to a benign effect for P796Q, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar classification, as no pathogenic claim is present. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.936162 | Disordered | 0.426363 | Uncertain | 0.427 | 0.900 | 0.875 | -6.575 | Likely Benign | 0.084 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -0.15 | Neutral | 0.325 | Benign | 0.103 | Benign | 4.27 | Benign | 0.01 | Affected | 0.1562 | 0.4117 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||||
| c.2387C>G | P796R 2D AIThe SynGAP1 P796R missense change is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as “Likely Benign.” Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.936162 | Disordered | 0.426363 | Uncertain | 0.427 | 0.900 | 0.875 | -7.053 | In-Between | 0.191 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.57 | Neutral | 0.002 | Benign | 0.002 | Benign | 4.28 | Benign | 0.01 | Affected | 0.1558 | 0.2934 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||
| c.2387C>T | P796L 2D AIThe SynGAP1 P796L missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is also benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.936162 | Disordered | 0.426363 | Uncertain | 0.427 | 0.900 | 0.875 | -5.442 | Likely Benign | 0.069 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -1.06 | Neutral | 0.325 | Benign | 0.182 | Benign | 4.24 | Benign | 0.01 | Affected | 0.2328 | 0.5712 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2389C>A | P797T 2D AIThe SynGAP1 missense variant P797T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.926919 | Disordered | 0.449970 | Uncertain | 0.561 | 0.902 | 0.875 | -6.554 | Likely Benign | 0.064 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -0.15 | Neutral | 0.901 | Possibly Damaging | 0.708 | Possibly Damaging | 4.23 | Benign | 0.24 | Tolerated | 0.1733 | 0.5830 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||||
| c.2389C>G | P797A 2D AIThe SynGAP1 missense variant P797A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence indicates that P797A is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.926919 | Disordered | 0.449970 | Uncertain | 0.561 | 0.902 | 0.875 | -5.548 | Likely Benign | 0.050 | Likely Benign | Likely Benign | 0.037 | Likely Benign | -0.33 | Neutral | 0.649 | Possibly Damaging | 0.535 | Possibly Damaging | 4.25 | Benign | 0.54 | Tolerated | 0.3704 | 0.4832 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||||||
| c.2389C>T | P797S 2D AIThe SynGAP1 missense variant P797S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect for P797S, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.926919 | Disordered | 0.449970 | Uncertain | 0.561 | 0.902 | 0.875 | -6.232 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -0.14 | Neutral | 0.901 | Possibly Damaging | 0.637 | Possibly Damaging | 4.25 | Benign | 0.38 | Tolerated | 0.3634 | 0.5228 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||||||
| c.238A>C | K80Q 2D AIThe SynGAP1 missense variant K80Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.477530 | Uncertain | 0.331 | 0.873 | 0.500 | -4.475 | Likely Benign | 0.662 | Likely Pathogenic | Likely Benign | 0.064 | Likely Benign | -0.81 | Neutral | 0.414 | Benign | 0.040 | Benign | 3.93 | Benign | 0.00 | Affected | 0.4373 | 0.1132 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||||
| c.238A>G | K80E 2D AIThe SynGAP1 K80E missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy consensus (SGM‑Consensus) is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yielding a “Likely Benign” classification. AlphaMissense‑Optimized returns an uncertain result, and Foldetta (which would combine FoldX‑MD and Rosetta outputs) has no available data for this variant. Overall, the balance of evidence favors a benign impact, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for K80E. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.477530 | Uncertain | 0.331 | 0.873 | 0.500 | -5.605 | Likely Benign | 0.943 | Likely Pathogenic | Ambiguous | 0.034 | Likely Benign | -0.91 | Neutral | 0.225 | Benign | 0.027 | Benign | 3.92 | Benign | 0.00 | Affected | 0.3850 | 0.0952 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2390C>A | P797Q 2D AIThe SynGAP1 missense variant P797Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.926919 | Disordered | 0.449970 | Uncertain | 0.561 | 0.902 | 0.875 | -6.201 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.04 | Neutral | 0.940 | Possibly Damaging | 0.801 | Possibly Damaging | 4.23 | Benign | 0.15 | Tolerated | 0.1636 | 0.4787 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||||
| c.2390C>G | P797R 2D AIThe SynGAP1 missense variant P797R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports “Likely Benign.” No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome; Foldetta results are not available. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.926919 | Disordered | 0.449970 | Uncertain | 0.561 | 0.902 | 0.875 | -5.642 | Likely Benign | 0.179 | Likely Benign | Likely Benign | 0.045 | Likely Benign | 0.00 | Neutral | 0.006 | Benign | 0.026 | Benign | 4.28 | Benign | 0.20 | Tolerated | 0.1549 | 0.3085 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||
| c.2390C>T | P797L 2D AIThe SynGAP1 missense variant P797L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.926919 | Disordered | 0.449970 | Uncertain | 0.561 | 0.902 | 0.875 | -5.631 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.033 | Likely Benign | -0.66 | Neutral | 0.818 | Possibly Damaging | 0.637 | Possibly Damaging | 4.23 | Benign | 0.40 | Tolerated | 0.2550 | 0.6538 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||
| c.2392C>A | P798T 2D AIThe SynGAP1 missense variant P798T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign effect for P798T, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.871313 | Disordered | 0.492709 | Uncertain | 0.426 | 0.899 | 0.875 | -5.926 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -0.38 | Neutral | 0.901 | Possibly Damaging | 0.534 | Possibly Damaging | 4.24 | Benign | 0.00 | Affected | 0.1293 | 0.4854 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||||||
| c.2392C>G | P798A 2D AIThe SynGAP1 missense variant P798A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas only polyPhen‑2 HumDiv and SIFT predict pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports benign, and the SGM‑Consensus also indicates benign. Foldetta results are not available, so they do not influence the assessment. Overall, the consensus of available predictions indicates that P798A is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.871313 | Disordered | 0.492709 | Uncertain | 0.426 | 0.899 | 0.875 | -4.841 | Likely Benign | 0.050 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.57 | Neutral | 0.790 | Possibly Damaging | 0.353 | Benign | 4.27 | Benign | 0.00 | Affected | 0.3060 | 0.4030 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||||||
| c.2392C>T | P798S 2D AIThe SynGAP1 missense variant P798S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.871313 | Disordered | 0.492709 | Uncertain | 0.426 | 0.899 | 0.875 | -5.382 | Likely Benign | 0.065 | Likely Benign | Likely Benign | 0.058 | Likely Benign | 0.14 | Neutral | 0.818 | Possibly Damaging | 0.353 | Benign | 4.28 | Benign | 0.00 | Affected | 0.2950 | 0.4425 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||||||
| c.2393C>A | P798Q 2D AIThe SynGAP1 missense variant P798Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for P798Q, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.871313 | Disordered | 0.492709 | Uncertain | 0.426 | 0.899 | 0.875 | -5.944 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.53 | Neutral | 0.988 | Probably Damaging | 0.780 | Possibly Damaging | 4.25 | Benign | 0.00 | Affected | 0.1290 | 0.3864 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||||||||||
Found 8751 rows. Show 800 rows per page. Page 4/11 « Previous | Next »