
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain and Structure information: based on WT protein | Annotated databases | Deep learning-based pathogenicity predictions | Folding stability-based pathogenicity predictions | Sequence/structure-based pathogenicity predictions | Phase Separation | Evolutionary/physical properties | Molecular Dynamics-based analysis | DOI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | IUPred2 | ANCHOR2 | AlphaFold | MobiDB | PhosphoSitePlus | ClinVar | gnomAD | ESM1b | AlphaMissense | FoldX | Rosetta | Foldetta | PremPS | REVEL | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PSMutPred | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | |||||||||||||||||||||||||||||||||||||||||||||
| Score | Prediction | Score | Prediction | pLDDT | disorder | disorder | LTP | HTP | KL | PTM | Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | IP RF | SP RF | Prediction | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.3026A>G | E1009G 2D  AIThe SynGAP1 missense variant E1009G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. In contrast, the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM‑Consensus indicates a likely pathogenic outcome; Foldetta results are unavailable. Overall, the preponderance of evidence from standard and high‑accuracy predictors points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | -2.758 | Likely Benign | 0.610 | Likely Pathogenic | Likely Benign | 0.123 | Likely Benign | -3.06 | Deleterious | 0.961 | Probably Damaging | 0.721 | Possibly Damaging | 2.36 | Pathogenic | 0.01 | Affected | 0.2800 | 0.6003 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.3026A>T | E1009V 2D  AIThe SynGAP1 missense variant E1009V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments give an uncertain result from AlphaMissense‑Optimized, a Likely Pathogenic verdict from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and no available data from Foldetta. Overall, the majority of evidence points to a deleterious effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | -3.660 | Likely Benign | 0.815 | Likely Pathogenic | Ambiguous | 0.156 | Likely Benign | -3.81 | Deleterious | 0.998 | Probably Damaging | 0.924 | Probably Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.1111 | 0.7580 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3027G>T | E1009D 2D  AIThe SynGAP1 missense variant E1009D is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | -2.958 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -0.37 | Neutral | 0.011 | Benign | 0.017 | Benign | 2.50 | Benign | 0.30 | Tolerated | 3.77 | 5 | 0.2077 | 0.4817 | 2 | 3 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.3028T>A | F1010I 2D  AIThe SynGAP1 missense variant F1010I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.741537 | Disordered | 0.912572 | Binding | 0.286 | 0.881 | 0.625 | -3.726 | Likely Benign | 0.664 | Likely Pathogenic | Likely Benign | 0.126 | Likely Benign | -1.74 | Neutral | 0.980 | Probably Damaging | 0.783 | Possibly Damaging | 2.57 | Benign | 0.05 | Affected | 0.2405 | 0.2464 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3028T>C | F1010L 2D  AIThe SynGAP1 missense variant F1010L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a Likely Benign classification, and AlphaMissense‑Optimized is currently uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.741537 | Disordered | 0.912572 | Binding | 0.286 | 0.881 | 0.625 | -1.982 | Likely Benign | 0.949 | Likely Pathogenic | Ambiguous | 0.099 | Likely Benign | -1.51 | Neutral | 0.910 | Possibly Damaging | 0.468 | Possibly Damaging | 2.75 | Benign | 0.62 | Tolerated | 0.2538 | 0.3245 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3028T>G | F1010V 2D  AIThe SynGAP1 missense variant F1010V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable. Overall, the balance of evidence from multiple independent predictors and the consensus analysis points to a benign classification for F1010V, with no conflict with ClinVar status (which has no entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.741537 | Disordered | 0.912572 | Binding | 0.286 | 0.881 | 0.625 | -2.482 | Likely Benign | 0.582 | Likely Pathogenic | Likely Benign | 0.113 | Likely Benign | -2.10 | Neutral | 0.961 | Probably Damaging | 0.721 | Possibly Damaging | 2.58 | Benign | 0.03 | Affected | 0.2351 | 0.2403 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||||||||||||
| c.3029T>A | F1010Y 2D  AIThe SynGAP1 missense variant F1010Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.741537 | Disordered | 0.912572 | Binding | 0.286 | 0.881 | 0.625 | -3.297 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -0.96 | Neutral | 0.031 | Benign | 0.064 | Benign | 2.64 | Benign | 0.05 | Affected | 0.1651 | 0.2514 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3037T>G | S1013A 2D  AIThe SynGAP1 missense variant S1013A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.899570 | Binding | 0.308 | 0.846 | 0.625 | -3.400 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.049 | Likely Benign | -0.89 | Neutral | 0.001 | Benign | 0.002 | Benign | 2.71 | Benign | 0.30 | Tolerated | 0.4728 | 0.5131 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3073C>G | Q1025E 2D  AIThe SynGAP1 missense variant Q1025E is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only polyPhen‑2 HumDiv predicts it as pathogenic. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -3.010 | Likely Benign | 0.254 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.60 | Neutral | 0.649 | Possibly Damaging | 0.353 | Benign | 2.79 | Benign | 1.00 | Tolerated | 0.1391 | 0.2269 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3100C>G | P1034A 2D  AIThe SynGAP1 missense variant P1034A is listed in ClinVar as Benign (ClinVar ID 1901716.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome, representing the sole discordant signal. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates a benign impact. This prediction aligns with the ClinVar status, with no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | Benign | 1 | -4.174 | Likely Benign | 0.178 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -2.44 | Neutral | 0.059 | Benign | 0.061 | Benign | 2.47 | Pathogenic | 0.06 | Tolerated | 3.77 | 5 | 0.3028 | 0.5622 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||
| c.3108G>C | Q1036H 2D  AIThe SynGAP1 missense variant Q1036H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -4.189 | Likely Benign | 0.536 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.18 | Neutral | 0.977 | Probably Damaging | 0.615 | Possibly Damaging | 2.50 | Benign | 0.29 | Tolerated | 3.77 | 5 | 0.1641 | 0.4835 | 0 | 3 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||
| c.3109A>C | I1037L 2D  AIThe SynGAP1 missense variant I1037L is not reported in ClinVar and is absent from gnomAD, indicating no known clinical or population evidence. Functional prediction tools uniformly favor a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity, and the only uncertain call comes from AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Benign.” High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that I1037L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -2.563 | Likely Benign | 0.448 | Ambiguous | Likely Benign | 0.074 | Likely Benign | -0.46 | Neutral | 0.421 | Benign | 0.128 | Benign | 2.82 | Benign | 0.81 | Tolerated | 0.0939 | 0.4302 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3109A>G | I1037V 2D  AIThe SynGAP1 missense variant I1037V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that I1037V is most likely benign, and this conclusion is not contradicted by ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -2.326 | Likely Benign | 0.461 | Ambiguous | Likely Benign | 0.085 | Likely Benign | -0.09 | Neutral | 0.421 | Benign | 0.128 | Benign | 2.76 | Benign | 0.93 | Tolerated | 0.1229 | 0.3985 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3109A>T | I1037F 2D  AIThe SynGAP1 missense variant I1037F is not reported in ClinVar and has no entries in gnomAD. Consensus from multiple in‑silico predictors shows a split: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy tools further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the majority of evidence points to a benign effect, and this assessment is consistent with the absence of a ClinVar pathogenic claim. Thus, the variant is most likely benign, and this conclusion does not contradict ClinVar, which has no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -3.517 | Likely Benign | 0.653 | Likely Pathogenic | Likely Benign | 0.115 | Likely Benign | -0.97 | Neutral | 0.977 | Probably Damaging | 0.632 | Possibly Damaging | 2.71 | Benign | 0.16 | Tolerated | 0.0653 | 0.3722 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.310C>A | R104S 2D  AIThe SynGAP1 missense variant R104S has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | -2.790 | Likely Benign | 0.771 | Likely Pathogenic | Likely Benign | 0.143 | Likely Benign | -0.23 | Neutral | 0.625 | Possibly Damaging | 0.118 | Benign | 4.13 | Benign | 0.00 | Affected | 0.2490 | 0.3806 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.310C>G | R104G 2D  AIThe SynGAP1 missense variant R104G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R104G, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | -2.373 | Likely Benign | 0.452 | Ambiguous | Likely Benign | 0.109 | Likely Benign | -0.90 | Neutral | 0.835 | Possibly Damaging | 0.165 | Benign | 4.03 | Benign | 0.00 | Affected | 0.3111 | 0.3625 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3110T>A | I1037N 2D  AIThe SynGAP1 missense variant I1037N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a Likely Benign classification, while AlphaMissense‑Optimized remains uncertain. Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -3.955 | Likely Benign | 0.832 | Likely Pathogenic | Ambiguous | 0.131 | Likely Benign | 1.56 | Neutral | 0.666 | Possibly Damaging | 0.211 | Benign | 2.81 | Benign | 0.34 | Tolerated | 0.1022 | 0.1140 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3110T>G | I1037S 2D  AIThe SynGAP1 missense variant I1037S is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which classifies the variant as Likely Benign. Only AlphaMissense‑Default predicts a pathogenic outcome, while AlphaMissense‑Optimized is uncertain and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Based on the overall consensus of the majority of tools, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -2.247 | Likely Benign | 0.935 | Likely Pathogenic | Ambiguous | 0.120 | Likely Benign | 0.43 | Neutral | 0.032 | Benign | 0.017 | Benign | 2.83 | Benign | 0.27 | Tolerated | 0.2634 | 0.1110 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3111C>G | I1037M 2D  AIThe SynGAP1 missense variant I1037M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Only two tools, polyPhen‑2 HumDiv and polyPhen‑2 HumVar, predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence supports a benign classification, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -3.849 | Likely Benign | 0.333 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -0.40 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 2.71 | Benign | 0.18 | Tolerated | 0.0775 | 0.3885 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3112A>C | T1038P 2D  AIThe SynGAP1 missense variant T1038P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (both HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -2.196 | Likely Benign | 0.390 | Ambiguous | Likely Benign | 0.116 | Likely Benign | -1.10 | Neutral | 0.965 | Probably Damaging | 0.611 | Possibly Damaging | 2.66 | Benign | 0.26 | Tolerated | 0.1856 | 0.4680 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3112A>G | T1038A 2D  AIThe SynGAP1 missense variant T1038A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, whereas only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign impact, and this is consistent with the lack of ClinVar evidence, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -3.544 | Likely Benign | 0.265 | Likely Benign | Likely Benign | 0.047 | Likely Benign | -0.79 | Neutral | 0.649 | Possibly Damaging | 0.209 | Benign | 2.81 | Benign | 0.15 | Tolerated | 0.3429 | 0.3983 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3112A>T | T1038S 2D  AIThe SynGAP1 missense variant T1038S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -2.693 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -0.17 | Neutral | 0.649 | Possibly Damaging | 0.209 | Benign | 2.98 | Benign | 0.74 | Tolerated | 0.2879 | 0.4035 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3113C>A | T1038N 2D  AIThe SynGAP1 missense variant T1038N is not reported in ClinVar and has no entries in gnomAD, indicating it has not been catalogued in these databases. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the substitution as benign, while the majority‑vote consensus from SGM (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, and AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign. No Foldetta stability analysis is available, so folding‑stability evidence is lacking. Overall, the preponderance of computational evidence supports a benign classification for T1038N, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this is not contradicted by ClinVar, which has no pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -3.837 | Likely Benign | 0.390 | Ambiguous | Likely Benign | 0.054 | Likely Benign | -1.36 | Neutral | 0.818 | Possibly Damaging | 0.355 | Benign | 2.68 | Benign | 0.10 | Tolerated | 0.1184 | 0.4490 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||||||||||||
| c.3107A>T | Q1036L 2D  AIThe SynGAP1 missense variant Q1036L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors a benign outcome, with two benign votes versus one pathogenic and one uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -4.389 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.092 | Likely Benign | -2.92 | Deleterious | 0.152 | Benign | 0.045 | Benign | 2.52 | Benign | 0.01 | Affected | 0.1069 | 0.5996 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3107A>G | Q1036R 2D  AIThe SynGAP1 missense variant Q1036R is listed in ClinVar (ID 4746139) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to a likely benign classification (3 benign vs. 1 pathogenic). High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | Uncertain | 1 | -3.816 | Likely Benign | 0.596 | Likely Pathogenic | Likely Benign | 0.083 | Likely Benign | -1.32 | Neutral | 0.292 | Benign | 0.071 | Benign | 2.54 | Benign | 0.04 | Affected | 0.1710 | 0.3071 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||
| c.3100C>T | P1034S 2D  AIThe SynGAP1 missense variant P1034S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for P1034S, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -3.730 | Likely Benign | 0.262 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -2.28 | Neutral | 0.011 | Benign | 0.015 | Benign | 2.44 | Pathogenic | 0.05 | Affected | 0.3044 | 0.5853 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3101C>A | P1034H 2D  AIThe SynGAP1 missense variant P1034H is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. three benign) lean toward pathogenicity, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -4.634 | Likely Benign | 0.540 | Ambiguous | Likely Benign | 0.083 | Likely Benign | -3.17 | Deleterious | 0.938 | Possibly Damaging | 0.750 | Possibly Damaging | 2.38 | Pathogenic | 0.02 | Affected | 0.1776 | 0.5451 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3101C>G | P1034R 2D  AIThe SynGAP1 P1034R variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic; Foldetta stability analysis is unavailable. Overall, the predictions are mixed, with a slight tilt toward pathogenicity due to the SGM Consensus result and the number of pathogenic calls. The variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -3.666 | Likely Benign | 0.676 | Likely Pathogenic | Likely Benign | 0.073 | Likely Benign | -3.04 | Deleterious | 0.002 | Benign | 0.005 | Benign | 2.40 | Pathogenic | 0.02 | Affected | 0.1366 | 0.4182 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3101C>T | P1034L 2D  AIThe SynGAP1 missense variant P1034L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default is uncertain. The high‑accuracy AlphaMissense‑Optimized model classifies the variant as benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign prediction (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates that P1034L is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -4.204 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.067 | Likely Benign | -3.24 | Deleterious | 0.001 | Benign | 0.005 | Benign | 2.53 | Benign | 0.01 | Affected | 0.2267 | 0.6937 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3103C>A | P1035T 2D  AIThe SynGAP1 missense variant P1035T is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. AlphaMissense‑Default remains uncertain, and no Foldetta stability assessment is available. High‑accuracy evidence from AlphaMissense‑Optimized and the SGM‑Consensus both support a benign classification, while the absence of a Foldetta result does not alter this view. Overall, the majority of predictions indicate a benign effect, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | Uncertain | 1 | -4.447 | Likely Benign | 0.426 | Ambiguous | Likely Benign | 0.087 | Likely Benign | -0.96 | Neutral | 0.901 | Possibly Damaging | 0.537 | Possibly Damaging | 2.72 | Benign | 0.23 | Tolerated | 3.77 | 5 | 0.1628 | 0.7220 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||
| c.3103C>G | P1035A 2D AIThe SynGAP1 missense variant P1035A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome, creating a single discordant signal. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -4.293 | Likely Benign | 0.241 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.02 | Neutral | 0.481 | Possibly Damaging | 0.222 | Benign | 2.74 | Benign | 0.41 | Tolerated | 0.3188 | 0.6022 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3103C>T | P1035S 2D AIThe SynGAP1 missense variant P1035S is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Only polyPhen‑2 HumDiv flags it as pathogenic, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign; Foldetta results are unavailable. Taken together, the preponderance of evidence points to a benign impact for P1035S, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -3.678 | Likely Benign | 0.341 | Ambiguous | Likely Benign | 0.059 | Likely Benign | -0.97 | Neutral | 0.818 | Possibly Damaging | 0.355 | Benign | 2.79 | Benign | 0.28 | Tolerated | 0.3257 | 0.6253 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3104C>A | P1035H 2D AIThe SynGAP1 missense variant P1035H is not reported in ClinVar and has no entries in gnomAD. Consensus predictions from multiple in‑silico tools are mixed: benign calls come from SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -5.333 | Likely Benign | 0.608 | Likely Pathogenic | Likely Benign | 0.058 | Likely Benign | -0.76 | Neutral | 0.997 | Probably Damaging | 0.889 | Possibly Damaging | 2.68 | Benign | 0.15 | Tolerated | 0.1893 | 0.5669 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3104C>G | P1035R 2D AIThe SynGAP1 missense variant P1035R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -4.534 | Likely Benign | 0.604 | Likely Pathogenic | Likely Benign | 0.119 | Likely Benign | 1.07 | Neutral | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 2.89 | Benign | 0.93 | Tolerated | 0.1424 | 0.4201 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3104C>T | P1035L 2D AIThe SynGAP1 missense variant P1035L is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.945666 | Disordered | 0.989572 | Binding | 0.300 | 0.756 | 0.625 | -5.694 | Likely Benign | 0.675 | Likely Pathogenic | Likely Benign | 0.071 | Likely Benign | -2.11 | Neutral | 0.970 | Probably Damaging | 0.728 | Possibly Damaging | 2.70 | Benign | 0.21 | Tolerated | 0.2444 | 0.7354 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3106C>A | Q1036K 2D  AIThe SynGAP1 missense variant Q1036K is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -3.757 | Likely Benign | 0.646 | Likely Pathogenic | Likely Benign | 0.079 | Likely Benign | -1.72 | Neutral | 0.011 | Benign | 0.005 | Benign | 2.58 | Benign | 0.05 | Affected | 0.2096 | 0.5220 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3106C>G | Q1036E 2D AIThe SynGAP1 missense variant Q1036E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome. When predictions are grouped by consensus, the benign group contains nine tools, while the pathogenic group contains one (SIFT). High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable, so they do not influence the assessment. Overall, the evidence strongly suggests the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -3.797 | Likely Benign | 0.313 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -1.22 | Neutral | 0.264 | Benign | 0.062 | Benign | 2.59 | Benign | 0.03 | Affected | 0.1629 | 0.3484 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3107A>C | Q1036P 2D  AIThe SynGAP1 missense variant Q1036P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a damaging or pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple prediction algorithms and the consensus score indicates that the variant is most likely benign, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -2.491 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -2.00 | Neutral | 0.802 | Possibly Damaging | 0.238 | Benign | 2.57 | Benign | 0.01 | Affected | 0.2018 | 0.5564 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3113C>G | T1038S 2D AIThe SynGAP1 missense variant T1038S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -2.693 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.102 | Likely Benign | -0.17 | Neutral | 0.649 | Possibly Damaging | 0.209 | Benign | 2.98 | Benign | 0.74 | Tolerated | 0.2879 | 0.4035 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3113C>T | T1038I 2D  AIThe SynGAP1 T1038I missense variant has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which classifies the variant as Likely Benign. Tools that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is Uncertain, and the Foldetta protein‑folding stability assessment is unavailable. Overall, the balance of evidence leans toward a benign interpretation, with one high‑accuracy tool inconclusive and no conflicting ClinVar annotation. Thus, the variant is most likely benign, and there is no ClinVar status that contradicts this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.938133 | Disordered | 0.982911 | Binding | 0.279 | 0.794 | 0.625 | -4.552 | Likely Benign | 0.877 | Likely Pathogenic | Ambiguous | 0.093 | Likely Benign | -2.02 | Neutral | 0.990 | Probably Damaging | 0.637 | Possibly Damaging | 2.69 | Benign | 0.04 | Affected | 0.1012 | 0.5036 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||||||||||||
| c.3115A>C | I1039L 2D AIThe SynGAP1 missense variant I1039L is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the consensus of all available predictions strongly supports a benign classification, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -2.035 | Likely Benign | 0.269 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -0.14 | Neutral | 0.264 | Benign | 0.048 | Benign | 2.82 | Benign | 0.98 | Tolerated | 0.1057 | 0.4605 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3122C>T | P1041L 2D AIThe SynGAP1 missense variant P1041L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also resolves to benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact. This conclusion does not contradict ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -4.901 | Likely Benign | 0.399 | Ambiguous | Likely Benign | 0.403 | Likely Benign | -3.14 | Deleterious | 0.905 | Possibly Damaging | 0.375 | Benign | 5.46 | Benign | 1.00 | Tolerated | 0.2357 | 0.6664 | -3 | -3 | 5.4 | 16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3124C>A | Q1042K 2D AIThe SynGAP1 missense variant Q1042K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -4.331 | Likely Benign | 0.498 | Ambiguous | Likely Benign | 0.304 | Likely Benign | -1.52 | Neutral | 0.224 | Benign | 0.091 | Benign | 5.44 | Benign | 0.13 | Tolerated | 0.2338 | 0.5202 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.3124C>G | Q1042E 2D AIThe SynGAP1 missense variant Q1042E is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -4.231 | Likely Benign | 0.262 | Likely Benign | Likely Benign | 0.267 | Likely Benign | -1.12 | Neutral | 0.224 | Benign | 0.077 | Benign | 5.44 | Benign | 0.15 | Tolerated | 0.2042 | 0.3276 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3125A>C | Q1042P 2D AIThe SynGAP1 missense variant Q1042P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -2.085 | Likely Benign | 0.060 | Likely Benign | Likely Benign | 0.461 | Likely Benign | -0.73 | Neutral | 0.586 | Possibly Damaging | 0.223 | Benign | 5.42 | Benign | 0.31 | Tolerated | 0.2336 | 0.5762 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3125A>T | Q1042L 2D AIThe SynGAP1 missense variant Q1042L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -3.796 | Likely Benign | 0.203 | Likely Benign | Likely Benign | 0.338 | Likely Benign | -2.47 | Neutral | 0.369 | Benign | 0.120 | Benign | 5.47 | Benign | 0.05 | Affected | 0.1469 | 0.6276 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3126G>C | Q1042H 2D AIThe SynGAP1 missense variant Q1042H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -4.258 | Likely Benign | 0.335 | Likely Benign | Likely Benign | 0.302 | Likely Benign | -1.54 | Neutral | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 5.42 | Benign | 0.03 | Affected | 0.2207 | 0.4671 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3126G>T | Q1042H 2D AIThe SynGAP1 missense variant Q1042H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -4.258 | Likely Benign | 0.335 | Likely Benign | Likely Benign | 0.302 | Likely Benign | -1.54 | Neutral | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 5.42 | Benign | 0.03 | Affected | 0.2207 | 0.4671 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3127A>G | R1043G 2D AIThe SynGAP1 missense variant R1043G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -2.168 | Likely Benign | 0.201 | Likely Benign | Likely Benign | 0.383 | Likely Benign | -3.17 | Deleterious | 0.130 | Benign | 0.049 | Benign | 5.94 | Benign | 0.00 | Affected | 0.3179 | 0.3697 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3127A>T | R1043W 2D  AIThe SynGAP1 missense variant R1043W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction; Foldetta results are unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -6.382 | Likely Benign | 0.523 | Ambiguous | Likely Benign | 0.444 | Likely Benign | -3.43 | Deleterious | 0.971 | Probably Damaging | 0.729 | Possibly Damaging | 5.38 | Benign | 0.00 | Affected | 0.1403 | 0.3696 | 2 | -3 | 3.6 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.3128G>A | R1043K 2D  AIThe SynGAP1 missense variant R1043K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar or gnomAD entries—there is no contradiction with existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -4.038 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.376 | Likely Benign | -1.41 | Neutral | 0.069 | Benign | 0.033 | Benign | 5.39 | Benign | 0.00 | Affected | 0.5141 | 0.4070 | Weaken | 3 | 2 | 0.6 | -28.01 | ||||||||||||||||||||||||||||||||||||||
| c.3128G>C | R1043T 2D  AIThe SynGAP1 missense variant R1043T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -3.928 | Likely Benign | 0.298 | Likely Benign | Likely Benign | 0.463 | Likely Benign | -1.77 | Neutral | 0.001 | Benign | 0.003 | Benign | 5.39 | Benign | 0.00 | Affected | 0.1975 | 0.5513 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||||||||||||
| c.3128G>T | R1043M 2D  AIThe SynGAP1 missense variant R1043M is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign, while polyPhen‑2 HumDiv and SIFT predict pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus also indicates Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for R1043M, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -4.800 | Likely Benign | 0.510 | Ambiguous | Likely Benign | 0.471 | Likely Benign | -1.98 | Neutral | 0.744 | Possibly Damaging | 0.229 | Benign | 5.38 | Benign | 0.00 | Affected | 0.1982 | 0.4468 | 0 | -1 | 6.4 | -24.99 | |||||||||||||||||||||||||||||||||||||||
| c.3129G>C | R1043S 2D AIThe SynGAP1 missense variant R1043S is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools cluster into two groups: benign predictions include PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from REVEL and SIFT. AlphaMissense‑Default is uncertain, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores benign, SGM‑Consensus is likely benign, and Foldetta results are unavailable. Taken together, the majority of evidence points to a benign impact for R1043S. This conclusion is consistent with the absence of a ClinVar assertion, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978672 | Disordered | 0.954069 | Binding | 0.299 | 0.853 | 0.625 | -3.223 | Likely Benign | 0.457 | Ambiguous | Likely Benign | 0.509 | Likely Pathogenic | -2.10 | Neutral | 0.036 | Benign | 0.018 | Benign | 5.42 | Benign | 0.00 | Affected | 3.77 | 5 | 0.2727 | 0.4628 | -1 | 0 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||
| c.3122C>G | P1041R 2D AIThe SynGAP1 missense variant P1041R is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The AlphaMissense‑Default result is uncertain, and no Foldetta stability data are available, so these are treated as unavailable. Overall, six tools support a benign classification while three support pathogenicity, and the high‑accuracy AlphaMissense‑Optimized and SGM Consensus both predict benign. Therefore, the variant is most likely benign based on the current predictions, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -4.808 | Likely Benign | 0.545 | Ambiguous | Likely Benign | 0.443 | Likely Benign | -3.33 | Deleterious | 0.986 | Probably Damaging | 0.787 | Possibly Damaging | 5.46 | Benign | 0.07 | Tolerated | 0.1401 | 0.4292 | 0 | -2 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||||||
| c.3122C>A | P1041H 2D AIThe SynGAP1 missense variant P1041H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, and Foldetta’s protein‑folding stability result is unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no conflict with ClinVar status because the variant has not been reported there. Thus, based on current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -5.312 | Likely Benign | 0.377 | Ambiguous | Likely Benign | 0.476 | Likely Benign | -3.32 | Deleterious | 0.999 | Probably Damaging | 0.917 | Probably Damaging | 5.45 | Benign | 0.03 | Affected | 0.1883 | 0.5434 | 0 | -2 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||||||
| c.3115A>G | I1039V 2D AIThe SynGAP1 missense variant I1039V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -2.455 | Likely Benign | 0.164 | Likely Benign | Likely Benign | 0.060 | Likely Benign | 0.21 | Neutral | 0.264 | Benign | 0.048 | Benign | 2.76 | Benign | 0.41 | Tolerated | 0.1405 | 0.4112 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3115A>T | I1039F 2D AIThe SynGAP1 missense variant I1039F is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign interpretation: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign, while the majority of other predictors (polyPhen‑2 HumDiv and HumVar) indicate pathogenic. When predictions are grouped by agreement, the benign‑oriented tools outnumber the pathogenic ones, and the single uncertain call from AlphaMissense‑Default does not alter this balance. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Benign. Foldetta data are unavailable. Overall, the computational evidence overwhelmingly supports a benign effect, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -2.872 | Likely Benign | 0.545 | Ambiguous | Likely Benign | 0.124 | Likely Benign | -1.16 | Neutral | 0.925 | Possibly Damaging | 0.510 | Possibly Damaging | 2.68 | Benign | 0.13 | Tolerated | 0.0710 | 0.4368 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3116T>A | I1039N 2D AIThe SynGAP1 missense variant I1039N has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -3.469 | Likely Benign | 0.951 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.24 | Neutral | 0.011 | Benign | 0.010 | Benign | 2.67 | Benign | 0.02 | Affected | 0.1151 | 0.1311 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3116T>G | I1039S 2D AIThe SynGAP1 missense variant I1039S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM all classify the substitution as benign. In contrast, SIFT and AlphaMissense‑Default predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. No Foldetta stability assessment is available. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -1.688 | Likely Benign | 0.829 | Likely Pathogenic | Ambiguous | 0.171 | Likely Benign | -0.20 | Neutral | 0.032 | Benign | 0.008 | Benign | 2.76 | Benign | 0.03 | Affected | 0.2917 | 0.1294 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.3117T>G | I1039M 2D AIThe SynGAP1 missense variant I1039M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for I1039M, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -3.444 | Likely Benign | 0.292 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.38 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 2.68 | Benign | 0.16 | Tolerated | 0.0926 | 0.4392 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3118G>A | G1040S 2D  AIThe SynGAP1 missense variant G1040S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence (six benign versus four pathogenic predictions) points to a benign impact for G1040S. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -2.179 | Likely Benign | 0.307 | Likely Benign | Likely Benign | 0.653 | Likely Pathogenic | -1.81 | Neutral | 0.827 | Possibly Damaging | 0.375 | Benign | -0.74 | Pathogenic | 0.02 | Affected | 0.2332 | 0.5298 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3118G>C | G1040R 2D  AIThe SynGAP1 missense variant G1040R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Benign predictions are limited to polyPhen‑2 HumVar and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus remains pathogenic. Foldetta, a protein‑folding stability predictor, has no available result for this residue. Taken together, the majority of evidence supports a pathogenic classification, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -2.901 | Likely Benign | 0.949 | Likely Pathogenic | Ambiguous | 0.704 | Likely Pathogenic | -3.00 | Deleterious | 0.463 | Possibly Damaging | 0.194 | Benign | -0.74 | Pathogenic | 0.00 | Affected | 0.0924 | 0.4415 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3118G>T | G1040C 2D  AIThe SynGAP1 missense variant G1040C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all predict pathogenicity, while ESM1b and AlphaMissense‑Optimized predict a benign outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized reports a benign prediction, whereas the SGM‑Consensus remains Likely Pathogenic; the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence from multiple in silico tools indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -6.272 | Likely Benign | 0.620 | Likely Pathogenic | Likely Benign | 0.744 | Likely Pathogenic | -3.04 | Deleterious | 0.999 | Probably Damaging | 0.917 | Probably Damaging | -0.74 | Pathogenic | 0.00 | Affected | 0.1155 | 0.4556 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3119G>A | G1040D 2D  AIThe SynGAP1 missense variant G1040D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only ESM1b, whereas the remaining tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic impact. The high‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus (a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it likely pathogenic, and the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence indicates that G1040D is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -4.154 | Likely Benign | 0.956 | Likely Pathogenic | Likely Pathogenic | 0.761 | Likely Pathogenic | -2.82 | Deleterious | 0.986 | Probably Damaging | 0.787 | Possibly Damaging | -0.74 | Pathogenic | 0.00 | Affected | 0.1677 | 0.2042 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.3119G>C | G1040A 2D  AIThe SynGAP1 missense variant G1040A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL and FATHMM, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign prediction (2 benign vs. 1 pathogenic). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation, as the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.964893 | Disordered | 0.973805 | Binding | 0.332 | 0.816 | 0.625 | -2.625 | Likely Benign | 0.409 | Ambiguous | Likely Benign | 0.606 | Likely Pathogenic | -1.65 | Neutral | 0.114 | Benign | 0.030 | Benign | -0.74 | Pathogenic | 0.30 | Tolerated | 0.3246 | 0.5331 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.311G>C | R104P 2D  AIThe SynGAP1 missense variant R104P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | -3.184 | Likely Benign | 0.510 | Ambiguous | Likely Benign | 0.200 | Likely Benign | -0.88 | Neutral | 0.947 | Possibly Damaging | 0.410 | Benign | 4.01 | Benign | 0.00 | Affected | 0.1759 | 0.4498 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.3121C>A | P1041T 2D AIThe SynGAP1 missense variant P1041T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -5.009 | Likely Benign | 0.187 | Likely Benign | Likely Benign | 0.361 | Likely Benign | -2.45 | Neutral | 0.051 | Benign | 0.025 | Benign | 5.50 | Benign | 0.10 | Tolerated | 0.1632 | 0.6699 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3121C>G | P1041A 2D AIThe SynGAP1 missense variant P1041A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.962114 | Disordered | 0.967463 | Binding | 0.345 | 0.833 | 0.625 | -4.597 | Likely Benign | 0.157 | Likely Benign | Likely Benign | 0.331 | Likely Benign | -2.73 | Deleterious | 0.798 | Possibly Damaging | 0.283 | Benign | 5.53 | Benign | 0.24 | Tolerated | 0.3154 | 0.5939 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3130C>G | P1044A 2D AIThe SynGAP1 missense variant P1044A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.979741 | Disordered | 0.952126 | Binding | 0.331 | 0.855 | 0.750 | -3.957 | Likely Benign | 0.059 | Likely Benign | Likely Benign | 0.359 | Likely Benign | -1.07 | Neutral | 0.059 | Benign | 0.061 | Benign | 5.50 | Benign | 0.18 | Tolerated | 0.3194 | 0.5850 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.3100C>A | P1034T 2D AIThe SynGAP1 missense variant P1034T is not reported in ClinVar and has no entry in gnomAD, indicating it is not catalogued in these databases. Prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only two tools predict a pathogenic outcome: SIFT and FATHMM. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions support a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.991713 | Binding | 0.343 | 0.752 | 0.625 | -4.367 | Likely Benign | 0.285 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -2.00 | Neutral | 0.126 | Benign | 0.096 | Benign | 2.44 | Pathogenic | 0.03 | Affected | 0.1518 | 0.6620 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.3074A>C | Q1025P 2D AIThe SynGAP1 missense variant Q1025P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -2.151 | Likely Benign | 0.211 | Likely Benign | Likely Benign | 0.153 | Likely Benign | -1.22 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.72 | Benign | 0.11 | Tolerated | 0.2211 | 0.5196 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.307G>C | G103R 2D AIThe SynGAP1 missense variant G103R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.384 | Likely Benign | 0.710 | Likely Pathogenic | Likely Benign | 0.100 | Likely Benign | 0.00 | Neutral | 0.949 | Possibly Damaging | 0.708 | Possibly Damaging | 4.27 | Benign | 0.00 | Affected | 0.1198 | 0.4362 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.307G>T | G103C 2D AIThe SynGAP1 missense variant G103C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect for G103C, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -5.681 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -1.12 | Neutral | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 4.17 | Benign | 0.00 | Affected | 0.1494 | 0.3767 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.3080A>C | N1027T 2D  AIThe SynGAP1 missense variant N1027T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus also reports Likely Benign; Foldetta results are not available. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is consistent with the absence of any ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -3.604 | Likely Benign | 0.199 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.71 | Neutral | 0.481 | Possibly Damaging | 0.220 | Benign | 2.75 | Benign | 0.13 | Tolerated | 0.1239 | 0.7188 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||||||||||
| c.3080A>G | N1027S 2D  AIThe SynGAP1 missense variant N1027S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -2.443 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.35 | Neutral | 0.068 | Benign | 0.039 | Benign | 2.77 | Benign | 0.50 | Tolerated | 0.3540 | 0.6874 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||||||||||
| c.3080A>T | N1027I 2D  AIThe SynGAP1 missense variant N1027I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -5.847 | Likely Benign | 0.751 | Likely Pathogenic | Likely Benign | 0.065 | Likely Benign | -2.36 | Neutral | 0.970 | Probably Damaging | 0.726 | Possibly Damaging | 2.71 | Benign | 0.02 | Affected | 0.0677 | 0.5724 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3081C>A | N1027K 2D  AIThe SynGAP1 missense variant N1027K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus result is benign; Foldetta predictions are unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -3.177 | Likely Benign | 0.841 | Likely Pathogenic | Ambiguous | 0.063 | Likely Benign | -0.64 | Neutral | 0.481 | Possibly Damaging | 0.220 | Benign | 2.81 | Benign | 0.65 | Tolerated | 0.1808 | 0.6079 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3081C>G | N1027K 2D AIThe SynGAP1 missense variant N1027K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and AlphaMissense‑Optimized is uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -3.177 | Likely Benign | 0.841 | Likely Pathogenic | Ambiguous | 0.063 | Likely Benign | -0.64 | Neutral | 0.481 | Possibly Damaging | 0.220 | Benign | 2.81 | Benign | 0.65 | Tolerated | 0.1808 | 0.6079 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||||
| c.3082C>A | L1028M 2D  AIThe SynGAP1 missense variant L1028M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for the variant, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -4.591 | Likely Benign | 0.229 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.07 | Neutral | 0.986 | Probably Damaging | 0.825 | Possibly Damaging | 2.70 | Benign | 0.18 | Tolerated | 0.0776 | 0.3124 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3082C>G | L1028V 2D 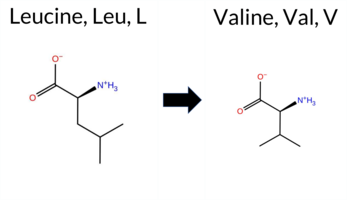 AIThe SynGAP1 missense variant L1028V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of computational evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -3.992 | Likely Benign | 0.217 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -0.68 | Neutral | 0.737 | Possibly Damaging | 0.376 | Benign | 2.74 | Benign | 0.26 | Tolerated | 0.1328 | 0.2715 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3083T>A | L1028Q 2D 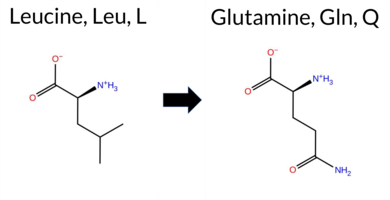 AIThe SynGAP1 missense variant L1028Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -2.718 | Likely Benign | 0.612 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -0.19 | Neutral | 0.986 | Probably Damaging | 0.825 | Possibly Damaging | 2.80 | Benign | 0.38 | Tolerated | 0.1110 | 0.1403 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3083T>C | L1028P 2D 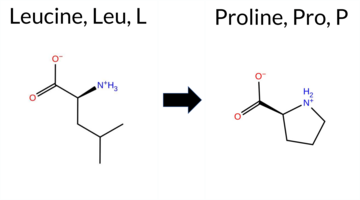 AIThe SynGAP1 missense variant L1028P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Taken together, the majority of evidence points to a benign impact for this variant, and there is no ClinVar annotation to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -3.080 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 0.215 | Likely Benign | -0.42 | Neutral | 0.004 | Benign | 0.010 | Benign | 2.70 | Benign | 0.25 | Tolerated | 0.2846 | 0.1763 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3083T>G | L1028R 2D 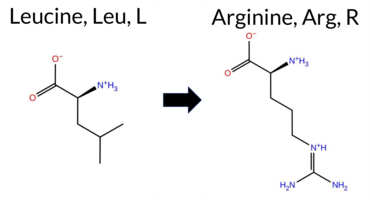 AIThe SynGAP1 missense variant L1028R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -2.204 | Likely Benign | 0.793 | Likely Pathogenic | Ambiguous | 0.167 | Likely Benign | -0.24 | Neutral | 0.960 | Probably Damaging | 0.761 | Possibly Damaging | 2.75 | Benign | 0.84 | Tolerated | 0.1328 | 0.1603 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3085C>A | Q1029K 2D AIThe SynGAP1 missense variant Q1029K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. AlphaMissense‑Default remains uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while Foldetta data is missing. Overall, the majority of evidence points to a benign impact for Q1029K, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.698 | Likely Benign | 0.516 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -1.18 | Neutral | 0.771 | Possibly Damaging | 0.482 | Possibly Damaging | 2.79 | Benign | 1.00 | Tolerated | 0.1656 | 0.4196 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||||||||||||
| c.307G>A | G103S 2D AIThe SynGAP1 missense variant G103S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.177 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -0.03 | Neutral | 0.565 | Possibly Damaging | 0.207 | Benign | 4.32 | Benign | 0.00 | Affected | 0.2816 | 0.4867 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.3079A>T | N1027Y 2D  AIThe SynGAP1 missense variant N1027Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -5.799 | Likely Benign | 0.626 | Likely Pathogenic | Likely Benign | 0.074 | Likely Benign | -2.15 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.70 | Benign | 0.03 | Affected | 0.0611 | 0.6133 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3074A>G | Q1025R 2D AIThe SynGAP1 missense variant Q1025R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (both HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -4.435 | Likely Benign | 0.480 | Ambiguous | Likely Benign | 0.101 | Likely Benign | -1.28 | Neutral | 0.818 | Possibly Damaging | 0.453 | Possibly Damaging | 2.72 | Benign | 0.10 | Tolerated | 0.1342 | 0.2656 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3074A>T | Q1025L 2D AIThe SynGAP1 missense variant Q1025L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -6.460 | Likely Benign | 0.463 | Ambiguous | Likely Benign | 0.117 | Likely Benign | -2.48 | Neutral | 0.901 | Possibly Damaging | 0.534 | Possibly Damaging | 2.70 | Benign | 0.05 | Affected | 0.0798 | 0.5497 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3075G>C | Q1025H 2D AIThe SynGAP1 missense variant Q1025H is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence supports a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -4.976 | Likely Benign | 0.405 | Ambiguous | Likely Benign | 0.051 | Likely Benign | -1.43 | Neutral | 0.014 | Benign | 0.012 | Benign | 2.68 | Benign | 0.05 | Affected | 0.1332 | 0.3852 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3075G>T | Q1025H 2D AIThe SynGAP1 missense variant Q1025H is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence supports a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -4.976 | Likely Benign | 0.405 | Ambiguous | Likely Benign | 0.051 | Likely Benign | -1.43 | Neutral | 0.014 | Benign | 0.012 | Benign | 2.68 | Benign | 0.05 | Affected | 0.1332 | 0.3852 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3076G>A | D1026N 2D  AIThe SynGAP1 D1026N variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and therefore reports a likely benign outcome. AlphaMissense‑Optimized also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -4.166 | Likely Benign | 0.608 | Likely Pathogenic | Likely Benign | 0.117 | Likely Benign | -2.07 | Neutral | 0.411 | Benign | 0.239 | Benign | 2.55 | Benign | 0.01 | Affected | 0.1240 | 0.5060 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3076G>T | D1026Y 2D  AIThe SynGAP1 missense variant D1026Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default all classify the variant as damaging. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -6.999 | Likely Benign | 0.892 | Likely Pathogenic | Ambiguous | 0.200 | Likely Benign | -3.08 | Deleterious | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 2.47 | Pathogenic | 0.00 | Affected | 0.0676 | 0.4936 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.3077A>C | D1026A 2D  AIThe SynGAP1 D1026A variant is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM, while those that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a tie, and Foldetta results are not available. Overall, the majority of standard tools favor a benign interpretation, and no ClinVar entry contradicts this assessment. Thus, the variant is most likely benign based on current predictions, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -4.211 | Likely Benign | 0.849 | Likely Pathogenic | Ambiguous | 0.070 | Likely Benign | -2.69 | Deleterious | 0.112 | Benign | 0.061 | Benign | 2.53 | Benign | 0.02 | Affected | 0.3392 | 0.5279 | 0 | -2 | 5.3 | -44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.3077A>G | D1026G 2D  AIThe SynGAP1 missense variant D1026G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign, two pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -4.125 | Likely Benign | 0.691 | Likely Pathogenic | Likely Benign | 0.098 | Likely Benign | -2.84 | Deleterious | 0.001 | Benign | 0.005 | Benign | 2.67 | Benign | 0.01 | Affected | 0.3377 | 0.5042 | 1 | -1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.3077A>T | D1026V 2D  AIThe SynGAP1 missense variant D1026V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Tools that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and therefore also indicates a likely pathogenic outcome. AlphaMissense‑Optimized is uncertain, and no Foldetta stability result is available, so it does not contribute evidence. Overall, the majority of reliable predictors classify the variant as pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -5.871 | Likely Benign | 0.900 | Likely Pathogenic | Ambiguous | 0.144 | Likely Benign | -3.13 | Deleterious | 0.004 | Benign | 0.004 | Benign | 2.48 | Pathogenic | 0.00 | Affected | 0.0957 | 0.5236 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3078C>A | D1026E 2D  AIThe SynGAP1 missense variant D1026E is listed in ClinVar (ID 4746138) as benign and is not reported in gnomAD. Across the spectrum of in‑silico predictors, every tool examined—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classifies the substitution as benign. No tool predicts pathogenicity. Grouping by agreement, all predictors fall into the benign category, with no pathogenic calls. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status remains unavailable. Overall, the computational evidence strongly supports a benign classification, fully consistent with the ClinVar designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | Benign | 1 | -3.431 | Likely Benign | 0.190 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.37 | Neutral | 0.001 | Benign | 0.005 | Benign | 2.73 | Benign | 0.56 | Tolerated | 0.1449 | 0.4783 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.3078C>G | D1026E 2D AIThe SynGAP1 missense variant D1026E is not reported in ClinVar and is absent from gnomAD. All available in silico predictors classify the change as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly supports a benign classification, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -3.431 | Likely Benign | 0.190 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -0.37 | Neutral | 0.001 | Benign | 0.005 | Benign | 2.73 | Benign | 0.56 | Tolerated | 0.1449 | 0.4783 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3079A>C | N1027H 2D  AIThe SynGAP1 missense variant N1027H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions, including the high‑accuracy tools, indicate that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -4.185 | Likely Benign | 0.193 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -1.44 | Neutral | 0.970 | Probably Damaging | 0.799 | Possibly Damaging | 2.74 | Benign | 0.07 | Tolerated | 0.1345 | 0.7176 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||||||||||
| c.3079A>G | N1027D 2D  AIThe SynGAP1 missense variant N1027D is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all score benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also classifies it as likely benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, and AlphaMissense‑Default remains uncertain. High‑accuracy tools reinforce the benign assessment: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence supports a benign classification, and this is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -2.891 | Likely Benign | 0.458 | Ambiguous | Likely Benign | 0.073 | Likely Benign | -1.27 | Neutral | 0.649 | Possibly Damaging | 0.353 | Benign | 2.74 | Benign | 0.27 | Tolerated | 0.1854 | 0.4004 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.3086A>C | Q1029P 2D AIThe SynGAP1 missense variant Q1029P is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available for this variant. Overall, the consensus of all available predictions strongly supports a benign impact, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -2.940 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.105 | Likely Benign | -1.23 | Neutral | 0.005 | Benign | 0.015 | Benign | 2.68 | Benign | 0.15 | Tolerated | 0.1966 | 0.4986 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||||||
| c.3086A>G | Q1029R 2D AIThe SynGAP1 missense variant Q1029R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (both HumDiv and HumVar) predict a pathogenic outcome. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign; Foldetta results are unavailable. Based on the preponderance of evidence from both general and high‑accuracy predictors, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.437 | Likely Benign | 0.420 | Ambiguous | Likely Benign | 0.073 | Likely Benign | -0.72 | Neutral | 0.961 | Probably Damaging | 0.677 | Possibly Damaging | 2.73 | Benign | 0.88 | Tolerated | 0.1377 | 0.2420 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||||||||||
| c.3086A>T | Q1029L 2D AIThe SynGAP1 missense variant Q1029L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a benign outcome (2 benign vs. 1 pathogenic, with one uncertain). AlphaMissense‑Default remains uncertain, and Foldetta results are unavailable. High‑accuracy predictions therefore point to a benign impact: AlphaMissense‑Optimized is benign, SGM Consensus is benign, and no Foldetta data are available. Overall, the computational evidence indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.984 | Likely Benign | 0.364 | Ambiguous | Likely Benign | 0.067 | Likely Benign | -2.65 | Deleterious | 0.891 | Possibly Damaging | 0.587 | Possibly Damaging | 2.70 | Benign | 0.16 | Tolerated | 0.0685 | 0.5866 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3092T>G | M1031R 2D  AIThe SynGAP1 missense variant M1031R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Taken together, the majority of evidence points to a benign impact for M1031R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -1.365 | Likely Benign | 0.673 | Likely Pathogenic | Likely Benign | 0.117 | Likely Benign | -0.85 | Neutral | 0.325 | Benign | 0.129 | Benign | 2.64 | Benign | 0.12 | Tolerated | 0.1368 | 0.1212 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||||||||||||
| c.3093G>A | M1031I 2D  AIThe SynGAP1 missense variant M1031I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a benign impact for M1031I. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -3.312 | Likely Benign | 0.739 | Likely Pathogenic | Likely Benign | 0.026 | Likely Benign | -0.99 | Neutral | 0.095 | Benign | 0.027 | Benign | 2.71 | Benign | 0.25 | Tolerated | 0.1128 | 0.3324 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3093G>C | M1031I 2D AIThe SynGAP1 missense variant M1031I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a benign impact for M1031I. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -3.312 | Likely Benign | 0.739 | Likely Pathogenic | Likely Benign | 0.025 | Likely Benign | -0.99 | Neutral | 0.095 | Benign | 0.027 | Benign | 2.71 | Benign | 0.25 | Tolerated | 0.1128 | 0.3324 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3093G>T | M1031I 2D AIThe SynGAP1 missense variant M1031I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the overwhelming majority of evidence points to a benign impact for M1031I. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -3.312 | Likely Benign | 0.739 | Likely Pathogenic | Likely Benign | 0.026 | Likely Benign | -0.99 | Neutral | 0.095 | Benign | 0.027 | Benign | 2.71 | Benign | 0.25 | Tolerated | 0.1128 | 0.3324 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3094C>A | L1032M 2D AIThe SynGAP1 missense variant L1032M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of any ClinVar pathogenic annotation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -4.353 | Likely Benign | 0.219 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -0.09 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 2.66 | Benign | 0.10 | Tolerated | 0.0935 | 0.4719 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3094C>G | L1032V 2D AIThe SynGAP1 missense variant L1032V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and polyPhen‑2 HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for the variant, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -4.123 | Likely Benign | 0.176 | Likely Benign | Likely Benign | 0.075 | Likely Benign | 0.16 | Neutral | 0.889 | Possibly Damaging | 0.514 | Possibly Damaging | 2.78 | Benign | 0.78 | Tolerated | 0.1637 | 0.4353 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3095T>A | L1032Q 2D AIThe SynGAP1 missense variant L1032Q is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar) and SIFT. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the aggregate evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -2.992 | Likely Benign | 0.467 | Ambiguous | Likely Benign | 0.134 | Likely Benign | -0.78 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 2.66 | Benign | 0.03 | Affected | 0.1217 | 0.1677 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3095T>C | L1032P 2D AIThe SynGAP1 missense variant L1032P has no ClinVar entry and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and the SGM‑Consensus score all classify the change as benign or likely benign. AlphaMissense‑Optimized also predicts a benign outcome, whereas SIFT uniquely flags the variant as pathogenic. The AlphaMissense‑Default assessment is uncertain, and no Foldetta stability data are available. High‑accuracy analyses reinforce the benign consensus: AlphaMissense‑Optimized reports a benign effect, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta results are missing, so they do not influence the interpretation. Overall, the majority of computational evidence supports a benign classification, which is consistent with the absence of a ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this conclusion does not contradict ClinVar status, as no ClinVar pathogenic claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -2.560 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.142 | Likely Benign | -0.71 | Neutral | 0.012 | Benign | 0.017 | Benign | 2.64 | Benign | 0.05 | Affected | 0.3024 | 0.2330 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.3095T>G | L1032R 2D AIThe SynGAP1 missense variant L1032R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -2.658 | Likely Benign | 0.707 | Likely Pathogenic | Likely Benign | 0.099 | Likely Benign | -1.03 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 2.66 | Benign | 0.02 | Affected | 0.1350 | 0.1919 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3097T>A | S1033T 2D  AIThe SynGAP1 missense variant S1033T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -3.702 | Likely Benign | 0.140 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -0.05 | Neutral | 0.568 | Possibly Damaging | 0.171 | Benign | 2.73 | Benign | 0.58 | Tolerated | 0.1379 | 0.6089 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3097T>C | S1033P 2D  AIThe SynGAP1 missense variant S1033P is not reported in ClinVar and is absent from gnomAD, indicating no documented population frequency. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are not available, so they do not influence the overall assessment. Based on the consensus of all available predictions, the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -2.046 | Likely Benign | 0.268 | Likely Benign | Likely Benign | 0.068 | Likely Benign | 0.05 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.68 | Benign | 0.27 | Tolerated | 0.1889 | 0.5486 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3097T>G | S1033A 2D AIThe SynGAP1 missense variant S1033A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions indicates that the variant is most likely benign, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -3.107 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.06 | Neutral | 0.220 | Benign | 0.085 | Benign | 2.81 | Benign | 1.00 | Tolerated | 0.4387 | 0.5282 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3098C>G | S1033C 2D  AIThe SynGAP1 missense variant S1033C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for S1033C, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -6.263 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.39 | Neutral | 0.992 | Probably Damaging | 0.750 | Possibly Damaging | 2.68 | Benign | 0.12 | Tolerated | 0.1031 | 0.5704 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.3092T>A | M1031K 2D  AIThe SynGAP1 missense variant M1031K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -1.940 | Likely Benign | 0.709 | Likely Pathogenic | Likely Benign | 0.114 | Likely Benign | -0.80 | Neutral | 0.325 | Benign | 0.098 | Benign | 2.66 | Benign | 0.18 | Tolerated | 0.1224 | 0.1412 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.3091A>T | M1031L 2D  AIThe SynGAP1 missense variant M1031L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -2.213 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.82 | Neutral | 0.044 | Benign | 0.018 | Benign | 2.84 | Benign | 0.33 | Tolerated | 0.1220 | 0.4050 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3087G>C | Q1029H 2D AIThe SynGAP1 missense variant Q1029H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar classification, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.984 | Likely Benign | 0.308 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.83 | Neutral | 0.989 | Probably Damaging | 0.879 | Possibly Damaging | 2.74 | Benign | 0.15 | Tolerated | 0.1268 | 0.4184 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3087G>T | Q1029H 2D AIThe SynGAP1 missense variant Q1029H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of the available predictions indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.984 | Likely Benign | 0.308 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.83 | Neutral | 0.989 | Probably Damaging | 0.879 | Possibly Damaging | 2.74 | Benign | 0.15 | Tolerated | 0.1268 | 0.4184 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3088C>A | H1030N 2D  AIThe SynGAP1 missense variant H1030N is not reported in ClinVar and is absent from gnomAD. Consensus prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized returns a benign prediction, and the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign classification, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -3.454 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.033 | Likely Benign | -0.88 | Neutral | 0.001 | Benign | 0.001 | Benign | 2.78 | Benign | 0.04 | Affected | 0.1553 | 0.3195 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.3088C>T | H1030Y 2D  AIThe SynGAP1 missense variant H1030Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for H1030Y, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -5.365 | Likely Benign | 0.268 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -1.73 | Neutral | 0.812 | Possibly Damaging | 0.298 | Benign | 2.73 | Benign | 0.01 | Affected | 0.0874 | 0.4236 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.3089A>C | H1030P 2D  AIThe SynGAP1 missense variant H1030P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of available predictions indicates that H1030P is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -2.185 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.154 | Likely Benign | -0.78 | Neutral | 0.812 | Possibly Damaging | 0.298 | Benign | 2.77 | Benign | 0.02 | Affected | 0.1872 | 0.4225 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3089A>G | H1030R 2D  AIThe SynGAP1 missense variant H1030R is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods likewise support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -2.312 | Likely Benign | 0.340 | Likely Benign | Likely Benign | 0.031 | Likely Benign | -1.08 | Neutral | 0.224 | Benign | 0.066 | Benign | 2.85 | Benign | 0.06 | Tolerated | 0.1872 | 0.2955 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||||||||||||
| c.3089A>T | H1030L 2D  AIThe SynGAP1 missense variant H1030L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that H1030L is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -3.513 | Likely Benign | 0.270 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -2.35 | Neutral | 0.224 | Benign | 0.120 | Benign | 2.76 | Benign | 0.02 | Affected | 0.0968 | 0.5710 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.308G>A | G103D 2D AIThe SynGAP1 missense variant G103D is not reported in ClinVar and has no entry in gnomAD. Consensus from multiple in‑silico predictors shows a predominance of benign calls: REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT each predict a pathogenic impact. The AlphaMissense‑Default tool remains uncertain, and no Foldetta stability assessment is available. High‑accuracy tools specifically highlight benign predictions: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta data are missing. Overall, the balance of evidence points to a benign effect for G103D, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.523 | Likely Benign | 0.477 | Ambiguous | Likely Benign | 0.110 | Likely Benign | -0.10 | Neutral | 0.949 | Possibly Damaging | 0.617 | Possibly Damaging | 4.26 | Benign | 0.00 | Affected | 0.2237 | 0.2896 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.308G>C | G103A 2D AIThe SynGAP1 missense variant G103A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.549 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -0.08 | Neutral | 0.008 | Benign | 0.008 | Benign | 4.31 | Benign | 0.00 | Affected | 0.3941 | 0.3784 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.308G>T | G103V 2D  AIThe SynGAP1 missense variant G103V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.687376 | Binding | 0.381 | 0.877 | 0.625 | -3.584 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.126 | Likely Benign | -0.96 | Neutral | 0.820 | Possibly Damaging | 0.376 | Benign | 4.22 | Benign | 0.00 | Affected | 0.1420 | 0.3404 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.3090C>A | H1030Q 2D  AIThe SynGAP1 missense variant H1030Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -2.548 | Likely Benign | 0.185 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.01 | Neutral | 0.004 | Benign | 0.004 | Benign | 2.88 | Benign | 0.53 | Tolerated | 0.1538 | 0.4001 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3090C>G | H1030Q 2D AIThe SynGAP1 missense variant H1030Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -2.548 | Likely Benign | 0.185 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.01 | Neutral | 0.004 | Benign | 0.004 | Benign | 2.88 | Benign | 0.53 | Tolerated | 0.1538 | 0.4001 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3091A>C | M1031L 2D AIThe SynGAP1 missense variant M1031L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995959 | Binding | 0.340 | 0.736 | 0.500 | -2.213 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.82 | Neutral | 0.044 | Benign | 0.018 | Benign | 2.84 | Benign | 0.33 | Tolerated | 0.1220 | 0.4050 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.3098C>T | S1033F 2D  AIThe SynGAP1 missense variant S1033F is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus is Likely Benign, and Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | -5.013 | Likely Benign | 0.555 | Ambiguous | Likely Benign | 0.022 | Likely Benign | -1.02 | Neutral | 0.440 | Benign | 0.185 | Benign | 2.70 | Benign | 0.05 | Affected | 0.0699 | 0.5546 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.2924C>G | T975S 2D AIThe SynGAP1 missense variant T975S is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the available predictions strongly suggest that T975S is most likely benign, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.969331 | Binding | 0.332 | 0.890 | 0.625 | Uncertain | 1 | -2.743 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.57 | Neutral | 0.059 | Benign | 0.061 | Benign | 4.16 | Benign | 0.20 | Tolerated | 0.3228 | 0.4366 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.2932C>A | P978T 2D AIThe SynGAP1 missense variant P978T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; the Foldetta stability analysis is not available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.975775 | Binding | 0.425 | 0.892 | 0.625 | -4.949 | Likely Benign | 0.217 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.23 | Neutral | 0.818 | Possibly Damaging | 0.453 | Possibly Damaging | 4.21 | Benign | 0.04 | Affected | 0.1817 | 0.7089 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2932C>G | P978A 2D AIThe SynGAP1 missense variant P978A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.975775 | Binding | 0.425 | 0.892 | 0.625 | -3.994 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -1.39 | Neutral | 0.008 | Benign | 0.010 | Benign | 4.30 | Benign | 0.07 | Tolerated | 0.3417 | 0.5891 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2932C>T | P978S 2D AIThe SynGAP1 missense variant P978S is listed in ClinVar (ID 3379672.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of predictions points to a benign effect, which is consistent with the ClinVar “Uncertain” status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.975775 | Binding | 0.425 | 0.892 | 0.625 | Uncertain | 1 | -3.913 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -1.07 | Neutral | 0.481 | Possibly Damaging | 0.220 | Benign | 4.22 | Benign | 0.48 | Tolerated | 0.3428 | 0.6001 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||
| c.2933C>A | P978Q 2D  AIThe SynGAP1 missense variant P978Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; the Foldetta stability analysis is not available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.975775 | Binding | 0.425 | 0.892 | 0.625 | -4.741 | Likely Benign | 0.305 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -1.72 | Neutral | 0.990 | Probably Damaging | 0.726 | Possibly Damaging | 4.15 | Benign | 0.01 | Affected | 0.1651 | 0.5699 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.2933C>G | P978R 2D AIThe SynGAP1 missense variant P978R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, all of which classify the variant as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple independent predictors indicates that P978R is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.975775 | Binding | 0.425 | 0.892 | 0.625 | -2.852 | Likely Benign | 0.487 | Ambiguous | Likely Benign | 0.105 | Likely Benign | -2.10 | Neutral | 0.970 | Probably Damaging | 0.726 | Possibly Damaging | 4.15 | Benign | 0.01 | Affected | 0.1530 | 0.4590 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2933C>T | P978L 2D AIThe SynGAP1 missense variant P978L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.975775 | Binding | 0.425 | 0.892 | 0.625 | -4.621 | Likely Benign | 0.386 | Ambiguous | Likely Benign | 0.092 | Likely Benign | -2.08 | Neutral | 0.818 | Possibly Damaging | 0.378 | Benign | 4.15 | Benign | 0.01 | Affected | 0.2326 | 0.6997 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2935T>A | F979I 2D AIThe SynGAP1 missense variant F979I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The AlphaMissense‑Default score is uncertain, and no Foldetta stability assessment is available. High‑accuracy methods reinforce the benign prediction: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta data are missing. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.977500 | Binding | 0.274 | 0.889 | 0.625 | -4.053 | Likely Benign | 0.512 | Ambiguous | Likely Benign | 0.163 | Likely Benign | -1.07 | Neutral | 0.925 | Possibly Damaging | 0.629 | Possibly Damaging | 4.19 | Benign | 0.06 | Tolerated | 0.2368 | 0.3009 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2935T>C | F979L 2D AIThe SynGAP1 missense variant F979L (ClinVar ID 1000410.0, status Uncertain, not found in gnomAD) has been evaluated by multiple in silico predictors. Benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM, while pathogenic predictions are reported by polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, whereas the SGM‑Consensus (majority vote) supports a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.977500 | Binding | 0.274 | 0.889 | 0.625 | Uncertain | 1 | -2.341 | Likely Benign | 0.870 | Likely Pathogenic | Ambiguous | 0.228 | Likely Benign | -1.00 | Neutral | 0.625 | Possibly Damaging | 0.430 | Benign | 4.22 | Benign | 0.73 | Tolerated | 4.32 | 2 | 0.2447 | 0.3876 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||
| c.2935T>G | F979V 2D AIThe SynGAP1 missense variant F979V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for F979V, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.977500 | Binding | 0.274 | 0.889 | 0.625 | -3.325 | Likely Benign | 0.434 | Ambiguous | Likely Benign | 0.199 | Likely Benign | -1.03 | Neutral | 0.925 | Possibly Damaging | 0.629 | Possibly Damaging | 4.21 | Benign | 0.04 | Affected | 0.2260 | 0.3176 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2936T>A | F979Y 2D AIThe SynGAP1 missense variant F979Y is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly supports a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.977500 | Binding | 0.274 | 0.889 | 0.625 | -3.420 | Likely Benign | 0.277 | Likely Benign | Likely Benign | 0.123 | Likely Benign | -0.26 | Neutral | 0.451 | Benign | 0.285 | Benign | 4.18 | Benign | 0.06 | Tolerated | 0.1451 | 0.2843 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2936T>C | F979S 2D  AIThe SynGAP1 missense variant F979S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and the AlphaMissense‑Optimized score also indicates a benign outcome. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists for F979S. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.977500 | Binding | 0.274 | 0.889 | 0.625 | -2.350 | Likely Benign | 0.718 | Likely Pathogenic | Likely Benign | 0.211 | Likely Benign | -0.05 | Neutral | 0.451 | Benign | 0.220 | Benign | 4.23 | Benign | 0.01 | Affected | 0.4397 | 0.0384 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||||||||||||
| c.2936T>G | F979C 2D  AIThe SynGAP1 missense variant F979C is not reported in ClinVar and has no gnomAD entry. Consensus from high‑accuracy predictors is benign: AlphaMissense‑Optimized scores it benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely benign. Other tools that agree with benign include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, did not return a result for this variant, so its stability impact is unavailable. Overall, the majority of evidence points to a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.977500 | Binding | 0.274 | 0.889 | 0.625 | -6.395 | Likely Benign | 0.589 | Likely Pathogenic | Likely Benign | 0.160 | Likely Benign | -0.94 | Neutral | 0.994 | Probably Damaging | 0.888 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 0.2646 | 0.2179 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||||||||||||
| c.2937C>A | F979L 2D AIThe SynGAP1 missense variant F979L has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM, while polyPhen‑2 HumDiv and AlphaMissense‑Default predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus remains Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.977500 | Binding | 0.274 | 0.889 | 0.625 | -2.341 | Likely Benign | 0.870 | Likely Pathogenic | Ambiguous | 0.328 | Likely Benign | -1.00 | Neutral | 0.625 | Possibly Damaging | 0.430 | Benign | 4.22 | Benign | 0.73 | Tolerated | 4.32 | 2 | 0.2447 | 0.3876 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||||
| c.2930C>T | A977V 2D  AIThe SynGAP1 missense variant A977V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for A977V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.975330 | Binding | 0.306 | 0.884 | 0.625 | -3.542 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -1.15 | Neutral | 0.818 | Possibly Damaging | 0.457 | Possibly Damaging | 4.01 | Benign | 0.01 | Affected | 0.1966 | 0.5727 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2930C>G | A977G 2D  AIThe SynGAP1 missense variant A977G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for A977G, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.975330 | Binding | 0.306 | 0.884 | 0.625 | -3.250 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.79 | Neutral | 0.965 | Probably Damaging | 0.702 | Possibly Damaging | 4.03 | Benign | 0.05 | Affected | 0.2180 | 0.4408 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2926T>A | F976I 2D AIThe SynGAP1 missense variant F976I has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.975061 | Binding | 0.311 | 0.894 | 0.625 | -4.595 | Likely Benign | 0.364 | Ambiguous | Likely Benign | 0.148 | Likely Benign | -1.16 | Neutral | 0.666 | Possibly Damaging | 0.265 | Benign | 4.16 | Benign | 0.21 | Tolerated | 0.2790 | 0.2714 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2926T>C | F976L 2D AIThe SynGAP1 missense variant F976L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which classifies the variant as “Likely Benign.” In contrast, AlphaMissense‑Default predicts a pathogenic effect, while AlphaMissense‑Optimized is uncertain. No Foldetta (FoldX‑MD/ Rosetta) stability result is available, so it does not contribute to the assessment. Overall, the majority of high‑confidence predictors indicate a benign impact, and this is consistent with the lack of ClinVar evidence. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.975061 | Binding | 0.311 | 0.894 | 0.625 | -2.432 | Likely Benign | 0.825 | Likely Pathogenic | Ambiguous | 0.237 | Likely Benign | -0.87 | Neutral | 0.264 | Benign | 0.102 | Benign | 4.20 | Benign | 0.53 | Tolerated | 4.32 | 2 | 0.2829 | 0.3082 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||||
| c.2926T>G | F976V 2D AIThe SynGAP1 missense variant F976V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.975061 | Binding | 0.311 | 0.894 | 0.625 | -3.328 | Likely Benign | 0.268 | Likely Benign | Likely Benign | 0.190 | Likely Benign | -1.32 | Neutral | 0.451 | Benign | 0.157 | Benign | 4.18 | Benign | 0.23 | Tolerated | 0.2696 | 0.2902 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2927T>A | F976Y 2D AIThe SynGAP1 missense variant F976Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.975061 | Binding | 0.311 | 0.894 | 0.625 | -3.902 | Likely Benign | 0.225 | Likely Benign | Likely Benign | 0.172 | Likely Benign | -0.50 | Neutral | 0.925 | Possibly Damaging | 0.529 | Possibly Damaging | 4.11 | Benign | 0.80 | Tolerated | 0.1942 | 0.2257 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2927T>C | F976S 2D AIThe SynGAP1 missense variant F976S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.975061 | Binding | 0.311 | 0.894 | 0.625 | -2.393 | Likely Benign | 0.432 | Ambiguous | Likely Benign | 0.217 | Likely Benign | -0.73 | Neutral | 0.292 | Benign | 0.102 | Benign | 4.16 | Benign | 0.49 | Tolerated | 0.4432 | 0.1134 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||||||||||||
| c.2927T>G | F976C 2D AIThe SynGAP1 missense variant F976C is not reported in ClinVar and is absent from gnomAD. In silico predictors cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus likewise indicates Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for F976C, and this conclusion is not in conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.975061 | Binding | 0.311 | 0.894 | 0.625 | -5.490 | Likely Benign | 0.490 | Ambiguous | Likely Benign | 0.103 | Likely Benign | -1.10 | Neutral | 0.977 | Probably Damaging | 0.840 | Possibly Damaging | 4.09 | Benign | 0.10 | Tolerated | 0.2961 | 0.2505 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||||||||||||
| c.2928T>G | F976L 2D AIThe SynGAP1 missense variant F976L is listed in ClinVar (ID 624245.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Only AlphaMissense‑Default predicts a pathogenic outcome. The high‑accuracy consensus predictor SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. AlphaMissense‑Optimized is inconclusive, and no Foldetta (FoldX‑MD/Rosetta stability) result is available. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar designation of uncertainty rather than a definitive pathogenic claim. Thus, the variant is most likely benign, and its predictions do not contradict the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.975061 | Binding | 0.311 | 0.894 | 0.625 | Uncertain | 1 | -2.432 | Likely Benign | 0.825 | Likely Pathogenic | Ambiguous | 0.212 | Likely Benign | -0.87 | Neutral | 0.264 | Benign | 0.102 | Benign | 4.20 | Benign | 0.53 | Tolerated | 4.32 | 2 | 0.2829 | 0.3082 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||
| c.2929G>A | A977T 2D  AIThe SynGAP1 missense variant A977T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.975330 | Binding | 0.306 | 0.884 | 0.625 | -3.814 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.106 | Likely Benign | -0.84 | Neutral | 0.965 | Probably Damaging | 0.782 | Possibly Damaging | 4.00 | Benign | 0.02 | Affected | 0.2155 | 0.6185 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2929G>C | A977P 2D  AIThe SynGAP1 missense variant A977P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for A977P, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.975330 | Binding | 0.306 | 0.884 | 0.625 | -2.665 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -1.12 | Neutral | 0.990 | Probably Damaging | 0.892 | Possibly Damaging | 3.94 | Benign | 0.02 | Affected | 0.2405 | 0.5212 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2929G>T | A977S 2D  AIThe SynGAP1 missense variant A977S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence indicates that A977S is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.975330 | Binding | 0.306 | 0.884 | 0.625 | -2.909 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -0.38 | Neutral | 0.965 | Probably Damaging | 0.702 | Possibly Damaging | 4.02 | Benign | 0.45 | Tolerated | 0.2820 | 0.5373 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.292C>A | H98N 2D AIThe SynGAP1 missense variant H98N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | -3.855 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -0.35 | Neutral | 0.115 | Benign | 0.012 | Benign | 4.24 | Benign | 0.00 | Affected | 0.2003 | 0.3759 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.292C>T | H98Y 2D AIThe SynGAP1 missense variant H98Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | -4.050 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.112 | Likely Benign | -0.86 | Neutral | 0.444 | Benign | 0.024 | Benign | 4.19 | Benign | 0.00 | Affected | 0.0950 | 0.5024 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.2930C>A | A977D 2D  AIThe SynGAP1 missense variant A977D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy methods give a consistent benign signal: AlphaMissense‑Optimized predicts benign, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely benign, while Foldetta’s protein‑folding stability assessment is unavailable. Overall, the majority of evidence points to a benign impact for A977D, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.882776 | Disordered | 0.975330 | Binding | 0.306 | 0.884 | 0.625 | -4.007 | Likely Benign | 0.717 | Likely Pathogenic | Likely Benign | 0.130 | Likely Benign | -1.15 | Neutral | 0.990 | Probably Damaging | 0.892 | Possibly Damaging | 3.96 | Benign | 0.01 | Affected | 0.2253 | 0.2651 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2937C>G | F979L 2D AIThe SynGAP1 missense variant F979L has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM, while polyPhen‑2 HumDiv and AlphaMissense‑Default predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus remains Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.977500 | Binding | 0.274 | 0.889 | 0.625 | -2.341 | Likely Benign | 0.870 | Likely Pathogenic | Ambiguous | 0.328 | Likely Benign | -1.00 | Neutral | 0.625 | Possibly Damaging | 0.430 | Benign | 4.22 | Benign | 0.73 | Tolerated | 4.32 | 2 | 0.2447 | 0.3876 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||||
| c.2938C>A | H980N 2D AIThe SynGAP1 missense variant H980N is not reported in ClinVar or gnomAD. Functional prediction tools largely agree on a benign effect. Benign calls come from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign classification. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus confirms a benign result; Foldetta data are unavailable, so no additional stability evidence is considered. Overall, the computational evidence indicates that H980N is most likely benign, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.974598 | Binding | 0.309 | 0.892 | 0.625 | -4.728 | Likely Benign | 0.291 | Likely Benign | Likely Benign | 0.070 | Likely Benign | -1.07 | Neutral | 0.451 | Benign | 0.209 | Benign | 4.17 | Benign | 0.00 | Affected | 0.2346 | 0.3638 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.2938C>G | H980D 2D  AIThe SynGAP1 missense variant H980D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.974598 | Binding | 0.309 | 0.892 | 0.625 | -5.489 | Likely Benign | 0.729 | Likely Pathogenic | Likely Benign | 0.117 | Likely Benign | -1.38 | Neutral | 0.451 | Benign | 0.265 | Benign | 4.18 | Benign | 0.00 | Affected | 0.2608 | 0.3066 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||||||||||||
| c.2944T>G | Y982D 2D  AIThe SynGAP1 missense variant Y982D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which collectively suggest a likely benign outcome. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and Foldetta (combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the balance of evidence leans toward a benign interpretation, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.966717 | Binding | 0.272 | 0.895 | 0.625 | -5.021 | Likely Benign | 0.952 | Likely Pathogenic | Ambiguous | 0.194 | Likely Benign | -1.29 | Neutral | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 3.87 | Benign | 0.00 | Affected | 0.3959 | 0.0545 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2945A>C | Y982S 2D  AIThe SynGAP1 missense variant Y982S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which collectively suggest a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default predict a pathogenic impact. High‑accuracy assessments show the SGM‑Consensus as Likely Benign, AlphaMissense‑Optimized as Uncertain, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.966717 | Binding | 0.272 | 0.895 | 0.625 | -2.919 | Likely Benign | 0.841 | Likely Pathogenic | Ambiguous | 0.131 | Likely Benign | -1.04 | Neutral | 0.965 | Probably Damaging | 0.783 | Possibly Damaging | 3.89 | Benign | 0.00 | Affected | 0.4556 | 0.1883 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||||||||||||
| c.2945A>T | Y982F 2D  AIThe SynGAP1 missense variant Y982F is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.966717 | Binding | 0.272 | 0.895 | 0.625 | -3.431 | Likely Benign | 0.208 | Likely Benign | Likely Benign | 0.154 | Likely Benign | -0.36 | Neutral | 0.006 | Benign | 0.009 | Benign | 4.63 | Benign | 0.00 | Affected | 0.2351 | 0.2980 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2947A>C | S983R 2D  AIThe SynGAP1 missense variant S983R is reported in ClinVar as “Not submitted” and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and also indicates a likely pathogenic outcome. AlphaMissense‑Optimized independently predicts pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for S983R, and this conclusion does not contradict any ClinVar annotation, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -4.733 | Likely Benign | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.156 | Likely Benign | -2.66 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1163 | 0.3740 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2947A>G | S983G 2D  AIThe SynGAP1 missense variant S983G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 vs 2). High‑accuracy methods show AlphaMissense‑Optimized as benign; the SGM Consensus is unavailable, and Foldetta results are not provided, so no folding‑stability evidence is available. Overall, the balance of evidence (five pathogenic vs four benign predictions) suggests the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -5.206 | Likely Benign | 0.638 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | -2.22 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 2.17 | Pathogenic | 0.00 | Affected | 0.2521 | 0.4480 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2947A>T | S983C 2D AIThe SynGAP1 missense variant S983C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools cluster into two groups: benign predictions come from REVEL and AlphaMissense‑Optimized, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, whereas the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points toward a pathogenic impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -7.083 | In-Between | 0.741 | Likely Pathogenic | Likely Benign | 0.162 | Likely Benign | -2.64 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1657 | 0.5298 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2948G>C | S983T 2D AIThe SynGAP1 missense variant S983T is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign). High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus and Foldetta (which would combine FoldX‑MD and Rosetta outputs) are unavailable. Overall, the balance of evidence leans toward pathogenicity, with five tools supporting a deleterious effect versus four supporting benign. This prediction does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -4.493 | Likely Benign | 0.588 | Likely Pathogenic | Likely Benign | 0.172 | Likely Benign | -1.65 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 2.08 | Pathogenic | 0.00 | Affected | 0.2042 | 0.5517 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2948G>T | S983I 2D  AIThe SynGAP1 missense variant S983I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). In silico predictors that agree on a benign effect are REVEL and ESM1b, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (3 pathogenic vs. 1 benign) is likely pathogenic. Foldetta results are unavailable. Overall, the preponderance of evidence indicates that S983I is most likely pathogenic, and this conclusion is not contradicted by the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -6.259 | Likely Benign | 0.968 | Likely Pathogenic | Likely Pathogenic | 0.190 | Likely Benign | -2.67 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.1380 | 0.4625 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2949C>A | S983R 2D AIThe SynGAP1 missense variant S983R is reported in ClinVar as “Not submitted” and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (3 pathogenic vs. 1 benign) is likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that S983R is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently lacks a classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -4.733 | Likely Benign | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.190 | Likely Benign | -2.66 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1163 | 0.3740 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2949C>G | S983R 2D AIThe SynGAP1 missense variant S983R is reported in ClinVar as “Not submitted” and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (3 pathogenic vs. 1 benign) is likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S983R is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently lacks a pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.707965 | Disordered | 0.960212 | Binding | 0.277 | 0.889 | 0.625 | -4.733 | Likely Benign | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.190 | Likely Benign | -2.66 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.03 | Pathogenic | 0.00 | Affected | 0.1163 | 0.3740 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.294T>A | H98Q 2D AIThe SynGAP1 missense variant H98Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | -2.749 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.47 | Neutral | 0.002 | Benign | 0.000 | Benign | 4.26 | Benign | 0.00 | Affected | 0.1831 | 0.4347 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.294T>G | H98Q 2D AIThe SynGAP1 missense variant H98Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | -2.749 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.47 | Neutral | 0.002 | Benign | 0.000 | Benign | 4.26 | Benign | 0.00 | Affected | 0.1831 | 0.4347 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2950A>C | K984Q 2D  AIThe SynGAP1 missense variant K984Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.951648 | Binding | 0.288 | 0.895 | 0.750 | -2.932 | Likely Benign | 0.612 | Likely Pathogenic | Likely Benign | 0.083 | Likely Benign | -0.67 | Neutral | 0.905 | Possibly Damaging | 0.637 | Possibly Damaging | 2.66 | Benign | 0.00 | Affected | 0.5054 | 0.1240 | Weaken | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||||||||||
| c.2944T>C | Y982H 2D  AIThe SynGAP1 missense variant Y982H has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus result is benign; Foldetta predictions are unavailable. Overall, the evidence is mixed, but the majority of consensus‑based and high‑accuracy tools lean toward a benign interpretation. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.966717 | Binding | 0.272 | 0.895 | 0.625 | -2.675 | Likely Benign | 0.893 | Likely Pathogenic | Ambiguous | 0.093 | Likely Benign | -0.63 | Neutral | 0.990 | Probably Damaging | 0.900 | Possibly Damaging | 3.92 | Benign | 0.00 | Affected | 0.2352 | 0.0545 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||||||||||||
| c.2944T>A | Y982N 2D  AIThe SynGAP1 Y982N variant has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, SGM‑Consensus is benign, and Foldetta results are unavailable. Overall, the majority of consensus‑based and high‑accuracy tools lean toward a benign interpretation. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar status, which currently has no entry for Y982N. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.966717 | Binding | 0.272 | 0.895 | 0.625 | -4.536 | Likely Benign | 0.881 | Likely Pathogenic | Ambiguous | 0.175 | Likely Benign | -1.14 | Neutral | 0.990 | Probably Damaging | 0.900 | Possibly Damaging | 3.88 | Benign | 0.00 | Affected | 0.2090 | 0.0545 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.2938C>T | H980Y 2D AIThe SynGAP1 missense variant H980Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for H980Y, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.974598 | Binding | 0.309 | 0.892 | 0.625 | -4.104 | Likely Benign | 0.299 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -1.12 | Neutral | 0.925 | Possibly Damaging | 0.529 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.1524 | 0.4706 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.2939A>C | H980P 2D AIThe SynGAP1 missense variant H980P is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools largely favor a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. In contrast, two tools—polyPhen‑2 HumDiv and SIFT—classify the change as pathogenic. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign and the SGM‑Consensus (majority vote) is likely benign; no Foldetta stability data are available. Overall, the preponderance of evidence points to a benign effect for H980P, and this conclusion is not in conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.974598 | Binding | 0.309 | 0.892 | 0.625 | -3.071 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.136 | Likely Benign | -1.70 | Neutral | 0.802 | Possibly Damaging | 0.432 | Benign | 4.15 | Benign | 0.00 | Affected | 0.2274 | 0.4301 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2939A>T | H980L 2D AIThe SynGAP1 missense variant H980L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.974598 | Binding | 0.309 | 0.892 | 0.625 | -1.984 | Likely Benign | 0.293 | Likely Benign | Likely Benign | 0.187 | Likely Benign | -1.98 | Neutral | 0.625 | Possibly Damaging | 0.265 | Benign | 4.16 | Benign | 0.00 | Affected | 0.1488 | 0.5538 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.293A>C | H98P 2D AIThe SynGAP1 missense variant H98P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar classification to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | -1.460 | Likely Benign | 0.069 | Likely Benign | Likely Benign | 0.275 | Likely Benign | -0.83 | Neutral | 0.659 | Possibly Damaging | 0.024 | Benign | 4.20 | Benign | 0.00 | Affected | 0.2062 | 0.4669 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.293A>G | H98R 2D AIThe SynGAP1 missense variant H98R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.631713 | Binding | 0.348 | 0.872 | 0.625 | -2.772 | Likely Benign | 0.180 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.39 | Neutral | 0.115 | Benign | 0.006 | Benign | 4.27 | Benign | 0.00 | Affected | 0.2271 | 0.3022 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||||||||||||
| c.2940T>A | H980Q 2D AIThe SynGAP1 missense variant H980Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for H980Q, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.974598 | Binding | 0.309 | 0.892 | 0.625 | -4.014 | Likely Benign | 0.385 | Ambiguous | Likely Benign | 0.090 | Likely Benign | -1.35 | Neutral | 0.802 | Possibly Damaging | 0.432 | Benign | 4.18 | Benign | 0.00 | Affected | 0.2236 | 0.3936 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2940T>G | H980Q 2D AIThe SynGAP1 missense variant H980Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for H980Q, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.974598 | Binding | 0.309 | 0.892 | 0.625 | -4.014 | Likely Benign | 0.385 | Ambiguous | Likely Benign | 0.090 | Likely Benign | -1.35 | Neutral | 0.802 | Possibly Damaging | 0.432 | Benign | 4.18 | Benign | 0.00 | Affected | 0.2236 | 0.3936 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2941G>A | G981S 2D AIThe SynGAP1 missense variant G981S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; the Foldetta stability analysis is not available for this variant. Overall, the majority of evidence—including the consensus and high‑accuracy tools—points to a benign effect. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.970320 | Binding | 0.275 | 0.897 | 0.625 | -2.749 | Likely Benign | 0.274 | Likely Benign | Likely Benign | 0.130 | Likely Benign | -0.57 | Neutral | 0.979 | Probably Damaging | 0.907 | Possibly Damaging | 3.87 | Benign | 0.00 | Affected | 0.2566 | 0.5470 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2941G>C | G981R 2D AIThe SynGAP1 missense variant G981R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is not available for this variant. Overall, the majority of evidence points toward a benign impact, and this conclusion does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely benign based on current predictive tools. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.970320 | Binding | 0.275 | 0.897 | 0.625 | -1.264 | Likely Benign | 0.953 | Likely Pathogenic | Ambiguous | 0.207 | Likely Benign | -2.11 | Neutral | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 3.74 | Benign | 0.00 | Affected | 0.0990 | 0.4776 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2941G>T | G981C 2D AIThe SynGAP1 missense variant G981C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for G981C, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.970320 | Binding | 0.275 | 0.897 | 0.625 | -5.646 | Likely Benign | 0.593 | Likely Pathogenic | Likely Benign | 0.228 | Likely Benign | -1.81 | Neutral | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 3.72 | Benign | 0.00 | Affected | 0.1398 | 0.4593 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2942G>A | G981D 2D AIThe SynGAP1 missense variant G981D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Taken together, the balance of evidence leans toward a benign interpretation; there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.970320 | Binding | 0.275 | 0.897 | 0.625 | -5.280 | Likely Benign | 0.945 | Likely Pathogenic | Ambiguous | 0.159 | Likely Benign | -1.64 | Neutral | 1.000 | Probably Damaging | 0.985 | Probably Damaging | 3.75 | Benign | 0.00 | Affected | 0.1961 | 0.2868 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2942G>C | G981A 2D AIThe SynGAP1 missense variant G981A is not reported in ClinVar and has no entry in gnomAD, indicating it is not catalogued in these databases. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.970320 | Binding | 0.275 | 0.897 | 0.625 | -3.374 | Likely Benign | 0.368 | Ambiguous | Likely Benign | 0.064 | Likely Benign | -0.95 | Neutral | 0.561 | Possibly Damaging | 0.376 | Benign | 3.95 | Benign | 0.00 | Affected | 0.3512 | 0.4925 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2942G>T | G981V 2D AIThe SynGAP1 missense variant G981V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for G981V, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.798249 | Disordered | 0.970320 | Binding | 0.275 | 0.897 | 0.625 | -3.873 | Likely Benign | 0.714 | Likely Pathogenic | Likely Benign | 0.156 | Likely Benign | -2.10 | Neutral | 0.997 | Probably Damaging | 0.958 | Probably Damaging | 3.75 | Benign | 0.00 | Affected | 0.1322 | 0.3861 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2950A>G | K984E 2D  AIThe SynGAP1 missense variant K984E has no ClinVar record and is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, PROVEAN, ESM1b, and FATHMM, while polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default predict it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. Overall, the majority of individual predictors lean toward benign, and the consensus score explicitly labels it benign, whereas a comparable number of tools predict pathogenicity. Based on the available evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.951648 | Binding | 0.288 | 0.895 | 0.750 | -4.909 | Likely Benign | 0.932 | Likely Pathogenic | Ambiguous | 0.086 | Likely Benign | -0.88 | Neutral | 0.798 | Possibly Damaging | 0.535 | Possibly Damaging | 2.71 | Benign | 0.00 | Affected | 0.4353 | 0.1200 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2923A>T | T975S 2D AIThe SynGAP1 missense variant T975S is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the predictions strongly suggest that T975S is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.969331 | Binding | 0.332 | 0.890 | 0.625 | -2.743 | Likely Benign | 0.068 | Likely Benign | Likely Benign | 0.127 | Likely Benign | -0.57 | Neutral | 0.059 | Benign | 0.061 | Benign | 4.16 | Benign | 0.20 | Tolerated | 0.3228 | 0.4366 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2890C>G | H964D 2D AIThe SynGAP1 missense variant H964D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic prediction comes from ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -11.061 | Likely Pathogenic | 0.207 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -0.45 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.18 | Benign | 0.24 | Tolerated | 0.2313 | 0.2923 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||||||||||||
| c.2898C>A | H966Q 2D AIThe SynGAP1 missense variant H966Q is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that H966Q is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -5.662 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.66 | Neutral | 0.748 | Possibly Damaging | 0.232 | Benign | 4.06 | Benign | 0.45 | Tolerated | 0.2058 | 0.3914 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2898C>G | H966Q 2D AIThe SynGAP1 missense variant H966Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -5.662 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -0.66 | Neutral | 0.748 | Possibly Damaging | 0.232 | Benign | 4.06 | Benign | 0.45 | Tolerated | 0.2058 | 0.3914 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.28C>G | R10G 2D AIThe SynGAP1 missense variant R10G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.513657 | Binding | 0.330 | 0.915 | 0.625 | -3.931 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.175 | Likely Benign | 0.48 | Neutral | 0.058 | Benign | 0.009 | Benign | 4.15 | Benign | 0.00 | Affected | 0.3796 | 0.4015 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2900G>C | R967P 2D AIThe SynGAP1 missense variant R967P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect for R967P, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.974374 | Disordered | 0.969686 | Binding | 0.340 | 0.888 | 0.750 | -2.506 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -0.76 | Neutral | 0.996 | Probably Damaging | 0.828 | Possibly Damaging | 4.14 | Benign | 0.17 | Tolerated | 0.2145 | 0.5533 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2902G>A | G968S 2D AIThe SynGAP1 missense variant G968S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -4.484 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -0.35 | Neutral | 0.058 | Benign | 0.023 | Benign | 4.22 | Benign | 0.37 | Tolerated | 0.2482 | 0.5699 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2902G>C | G968R 2D AIThe SynGAP1 missense variant G968R is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the evidence strongly supports a benign classification, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -4.492 | Likely Benign | 0.332 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -0.63 | Neutral | 0.005 | Benign | 0.012 | Benign | 4.19 | Benign | 0.08 | Tolerated | 0.0972 | 0.5017 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2902G>T | G968C 2D AIThe SynGAP1 missense variant G968C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -8.441 | Likely Pathogenic | 0.117 | Likely Benign | Likely Benign | 0.170 | Likely Benign | -0.99 | Neutral | 0.992 | Probably Damaging | 0.820 | Possibly Damaging | 4.12 | Benign | 0.07 | Tolerated | 0.1294 | 0.4768 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.2903G>C | G968A 2D AIThe SynGAP1 missense variant G968A is predicted to be benign by all available in‑silico tools. Consensus predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the change as benign. No tools predict pathogenicity, so the pathogenic group is empty. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized reports benign, and the SGM‑Consensus indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Based on the unanimous benign predictions and the absence of ClinVar evidence, the variant is most likely benign and does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -4.721 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -0.33 | Neutral | 0.245 | Benign | 0.140 | Benign | 4.24 | Benign | 0.68 | Tolerated | 0.3401 | 0.5154 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2903G>T | G968V 2D AIThe SynGAP1 missense variant G968V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -6.152 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.239 | Likely Benign | -1.08 | Neutral | 0.918 | Possibly Damaging | 0.525 | Possibly Damaging | 4.19 | Benign | 0.11 | Tolerated | 0.1252 | 0.4036 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2905G>A | G969R 2D AIThe SynGAP1 missense variant G969R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.956572 | Binding | 0.405 | 0.898 | 0.750 | -4.783 | Likely Benign | 0.316 | Likely Benign | Likely Benign | 0.152 | Likely Benign | -0.70 | Neutral | 0.611 | Possibly Damaging | 0.305 | Benign | 4.20 | Benign | 0.01 | Affected | 0.1039 | 0.5473 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2905G>C | G969R 2D AIThe SynGAP1 missense variant G969R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.956572 | Binding | 0.405 | 0.898 | 0.750 | -4.783 | Likely Benign | 0.316 | Likely Benign | Likely Benign | 0.152 | Likely Benign | -0.70 | Neutral | 0.611 | Possibly Damaging | 0.305 | Benign | 4.20 | Benign | 0.01 | Affected | 0.1039 | 0.5473 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2906G>A | G969E 2D  AIThe SynGAP1 missense variant G969E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.956572 | Binding | 0.405 | 0.898 | 0.750 | -4.721 | Likely Benign | 0.167 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -0.10 | Neutral | 0.611 | Possibly Damaging | 0.171 | Benign | 4.28 | Benign | 0.01 | Affected | 0.1663 | 0.5399 | 0 | -2 | -3.1 | 72.06 | |||||||||||||||||||||||||||||||||||||||
| c.2906G>C | G969A 2D AIThe SynGAP1 missense variant G969A is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity, so the pathogenic‑prediction group is empty. High‑accuracy assessments corroborate this benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are not available, so they do not influence the interpretation. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.956572 | Binding | 0.405 | 0.898 | 0.750 | -4.693 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.120 | Likely Benign | -0.36 | Neutral | 0.393 | Benign | 0.096 | Benign | 4.25 | Benign | 0.51 | Tolerated | 0.3414 | 0.5096 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2897A>T | H966L 2D AIThe SynGAP1 missense variant H966L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. No pathogenic predictions are present among the evaluated algorithms. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) reports likely benign. Foldetta results are not available for this variant. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -6.119 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -1.74 | Neutral | 0.174 | Benign | 0.062 | Benign | 4.05 | Benign | 0.35 | Tolerated | 0.1514 | 0.5316 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2897A>C | H966P 2D AIThe SynGAP1 missense variant H966P is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -5.831 | Likely Benign | 0.064 | Likely Benign | Likely Benign | 0.279 | Likely Benign | -0.27 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.01 | Benign | 0.72 | Tolerated | 0.2168 | 0.4301 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2890C>T | H964Y 2D AIThe SynGAP1 missense variant H964Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, SGM‑Consensus indicates likely benign, and the Foldetta stability analysis is unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.385 | In-Between | 0.139 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -1.13 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.11 | Benign | 0.02 | Affected | 0.1422 | 0.4363 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.2891A>C | H964P 2D AIThe SynGAP1 missense variant H964P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign, while the only pathogenic call comes from SIFT. ESM1b is uncertain, and the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign. Foldetta stability analysis is unavailable. Overall, the collective evidence points to a benign impact for H964P, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.466 | In-Between | 0.063 | Likely Benign | Likely Benign | 0.156 | Likely Benign | -0.34 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.12 | Benign | 0.04 | Affected | 0.1978 | 0.4301 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2891A>G | H964R 2D AIThe SynGAP1 missense variant H964R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and ESM1b. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (3 benign vs. 1 pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -8.275 | Likely Pathogenic | 0.163 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -0.68 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.19 | Benign | 0.05 | Affected | 0.2237 | 0.3467 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||||||||||||
| c.2891A>T | H964L 2D AIThe SynGAP1 missense variant H964L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.568 | In-Between | 0.092 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.20 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.15 | Benign | 0.02 | Affected | 0.1409 | 0.5195 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2892C>A | H964Q 2D AIThe SynGAP1 missense variant H964Q is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. The only tool with an uncertain call is ESM1b, and no pathogenic predictions are reported. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.279 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.18 | Benign | 0.07 | Tolerated | 0.1969 | 0.3593 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2892C>G | H964Q 2D AIThe SynGAP1 missense variant H964Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only uncertain result comes from ESM1b. The high‑accuracy consensus methods also support a benign classification: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the evidence overwhelmingly indicates that H964Q is most likely benign, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.279 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.18 | Benign | 0.07 | Tolerated | 0.1969 | 0.3593 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2893C>A | H965N 2D AIThe SynGAP1 missense variant H965N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Across the available in‑silico predictors, the majority (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a benign effect, while no tool predicts pathogenicity. ESM1b is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -7.605 | In-Between | 0.082 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -0.50 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.09 | Benign | 0.80 | Tolerated | 0.2360 | 0.3838 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.2893C>T | H965Y 2D AIThe SynGAP1 missense variant H965Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as tolerated. The consensus score from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” No tool predicts pathogenicity, and the single uncertain result from ESM1b does not alter the overall benign consensus. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta data are unavailable. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -7.121 | In-Between | 0.133 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -1.02 | Neutral | 0.327 | Benign | 0.147 | Benign | 4.03 | Benign | 0.25 | Tolerated | 0.1582 | 0.4906 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.2894A>C | H965P 2D AIThe SynGAP1 missense variant H965P is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. No tool predicts pathogenicity. The high‑accuracy consensus methods also support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Overall, the variant is most likely benign, and this prediction does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -6.835 | Likely Benign | 0.059 | Likely Benign | Likely Benign | 0.245 | Likely Benign | -0.69 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.06 | Benign | 0.19 | Tolerated | 0.2342 | 0.4901 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2894A>T | H965L 2D AIThe SynGAP1 missense variant H965L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect. Benign predictors include REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool in the dataset returned a pathogenic prediction. Consensus predictors such as SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classify the variant as Likely Benign. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus is Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -6.708 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -1.66 | Neutral | 0.033 | Benign | 0.018 | Benign | 4.06 | Benign | 1.00 | Tolerated | 0.1606 | 0.5338 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2895C>A | H965Q 2D AIThe SynGAP1 missense variant H965Q is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -6.447 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -0.71 | Neutral | 0.138 | Benign | 0.105 | Benign | 4.09 | Benign | 0.28 | Tolerated | 0.2257 | 0.3936 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2895C>G | H965Q 2D AIThe SynGAP1 missense variant H965Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect. Consensus predictors such as SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classify the variant as Likely Benign. Individual algorithms—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—all predict a benign outcome. No tool in the dataset returned a pathogenic prediction. High‑accuracy methods: AlphaMissense‑Optimized is benign; SGM‑Consensus is Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -6.447 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -0.71 | Neutral | 0.138 | Benign | 0.105 | Benign | 4.09 | Benign | 0.28 | Tolerated | 0.2257 | 0.3936 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.2896C>A | H966N 2D AIThe SynGAP1 missense variant H966N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while ESM1b is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that H966N is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -7.579 | In-Between | 0.085 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -0.84 | Neutral | 0.748 | Possibly Damaging | 0.232 | Benign | 4.06 | Benign | 0.89 | Tolerated | 0.2153 | 0.3788 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.2906G>T | G969V 2D AIThe SynGAP1 missense variant G969V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.956572 | Binding | 0.405 | 0.898 | 0.750 | -5.946 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.92 | Neutral | 0.761 | Possibly Damaging | 0.239 | Benign | 4.18 | Benign | 0.01 | Affected | 0.1497 | 0.3847 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2908G>A | E970K 2D  AIThe SynGAP1 missense variant E970K is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign interpretation, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.951925 | Disordered | 0.953422 | Binding | 0.342 | 0.902 | 0.750 | -3.344 | Likely Benign | 0.303 | Likely Benign | Likely Benign | 0.102 | Likely Benign | -0.24 | Neutral | 0.078 | Benign | 0.042 | Benign | 4.18 | Benign | 0.17 | Tolerated | 0.3372 | 0.7361 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2909A>C | E970A 2D  AIThe SynGAP1 missense variant E970A is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the consensus of all predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.951925 | Disordered | 0.953422 | Binding | 0.342 | 0.902 | 0.750 | -1.704 | Likely Benign | 0.156 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.89 | Neutral | 0.069 | Benign | 0.018 | Benign | 4.15 | Benign | 0.19 | Tolerated | 0.3679 | 0.6798 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2917G>T | G973W 2D  AIThe SynGAP1 missense variant G973W is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized reports Benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for G973W, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.959498 | Binding | 0.390 | 0.897 | 0.625 | -6.896 | Likely Benign | 0.329 | Likely Benign | Likely Benign | 0.105 | Likely Benign | -1.53 | Neutral | 0.983 | Probably Damaging | 0.813 | Possibly Damaging | 4.10 | Benign | 0.00 | Affected | 0.0846 | 0.4046 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.2918G>A | G973E 2D AIThe SynGAP1 missense variant G973E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions point to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.959498 | Binding | 0.390 | 0.897 | 0.625 | -3.712 | Likely Benign | 0.210 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -0.72 | Neutral | 0.001 | Benign | 0.003 | Benign | 4.19 | Benign | 0.01 | Affected | 0.1463 | 0.4258 | 0 | -2 | -3.1 | 72.06 | |||||||||||||||||||||||||||||||||||||||
| c.2918G>T | G973V 2D AIThe SynGAP1 missense variant G973V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. Grouping by consensus, the benign‑predicting tools outnumber the single pathogenic prediction. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign classification. Foldetta results are unavailable, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.959498 | Binding | 0.390 | 0.897 | 0.625 | -4.688 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -0.76 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.18 | Benign | 0.01 | Affected | 0.1392 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.291G>C | E97D 2D AIThe SynGAP1 missense variant E97D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the overall assessment. Overall, the majority of evidence points to the variant being most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.609018 | Binding | 0.340 | 0.867 | 0.625 | -3.239 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -0.49 | Neutral | 0.880 | Possibly Damaging | 0.636 | Possibly Damaging | 4.12 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1987 | 0.5559 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||
| c.2920G>A | D974N 2D AIThe SynGAP1 missense variant D974N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect. Consensus predictors such as SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and high‑accuracy methods including AlphaMissense‑Optimized all classify the variant as benign. Additional in silico assessments—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—also predict a benign outcome. No tool in the dataset suggests pathogenicity. Protein‑stability analysis via Foldetta is unavailable for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -4.051 | Likely Benign | 0.145 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.35 | Neutral | 0.144 | Benign | 0.085 | Benign | 4.20 | Benign | 0.09 | Tolerated | 0.1972 | 0.7698 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.2920G>C | D974H 2D  AIThe SynGAP1 missense variant D974H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -3.034 | Likely Benign | 0.333 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.95 | Neutral | 0.744 | Possibly Damaging | 0.382 | Benign | 4.14 | Benign | 0.02 | Affected | 0.2249 | 0.7803 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.2920G>T | D974Y 2D AIThe SynGAP1 missense variant D974Y is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the majority of computational evidence points to a benign impact, which is consistent with the absence of ClinVar pathogenic classification and gnomAD observations. Thus, the variant is most likely benign, and this assessment does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -4.290 | Likely Benign | 0.384 | Ambiguous | Likely Benign | 0.130 | Likely Benign | -1.85 | Neutral | 0.716 | Possibly Damaging | 0.284 | Benign | 4.13 | Benign | 0.01 | Affected | 0.1072 | 0.6627 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2921A>C | D974A 2D AIThe SynGAP1 missense variant D974A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of available predictions indicates that D974A is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -2.619 | Likely Benign | 0.224 | Likely Benign | Likely Benign | 0.242 | Likely Benign | -0.96 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.22 | Benign | 0.04 | Affected | 0.3398 | 0.7129 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2921A>G | D974G 2D AIThe SynGAP1 missense variant D974G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -1.638 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.203 | Likely Benign | -0.72 | Neutral | 0.036 | Benign | 0.026 | Benign | 4.18 | Benign | 0.06 | Tolerated | 0.3379 | 0.6771 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.2921A>T | D974V 2D AIThe SynGAP1 missense variant D974V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective predictions strongly suggest that D974V is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -3.629 | Likely Benign | 0.351 | Ambiguous | Likely Benign | 0.211 | Likely Benign | -1.49 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.16 | Benign | 0.01 | Affected | 0.1449 | 0.7286 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2922C>A | D974E 2D AIThe SynGAP1 missense variant D974E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -3.617 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -0.63 | Neutral | 0.036 | Benign | 0.026 | Benign | 4.26 | Benign | 0.05 | Affected | 0.2119 | 0.7516 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2922C>G | D974E 2D AIThe SynGAP1 missense variant D974E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -3.617 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -0.63 | Neutral | 0.036 | Benign | 0.026 | Benign | 4.26 | Benign | 0.05 | Affected | 0.2119 | 0.7516 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2923A>C | T975P 2D AIThe SynGAP1 missense variant T975P has no ClinVar entry and is not reported in gnomAD. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.969331 | Binding | 0.332 | 0.890 | 0.625 | -2.181 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.286 | Likely Benign | -1.21 | Neutral | 0.000 | Benign | 0.002 | Benign | 4.14 | Benign | 0.06 | Tolerated | 0.2047 | 0.4758 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2917G>C | G973R 2D AIThe SynGAP1 missense variant G973R is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta results are not available for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.959498 | Binding | 0.390 | 0.897 | 0.625 | -3.950 | Likely Benign | 0.329 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -0.37 | Neutral | 0.001 | Benign | 0.003 | Benign | 4.16 | Benign | 0.01 | Affected | 0.0930 | 0.4532 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2917G>A | G973R 2D AIThe SynGAP1 missense variant G973R is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.959498 | Binding | 0.390 | 0.897 | 0.625 | -3.950 | Likely Benign | 0.329 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -0.37 | Neutral | 0.001 | Benign | 0.003 | Benign | 4.16 | Benign | 0.01 | Affected | 0.0930 | 0.4532 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.2909A>G | E970G 2D AIThe SynGAP1 missense variant E970G is listed in ClinVar (ID 2013677.0) as Benign and is not reported in gnomAD. All available in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant. Overall, the computational evidence overwhelmingly supports a benign classification, which is consistent with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.951925 | Disordered | 0.953422 | Binding | 0.342 | 0.902 | 0.750 | Benign | 1 | -0.167 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -0.93 | Neutral | 0.144 | Benign | 0.058 | Benign | 4.09 | Benign | 0.10 | Tolerated | 4.32 | 2 | 0.2701 | 0.5833 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||
| c.2909A>T | E970V 2D AIThe SynGAP1 missense variant E970V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign interpretation, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.951925 | Disordered | 0.953422 | Binding | 0.342 | 0.902 | 0.750 | -2.791 | Likely Benign | 0.208 | Likely Benign | Likely Benign | 0.245 | Likely Benign | -1.08 | Neutral | 0.002 | Benign | 0.002 | Benign | 4.11 | Benign | 0.08 | Tolerated | 0.1990 | 0.7037 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.290A>C | E97A 2D AIThe SynGAP1 missense variant E97A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also likely benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for E97A, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.609018 | Binding | 0.340 | 0.867 | 0.625 | -3.091 | Likely Benign | 0.374 | Ambiguous | Likely Benign | 0.098 | Likely Benign | -0.58 | Neutral | 0.880 | Possibly Damaging | 0.636 | Possibly Damaging | 4.16 | Benign | 0.00 | Affected | 0.4505 | 0.7728 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.290A>G | E97G 2D AIThe SynGAP1 missense variant E97G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.609018 | Binding | 0.340 | 0.867 | 0.625 | -2.752 | Likely Benign | 0.282 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -1.03 | Neutral | 0.947 | Possibly Damaging | 0.727 | Possibly Damaging | 4.07 | Benign | 0.00 | Affected | 0.3286 | 0.6569 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.2910G>C | E970D 2D AIThe SynGAP1 missense variant E970D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification—there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.951925 | Disordered | 0.953422 | Binding | 0.342 | 0.902 | 0.750 | -3.381 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.44 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.14 | Benign | 0.33 | Tolerated | 0.2729 | 0.4610 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2910G>T | E970D 2D AIThe SynGAP1 missense variant E970D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.951925 | Disordered | 0.953422 | Binding | 0.342 | 0.902 | 0.750 | -3.381 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.44 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.14 | Benign | 0.33 | Tolerated | 0.2729 | 0.4610 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2911C>A | P971T 2D AIThe SynGAP1 missense variant P971T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.951523 | Binding | 0.545 | 0.905 | 0.625 | -5.627 | Likely Benign | 0.053 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -0.95 | Neutral | 0.001 | Benign | 0.003 | Benign | 3.96 | Benign | 0.00 | Affected | 0.1388 | 0.5888 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2911C>G | P971A 2D AIThe SynGAP1 missense variant P971A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.951523 | Binding | 0.545 | 0.905 | 0.625 | -4.244 | Likely Benign | 0.049 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.51 | Neutral | 0.000 | Benign | 0.002 | Benign | 4.05 | Benign | 0.00 | Affected | 0.3100 | 0.5282 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2912C>G | P971R 2D AIThe SynGAP1 missense variant P971R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.951523 | Binding | 0.545 | 0.905 | 0.625 | -4.407 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -1.01 | Neutral | 0.453 | Possibly Damaging | 0.078 | Benign | 3.91 | Benign | 0.00 | Affected | 0.1306 | 0.3818 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2912C>T | P971L 2D AIThe SynGAP1 missense variant P971L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.951523 | Binding | 0.545 | 0.905 | 0.625 | -4.892 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -1.57 | Neutral | 0.144 | Benign | 0.026 | Benign | 3.93 | Benign | 0.00 | Affected | 0.2046 | 0.5985 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2914C>A | P972T 2D AIThe SynGAP1 missense variant P972T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.954150 | Binding | 0.472 | 0.904 | 0.625 | -5.144 | Likely Benign | 0.057 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -1.01 | Neutral | 0.078 | Benign | 0.042 | Benign | 4.29 | Benign | 0.03 | Affected | 0.1802 | 0.5726 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2915C>A | P972H 2D AIThe SynGAP1 missense variant P972H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools (polyPhen‑2 HumDiv and SIFT) predict pathogenicity, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign classification, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.954150 | Binding | 0.472 | 0.904 | 0.625 | -4.791 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.049 | Likely Benign | -1.62 | Neutral | 0.589 | Possibly Damaging | 0.229 | Benign | 4.19 | Benign | 0.02 | Affected | 0.1930 | 0.4798 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2915C>T | P972L 2D AIThe SynGAP1 missense variant P972L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.954150 | Binding | 0.472 | 0.904 | 0.625 | -4.399 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.020 | Likely Benign | -1.73 | Neutral | 0.036 | Benign | 0.026 | Benign | 4.24 | Benign | 0.02 | Affected | 0.2107 | 0.5447 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2923A>G | T975A 2D AIThe SynGAP1 missense change T975A is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the computational evidence strongly supports a benign classification, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.871313 | Disordered | 0.969331 | Binding | 0.332 | 0.890 | 0.625 | -2.866 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.110 | Likely Benign | -0.71 | Neutral | 0.000 | Benign | 0.002 | Benign | 4.19 | Benign | 0.18 | Tolerated | 0.3821 | 0.4275 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2951A>C | K984T 2D  AIThe SynGAP1 missense variant K984T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the predictions are mixed; however, the SGM‑Consensus and the majority of benign‑predicting tools lean toward a benign interpretation. This assessment does not contradict ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.951648 | Binding | 0.288 | 0.895 | 0.750 | -3.429 | Likely Benign | 0.854 | Likely Pathogenic | Ambiguous | 0.087 | Likely Benign | -1.10 | Neutral | 0.951 | Possibly Damaging | 0.708 | Possibly Damaging | 2.71 | Benign | 0.00 | Affected | 0.2441 | 0.3463 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||||||||||||
| c.2981A>C | K994T 2D AIThe SynGAP1 missense variant K994T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Taken together, the majority of evidence points to a benign effect. There is no conflict with ClinVar status, as no ClinVar classification exists for this variant. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.930054 | Binding | 0.289 | 0.912 | 0.750 | -2.346 | Likely Benign | 0.226 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.22 | Neutral | 0.144 | Benign | 0.085 | Benign | 4.09 | Benign | 0.01 | Affected | 0.2503 | 0.4248 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||||||||||||
| c.2989G>A | A997T 2D AIThe SynGAP1 missense variant A997T is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (derived from the four high‑accuracy predictors) is benign; Foldetta results are unavailable. Taken together, the majority of computational evidence indicates a benign effect, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.948624 | Binding | 0.273 | 0.901 | 0.500 | Uncertain | 1 | -4.102 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -0.62 | Neutral | 0.224 | Benign | 0.120 | Benign | 4.17 | Benign | 0.00 | Affected | 4.32 | 4 | 0.1582 | 0.7182 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||
| c.2989G>C | A997P 2D AIThe SynGAP1 missense variant A997P is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.948624 | Binding | 0.273 | 0.901 | 0.500 | -2.014 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -1.17 | Neutral | 0.001 | Benign | 0.003 | Benign | 4.11 | Benign | 0.00 | Affected | 0.1938 | 0.5294 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2989G>T | A997S 2D AIThe SynGAP1 missense variant A997S is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.948624 | Binding | 0.273 | 0.901 | 0.500 | -3.362 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.63 | Neutral | 0.224 | Benign | 0.066 | Benign | 4.18 | Benign | 0.00 | Affected | 0.2661 | 0.5694 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.298T>A | Y100N 2D AIThe SynGAP1 missense variant Y100N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -2.579 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -0.66 | Neutral | 0.675 | Possibly Damaging | 0.099 | Benign | 4.23 | Benign | 0.00 | Affected | 0.2487 | 0.0373 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.298T>G | Y100D 2D AIThe SynGAP1 missense variant Y100D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -1.269 | Likely Benign | 0.215 | Likely Benign | Likely Benign | 0.233 | Likely Benign | -1.01 | Neutral | 0.003 | Benign | 0.003 | Benign | 4.22 | Benign | 0.00 | Affected | 0.4465 | 0.0373 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.2990C>A | A997D 2D AIThe SynGAP1 missense variant A997D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta results are not available for this variant. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of any ClinVar pathogenic annotation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.948624 | Binding | 0.273 | 0.901 | 0.500 | -5.251 | Likely Benign | 0.319 | Likely Benign | Likely Benign | 0.131 | Likely Benign | -1.09 | Neutral | 0.411 | Benign | 0.120 | Benign | 4.15 | Benign | 0.00 | Affected | 0.1815 | 0.2143 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2990C>G | A997G 2D AIThe SynGAP1 missense variant A997G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.948624 | Binding | 0.273 | 0.901 | 0.500 | -3.424 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.82 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.13 | Benign | 0.00 | Affected | 0.2145 | 0.4741 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2990C>T | A997V 2D AIThe SynGAP1 missense variant A997V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The prediction is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.948624 | Binding | 0.273 | 0.901 | 0.500 | -4.504 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.052 | Likely Benign | -1.01 | Neutral | 0.369 | Benign | 0.120 | Benign | 4.15 | Benign | 0.00 | Affected | 0.1232 | 0.6335 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2992G>A | A998T 2D AIThe SynGAP1 missense variant A998T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.759478 | Disordered | 0.951758 | Binding | 0.318 | 0.902 | 0.500 | -3.909 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -0.97 | Neutral | 0.611 | Possibly Damaging | 0.321 | Benign | 4.11 | Benign | 0.00 | Affected | 0.1620 | 0.6994 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2992G>C | A998P 2D AIThe SynGAP1 missense variant A998P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of evidence, including the high‑accuracy tools, points to a benign effect for A998P. This conclusion does not contradict ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.759478 | Disordered | 0.951758 | Binding | 0.318 | 0.902 | 0.500 | -2.220 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.171 | Likely Benign | -0.40 | Neutral | 0.971 | Probably Damaging | 0.690 | Possibly Damaging | 4.31 | Benign | 0.00 | Affected | 0.1924 | 0.5106 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2992G>T | A998S 2D AIThe SynGAP1 missense variant A998S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is therefore most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.759478 | Disordered | 0.951758 | Binding | 0.318 | 0.902 | 0.500 | -2.893 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.022 | Likely Benign | -0.42 | Neutral | 0.611 | Possibly Damaging | 0.237 | Benign | 4.14 | Benign | 0.00 | Affected | 0.2674 | 0.5506 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2993C>A | A998D 2D AIThe SynGAP1 missense variant A998D is not reported in ClinVar and has no entry in gnomAD. Consensus from multiple in‑silico predictors shows a split: benign calls come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default remains uncertain. The high‑accuracy assessment indicates that AlphaMissense‑Optimized predicts a benign effect, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports a likely benign outcome, and Foldetta data are unavailable. Overall, the majority of robust predictors lean toward a benign impact. Therefore, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.759478 | Disordered | 0.951758 | Binding | 0.318 | 0.902 | 0.500 | -5.481 | Likely Benign | 0.365 | Ambiguous | Likely Benign | 0.122 | Likely Benign | -1.55 | Neutral | 0.971 | Probably Damaging | 0.690 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 0.1754 | 0.2143 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2993C>G | A998G 2D AIThe SynGAP1 missense variant A998G is not reported in ClinVar or gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the available predictions points to a benign impact, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.759478 | Disordered | 0.951758 | Binding | 0.318 | 0.902 | 0.500 | -3.173 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.032 | Likely Benign | -1.29 | Neutral | 0.761 | Possibly Damaging | 0.396 | Benign | 4.13 | Benign | 0.00 | Affected | 0.2234 | 0.4741 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2987C>T | P996L 2D AIThe SynGAP1 missense variant P996L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect; there is no conflict with ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.775545 | Disordered | 0.942262 | Binding | 0.312 | 0.900 | 0.750 | -5.302 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -1.65 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.25 | Benign | 0.02 | Affected | 0.2057 | 0.6631 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2987C>G | P996R 2D AIThe SynGAP1 missense variant P996R is listed in ClinVar (ID 2808854.0) as benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates a benign impact. This prediction aligns with the ClinVar benign classification and does not contradict the existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.775545 | Disordered | 0.942262 | Binding | 0.312 | 0.900 | 0.750 | Benign | 1 | -4.457 | Likely Benign | 0.141 | Likely Benign | Likely Benign | 0.040 | Likely Benign | -1.04 | Neutral | 0.144 | Benign | 0.085 | Benign | 4.26 | Benign | 0.01 | Affected | 4.32 | 4 | 0.1297 | 0.3385 | -2 | 0 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||
| c.2981A>G | K994R 2D  AIThe SynGAP1 missense variant K994R is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.930054 | Binding | 0.289 | 0.912 | 0.750 | -1.990 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.071 | Likely Benign | 0.02 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.21 | Benign | 0.47 | Tolerated | 0.5064 | 0.1657 | Weaken | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||||||||||
| c.2981A>T | K994M 2D  AIThe SynGAP1 missense variant K994M is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Pathogenicity is suggested only by polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus (SGM‑Consensus) classifies the variant as likely benign, and AlphaMissense‑Optimized also predicts benign. No Foldetta stability analysis is available. Overall, the majority of evidence points to a benign impact, and this assessment does not conflict with ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.930054 | Binding | 0.289 | 0.912 | 0.750 | -2.974 | Likely Benign | 0.424 | Ambiguous | Likely Benign | 0.057 | Likely Benign | -1.21 | Neutral | 0.589 | Possibly Damaging | 0.187 | Benign | 4.04 | Benign | 0.00 | Affected | 0.1612 | 0.4395 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||||||||||||
| c.2982G>C | K994N 2D  AIThe SynGAP1 missense variant K994N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic call comes from SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, SGM‑Consensus confirms a Likely Benign status, and Foldetta data are unavailable. Taken together, the preponderance of evidence indicates that K994N is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.930054 | Binding | 0.289 | 0.912 | 0.750 | -3.724 | Likely Benign | 0.519 | Ambiguous | Likely Benign | 0.011 | Likely Benign | -0.73 | Neutral | 0.255 | Benign | 0.113 | Benign | 4.09 | Benign | 0.01 | Affected | 0.4010 | 0.2376 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.2982G>T | K994N 2D AIThe SynGAP1 missense variant K994N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic call comes from SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, SGM‑Consensus confirms a Likely Benign status, and Foldetta data are unavailable. Taken together, the preponderance of evidence indicates that K994N is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.930054 | Binding | 0.289 | 0.912 | 0.750 | -3.724 | Likely Benign | 0.519 | Ambiguous | Likely Benign | 0.009 | Likely Benign | -0.73 | Neutral | 0.255 | Benign | 0.113 | Benign | 4.09 | Benign | 0.01 | Affected | 0.4010 | 0.2376 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||||
| c.2983C>A | P995T 2D AIThe SynGAP1 missense variant P995T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect for P995T, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.935305 | Binding | 0.338 | 0.902 | 0.750 | -5.148 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.034 | Likely Benign | -1.15 | Neutral | 0.224 | Benign | 0.096 | Benign | 4.19 | Benign | 0.00 | Affected | 0.1467 | 0.5667 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2983C>G | P995A 2D AIThe SynGAP1 missense variant P995A is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of evidence points to a benign impact for P995A, and this conclusion is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.935305 | Binding | 0.338 | 0.902 | 0.750 | -4.465 | Likely Benign | 0.051 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -1.02 | Neutral | 0.001 | Benign | 0.008 | Benign | 4.22 | Benign | 0.00 | Affected | 0.3287 | 0.4841 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2983C>T | P995S 2D AIThe SynGAP1 missense variant P995S is listed in ClinVar (ID 436929.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). The only tool that predicts a pathogenic outcome is SIFT. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates “Likely Benign.” No Foldetta (FoldX‑MD/Rosetta) stability result is available, so it does not influence the assessment. Overall, the majority of predictions, including the high‑accuracy tools, point to a benign effect, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.935305 | Binding | 0.338 | 0.902 | 0.750 | Uncertain | 1 | -4.457 | Likely Benign | 0.071 | Likely Benign | Likely Benign | 0.042 | Likely Benign | -1.03 | Neutral | 0.011 | Benign | 0.015 | Benign | 4.24 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3204 | 0.5236 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||
| c.2984C>A | P995H 2D AIThe SynGAP1 missense variant P995H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P995H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.935305 | Binding | 0.338 | 0.902 | 0.750 | -5.347 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.75 | Neutral | 0.832 | Possibly Damaging | 0.600 | Possibly Damaging | 4.16 | Benign | 0.00 | Affected | 0.1618 | 0.4571 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2984C>G | P995R 2D AIThe SynGAP1 missense variant P995R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv and SIFT predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.935305 | Binding | 0.338 | 0.902 | 0.750 | -4.605 | Likely Benign | 0.141 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -1.06 | Neutral | 0.586 | Possibly Damaging | 0.304 | Benign | 4.18 | Benign | 0.00 | Affected | 0.1370 | 0.3424 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2984C>T | P995L 2D AIThe SynGAP1 missense variant P995L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta results are not available for this variant. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.935305 | Binding | 0.338 | 0.902 | 0.750 | -4.948 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -1.03 | Neutral | 0.411 | Benign | 0.096 | Benign | 4.16 | Benign | 0.00 | Affected | 0.2294 | 0.6324 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.2986C>A | P996T 2D AIThe SynGAP1 missense variant P996T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.775545 | Disordered | 0.942262 | Binding | 0.312 | 0.900 | 0.750 | -5.138 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -0.71 | Neutral | 0.036 | Benign | 0.039 | Benign | 4.26 | Benign | 0.03 | Affected | 0.1418 | 0.6163 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2986C>G | P996A 2D AIThe SynGAP1 missense variant P996A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools that assess sequence conservation and structural impact uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the collective evidence strongly supports a benign classification, and this conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.775545 | Disordered | 0.942262 | Binding | 0.312 | 0.900 | 0.750 | -4.596 | Likely Benign | 0.045 | Likely Benign | Likely Benign | 0.086 | Likely Benign | -0.40 | Neutral | 0.000 | Benign | 0.002 | Benign | 4.30 | Benign | 0.86 | Tolerated | 0.3242 | 0.4961 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2987C>A | P996H 2D AIThe SynGAP1 missense variant P996H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.775545 | Disordered | 0.942262 | Binding | 0.312 | 0.900 | 0.750 | -5.554 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -1.19 | Neutral | 0.001 | Benign | 0.003 | Benign | 4.25 | Benign | 0.00 | Affected | 0.1585 | 0.5160 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2993C>T | A998V 2D AIThe SynGAP1 missense variant A998V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.759478 | Disordered | 0.951758 | Binding | 0.318 | 0.902 | 0.500 | -4.795 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -1.09 | Neutral | 0.245 | Benign | 0.138 | Benign | 4.09 | Benign | 0.00 | Affected | 0.1281 | 0.6147 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2995T>A | S999T 2D AIThe SynGAP1 missense variant S999T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.950682 | Binding | 0.262 | 0.897 | 0.625 | -3.961 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -0.94 | Neutral | 0.625 | Possibly Damaging | 0.266 | Benign | 2.69 | Benign | 0.04 | Affected | 0.1469 | 0.6651 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2995T>C | S999P 2D AIThe SynGAP1 missense variant S999P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for S999P, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.950682 | Binding | 0.262 | 0.897 | 0.625 | -2.279 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -1.05 | Neutral | 0.966 | Probably Damaging | 0.773 | Possibly Damaging | 2.65 | Benign | 0.04 | Affected | 0.1963 | 0.6268 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.3001C>A | L1001I 2D  AIThe SynGAP1 missense variant L1001I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy consensus methods reinforce the benign assessment: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools points to a benign effect, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | -4.486 | Likely Benign | 0.076 | Likely Benign | Likely Benign | 0.034 | Likely Benign | -0.17 | Neutral | 0.022 | Benign | 0.018 | Benign | 2.69 | Benign | 0.00 | Affected | 0.0994 | 0.3264 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.3001C>G | L1001V 2D AIThe SynGAP1 missense variant L1001V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the available predictions points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | -3.865 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -0.20 | Neutral | 0.022 | Benign | 0.008 | Benign | 2.71 | Benign | 0.00 | Affected | 0.1569 | 0.2714 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3001C>T | L1001F 2D  AIThe SynGAP1 missense variant L1001F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while the SGM‑Consensus (derived from the majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (majority vote of the four high‑confidence tools) is benign; Foldetta data are unavailable. Overall, the preponderance of evidence supports a benign classification for L1001F, and this conclusion does not conflict with ClinVar, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | -4.712 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -0.85 | Neutral | 0.934 | Possibly Damaging | 0.617 | Possibly Damaging | 2.65 | Benign | 0.00 | Affected | 0.0703 | 0.2876 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.3002T>A | L1001H 2D  AIThe SynGAP1 missense variant L1001H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | -3.119 | Likely Benign | 0.258 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -1.02 | Neutral | 0.997 | Probably Damaging | 0.870 | Possibly Damaging | 2.64 | Benign | 0.00 | Affected | 0.1141 | 0.0958 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3002T>C | L1001P 2D AIThe SynGAP1 missense variant L1001P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence—including high‑accuracy tools—points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | Uncertain | 1 | -3.071 | Likely Benign | 0.209 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -1.02 | Neutral | 0.966 | Probably Damaging | 0.690 | Possibly Damaging | 2.65 | Benign | 0.00 | Affected | 4.32 | 4 | 0.3322 | 0.0929 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||
| c.3002T>G | L1001R 2D AIThe SynGAP1 missense variant L1001R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | -2.285 | Likely Benign | 0.320 | Likely Benign | Likely Benign | 0.091 | Likely Benign | -1.09 | Neutral | 0.966 | Probably Damaging | 0.708 | Possibly Damaging | 2.67 | Benign | 0.00 | Affected | 0.1299 | 0.0832 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.3004C>A | H1002N 2D AIThe SynGAP1 missense variant H1002N is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The AlphaMissense‑Default score is uncertain, and no Foldetta stability assessment is available. High‑accuracy methods reinforce the benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign, with no Foldetta data to contradict. Overall, the preponderance of evidence points to a benign effect for H1002N, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.953758 | Binding | 0.285 | 0.900 | 0.500 | -5.622 | Likely Benign | 0.466 | Ambiguous | Likely Benign | 0.076 | Likely Benign | -1.41 | Neutral | 0.801 | Possibly Damaging | 0.596 | Possibly Damaging | 2.76 | Benign | 1.00 | Tolerated | 0.2084 | 0.3057 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.3004C>G | H1002D 2D AIThe SynGAP1 missense variant H1002D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.953758 | Binding | 0.285 | 0.900 | 0.500 | -6.511 | Likely Benign | 0.852 | Likely Pathogenic | Ambiguous | 0.218 | Likely Benign | -2.09 | Neutral | 0.891 | Possibly Damaging | 0.673 | Possibly Damaging | 2.75 | Benign | 0.89 | Tolerated | 0.2597 | 0.2335 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||||||||||||
| c.3004C>T | H1002Y 2D AIThe SynGAP1 missense variant H1002Y is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors indicates a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all score the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also classifies it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The AlphaMissense‑Default tool remains uncertain, and no Foldetta stability assessment is available. High‑accuracy methods that are available—AlphaMissense‑Optimized and the SGM‑Consensus—both support a benign interpretation. Therefore, the variant is most likely benign based on the current predictive evidence, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.953758 | Binding | 0.285 | 0.900 | 0.500 | -6.159 | Likely Benign | 0.489 | Ambiguous | Likely Benign | 0.181 | Likely Benign | -1.85 | Neutral | 0.961 | Probably Damaging | 0.808 | Possibly Damaging | 2.74 | Benign | 0.14 | Tolerated | 0.1243 | 0.4427 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.3005A>C | H1002P 2D AIThe SynGAP1 missense variant H1002P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus likewise indicates Likely Benign. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the majority of predictions and the high‑accuracy consensus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.953758 | Binding | 0.285 | 0.900 | 0.500 | -4.616 | Likely Benign | 0.242 | Likely Benign | Likely Benign | 0.213 | Likely Benign | -2.02 | Neutral | 0.989 | Probably Damaging | 0.874 | Possibly Damaging | 2.77 | Benign | 0.28 | Tolerated | 0.2015 | 0.3738 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.3005A>T | H1002L 2D AIThe SynGAP1 H1002L missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and the absence of the variant in population databases, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.953758 | Binding | 0.285 | 0.900 | 0.500 | -6.448 | Likely Benign | 0.556 | Ambiguous | Likely Benign | 0.157 | Likely Benign | -3.12 | Deleterious | 0.801 | Possibly Damaging | 0.602 | Possibly Damaging | 2.79 | Benign | 0.13 | Tolerated | 0.1296 | 0.5088 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3006T>A | H1002Q 2D AIThe SynGAP1 missense variant H1002Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.953758 | Binding | 0.285 | 0.900 | 0.500 | -5.071 | Likely Benign | 0.650 | Likely Pathogenic | Likely Benign | 0.140 | Likely Benign | -1.83 | Neutral | 0.801 | Possibly Damaging | 0.602 | Possibly Damaging | 2.77 | Benign | 0.23 | Tolerated | 0.1927 | 0.3510 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3006T>G | H1002Q 2D AIThe SynGAP1 missense variant H1002Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this conclusion, so the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.953758 | Binding | 0.285 | 0.900 | 0.500 | -5.071 | Likely Benign | 0.650 | Likely Pathogenic | Likely Benign | 0.140 | Likely Benign | -1.83 | Neutral | 0.801 | Possibly Damaging | 0.602 | Possibly Damaging | 2.77 | Benign | 0.23 | Tolerated | 0.1927 | 0.3510 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.3000C>G | I1000M 2D AIThe SynGAP1 missense variant I1000M is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | -3.541 | Likely Benign | 0.161 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -0.32 | Neutral | 0.437 | Benign | 0.108 | Benign | 2.70 | Benign | 0.17 | Tolerated | 0.0769 | 0.3104 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.29G>T | R10L 2D  AIThe SynGAP1 missense variant R10L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta results are not available for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.513657 | Binding | 0.330 | 0.915 | 0.625 | -3.269 | Likely Benign | 0.244 | Likely Benign | Likely Benign | 0.143 | Likely Benign | 0.09 | Neutral | 0.058 | Benign | 0.009 | Benign | 4.21 | Benign | 0.00 | Affected | 0.2255 | 0.5261 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2995T>G | S999A 2D AIThe SynGAP1 missense variant S999A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.950682 | Binding | 0.262 | 0.897 | 0.625 | -3.719 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -0.83 | Neutral | 0.005 | Benign | 0.016 | Benign | 2.71 | Benign | 0.81 | Tolerated | 0.4516 | 0.5434 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2996C>A | S999Y 2D  AIThe SynGAP1 missense variant S999Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S999Y, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.950682 | Binding | 0.262 | 0.897 | 0.625 | -6.446 | Likely Benign | 0.346 | Ambiguous | Likely Benign | 0.069 | Likely Benign | -1.74 | Neutral | 0.934 | Possibly Damaging | 0.559 | Possibly Damaging | 2.64 | Benign | 0.00 | Affected | 0.0881 | 0.6249 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||||
| c.2996C>G | S999C 2D AIThe SynGAP1 missense variant S999C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S999C, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.950682 | Binding | 0.262 | 0.897 | 0.625 | -7.751 | In-Between | 0.139 | Likely Benign | Likely Benign | 0.049 | Likely Benign | -1.68 | Neutral | 0.991 | Probably Damaging | 0.873 | Possibly Damaging | 2.63 | Benign | 0.01 | Affected | 0.1131 | 0.6212 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2996C>T | S999F 2D AIThe SynGAP1 missense variant S999F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized independently predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect; this is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.950682 | Binding | 0.262 | 0.897 | 0.625 | -6.206 | Likely Benign | 0.368 | Ambiguous | Likely Benign | 0.072 | Likely Benign | -1.79 | Neutral | 0.966 | Probably Damaging | 0.837 | Possibly Damaging | 2.64 | Benign | 0.01 | Affected | 0.0865 | 0.6337 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.2998A>C | I1000L 2D AIThe SynGAP1 missense variant I1000L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | -2.103 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -0.34 | Neutral | 0.211 | Benign | 0.108 | Benign | 2.76 | Benign | 0.70 | Tolerated | 0.0916 | 0.4034 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.2998A>G | I1000V 2D AIThe SynGAP1 missense variant I1000V is listed in ClinVar (ID 2572013.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Functional prediction tools that assess evolutionary conservation and structural impact (REVEL, SIFT, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default) all converge on a benign outcome. No tool in the dataset predicts pathogenicity. High‑accuracy predictors reinforce this consensus: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | Uncertain | 2 | -4.102 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.086 | Likely Benign | -0.20 | Neutral | 0.437 | Benign | 0.170 | Benign | 2.76 | Benign | 0.81 | Tolerated | 4.32 | 4 | 0.1220 | 0.3404 | 3 | 4 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||
| c.2998A>T | I1000F 2D AIThe SynGAP1 missense variant I1000F is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of predictions indicates that I1000F is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | -3.801 | Likely Benign | 0.201 | Likely Benign | Likely Benign | 0.147 | Likely Benign | -1.02 | Neutral | 0.968 | Probably Damaging | 0.713 | Possibly Damaging | 2.70 | Benign | 0.13 | Tolerated | 0.0594 | 0.3259 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.2999T>A | I1000N 2D AIThe SynGAP1 missense variant I1000N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | -5.246 | Likely Benign | 0.677 | Likely Pathogenic | Likely Benign | 0.145 | Likely Benign | -0.82 | Neutral | 0.995 | Probably Damaging | 0.913 | Probably Damaging | 2.72 | Benign | 0.16 | Tolerated | 0.1001 | 0.0900 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2999T>C | I1000T 2D  AIThe SynGAP1 missense variant I1000T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and there is no ClinVar status to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | -4.748 | Likely Benign | 0.721 | Likely Pathogenic | Likely Benign | 0.131 | Likely Benign | -0.87 | Neutral | 0.896 | Possibly Damaging | 0.596 | Possibly Damaging | 2.78 | Benign | 0.29 | Tolerated | 0.1045 | 0.1594 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||||||||||||
| c.2999T>G | I1000S 2D AIThe SynGAP1 missense variant I1000S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence (seven benign vs. three pathogenic predictions) supports a benign classification. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | -3.694 | Likely Benign | 0.587 | Likely Pathogenic | Likely Benign | 0.151 | Likely Benign | -0.38 | Neutral | 0.946 | Possibly Damaging | 0.673 | Possibly Damaging | 2.80 | Benign | 0.19 | Tolerated | 0.2501 | 0.1270 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.299A>C | Y100S 2D AIThe SynGAP1 missense variant Y100S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -1.217 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.182 | Likely Benign | -0.14 | Neutral | 0.675 | Possibly Damaging | 0.175 | Benign | 4.31 | Benign | 0.00 | Affected | 0.5066 | 0.1600 | Weaken | -3 | -2 | 0.5 | -76.10 | ||||||||||||||||||||||||||||||||||||||
| c.299A>G | Y100C 2D  AIThe SynGAP1 missense variant Y100C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -3.789 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.172 | Likely Benign | -0.88 | Neutral | 0.994 | Probably Damaging | 0.816 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.3073 | 0.2213 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||||||||||||
| c.299A>T | Y100F 2D AIThe SynGAP1 missense variant Y100F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -3.056 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -0.50 | Neutral | 0.928 | Possibly Damaging | 0.222 | Benign | 4.21 | Benign | 0.00 | Affected | 0.2784 | 0.3087 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.3007A>C | S1003R 2D AIThe SynGAP1 missense variant S1003R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the majority of evaluated tools (six pathogenic vs. three benign) indicate a pathogenic impact. This prediction aligns with the lack of ClinVar annotation and does not contradict any existing clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.947349 | Binding | 0.272 | 0.901 | 0.625 | -5.113 | Likely Benign | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.110 | Likely Benign | -1.88 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.48 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1151 | 0.3746 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||||
| c.2980A>C | K994Q 2D AIThe SynGAP1 missense variant K994Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.862302 | Disordered | 0.930054 | Binding | 0.289 | 0.912 | 0.750 | -1.947 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.105 | Likely Benign | -0.45 | Neutral | 0.002 | Benign | 0.004 | Benign | 4.16 | Benign | 0.03 | Affected | 0.4975 | 0.1889 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||||||||||||
| c.2951A>T | K984M 2D AIThe SynGAP1 missense variant K984M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.951648 | Binding | 0.288 | 0.895 | 0.750 | -4.761 | Likely Benign | 0.934 | Likely Pathogenic | Ambiguous | 0.141 | Likely Benign | -1.82 | Neutral | 0.995 | Probably Damaging | 0.944 | Probably Damaging | 2.60 | Benign | 0.00 | Affected | 0.1576 | 0.4168 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||||||||||||
| c.2958G>T | E986D 2D AIThe SynGAP1 missense variant E986D is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs two benign votes) and Foldetta results are unavailable. Overall, the majority of conventional tools lean toward pathogenicity, but the single high‑accuracy benign prediction and the inconclusive consensus leave the variant’s impact uncertain. There is no contradiction with ClinVar status, as the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.001 | Likely Benign | 0.628 | Likely Pathogenic | Likely Benign | 0.129 | Likely Benign | -1.22 | Neutral | 0.974 | Probably Damaging | 0.715 | Possibly Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.2081 | 0.5364 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2959G>A | D987N 2D AIThe SynGAP1 D987N missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -5.035 | Likely Benign | 0.698 | Likely Pathogenic | Likely Benign | 0.126 | Likely Benign | -1.42 | Neutral | 0.943 | Possibly Damaging | 0.755 | Possibly Damaging | 2.41 | Pathogenic | 0.49 | Tolerated | 0.1331 | 0.7427 | 2 | 1 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2959G>C | D987H 2D AIThe SynGAP1 missense variant D987H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and the Foldetta stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus result, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -5.580 | Likely Benign | 0.925 | Likely Pathogenic | Ambiguous | 0.249 | Likely Benign | -3.16 | Deleterious | 0.998 | Probably Damaging | 0.951 | Probably Damaging | 2.35 | Pathogenic | 0.02 | Affected | 0.1553 | 0.7629 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.2959G>T | D987Y 2D AIThe SynGAP1 missense variant D987Y is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as likely pathogenic (3 pathogenic vs 1 benign). AlphaMissense‑Optimized returns an uncertain result, and Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -6.208 | Likely Benign | 0.831 | Likely Pathogenic | Ambiguous | 0.274 | Likely Benign | -4.41 | Deleterious | 0.999 | Probably Damaging | 0.966 | Probably Damaging | 2.33 | Pathogenic | 0.01 | Affected | 0.0632 | 0.6818 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.295G>A | E99K 2D AIThe SynGAP1 E99K missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) also as benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar reporting, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.645246 | Binding | 0.325 | 0.874 | 0.500 | -4.746 | Likely Benign | 0.678 | Likely Pathogenic | Likely Benign | 0.071 | Likely Benign | -0.88 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.14 | Benign | 0.00 | Affected | 0.2752 | 0.8149 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2960A>C | D987A 2D AIThe SynGAP1 D987A missense variant is not reported in ClinVar and has no gnomAD entry. Consensus prediction tools that classify the change as benign include REVEL and ESM1b, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) and the SGM‑Consensus score (Likely Pathogenic) indicate a pathogenic effect. Grouping by agreement, benign predictions are limited to two tools, while pathogenic predictions are supported by seven distinct algorithms. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized returns an uncertain result, SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a Likely Pathogenic classification, and the protein‑folding stability method Foldetta is unavailable for this variant. Overall, the preponderance of evidence points to a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -4.880 | Likely Benign | 0.853 | Likely Pathogenic | Ambiguous | 0.261 | Likely Benign | -3.72 | Deleterious | 0.943 | Possibly Damaging | 0.686 | Possibly Damaging | 2.39 | Pathogenic | 0.02 | Affected | 0.3930 | 0.6846 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.2960A>G | D987G 2D AIThe SynGAP1 missense variant D987G (ClinVar ID 1061058.0) is listed as ClinVar status Uncertain and is not reported in gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, SIFT, and ESM1b, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates majority votes from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further indicate that AlphaMissense‑Optimized is uncertain, while Foldetta data are unavailable. Overall, the majority of evidence points toward a pathogenic effect, aligning with the SGM‑Consensus but contradicting the ClinVar Uncertain designation. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment is in conflict with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | Uncertain | 1 | -4.782 | Likely Benign | 0.849 | Likely Pathogenic | Ambiguous | 0.234 | Likely Benign | -2.79 | Deleterious | 0.943 | Possibly Damaging | 0.808 | Possibly Damaging | 2.45 | Pathogenic | 0.07 | Tolerated | 4.32 | 2 | 0.3835 | 0.6710 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||
| c.2960A>T | D987V 2D AIThe SynGAP1 missense variant D987V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that D987V is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -4.647 | Likely Benign | 0.857 | Likely Pathogenic | Ambiguous | 0.389 | Likely Benign | -4.01 | Deleterious | 0.992 | Probably Damaging | 0.913 | Probably Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.0907 | 0.7202 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2961C>G | D987E 2D AIThe SynGAP1 missense variant D987E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus indicates Likely Benign, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for D987E, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -4.406 | Likely Benign | 0.425 | Ambiguous | Likely Benign | 0.124 | Likely Benign | -1.36 | Neutral | 0.049 | Benign | 0.044 | Benign | 2.64 | Benign | 0.04 | Affected | 4.32 | 2 | 0.1586 | 0.7239 | 2 | 3 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.2962C>A | L988I 2D AIThe SynGAP1 missense variant L988I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.918781 | Binding | 0.360 | 0.913 | 0.750 | -4.226 | Likely Benign | 0.204 | Likely Benign | Likely Benign | 0.120 | Likely Benign | -1.08 | Neutral | 0.924 | Possibly Damaging | 0.652 | Possibly Damaging | 2.70 | Benign | 0.00 | Affected | 0.1034 | 0.4143 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.2962C>G | L988V 2D AIThe SynGAP1 missense variant L988V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for L988V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.918781 | Binding | 0.360 | 0.913 | 0.750 | -3.626 | Likely Benign | 0.226 | Likely Benign | Likely Benign | 0.096 | Likely Benign | -1.42 | Neutral | 0.856 | Possibly Damaging | 0.474 | Possibly Damaging | 2.73 | Benign | 0.00 | Affected | 0.1652 | 0.3793 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2963T>A | L988H 2D AIThe SynGAP1 missense variant L988H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2). Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs four benign) lean toward pathogenicity. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.918781 | Binding | 0.360 | 0.913 | 0.750 | -4.779 | Likely Benign | 0.733 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -2.70 | Deleterious | 0.998 | Probably Damaging | 0.947 | Probably Damaging | 2.62 | Benign | 0.00 | Affected | 0.1179 | 0.1414 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2963T>C | L988P 2D AIThe SynGAP1 missense variant L988P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward pathogenicity. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.918781 | Binding | 0.360 | 0.913 | 0.750 | -3.848 | Likely Benign | 0.703 | Likely Pathogenic | Likely Benign | 0.216 | Likely Benign | -2.96 | Deleterious | 0.977 | Probably Damaging | 0.900 | Possibly Damaging | 2.62 | Benign | 0.00 | Affected | 0.3282 | 0.1698 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2958G>C | E986D 2D AIThe SynGAP1 missense variant E986D is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs two benign votes) and Foldetta results are unavailable. Overall, the majority of conventional tools lean toward pathogenicity, but the single high‑accuracy benign prediction and the inconclusive consensus leave the variant’s impact uncertain. There is no contradiction with ClinVar status, as the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.001 | Likely Benign | 0.628 | Likely Pathogenic | Likely Benign | 0.129 | Likely Benign | -1.22 | Neutral | 0.974 | Probably Damaging | 0.715 | Possibly Damaging | 2.32 | Pathogenic | 0.00 | Affected | 0.2081 | 0.5364 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.2957A>T | E986V 2D AIThe SynGAP1 E986V missense variant is not reported in ClinVar and has no gnomAD entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b, whereas pathogenic predictions arise from PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (benign), FATHMM (pathogenic), and PROVEAN (pathogenic)—also indicates pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a pathogenic effect for E986V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.511 | Likely Benign | 0.965 | Likely Pathogenic | Likely Pathogenic | 0.220 | Likely Benign | -3.48 | Deleterious | 0.018 | Benign | 0.028 | Benign | 2.10 | Pathogenic | 0.00 | Affected | 0.1146 | 0.7960 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2952G>T | K984N 2D AISynGAP1 missense variant K984N has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is Uncertain, SGM‑Consensus is Likely Benign, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.703578 | Disordered | 0.951648 | Binding | 0.288 | 0.895 | 0.750 | -3.020 | Likely Benign | 0.955 | Likely Pathogenic | Ambiguous | 0.091 | Likely Benign | 0.52 | Neutral | 0.951 | Possibly Damaging | 0.637 | Possibly Damaging | 2.89 | Benign | 0.00 | Affected | 4.32 | 1 | 0.4138 | 0.1677 | 0 | 1 | 0.4 | -14.07 | |||||||||||||||||||||||||||||||||||||
| c.2953A>C | S985R 2D AIThe SynGAP1 missense variant S985R has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign). Those that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains benign; Foldetta, a protein‑folding stability method, has no available result for this variant. Consequently, the evidence is split evenly between benign and pathogenic predictions, with no contradiction to ClinVar status. The variant’s impact remains uncertain, and no definitive benign or pathogenic classification can be assigned based solely on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.720929 | Disordered | 0.941547 | Binding | 0.302 | 0.896 | 0.750 | -6.148 | Likely Benign | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.174 | Likely Benign | -2.40 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.52 | Benign | 0.00 | Affected | 0.1213 | 0.3905 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2953A>G | S985G 2D AIThe SynGAP1 missense variant S985G is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT). The AlphaMissense‑Default score is uncertain, and Foldetta stability analysis is not available. High‑accuracy assessments show AlphaMissense‑Optimized predicting a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. Foldetta, which integrates FoldX‑MD and Rosetta outputs, has no reported result for this variant. Overall, the majority of reliable predictors and the SGM‑Consensus support a benign classification. The variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.720929 | Disordered | 0.941547 | Binding | 0.302 | 0.896 | 0.750 | -6.151 | Likely Benign | 0.546 | Ambiguous | Likely Benign | 0.132 | Likely Benign | -2.29 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 2.51 | Benign | 0.00 | Affected | 0.2684 | 0.4540 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.2953A>T | S985C 2D AIThe SynGAP1 missense variant S985C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and PROVEAN, whereas the majority of tools predict a pathogenic impact: polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all classify the variant as pathogenic. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is classified as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple in silico predictors indicates that S985C is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.720929 | Disordered | 0.941547 | Binding | 0.302 | 0.896 | 0.750 | -8.918 | Likely Pathogenic | 0.860 | Likely Pathogenic | Ambiguous | 0.147 | Likely Benign | -2.49 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.48 | Pathogenic | 0.00 | Affected | 0.1531 | 0.5395 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2954G>A | S985N 2D  AIThe SynGAP1 missense variant S985N is listed in ClinVar (ID 2087879.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. Separately, the high‑accuracy AlphaMissense‑Optimized result is “Uncertain,” and the Foldetta protein‑folding stability assessment is unavailable. Based on the overall distribution of predictions, the variant is most likely benign; this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.720929 | Disordered | 0.941547 | Binding | 0.302 | 0.896 | 0.750 | Uncertain | 1 | -6.979 | Likely Benign | 0.845 | Likely Pathogenic | Ambiguous | 0.088 | Likely Benign | -1.68 | Neutral | 0.991 | Probably Damaging | 0.988 | Probably Damaging | 2.65 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1812 | 0.4822 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||||||||
| c.2954G>C | S985T 2D AIThe SynGAP1 missense variant S985T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S985T, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.720929 | Disordered | 0.941547 | Binding | 0.302 | 0.896 | 0.750 | -5.658 | Likely Benign | 0.475 | Ambiguous | Likely Benign | 0.153 | Likely Benign | -1.62 | Neutral | 0.979 | Probably Damaging | 0.982 | Probably Damaging | 2.54 | Benign | 0.00 | Affected | 0.1889 | 0.5844 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2954G>T | S985I 2D AIThe SynGAP1 missense variant S985I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. ESM1b remains uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that S985I is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.720929 | Disordered | 0.941547 | Binding | 0.302 | 0.896 | 0.750 | -7.858 | In-Between | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.131 | Likely Benign | -2.78 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 2.50 | Benign | 0.00 | Affected | 0.1367 | 0.5206 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2955T>A | S985R 2D AIThe SynGAP1 missense variant S985R has no ClinVar entry and is not reported in gnomAD. Consensus predictions from multiple in‑silico tools are split: benign calls come from REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) which reports a likely benign outcome. Pathogenic calls arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments are mixed: AlphaMissense‑Optimized predicts pathogenic, whereas the SGM‑Consensus (a majority‑vote aggregator) predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the predictions are evenly divided, leaving the variant’s clinical significance uncertain; it does not contradict any ClinVar status because none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.720929 | Disordered | 0.941547 | Binding | 0.302 | 0.896 | 0.750 | -6.148 | Likely Benign | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.203 | Likely Benign | -2.40 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.52 | Benign | 0.00 | Affected | 0.1213 | 0.3905 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2955T>G | S985R 2D AISynGAP1 missense variant S985R has no ClinVar record and is not present in gnomAD. Computational predictions are mixed: benign calls come from REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign). Pathogenic calls come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy tools give a split result: AlphaMissense‑Optimized predicts pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely benign; Foldetta data are unavailable. Consequently, the variant’s predicted impact is ambiguous, with an equal number of benign and pathogenic scores. No ClinVar evidence contradicts these predictions. Thus, the variant is best classified as of uncertain significance, with no clear bias toward benign or pathogenic status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.720929 | Disordered | 0.941547 | Binding | 0.302 | 0.896 | 0.750 | -6.148 | Likely Benign | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.203 | Likely Benign | -2.40 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.52 | Benign | 0.00 | Affected | 0.1213 | 0.3905 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.2956G>A | E986K 2D AIThe SynGAP1 missense variant E986K is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are REVEL and PROVEAN, while those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The remaining tools, ESM1b and AlphaMissense‑Optimized, return uncertain results and are not considered evidence for either side. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a pathogenic verdict (2 pathogenic vs. 1 benign, with one uncertain). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑accuracy predictors classify the variant as pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -7.174 | In-Between | 0.950 | Likely Pathogenic | Ambiguous | 0.164 | Likely Benign | -2.19 | Neutral | 0.924 | Possibly Damaging | 0.722 | Possibly Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.2760 | 0.7973 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.2956G>C | E986Q 2D  AIThe SynGAP1 missense variant E986Q is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward a pathogenic interpretation. This assessment does not contradict ClinVar status, as the variant is not yet catalogued there. Thus, based on current computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.471 | Likely Benign | 0.839 | Likely Pathogenic | Ambiguous | 0.160 | Likely Benign | -1.66 | Neutral | 0.974 | Probably Damaging | 0.842 | Possibly Damaging | 2.14 | Pathogenic | 0.00 | Affected | 0.1700 | 0.7589 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2957A>C | E986A 2D AIThe SynGAP1 missense variant E986A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, and ESM1b, whereas tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split; Foldetta stability analysis is unavailable. Overall, the evidence is evenly divided, with no clear majority. Based on the current predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.653 | Likely Benign | 0.895 | Likely Pathogenic | Ambiguous | 0.160 | Likely Benign | -2.32 | Neutral | 0.552 | Possibly Damaging | 0.388 | Benign | 2.14 | Pathogenic | 0.00 | Affected | 0.4207 | 0.7733 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.2957A>G | E986G 2D AIThe SynGAP1 missense variant E986G is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas seven tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this assessment does not conflict with the ClinVar status, which currently contains no classification for E986G. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -5.584 | Likely Benign | 0.834 | Likely Pathogenic | Ambiguous | 0.219 | Likely Benign | -3.14 | Deleterious | 0.924 | Possibly Damaging | 0.784 | Possibly Damaging | 2.13 | Pathogenic | 0.00 | Affected | 0.2866 | 0.6225 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.2963T>G | L988R 2D AIThe SynGAP1 missense variant L988R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.918781 | Binding | 0.360 | 0.913 | 0.750 | -4.412 | Likely Benign | 0.681 | Likely Pathogenic | Likely Benign | 0.202 | Likely Benign | -2.39 | Neutral | 0.954 | Possibly Damaging | 0.867 | Possibly Damaging | 2.69 | Benign | 0.00 | Affected | 0.1257 | 0.1088 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2965T>A | S989T 2D AIThe SynGAP1 missense variant S989T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign outcome. No Foldetta stability data are available. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.908835 | Binding | 0.296 | 0.911 | 0.750 | -3.995 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -1.48 | Neutral | 0.770 | Possibly Damaging | 0.396 | Benign | 2.66 | Benign | 0.00 | Affected | 0.1183 | 0.5377 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2965T>C | S989P 2D AIThe SynGAP1 missense variant S989P is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign; Foldetta results are unavailable. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.908835 | Binding | 0.296 | 0.911 | 0.750 | -2.688 | Likely Benign | 0.209 | Likely Benign | Likely Benign | 0.147 | Likely Benign | -2.48 | Neutral | 0.010 | Benign | 0.015 | Benign | 2.67 | Benign | 0.00 | Affected | 0.1690 | 0.4941 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2972G>A | G991E 2D AIThe SynGAP1 missense variant G991E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic outcome. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized independently predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.911393 | Binding | 0.286 | 0.920 | 0.750 | -4.729 | Likely Benign | 0.341 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.30 | Neutral | 0.846 | Possibly Damaging | 0.697 | Possibly Damaging | 4.13 | Benign | 0.01 | Affected | 0.1480 | 0.4277 | 0 | -2 | -3.1 | 72.06 | |||||||||||||||||||||||||||||||||||||||
| c.2972G>T | G991V 2D AIThe SynGAP1 missense variant G991V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.911393 | Binding | 0.286 | 0.920 | 0.750 | -4.129 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.064 | Likely Benign | -1.61 | Neutral | 0.440 | Benign | 0.253 | Benign | 4.16 | Benign | 0.01 | Affected | 0.1148 | 0.3961 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2974G>C | V992L 2D  AIThe SynGAP1 missense variant V992L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.921728 | Binding | 0.331 | 0.917 | 0.750 | -2.333 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.060 | Likely Benign | -0.50 | Neutral | 0.086 | Benign | 0.038 | Benign | 4.24 | Benign | 0.16 | Tolerated | 0.1211 | 0.5202 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2974G>T | V992F 2D  AIThe SynGAP1 missense variant V992F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and there is no conflict with ClinVar status, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.921728 | Binding | 0.331 | 0.917 | 0.750 | -3.131 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -1.25 | Neutral | 0.680 | Possibly Damaging | 0.356 | Benign | 4.17 | Benign | 0.04 | Affected | 0.0832 | 0.4083 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2975T>A | V992D 2D  AIThe SynGAP1 missense variant V992D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only SIFT predicts pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates that V992D is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.921728 | Binding | 0.331 | 0.917 | 0.750 | -3.976 | Likely Benign | 0.306 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -1.24 | Neutral | 0.302 | Benign | 0.158 | Benign | 4.21 | Benign | 0.05 | Affected | 0.1645 | 0.1056 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2975T>C | V992A 2D  AIThe SynGAP1 missense variant V992A is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a benign outcome: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. No tool in the dataset indicates a pathogenic effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.921728 | Binding | 0.331 | 0.917 | 0.750 | -2.607 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -0.08 | Neutral | 0.000 | Benign | 0.003 | Benign | 4.29 | Benign | 0.41 | Tolerated | 0.3041 | 0.2668 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.2975T>G | V992G 2D  AIThe SynGAP1 missense variant V992G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). All available in silico predictors classify the variant as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity, so the benign group includes every listed predictor, while the pathogenic group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.754692 | Disordered | 0.921728 | Binding | 0.331 | 0.917 | 0.750 | -1.906 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -0.28 | Neutral | 0.056 | Benign | 0.086 | Benign | 4.21 | Benign | 0.16 | Tolerated | 0.2114 | 0.2612 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2977C>A | P993T 2D AIThe SynGAP1 missense variant P993T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.923979 | Binding | 0.319 | 0.908 | 0.750 | -4.444 | Likely Benign | 0.061 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -0.65 | Neutral | 0.001 | Benign | 0.010 | Benign | 4.32 | Benign | 0.04 | Affected | 0.1603 | 0.5789 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.2977C>G | P993A 2D AIThe SynGAP1 missense variant P993A is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign. Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.923979 | Binding | 0.319 | 0.908 | 0.750 | -4.030 | Likely Benign | 0.052 | Likely Benign | Likely Benign | 0.036 | Likely Benign | -0.46 | Neutral | 0.001 | Benign | 0.006 | Benign | 4.26 | Benign | 0.07 | Tolerated | 0.3487 | 0.4991 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.2978C>A | P993H 2D AIThe SynGAP1 missense variant P993H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P993H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.923979 | Binding | 0.319 | 0.908 | 0.750 | -4.535 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -0.93 | Neutral | 0.938 | Possibly Damaging | 0.819 | Possibly Damaging | 4.11 | Benign | 0.00 | Affected | 0.1799 | 0.4591 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.2978C>G | P993R 2D AIThe SynGAP1 missense variant P993R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.923979 | Binding | 0.319 | 0.908 | 0.750 | -3.508 | Likely Benign | 0.158 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.85 | Neutral | 0.586 | Possibly Damaging | 0.478 | Possibly Damaging | 4.14 | Benign | 0.01 | Affected | 0.1469 | 0.3122 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.2978C>T | P993L 2D AIThe SynGAP1 missense variant P993L is reported in ClinVar as “Not listed” and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which contains no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.923979 | Binding | 0.319 | 0.908 | 0.750 | -3.581 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.028 | Likely Benign | -1.37 | Neutral | 0.224 | Benign | 0.138 | Benign | 4.14 | Benign | 0.01 | Affected | 0.2299 | 0.6697 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.297A>C | E99D 2D AIThe SynGAP1 missense variant E99D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that E99D is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.645246 | Binding | 0.325 | 0.874 | 0.500 | -3.323 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -0.78 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.09 | Benign | 0.00 | Affected | 0.2360 | 0.5200 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2971G>T | G991W 2D AIThe SynGAP1 missense variant G991W is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.911393 | Binding | 0.286 | 0.920 | 0.750 | -6.281 | Likely Benign | 0.336 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -2.32 | Neutral | 0.997 | Probably Damaging | 0.975 | Probably Damaging | 4.07 | Benign | 0.00 | Affected | 0.0745 | 0.3654 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||||
| c.2971G>C | G991R 2D AIThe SynGAP1 missense variant G991R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of evidence points to a benign effect for G991R, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.911393 | Binding | 0.286 | 0.920 | 0.750 | -3.934 | Likely Benign | 0.411 | Ambiguous | Likely Benign | 0.106 | Likely Benign | -1.20 | Neutral | 0.984 | Probably Damaging | 0.772 | Possibly Damaging | 4.11 | Benign | 0.01 | Affected | 4.32 | 2 | 0.0956 | 0.4181 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.2965T>G | S989A 2D AIThe SynGAP1 missense variant S989A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.908835 | Binding | 0.296 | 0.911 | 0.750 | -3.495 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -1.15 | Neutral | 0.580 | Possibly Damaging | 0.253 | Benign | 2.68 | Benign | 0.00 | Affected | 0.4580 | 0.4221 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2966C>A | S989Y 2D AIThe SynGAP1 missense variant S989Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. High‑accuracy predictions therefore indicate a benign outcome: AlphaMissense‑Optimized is benign, the SGM Consensus is benign, and Foldetta data are unavailable. Consequently, the variant is most likely benign based on the current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.795062 | Disordered | 0.908835 | Binding | 0.296 | 0.911 | 0.750 | -4.774 | Likely Benign | 0.471 | Ambiguous | Likely Benign | 0.103 | Likely Benign | -3.32 | Deleterious | 0.986 | Probably Damaging | 0.876 | Possibly Damaging | 2.60 | Benign | 0.00 | Affected | 0.0611 | 0.4815 | -3 | -2 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||||||
| c.2966C>G | S989C 2D AIThe SynGAP1 missense variant S989C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.908835 | Binding | 0.296 | 0.911 | 0.750 | -5.889 | Likely Benign | 0.160 | Likely Benign | Likely Benign | 0.080 | Likely Benign | -2.88 | Deleterious | 0.996 | Probably Damaging | 0.905 | Possibly Damaging | 2.59 | Benign | 0.00 | Affected | 0.0828 | 0.5202 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2966C>T | S989F 2D AIThe SynGAP1 missense variant S989F is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, and Foldetta results are unavailable. Overall, the majority of reliable predictors and the consensus high‑accuracy tools indicate a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.795062 | Disordered | 0.908835 | Binding | 0.296 | 0.911 | 0.750 | -4.115 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.109 | Likely Benign | -3.35 | Deleterious | 0.986 | Probably Damaging | 0.876 | Possibly Damaging | 2.60 | Benign | 0.00 | Affected | 0.0598 | 0.5103 | -3 | -2 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||||||
| c.2968T>A | S990T 2D AIThe SynGAP1 missense variant S990T is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.902387 | Binding | 0.301 | 0.919 | 0.750 | -4.439 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.026 | Likely Benign | -0.06 | Neutral | 0.001 | Benign | 0.007 | Benign | 2.89 | Benign | 1.00 | Tolerated | 0.1524 | 0.5598 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2968T>C | S990P 2D AIThe SynGAP1 missense variant S990P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of any ClinVar pathogenic annotation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.902387 | Binding | 0.301 | 0.919 | 0.750 | -3.150 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -1.95 | Neutral | 0.586 | Possibly Damaging | 0.377 | Benign | 2.74 | Benign | 0.01 | Affected | 0.1910 | 0.5345 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.2968T>G | S990A 2D AIThe SynGAP1 missense variant S990A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.902387 | Binding | 0.301 | 0.919 | 0.750 | -3.554 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.020 | Likely Benign | -0.51 | Neutral | 0.001 | Benign | 0.004 | Benign | 2.91 | Benign | 0.03 | Affected | 0.4537 | 0.4509 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.2969C>A | S990Y 2D AIThe SynGAP1 missense variant S990Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Taken together, the majority of evidence points to a benign impact. The predictions do not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.902387 | Binding | 0.301 | 0.919 | 0.750 | -4.272 | Likely Benign | 0.314 | Likely Benign | Likely Benign | 0.131 | Likely Benign | -2.52 | Deleterious | 0.832 | Possibly Damaging | 0.500 | Possibly Damaging | 2.74 | Benign | 0.00 | Affected | 0.0893 | 0.5832 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||||
| c.2969C>G | S990C 2D AIThe SynGAP1 missense variant S990C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for S990C, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.902387 | Binding | 0.301 | 0.919 | 0.750 | -5.753 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.112 | Likely Benign | -1.91 | Neutral | 0.938 | Possibly Damaging | 0.690 | Possibly Damaging | 2.73 | Benign | 0.01 | Affected | 0.1041 | 0.5823 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.2969C>T | S990F 2D AIThe SynGAP1 missense variant S990F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for S990F. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.902387 | Binding | 0.301 | 0.919 | 0.750 | -4.253 | Likely Benign | 0.290 | Likely Benign | Likely Benign | 0.107 | Likely Benign | -2.65 | Deleterious | 0.710 | Possibly Damaging | 0.272 | Benign | 2.75 | Benign | 0.00 | Affected | 0.0814 | 0.6021 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.296A>C | E99A 2D AIThe SynGAP1 E99A missense change is not reported in ClinVar and has no entry in gnomAD. Consensus‑based predictors cluster around a benign interpretation: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all score the variant as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” verdict. AlphaMissense‑Default remains uncertain, and no Foldetta stability assessment is available. High‑accuracy tools therefore converge on a benign prediction: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta data are missing. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.645246 | Binding | 0.325 | 0.874 | 0.500 | -3.173 | Likely Benign | 0.347 | Ambiguous | Likely Benign | 0.127 | Likely Benign | -1.49 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.07 | Benign | 0.00 | Affected | 0.4245 | 0.7548 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.296A>G | E99G 2D AIThe SynGAP1 missense variant E99G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.645246 | Binding | 0.325 | 0.874 | 0.500 | -3.031 | Likely Benign | 0.259 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -1.69 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.04 | Benign | 0.00 | Affected | 0.3048 | 0.6399 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.296A>T | E99V 2D AIThe SynGAP1 missense variant E99V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus likewise reports Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign effect for E99V, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.645246 | Binding | 0.325 | 0.874 | 0.500 | -3.628 | Likely Benign | 0.544 | Ambiguous | Likely Benign | 0.208 | Likely Benign | -1.69 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.02 | Benign | 0.00 | Affected | 0.1109 | 0.8175 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.297A>T | E99D 2D AIThe SynGAP1 missense variant E99D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that E99D is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.645246 | Binding | 0.325 | 0.874 | 0.500 | -3.323 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -0.78 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.09 | Benign | 0.00 | Affected | 0.2360 | 0.5200 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.3637A>C | N1213H 2D AIThe SynGAP1 missense variant N1213H is predicted to be benign by the majority of in‑silico tools. Benign predictions come from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT classifies the change as pathogenic, while the consensus score from the SGM framework (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized reports benign, and the SGM Consensus also reports likely benign. Foldetta, a protein‑folding stability predictor, has no available result for this variant. The variant is not listed in ClinVar and has no entry in gnomAD, so there is no external evidence to contradict the computational predictions. Based on the collective predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.580690 | Disordered | 0.521638 | Binding | 0.888 | 0.561 | 0.500 | -5.358 | Likely Benign | 0.147 | Likely Benign | Likely Benign | 0.036 | Likely Benign | -1.23 | Neutral | 0.015 | Benign | 0.028 | Benign | 2.69 | Benign | 0.02 | Affected | 0.0883 | 0.4516 | 2 | 1 | 0.3 | 23.04 | ||||||||||||||||||||||||||||||||||||||
| c.686A>C | K229T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K229T is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include Rosetta, Foldetta, premPS, and FATHMM. Those that predict pathogenicity are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus. High‑accuracy methods give a pathogenic result from AlphaMissense‑Optimized, a likely pathogenic verdict from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and a benign outcome from Foldetta. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.179055 | Structured | 0.310912 | Uncertain | 0.843 | 0.306 | 0.000 | -12.639 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.83 | Ambiguous | 0.0 | -0.08 | Likely Benign | 0.38 | Likely Benign | 0.00 | Likely Benign | 0.813 | Likely Pathogenic | -4.77 | Deleterious | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 5.86 | Benign | 0.01 | Affected | 0.1890 | 0.3029 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.691T>G | F231V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F231V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are polyPhen‑2 HumVar and FATHMM. All other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely pathogenic status; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also indicates pathogenicity. Taken together, the evidence overwhelmingly points to a pathogenic effect for F231V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.306467 | Uncertain | 0.895 | 0.300 | 0.000 | -13.201 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.16 | Destabilizing | 0.3 | 2.30 | Destabilizing | 2.23 | Destabilizing | 1.13 | Destabilizing | 0.910 | Likely Pathogenic | -5.90 | Deleterious | 0.759 | Possibly Damaging | 0.328 | Benign | 5.72 | Benign | 0.00 | Affected | 0.2212 | 0.3030 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.692T>A | F231Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F231Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on benign effects include Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict pathogenicity are REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX and premPS are inconclusive. High‑accuracy methods give AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of standard tools lean benign, but the two most accurate predictors and the consensus vote favor pathogenicity. Thus, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, which currently has no classification for F231Y. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.306467 | Uncertain | 0.895 | 0.300 | 0.000 | -9.831 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.69 | Ambiguous | 0.1 | 0.00 | Likely Benign | 0.35 | Likely Benign | 0.85 | Ambiguous | 0.687 | Likely Pathogenic | -2.60 | Deleterious | 0.437 | Benign | 0.079 | Benign | 5.50 | Benign | 0.12 | Tolerated | 0.1355 | 0.2732 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.692T>C | F231S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F231S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are polyPhen‑2 HumVar and FATHMM; all other evaluated algorithms—including REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.306467 | Uncertain | 0.895 | 0.300 | 0.000 | -14.655 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.89 | Destabilizing | 0.4 | 4.22 | Destabilizing | 3.56 | Destabilizing | 2.22 | Destabilizing | 0.909 | Likely Pathogenic | -6.92 | Deleterious | 0.608 | Possibly Damaging | 0.205 | Benign | 5.48 | Benign | 0.00 | Affected | 0.4297 | 0.0544 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.692T>G | F231C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F231C is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity largely agree: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a deleterious effect, while only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among the majority of tools and the high‑accuracy methods, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.306467 | Uncertain | 0.895 | 0.300 | 0.000 | -13.315 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.26 | Destabilizing | 0.3 | 2.82 | Destabilizing | 2.54 | Destabilizing | 2.04 | Destabilizing | 0.937 | Likely Pathogenic | -6.89 | Deleterious | 0.992 | Probably Damaging | 0.707 | Possibly Damaging | 5.49 | Benign | 0.00 | Affected | 0.2527 | 0.1612 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.693T>A | F231L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F231L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that agree on a pathogenic effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; the SGM‑Consensus score is “Likely Pathogenic.” Predictions that are uncertain or inconclusive are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points to a pathogenic impact for F231L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.306467 | Uncertain | 0.895 | 0.300 | 0.000 | -9.482 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 1.07 | Ambiguous | 0.3 | 1.26 | Ambiguous | 1.17 | Ambiguous | 0.85 | Ambiguous | 0.662 | Likely Pathogenic | -5.08 | Deleterious | 0.390 | Benign | 0.142 | Benign | 6.04 | Benign | 0.01 | Affected | 0.2338 | 0.3794 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.693T>G | F231L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F231L is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar and FATHMM, whereas REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default and AlphaMissense‑Optimized all predict a pathogenic effect. The SGM‑Consensus score is labeled Likely Pathogenic, and the remaining tools (FoldX, Rosetta, Foldetta, premPS) are inconclusive. High‑accuracy methods give a pathogenic verdict from AlphaMissense‑Optimized, a Likely Pathogenic result from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and an uncertain outcome from Foldetta. Overall, the majority of evidence points to a pathogenic impact for F231L, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.306467 | Uncertain | 0.895 | 0.300 | 0.000 | -9.482 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 1.07 | Ambiguous | 0.3 | 1.26 | Ambiguous | 1.17 | Ambiguous | 0.85 | Ambiguous | 0.661 | Likely Pathogenic | -5.08 | Deleterious | 0.390 | Benign | 0.142 | Benign | 6.04 | Benign | 0.01 | Affected | 0.2338 | 0.3794 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.694G>C | A232P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A232P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, SIFT, and FATHMM. Tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain or inconclusive results come from Rosetta, premPS, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome, and Foldetta remains uncertain. Overall, the majority of evaluated tools predict a pathogenic impact, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.254060 | Structured | 0.307228 | Uncertain | 0.878 | 0.305 | 0.000 | -12.697 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.05 | Likely Benign | 0.8 | 1.25 | Ambiguous | 0.65 | Ambiguous | 0.71 | Ambiguous | 0.748 | Likely Pathogenic | -2.75 | Deleterious | 0.917 | Possibly Damaging | 0.502 | Possibly Damaging | 5.78 | Benign | 0.06 | Tolerated | 0.2163 | 0.5259 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.694G>T | A232S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A232S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Across the evaluated in‑silico predictors, all tools except AlphaMissense‑Default converge on a benign interpretation: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all predict benign. AlphaMissense‑Default is uncertain, while AlphaMissense‑Optimized predicts benign. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.254060 | Structured | 0.307228 | Uncertain | 0.878 | 0.305 | 0.000 | -3.264 | Likely Benign | 0.344 | Ambiguous | Likely Benign | 0.23 | Likely Benign | 0.1 | -0.23 | Likely Benign | 0.00 | Likely Benign | -0.20 | Likely Benign | 0.378 | Likely Benign | 0.54 | Neutral | 0.057 | Benign | 0.025 | Benign | 5.95 | Benign | 1.00 | Tolerated | 0.2777 | 0.5300 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.695C>A | A232D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A232D is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect comprise REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus. Uncertain or inconclusive results are reported for Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as pathogenic, while Foldetta remains uncertain. Overall, the majority of available predictions support a pathogenic impact. Thus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.254060 | Structured | 0.307228 | Uncertain | 0.878 | 0.305 | 0.000 | -13.956 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.14 | Likely Benign | 0.2 | 1.55 | Ambiguous | 0.85 | Ambiguous | 0.77 | Ambiguous | 0.725 | Likely Pathogenic | -2.50 | Deleterious | 0.845 | Possibly Damaging | 0.348 | Benign | 5.78 | Benign | 0.02 | Affected | 0.2066 | 0.2896 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.695C>G | A232G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A232G missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the change as benign include REVEL, FoldX, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Those that predict pathogenicity are polyPhen‑2 HumDiv and AlphaMissense‑Default. Uncertain or inconclusive results come from Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta as Uncertain. Overall, the preponderance of evidence points to a benign effect; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.254060 | Structured | 0.307228 | Uncertain | 0.878 | 0.305 | 0.000 | -4.924 | Likely Benign | 0.835 | Likely Pathogenic | Ambiguous | 0.43 | Likely Benign | 0.1 | 1.43 | Ambiguous | 0.93 | Ambiguous | 0.53 | Ambiguous | 0.453 | Likely Benign | -1.39 | Neutral | 0.608 | Possibly Damaging | 0.172 | Benign | 5.81 | Benign | 0.11 | Tolerated | 0.2012 | 0.4724 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.697T>A | C233S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C233S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta give uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors pathogenicity (3 pathogenic vs 1 benign). Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.239899 | Structured | 0.306787 | Uncertain | 0.868 | 0.322 | 0.000 | -10.862 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.61 | Ambiguous | 0.1 | 1.25 | Ambiguous | 0.93 | Ambiguous | 1.50 | Destabilizing | 0.764 | Likely Pathogenic | -8.89 | Deleterious | 0.421 | Benign | 0.080 | Benign | 5.79 | Benign | 0.03 | Affected | 0.4528 | 0.2833 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.697T>C | C233R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C233R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. In contrast, the majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Based on the overall consensus of the majority of tools and the high‑accuracy methods, the variant is most likely pathogenic, which is consistent with the lack of ClinVar annotation and gnomAD presence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.239899 | Structured | 0.306787 | Uncertain | 0.868 | 0.322 | 0.000 | -16.789 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.79 | Destabilizing | 3.4 | 2.17 | Destabilizing | 2.98 | Destabilizing | 1.75 | Destabilizing | 0.830 | Likely Pathogenic | -10.68 | Deleterious | 0.002 | Benign | 0.002 | Benign | 5.71 | Benign | 0.01 | Affected | 0.1733 | 0.2212 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.697T>G | C233G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C233G is not reported in ClinVar (ClinVar ID = None) and has no entry in gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include polyPhen‑2 HumVar, SIFT, and FATHMM. Tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta are inconclusive and are treated as unavailable. High‑accuracy predictions: AlphaMissense‑Optimized reports Pathogenic; SGM‑Consensus reports Likely Pathogenic; Foldetta is Uncertain. Overall, the majority of available predictions indicate a pathogenic impact. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.239899 | Structured | 0.306787 | Uncertain | 0.868 | 0.322 | 0.000 | -14.155 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 1.29 | Ambiguous | 0.1 | 1.01 | Ambiguous | 1.15 | Ambiguous | 1.31 | Destabilizing | 0.849 | Likely Pathogenic | -10.68 | Deleterious | 0.596 | Possibly Damaging | 0.107 | Benign | 5.79 | Benign | 0.09 | Tolerated | 0.3317 | 0.3570 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.691T>C | F231L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F231L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that agree on a pathogenic effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; the SGM‑Consensus score is “Likely Pathogenic.” Predictions that are uncertain or inconclusive are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points to a pathogenic impact for F231L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.306467 | Uncertain | 0.895 | 0.300 | 0.000 | -9.482 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 1.07 | Ambiguous | 0.3 | 1.26 | Ambiguous | 1.17 | Ambiguous | 0.85 | Ambiguous | 0.801 | Likely Pathogenic | -5.08 | Deleterious | 0.390 | Benign | 0.142 | Benign | 6.04 | Benign | 0.01 | Affected | 0.2338 | 0.3794 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.691T>A | F231I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F231I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include polyPhen‑2 HumVar and FATHMM, whereas the majority of tools predict a pathogenic impact: REVEL, SIFT, PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta’s stability prediction is uncertain and thus not considered evidence. No other tools provide definitive pathogenic or benign conclusions. Based on the preponderance of pathogenic predictions and the lack of contrary evidence, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.306467 | Uncertain | 0.895 | 0.300 | 0.000 | -13.827 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.16 | Ambiguous | 0.4 | 1.65 | Ambiguous | 1.41 | Ambiguous | 0.94 | Ambiguous | 0.894 | Likely Pathogenic | -5.01 | Deleterious | 0.759 | Possibly Damaging | 0.328 | Benign | 5.76 | Benign | 0.00 | Affected | 0.2122 | 0.2813 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.686A>T | K229I 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant K229I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from FoldX, Foldetta, premPS, and FATHMM, whereas pathogenic predictions are returned by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports benign. No prediction is inconclusive. Overall, the majority of tools, including the high‑accuracy ones, lean toward pathogenicity, and this does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.179055 | Structured | 0.310912 | Uncertain | 0.843 | 0.306 | 0.000 | -15.276 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | -0.03 | Likely Benign | 0.1 | -0.63 | Ambiguous | -0.33 | Likely Benign | -0.19 | Likely Benign | 0.833 | Likely Pathogenic | -6.52 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 5.92 | Benign | 0.00 | Affected | 0.1202 | 0.3711 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.687A>C | K229N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K229N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. Four tools (FoldX, Rosetta, Foldetta, premPS) return uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect. The variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.179055 | Structured | 0.310912 | Uncertain | 0.843 | 0.306 | 0.000 | -13.620 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.82 | Ambiguous | 0.1 | 0.64 | Ambiguous | 0.73 | Ambiguous | 0.61 | Ambiguous | 0.590 | Likely Pathogenic | -3.79 | Deleterious | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 5.83 | Benign | 0.01 | Affected | 0.3682 | 0.1013 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.687A>T | K229N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K229N missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining nine tools—REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is labeled Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is uncertain and therefore not considered evidence for or against pathogenicity. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.179055 | Structured | 0.310912 | Uncertain | 0.843 | 0.306 | 0.000 | -13.620 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.82 | Ambiguous | 0.1 | 0.64 | Ambiguous | 0.73 | Ambiguous | 0.61 | Ambiguous | 0.588 | Likely Pathogenic | -3.79 | Deleterious | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 5.83 | Benign | 0.01 | Affected | 0.3682 | 0.1013 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.688T>A | C230S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C230S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict a pathogenic effect are REVEL, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as benign. With an equal split of 7 benign versus 7 pathogenic predictions overall, the higher‑confidence tools lean toward pathogenicity. Therefore, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.268042 | Structured | 0.308076 | Uncertain | 0.870 | 0.308 | 0.000 | -9.994 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.42 | Likely Benign | 0.1 | 0.05 | Likely Benign | 0.24 | Likely Benign | 1.08 | Destabilizing | 0.859 | Likely Pathogenic | -8.24 | Deleterious | 0.421 | Benign | 0.115 | Benign | 5.91 | Benign | 0.07 | Tolerated | 0.4926 | 0.1822 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.688T>C | C230R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C230R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from FoldX and FATHMM, while pathogenic predictions are made by REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are reported by Rosetta and Foldetta. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts a pathogenic effect; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic; Foldetta remains inconclusive. Overall, the preponderance of evidence points to a pathogenic impact for C230R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.268042 | Structured | 0.308076 | Uncertain | 0.870 | 0.308 | 0.000 | -12.478 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | -0.48 | Likely Benign | 0.0 | -1.61 | Ambiguous | -1.05 | Ambiguous | 1.12 | Destabilizing | 0.905 | Likely Pathogenic | -9.88 | Deleterious | 0.838 | Possibly Damaging | 0.548 | Possibly Damaging | 5.91 | Benign | 0.01 | Affected | 0.1744 | 0.1637 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.688T>G | C230G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C230G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reached a consensus fall into two groups: benign predictions come from polyPhen‑2 (HumDiv and HumVar) and FATHMM, while pathogenic predictions are made by REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Uncertain or inconclusive results from FoldX, Rosetta, Foldetta, and premPS are treated as unavailable and do not influence the overall assessment. High‑accuracy methods specifically indicate pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also Likely Pathogenic; Foldetta remains uncertain. Based on the aggregate predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.268042 | Structured | 0.308076 | Uncertain | 0.870 | 0.308 | 0.000 | -10.888 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.72 | Ambiguous | 0.1 | 0.86 | Ambiguous | 0.79 | Ambiguous | 0.81 | Ambiguous | 0.775 | Likely Pathogenic | -9.95 | Deleterious | 0.001 | Benign | 0.004 | Benign | 5.85 | Benign | 0.03 | Affected | 0.3313 | 0.2574 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.689G>A | C230Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C230Y is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that indicate a benign effect include FoldX, Rosetta, premPS, SIFT, and FATHMM, whereas those that predict a pathogenic effect comprise REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score is “Likely Pathogenic,” and the Foldetta stability assessment is “Uncertain.” High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.268042 | Structured | 0.308076 | Uncertain | 0.870 | 0.308 | 0.000 | -8.978 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | -0.03 | Likely Benign | 0.1 | -2.24 | Stabilizing | -1.14 | Ambiguous | 0.00 | Likely Benign | 0.816 | Likely Pathogenic | -8.46 | Deleterious | 0.940 | Possibly Damaging | 0.641 | Possibly Damaging | 6.17 | Benign | 0.14 | Tolerated | 0.1159 | 0.3655 | 0 | -2 | -3.8 | 60.04 | |||||||||||||||||||||||||||||
| c.689G>C | C230S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant C230S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools cluster into two groups: benign calls come from FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM; pathogenic calls come from REVEL, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as benign. No prediction or stability result is missing. Overall, the predictions are evenly split between benign and pathogenic, leaving the variant’s functional impact uncertain. This uncertainty does not contradict ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.268042 | Structured | 0.308076 | Uncertain | 0.870 | 0.308 | 0.000 | -9.994 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.42 | Likely Benign | 0.1 | 0.05 | Likely Benign | 0.24 | Likely Benign | 1.08 | Destabilizing | 0.844 | Likely Pathogenic | -8.24 | Deleterious | 0.421 | Benign | 0.115 | Benign | 5.91 | Benign | 0.07 | Tolerated | 0.4926 | 0.1822 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.689G>T | C230F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C230F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, and FATHMM, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Foldetta and premPS give uncertain results. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; Foldetta remains uncertain. Overall, the preponderance of evidence indicates that C230F is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.268042 | Structured | 0.308076 | Uncertain | 0.870 | 0.308 | 0.000 | -11.134 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | -0.09 | Likely Benign | 0.1 | -2.03 | Stabilizing | -1.06 | Ambiguous | 0.51 | Ambiguous | 0.840 | Likely Pathogenic | -8.73 | Deleterious | 0.940 | Possibly Damaging | 0.641 | Possibly Damaging | 5.96 | Benign | 0.03 | Affected | 0.1329 | 0.4173 | -4 | -2 | 0.3 | 44.04 | |||||||||||||||||||||||||||||
| c.68A>C | D23A 2D AIThe SynGAP1 D23A missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic versus four benign) suggest a pathogenic impact. This conclusion does not contradict ClinVar status, as the variant has no existing ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.557691 | Disordered | 0.440341 | Uncertain | 0.369 | 0.892 | 0.375 | -2.732 | Likely Benign | 0.777 | Likely Pathogenic | Likely Benign | 0.112 | Likely Benign | -2.57 | Deleterious | 0.909 | Possibly Damaging | 0.539 | Possibly Damaging | 3.52 | Benign | 0.00 | Affected | 0.4613 | 0.8203 | 0 | -2 | 5.3 | -44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.68A>G | D23G 2D AIThe SynGAP1 missense variant D23G is listed in ClinVar (ID 3644551.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a benign impact, which does not contradict the ClinVar “Uncertain” classification but leans toward a benign interpretation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.557691 | Disordered | 0.440341 | Uncertain | 0.369 | 0.892 | 0.375 | Uncertain | 1 | -2.622 | Likely Benign | 0.684 | Likely Pathogenic | Likely Benign | 0.100 | Likely Benign | -2.45 | Neutral | 0.805 | Possibly Damaging | 0.539 | Possibly Damaging | 3.50 | Benign | 0.00 | Affected | 0.4682 | 0.7632 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||
| c.68A>T | D23V 2D AIThe SynGAP1 D23V missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie and thus unavailable; Foldetta predictions are not provided. Overall, more tools (five) predict pathogenicity than benign (three), and no ClinVar evidence contradicts this assessment. Therefore, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.557691 | Disordered | 0.440341 | Uncertain | 0.369 | 0.892 | 0.375 | -3.244 | Likely Benign | 0.842 | Likely Pathogenic | Ambiguous | 0.137 | Likely Benign | -2.72 | Deleterious | 0.972 | Probably Damaging | 0.804 | Possibly Damaging | 3.48 | Benign | 0.00 | Affected | 0.1515 | 0.8367 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.690C>G | C230W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C230W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions are made by FATHMM and FoldX, whereas the majority of other in silico predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic effect. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, SGM‑Consensus confirms a likely pathogenic outcome, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Overall, the preponderance of evidence points to a pathogenic impact for C230W, and this conclusion is not contradicted by any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.268042 | Structured | 0.308076 | Uncertain | 0.870 | 0.308 | 0.000 | -11.022 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | -0.21 | Likely Benign | 0.0 | -1.87 | Ambiguous | -1.04 | Ambiguous | 0.74 | Ambiguous | 0.739 | Likely Pathogenic | -8.79 | Deleterious | 0.983 | Probably Damaging | 0.841 | Possibly Damaging | 5.84 | Benign | 0.01 | Affected | 0.1589 | 0.3663 | -8 | -2 | -3.4 | 83.07 | |||||||||||||||||||||||||||||
| c.698G>A | C233Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C233Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are Rosetta and FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact; premPS is uncertain. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic (3 pathogenic vs. 1 benign); and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. Thus, the variant is most likely pathogenic based on the aggregate predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.239899 | Structured | 0.306787 | Uncertain | 0.868 | 0.322 | 0.000 | -17.893 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 13.15 | Destabilizing | 4.6 | 0.04 | Likely Benign | 6.60 | Destabilizing | 0.71 | Ambiguous | 0.904 | Likely Pathogenic | -9.79 | Deleterious | 0.940 | Possibly Damaging | 0.459 | Possibly Damaging | 5.71 | Benign | 0.00 | Affected | 4.29 | 391 | 0.1324 | 0.5284 | 0 | -2 | -3.8 | 60.04 | 248.9 | -63.0 | 0.0 | 0.3 | 0.0 | 0.4 | X | X | Potentially Pathogenic | The introduced residue Tyr233 is located in a β-α loop between an anti-parallel β sheet strand (res. Gly227-Ala232) and an α helix (residues Ala236-Val250). Although the thiol group of a cysteine side chain is not a strong hydrogen bond acceptor or donor, it facilitates hydrogen bonding between Cys233 and the backbone carbonyl of Ala232 in the WT simulations. In the variant simulations, the bulky phenol ring of the Tyr233 side chain stacks with the indole ring of the Trp242 side chain. This interaction could alter the tertiary assembly of the β sheet and α helix due to the residue swap. Indeed, in the second replica simulation, the protein structure begins to unfold to accommodate the introduced Tyr233 side chain. | 10.1016/j.ajhg.2020.11.011 | ||||||||||||||||
| c.698G>C | C233S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C233S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM, while pathogenic predictions arise from SGM‑Consensus, REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, yields an uncertain result and is treated as unavailable evidence. Overall, the preponderance of evidence points to a pathogenic impact for C233S, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.239899 | Structured | 0.306787 | Uncertain | 0.868 | 0.322 | 0.000 | -10.862 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.61 | Ambiguous | 0.1 | 1.25 | Ambiguous | 0.93 | Ambiguous | 1.50 | Destabilizing | 0.830 | Likely Pathogenic | -8.89 | Deleterious | 0.421 | Benign | 0.080 | Benign | 5.79 | Benign | 0.03 | Affected | 0.4528 | 0.2833 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.698G>T | C233F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C233F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include Rosetta, premPS, and FATHMM, whereas the remaining tools—REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, SGM‑Consensus, and Foldetta—predict a pathogenic or likely pathogenic impact. High‑accuracy methods give consistent results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Overall, the majority of evidence supports a pathogenic effect, and this conclusion does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.239899 | Structured | 0.306787 | Uncertain | 0.868 | 0.322 | 0.000 | -14.564 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 9.36 | Destabilizing | 3.0 | -0.19 | Likely Benign | 4.59 | Destabilizing | 0.37 | Likely Benign | 0.901 | Likely Pathogenic | -9.79 | Deleterious | 0.940 | Possibly Damaging | 0.459 | Possibly Damaging | 5.73 | Benign | 0.00 | Affected | 0.1524 | 0.5231 | -4 | -2 | 0.3 | 44.04 | |||||||||||||||||||||||||||||
| c.707C>G | A236G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A236G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, premPS, and PROVEAN. Four tools (FoldX, Rosetta, Foldetta, and ESM1b) return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also favors benign, while Foldetta remains uncertain. Overall, the majority of available predictions support a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.185198 | Structured | 0.329926 | Uncertain | 0.775 | 0.330 | 0.000 | -7.412 | In-Between | 0.162 | Likely Benign | Likely Benign | 0.92 | Ambiguous | 0.1 | 1.15 | Ambiguous | 1.04 | Ambiguous | 1.09 | Destabilizing | 0.615 | Likely Pathogenic | -3.35 | Deleterious | 0.067 | Benign | 0.028 | Benign | 5.74 | Benign | 0.07 | Tolerated | 0.2200 | 0.3899 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||
| c.709G>A | A237T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A237T missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include polyPhen‑2 (HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv), and ESM1b. Four tools (FoldX, Rosetta, Foldetta, premPS) return uncertain results. High‑accuracy methods give the following: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split; Foldetta also yields an inconclusive stability assessment. Overall, the majority of evidence leans toward a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.200174 | Structured | 0.334699 | Uncertain | 0.719 | 0.352 | 0.000 | -8.664 | Likely Pathogenic | 0.213 | Likely Benign | Likely Benign | 0.74 | Ambiguous | 0.3 | 0.55 | Ambiguous | 0.65 | Ambiguous | 0.71 | Ambiguous | 0.539 | Likely Pathogenic | -2.66 | Deleterious | 0.900 | Possibly Damaging | 0.348 | Benign | 5.80 | Benign | 0.06 | Tolerated | 0.0975 | 0.5737 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||
| c.709G>C | A237P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A237P is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include FoldX and FATHMM, whereas the majority of tools predict a pathogenic impact: REVEL, SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, Rosetta, and Foldetta. High‑accuracy assessments further support pathogenicity: the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, Foldetta is pathogenic, and AlphaMissense‑Optimized is uncertain (treated as unavailable). Overall, the preponderance of evidence indicates that A237P is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar status is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.200174 | Structured | 0.334699 | Uncertain | 0.719 | 0.352 | 0.000 | -9.119 | Likely Pathogenic | 0.827 | Likely Pathogenic | Ambiguous | 0.10 | Likely Benign | 0.5 | 4.20 | Destabilizing | 2.15 | Destabilizing | 1.04 | Destabilizing | 0.752 | Likely Pathogenic | -3.68 | Deleterious | 0.995 | Probably Damaging | 0.832 | Possibly Damaging | 5.77 | Benign | 0.03 | Affected | 0.1593 | 0.3724 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.709G>T | A237S 2D AIThe SynGAP1 missense variant A237S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and FATHMM. No tool predicts a pathogenic outcome; the only inconclusive results come from premPS and ESM1b, which are treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.200174 | Structured | 0.334699 | Uncertain | 0.719 | 0.352 | 0.000 | -7.696 | In-Between | 0.112 | Likely Benign | Likely Benign | 0.48 | Likely Benign | 0.6 | 0.34 | Likely Benign | 0.41 | Likely Benign | 0.61 | Ambiguous | 0.421 | Likely Benign | -1.41 | Neutral | 0.259 | Benign | 0.048 | Benign | 5.82 | Benign | 0.31 | Tolerated | 0.2209 | 0.4357 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.70G>C | V24L 2D AIThe SynGAP1 missense variant V24L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect for V24L, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.438970 | Uncertain | 0.382 | 0.890 | 0.500 | -2.590 | Likely Benign | 0.320 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -0.62 | Neutral | 0.043 | Benign | 0.011 | Benign | 3.94 | Benign | 0.00 | Affected | 0.1102 | 0.5226 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.70G>T | V24L 2D AIThe SynGAP1 missense variant V24L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect for V24L, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.438970 | Uncertain | 0.382 | 0.890 | 0.500 | -2.590 | Likely Benign | 0.320 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -0.62 | Neutral | 0.043 | Benign | 0.011 | Benign | 3.94 | Benign | 0.00 | Affected | 0.1102 | 0.5226 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.710C>A | A237D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A237D is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, while the remaining tools—SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict pathogenicity. Two tools, FoldX and AlphaMissense‑Optimized, return uncertain results. High‑accuracy assessments further support a deleterious impact: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) favors pathogenicity, AlphaMissense‑Optimized is uncertain, and Foldetta predicts a destabilizing, pathogenic effect. Overall, the evidence overwhelmingly indicates that the variant is pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.200174 | Structured | 0.334699 | Uncertain | 0.719 | 0.352 | 0.000 | -8.880 | Likely Pathogenic | 0.921 | Likely Pathogenic | Ambiguous | 1.23 | Ambiguous | 0.4 | 3.49 | Destabilizing | 2.36 | Destabilizing | 1.20 | Destabilizing | 0.769 | Likely Pathogenic | -3.97 | Deleterious | 0.969 | Probably Damaging | 0.704 | Possibly Damaging | 5.88 | Benign | 0.05 | Affected | 0.1419 | 0.2302 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.710C>G | A237G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A237G is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Given the balance of evidence, the majority of high‑confidence predictions lean toward a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.200174 | Structured | 0.334699 | Uncertain | 0.719 | 0.352 | 0.000 | -6.647 | Likely Benign | 0.316 | Likely Benign | Likely Benign | 1.00 | Ambiguous | 0.0 | 2.01 | Destabilizing | 1.51 | Ambiguous | 1.14 | Destabilizing | 0.538 | Likely Pathogenic | -2.91 | Deleterious | 0.900 | Possibly Damaging | 0.430 | Benign | 5.81 | Benign | 0.05 | Affected | 0.1809 | 0.2910 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.710C>T | A237V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A237V is not reported in ClinVar and is absent from gnomAD. Standard in‑silico predictors are divided: benign calls come from premPS, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic calls come from REVEL, PROVEAN, polyPhen‑2 HumDiv, and ESM1b. The remaining tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Default) are uncertain. High‑accuracy assessments give a benign result from AlphaMissense‑Optimized, a pathogenic outcome from the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and an uncertain result from Foldetta. Because the majority of conventional predictors and the optimized AlphaMissense model favor benign, the variant is most likely benign, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.200174 | Structured | 0.334699 | Uncertain | 0.719 | 0.352 | 0.000 | -8.927 | Likely Pathogenic | 0.397 | Ambiguous | Likely Benign | 0.87 | Ambiguous | 0.4 | 0.97 | Ambiguous | 0.92 | Ambiguous | 0.18 | Likely Benign | 0.565 | Likely Pathogenic | -3.26 | Deleterious | 0.900 | Possibly Damaging | 0.430 | Benign | 5.87 | Benign | 0.10 | Tolerated | 0.0771 | 0.4853 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||
| c.712G>A | E238K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E238K missense change is not reported in ClinVar and is absent from gnomAD. In silico predictors cluster into two groups: a single benign call from FATHMM, and a consensus of pathogenic predictions from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Four tools (FoldX, Rosetta, Foldetta, premPS) returned uncertain results and are not considered evidence. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, while Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E238K. This conclusion is not contradicted by ClinVar, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.194234 | Structured | 0.332638 | Uncertain | 0.796 | 0.326 | 0.000 | -13.475 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.56 | Ambiguous | 0.4 | 1.83 | Ambiguous | 1.20 | Ambiguous | 0.83 | Ambiguous | 0.858 | Likely Pathogenic | -3.63 | Deleterious | 0.995 | Probably Damaging | 0.695 | Possibly Damaging | 5.46 | Benign | 0.01 | Affected | 4.29 | 391 | 0.2812 | 0.5524 | 0 | 1 | -0.4 | -0.94 | 209.0 | 55.9 | 0.0 | 0.0 | -0.1 | 0.0 | X | Potentially Pathogenic | The negatively charged residue Glu238, located in an α helix (res. Ala236-Val250), is replaced by the positively charged residue Lys238. This charge reversal removes the periodically formed salt bridge between the carboxylate group of Glu238 and the guanidinium group of Arg234 observed in the WT simulations. In the variant simulations, both Lys238 and Arg234 form alternative salt bridges with the carboxylate group of Glu680 in the GAP domain loop. Although not visible in the simulations, the absence of the Glu238-Arg234 salt bridge could weaken the integrity of the α helix (residues Ala236-Val250) and potentially affect the tertiary assembly between the PH and GAP domains. | ||||||||||||||||||
| c.712G>C | E238Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E238Q missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two consensus groups: benign predictions come from SIFT and FATHMM, while pathogenic predictions are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Stability‑based methods (FoldX, Rosetta, premPS) and Foldetta give uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts Pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.194234 | Structured | 0.332638 | Uncertain | 0.796 | 0.326 | 0.000 | -11.476 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.63 | Ambiguous | 0.5 | 0.59 | Ambiguous | 0.61 | Ambiguous | 0.52 | Ambiguous | 0.723 | Likely Pathogenic | -2.72 | Deleterious | 0.996 | Probably Damaging | 0.891 | Possibly Damaging | 5.44 | Benign | 0.06 | Tolerated | 0.1372 | 0.5412 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.713A>C | E238A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E238A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: FATHMM predicts benign, whereas the remaining evaluated algorithms (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. Four methods (FoldX, Rosetta, Foldetta, premPS) return uncertain results and are treated as unavailable. High‑accuracy assessments further support a damaging outcome: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta is uncertain. Overall, the preponderance of evidence indicates that E238A is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.194234 | Structured | 0.332638 | Uncertain | 0.796 | 0.326 | 0.000 | -13.252 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.91 | Ambiguous | 0.3 | 1.89 | Ambiguous | 1.40 | Ambiguous | 0.57 | Ambiguous | 0.879 | Likely Pathogenic | -5.44 | Deleterious | 0.970 | Probably Damaging | 0.681 | Possibly Damaging | 5.44 | Benign | 0.04 | Affected | 0.4164 | 0.5610 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.713A>T | E238V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E238V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include premPS and FATHMM, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta give uncertain results. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E238V. This conclusion is not contradicted by ClinVar, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.194234 | Structured | 0.332638 | Uncertain | 0.796 | 0.326 | 0.000 | -14.329 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.88 | Ambiguous | 0.4 | 0.75 | Ambiguous | 0.82 | Ambiguous | 0.45 | Likely Benign | 0.890 | Likely Pathogenic | -6.35 | Deleterious | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 5.42 | Benign | 0.00 | Affected | 0.0848 | 0.6093 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.706G>T | A236S 2D AIThe SynGAP1 A236S variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include FoldX, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Predictions that are inconclusive or uncertain are Rosetta, Foldetta, premPS, and ESM1b. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact for A236S, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.185198 | Structured | 0.329926 | Uncertain | 0.775 | 0.330 | 0.000 | -7.845 | In-Between | 0.106 | Likely Benign | Likely Benign | 0.40 | Likely Benign | 0.5 | 1.24 | Ambiguous | 0.82 | Ambiguous | 0.65 | Ambiguous | 0.648 | Likely Pathogenic | -2.30 | Neutral | 0.948 | Possibly Damaging | 0.588 | Possibly Damaging | 5.83 | Benign | 0.13 | Tolerated | 0.2641 | 0.5547 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.706G>C | A236P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A236P missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Foldetta, SIFT, FATHMM, and AlphaMissense‑Optimized, whereas those that predict a pathogenic outcome are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and AlphaMissense‑Default; the remaining tools (FoldX, Rosetta, premPS, ESM1b) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. No prediction or stability result is missing or inconclusive. Overall, the majority of individual tools lean toward pathogenicity, but the two most reliable high‑accuracy methods (AlphaMissense‑Optimized and Foldetta) suggest a benign effect, while the SGM Consensus indicates pathogenicity. Given the mixed evidence, the variant is most likely benign based on the strongest high‑accuracy predictions, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.185198 | Structured | 0.329926 | Uncertain | 0.775 | 0.330 | 0.000 | -7.932 | In-Between | 0.621 | Likely Pathogenic | Likely Benign | -0.99 | Ambiguous | 0.1 | 1.44 | Ambiguous | 0.23 | Likely Benign | 0.67 | Ambiguous | 0.862 | Likely Pathogenic | -4.31 | Deleterious | 0.995 | Probably Damaging | 0.880 | Possibly Damaging | 5.78 | Benign | 0.12 | Tolerated | 0.1980 | 0.4897 | 1 | -1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||
| c.699T>G | C233W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C233W is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: pathogenic calls come from SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a benign outcome, while Rosetta and premPS are uncertain. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Taken together, the overwhelming majority of evidence indicates that C233W is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.239899 | Structured | 0.306787 | Uncertain | 0.868 | 0.322 | 0.000 | -17.899 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 14.72 | Destabilizing | 6.6 | 1.07 | Ambiguous | 7.90 | Destabilizing | 0.54 | Ambiguous | 0.772 | Likely Pathogenic | -9.79 | Deleterious | 0.983 | Probably Damaging | 0.715 | Possibly Damaging | 5.71 | Benign | 0.00 | Affected | 0.1727 | 0.5292 | -8 | -2 | -3.4 | 83.07 | |||||||||||||||||||||||||||||
| c.69T>A | D23E 2D AIThe SynGAP1 D23E missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.557691 | Disordered | 0.440341 | Uncertain | 0.369 | 0.892 | 0.375 | -3.329 | Likely Benign | 0.565 | Likely Pathogenic | Likely Benign | 0.084 | Likely Benign | -1.35 | Neutral | 0.643 | Possibly Damaging | 0.417 | Benign | 3.59 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2420 | 0.8363 | 2 | 3 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||
| c.6C>G | S2R 2D AIThe SynGAP1 missense variant S2R is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the preponderance of evidence from multiple prediction tools and high‑accuracy methods indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.543646 | Binding | 0.382 | 0.922 | 0.750 | -3.684 | Likely Benign | 0.426 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -0.44 | Neutral | 0.117 | Benign | 0.008 | Benign | 4.05 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0996 | 0.4503 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.700C>G | R234G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R234G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that agree on a pathogenic effect are REVEL, PROVEAN, and AlphaMissense‑Default. The remaining tools (FoldX, Rosetta, Foldetta, premPS, ESM1b, and AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy methods show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of individual predictors lean toward pathogenicity, and the high‑accuracy consensus also supports a pathogenic classification. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.239899 | Structured | 0.311558 | Uncertain | 0.804 | 0.322 | 0.000 | -7.163 | In-Between | 0.923 | Likely Pathogenic | Ambiguous | 1.50 | Ambiguous | 0.2 | 0.88 | Ambiguous | 1.19 | Ambiguous | 0.61 | Ambiguous | 0.724 | Likely Pathogenic | -4.31 | Deleterious | 0.276 | Benign | 0.103 | Benign | 5.82 | Benign | 0.12 | Tolerated | 0.3194 | 0.3426 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||
| c.701G>C | R234P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R234P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include premPS and FATHMM, whereas the majority of evaluated algorithms (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict a pathogenic impact. Uncertain results are reported by FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.239899 | Structured | 0.311558 | Uncertain | 0.804 | 0.322 | 0.000 | -10.126 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | 1.33 | Ambiguous | 0.6 | 1.36 | Ambiguous | 1.35 | Ambiguous | 0.13 | Likely Benign | 0.826 | Likely Pathogenic | -4.43 | Deleterious | 0.929 | Possibly Damaging | 0.519 | Possibly Damaging | 5.93 | Benign | 0.04 | Affected | 0.1972 | 0.4336 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.701G>T | R234L 2D AIThe SynGAP1 missense variant R234L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, polyPhen‑2 HumVar, SIFT, and FATHMM, while those that agree on a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy methods show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of predictions (7 pathogenic vs. 4 benign) and the pathogenic consensus from the high‑accuracy SGM‑Consensus suggest that the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.239899 | Structured | 0.311558 | Uncertain | 0.804 | 0.322 | 0.000 | -11.153 | Likely Pathogenic | 0.935 | Likely Pathogenic | Ambiguous | 1.38 | Ambiguous | 0.9 | 0.50 | Ambiguous | 0.94 | Ambiguous | 0.20 | Likely Benign | 0.734 | Likely Pathogenic | -4.64 | Deleterious | 0.649 | Possibly Damaging | 0.199 | Benign | 5.78 | Benign | 0.11 | Tolerated | 0.1846 | 0.4783 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.703T>A | S235T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S235T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome, while FoldX, Rosetta, and AlphaMissense‑Default are uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. No prediction or folding result is missing or inconclusive. Based on the overall consensus of the available tools, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.250310 | Structured | 0.319150 | Uncertain | 0.743 | 0.331 | 0.000 | -9.422 | Likely Pathogenic | 0.451 | Ambiguous | Likely Benign | 0.80 | Ambiguous | 0.0 | -0.54 | Ambiguous | 0.13 | Likely Benign | 0.21 | Likely Benign | 0.453 | Likely Benign | -2.02 | Neutral | 0.057 | Benign | 0.020 | Benign | 5.75 | Benign | 0.13 | Tolerated | 0.1674 | 0.6337 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||
| c.703T>C | S235P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S235P is listed in ClinVar as Pathogenic (ClinVar ID 1067856.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect are polyPhen‑2 HumVar and FATHMM; all other evaluated algorithms—including REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports pathogenic. No predictions or stability results are missing or inconclusive. **Based on the collective predictions, the variant is most likely pathogenic, and this conclusion aligns with its ClinVar status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.250310 | Structured | 0.319150 | Uncertain | 0.743 | 0.331 | 0.000 | Likely Pathogenic | 1 | -14.857 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.02 | Destabilizing | 0.1 | 6.91 | Destabilizing | 5.47 | Destabilizing | 1.23 | Destabilizing | 0.870 | Likely Pathogenic | -4.24 | Deleterious | 0.917 | Possibly Damaging | 0.446 | Benign | 5.47 | Benign | 0.01 | Affected | 3.40 | 14 | 0.2432 | 0.5307 | 1 | -1 | -0.8 | 10.04 | 201.5 | 17.0 | 0.1 | 0.0 | -0.6 | 0.0 | X | Potentially Pathogenic | In the WT, the hydroxyl group of Ser235, located in a β-α loop between an anti-parallel β sheet strand (res. Gly227-Phe231) and an α helix (residues Ala236-Val250), forms hydrogen bonds with the GAP domain loop residue Glu680 and with the backbone amide groups of Ala237 and Glu238 from the α helix. In the variant simulations, the pyrrolidine ring of Pro235 cannot stabilize the α helix end or maintain tertiary bonding interactions between the PH and GAP domains via hydrogen bonding as effectively as serine. | ||||||||||||||||
| c.703T>G | S235A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S235A has no ClinVar entry and is not present in gnomAD. Prediction tools that indicate benign effects include Rosetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. FoldX and Foldetta return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as pathogenic; Foldetta remains uncertain. Overall, the balance of evidence leans toward a benign interpretation, but the SGM Consensus introduces a pathogenic signal, resulting in a conflicting prediction profile. This assessment does not contradict ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.250310 | Structured | 0.319150 | Uncertain | 0.743 | 0.331 | 0.000 | -10.861 | Likely Pathogenic | 0.707 | Likely Pathogenic | Likely Benign | 1.92 | Ambiguous | 0.1 | 0.19 | Likely Benign | 1.06 | Ambiguous | 0.48 | Likely Benign | 0.646 | Likely Pathogenic | -2.52 | Deleterious | 0.002 | Benign | 0.004 | Benign | 5.49 | Benign | 0.01 | Affected | 0.5168 | 0.4563 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||
| c.704C>A | S235Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S235Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include FATHMM and Rosetta, whereas the majority of other in silico predictors (REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) classify the variant as pathogenic. Two tools give inconclusive results: Foldetta (protein‑folding stability) and premPS. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic,” and Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for S235Y, and this conclusion is not contradicted by any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.250310 | Structured | 0.319150 | Uncertain | 0.743 | 0.331 | 0.000 | -15.842 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 3.53 | Destabilizing | 1.8 | 0.43 | Likely Benign | 1.98 | Ambiguous | 0.66 | Ambiguous | 0.887 | Likely Pathogenic | -5.14 | Deleterious | 0.971 | Probably Damaging | 0.641 | Possibly Damaging | 5.45 | Benign | 0.00 | Affected | 0.0733 | 0.5269 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||
| c.704C>G | S235C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S235C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FATHMM and Rosetta, whereas a majority of tools (REVEL, SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. Results from high‑accuracy methods are mixed: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta is uncertain. Because the uncertain predictions are treated as unavailable, the overall evidence still favors pathogenicity. Thus, the variant is most likely pathogenic, and this assessment does not contradict the current ClinVar status, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.250310 | Structured | 0.319150 | Uncertain | 0.743 | 0.331 | 0.000 | -11.350 | Likely Pathogenic | 0.800 | Likely Pathogenic | Ambiguous | 1.74 | Ambiguous | 0.1 | 0.42 | Likely Benign | 1.08 | Ambiguous | 0.76 | Ambiguous | 0.878 | Likely Pathogenic | -4.24 | Deleterious | 0.977 | Probably Damaging | 0.620 | Possibly Damaging | 5.45 | Benign | 0.00 | Affected | 0.1284 | 0.6015 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.704C>T | S235F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S235F is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include FATHMM and Rosetta, whereas the majority of other in silico predictors (REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) classify the variant as pathogenic. Two tools give inconclusive results: Foldetta (protein‑folding stability) and premPS. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic,” and Foldetta remains uncertain. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.250310 | Structured | 0.319150 | Uncertain | 0.743 | 0.331 | 0.000 | -14.456 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 3.03 | Destabilizing | 1.7 | 0.46 | Likely Benign | 1.75 | Ambiguous | 0.56 | Ambiguous | 0.919 | Likely Pathogenic | -5.14 | Deleterious | 0.917 | Possibly Damaging | 0.548 | Possibly Damaging | 5.45 | Benign | 0.00 | Affected | 0.0617 | 0.5393 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||
| c.706G>A | A236T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A236T missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Predictions that are uncertain or inconclusive are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a 2‑vs‑2 tie and thus unavailable. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also uncertain. Overall, six tools favor pathogenicity versus three favor benign, and no high‑confidence consensus tool contradicts this trend. Therefore, the variant is most likely pathogenic, and this assessment does not conflict with any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.185198 | Structured | 0.329926 | Uncertain | 0.775 | 0.330 | 0.000 | -8.319 | Likely Pathogenic | 0.240 | Likely Benign | Likely Benign | 0.73 | Ambiguous | 0.2 | 1.58 | Ambiguous | 1.16 | Ambiguous | 0.83 | Ambiguous | 0.811 | Likely Pathogenic | -3.35 | Deleterious | 0.982 | Probably Damaging | 0.747 | Possibly Damaging | 5.79 | Benign | 0.03 | Affected | 0.1342 | 0.6926 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||
| c.714A>C | E238D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E238D missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact; the remaining methods (FoldX, Rosetta, Foldetta, premPS, ESM1b) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect. The variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status because no ClinVar record exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.194234 | Structured | 0.332638 | Uncertain | 0.796 | 0.326 | 0.000 | -7.861 | In-Between | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.25 | Ambiguous | 0.4 | 1.72 | Ambiguous | 1.49 | Ambiguous | 0.78 | Ambiguous | 0.691 | Likely Pathogenic | -2.72 | Deleterious | 0.868 | Possibly Damaging | 0.504 | Possibly Damaging | 5.57 | Benign | 0.05 | Affected | 0.1975 | 0.3619 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.685A>G | K229E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K229E is not reported in ClinVar (ClinVar status: not reported) and is absent from gnomAD (gnomAD ID: none). Prediction tools that classify the variant as benign include FoldX, Rosetta, premPS, SIFT, and FATHMM. Tools that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall distribution of predictions, the variant is most likely pathogenic; this conclusion does not contradict the ClinVar status, which currently has no entry for K229E. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.179055 | Structured | 0.310912 | Uncertain | 0.843 | 0.306 | 0.000 | -12.828 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.47 | Likely Benign | 0.0 | 0.24 | Likely Benign | 0.36 | Likely Benign | 0.18 | Likely Benign | 0.816 | Likely Pathogenic | -3.10 | Deleterious | 0.993 | Probably Damaging | 0.971 | Probably Damaging | 5.84 | Benign | 0.09 | Tolerated | 0.3915 | 0.0793 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.654C>A | F218L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F218L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Tools that agree on a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), Rosetta, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX and Foldetta are inconclusive and are treated as unavailable. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also predicts Likely Pathogenic; Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic impact. There is no ClinVar annotation to contradict this assessment, so the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.281712 | Structured | 0.408725 | Uncertain | 0.848 | 0.272 | 0.000 | -8.861 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.96 | Ambiguous | 0.4 | 2.36 | Destabilizing | 1.66 | Ambiguous | 1.08 | Destabilizing | 0.477 | Likely Benign | -3.80 | Deleterious | 0.158 | Benign | 0.025 | Benign | 5.90 | Benign | 0.15 | Tolerated | 0.2331 | 0.3241 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.65G>T | R22I 2D  AIThe SynGAP1 missense variant R22I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.538167 | Disordered | 0.441505 | Uncertain | 0.377 | 0.891 | 0.500 | -4.849 | Likely Benign | 0.692 | Likely Pathogenic | Likely Benign | 0.118 | Likely Benign | 0.06 | Neutral | 0.676 | Possibly Damaging | 0.308 | Benign | 4.20 | Benign | 0.00 | Affected | 0.2120 | 0.5048 | -2 | -3 | 9.0 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.660T>A | F220L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F220L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. All other evaluated predictors—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—indicate a pathogenic effect. The SGM‑Consensus result is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN (3 pathogenic, 1 benign), thus supporting a pathogenic classification. High‑accuracy assessments further corroborate this: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.219301 | Structured | 0.429422 | Uncertain | 0.898 | 0.295 | 0.000 | -9.601 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.08 | Destabilizing | 0.1 | 2.24 | Destabilizing | 2.16 | Destabilizing | 1.33 | Destabilizing | 0.752 | Likely Pathogenic | -4.95 | Deleterious | 0.003 | Benign | 0.005 | Benign | 4.24 | Benign | 0.02 | Affected | 0.2589 | 0.4108 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.660T>G | F220L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F220L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. All other evaluated predictors—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—indicate a pathogenic effect. The SGM‑Consensus result is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN (3 pathogenic, 1 benign), thus supporting a pathogenic classification. High‑accuracy assessments further corroborate this: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.219301 | Structured | 0.429422 | Uncertain | 0.898 | 0.295 | 0.000 | -9.601 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.08 | Destabilizing | 0.1 | 2.24 | Destabilizing | 2.16 | Destabilizing | 1.33 | Destabilizing | 0.752 | Likely Pathogenic | -4.95 | Deleterious | 0.003 | Benign | 0.005 | Benign | 4.24 | Benign | 0.02 | Affected | 0.2589 | 0.4108 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.661G>A | E221K 2D AIThe SynGAP1 E221K missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Rosetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus. FoldX and Foldetta give uncertain results. High‑accuracy methods show AlphaMissense‑Optimized as pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also predicts pathogenic (3 pathogenic vs 1 benign). Foldetta remains uncertain. Overall, the majority of reliable predictors indicate a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.413334 | Uncertain | 0.891 | 0.283 | 0.000 | -12.331 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | -1.01 | Ambiguous | 0.4 | -0.18 | Likely Benign | -0.60 | Ambiguous | 0.19 | Likely Benign | 0.815 | Likely Pathogenic | -2.92 | Deleterious | 0.131 | Benign | 0.058 | Benign | 5.92 | Benign | 0.02 | Affected | 0.2491 | 0.8002 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.661G>C | E221Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E221Q missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Two tools predict a pathogenic outcome: ESM1b and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs two benign), and Foldetta predicts a benign effect. Overall, the majority of evidence (10 benign vs 2 pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.127496 | Structured | 0.413334 | Uncertain | 0.891 | 0.283 | 0.000 | -8.243 | Likely Pathogenic | 0.825 | Likely Pathogenic | Ambiguous | -0.26 | Likely Benign | 0.1 | -0.33 | Likely Benign | -0.30 | Likely Benign | 0.12 | Likely Benign | 0.455 | Likely Benign | -1.64 | Neutral | 0.028 | Benign | 0.015 | Benign | 6.13 | Benign | 0.23 | Tolerated | 0.1207 | 0.7690 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.662A>C | E221A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E221A missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as benign. Overall, the predictions are split, but the two high‑confidence pathogenic calls outweigh the single high‑confidence benign call, suggesting the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.413334 | Uncertain | 0.891 | 0.283 | 0.000 | -12.906 | Likely Pathogenic | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.31 | Likely Benign | 0.0 | -0.11 | Likely Benign | 0.10 | Likely Benign | 0.52 | Ambiguous | 0.796 | Likely Pathogenic | -4.70 | Deleterious | 0.231 | Benign | 0.081 | Benign | 5.82 | Benign | 0.01 | Affected | 0.3486 | 0.7335 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.662A>G | E221G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E221G missense variant is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumVar and FATHMM, while the majority of other in silico predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) indicate a pathogenic impact; FoldX, Rosetta, Foldetta, and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Based on the collective evidence, the variant is most likely pathogenic, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.413334 | Uncertain | 0.891 | 0.283 | 0.000 | Uncertain | 1 | -12.221 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.40 | Ambiguous | 0.1 | 1.74 | Ambiguous | 1.57 | Ambiguous | 0.71 | Ambiguous | 0.863 | Likely Pathogenic | -5.56 | Deleterious | 0.596 | Possibly Damaging | 0.201 | Benign | 5.79 | Benign | 0.00 | Affected | 0.2611 | 0.6370 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||
| c.662A>T | E221V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E221V missense variant is reported in ClinVar as Pathogenic (ClinVar ID 2413181.0) and is not found in gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Benign predictions are limited to premPS, polyPhen‑2 HumVar, and FATHMM. Uncertain results are reported by FoldX, Rosetta, and Foldetta. High‑accuracy assessments reinforce the pathogenic interpretation: AlphaMissense‑Optimized predicts Pathogenic, the SGM‑Consensus also indicates Likely Pathogenic, while Foldetta remains Uncertain. Taken together, the preponderance of evidence supports a pathogenic effect for E221V, and this conclusion aligns with the ClinVar classification, showing no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.413334 | Uncertain | 0.891 | 0.283 | 0.000 | Likely Pathogenic | 1 | -14.954 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | -0.66 | Ambiguous | 0.2 | -0.89 | Ambiguous | -0.78 | Ambiguous | 0.49 | Likely Benign | 0.875 | Likely Pathogenic | -5.54 | Deleterious | 0.596 | Possibly Damaging | 0.203 | Benign | 5.86 | Benign | 0.00 | Affected | 3.41 | 13 | 0.0806 | 0.8138 | -2 | -2 | 7.7 | -29.98 | 234.5 | 50.6 | 0.0 | 0.0 | -0.4 | 0.2 | X | Uncertain | The introduced residue Val221 is located on the outer surface of an anti-parallel β sheet strand (res. Cys219-Thr224). Unlike the carboxylate group of Glu221, Val221 cannot form hydrogen bonds with Thr223 or a salt bridge with the amino group of the Lys207 side chain. Despite this, the WT simulations containing Glu221 do not show significant differences compared to the variant simulations. However, since the model ends abruptly at the N-terminus, no definite conclusions can be drawn from the simulations. | ||||||||||||||||
| c.663G>C | E221D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E221D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that agree on a pathogenic effect are REVEL, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Four tools (FoldX, Rosetta, Foldetta, premPS) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs 2 benign), and Foldetta remains uncertain. Overall, the majority of available predictions lean toward pathogenicity, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.127496 | Structured | 0.413334 | Uncertain | 0.891 | 0.283 | 0.000 | -12.237 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.59 | Ambiguous | 0.1 | 1.07 | Ambiguous | 1.33 | Ambiguous | 0.84 | Ambiguous | 0.750 | Likely Pathogenic | -2.43 | Neutral | 0.421 | Benign | 0.107 | Benign | 5.80 | Benign | 0.02 | Affected | 0.1755 | 0.5228 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.663G>T | E221D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E221D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that agree on a pathogenic effect are REVEL, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Four tools (FoldX, Rosetta, Foldetta, premPS) returned uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2), and Foldetta is uncertain. Overall, the majority of available predictions lean toward pathogenicity, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.127496 | Structured | 0.413334 | Uncertain | 0.891 | 0.283 | 0.000 | -12.237 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.59 | Ambiguous | 0.1 | 1.07 | Ambiguous | 1.33 | Ambiguous | 0.84 | Ambiguous | 0.750 | Likely Pathogenic | -2.43 | Neutral | 0.421 | Benign | 0.107 | Benign | 5.80 | Benign | 0.02 | Affected | 0.1755 | 0.5228 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.664G>A | V222I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 V222I missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, premPS, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The high‑accuracy consensus methods give a benign signal: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, while Foldetta’s stability assessment is uncertain. Overall, the majority of evidence points to a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.116183 | Structured | 0.402706 | Uncertain | 0.885 | 0.310 | 0.125 | -6.347 | Likely Benign | 0.604 | Likely Pathogenic | Likely Benign | 0.21 | Likely Benign | 0.2 | 1.45 | Ambiguous | 0.83 | Ambiguous | 0.09 | Likely Benign | 0.612 | Likely Pathogenic | -0.87 | Neutral | 0.984 | Probably Damaging | 0.956 | Probably Damaging | 5.77 | Benign | 0.08 | Tolerated | 0.0637 | 0.3574 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||
| c.664G>C | V222L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V222L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, FATHMM, and Foldetta, whereas those that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools (premPS and ESM1b) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic)—also predicts pathogenic; Foldetta, a protein‑folding stability method, predicts benign. Overall, the majority of evidence (seven pathogenic vs. four benign) points to a pathogenic effect. The variant’s predicted pathogenicity is not contradicted by any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.116183 | Structured | 0.402706 | Uncertain | 0.885 | 0.310 | 0.125 | -7.626 | In-Between | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.00 | Likely Benign | 0.3 | 0.06 | Likely Benign | 0.03 | Likely Benign | 0.60 | Ambiguous | 0.617 | Likely Pathogenic | -2.59 | Deleterious | 0.984 | Probably Damaging | 0.956 | Probably Damaging | 5.49 | Benign | 0.05 | Affected | 0.0863 | 0.4349 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||
| c.664G>T | V222L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V222L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, FATHMM, and Foldetta, whereas those that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools (premPS and ESM1b) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic)—also predicts pathogenic; Foldetta, a protein‑folding stability method, predicts benign. Overall, the majority of evidence (seven pathogenic vs. four benign) points to a pathogenic effect. The variant’s predicted pathogenicity is not contradicted by any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.116183 | Structured | 0.402706 | Uncertain | 0.885 | 0.310 | 0.125 | -7.626 | In-Between | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.00 | Likely Benign | 0.3 | 0.06 | Likely Benign | 0.03 | Likely Benign | 0.60 | Ambiguous | 0.617 | Likely Pathogenic | -2.59 | Deleterious | 0.984 | Probably Damaging | 0.956 | Probably Damaging | 5.49 | Benign | 0.05 | Affected | 0.0863 | 0.4349 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||
| c.65G>C | R22T 2D AIThe SynGAP1 missense variant R22T is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools show a split: benign calls from REVEL, PROVEAN, polyPhen2_HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls come from polyPhen2_HumDiv and SIFT. The majority‑vote SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect. High‑accuracy predictors further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is benign; Foldetta predictions are not available. Given the preponderance of benign predictions and the lack of pathogenic evidence, the variant is most likely benign. This assessment aligns with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.538167 | Disordered | 0.441505 | Uncertain | 0.377 | 0.891 | 0.500 | -4.079 | Likely Benign | 0.510 | Ambiguous | Likely Benign | 0.146 | Likely Benign | -0.21 | Neutral | 0.462 | Possibly Damaging | 0.227 | Benign | 4.23 | Benign | 0.00 | Affected | 0.2140 | 0.5599 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||||||||||||
| c.659T>G | F220C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F220C is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include only FATHMM. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the overwhelming agreement among these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.219301 | Structured | 0.429422 | Uncertain | 0.898 | 0.295 | 0.000 | -12.948 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.67 | Destabilizing | 0.0 | 5.03 | Destabilizing | 4.35 | Destabilizing | 2.22 | Destabilizing | 0.941 | Likely Pathogenic | -6.72 | Deleterious | 0.994 | Probably Damaging | 0.753 | Possibly Damaging | 4.03 | Benign | 0.00 | Affected | 0.2264 | 0.2012 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.654C>G | F218L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F218L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Tools that agree on a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), Rosetta, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX and Foldetta are inconclusive and are treated as unavailable. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also predicts Likely Pathogenic; Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic impact. There is no ClinVar annotation to contradict this assessment, so the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.281712 | Structured | 0.408725 | Uncertain | 0.848 | 0.272 | 0.000 | -8.861 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.96 | Ambiguous | 0.4 | 2.36 | Destabilizing | 1.66 | Ambiguous | 1.08 | Destabilizing | 0.477 | Likely Benign | -3.80 | Deleterious | 0.158 | Benign | 0.025 | Benign | 5.90 | Benign | 0.15 | Tolerated | 0.2331 | 0.3241 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.655T>A | C219S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C219S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign calls come from FoldX, polyPhen‑2 HumVar, and FATHMM, while pathogenic predictions are made by SGM‑Consensus (Likely Pathogenic), REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates pathogenicity, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive. With the majority of evidence pointing to a damaging effect and no ClinVar annotation to contradict, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.254060 | Structured | 0.426845 | Uncertain | 0.903 | 0.279 | 0.000 | -12.962 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.09 | Likely Benign | 0.4 | 1.53 | Ambiguous | 0.81 | Ambiguous | 1.56 | Destabilizing | 0.892 | Likely Pathogenic | -8.35 | Deleterious | 0.900 | Possibly Damaging | 0.380 | Benign | 5.95 | Benign | 0.04 | Affected | 0.3568 | 0.1454 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.655T>C | C219R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C219R is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, while the remaining 13 tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts Pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.254060 | Structured | 0.426845 | Uncertain | 0.903 | 0.279 | 0.000 | -15.727 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.00 | Destabilizing | 1.5 | 2.20 | Destabilizing | 2.60 | Destabilizing | 1.33 | Destabilizing | 0.904 | Likely Pathogenic | -9.90 | Deleterious | 0.969 | Probably Damaging | 0.680 | Possibly Damaging | 5.80 | Benign | 0.00 | Affected | 0.1381 | 0.1837 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.655T>G | C219G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C219G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the remaining 11 tools—including REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic impact. High‑accuracy assessments further support this view: AlphaMissense‑Optimized returns a pathogenic score, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates pathogenicity, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is inconclusive. Taken together, the overwhelming majority of evidence points to a pathogenic effect for C219G. This conclusion aligns with the absence of a ClinVar entry and does not contradict any existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.254060 | Structured | 0.426845 | Uncertain | 0.903 | 0.279 | 0.000 | -15.072 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.72 | Ambiguous | 0.3 | 1.98 | Ambiguous | 1.35 | Ambiguous | 1.18 | Destabilizing | 0.900 | Likely Pathogenic | -10.09 | Deleterious | 0.900 | Possibly Damaging | 0.461 | Possibly Damaging | 5.83 | Benign | 0.02 | Affected | 0.2159 | 0.2169 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.656G>A | C219Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C219Y is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that classify the variant as benign include only FATHMM. All other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic effect. High‑accuracy assessments further support this view: AlphaMissense‑Optimized returns a pathogenic prediction; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Based on the preponderance of evidence, the variant is most likely pathogenic, a conclusion that does not contradict its ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.254060 | Structured | 0.426845 | Uncertain | 0.903 | 0.279 | 0.000 | Uncertain | 1 | -14.797 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 8.98 | Destabilizing | 2.0 | 2.84 | Destabilizing | 5.91 | Destabilizing | 0.55 | Ambiguous | 0.812 | Likely Pathogenic | -9.09 | Deleterious | 0.990 | Probably Damaging | 0.758 | Possibly Damaging | 5.81 | Benign | 0.00 | Affected | 0.0772 | 0.3285 | 0 | -2 | -3.8 | 60.04 | |||||||||||||||||||||||||||
| c.656G>C | C219S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C219S is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include FoldX, polyPhen‑2 HumVar, and FATHMM, whereas the majority of tools predict it to be pathogenic: SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates pathogenicity, and Foldetta’s stability analysis is inconclusive (treated as unavailable). Taken together, the preponderance of evidence from both general and high‑accuracy predictors points to a pathogenic impact for C219S. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.254060 | Structured | 0.426845 | Uncertain | 0.903 | 0.279 | 0.000 | -12.962 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.09 | Likely Benign | 0.4 | 1.53 | Ambiguous | 0.81 | Ambiguous | 1.56 | Destabilizing | 0.817 | Likely Pathogenic | -8.35 | Deleterious | 0.900 | Possibly Damaging | 0.380 | Benign | 5.95 | Benign | 0.04 | Affected | 0.3568 | 0.1454 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.656G>T | C219F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C219F is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are premPS and FATHMM, while the remaining 13 tools—including SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized reports pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among the majority of tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.254060 | Structured | 0.426845 | Uncertain | 0.903 | 0.279 | 0.000 | -14.322 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 7.31 | Destabilizing | 1.9 | 2.45 | Destabilizing | 4.88 | Destabilizing | 0.47 | Likely Benign | 0.814 | Likely Pathogenic | -9.18 | Deleterious | 0.940 | Possibly Damaging | 0.505 | Possibly Damaging | 5.82 | Benign | 0.00 | Affected | 0.0959 | 0.3804 | -4 | -2 | 0.3 | 44.04 | |||||||||||||||||||||||||||||
| c.657T>G | C219W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C219W is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the majority of tools (SGM‑Consensus, REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; Rosetta and premPS are inconclusive. High‑accuracy methods give consistent pathogenic results: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Taken together, the overwhelming evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this assessment does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.254060 | Structured | 0.426845 | Uncertain | 0.903 | 0.279 | 0.000 | -16.565 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 10.10 | Destabilizing | 3.6 | 1.20 | Ambiguous | 5.65 | Destabilizing | 0.52 | Ambiguous | 0.706 | Likely Pathogenic | -9.23 | Deleterious | 0.997 | Probably Damaging | 0.808 | Possibly Damaging | 5.77 | Benign | 0.00 | Affected | 0.1131 | 0.3285 | -8 | -2 | -3.4 | 83.07 | |||||||||||||||||||||||||||||
| c.658T>A | F220I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F220I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic effect. The SGM‑Consensus result is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yielding 3 pathogenic versus 1 benign calls, thus supporting a pathogenic classification. High‑accuracy tools specifically report pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM‑Consensus (majority vote) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.219301 | Structured | 0.429422 | Uncertain | 0.898 | 0.295 | 0.000 | -12.041 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.11 | Destabilizing | 0.1 | 2.35 | Destabilizing | 2.73 | Destabilizing | 1.03 | Destabilizing | 0.921 | Likely Pathogenic | -4.98 | Deleterious | 0.300 | Benign | 0.098 | Benign | 4.06 | Benign | 0.01 | Affected | 0.2292 | 0.3127 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.658T>C | F220L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F220L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. All other evaluated predictors—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—indicate a pathogenic effect. The SGM‑Consensus result is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN (3 pathogenic, 1 benign), thus supporting a pathogenic classification. High‑accuracy assessments further corroborate this: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.219301 | Structured | 0.429422 | Uncertain | 0.898 | 0.295 | 0.000 | -9.601 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.08 | Destabilizing | 0.1 | 2.24 | Destabilizing | 2.16 | Destabilizing | 1.33 | Destabilizing | 0.850 | Likely Pathogenic | -4.95 | Deleterious | 0.003 | Benign | 0.005 | Benign | 4.24 | Benign | 0.02 | Affected | 0.2589 | 0.4108 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.658T>G | F220V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F220V is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic effect. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.219301 | Structured | 0.429422 | Uncertain | 0.898 | 0.295 | 0.000 | -11.599 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.83 | Destabilizing | 0.1 | 4.11 | Destabilizing | 3.97 | Destabilizing | 1.56 | Destabilizing | 0.911 | Likely Pathogenic | -5.81 | Deleterious | 0.075 | Benign | 0.015 | Benign | 4.04 | Benign | 0.01 | Affected | 0.2351 | 0.3315 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.659T>A | F220Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F220Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta, FATHMM, and polyPhen‑2 HumVar. In contrast, the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is classified as pathogenic, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain and therefore not considered evidence. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that F220Y is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.219301 | Structured | 0.429422 | Uncertain | 0.898 | 0.295 | 0.000 | -12.541 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 2.25 | Destabilizing | 0.1 | 0.35 | Likely Benign | 1.30 | Ambiguous | 1.14 | Destabilizing | 0.879 | Likely Pathogenic | -2.54 | Deleterious | 0.928 | Possibly Damaging | 0.395 | Benign | 4.07 | Benign | 0.00 | Affected | 0.1479 | 0.2661 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.659T>C | F220S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F220S is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: all available scores (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, AlphaMissense‑Default) indicate pathogenicity, while only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also labels it pathogenic. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.219301 | Structured | 0.429422 | Uncertain | 0.898 | 0.295 | 0.000 | -15.258 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 4.06 | Destabilizing | 0.1 | 4.67 | Destabilizing | 4.37 | Destabilizing | 1.44 | Destabilizing | 0.954 | Likely Pathogenic | -6.67 | Deleterious | 0.928 | Possibly Damaging | 0.477 | Possibly Damaging | 4.03 | Benign | 0.01 | Affected | 0.3813 | 0.1143 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.665T>A | V222E 2D 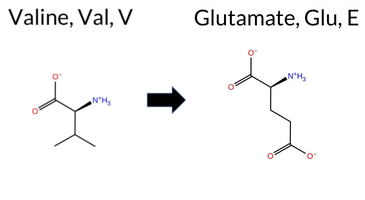 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V222E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is consistent with the lack of ClinVar reporting (i.e., no contradictory evidence). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.116183 | Structured | 0.402706 | Uncertain | 0.885 | 0.310 | 0.125 | -18.017 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.04 | Destabilizing | 0.2 | 3.46 | Destabilizing | 3.75 | Destabilizing | 1.64 | Destabilizing | 0.960 | Likely Pathogenic | -5.20 | Deleterious | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 5.20 | Benign | 0.00 | Affected | 0.0833 | 0.1495 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.665T>C | V222A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V222A missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity largely agree: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. Only FATHMM predicts a benign outcome. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No predictions are missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.116183 | Structured | 0.402706 | Uncertain | 0.885 | 0.310 | 0.125 | -10.248 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.89 | Destabilizing | 0.3 | 2.98 | Destabilizing | 2.94 | Destabilizing | 2.06 | Destabilizing | 0.894 | Likely Pathogenic | -3.50 | Deleterious | 0.984 | Probably Damaging | 0.956 | Probably Damaging | 5.26 | Benign | 0.04 | Affected | 0.2386 | 0.2081 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.665T>G | V222G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V222G resides in the PH domain. ClinVar has no entry for this variant, and it is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining eleven tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) all predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Thus, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict the ClinVar status, which currently contains no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.116183 | Structured | 0.402706 | Uncertain | 0.885 | 0.310 | 0.125 | -14.857 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 4.54 | Destabilizing | 0.4 | 5.25 | Destabilizing | 4.90 | Destabilizing | 2.05 | Destabilizing | 0.962 | Likely Pathogenic | -6.05 | Deleterious | 0.987 | Probably Damaging | 0.998 | Probably Damaging | 5.21 | Benign | 0.00 | Affected | 0.1678 | 0.1996 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.676T>G | S226A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S226A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the variant as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus predicts likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.129801 | Structured | 0.334959 | Uncertain | 0.800 | 0.324 | 0.250 | -4.665 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.06 | Likely Benign | 0.0 | 0.25 | Likely Benign | 0.16 | Likely Benign | -0.02 | Likely Benign | 0.339 | Likely Benign | -1.64 | Neutral | 0.123 | Benign | 0.050 | Benign | 5.76 | Benign | 0.06 | Tolerated | 0.4812 | 0.5599 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.677C>T | S226L 2D 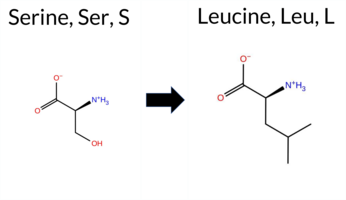 AIThe SynGAP1 missense variant S226L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, PROVEAN, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus (majority vote) also indicates benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts benign. No prediction or folding result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.129801 | Structured | 0.334959 | Uncertain | 0.800 | 0.324 | 0.250 | -6.098 | Likely Benign | 0.239 | Likely Benign | Likely Benign | -0.12 | Likely Benign | 0.5 | -0.14 | Likely Benign | -0.13 | Likely Benign | 0.36 | Likely Benign | 0.578 | Likely Pathogenic | -3.87 | Deleterious | 0.437 | Benign | 0.048 | Benign | 5.73 | Benign | 0.03 | Affected | 0.1525 | 0.5907 | -3 | -2 | 4.6 | 26.08 | |||||||||||||||||||||||||||||
| c.679G>A | G227R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G227R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. In contrast, the majority of other in silico predictors classify the variant as pathogenic: SGM‑Consensus, REVEL, FoldX, Rosetta, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized (premPS is inconclusive). High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a pathogenic effect. Overall, the preponderance of evidence points to a pathogenic classification for G227R, with no conflict from ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.106997 | Structured | 0.329995 | Uncertain | 0.800 | 0.329 | 0.250 | -9.776 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.31 | Destabilizing | 0.3 | 5.29 | Destabilizing | 3.80 | Destabilizing | 0.90 | Ambiguous | 0.765 | Likely Pathogenic | -6.49 | Deleterious | 0.020 | Benign | 0.018 | Benign | 6.02 | Benign | 0.01 | Affected | 3.43 | 12 | 0.0997 | 0.4195 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||
| c.67G>A | D23N 2D AIThe SynGAP1 D23N missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.557691 | Disordered | 0.440341 | Uncertain | 0.369 | 0.892 | 0.375 | -3.918 | Likely Benign | 0.719 | Likely Pathogenic | Likely Benign | 0.077 | Likely Benign | -1.88 | Neutral | 0.805 | Possibly Damaging | 0.539 | Possibly Damaging | 3.52 | Benign | 0.00 | Affected | 0.2236 | 0.8784 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.67G>C | D23H 2D AIThe SynGAP1 D23H missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are unavailable. Overall, the majority of evidence points toward a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current predictive data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.557691 | Disordered | 0.440341 | Uncertain | 0.369 | 0.892 | 0.375 | -3.801 | Likely Benign | 0.867 | Likely Pathogenic | Ambiguous | 0.103 | Likely Benign | -2.46 | Neutral | 0.972 | Probably Damaging | 0.861 | Possibly Damaging | 3.47 | Benign | 0.00 | Affected | 0.2644 | 0.9017 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.680G>C | G227A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G227A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that agree on a pathogenic effect are REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, and AlphaMissense‑Default. Predictions that remain uncertain are premPS, ESM1b, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic majority, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the overall consensus of the majority of tools and the high‑accuracy predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.106997 | Structured | 0.329995 | Uncertain | 0.800 | 0.329 | 0.250 | -7.344 | In-Between | 0.920 | Likely Pathogenic | Ambiguous | 2.45 | Destabilizing | 0.4 | 5.14 | Destabilizing | 3.80 | Destabilizing | 0.78 | Ambiguous | 0.682 | Likely Pathogenic | -5.08 | Deleterious | 0.097 | Benign | 0.023 | Benign | 5.71 | Benign | 0.04 | Affected | 0.3987 | 0.5221 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||
| c.680G>T | G227V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G227V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while the remaining twelve tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The high‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.106997 | Structured | 0.329995 | Uncertain | 0.800 | 0.329 | 0.250 | -9.329 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 3.57 | Destabilizing | 0.4 | 6.24 | Destabilizing | 4.91 | Destabilizing | 0.78 | Ambiguous | 0.839 | Likely Pathogenic | -7.58 | Deleterious | 0.952 | Possibly Damaging | 0.521 | Possibly Damaging | 5.67 | Benign | 0.01 | Affected | 0.1130 | 0.4703 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.682A>C | T228P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T228P is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include FATHMM, Rosetta, and premPS, whereas the majority of tools predict a pathogenic impact: REVEL, SIFT, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is inconclusive, but the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also inconclusive. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates a pathogenic effect. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.104810 | Structured | 0.321733 | Uncertain | 0.829 | 0.316 | 0.125 | -8.559 | Likely Pathogenic | 0.939 | Likely Pathogenic | Ambiguous | 1.65 | Ambiguous | 0.6 | 0.47 | Likely Benign | 1.06 | Ambiguous | 0.21 | Likely Benign | 0.690 | Likely Pathogenic | -3.54 | Deleterious | 0.984 | Probably Damaging | 0.690 | Possibly Damaging | 5.64 | Benign | 0.02 | Affected | 0.2207 | 0.6360 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.682A>G | T228A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T228A variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and the SGM‑Consensus score (which is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are REVEL, SIFT, and AlphaMissense‑Default. Predictions that are inconclusive or uncertain are Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as benign (majority of its constituent predictors are benign), and Foldetta as uncertain. Overall, the balance of evidence favors a benign interpretation; this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.104810 | Structured | 0.321733 | Uncertain | 0.829 | 0.316 | 0.125 | -5.190 | Likely Benign | 0.793 | Likely Pathogenic | Ambiguous | 0.44 | Likely Benign | 0.0 | 0.82 | Ambiguous | 0.63 | Ambiguous | 0.58 | Ambiguous | 0.520 | Likely Pathogenic | -2.30 | Neutral | 0.363 | Benign | 0.138 | Benign | 5.63 | Benign | 0.04 | Affected | 0.4072 | 0.5014 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.682A>T | T228S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T228S missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while Rosetta and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a benign stability change. Taken together, the overwhelming majority of evidence indicates a benign impact for T228S. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.104810 | Structured | 0.321733 | Uncertain | 0.829 | 0.316 | 0.125 | -2.028 | Likely Benign | 0.493 | Ambiguous | Likely Benign | 0.32 | Likely Benign | 0.1 | 0.63 | Ambiguous | 0.48 | Likely Benign | 0.05 | Likely Benign | 0.458 | Likely Benign | -0.50 | Neutral | 0.734 | Possibly Damaging | 0.138 | Benign | 5.65 | Benign | 1.00 | Tolerated | 0.3395 | 0.5107 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.683C>A | T228K 2D 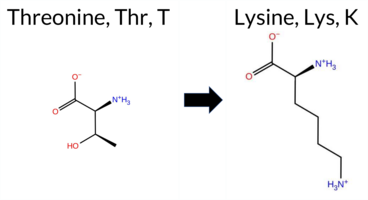 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T228K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Foldetta, and FATHMM, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools give uncertain results (Rosetta and premPS). High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Overall, the balance of evidence favors a pathogenic classification, and this conclusion does not contradict any existing ClinVar annotation because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.104810 | Structured | 0.321733 | Uncertain | 0.829 | 0.316 | 0.125 | -9.143 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 0.03 | Likely Benign | 0.1 | 0.87 | Ambiguous | 0.45 | Likely Benign | 0.70 | Ambiguous | 0.676 | Likely Pathogenic | -3.00 | Deleterious | 0.906 | Possibly Damaging | 0.521 | Possibly Damaging | 5.60 | Benign | 0.02 | Affected | 0.1322 | 0.3590 | 0 | -1 | -3.2 | 27.07 | |||||||||||||||||||||||||||||
| c.683C>G | T228R 2D 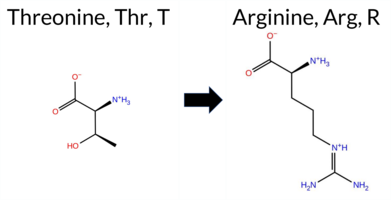 3DClick to see structure in 3D Viewer AIThe SynGAP1 T228R missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Foldetta, and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain predictions come from Rosetta, premPS, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic impact. This conclusion is not contradicted by any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.104810 | Structured | 0.321733 | Uncertain | 0.829 | 0.316 | 0.125 | -7.218 | In-Between | 0.965 | Likely Pathogenic | Likely Pathogenic | -0.23 | Likely Benign | 0.1 | 0.69 | Ambiguous | 0.23 | Likely Benign | 0.67 | Ambiguous | 0.693 | Likely Pathogenic | -3.40 | Deleterious | 0.952 | Possibly Damaging | 0.694 | Possibly Damaging | 5.60 | Benign | 0.01 | Affected | 0.1108 | 0.3405 | -1 | -1 | -3.8 | 55.08 | ||||||||||||||||||||||||||||||
| c.683C>T | T228I 2D AIThe SynGAP1 missense variant T228I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, SIFT, and FATHMM, while those that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. With eight tools favoring pathogenicity versus six favoring benign, and two of the three high‑accuracy methods supporting pathogenicity, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, which currently contains no entry for T228I. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.104810 | Structured | 0.321733 | Uncertain | 0.829 | 0.316 | 0.125 | -8.605 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.02 | Likely Benign | 0.6 | 0.21 | Likely Benign | 0.12 | Likely Benign | 0.23 | Likely Benign | 0.692 | Likely Pathogenic | -3.99 | Deleterious | 0.984 | Probably Damaging | 0.690 | Possibly Damaging | 5.65 | Benign | 0.06 | Tolerated | 0.0951 | 0.7051 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.676T>C | S226P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S226P is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools (REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus) all predict a pathogenic or likely pathogenic outcome. Uncertain results are reported by FoldX, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is inconclusive, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic effect. Overall, the preponderance of evidence indicates that S226P is most likely pathogenic, and this conclusion is not contradicted by ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.129801 | Structured | 0.334959 | Uncertain | 0.800 | 0.324 | 0.250 | -11.030 | Likely Pathogenic | 0.933 | Likely Pathogenic | Ambiguous | 1.51 | Ambiguous | 1.1 | 5.29 | Destabilizing | 3.40 | Destabilizing | 0.57 | Ambiguous | 0.776 | Likely Pathogenic | -3.22 | Deleterious | 0.971 | Probably Damaging | 0.543 | Possibly Damaging | 5.74 | Benign | 0.04 | Affected | 0.2837 | 0.7038 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.676T>A | S226T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S226T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while FoldX is uncertain and therefore treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.129801 | Structured | 0.334959 | Uncertain | 0.800 | 0.324 | 0.250 | -5.066 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.53 | Ambiguous | 0.2 | 0.12 | Likely Benign | 0.33 | Likely Benign | -0.10 | Likely Benign | 0.326 | Likely Benign | -1.47 | Neutral | 0.390 | Benign | 0.079 | Benign | 5.75 | Benign | 0.05 | Affected | 0.1991 | 0.6681 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.667A>C | T223P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T223P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include FoldX, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as Uncertain. No other high‑accuracy tools provide a definitive prediction. Overall, the majority of evidence points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.070400 | Structured | 0.382605 | Uncertain | 0.867 | 0.316 | 0.125 | -13.707 | Likely Pathogenic | 0.974 | Likely Pathogenic | Likely Pathogenic | 0.42 | Likely Benign | 0.3 | 3.45 | Destabilizing | 1.94 | Ambiguous | 0.67 | Ambiguous | 0.898 | Likely Pathogenic | -4.54 | Deleterious | 0.838 | Possibly Damaging | 0.367 | Benign | 5.72 | Benign | 0.01 | Affected | 0.1464 | 0.3728 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.668C>A | T223K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T223K missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. The remaining tools, premPS and AlphaMissense‑Optimized, are uncertain and therefore treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the balance of evidence leans toward a benign impact, with no contradiction to the ClinVar status (which is currently unreported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.070400 | Structured | 0.382605 | Uncertain | 0.867 | 0.316 | 0.125 | -12.084 | Likely Pathogenic | 0.853 | Likely Pathogenic | Ambiguous | -0.30 | Likely Benign | 0.1 | 0.42 | Likely Benign | 0.06 | Likely Benign | 0.93 | Ambiguous | 0.810 | Likely Pathogenic | -4.60 | Deleterious | 0.267 | Benign | 0.086 | Benign | 5.78 | Benign | 0.01 | Affected | 0.0793 | 0.2462 | 0 | -1 | -3.2 | 27.07 | |||||||||||||||||||||||||||||
| c.668C>G | T223R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T223R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a mixed profile: benign calls come from FoldX, Rosetta, Foldetta, polyPhen‑2 HumVar, SIFT, and FATHMM, while pathogenic calls arise from REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy consensus (SGM Consensus) favors pathogenicity (3/4 votes pathogenic), whereas Foldetta predicts benign stability. Because the majority of individual predictors lean benign and the high‑accuracy consensus is split, the overall assessment remains inconclusive. The variant is most likely benign, but the presence of several pathogenic predictions and the SGM Consensus result indicates that pathogenicity cannot be ruled out. This conclusion does not contradict ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.070400 | Structured | 0.382605 | Uncertain | 0.867 | 0.316 | 0.125 | -12.079 | Likely Pathogenic | 0.794 | Likely Pathogenic | Ambiguous | -0.36 | Likely Benign | 0.1 | -0.27 | Likely Benign | -0.32 | Likely Benign | 0.75 | Ambiguous | 0.827 | Likely Pathogenic | -4.62 | Deleterious | 0.561 | Possibly Damaging | 0.178 | Benign | 5.73 | Benign | 0.06 | Tolerated | 0.0708 | 0.2407 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.668C>T | T223I 2D AIThe SynGAP1 T223I variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that clearly indicate benign impact include FoldX, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict pathogenicity are REVEL and PROVEAN. Predictions that are inconclusive (Rosetta, Foldetta, AlphaMissense‑Default) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, while Foldetta remains uncertain. Overall, the majority of definitive predictions support a benign effect. Thus, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.070400 | Structured | 0.382605 | Uncertain | 0.867 | 0.316 | 0.125 | -6.543 | Likely Benign | 0.356 | Ambiguous | Likely Benign | -0.24 | Likely Benign | 0.8 | -0.99 | Ambiguous | -0.62 | Ambiguous | 0.30 | Likely Benign | 0.640 | Likely Pathogenic | -4.09 | Deleterious | 0.010 | Benign | 0.005 | Benign | 5.93 | Benign | 0.07 | Tolerated | 0.0569 | 0.5393 | 0 | -1 | 5.2 | 12.05 | ||||||||||||||||||||||||||||||
| c.66A>C | R22S 2D AIThe SynGAP1 missense variant R22S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; a Foldetta stability prediction is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.538167 | Disordered | 0.441505 | Uncertain | 0.377 | 0.891 | 0.500 | -3.419 | Likely Benign | 0.602 | Likely Pathogenic | Likely Benign | 0.190 | Likely Benign | 0.01 | Neutral | 0.462 | Possibly Damaging | 0.227 | Benign | 4.28 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3252 | 0.5187 | -1 | 0 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||
| c.670A>C | T224P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T224P is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are SIFT and FATHMM, while the remaining tools (REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b) all predict a pathogenic impact; premPS is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.104810 | Structured | 0.360921 | Uncertain | 0.848 | 0.315 | 0.125 | -11.812 | Likely Pathogenic | 0.960 | Likely Pathogenic | Likely Pathogenic | 3.63 | Destabilizing | 0.4 | 2.68 | Destabilizing | 3.16 | Destabilizing | 0.57 | Ambiguous | 0.765 | Likely Pathogenic | -3.65 | Deleterious | 0.971 | Probably Damaging | 0.543 | Possibly Damaging | 5.56 | Benign | 0.23 | Tolerated | 0.2355 | 0.6063 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.670A>T | T224S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T224S missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 (HumDiv) predicts a pathogenic outcome; the remaining tools (Foldetta, premPS, AlphaMissense‑Default, Rosetta) give uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as uncertain. Overall, the preponderance of evidence points to a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.104810 | Structured | 0.360921 | Uncertain | 0.848 | 0.315 | 0.125 | -5.928 | Likely Benign | 0.558 | Ambiguous | Likely Benign | 0.29 | Likely Benign | 0.1 | 0.70 | Ambiguous | 0.50 | Ambiguous | 0.71 | Ambiguous | 0.412 | Likely Benign | -2.09 | Neutral | 0.608 | Possibly Damaging | 0.079 | Benign | 5.56 | Benign | 0.66 | Tolerated | 0.3498 | 0.4994 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.671C>A | T224N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T224N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict a pathogenic effect are premPS, polyPhen‑2 HumDiv, and AlphaMissense‑Default. Uncertain or inconclusive results come from FoldX, ESM1b, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and gnomAD presence, indicating no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.104810 | Structured | 0.360921 | Uncertain | 0.848 | 0.315 | 0.125 | -7.342 | In-Between | 0.814 | Likely Pathogenic | Ambiguous | 0.66 | Ambiguous | 0.2 | -0.06 | Likely Benign | 0.30 | Likely Benign | 1.15 | Destabilizing | 0.370 | Likely Benign | -2.40 | Neutral | 0.845 | Possibly Damaging | 0.368 | Benign | 5.54 | Benign | 0.34 | Tolerated | 0.1460 | 0.5079 | 0 | 0 | -2.8 | 13.00 | ||||||||||||||||||||||||||||||
| c.671C>G | T224S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T224S missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 (HumDiv) predicts a pathogenic outcome; the remaining tools (Foldetta, premPS, AlphaMissense‑Default, Rosetta) give uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as uncertain. Overall, the preponderance of evidence points to a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.104810 | Structured | 0.360921 | Uncertain | 0.848 | 0.315 | 0.125 | -5.928 | Likely Benign | 0.558 | Ambiguous | Likely Benign | 0.29 | Likely Benign | 0.1 | 0.70 | Ambiguous | 0.50 | Ambiguous | 0.71 | Ambiguous | 0.374 | Likely Benign | -2.09 | Neutral | 0.608 | Possibly Damaging | 0.079 | Benign | 5.56 | Benign | 0.66 | Tolerated | 0.3498 | 0.4994 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.671C>T | T224I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T224I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumVar, SIFT, and FATHMM. Tools that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as benign. With an equal split of benign and pathogenic calls overall, the two high‑accuracy pathogenic predictions outweigh the single high‑accuracy benign prediction, indicating that the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as no ClinVar assertion is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.104810 | Structured | 0.360921 | Uncertain | 0.848 | 0.315 | 0.125 | -8.831 | Likely Pathogenic | 0.973 | Likely Pathogenic | Likely Pathogenic | 0.49 | Likely Benign | 0.2 | 0.35 | Likely Benign | 0.42 | Likely Benign | 0.02 | Likely Benign | 0.657 | Likely Pathogenic | -3.47 | Deleterious | 0.608 | Possibly Damaging | 0.154 | Benign | 5.58 | Benign | 0.38 | Tolerated | 0.1015 | 0.7225 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.673T>A | S225T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S225T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also benign; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. No pathogenic predictions are present. Therefore, based on the available computational evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.150080 | Structured | 0.344191 | Uncertain | 0.817 | 0.319 | 0.250 | -6.351 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.64 | Ambiguous | 0.4 | 0.59 | Ambiguous | 0.62 | Ambiguous | 0.03 | Likely Benign | 0.244 | Likely Benign | -1.98 | Neutral | 0.118 | Benign | 0.049 | Benign | 5.73 | Benign | 0.55 | Tolerated | 0.1419 | 0.7395 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.673T>C | S225P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S225P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split opinion: benign calls come from premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls arise from REVEL, FoldX, PROVEAN, and ESM1b. Two tools—Rosetta and Foldetta—return uncertain results and are treated as unavailable. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign, whereas the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta remains unavailable. Overall, the majority of evidence leans toward a benign effect, and this conclusion does not conflict with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.150080 | Structured | 0.344191 | Uncertain | 0.817 | 0.319 | 0.250 | -8.084 | Likely Pathogenic | 0.156 | Likely Benign | Likely Benign | 2.22 | Destabilizing | 1.8 | 1.14 | Ambiguous | 1.68 | Ambiguous | 0.15 | Likely Benign | 0.503 | Likely Pathogenic | -3.31 | Deleterious | 0.396 | Benign | 0.133 | Benign | 5.91 | Benign | 0.17 | Tolerated | 0.2128 | 0.6946 | 1 | -1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||
| c.673T>G | S225A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S225A is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. The high‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also benign. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive (uncertain) and therefore does not alter the overall benign assessment. Consequently, the variant is most likely benign based on the available predictions, and this conclusion is not contradicted by ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.150080 | Structured | 0.344191 | Uncertain | 0.817 | 0.319 | 0.250 | -3.752 | Likely Benign | 0.052 | Likely Benign | Likely Benign | 0.31 | Likely Benign | 0.3 | 0.79 | Ambiguous | 0.55 | Ambiguous | 0.11 | Likely Benign | 0.201 | Likely Benign | -1.52 | Neutral | 0.001 | Benign | 0.001 | Benign | 5.79 | Benign | 0.58 | Tolerated | 0.4840 | 0.5897 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.685A>C | K229Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K229Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from FoldX, Rosetta, FATHMM, and Foldetta; pathogenic predictions arise from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a pathogenic bias: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the preponderance of evidence indicates the variant is most likely pathogenic, and this assessment does not conflict with ClinVar status, which currently has no entry for K229Q. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.179055 | Structured | 0.310912 | Uncertain | 0.843 | 0.306 | 0.000 | -9.606 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.41 | Likely Benign | 0.0 | -0.05 | Likely Benign | 0.18 | Likely Benign | 0.55 | Ambiguous | 0.813 | Likely Pathogenic | -3.03 | Deleterious | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 5.84 | Benign | 0.02 | Affected | 0.4456 | 0.1057 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.714A>T | E238D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E238D missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact; the remaining methods (FoldX, Rosetta, Foldetta, premPS, ESM1b) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect. The variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status because no ClinVar record exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.194234 | Structured | 0.332638 | Uncertain | 0.796 | 0.326 | 0.000 | -7.861 | In-Between | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.25 | Ambiguous | 0.4 | 1.72 | Ambiguous | 1.49 | Ambiguous | 0.78 | Ambiguous | 0.691 | Likely Pathogenic | -2.72 | Deleterious | 0.868 | Possibly Damaging | 0.504 | Possibly Damaging | 5.57 | Benign | 0.05 | Affected | 0.1975 | 0.3619 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.741G>T | Q247H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q247H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, ESM1b, FATHMM, AlphaMissense‑Optimized, and PROVEAN. Those that predict a pathogenic impact are REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default, while premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as benign. No prediction or folding stability result is missing or inconclusive. Based on the overall balance of evidence—seven benign versus five pathogenic predictions, and all high‑accuracy tools indicating benign—the variant is most likely benign. This conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.490133 | Structured | 0.283012 | Uncertain | 0.822 | 0.339 | 0.250 | -6.239 | Likely Benign | 0.583 | Likely Pathogenic | Likely Benign | 0.37 | Likely Benign | 0.2 | 0.02 | Likely Benign | 0.20 | Likely Benign | 0.58 | Ambiguous | 0.579 | Likely Pathogenic | -2.10 | Neutral | 0.995 | Probably Damaging | 0.854 | Possibly Damaging | 5.71 | Benign | 0.02 | Affected | 0.0932 | 0.2558 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.749T>G | V250G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V250G missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (both HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default, while only FATHMM predicts a benign outcome. Uncertain results are reported by FoldX and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, AlphaMissense‑Optimized remains uncertain, and Foldetta is pathogenic. Taken together, the overwhelming majority of evidence indicates a pathogenic effect. This conclusion is consistent with the absence of a ClinVar classification; there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.447574 | Structured | 0.244075 | Uncertain | 0.778 | 0.324 | 0.125 | -11.255 | Likely Pathogenic | 0.917 | Likely Pathogenic | Ambiguous | 1.65 | Ambiguous | 0.3 | 3.18 | Destabilizing | 2.42 | Destabilizing | 2.08 | Destabilizing | 0.900 | Likely Pathogenic | -5.90 | Deleterious | 0.879 | Possibly Damaging | 0.997 | Probably Damaging | 5.75 | Benign | 0.00 | Affected | 0.1884 | 0.1996 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.74G>C | R25P 2D AIThe SynGAP1 missense variant R25P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT uniformly predict a pathogenic outcome. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.461924 | Structured | 0.438941 | Uncertain | 0.373 | 0.890 | 0.375 | -3.394 | Likely Benign | 0.424 | Ambiguous | Likely Benign | 0.155 | Likely Benign | -1.56 | Neutral | 0.841 | Possibly Damaging | 0.809 | Possibly Damaging | 3.94 | Benign | 0.00 | Affected | 0.2217 | 0.4571 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.74G>T | R25L 2D AIThe SynGAP1 missense variant R25L is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors shows a predominance of benign calls: REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized all predict a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT each predict a pathogenic impact. AlphaMissense‑Default remains uncertain, and no Foldetta stability assessment is available. High‑accuracy tools specifically highlight a benign prediction: AlphaMissense‑Optimized is benign, the SGM‑Consensus is likely benign, and Foldetta data are missing. Taken together, the majority of robust predictors and the consensus analysis support a benign classification for R25L. This conclusion is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.461924 | Structured | 0.438941 | Uncertain | 0.373 | 0.890 | 0.375 | -3.443 | Likely Benign | 0.484 | Ambiguous | Likely Benign | 0.121 | Likely Benign | -1.59 | Neutral | 0.686 | Possibly Damaging | 0.630 | Possibly Damaging | 3.97 | Benign | 0.00 | Affected | 0.2045 | 0.4792 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.751A>C | K251Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K251Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and polyPhen‑2 HumVar, while premPS and AlphaMissense‑Default are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Taken together, the majority of evidence supports a benign classification for K251Q, and this conclusion does not contradict the absence of a ClinVar entry. Based on the aggregate predictions, K251Q is most likely benign, and this is consistent with its absence from ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.447574 | Structured | 0.226632 | Uncertain | 0.758 | 0.312 | 0.125 | -5.869 | Likely Benign | 0.362 | Ambiguous | Likely Benign | 0.36 | Likely Benign | 0.1 | 0.42 | Likely Benign | 0.39 | Likely Benign | -0.62 | Ambiguous | 0.465 | Likely Benign | 0.55 | Neutral | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 5.86 | Benign | 0.63 | Tolerated | 0.4466 | 0.1085 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.751A>G | K251E 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K251E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include premPS, PROVEAN, SIFT, and FATHMM, while those that agree on a pathogenic effect are REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The remaining tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain results. High‑accuracy methods are likewise inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a tie, and Foldetta is uncertain. Overall, the majority of available predictions lean toward pathogenicity, with a small but notable benign signal. Therefore, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.447574 | Structured | 0.226632 | Uncertain | 0.758 | 0.312 | 0.125 | -12.812 | Likely Pathogenic | 0.906 | Likely Pathogenic | Ambiguous | 0.88 | Ambiguous | 0.2 | 0.99 | Ambiguous | 0.94 | Ambiguous | 0.40 | Likely Benign | 0.571 | Likely Pathogenic | -0.54 | Neutral | 0.970 | Probably Damaging | 0.584 | Possibly Damaging | 5.80 | Benign | 0.46 | Tolerated | 0.3929 | 0.0860 | 0 | 1 | 0.4 | 0.94 | ||||||||||||||||||||||||||||||
| c.752A>C | K251T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K251T is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include premPS, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Tools with uncertain or inconclusive results are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) supports a pathogenic prediction, while AlphaMissense‑Optimized and Foldetta remain uncertain. Overall, the balance of evidence favors a pathogenic effect for K251T, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.447574 | Structured | 0.226632 | Uncertain | 0.758 | 0.312 | 0.125 | -10.552 | Likely Pathogenic | 0.841 | Likely Pathogenic | Ambiguous | 1.21 | Ambiguous | 0.4 | 0.50 | Ambiguous | 0.86 | Ambiguous | 0.46 | Likely Benign | 0.742 | Likely Pathogenic | -2.72 | Deleterious | 0.970 | Probably Damaging | 0.749 | Possibly Damaging | 5.76 | Benign | 0.28 | Tolerated | 0.2353 | 0.2852 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.752A>G | K251R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K251R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Rosetta and Foldetta give uncertain results. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta as Uncertain. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.447574 | Structured | 0.226632 | Uncertain | 0.758 | 0.312 | 0.125 | -4.355 | Likely Benign | 0.123 | Likely Benign | Likely Benign | -0.37 | Likely Benign | 0.0 | -0.64 | Ambiguous | -0.51 | Ambiguous | 0.14 | Likely Benign | 0.531 | Likely Pathogenic | -0.61 | Neutral | 0.970 | Probably Damaging | 0.681 | Possibly Damaging | 5.92 | Benign | 0.34 | Tolerated | 0.4756 | 0.1049 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.752A>T | K251M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K251M missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, premPS, PROVEAN, and FATHMM. Those that predict pathogenicity are REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta predicts a benign effect. Overall, the evidence is evenly split between benign and pathogenic predictions, with the most reliable high‑accuracy tools leaning toward a benign outcome. Thus, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.447574 | Structured | 0.226632 | Uncertain | 0.758 | 0.312 | 0.125 | -10.678 | Likely Pathogenic | 0.796 | Likely Pathogenic | Ambiguous | 0.14 | Likely Benign | 0.1 | 0.10 | Likely Benign | 0.12 | Likely Benign | 0.05 | Likely Benign | 0.751 | Likely Pathogenic | -2.36 | Neutral | 0.999 | Probably Damaging | 0.970 | Probably Damaging | 5.73 | Benign | 0.05 | Affected | 0.1271 | 0.3475 | 0 | -1 | 5.8 | 3.02 | ||||||||||||||||||||||||||||||
| c.753G>C | K251N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K251N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM (nine tools). Only AlphaMissense‑Default predicts a pathogenic outcome, while ESM1b and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign consensus; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Overall, the preponderance of evidence indicates that K251N is most likely benign, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.447574 | Structured | 0.226632 | Uncertain | 0.758 | 0.312 | 0.125 | -7.978 | In-Between | 0.930 | Likely Pathogenic | Ambiguous | 0.29 | Likely Benign | 0.1 | 0.21 | Likely Benign | 0.25 | Likely Benign | 0.32 | Likely Benign | 0.298 | Likely Benign | -1.29 | Neutral | 0.384 | Benign | 0.070 | Benign | 5.73 | Benign | 0.39 | Tolerated | 0.3848 | 0.1196 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||
| c.753G>T | K251N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K251N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM (nine tools). Only AlphaMissense‑Default predicts a pathogenic outcome, while ESM1b and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign consensus; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Overall, the preponderance of evidence indicates that K251N is most likely benign, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.447574 | Structured | 0.226632 | Uncertain | 0.758 | 0.312 | 0.125 | -7.978 | In-Between | 0.930 | Likely Pathogenic | Ambiguous | 0.29 | Likely Benign | 0.1 | 0.21 | Likely Benign | 0.25 | Likely Benign | 0.32 | Likely Benign | 0.298 | Likely Benign | -1.29 | Neutral | 0.384 | Benign | 0.070 | Benign | 5.73 | Benign | 0.39 | Tolerated | 0.3848 | 0.1196 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||
| c.754C>A | P252T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P252T is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized and the SGM‑Consensus score (Likely Pathogenic). Four tools (FoldX, Rosetta, Foldetta, premPS) give uncertain results and are treated as unavailable. High‑accuracy methods give the following: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic; Foldetta’s stability assessment is uncertain. Overall, the majority of evidence points to a pathogenic effect, and this is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.461924 | Structured | 0.211606 | Uncertain | 0.753 | 0.304 | 0.250 | -11.824 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.24 | Ambiguous | 0.1 | 1.89 | Ambiguous | 1.57 | Ambiguous | 0.71 | Ambiguous | 0.828 | Likely Pathogenic | -7.35 | Deleterious | 0.384 | Benign | 0.177 | Benign | 5.78 | Benign | 0.01 | Affected | 0.1514 | 0.4842 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||
| c.754C>G | P252A 2D 3DClick to see structure in 3D Viewer AISynGAP1 P252A is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: a single benign call from FATHMM, and a majority of pathogenic calls from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Uncertain results are reported by FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for P252A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.461924 | Structured | 0.211606 | Uncertain | 0.753 | 0.304 | 0.250 | -10.587 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.97 | Ambiguous | 0.1 | 1.74 | Ambiguous | 1.36 | Ambiguous | 0.69 | Ambiguous | 0.831 | Likely Pathogenic | -7.35 | Deleterious | 0.941 | Possibly Damaging | 0.607 | Possibly Damaging | 5.87 | Benign | 0.03 | Affected | 0.3490 | 0.4161 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.754C>T | P252S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P252S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, while the majority of tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. Predictions that are uncertain (FoldX, Rosetta, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain. Overall, the evidence strongly favors a pathogenic effect for P252S, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.461924 | Structured | 0.211606 | Uncertain | 0.753 | 0.304 | 0.250 | -10.926 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.76 | Ambiguous | 0.1 | 1.81 | Ambiguous | 1.29 | Ambiguous | 0.79 | Ambiguous | 0.840 | Likely Pathogenic | -7.35 | Deleterious | 0.941 | Possibly Damaging | 0.531 | Possibly Damaging | 5.79 | Benign | 0.05 | Affected | 0.3508 | 0.4361 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||
| c.749T>C | V250A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V250A missense change is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a pathogenic effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default all indicate pathogenicity, whereas ESM1b and FATHMM predict a benign outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) and AlphaMissense‑Optimized return uncertain results and are treated as unavailable. High‑accuracy assessments are likewise inconclusive: AlphaMissense‑Optimized is uncertain, the SGM consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is balanced and therefore unavailable, and Foldetta is uncertain. Overall, the preponderance of evidence points to a pathogenic effect for V250A, and this assessment does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.447574 | Structured | 0.244075 | Uncertain | 0.778 | 0.324 | 0.125 | -6.385 | Likely Benign | 0.852 | Likely Pathogenic | Ambiguous | 0.82 | Ambiguous | 0.1 | 1.22 | Ambiguous | 1.02 | Ambiguous | 1.48 | Destabilizing | 0.818 | Likely Pathogenic | -3.11 | Deleterious | 0.930 | Possibly Damaging | 0.584 | Possibly Damaging | 5.82 | Benign | 0.02 | Affected | 0.2491 | 0.2151 | 0 | 0 | -2.4 | -28.05 | ||||||||||||||||||||||||||||||
| c.749T>A | V250E 2D 3DClick to see structure in 3D Viewer AISynGAP1 variant V250E is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, whereas the remaining 12 tools (SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify the change as pathogenic. The high‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates pathogenic. No prediction or stability result is inconclusive. Consequently, the variant is most likely pathogenic, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.447574 | Structured | 0.244075 | Uncertain | 0.778 | 0.324 | 0.125 | -15.723 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 1.85 | Ambiguous | 0.1 | 3.34 | Destabilizing | 2.60 | Destabilizing | 2.01 | Destabilizing | 0.906 | Likely Pathogenic | -5.13 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 5.74 | Benign | 0.00 | Affected | 0.0787 | 0.1564 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.742C>G | R248G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R248G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM. All other evaluated predictors—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.267126 | Uncertain | 0.781 | 0.346 | 0.250 | -13.413 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.23 | Destabilizing | 0.7 | 2.67 | Destabilizing | 2.45 | Destabilizing | 1.33 | Destabilizing | 0.770 | Likely Pathogenic | -6.07 | Deleterious | 0.958 | Probably Damaging | 0.502 | Possibly Damaging | 5.70 | Benign | 0.00 | Affected | 0.3011 | 0.3385 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.742C>T | R248W 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R248W is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from Rosetta, Foldetta, and FATHMM, while pathogenic predictions are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Uncertain results from FoldX and premPS are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. Overall, the majority of evidence points to a pathogenic effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.267126 | Uncertain | 0.781 | 0.346 | 0.250 | Uncertain | 1 | -11.647 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 1.17 | Ambiguous | 0.3 | -0.20 | Likely Benign | 0.49 | Likely Benign | 0.89 | Ambiguous | 0.699 | Likely Pathogenic | -6.98 | Deleterious | 1.000 | Probably Damaging | 0.948 | Probably Damaging | 5.62 | Benign | 0.00 | Affected | 3.41 | 14 | 0.1124 | 0.4037 | 2 | -3 | 3.6 | 30.03 | 266.4 | 42.3 | 0.0 | 0.0 | 0.3 | 0.1 | X | Potentially Pathogenic | The guanidinium group of Arg248, located on an α helix (res. Ala236-Val250), forms two very stable salt bridges with Asp255 (from a short α helical section, res. Lys254-Asn256) and Glu244 (from a nearby loop) in the WT simulations. In the variant simulations, the indole group of Trp248 cannot form any salt bridges, which could negatively affect the tertiary structure assembly of the PH domain. Instead, in the variant simulations, the indole ring of Trp248 stacks against Pro252, which makes a turn after the α helix. | ||||||||||||||||
| c.743G>C | R248P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R248P is listed in ClinVar as Pathogenic (ClinVar ID 1065478.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized returns a pathogenic score, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic effect. Based on the overwhelming consensus of pathogenic predictions and the high‑accuracy tool results, the variant is most likely pathogenic, which aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.267126 | Uncertain | 0.781 | 0.346 | 0.250 | Likely Pathogenic | 1 | -10.751 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.09 | Destabilizing | 0.6 | 8.87 | Destabilizing | 5.98 | Destabilizing | 1.21 | Destabilizing | 0.848 | Likely Pathogenic | -5.97 | Deleterious | 0.998 | Probably Damaging | 0.878 | Possibly Damaging | 5.64 | Benign | 0.00 | Affected | 3.41 | 14 | 0.1943 | 0.4528 | 0 | -2 | 2.9 | -59.07 | 223.8 | 126.6 | 0.0 | 0.0 | -0.2 | 0.1 | X | X | Potentially Pathogenic | The guanidinium group of Arg248, located on an α helix (residues Ala236-Val250), forms two very stable salt bridges with Asp255 (from a short α helical section, res. Lys254-Asn256) and Glu244 (from a nearby loop) in the WT simulations. In the variant simulations, the pyrrolidine side chain of Pro248 cannot form any salt bridges, which could negatively affect the tertiary structure assembly of the PH domain. Additionally, Pro248 lacks a free amide group needed for hydrogen bonding with the backbone carbonyl group of Asn245, disrupting the continuity of the α helix. | |||||||||||||||
| c.743G>T | R248L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R248L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta and FATHMM, whereas the majority of algorithms predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Predictions labeled uncertain are FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the consensus of the available evidence points to a pathogenic effect for R248L, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.267126 | Uncertain | 0.781 | 0.346 | 0.250 | -12.110 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 1.13 | Ambiguous | 0.5 | 0.01 | Likely Benign | 0.57 | Ambiguous | 0.67 | Ambiguous | 0.825 | Likely Pathogenic | -6.06 | Deleterious | 0.979 | Probably Damaging | 0.680 | Possibly Damaging | 5.66 | Benign | 0.03 | Affected | 0.1616 | 0.4741 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.745G>A | A249T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A249T is listed in ClinVar (ID 1031675.0) with an uncertain significance annotation and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, and FATHMM, whereas polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default predict a pathogenic outcome. Predictions that are inconclusive are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta (combining FoldX‑MD and Rosetta) as uncertain. Overall, the balance of evidence favors a benign interpretation, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.505461 | Disordered | 0.255452 | Uncertain | 0.810 | 0.336 | 0.125 | Uncertain | 1 | -3.564 | Likely Benign | 0.805 | Likely Pathogenic | Ambiguous | 1.50 | Ambiguous | 0.6 | 1.39 | Ambiguous | 1.45 | Ambiguous | 0.30 | Likely Benign | 0.487 | Likely Benign | -0.96 | Neutral | 0.990 | Probably Damaging | 0.815 | Possibly Damaging | 5.65 | Benign | 0.40 | Tolerated | 3.39 | 15 | 0.0909 | 0.4972 | 1 | 0 | -2.5 | 30.03 | 214.5 | -43.3 | 0.0 | 0.0 | 0.5 | 0.2 | X | Potentially Benign | The methyl group of Ala249, located on the surface of an α helix (res. Ala236-Val250) facing an anti-parallel β sheet strand (res. Ile205-Val209), packs against nearby hydrophobic residues such as Leu200, Leu246, and Val250. In the variant simulations, the hydroxyl group of Thr249, which is not suitable for hydrophobic packing, forms a stable hydrogen bond with the backbone carbonyl of Asn245 in the same helix. Although this interaction could theoretically weaken the structural integrity of the α helix, this destabilizing effect is not observed in the variant simulations. | ||||||||||||||||
| c.745G>C | A249P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A249P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is therefore pathogenic (3 pathogenic vs. 1 benign). High‑accuracy assessments are consistent: AlphaMissense‑Optimized indicates pathogenicity; the SGM‑Consensus (majority vote) is pathogenic; and Foldetta, integrating FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. **Based on the collective predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.505461 | Disordered | 0.255452 | Uncertain | 0.810 | 0.336 | 0.125 | -10.727 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.76 | Destabilizing | 0.4 | 8.40 | Destabilizing | 5.58 | Destabilizing | 1.04 | Destabilizing | 0.756 | Likely Pathogenic | -3.32 | Deleterious | 0.176 | Benign | 0.039 | Benign | 5.57 | Benign | 0.03 | Affected | 0.1429 | 0.3537 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.745G>T | A249S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A249S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. Protein‑stability assessments are mixed: FoldX indicates a benign effect, while Rosetta and Foldetta are uncertain. High‑accuracy predictions show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta as Uncertain. Overall, the preponderance of evidence—including the high‑accuracy tools—suggests that A249S is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.505461 | Disordered | 0.255452 | Uncertain | 0.810 | 0.336 | 0.125 | -5.707 | Likely Benign | 0.634 | Likely Pathogenic | Likely Benign | 0.48 | Likely Benign | 0.3 | 0.84 | Ambiguous | 0.66 | Ambiguous | 0.33 | Likely Benign | 0.465 | Likely Benign | -1.40 | Neutral | 0.990 | Probably Damaging | 0.681 | Possibly Damaging | 5.63 | Benign | 0.19 | Tolerated | 0.1962 | 0.3393 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.746C>A | A249E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A249E has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are SIFT and FATHMM. The majority of other in‑silico predictors (SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) classify the variant as pathogenic; FoldX is uncertain and is not counted as evidence for either side. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the preponderance of pathogenic predictions and the absence of benign evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.505461 | Disordered | 0.255452 | Uncertain | 0.810 | 0.336 | 0.125 | -13.519 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 1.60 | Ambiguous | 1.2 | 2.59 | Destabilizing | 2.10 | Destabilizing | 1.02 | Destabilizing | 0.818 | Likely Pathogenic | -3.29 | Deleterious | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 5.64 | Benign | 0.09 | Tolerated | 0.0799 | 0.1509 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||
| c.746C>G | A249G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A249G is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: pathogenic calls come from SGM‑Consensus (Likely Pathogenic), REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Only FATHMM predicts a benign outcome. Uncertain results are reported by FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show SGM‑Consensus as Likely Pathogenic, while AlphaMissense‑Optimized and Foldetta remain inconclusive. Overall, the preponderance of evidence supports a pathogenic classification for A249G, and this conclusion does not conflict with ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.505461 | Disordered | 0.255452 | Uncertain | 0.810 | 0.336 | 0.125 | -9.678 | Likely Pathogenic | 0.947 | Likely Pathogenic | Ambiguous | 1.04 | Ambiguous | 0.2 | 2.02 | Destabilizing | 1.53 | Ambiguous | 1.19 | Destabilizing | 0.607 | Likely Pathogenic | -2.97 | Deleterious | 0.990 | Probably Damaging | 0.760 | Possibly Damaging | 5.59 | Benign | 0.04 | Affected | 0.1667 | 0.2675 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.746C>T | A249V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A249V missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include premPS, PROVEAN, and FATHMM, while those that agree on a pathogenic effect are REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy methods are likewise inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta is uncertain. Consequently, no high‑accuracy tool provides a definitive pathogenic or benign verdict. Overall, the majority of available predictions (seven pathogenic vs. three benign) lean toward a pathogenic interpretation. This assessment does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.505461 | Disordered | 0.255452 | Uncertain | 0.810 | 0.336 | 0.125 | -9.417 | Likely Pathogenic | 0.856 | Likely Pathogenic | Ambiguous | 1.48 | Ambiguous | 0.6 | 0.51 | Ambiguous | 1.00 | Ambiguous | 0.41 | Likely Benign | 0.652 | Likely Pathogenic | -2.47 | Neutral | 0.990 | Probably Damaging | 0.760 | Possibly Damaging | 5.76 | Benign | 0.03 | Affected | 0.0737 | 0.4677 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||
| c.748G>A | V250I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V250I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). All evaluated in silico predictors classify the change as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). The high‑accuracy tools likewise predict a benign effect: AlphaMissense‑Optimized is benign, the SGM Consensus is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No pathogenic predictions are present. Therefore, based on the available computational evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.447574 | Structured | 0.244075 | Uncertain | 0.778 | 0.324 | 0.125 | -6.696 | Likely Benign | 0.085 | Likely Benign | Likely Benign | -0.44 | Likely Benign | 0.1 | -0.01 | Likely Benign | -0.23 | Likely Benign | 0.35 | Likely Benign | 0.362 | Likely Benign | -0.79 | Neutral | 0.384 | Benign | 0.070 | Benign | 6.09 | Benign | 0.13 | Tolerated | 0.0571 | 0.3644 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||
| c.748G>C | V250L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V250L variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include FoldX, PROVEAN, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar. Those that predict pathogenicity are REVEL, polyPhen2_HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. Tools with inconclusive results—Foldetta and premPS—are noted as uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is ambiguous (two pathogenic vs. two benign votes), and Foldetta remains uncertain. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic effect, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.447574 | Structured | 0.244075 | Uncertain | 0.778 | 0.324 | 0.125 | -11.649 | Likely Pathogenic | 0.587 | Likely Pathogenic | Likely Benign | -0.23 | Likely Benign | 0.1 | 2.47 | Destabilizing | 1.12 | Ambiguous | 0.66 | Ambiguous | 0.690 | Likely Pathogenic | -2.47 | Neutral | 0.767 | Possibly Damaging | 0.344 | Benign | 5.81 | Benign | 0.04 | Affected | 0.0698 | 0.4229 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||
| c.748G>T | V250L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V250L variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include FoldX, PROVEAN, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar. Those that predict pathogenicity are REVEL, polyPhen2_HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. Tools with inconclusive results—Foldetta and premPS—are noted as uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is ambiguous (two pathogenic vs. two benign votes), and Foldetta remains uncertain. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic effect, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.447574 | Structured | 0.244075 | Uncertain | 0.778 | 0.324 | 0.125 | -11.649 | Likely Pathogenic | 0.587 | Likely Pathogenic | Likely Benign | -0.23 | Likely Benign | 0.1 | 2.47 | Destabilizing | 1.12 | Ambiguous | 0.66 | Ambiguous | 0.690 | Likely Pathogenic | -2.47 | Neutral | 0.767 | Possibly Damaging | 0.344 | Benign | 5.81 | Benign | 0.04 | Affected | 0.0698 | 0.4229 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||
| c.755C>A | P252H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P252H missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, while the remaining evaluated algorithms (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. No other high‑confidence stability predictions are available. Overall, the preponderance of evidence indicates that P252H is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.461924 | Structured | 0.211606 | Uncertain | 0.753 | 0.304 | 0.250 | -11.991 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.06 | Ambiguous | 0.2 | 1.96 | Ambiguous | 1.51 | Ambiguous | 0.82 | Ambiguous | 0.845 | Likely Pathogenic | -8.27 | Deleterious | 0.999 | Probably Damaging | 0.936 | Probably Damaging | 5.76 | Benign | 0.00 | Affected | 0.1592 | 0.4128 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||
| c.755C>G | P252R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P252R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Computational predictors that classify the variant as benign are FoldX and FATHMM, whereas the majority of tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. Predictions that are inconclusive are Rosetta, Foldetta, and premPS. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; Foldetta remains uncertain. Overall, the preponderance of evidence indicates that P252R is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.461924 | Structured | 0.211606 | Uncertain | 0.753 | 0.304 | 0.250 | -14.648 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.40 | Likely Benign | 0.1 | 1.23 | Ambiguous | 0.82 | Ambiguous | 0.81 | Ambiguous | 0.890 | Likely Pathogenic | -8.27 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 5.80 | Benign | 0.00 | Affected | 0.1413 | 0.2848 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.755C>T | P252L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P252L missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify the variant as benign are premPS and FATHMM, while the majority of other in silico predictors (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a pathogenic effect. Predictions from FoldX, Rosetta, and Foldetta are uncertain and therefore do not contribute to the overall assessment. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.461924 | Structured | 0.211606 | Uncertain | 0.753 | 0.304 | 0.250 | -10.181 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.93 | Ambiguous | 0.0 | 1.56 | Ambiguous | 1.25 | Ambiguous | 0.47 | Likely Benign | 0.794 | Likely Pathogenic | -9.19 | Deleterious | 0.991 | Probably Damaging | 0.781 | Possibly Damaging | 5.81 | Benign | 0.00 | Affected | 0.2027 | 0.6475 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.763G>T | D255Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D255Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include premPS and FATHMM, while the majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Predictions from FoldX, Rosetta, and Foldetta are uncertain and therefore not used as evidence. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as Uncertain. Based on the overall consensus of the available predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.219132 | Uncertain | 0.801 | 0.273 | 0.250 | -13.180 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 1.25 | Ambiguous | 0.2 | 0.96 | Ambiguous | 1.11 | Ambiguous | 0.13 | Likely Benign | 0.832 | Likely Pathogenic | -7.77 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 5.73 | Benign | 0.00 | Affected | 0.0464 | 0.5178 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||
| c.764A>C | D255A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D255A missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include premPS and FATHMM, while the majority of tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus “Likely Pathogenic” vote) predict a pathogenic impact. Predictions from FoldX, Rosetta, and Foldetta are uncertain and therefore do not contribute evidence. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.219132 | Uncertain | 0.801 | 0.273 | 0.250 | -11.789 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.48 | Ambiguous | 0.2 | 1.04 | Ambiguous | 1.26 | Ambiguous | 0.35 | Likely Benign | 0.829 | Likely Pathogenic | -6.85 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 5.80 | Benign | 0.01 | Affected | 0.3363 | 0.4805 | 0 | -2 | 5.3 | -44.01 | ||||||||||||||||||||||||||||||
| c.764A>G | D255G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D255G missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include premPS and FATHMM, while the majority of tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict a pathogenic impact. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive results and are therefore not used as evidence. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain. Based on the collective predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.219132 | Uncertain | 0.801 | 0.273 | 0.250 | -12.652 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.72 | Ambiguous | 0.1 | 1.34 | Ambiguous | 1.53 | Ambiguous | 0.31 | Likely Benign | 0.885 | Likely Pathogenic | -6.13 | Deleterious | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 5.86 | Benign | 0.01 | Affected | 0.3211 | 0.5185 | 1 | -1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||
| c.764A>T | D255V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D255V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from premPS and FATHMM, while pathogenic predictions are made by SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are reported by FoldX, Rosetta, and Foldetta. High‑accuracy methods reinforce the pathogenic interpretation: AlphaMissense‑Optimized predicts Pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains Uncertain. Overall, the consensus of available predictions points to a pathogenic effect for D255V, and this conclusion is consistent with the lack of ClinVar annotation (no contradiction). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.219132 | Uncertain | 0.801 | 0.273 | 0.250 | -13.575 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.76 | Ambiguous | 0.2 | 1.07 | Ambiguous | 1.42 | Ambiguous | 0.16 | Likely Benign | 0.895 | Likely Pathogenic | -7.77 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 5.75 | Benign | 0.00 | Affected | 0.0684 | 0.5162 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||
| c.765C>A | D255E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change D255E is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include premPS, SIFT, ESM1b, and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta is also inconclusive. Overall, the majority of available predictions (seven pathogenic vs. four benign) point to a likely pathogenic impact. This conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.501700 | Disordered | 0.219132 | Uncertain | 0.801 | 0.273 | 0.250 | -6.876 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.80 | Ambiguous | 0.3 | 0.90 | Ambiguous | 0.85 | Ambiguous | 0.15 | Likely Benign | 0.508 | Likely Pathogenic | -2.98 | Deleterious | 0.994 | Probably Damaging | 0.978 | Probably Damaging | 5.89 | Benign | 0.08 | Tolerated | 0.1365 | 0.5067 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||
| c.765C>G | D255E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D255E is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include premPS, SIFT, ESM1b, and FATHMM, whereas those that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. The remaining tools (FoldX, Rosetta, Foldetta) provide uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta is also inconclusive. Overall, the majority of available predictions (7 pathogenic vs. 4 benign) indicate a pathogenic effect. Thus, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.501700 | Disordered | 0.219132 | Uncertain | 0.801 | 0.273 | 0.250 | -6.876 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.80 | Ambiguous | 0.3 | 0.90 | Ambiguous | 0.85 | Ambiguous | 0.15 | Likely Benign | 0.509 | Likely Pathogenic | -2.98 | Deleterious | 0.994 | Probably Damaging | 0.978 | Probably Damaging | 5.89 | Benign | 0.08 | Tolerated | 0.1365 | 0.5067 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||
| c.766A>C | N256H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256H is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Rosetta, Foldetta, premPS, and FATHMM. Those that predict pathogenicity are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). The high‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, whereas Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | -8.090 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.56 | Ambiguous | 0.3 | 0.26 | Likely Benign | 0.41 | Likely Benign | 0.08 | Likely Benign | 0.698 | Likely Pathogenic | -4.06 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 5.82 | Benign | 0.05 | Affected | 0.1233 | 0.5646 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.766A>G | N256D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N256D is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include Rosetta, Foldetta, premPS, and FATHMM, while the majority of tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict a pathogenic impact. FoldX is uncertain and therefore not considered evidence for either side. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as benign. Taken together, the overall consensus leans toward a pathogenic effect, with 10 tools supporting pathogenicity versus 4 supporting benignity. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | -12.478 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.69 | Ambiguous | 0.3 | 0.21 | Likely Benign | 0.45 | Likely Benign | 0.44 | Likely Benign | 0.701 | Likely Pathogenic | -4.36 | Deleterious | 0.997 | Probably Damaging | 0.980 | Probably Damaging | 5.81 | Benign | 0.03 | Affected | 0.1890 | 0.3514 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.766A>T | N256Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that classify the variant as benign include FoldX, Foldetta, premPS, and FATHMM, whereas the majority of tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—label it pathogenic. The high‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the preponderance of evidence from standard predictors and the two high‑accuracy pathogenic calls suggests that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | -10.881 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.17 | Likely Benign | 0.2 | -0.80 | Ambiguous | -0.32 | Likely Benign | 0.30 | Likely Benign | 0.868 | Likely Pathogenic | -6.85 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 5.83 | Benign | 0.00 | Affected | 0.0493 | 0.5881 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.767A>C | N256T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split opinion: benign predictions come from Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). The high‑accuracy AlphaMissense‑Optimized model classifies the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, while the Foldetta stability assessment reports a benign effect. No prediction or stability result is missing or inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | -12.212 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.88 | Ambiguous | 0.1 | -0.18 | Likely Benign | 0.35 | Likely Benign | 0.49 | Likely Benign | 0.819 | Likely Pathogenic | -5.25 | Deleterious | 0.997 | Probably Damaging | 0.980 | Probably Damaging | 5.85 | Benign | 0.01 | Affected | 0.1249 | 0.5848 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.767A>G | N256S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256S is listed in ClinVar as Pathogenic (ClinVar ID 2584352.0) and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from FoldX, Rosetta, Foldetta, premPS, and FATHMM, while pathogenic calls come from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy subset gives AlphaMissense‑Optimized as Uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of predictions support a pathogenic effect, aligning with the ClinVar classification. Therefore, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | Likely Pathogenic | 1 | -10.640 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 0.31 | Likely Benign | 0.2 | 0.36 | Likely Benign | 0.34 | Likely Benign | 0.48 | Likely Benign | 0.707 | Likely Pathogenic | -4.33 | Deleterious | 0.997 | Probably Damaging | 0.970 | Probably Damaging | 5.87 | Benign | 0.02 | Affected | 3.39 | 15 | 0.3024 | 0.5864 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||
| c.767A>T | N256I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta, premPS, and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy methods give a pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | -14.050 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.64 | Ambiguous | 0.4 | 0.45 | Likely Benign | 0.55 | Ambiguous | 0.31 | Likely Benign | 0.849 | Likely Pathogenic | -7.91 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 5.87 | Benign | 0.00 | Affected | 0.0596 | 0.6260 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.768C>A | N256K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256K is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, and FATHMM. Those that predict pathogenicity comprise SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools favor a pathogenic effect, whereas a minority suggest benign. Thus, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | -13.814 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.22 | Likely Benign | 0.3 | -0.10 | Likely Benign | 0.06 | Likely Benign | 0.55 | Ambiguous | 0.569 | Likely Pathogenic | -5.02 | Deleterious | 0.997 | Probably Damaging | 0.986 | Probably Damaging | 5.90 | Benign | 0.01 | Affected | 0.1702 | 0.5240 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.763G>C | D255H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D255H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from premPS and FATHMM, while pathogenic predictions are made by SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are reported by FoldX, Rosetta, and Foldetta. High‑accuracy methods reinforce the pathogenic interpretation: AlphaMissense‑Optimized predicts Pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains Uncertain. Overall, the consensus of the available predictions points to a pathogenic effect for D255H, and this conclusion is consistent with the lack of ClinVar annotation (no contradiction). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.219132 | Uncertain | 0.801 | 0.273 | 0.250 | -14.645 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.72 | Ambiguous | 0.2 | 1.17 | Ambiguous | 1.45 | Ambiguous | 0.44 | Likely Benign | 0.808 | Likely Pathogenic | -5.93 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 5.76 | Benign | 0.00 | Affected | 0.1447 | 0.5211 | 1 | -1 | 0.3 | 22.05 | ||||||||||||||||||||||||||||||
| c.763G>A | D255N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D255N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include premPS and FATHMM, whereas the majority of algorithms—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. FoldX, Rosetta, and Foldetta provide uncertain results. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta remains inconclusive. Overall, the consensus of the available predictions points to a pathogenic effect for D255N, and this conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.219132 | Uncertain | 0.801 | 0.273 | 0.250 | -12.251 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.86 | Ambiguous | 0.1 | 1.48 | Ambiguous | 1.17 | Ambiguous | 0.38 | Likely Benign | 0.682 | Likely Pathogenic | -4.30 | Deleterious | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 5.84 | Benign | 0.01 | Affected | 0.1117 | 0.4774 | 2 | 1 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.757A>C | N253H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N253H is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include FoldX, FATHMM, and premPS, whereas a larger group predicts pathogenicity: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show that the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome, while AlphaMissense‑Optimized and Foldetta provide inconclusive results and are treated as unavailable. No contradictory evidence is present in ClinVar. Overall, the preponderance of evidence from multiple in‑silico predictors points to a pathogenic effect for the variant, and this conclusion does not conflict with the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.201744 | Uncertain | 0.771 | 0.298 | 0.250 | -12.199 | Likely Pathogenic | 0.899 | Likely Pathogenic | Ambiguous | 0.31 | Likely Benign | 0.1 | 0.76 | Ambiguous | 0.54 | Ambiguous | -0.06 | Likely Benign | 0.832 | Likely Pathogenic | -4.39 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 5.51 | Benign | 0.01 | Affected | 0.1936 | 0.8033 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.757A>G | N253D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N253D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on benign effects include FoldX, Rosetta, Foldetta, premPS, SIFT, and FATHMM, while those that predict pathogenicity are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicating likely pathogenic, whereas Foldetta predicts a benign effect on protein stability. Overall, the majority of tools (8/15) favor a pathogenic outcome, with no conflict with ClinVar status because the variant is not yet catalogued. Thus, the variant is most likely pathogenic based on current computational predictions, and this assessment does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.201744 | Uncertain | 0.771 | 0.298 | 0.250 | -14.105 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.25 | Likely Benign | 0.0 | 0.37 | Likely Benign | 0.31 | Likely Benign | 0.09 | Likely Benign | 0.742 | Likely Pathogenic | -4.13 | Deleterious | 0.993 | Probably Damaging | 0.971 | Probably Damaging | 5.60 | Benign | 0.09 | Tolerated | 0.2248 | 0.5186 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.757A>T | N253Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N253Y is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect include FoldX, FATHMM, and premPS, whereas a larger set—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Because the majority of evidence points to a deleterious impact, the variant is most likely pathogenic; this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.201744 | Uncertain | 0.771 | 0.298 | 0.250 | -14.749 | Likely Pathogenic | 0.920 | Likely Pathogenic | Ambiguous | 0.27 | Likely Benign | 0.1 | 1.13 | Ambiguous | 0.70 | Ambiguous | 0.29 | Likely Benign | 0.896 | Likely Pathogenic | -7.01 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 5.55 | Benign | 0.01 | Affected | 0.0642 | 0.7055 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.758A>C | N253T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N253T is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include premPS, SIFT, and FATHMM, whereas the majority of tools (SGM‑Consensus, REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default) predict a pathogenic outcome. High‑accuracy methods give a consistent pathogenic signal: the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta) also predicts pathogenic. AlphaMissense‑Optimized is uncertain and is treated as unavailable. Overall, the preponderance of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.201744 | Uncertain | 0.771 | 0.298 | 0.250 | -11.656 | Likely Pathogenic | 0.929 | Likely Pathogenic | Ambiguous | 1.96 | Ambiguous | 0.3 | 2.67 | Destabilizing | 2.32 | Destabilizing | 0.16 | Likely Benign | 0.739 | Likely Pathogenic | -5.01 | Deleterious | 0.993 | Probably Damaging | 0.971 | Probably Damaging | 5.54 | Benign | 0.06 | Tolerated | 0.1640 | 0.8665 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.759C>A | N253K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N253K resides in the PH domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Functional prediction tools that classify the variant as benign include FoldX, Foldetta, premPS, and FATHMM. In contrast, the majority of predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—label it pathogenic or likely pathogenic. The high‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus also indicates likely pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the preponderance of evidence from standard and high‑accuracy predictors points to a pathogenic effect. This assessment is not contradicted by ClinVar, which currently contains no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.201744 | Uncertain | 0.771 | 0.298 | 0.250 | -15.834 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.05 | Likely Benign | 0.0 | 0.90 | Ambiguous | 0.48 | Likely Benign | 0.17 | Likely Benign | 0.663 | Likely Pathogenic | -5.21 | Deleterious | 0.993 | Probably Damaging | 0.979 | Probably Damaging | 5.53 | Benign | 0.03 | Affected | 0.2640 | 0.6592 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.759C>G | N253K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N253K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools fall into two groups: benign predictions come from FoldX, FATHMM, premPS, and Foldetta, while pathogenic predictions are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Benign. Overall, the majority of tools predict pathogenicity, and the two most reliable predictors also lean pathogenic, whereas only one high‑accuracy tool suggests benign. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.201744 | Uncertain | 0.771 | 0.298 | 0.250 | -15.834 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.05 | Likely Benign | 0.0 | 0.90 | Ambiguous | 0.48 | Likely Benign | 0.17 | Likely Benign | 0.663 | Likely Pathogenic | -5.21 | Deleterious | 0.993 | Probably Damaging | 0.979 | Probably Damaging | 5.53 | Benign | 0.03 | Affected | 0.2640 | 0.6592 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.760A>C | K254Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K254Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Foldetta, SIFT, and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). Two tools, Rosetta and premPS, give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. No prediction or folding stability result is missing or inconclusive. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.207751 | Uncertain | 0.799 | 0.285 | 0.375 | -12.332 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.13 | Likely Benign | 0.1 | -0.61 | Ambiguous | -0.24 | Likely Benign | 0.90 | Ambiguous | 0.737 | Likely Pathogenic | -3.04 | Deleterious | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 5.91 | Benign | 0.11 | Tolerated | 0.3749 | 0.1483 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.760A>G | K254E 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K254E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: FATHMM is the sole benign predictor, while the remaining 13 tools (REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and SGM‑Consensus) predict pathogenicity; FoldX and Foldetta are uncertain. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized indicates pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) remains uncertain. Overall, the preponderance of evidence supports a pathogenic classification for K254E, and this conclusion does not conflict with ClinVar, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.207751 | Uncertain | 0.799 | 0.285 | 0.375 | -14.745 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.72 | Ambiguous | 0.1 | 2.34 | Destabilizing | 1.53 | Ambiguous | 1.17 | Destabilizing | 0.860 | Likely Pathogenic | -3.27 | Deleterious | 0.970 | Probably Damaging | 0.584 | Possibly Damaging | 5.87 | Benign | 0.05 | Affected | 0.3224 | 0.1352 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.761A>C | K254T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K254T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, PROVEAN, both polyPhen‑2 HumDiv and HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). Benign predictions are limited to SIFT and FATHMM. Uncertain results are reported by FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, while Foldetta’s stability analysis is inconclusive. Overall, the majority of evidence points to a pathogenic effect for K254T, and this conclusion does not conflict with ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.207751 | Uncertain | 0.799 | 0.285 | 0.375 | -13.793 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.51 | Ambiguous | 0.0 | 1.92 | Ambiguous | 1.22 | Ambiguous | 0.62 | Ambiguous | 0.883 | Likely Pathogenic | -5.14 | Deleterious | 0.970 | Probably Damaging | 0.749 | Possibly Damaging | 5.88 | Benign | 0.06 | Tolerated | 0.1825 | 0.2562 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.761A>G | K254R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K254R missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. The remaining tools—Rosetta, Foldetta, premPS, and AlphaMissense‑Default—return uncertain or inconclusive results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority (2 benign vs. 1 pathogenic, 1 uncertain); Foldetta is uncertain. Taken together, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.513880 | Disordered | 0.207751 | Uncertain | 0.799 | 0.285 | 0.375 | -11.064 | Likely Pathogenic | 0.528 | Ambiguous | Likely Benign | -0.44 | Likely Benign | 0.1 | -1.13 | Ambiguous | -0.79 | Ambiguous | 0.76 | Ambiguous | 0.604 | Likely Pathogenic | -2.36 | Neutral | 0.970 | Probably Damaging | 0.681 | Possibly Damaging | 5.81 | Benign | 0.09 | Tolerated | 0.4070 | 0.1658 | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||
| c.761A>T | K254M 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K254M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show discordant results: benign predictions come from FoldX, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions are reported by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further highlight the conflict: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. No prediction or stability result is missing or inconclusive. Overall, the majority of tools and the high‑accuracy consensus lean toward a pathogenic effect, and this assessment does not contradict ClinVar status, which currently has no entry for it. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.207751 | Uncertain | 0.799 | 0.285 | 0.375 | -12.832 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | -0.35 | Likely Benign | 0.5 | 0.48 | Likely Benign | 0.07 | Likely Benign | 0.23 | Likely Benign | 0.864 | Likely Pathogenic | -5.08 | Deleterious | 0.999 | Probably Damaging | 0.970 | Probably Damaging | 5.78 | Benign | 0.00 | Affected | 0.1073 | 0.3562 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.762G>C | K254N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K254N is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools that classify the variant as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The majority of other in silico predictors—REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—indicate a pathogenic effect. Stability‑based methods FoldX, Rosetta, and Foldetta returned uncertain results and are therefore not considered evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as unavailable. Overall, the preponderance of evidence supports a pathogenic classification, which contradicts the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.207751 | Uncertain | 0.799 | 0.285 | 0.375 | Uncertain | 1 | -13.306 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.73 | Ambiguous | 0.2 | 1.87 | Ambiguous | 1.30 | Ambiguous | 1.19 | Destabilizing | 0.757 | Likely Pathogenic | -4.23 | Deleterious | 0.384 | Benign | 0.070 | Benign | 5.93 | Benign | 0.01 | Affected | 3.39 | 15 | 0.3200 | 0.1488 | 1 | 0 | 0.4 | -14.07 | 215.3 | -21.0 | -1.0 | 1.7 | 0.2 | 0.3 | X | Potentially Pathogenic | The amino group of Lys254, located in an α-β loop connecting the PH and C2 domains (res. Lys251-Arg258), forms salt bridges with the carboxylate groups of Glu244 and Asp684. Since the neutral carboxamide group of the Asn254 side chain cannot form salt bridges with acidic residues, the residue swap potentially weakens the tertiary structure assembly and/or influences the loop positioning. Regardless, in both the variant and WT simulations, all hydrogen bonds formed by the residue’s side chain were broken, and the residue rotated outwards. The partially α helical conformation of the loop, which extends to a nearby α helix (res. Met414-Asn426), is dynamic, making it unclear if the mutation affects it. | ||||||||||||||||
| c.762G>T | K254N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K254N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic” (3 pathogenic vs. 1 benign). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No evidence from FoldX or Rosetta alone is conclusive. Based on the overall pattern of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not currently catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.207751 | Uncertain | 0.799 | 0.285 | 0.375 | -13.306 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.73 | Ambiguous | 0.2 | 1.87 | Ambiguous | 1.30 | Ambiguous | 1.19 | Destabilizing | 0.757 | Likely Pathogenic | -4.23 | Deleterious | 0.384 | Benign | 0.070 | Benign | 5.93 | Benign | 0.01 | Affected | 3.39 | 15 | 0.3200 | 0.1488 | 1 | 0 | 0.4 | -14.07 | 215.3 | -21.0 | -1.0 | 1.7 | 0.2 | 0.3 | X | Potentially Pathogenic | The amino group of Lys254, located in an α-β loop connecting the PH and C2 domains (res. Lys251-Arg258), forms salt bridges with the carboxylate groups of Glu244 and Asp684. Since the neutral carboxamide group of the Asn254 side chain cannot form salt bridges with acidic residues, the residue swap potentially weakens the tertiary structure assembly and/or influences the loop positioning. Regardless, in both the variant and WT simulations, all hydrogen bonds formed by the residue’s side chain were broken, and the residue rotated outwards. The partially α helical conformation of the loop, which extends to a nearby α helix (res. Met414-Asn426), is dynamic, making it unclear if the mutation affects it. | ||||||||||||||||||
| c.768C>G | N256K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256K is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, and FATHMM. Those that predict pathogenicity comprise SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools support a pathogenic effect, whereas a minority suggest benign. Thus, the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | -13.814 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.22 | Likely Benign | 0.3 | -0.10 | Likely Benign | 0.06 | Likely Benign | 0.55 | Ambiguous | 0.569 | Likely Pathogenic | -5.02 | Deleterious | 0.997 | Probably Damaging | 0.986 | Probably Damaging | 5.90 | Benign | 0.01 | Affected | 0.1702 | 0.5240 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.741G>C | Q247H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q247H is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include SGM‑Consensus, FoldX, Rosetta, Foldetta, ESM1b, FATHMM, AlphaMissense‑Optimized, and PROVEAN. Tools that predict a pathogenic outcome are REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. No predictions are missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.490133 | Structured | 0.283012 | Uncertain | 0.822 | 0.339 | 0.250 | -6.239 | Likely Benign | 0.583 | Likely Pathogenic | Likely Benign | 0.37 | Likely Benign | 0.2 | 0.02 | Likely Benign | 0.20 | Likely Benign | 0.58 | Ambiguous | 0.579 | Likely Pathogenic | -2.10 | Neutral | 0.995 | Probably Damaging | 0.854 | Possibly Damaging | 5.71 | Benign | 0.02 | Affected | 0.0932 | 0.2558 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.715A>G | R239G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R239G missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to FATHMM, whereas the remaining twelve tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts a pathogenic effect. Based on the collective predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.170161 | Structured | 0.336504 | Uncertain | 0.854 | 0.319 | 0.000 | -12.133 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 4.05 | Destabilizing | 0.1 | 3.41 | Destabilizing | 3.73 | Destabilizing | 1.33 | Destabilizing | 0.912 | Likely Pathogenic | -6.26 | Deleterious | 0.982 | Probably Damaging | 0.533 | Possibly Damaging | 5.62 | Benign | 0.02 | Affected | 0.3415 | 0.3154 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.721A>C | K241Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K241Q missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include FoldX, Rosetta, FATHMM, and Foldetta, whereas the majority of tools predict it to be pathogenic: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, nine tools predict pathogenicity versus four predicting benign, with no ClinVar evidence to contradict these findings. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.196879 | Structured | 0.349250 | Uncertain | 0.797 | 0.347 | 0.000 | -10.593 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.33 | Likely Benign | 0.0 | 0.46 | Likely Benign | 0.40 | Likely Benign | 0.63 | Ambiguous | 0.767 | Likely Pathogenic | -3.33 | Deleterious | 0.995 | Probably Damaging | 0.914 | Probably Damaging | 5.83 | Benign | 0.04 | Affected | 0.4225 | 0.1527 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.721A>G | K241E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K241E missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include SIFT and FATHMM, whereas a majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments further support a deleterious interpretation: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains uncertain. Overall, the preponderance of evidence from multiple independent predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.196879 | Structured | 0.349250 | Uncertain | 0.797 | 0.347 | 0.000 | -12.582 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.75 | Ambiguous | 0.1 | 1.36 | Ambiguous | 1.06 | Ambiguous | 0.68 | Ambiguous | 0.878 | Likely Pathogenic | -3.43 | Deleterious | 0.982 | Probably Damaging | 0.679 | Possibly Damaging | 6.00 | Benign | 0.10 | Tolerated | 0.3610 | 0.1179 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.722A>C | K241T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K241T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Benign predictions are reported by Rosetta and FATHMM. Two tools (Foldetta and premPS) give uncertain results. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenic; Foldetta remains uncertain. Overall, the consensus of the majority of evidence points to a pathogenic effect for K241T, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.196879 | Structured | 0.349250 | Uncertain | 0.797 | 0.347 | 0.000 | -10.911 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 2.00 | Destabilizing | 0.4 | 0.06 | Likely Benign | 1.03 | Ambiguous | 0.60 | Ambiguous | 0.853 | Likely Pathogenic | -5.14 | Deleterious | 0.995 | Probably Damaging | 0.747 | Possibly Damaging | 5.74 | Benign | 0.03 | Affected | 0.2021 | 0.3951 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.722A>G | K241R 2D AIThe SynGAP1 missense variant K241R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.196879 | Structured | 0.349250 | Uncertain | 0.797 | 0.347 | 0.000 | -4.615 | Likely Benign | 0.444 | Ambiguous | Likely Benign | -0.23 | Likely Benign | 0.5 | -0.03 | Likely Benign | -0.13 | Likely Benign | 0.43 | Likely Benign | 0.602 | Likely Pathogenic | -2.12 | Neutral | 0.982 | Probably Damaging | 0.679 | Possibly Damaging | 5.89 | Benign | 0.14 | Tolerated | 0.4571 | 0.1242 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.722A>T | K241I 2D 3DClick to see structure in 3D Viewer AISynGAP1 K241I is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Rosetta, premPS, and FATHMM. Those that predict a damaging effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). FoldX and Foldetta return uncertain results. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized reports pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, while Foldetta remains inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.196879 | Structured | 0.349250 | Uncertain | 0.797 | 0.347 | 0.000 | -14.370 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.31 | Ambiguous | 0.5 | 0.28 | Likely Benign | 0.80 | Ambiguous | 0.41 | Likely Benign | 0.852 | Likely Pathogenic | -6.96 | Deleterious | 0.985 | Probably Damaging | 0.704 | Possibly Damaging | 5.71 | Benign | 0.01 | Affected | 0.1176 | 0.3868 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.723A>C | K241N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K241N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.196879 | Structured | 0.349250 | Uncertain | 0.797 | 0.347 | 0.000 | -10.198 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.92 | Ambiguous | 0.1 | 1.13 | Ambiguous | 1.03 | Ambiguous | 0.89 | Ambiguous | 0.663 | Likely Pathogenic | -4.23 | Deleterious | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 5.81 | Benign | 0.03 | Affected | 0.3426 | 0.1872 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.723A>T | K241N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K241N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are reported for FoldX, Rosetta, Foldetta, and premPS and are not used as evidence. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain. Based on the overall consensus of the available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.196879 | Structured | 0.349250 | Uncertain | 0.797 | 0.347 | 0.000 | -10.198 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.92 | Ambiguous | 0.1 | 1.13 | Ambiguous | 1.03 | Ambiguous | 0.89 | Ambiguous | 0.663 | Likely Pathogenic | -4.23 | Deleterious | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 5.81 | Benign | 0.03 | Affected | 0.3426 | 0.1872 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.724T>A | W242R 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant W242R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that assess evolutionary conservation and protein function uniformly indicate a deleterious effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool predicts a benign outcome; the only inconclusive results come from FoldX, Rosetta, and Foldetta, which are treated as unavailable evidence. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta remains uncertain. Consequently, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.328603 | Structured | 0.352582 | Uncertain | 0.847 | 0.341 | 0.000 | -11.948 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.85 | Ambiguous | 0.5 | 1.15 | Ambiguous | 1.00 | Ambiguous | 1.34 | Destabilizing | 0.859 | Likely Pathogenic | -12.71 | Deleterious | 0.995 | Probably Damaging | 0.854 | Possibly Damaging | 1.52 | Pathogenic | 0.00 | Affected | 3.40 | 14 | 0.4206 | 0.0771 | -3 | 2 | -3.6 | -30.03 | |||||||||||||||||||||||||||
| c.724T>G | W242G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W242G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD, so no population frequency data are available. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while the single uncertain call from premPS is treated as unavailable. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the unanimous pathogenic predictions and the lack of benign evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.328603 | Structured | 0.352582 | Uncertain | 0.847 | 0.341 | 0.000 | -14.319 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.70 | Destabilizing | 0.3 | 2.48 | Destabilizing | 2.59 | Destabilizing | 0.83 | Ambiguous | 0.883 | Likely Pathogenic | -11.80 | Deleterious | 0.900 | Possibly Damaging | 0.452 | Possibly Damaging | 1.52 | Pathogenic | 0.00 | Affected | 0.4378 | 0.1851 | -7 | -2 | 0.5 | -129.16 | |||||||||||||||||||||||||||||
| c.725G>C | W242S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W242S, located in the PH domain, is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool predicts a benign outcome. High‑accuracy assessments further support a harmful impact: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic,” while Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Overall, the consensus of available predictions indicates that W242S is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.328603 | Structured | 0.352582 | Uncertain | 0.847 | 0.341 | 0.000 | -14.674 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 1.96 | Ambiguous | 0.2 | 1.97 | Ambiguous | 1.97 | Ambiguous | 0.83 | Ambiguous | 0.797 | Likely Pathogenic | -12.71 | Deleterious | 0.900 | Possibly Damaging | 0.535 | Possibly Damaging | 1.52 | Pathogenic | 0.00 | Affected | 0.4424 | 0.1418 | -2 | -3 | 0.1 | -99.14 | |||||||||||||||||||||||||||||
| c.725G>T | W242L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W242L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions (FoldX, Rosetta, Foldetta, premPS) and pathogenic predictions (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Pathogenic). High‑accuracy assessments further support this dichotomy: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a benign effect. Overall, the majority of evidence points toward a pathogenic impact for W242L. This conclusion is consistent with the absence of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.328603 | Structured | 0.352582 | Uncertain | 0.847 | 0.341 | 0.000 | -12.297 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | -0.29 | Likely Benign | 0.3 | 0.12 | Likely Benign | -0.09 | Likely Benign | 0.44 | Likely Benign | 0.799 | Likely Pathogenic | -11.80 | Deleterious | 0.948 | Possibly Damaging | 0.533 | Possibly Damaging | 1.52 | Pathogenic | 0.01 | Affected | 0.2370 | 0.2986 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||
| c.726G>C | W242C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 W242C missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that are uncertain or inconclusive are FoldX, Rosetta, Foldetta, and premPS, and no tools predict a benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain. Based on the collective predictions, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as ClinVar has not yet classified the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.328603 | Structured | 0.352582 | Uncertain | 0.847 | 0.341 | 0.000 | -13.117 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 1.77 | Ambiguous | 0.4 | 1.62 | Ambiguous | 1.70 | Ambiguous | 0.63 | Ambiguous | 0.889 | Likely Pathogenic | -11.80 | Deleterious | 0.999 | Probably Damaging | 0.887 | Possibly Damaging | 1.52 | Pathogenic | 0.00 | Affected | 0.3626 | 0.1617 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||||
| c.726G>T | W242C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W242C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that are uncertain or inconclusive are FoldX, Rosetta, Foldetta, and premPS, and no tools predict a benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain. Based on the collective predictions, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as ClinVar has not yet classified the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.328603 | Structured | 0.352582 | Uncertain | 0.847 | 0.341 | 0.000 | -13.117 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 1.77 | Ambiguous | 0.4 | 1.62 | Ambiguous | 1.70 | Ambiguous | 0.63 | Ambiguous | 0.889 | Likely Pathogenic | -11.80 | Deleterious | 0.999 | Probably Damaging | 0.887 | Possibly Damaging | 1.52 | Pathogenic | 0.00 | Affected | 0.3626 | 0.1617 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||||
| c.720C>G | D240E 2D 3DClick to see structure in 3D Viewer AISynGAP1 D240E is a missense change in the PH domain. No ClinVar entry exists for this variant, and it is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from SIFT, FoldX, premPS, and FATHMM, while pathogenic predictions arise from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Tools with inconclusive results—AlphaMissense‑Optimized, Rosetta, and Foldetta—are treated as unavailable. High‑accuracy assessments show the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifying the variant as Likely Pathogenic, whereas AlphaMissense‑Optimized and Foldetta provide no definitive verdict. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.343480 | Uncertain | 0.822 | 0.333 | 0.000 | -8.619 | Likely Pathogenic | 0.796 | Likely Pathogenic | Ambiguous | 0.49 | Likely Benign | 0.1 | 0.51 | Ambiguous | 0.50 | Ambiguous | 0.28 | Likely Benign | 0.713 | Likely Pathogenic | -3.43 | Deleterious | 0.984 | Probably Damaging | 0.967 | Probably Damaging | 5.84 | Benign | 0.10 | Tolerated | 0.1155 | 0.5066 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.720C>A | D240E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D240E missense variant is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include FoldX, premPS, SIFT, and FATHMM, while those that agree on a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Tools with uncertain or inconclusive results are Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evaluated predictors (seven pathogenic vs. four benign) lean toward a pathogenic interpretation. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.343480 | Uncertain | 0.822 | 0.333 | 0.000 | -8.619 | Likely Pathogenic | 0.796 | Likely Pathogenic | Ambiguous | 0.49 | Likely Benign | 0.1 | 0.51 | Ambiguous | 0.50 | Ambiguous | 0.28 | Likely Benign | 0.713 | Likely Pathogenic | -3.43 | Deleterious | 0.984 | Probably Damaging | 0.967 | Probably Damaging | 5.84 | Benign | 0.10 | Tolerated | 0.1155 | 0.5066 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.716G>A | R239K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R239K missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are polyPhen‑2 HumVar and FATHMM, while the majority of other in silico predictors (SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default) indicate a pathogenic impact. High‑accuracy assessments show that AlphaMissense‑Optimized is uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Because uncertain or unavailable results are not taken as evidence for or against pathogenicity, the overall evidence still leans toward a deleterious effect. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.170161 | Structured | 0.336504 | Uncertain | 0.854 | 0.319 | 0.000 | -12.492 | Likely Pathogenic | 0.897 | Likely Pathogenic | Ambiguous | 1.93 | Ambiguous | 0.2 | 1.62 | Ambiguous | 1.78 | Ambiguous | 1.41 | Destabilizing | 0.719 | Likely Pathogenic | -2.52 | Deleterious | 0.882 | Possibly Damaging | 0.428 | Benign | 5.78 | Benign | 0.03 | Affected | 0.5222 | 0.4000 | Weaken | 3 | 2 | 0.6 | -28.01 | ||||||||||||||||||||||||||||
| c.716G>T | R239I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R239I is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, while the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS (uncertain), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. No predictions are missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.170161 | Structured | 0.336504 | Uncertain | 0.854 | 0.319 | 0.000 | -19.414 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.32 | Destabilizing | 0.5 | 2.53 | Destabilizing | 3.43 | Destabilizing | 0.83 | Ambiguous | 0.890 | Likely Pathogenic | -7.16 | Deleterious | 0.985 | Probably Damaging | 0.724 | Possibly Damaging | 5.69 | Benign | 0.00 | Affected | 0.1480 | 0.3985 | -2 | -3 | 9.0 | -43.03 | |||||||||||||||||||||||||||||
| c.717A>C | R239S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R239S missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumVar and FATHMM. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.170161 | Structured | 0.336504 | Uncertain | 0.854 | 0.319 | 0.000 | -13.418 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.67 | Destabilizing | 0.1 | 2.94 | Destabilizing | 3.31 | Destabilizing | 1.26 | Destabilizing | 0.813 | Likely Pathogenic | -5.31 | Deleterious | 0.900 | Possibly Damaging | 0.376 | Benign | 5.64 | Benign | 0.02 | Affected | 0.3160 | 0.3788 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||
| c.717A>T | R239S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R239S missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumVar and FATHMM. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.170161 | Structured | 0.336504 | Uncertain | 0.854 | 0.319 | 0.000 | -13.418 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.67 | Destabilizing | 0.1 | 2.94 | Destabilizing | 3.31 | Destabilizing | 1.26 | Destabilizing | 0.813 | Likely Pathogenic | -5.31 | Deleterious | 0.900 | Possibly Damaging | 0.376 | Benign | 5.64 | Benign | 0.02 | Affected | 0.3160 | 0.3788 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||
| c.718G>A | D240N 2D AIThe SynGAP1 missense variant D240N is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Benign predictions are provided by FoldX, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions come from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus. High‑accuracy methods give a split: AlphaMissense‑Optimized predicts benign, SGM‑Consensus predicts pathogenic, and Foldetta predicts benign. Overall, the majority of tools favor a benign effect, and this consensus does not contradict the ClinVar uncertain status. Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.343480 | Uncertain | 0.822 | 0.333 | 0.000 | Uncertain | 1 | -12.942 | Likely Pathogenic | 0.755 | Likely Pathogenic | Likely Benign | 0.22 | Likely Benign | 0.9 | 0.47 | Likely Benign | 0.35 | Likely Benign | 0.37 | Likely Benign | 0.701 | Likely Pathogenic | -4.37 | Deleterious | 0.993 | Probably Damaging | 0.984 | Probably Damaging | 5.88 | Benign | 0.01 | Affected | 0.0993 | 0.4973 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||
| c.718G>C | D240H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D240H missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include premPS and FATHMM, whereas the majority of tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; FoldX, Rosetta, and Foldetta are uncertain. High‑accuracy methods give a pathogenic verdict: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta remains uncertain. Overall, the consensus of the available predictions indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.343480 | Uncertain | 0.822 | 0.333 | 0.000 | -14.551 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.14 | Ambiguous | 0.0 | 0.61 | Ambiguous | 0.88 | Ambiguous | 0.19 | Likely Benign | 0.865 | Likely Pathogenic | -6.12 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 5.78 | Benign | 0.00 | Affected | 0.1223 | 0.5398 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.718G>T | D240Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D240Y missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from FoldX, premPS, and FATHMM; pathogenic predictions come from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive and therefore not considered evidence. Taken together, the overwhelming majority of reliable predictors indicate a pathogenic effect for D240Y. This conclusion is consistent with the lack of a ClinVar classification, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.343480 | Uncertain | 0.822 | 0.333 | 0.000 | -16.890 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.29 | Likely Benign | 0.3 | 1.07 | Ambiguous | 0.68 | Ambiguous | -0.01 | Likely Benign | 0.899 | Likely Pathogenic | -7.90 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 5.77 | Benign | 0.00 | Affected | 0.0439 | 0.4932 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.719A>C | D240A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D240A missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are premPS and FATHMM, while the remaining pathogenic‑predicting tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently flag the variant as deleterious. Tools with uncertain outcomes (FoldX, Rosetta, Foldetta) provide no definitive guidance. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates pathogenicity, and Foldetta remains inconclusive. Overall, the preponderance of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.343480 | Uncertain | 0.822 | 0.333 | 0.000 | -12.935 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.75 | Ambiguous | 0.1 | 0.99 | Ambiguous | 0.87 | Ambiguous | 0.22 | Likely Benign | 0.872 | Likely Pathogenic | -7.03 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 5.80 | Benign | 0.05 | Affected | 0.3317 | 0.4992 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.719A>G | D240G 2D AIThe SynGAP1 missense variant D240G is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Benign predictions are provided by premPS and FATHMM, whereas pathogenic predictions are made by REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta. FoldX‑MD is inconclusive, and AlphaMissense‑Optimized is uncertain. High‑accuracy methods show that AlphaMissense‑Optimized is inconclusive, SGM Consensus predicts pathogenic, and Foldetta predicts pathogenic. Overall, the majority of evidence points to a pathogenic effect, which is consistent with the ClinVar uncertain status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.343480 | Uncertain | 0.822 | 0.333 | 0.000 | Uncertain | 1 | -12.825 | Likely Pathogenic | 0.951 | Likely Pathogenic | Ambiguous | 1.85 | Ambiguous | 0.1 | 2.72 | Destabilizing | 2.29 | Destabilizing | 0.24 | Likely Benign | 0.912 | Likely Pathogenic | -6.19 | Deleterious | 0.993 | Probably Damaging | 0.984 | Probably Damaging | 5.79 | Benign | 0.01 | Affected | 0.3474 | 0.4989 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||
| c.719A>T | D240V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D240V missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta, FATHMM, and premPS, whereas the majority of algorithms—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score—classify the change as pathogenic or likely pathogenic. FoldX and Foldetta return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for D240V. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.343480 | Uncertain | 0.822 | 0.333 | 0.000 | -15.095 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 1.32 | Ambiguous | 0.1 | 0.28 | Likely Benign | 0.80 | Ambiguous | 0.11 | Likely Benign | 0.894 | Likely Pathogenic | -7.94 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 5.82 | Benign | 0.00 | Affected | 0.0610 | 0.4949 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.71T>A | V24E 2D AIThe SynGAP1 missense variant V24E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, the SGM‑Consensus also indicates Likely Benign, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.438970 | Uncertain | 0.382 | 0.890 | 0.500 | -3.397 | Likely Benign | 0.517 | Ambiguous | Likely Benign | 0.171 | Likely Benign | -1.73 | Neutral | 0.198 | Benign | 0.074 | Benign | 3.77 | Benign | 0.00 | Affected | 0.1133 | 0.2415 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||||||||||||
| c.71T>C | V24A 2D AIThe SynGAP1 missense variant V24A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only SIFT predicts pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of tools, including the high‑accuracy methods, points to a benign impact for V24A. This prediction is consistent with the lack of ClinVar evidence and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.438970 | Uncertain | 0.382 | 0.890 | 0.500 | -2.980 | Likely Benign | 0.299 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -1.09 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.83 | Benign | 0.00 | Affected | 0.3117 | 0.3350 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.71T>G | V24G 2D AIThe SynGAP1 missense variant V24G is not reported in ClinVar (ClinVar status: not present) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.541878 | Disordered | 0.438970 | Uncertain | 0.382 | 0.890 | 0.500 | -2.673 | Likely Benign | 0.256 | Likely Benign | Likely Benign | 0.178 | Likely Benign | -1.67 | Neutral | 0.026 | Benign | 0.049 | Benign | 3.77 | Benign | 0.00 | Affected | 0.2081 | 0.3105 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.727A>C | I243L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I243L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from AlphaMissense‑Default and ESM1b, which are treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields benign; and Foldetta also predicts benign. Overall, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.363090 | Structured | 0.344471 | Uncertain | 0.842 | 0.347 | 0.000 | -7.890 | In-Between | 0.524 | Ambiguous | Likely Benign | -0.03 | Likely Benign | 0.2 | -0.35 | Likely Benign | -0.19 | Likely Benign | 0.32 | Likely Benign | 0.380 | Likely Benign | -0.33 | Neutral | 0.048 | Benign | 0.039 | Benign | 5.74 | Benign | 0.19 | Tolerated | 0.0626 | 0.2891 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.727A>G | I243V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I243V is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. FoldX and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.363090 | Structured | 0.344471 | Uncertain | 0.842 | 0.347 | 0.000 | -8.237 | Likely Pathogenic | 0.314 | Likely Benign | Likely Benign | 1.09 | Ambiguous | 0.1 | 0.19 | Likely Benign | 0.64 | Ambiguous | 0.39 | Likely Benign | 0.445 | Likely Benign | -0.39 | Neutral | 0.617 | Possibly Damaging | 0.140 | Benign | 5.71 | Benign | 0.15 | Tolerated | 0.1019 | 0.2591 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||
| c.727A>T | I243F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I243F missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, and FATHMM. Tools that agree on a pathogenic effect include REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give inconclusive results: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie and thus unavailable; Foldetta is uncertain. Overall, the majority of available predictions (7 pathogenic vs. 3 benign) indicate a likely pathogenic impact. This assessment does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.363090 | Structured | 0.344471 | Uncertain | 0.842 | 0.347 | 0.000 | -12.559 | Likely Pathogenic | 0.934 | Likely Pathogenic | Ambiguous | 2.92 | Destabilizing | 2.5 | 0.53 | Ambiguous | 1.73 | Ambiguous | 0.42 | Likely Benign | 0.793 | Likely Pathogenic | -2.21 | Neutral | 0.985 | Probably Damaging | 0.724 | Possibly Damaging | 5.53 | Benign | 0.02 | Affected | 0.0409 | 0.2205 | 1 | 0 | -1.7 | 34.02 | ||||||||||||||||||||||||||||||
| c.734A>G | N245S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N245S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence (8 benign vs. 5 pathogenic) supports a benign classification. This conclusion does not contradict ClinVar, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -6.792 | Likely Benign | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.46 | Likely Benign | 0.1 | -0.16 | Likely Benign | 0.15 | Likely Benign | 0.42 | Likely Benign | 0.524 | Likely Pathogenic | -3.75 | Deleterious | 0.787 | Possibly Damaging | 0.404 | Benign | 5.90 | Benign | 0.07 | Tolerated | 0.3557 | 0.7229 | 1 | 1 | 2.7 | -27.03 | ||||||||||||||||||||||||||||||
| c.734A>T | N245I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N245I is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include Rosetta, Foldetta, and FATHMM, while those that predict a pathogenic effect comprise REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as the variant is currently unreported in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -14.527 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | -0.56 | Ambiguous | 0.1 | 0.04 | Likely Benign | -0.26 | Likely Benign | 0.60 | Ambiguous | 0.831 | Likely Pathogenic | -7.46 | Deleterious | 0.995 | Probably Damaging | 0.832 | Possibly Damaging | 5.88 | Benign | 0.00 | Affected | 0.0672 | 0.6999 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.735T>A | N245K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N245K is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, FoldX, Rosetta, and FATHMM, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM, aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and yields a majority pathogenic verdict. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, classifies the variant as benign. Overall, the majority of individual predictors and the SGM consensus favor a pathogenic effect, while Foldetta and a subset of benign tools suggest stability preservation. Given the predominance of pathogenic predictions and the absence of ClinVar annotation, the variant is most likely pathogenic and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -12.110 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | -0.32 | Likely Benign | 0.1 | 0.01 | Likely Benign | -0.16 | Likely Benign | 0.83 | Ambiguous | 0.474 | Likely Benign | -4.86 | Deleterious | 0.948 | Possibly Damaging | 0.588 | Possibly Damaging | 5.88 | Benign | 0.01 | Affected | 0.2271 | 0.5645 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.735T>G | N245K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N245K is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, FoldX, Rosetta, and FATHMM, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic, the SGM Consensus also indicates Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a Benign effect. Overall, the preponderance of evidence—including the high‑accuracy tools—points to a pathogenic impact for N245K, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -12.110 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | -0.32 | Likely Benign | 0.1 | 0.01 | Likely Benign | -0.16 | Likely Benign | 0.83 | Ambiguous | 0.474 | Likely Benign | -4.86 | Deleterious | 0.948 | Possibly Damaging | 0.588 | Possibly Damaging | 5.88 | Benign | 0.01 | Affected | 0.2271 | 0.5645 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.736C>A | L246M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L246M has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include PROVEAN and FATHMM, while a majority (REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) are uncertain. High‑accuracy methods are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta is uncertain. Thus no high‑accuracy tool provides a definitive verdict. Overall, the balance of evidence favors a pathogenic classification, and this assessment does not contradict any ClinVar status because no ClinVar record exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.472492 | Structured | 0.302312 | Uncertain | 0.859 | 0.364 | 0.000 | -11.386 | Likely Pathogenic | 0.922 | Likely Pathogenic | Ambiguous | 0.65 | Ambiguous | 0.2 | 0.76 | Ambiguous | 0.71 | Ambiguous | 0.87 | Ambiguous | 0.661 | Likely Pathogenic | -1.79 | Neutral | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 4.72 | Benign | 0.01 | Affected | 0.0710 | 0.3511 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.737T>A | L246Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L246Q missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining tools—SGM‑Consensus, REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact; Rosetta is uncertain and is not grouped. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Taken together, the overwhelming majority of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.472492 | Structured | 0.302312 | Uncertain | 0.859 | 0.364 | 0.000 | -15.420 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.82 | Destabilizing | 0.3 | 1.79 | Ambiguous | 2.31 | Destabilizing | 1.69 | Destabilizing | 0.921 | Likely Pathogenic | -5.43 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 4.67 | Benign | 0.00 | Affected | 0.1092 | 0.0758 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.737T>C | L246P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L246P is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: the single benign prediction comes from FATHMM, while all other evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.472492 | Structured | 0.302312 | Uncertain | 0.859 | 0.364 | 0.000 | -16.581 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.42 | Destabilizing | 0.3 | 8.72 | Destabilizing | 7.57 | Destabilizing | 1.79 | Destabilizing | 0.944 | Likely Pathogenic | -6.30 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 4.67 | Benign | 0.00 | Affected | 0.3731 | 0.1582 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.737T>G | L246R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L246R missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.472492 | Structured | 0.302312 | Uncertain | 0.859 | 0.364 | 0.000 | -13.849 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.94 | Destabilizing | 0.8 | 2.77 | Destabilizing | 3.36 | Destabilizing | 1.72 | Destabilizing | 0.925 | Likely Pathogenic | -5.43 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 4.67 | Benign | 0.00 | Affected | 0.1266 | 0.0600 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.739C>A | Q247K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q247K (PH domain) is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, premPS, PROVEAN, SIFT, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that predict a pathogenic effect are REVEL, polyPhen2_HumDiv, and ESM1b. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a benign outcome (2 benign vs. 1 pathogenic). High‑accuracy methods all support benignity: AlphaMissense‑Optimized is benign, the SGM Consensus is benign, and Foldetta (combining FoldX‑MD and Rosetta) is benign. Uncertain results from AlphaMissense‑Default and Rosetta are treated as unavailable. Overall, the collective evidence points to a benign impact for Q247K, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.490133 | Structured | 0.283012 | Uncertain | 0.822 | 0.339 | 0.250 | -10.377 | Likely Pathogenic | 0.502 | Ambiguous | Likely Benign | -0.28 | Likely Benign | 0.1 | 0.74 | Ambiguous | 0.23 | Likely Benign | -0.13 | Likely Benign | 0.529 | Likely Pathogenic | -0.44 | Neutral | 0.787 | Possibly Damaging | 0.351 | Benign | 5.89 | Benign | 0.13 | Tolerated | 0.1366 | 0.2758 | 1 | 1 | -0.4 | 0.04 | ||||||||||||||||||||||||||||||
| c.739C>G | Q247E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q247E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and FATHMM. Tools that predict a pathogenic effect are premPS, SIFT, and ESM1b. AlphaMissense‑Default is uncertain, while AlphaMissense‑Optimized predicts benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a benign consensus (2 benign vs. 1 pathogenic, with the uncertain result treated as unavailable). High‑accuracy assessments confirm benignity: AlphaMissense‑Optimized is benign, Foldetta (combining FoldX‑MD and Rosetta outputs) is benign, and the SGM Consensus is benign. Overall, the collective evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.490133 | Structured | 0.283012 | Uncertain | 0.822 | 0.339 | 0.250 | -13.564 | Likely Pathogenic | 0.373 | Ambiguous | Likely Benign | 0.42 | Likely Benign | 0.1 | -0.26 | Likely Benign | 0.08 | Likely Benign | 1.04 | Destabilizing | 0.474 | Likely Benign | -1.46 | Neutral | 0.128 | Benign | 0.039 | Benign | 5.80 | Benign | 0.03 | Affected | 0.1055 | 0.1476 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.73C>G | R25G 2D AIThe SynGAP1 missense variant R25G is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors shows a split: benign calls come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls arise from polyPhen‑2 (HumDiv and HumVar) and SIFT. AlphaMissense‑Default remains uncertain. When high‑accuracy tools are considered separately, AlphaMissense‑Optimized predicts a benign effect, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome, and Foldetta data are unavailable. Overall, the majority of robust predictors lean toward a benign interpretation. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.461924 | Structured | 0.438941 | Uncertain | 0.373 | 0.890 | 0.375 | -3.533 | Likely Benign | 0.373 | Ambiguous | Likely Benign | 0.122 | Likely Benign | -1.69 | Neutral | 0.686 | Possibly Damaging | 0.630 | Possibly Damaging | 3.95 | Benign | 0.00 | Affected | 0.3621 | 0.3572 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.740A>C | Q247P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q247P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic or likely pathogenic impact; premPS is uncertain and therefore treated as unavailable. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.490133 | Structured | 0.283012 | Uncertain | 0.822 | 0.339 | 0.250 | -14.361 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.43 | Destabilizing | 0.2 | 7.83 | Destabilizing | 5.63 | Destabilizing | 0.81 | Ambiguous | 0.770 | Likely Pathogenic | -3.56 | Deleterious | 0.995 | Probably Damaging | 0.795 | Possibly Damaging | 5.69 | Benign | 0.02 | Affected | 0.1768 | 0.3631 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||
| c.740A>G | Q247R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q247R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Uncertain predictions come from premPS and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or folding stability result is missing or inconclusive. Based on the overall consensus of the majority of tools and the high‑accuracy methods, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.490133 | Structured | 0.283012 | Uncertain | 0.822 | 0.339 | 0.250 | -4.022 | Likely Benign | 0.442 | Ambiguous | Likely Benign | -0.38 | Likely Benign | 0.0 | 0.21 | Likely Benign | -0.09 | Likely Benign | -0.91 | Ambiguous | 0.471 | Likely Benign | 1.22 | Neutral | 0.948 | Possibly Damaging | 0.533 | Possibly Damaging | 5.91 | Benign | 1.00 | Tolerated | 0.1196 | 0.1324 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||
| c.734A>C | N245T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N245T is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include FATHMM, Rosetta, and Foldetta, whereas the majority of tools predict a pathogenic impact: REVEL, SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support this dichotomy: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic, while Foldetta predicts a benign effect. No predictions are missing or inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for the variant, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -11.955 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.52 | Ambiguous | 0.1 | 0.04 | Likely Benign | 0.28 | Likely Benign | 0.76 | Ambiguous | 0.794 | Likely Pathogenic | -4.79 | Deleterious | 0.948 | Possibly Damaging | 0.588 | Possibly Damaging | 5.87 | Benign | 0.01 | Affected | 0.1397 | 0.7734 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.733A>T | N245Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N245Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from FoldX, Rosetta, Foldetta, and FATHMM, whereas pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a pathogenic majority (3/4). AlphaMissense‑Optimized independently predicts pathogenicity, while Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts benign. premPS remains uncertain. Overall, the majority of evidence points toward a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Thus, the variant is most likely pathogenic, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -12.476 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | -0.36 | Likely Benign | 0.1 | 0.11 | Likely Benign | -0.13 | Likely Benign | 0.59 | Ambiguous | 0.855 | Likely Pathogenic | -6.68 | Deleterious | 0.995 | Probably Damaging | 0.880 | Possibly Damaging | 5.86 | Benign | 0.00 | Affected | 0.0706 | 0.6771 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.728T>A | I243N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant I243N is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into three groups: benign (FATHMM), pathogenic (SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized), and uncertain (FoldX, Rosetta, Foldetta). High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.363090 | Structured | 0.344471 | Uncertain | 0.842 | 0.347 | 0.000 | -14.784 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 1.90 | Ambiguous | 0.2 | 1.43 | Ambiguous | 1.67 | Ambiguous | 1.76 | Destabilizing | 0.811 | Likely Pathogenic | -4.46 | Deleterious | 0.995 | Probably Damaging | 0.854 | Possibly Damaging | 5.49 | Benign | 0.00 | Affected | 0.0899 | 0.0212 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.728T>C | I243T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I243T is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all classify the variant as pathogenic. Only FATHMM predicts a benign outcome, while Foldetta, AlphaMissense‑Optimized, and Rosetta return uncertain results, which are treated as unavailable evidence. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence indicates that I243T is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.363090 | Structured | 0.344471 | Uncertain | 0.842 | 0.347 | 0.000 | -9.102 | Likely Pathogenic | 0.922 | Likely Pathogenic | Ambiguous | 2.15 | Destabilizing | 0.2 | 1.52 | Ambiguous | 1.84 | Ambiguous | 1.72 | Destabilizing | 0.816 | Likely Pathogenic | -3.06 | Deleterious | 0.982 | Probably Damaging | 0.702 | Possibly Damaging | 5.55 | Benign | 0.01 | Affected | 0.1027 | 0.0541 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.728T>G | I243S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I243S is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity largely agree: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. Only FATHMM predicts a benign outcome. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No predictions are missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.363090 | Structured | 0.344471 | Uncertain | 0.842 | 0.347 | 0.000 | -14.097 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 2.52 | Destabilizing | 0.2 | 2.22 | Destabilizing | 2.37 | Destabilizing | 1.71 | Destabilizing | 0.802 | Likely Pathogenic | -3.55 | Deleterious | 0.995 | Probably Damaging | 0.795 | Possibly Damaging | 5.52 | Benign | 0.00 | Affected | 0.2658 | 0.0600 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.729T>G | I243M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I243M has no ClinVar entry (ClinVar status: not reported) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign”; and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, also predicts a benign effect. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates that the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.363090 | Structured | 0.344471 | Uncertain | 0.842 | 0.347 | 0.000 | -4.305 | Likely Benign | 0.175 | Likely Benign | Likely Benign | 0.09 | Likely Benign | 0.1 | 0.16 | Likely Benign | 0.13 | Likely Benign | -0.01 | Likely Benign | 0.474 | Likely Benign | 0.58 | Neutral | 0.985 | Probably Damaging | 0.798 | Possibly Damaging | 5.61 | Benign | 0.94 | Tolerated | 0.0553 | 0.2200 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||
| c.730G>A | E244K 2D AISynGAP1 missense variant E244K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from FoldX, Rosetta, Foldetta, SIFT, and FATHMM, whereas pathogenic predictions are reported by REVEL, PROVEAN, both polyPhen‑2 HumDiv and HumVar, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (Likely Pathogenic). The high‑accuracy methods give a clearer picture: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus also indicates likely pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a benign effect. With the majority of consensus tools leaning toward pathogenic and the two high‑accuracy pathogenic predictions outweighing the benign Foldetta result, the variant is most likely pathogenic. This assessment does not contradict any ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.450668 | Structured | 0.329406 | Uncertain | 0.778 | 0.360 | 0.000 | -13.975 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.40 | Likely Benign | 1.0 | 0.45 | Likely Benign | 0.43 | Likely Benign | 0.80 | Ambiguous | 0.841 | Likely Pathogenic | -3.37 | Deleterious | 0.990 | Probably Damaging | 0.760 | Possibly Damaging | 5.82 | Benign | 0.07 | Tolerated | 0.1887 | 0.6177 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.730G>C | E244Q 2D AIThe SynGAP1 E244Q missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Foldetta, premPS, PROVEAN, and FATHMM, while those that agree on a pathogenic effect are REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Two tools give uncertain results: Rosetta and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, six tools predict pathogenicity versus five predicting benignity, with no ClinVar evidence to contradict these findings. Thus, the variant is most likely pathogenic based on the current predictive landscape. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.450668 | Structured | 0.329406 | Uncertain | 0.778 | 0.360 | 0.000 | -10.245 | Likely Pathogenic | 0.928 | Likely Pathogenic | Ambiguous | 0.23 | Likely Benign | 0.9 | -0.77 | Ambiguous | -0.27 | Likely Benign | 0.40 | Likely Benign | 0.695 | Likely Pathogenic | -2.49 | Neutral | 0.990 | Probably Damaging | 0.815 | Possibly Damaging | 5.78 | Benign | 0.05 | Affected | 0.1016 | 0.5610 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.731A>C | E244A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E244A missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from FoldX, Rosetta, and FATHMM, while pathogenic predictions are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign stability. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.450668 | Structured | 0.329406 | Uncertain | 0.778 | 0.360 | 0.000 | -11.227 | Likely Pathogenic | 0.969 | Likely Pathogenic | Likely Pathogenic | 0.39 | Likely Benign | 0.1 | -0.45 | Likely Benign | -0.03 | Likely Benign | 0.82 | Ambiguous | 0.865 | Likely Pathogenic | -4.99 | Deleterious | 0.970 | Probably Damaging | 0.584 | Possibly Damaging | 5.74 | Benign | 0.01 | Affected | 0.2672 | 0.4926 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.731A>G | E244G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E244G missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this view: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome. Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, yields an uncertain result and is treated as unavailable evidence. Overall, the preponderance of computational evidence points to the variant being most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.450668 | Structured | 0.329406 | Uncertain | 0.778 | 0.360 | 0.000 | -10.452 | Likely Pathogenic | 0.958 | Likely Pathogenic | Likely Pathogenic | 1.28 | Ambiguous | 0.0 | 0.74 | Ambiguous | 1.01 | Ambiguous | 1.01 | Destabilizing | 0.900 | Likely Pathogenic | -5.67 | Deleterious | 0.990 | Probably Damaging | 0.815 | Possibly Damaging | 5.73 | Benign | 0.01 | Affected | 0.2266 | 0.5252 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.731A>T | E244V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E244V missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. Four tools (FoldX, Rosetta, Foldetta, premPS) returned uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also predicts pathogenicity; Foldetta remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.450668 | Structured | 0.329406 | Uncertain | 0.778 | 0.360 | 0.000 | -12.118 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.04 | Ambiguous | 0.1 | 1.02 | Ambiguous | 1.03 | Ambiguous | 0.56 | Ambiguous | 0.925 | Likely Pathogenic | -5.90 | Deleterious | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 5.68 | Benign | 0.00 | Affected | 0.0636 | 0.6073 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.732G>C | E244D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E244D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX and FATHMM, whereas a majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are reported by Rosetta, Foldetta, premPS, and ESM1b. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta remains uncertain. Overall, the balance of evidence favors a pathogenic classification for E244D, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.450668 | Structured | 0.329406 | Uncertain | 0.778 | 0.360 | 0.000 | -7.839 | In-Between | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.46 | Likely Benign | 0.1 | 1.61 | Ambiguous | 1.04 | Ambiguous | 0.93 | Ambiguous | 0.730 | Likely Pathogenic | -2.53 | Deleterious | 0.976 | Probably Damaging | 0.675 | Possibly Damaging | 5.78 | Benign | 0.03 | Affected | 0.1740 | 0.3783 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.732G>T | E244D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E244D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX and FATHMM, whereas a majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results come from Rosetta, Foldetta, premPS, and ESM1b. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta remains uncertain. Overall, the balance of evidence points to a likely pathogenic effect for E244D, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.450668 | Structured | 0.329406 | Uncertain | 0.778 | 0.360 | 0.000 | -7.839 | In-Between | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.46 | Likely Benign | 0.1 | 1.61 | Ambiguous | 1.04 | Ambiguous | 0.93 | Ambiguous | 0.730 | Likely Pathogenic | -2.53 | Deleterious | 0.976 | Probably Damaging | 0.675 | Possibly Damaging | 5.78 | Benign | 0.03 | Affected | 0.1740 | 0.3783 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.733A>C | N245H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N245H is not reported in ClinVar and is absent from gnomAD. Among in‑silico predictors, benign calls come from FoldX, Rosetta, and FATHMM, whereas pathogenic calls are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. The overall pattern of predictions leans toward pathogenicity, with a majority of tools indicating a deleterious effect. Thus, the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -11.536 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.23 | Likely Benign | 0.1 | -0.20 | Likely Benign | 0.02 | Likely Benign | 0.55 | Ambiguous | 0.825 | Likely Pathogenic | -4.15 | Deleterious | 0.995 | Probably Damaging | 0.880 | Possibly Damaging | 5.84 | Benign | 0.01 | Affected | 0.1437 | 0.7271 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.733A>G | N245D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N245D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, SIFT, and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools, premPS and ESM1b, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions (six pathogenic vs five benign) and the consensus of high‑accuracy methods lean toward a pathogenic interpretation. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -7.847 | In-Between | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.27 | Likely Benign | 0.0 | 0.09 | Likely Benign | 0.18 | Likely Benign | 0.79 | Ambiguous | 0.712 | Likely Pathogenic | -3.62 | Deleterious | 0.982 | Probably Damaging | 0.679 | Possibly Damaging | 5.84 | Benign | 0.13 | Tolerated | 0.1953 | 0.4599 | 2 | 1 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.740A>T | Q247L 2D AIThe SynGAP1 missense variant Q247L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The remaining tools—FoldX, Rosetta, Foldetta, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence (seven pathogenic versus three benign predictions) points to a pathogenic impact, and this conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.490133 | Structured | 0.283012 | Uncertain | 0.822 | 0.339 | 0.250 | -10.554 | Likely Pathogenic | 0.352 | Ambiguous | Likely Benign | -0.86 | Ambiguous | 0.5 | -1.14 | Ambiguous | -1.00 | Ambiguous | 0.38 | Likely Benign | 0.687 | Likely Pathogenic | -3.89 | Deleterious | 0.982 | Probably Damaging | 0.628 | Possibly Damaging | 5.70 | Benign | 0.02 | Affected | 0.0573 | 0.4129 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||
| c.570C>G | S190R 2D AIThe SynGAP1 missense variant S190R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts Pathogenic, and the SGM‑Consensus (majority vote) also indicates Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from both general and high‑accuracy prediction tools indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar annotation exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.422041 | Structured | 0.428613 | Uncertain | 0.338 | 0.615 | 0.250 | -10.137 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.146 | Likely Benign | -2.92 | Deleterious | 0.917 | Possibly Damaging | 0.446 | Benign | 4.07 | Benign | 0.04 | Affected | 0.0809 | 0.4059 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.577G>C | A193P 2D AIThe SynGAP1 missense variant A193P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are REVEL and FATHMM, while the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized model classifies the variant as pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic), also yields a pathogenic consensus. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple independent predictors indicates that A193P is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.429200 | Structured | 0.428386 | Uncertain | 0.310 | 0.577 | 0.125 | -7.293 | In-Between | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.267 | Likely Benign | -2.70 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 3.99 | Benign | 0.05 | Affected | 0.1855 | 0.5601 | 1 | -1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||||||||||||
| c.577G>T | A193S 2D AIThe SynGAP1 missense variant A193S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.428386 | Uncertain | 0.310 | 0.577 | 0.125 | -2.408 | Likely Benign | 0.533 | Ambiguous | Likely Benign | 0.180 | Likely Benign | -1.62 | Neutral | 0.990 | Probably Damaging | 0.760 | Possibly Damaging | 4.04 | Benign | 0.04 | Affected | 0.2595 | 0.5852 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.578C>A | A193D 2D AIThe SynGAP1 missense variant A193D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are REVEL and FATHMM, while the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized model classifies the variant as pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic), also yields a pathogenic consensus. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that A193D is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.429200 | Structured | 0.428386 | Uncertain | 0.310 | 0.577 | 0.125 | -7.488 | In-Between | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.217 | Likely Benign | -3.46 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 3.99 | Benign | 0.01 | Affected | 0.1688 | 0.1835 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||||||||
| c.578C>G | A193G 2D AIThe SynGAP1 missense variant A193G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, more tools predict pathogenicity (5) than benignity (3), and no ClinVar evidence contradicts this assessment. Therefore, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.429200 | Structured | 0.428386 | Uncertain | 0.310 | 0.577 | 0.125 | -4.822 | Likely Benign | 0.835 | Likely Pathogenic | Ambiguous | 0.136 | Likely Benign | -2.61 | Deleterious | 0.990 | Probably Damaging | 0.760 | Possibly Damaging | 4.00 | Benign | 0.01 | Affected | 0.2283 | 0.4999 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.578C>T | A193V 2D AIThe SynGAP1 A193V missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.428386 | Uncertain | 0.310 | 0.577 | 0.125 | -3.548 | Likely Benign | 0.744 | Likely Pathogenic | Likely Benign | 0.177 | Likely Benign | 0.52 | Neutral | 0.767 | Possibly Damaging | 0.344 | Benign | 4.30 | Benign | 1.00 | Tolerated | 0.1169 | 0.6775 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||||||||||||
| c.580G>A | E194K 2D AIThe SynGAP1 missense variant E194K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.418646 | Structured | 0.430723 | Uncertain | 0.346 | 0.551 | 0.125 | -13.294 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.259 | Likely Benign | -2.53 | Deleterious | 0.734 | Possibly Damaging | 0.321 | Benign | 4.04 | Benign | 0.01 | Affected | 0.2231 | 0.5152 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.580G>C | E194Q 2D AIThe SynGAP1 missense variant E194Q is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM. Tools that agree on a pathogenic effect include polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool rates the variant as uncertain, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, more evidence supports a pathogenic classification (5 pathogenic vs. 3 benign predictions). Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.418646 | Structured | 0.430723 | Uncertain | 0.346 | 0.551 | 0.125 | -8.902 | Likely Pathogenic | 0.955 | Likely Pathogenic | Ambiguous | 0.140 | Likely Benign | -1.97 | Neutral | 0.849 | Possibly Damaging | 0.478 | Possibly Damaging | 4.00 | Benign | 0.02 | Affected | 0.1043 | 0.4954 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||||||||
| c.581A>C | E194A 2D AIThe SynGAP1 missense variant E194A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that agree on a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. The high‑accuracy assessment shows AlphaMissense‑Optimized predicts pathogenicity. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic), also indicates pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions—including the high‑accuracy tools—suggest that E194A is likely pathogenic, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.418646 | Structured | 0.430723 | Uncertain | 0.346 | 0.551 | 0.125 | -7.101 | In-Between | 0.958 | Likely Pathogenic | Likely Pathogenic | 0.235 | Likely Benign | -3.75 | Deleterious | 0.009 | Benign | 0.012 | Benign | 3.99 | Benign | 0.01 | Affected | 0.4331 | 0.5079 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.581A>G | E194G 2D AIThe SynGAP1 missense variant E194G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts Pathogenic, and the SGM‑Consensus also indicates Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.418646 | Structured | 0.430723 | Uncertain | 0.346 | 0.551 | 0.125 | -9.136 | Likely Pathogenic | 0.961 | Likely Pathogenic | Likely Pathogenic | 0.316 | Likely Benign | -4.47 | Deleterious | 0.580 | Possibly Damaging | 0.196 | Benign | 3.96 | Benign | 0.01 | Affected | 0.3633 | 0.4616 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.581A>T | E194V 2D AIThe SynGAP1 missense variant E194V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. No Foldetta stability analysis is available for this variant. Overall, the preponderance of computational evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.418646 | Structured | 0.430723 | Uncertain | 0.346 | 0.551 | 0.125 | -10.261 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.284 | Likely Benign | -4.67 | Deleterious | 0.580 | Possibly Damaging | 0.254 | Benign | 3.94 | Benign | 0.00 | Affected | 0.0623 | 0.5469 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.582G>C | E194D 2D AIThe SynGAP1 missense variant E194D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” AlphaMissense‑Optimized is uncertain, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.418646 | Structured | 0.430723 | Uncertain | 0.346 | 0.551 | 0.125 | -6.310 | Likely Benign | 0.883 | Likely Pathogenic | Ambiguous | 0.079 | Likely Benign | -2.07 | Neutral | 0.849 | Possibly Damaging | 0.301 | Benign | 4.01 | Benign | 0.04 | Affected | 0.1969 | 0.2779 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.582G>T | E194D 2D AIThe SynGAP1 missense variant E194D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a “Likely Benign” classification, while AlphaMissense‑Optimized is uncertain. Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.418646 | Structured | 0.430723 | Uncertain | 0.346 | 0.551 | 0.125 | -6.310 | Likely Benign | 0.883 | Likely Pathogenic | Ambiguous | 0.079 | Likely Benign | -2.07 | Neutral | 0.849 | Possibly Damaging | 0.301 | Benign | 4.01 | Benign | 0.04 | Affected | 0.1969 | 0.2779 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.583G>A | A195T 2D AIThe SynGAP1 missense variant A195T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The remaining tools, ESM1b and AlphaMissense‑Optimized, return uncertain results. High‑accuracy assessment shows that the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans benign (2 benign vs. 1 pathogenic, 1 uncertain). Foldetta, which would provide a protein‑folding stability estimate, has no available output for this variant. Overall, the preponderance of evidence indicates that A195T is most likely benign, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.346032 | Structured | 0.430388 | Uncertain | 0.363 | 0.533 | 0.125 | -7.060 | In-Between | 0.883 | Likely Pathogenic | Ambiguous | 0.085 | Likely Benign | -2.09 | Neutral | 0.970 | Probably Damaging | 0.681 | Possibly Damaging | 4.08 | Benign | 0.14 | Tolerated | 0.0937 | 0.6006 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.577G>A | A193T 2D AIThe SynGAP1 missense variant A193T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.428386 | Uncertain | 0.310 | 0.577 | 0.125 | -4.973 | Likely Benign | 0.789 | Likely Pathogenic | Ambiguous | 0.208 | Likely Benign | -1.36 | Neutral | 0.990 | Probably Damaging | 0.815 | Possibly Damaging | 4.07 | Benign | 0.21 | Tolerated | 0.1452 | 0.7231 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.575C>T | A192V 2D AIThe SynGAP1 missense variant A192V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b remains uncertain. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic majority (2 pathogenic vs. 1 benign, 1 uncertain). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that A192V is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.422041 | Structured | 0.428195 | Uncertain | 0.321 | 0.589 | 0.125 | -7.464 | In-Between | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.172 | Likely Benign | -2.66 | Deleterious | 0.996 | Probably Damaging | 0.877 | Possibly Damaging | 4.01 | Benign | 0.11 | Tolerated | 0.0904 | 0.5066 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||||||||
| c.571A>C | S191R 2D AIThe SynGAP1 missense variant S191R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; the Foldetta protein‑folding stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.429200 | Structured | 0.428475 | Uncertain | 0.322 | 0.615 | 0.125 | -10.046 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.324 | Likely Benign | -3.82 | Deleterious | 0.838 | Possibly Damaging | 0.367 | Benign | 3.78 | Benign | 0.01 | Affected | 0.0973 | 0.4344 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.571A>G | S191G 2D AIThe SynGAP1 missense variant S191G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also leans toward benign (two benign versus two uncertain). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S191G is most likely benign, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.429200 | Structured | 0.428475 | Uncertain | 0.322 | 0.615 | 0.125 | -7.161 | In-Between | 0.506 | Ambiguous | Likely Benign | 0.173 | Likely Benign | -2.43 | Neutral | 0.131 | Benign | 0.058 | Benign | 3.77 | Benign | 0.03 | Affected | 0.2893 | 0.5335 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.571A>T | S191C 2D AIThe SynGAP1 missense variant S191C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic)—leans pathogenic. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of tools and the SGM Consensus predict a pathogenic impact, so the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.429200 | Structured | 0.428475 | Uncertain | 0.322 | 0.615 | 0.125 | -7.009 | In-Between | 0.709 | Likely Pathogenic | Likely Benign | 0.288 | Likely Benign | -3.39 | Deleterious | 0.983 | Probably Damaging | 0.635 | Possibly Damaging | 3.73 | Benign | 0.00 | Affected | 0.1064 | 0.7134 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||||||||
| c.572G>A | S191N 2D AIThe SynGAP1 missense variant S191N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. Two tools report an uncertain outcome: ESM1b and AlphaMissense‑Optimized. High‑accuracy assessment shows that the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans toward benign (2 benign vs. 1 pathogenic). AlphaMissense‑Optimized remains uncertain, and Foldetta folding‑stability analysis is unavailable. Overall, the preponderance of evidence points to a benign impact for S191N, and this conclusion does not contradict any ClinVar annotation, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.429200 | Structured | 0.428475 | Uncertain | 0.322 | 0.615 | 0.125 | -7.887 | In-Between | 0.830 | Likely Pathogenic | Ambiguous | 0.148 | Likely Benign | -2.21 | Neutral | 0.596 | Possibly Damaging | 0.260 | Benign | 3.78 | Benign | 0.03 | Affected | 0.1366 | 0.5898 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||||||||||
| c.572G>C | S191T 2D AIThe SynGAP1 missense variant S191T is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools overwhelmingly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score benign, while the majority‑vote SGM‑Consensus also classifies it as likely benign. Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy assessments corroborate the benign consensus: AlphaMissense‑Optimized returns a benign prediction and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports likely benign. Foldetta stability analysis is not available for this variant. Overall, the preponderance of evidence supports a benign classification, and this assessment does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.428475 | Uncertain | 0.322 | 0.615 | 0.125 | -7.315 | In-Between | 0.319 | Likely Benign | Likely Benign | 0.150 | Likely Benign | -2.34 | Neutral | 0.231 | Benign | 0.081 | Benign | 3.81 | Benign | 0.03 | Affected | 0.1466 | 0.7405 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.572G>T | S191I 2D AIThe SynGAP1 missense variant S191I is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split opinion: benign predictions come from REVEL, polyPhen‑2 (HumDiv and HumVar), and FATHMM, while pathogenic predictions arise from PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. When the predictions are grouped by consensus, four tools favor benign and four favor pathogenic. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized returns an uncertain result, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Taken together, the majority of evidence, including the SGM Consensus, points to a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.429200 | Structured | 0.428475 | Uncertain | 0.322 | 0.615 | 0.125 | -11.271 | Likely Pathogenic | 0.927 | Likely Pathogenic | Ambiguous | 0.283 | Likely Benign | -4.51 | Deleterious | 0.421 | Benign | 0.086 | Benign | 3.76 | Benign | 0.00 | Affected | 0.0954 | 0.6842 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.573T>A | S191R 2D AIThe SynGAP1 missense variant S191R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; the Foldetta protein‑folding stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.429200 | Structured | 0.428475 | Uncertain | 0.322 | 0.615 | 0.125 | -10.046 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.191 | Likely Benign | -3.82 | Deleterious | 0.838 | Possibly Damaging | 0.367 | Benign | 3.78 | Benign | 0.01 | Affected | 0.0973 | 0.4344 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.573T>G | S191R 2D AIThe SynGAP1 missense variant S191R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of computational evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.429200 | Structured | 0.428475 | Uncertain | 0.322 | 0.615 | 0.125 | -10.046 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.191 | Likely Benign | -3.82 | Deleterious | 0.838 | Possibly Damaging | 0.367 | Benign | 3.78 | Benign | 0.01 | Affected | 0.0973 | 0.4344 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.574G>A | A192T 2D AIThe SynGAP1 missense variant A192T has no ClinVar record (ClinVar status: not reported) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a benign outcome (2 benign vs. 1 pathogenic, 1 uncertain). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM Consensus remains benign; Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.422041 | Structured | 0.428195 | Uncertain | 0.321 | 0.589 | 0.125 | -7.562 | In-Between | 0.958 | Likely Pathogenic | Likely Pathogenic | 0.089 | Likely Benign | -2.46 | Neutral | 0.978 | Probably Damaging | 0.714 | Possibly Damaging | 3.96 | Benign | 0.34 | Tolerated | 0.1148 | 0.5343 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.574G>C | A192P 2D AIThe SynGAP1 missense variant A192P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from both general and high‑accuracy prediction tools points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.422041 | Structured | 0.428195 | Uncertain | 0.321 | 0.589 | 0.125 | -9.795 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.245 | Likely Benign | -3.35 | Deleterious | 0.999 | Probably Damaging | 0.946 | Probably Damaging | 3.90 | Benign | 0.02 | Affected | 0.1658 | 0.3719 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||||||
| c.574G>T | A192S 2D AIThe SynGAP1 missense variant A192S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default; ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.422041 | Structured | 0.428195 | Uncertain | 0.321 | 0.589 | 0.125 | -7.969 | In-Between | 0.757 | Likely Pathogenic | Likely Benign | 0.118 | Likely Benign | -2.01 | Neutral | 0.633 | Possibly Damaging | 0.171 | Benign | 3.98 | Benign | 0.12 | Tolerated | 0.2365 | 0.3764 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||||||||||||
| c.575C>A | A192E 2D AIThe SynGAP1 missense variant A192E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. The protein‑folding stability method Foldetta has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.422041 | Structured | 0.428195 | Uncertain | 0.321 | 0.589 | 0.125 | -12.188 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.268 | Likely Benign | -3.52 | Deleterious | 0.997 | Probably Damaging | 0.888 | Possibly Damaging | 3.93 | Benign | 0.01 | Affected | 0.0939 | 0.1628 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.575C>G | A192G 2D AIThe SynGAP1 missense variant A192G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all indicate pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. AlphaMissense‑Optimized returns an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. Overall, the preponderance of evidence from multiple in silico tools points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.422041 | Structured | 0.428195 | Uncertain | 0.321 | 0.589 | 0.125 | -9.003 | Likely Pathogenic | 0.884 | Likely Pathogenic | Ambiguous | 0.133 | Likely Benign | -3.00 | Deleterious | 0.989 | Probably Damaging | 0.621 | Possibly Damaging | 3.91 | Benign | 0.02 | Affected | 0.2081 | 0.2857 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.583G>C | A195P 2D AIThe SynGAP1 missense variant A195P is listed in ClinVar as Pathogenic (ClinVar ID 375527.0) and is not reported in gnomAD. Prediction tools that indicate a benign effect are REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as likely pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence supports a pathogenic classification, which aligns with the ClinVar status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.346032 | Structured | 0.430388 | Uncertain | 0.363 | 0.533 | 0.125 | Likely Pathogenic | 1 | -9.715 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.152 | Likely Benign | -3.03 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 4.00 | Benign | 0.04 | Affected | 3.54 | 6 | 0.1403 | 0.4402 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||||||||
| c.584C>A | A195E 2D AIThe SynGAP1 missense variant A195E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.346032 | Structured | 0.430388 | Uncertain | 0.363 | 0.533 | 0.125 | -10.909 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.237 | Likely Benign | -3.20 | Deleterious | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 4.12 | Benign | 0.02 | Affected | 0.0855 | 0.1877 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.584C>G | A195G 2D AIThe SynGAP1 missense variant A195G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and FATHMM, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. Two tools give uncertain results: ESM1b and AlphaMissense‑Optimized. High‑accuracy assessment shows that AlphaMissense‑Optimized remains uncertain, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic)—favors a pathogenic outcome. Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic impact for A195G, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.346032 | Structured | 0.430388 | Uncertain | 0.363 | 0.533 | 0.125 | -7.186 | In-Between | 0.907 | Likely Pathogenic | Ambiguous | 0.146 | Likely Benign | -2.64 | Deleterious | 0.990 | Probably Damaging | 0.760 | Possibly Damaging | 4.01 | Benign | 0.05 | Affected | 0.1694 | 0.3388 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.592C>A | L198I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L198I is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Uncertain or inconclusive results come from AlphaMissense‑Default, FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, while Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.444081 | Structured | 0.431715 | Uncertain | 0.572 | 0.485 | 0.125 | -10.411 | Likely Pathogenic | 0.563 | Ambiguous | Likely Benign | 0.59 | Ambiguous | 0.1 | 0.70 | Ambiguous | 0.65 | Ambiguous | 0.10 | Likely Benign | 0.234 | Likely Benign | -1.72 | Neutral | 0.990 | Probably Damaging | 0.760 | Possibly Damaging | 3.43 | Benign | 0.00 | Affected | 0.0921 | 0.3307 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||
| c.592C>G | L198V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L198V variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled Likely Pathogenic. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (3 pathogenic vs. 1 benign) indicates pathogenicity; Foldetta remains uncertain. Overall, the majority of evaluated tools predict a pathogenic impact, and this is not contradicted by any ClinVar annotation (none exists). Thus, the variant is most likely pathogenic based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.444081 | Structured | 0.431715 | Uncertain | 0.572 | 0.485 | 0.125 | -9.343 | Likely Pathogenic | 0.747 | Likely Pathogenic | Likely Benign | 1.10 | Ambiguous | 0.2 | 1.24 | Ambiguous | 1.17 | Ambiguous | 0.34 | Likely Benign | 0.265 | Likely Benign | -2.54 | Deleterious | 0.990 | Probably Damaging | 0.675 | Possibly Damaging | 3.44 | Benign | 0.00 | Affected | 0.1520 | 0.3017 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||
| c.592C>T | L198F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L198F has no ClinVar entry and is absent from gnomAD. Functional prediction tools split into two consensus groups: benign predictions come from REVEL, Rosetta, premPS, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments are mixed: AlphaMissense‑Optimized and Foldetta report uncertain results, whereas the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect. No evidence from ClinVar contradicts these findings. Overall, the preponderance of pathogenic predictions and the SGM‑Consensus designation suggest that the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.444081 | Structured | 0.431715 | Uncertain | 0.572 | 0.485 | 0.125 | -10.602 | Likely Pathogenic | 0.930 | Likely Pathogenic | Ambiguous | 1.03 | Ambiguous | 0.2 | 0.27 | Likely Benign | 0.65 | Ambiguous | 0.00 | Likely Benign | 0.282 | Likely Benign | -3.33 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 3.39 | Benign | 0.00 | Affected | 0.0586 | 0.3112 | 2 | 0 | -1.0 | 34.02 | ||||||||||||||||||||||||||||||
| c.593T>A | L198H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L198H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict pathogenicity: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for L198H. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.444081 | Structured | 0.431715 | Uncertain | 0.572 | 0.485 | 0.125 | -15.650 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.09 | Ambiguous | 0.2 | 1.04 | Ambiguous | 1.07 | Ambiguous | 0.74 | Ambiguous | 0.419 | Likely Benign | -5.91 | Deleterious | 0.999 | Probably Damaging | 0.936 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.1057 | 0.0519 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||
| c.593T>C | L198P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L198P has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and FATHMM, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX, Foldetta, and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points toward a deleterious effect. Thus, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.444081 | Structured | 0.431715 | Uncertain | 0.572 | 0.485 | 0.125 | -12.250 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.55 | Ambiguous | 0.3 | 0.40 | Likely Benign | 0.98 | Ambiguous | 0.65 | Ambiguous | 0.491 | Likely Benign | -5.87 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.3882 | 0.1582 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||
| c.593T>G | L198R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L198R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence from multiple in‑silico predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.444081 | Structured | 0.431715 | Uncertain | 0.572 | 0.485 | 0.125 | -15.992 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.74 | Ambiguous | 0.2 | 1.56 | Ambiguous | 1.15 | Ambiguous | 0.69 | Ambiguous | 0.410 | Likely Benign | -4.98 | Deleterious | 0.982 | Probably Damaging | 0.648 | Possibly Damaging | 3.34 | Benign | 0.00 | Affected | 0.1180 | 0.0719 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||
| c.595A>C | N199H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N199H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No predictions are missing or inconclusive. Overall, the majority of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which contains no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -6.387 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.17 | Likely Benign | 0.1 | 0.15 | Likely Benign | 0.16 | Likely Benign | 0.20 | Likely Benign | 0.064 | Likely Benign | -1.99 | Neutral | 0.952 | Possibly Damaging | 0.496 | Possibly Damaging | 4.17 | Benign | 0.08 | Tolerated | 0.0879 | 0.5635 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.595A>G | N199D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N199D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach a consensus classify the variant as benign: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. The only inconclusive results come from ESM1b and Rosetta, which are treated as unavailable. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a benign outcome. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -7.874 | In-Between | 0.338 | Likely Benign | Likely Benign | -0.14 | Likely Benign | 0.1 | -0.67 | Ambiguous | -0.41 | Likely Benign | 0.13 | Likely Benign | 0.028 | Likely Benign | -1.41 | Neutral | 0.276 | Benign | 0.062 | Benign | 4.22 | Benign | 0.35 | Tolerated | 0.1516 | 0.3514 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.595A>T | N199Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N199Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumVar), FATHMM, and AlphaMissense‑Optimized; pathogenic predictions arise from SIFT, PROVEAN, polyPhen‑2 (HumDiv), and ESM1b. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates benign stability. Overall, the majority of conventional tools (8 benign vs. 4 pathogenic) favor a benign effect, and this interpretation does not conflict with the absence of ClinVar annotation. Therefore, the variant is most likely benign based on the current predictive evidence, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -8.575 | Likely Pathogenic | 0.391 | Ambiguous | Likely Benign | -0.06 | Likely Benign | 0.1 | 0.14 | Likely Benign | 0.04 | Likely Benign | 0.20 | Likely Benign | 0.097 | Likely Benign | -3.12 | Deleterious | 0.952 | Possibly Damaging | 0.395 | Benign | 4.15 | Benign | 0.04 | Affected | 0.0389 | 0.5630 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||
| c.596A>C | N199T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N199T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify it as benign or likely benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM Consensus predicts likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -4.123 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.16 | Likely Benign | 0.2 | -0.10 | Likely Benign | 0.03 | Likely Benign | -0.04 | Likely Benign | 0.025 | Likely Benign | -0.82 | Neutral | 0.002 | Benign | 0.003 | Benign | 4.24 | Benign | 0.10 | Tolerated | 0.0862 | 0.6186 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.596A>G | N199S 2D AIThe SynGAP1 missense variant N199S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). All available in‑silico predictors classify the variant as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts pathogenicity. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus (majority vote) indicates likely benign; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Overall, the variant is most likely benign, and this prediction does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -0.268 | Likely Benign | 0.079 | Likely Benign | Likely Benign | 0.08 | Likely Benign | 0.4 | -0.08 | Likely Benign | 0.00 | Likely Benign | -0.44 | Likely Benign | 0.040 | Likely Benign | 0.09 | Neutral | 0.160 | Benign | 0.045 | Benign | 4.36 | Benign | 0.57 | Tolerated | 0.2601 | 0.6023 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||
| c.596A>T | N199I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N199I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields an equal split of benign and pathogenic calls. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -8.299 | Likely Pathogenic | 0.328 | Likely Benign | Likely Benign | 0.27 | Likely Benign | 0.1 | -0.11 | Likely Benign | 0.08 | Likely Benign | 0.20 | Likely Benign | 0.066 | Likely Benign | -3.27 | Deleterious | 0.316 | Benign | 0.045 | Benign | 4.16 | Benign | 0.01 | Affected | 0.0450 | 0.6009 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||
| c.597C>A | N199K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N199K is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized all classify the substitution as benign. Only ESM1b and AlphaMissense‑Default predict pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the preponderance of evidence supports a benign impact, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | Uncertain | 1 | -8.198 | Likely Pathogenic | 0.686 | Likely Pathogenic | Likely Benign | -0.19 | Likely Benign | 0.1 | 0.03 | Likely Benign | -0.08 | Likely Benign | 0.33 | Likely Benign | 0.024 | Likely Benign | -1.48 | Neutral | 0.276 | Benign | 0.083 | Benign | 4.27 | Benign | 0.13 | Tolerated | 3.47 | 9 | 0.1399 | 0.5218 | 1 | 0 | -0.4 | 14.07 | 207.8 | 21.5 | -0.1 | 1.5 | 0.1 | 0.0 | X | Uncertain | Asn199, located in the N-terminal loop before the first anti-parallel β sheet strand (res. Ile205-Pro208), is replaced by a positively charged lysine. On the protein surface, both the carboxamide group of Asn199 and the amino group of Lys199 side chains can form hydrogen bonds with the backbone carbonyl groups of residues (e.g., Ala249) at the end of an α helix (res. Ala236-Lys251). However, since the model ends abruptly at the N-terminus, no definite conclusions can be drawn from the simulations. | |||||||||||||||||
| c.591G>T | E197D 2D AIThe SynGAP1 missense variant E197D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions strongly supports a benign classification, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | 0.784 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.066 | Likely Benign | 1.81 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.66 | Benign | 1.00 | Tolerated | 0.1579 | 0.3345 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.591G>C | E197D 2D AIThe SynGAP1 missense variant E197D is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions strongly supports a benign classification, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | 0.784 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.066 | Likely Benign | 1.81 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.66 | Benign | 1.00 | Tolerated | 0.1579 | 0.3345 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.584C>T | A195V 2D AIThe SynGAP1 missense variant A195V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN and AlphaMissense‑Default. High‑accuracy methods give no definitive verdict: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 pathogenic vs. 2 benign votes), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.346032 | Structured | 0.430388 | Uncertain | 0.363 | 0.533 | 0.125 | -5.830 | Likely Benign | 0.924 | Likely Pathogenic | Ambiguous | 0.210 | Likely Benign | -2.63 | Deleterious | 0.384 | Benign | 0.070 | Benign | 4.05 | Benign | 0.12 | Tolerated | 0.0782 | 0.5560 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||||||||
| c.586T>A | L196M 2D AIThe SynGAP1 missense variant L196M is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool rates the variant as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta (combining FoldX‑MD and Rosetta outputs) has no available result. Overall, more tools predict pathogenicity (5) than benignity (3), and the high‑accuracy predictions do not overturn this trend. Therefore, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.284882 | Structured | 0.429452 | Uncertain | 0.489 | 0.521 | 0.125 | -8.957 | Likely Pathogenic | 0.902 | Likely Pathogenic | Ambiguous | 0.094 | Likely Benign | -1.48 | Neutral | 0.971 | Probably Damaging | 0.691 | Possibly Damaging | 3.64 | Benign | 0.01 | Affected | 0.0689 | 0.2786 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||||||
| c.586T>G | L196V 2D AIThe SynGAP1 missense variant L196V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, the majority of evidence (five benign vs three pathogenic) points to a benign impact. This conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.284882 | Structured | 0.429452 | Uncertain | 0.489 | 0.521 | 0.125 | -8.702 | Likely Pathogenic | 0.898 | Likely Pathogenic | Ambiguous | 0.106 | Likely Benign | -2.31 | Neutral | 0.243 | Benign | 0.097 | Benign | 3.71 | Benign | 0.01 | Affected | 0.1436 | 0.2850 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.587T>C | L196S 2D  AIThe SynGAP1 missense variant L196S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is Likely Pathogenic, while Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.284882 | Structured | 0.429452 | Uncertain | 0.489 | 0.521 | 0.125 | -9.987 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.353 | Likely Benign | -4.75 | Deleterious | 0.437 | Benign | 0.186 | Benign | 3.65 | Benign | 0.00 | Affected | 0.3473 | 0.0736 | -3 | -2 | -4.6 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.587T>G | L196W 2D  AIThe SynGAP1 missense variant L196W has no ClinVar entry (ClinVar status: not reported) and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for this variant, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.284882 | Structured | 0.429452 | Uncertain | 0.489 | 0.521 | 0.125 | -12.148 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.293 | Likely Benign | -4.91 | Deleterious | 0.992 | Probably Damaging | 0.869 | Possibly Damaging | 3.58 | Benign | 0.00 | Affected | 0.0551 | 0.2718 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||||||||||||
| c.588G>C | L196F 2D AIThe SynGAP1 missense variant L196F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.284882 | Structured | 0.429452 | Uncertain | 0.489 | 0.521 | 0.125 | -10.245 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.177 | Likely Benign | -3.17 | Deleterious | 0.917 | Possibly Damaging | 0.502 | Possibly Damaging | 3.64 | Benign | 0.01 | Affected | 0.0557 | 0.2756 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.588G>T | L196F 2D AIThe SynGAP1 missense variant L196F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the collective predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.284882 | Structured | 0.429452 | Uncertain | 0.489 | 0.521 | 0.125 | -10.245 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.177 | Likely Benign | -3.17 | Deleterious | 0.917 | Possibly Damaging | 0.502 | Possibly Damaging | 3.64 | Benign | 0.01 | Affected | 0.0557 | 0.2756 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.589G>A | E197K 2D AIThe SynGAP1 missense variant E197K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 (HumDiv and HumVar) and FATHMM, whereas pathogenic calls arise from PROVEAN, SIFT, ESM1b, AlphaMissense‑Default and AlphaMissense‑Optimized. Grouping by consensus, the majority of high‑confidence predictors (AlphaMissense‑Optimized, SGM‑Consensus, PROVEAN, SIFT, ESM1b) indicate pathogenicity, while a minority (REVEL, polyPhen‑2, FATHMM) suggest benign impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM and PROVEAN, is classified as Likely Pathogenic. AlphaMissense‑Optimized also predicts Pathogenic. Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for E197K, and this assessment does not conflict with ClinVar, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | -11.045 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 0.198 | Likely Benign | -2.50 | Deleterious | 0.118 | Benign | 0.037 | Benign | 4.09 | Benign | 0.02 | Affected | 0.1824 | 0.5890 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.589G>C | E197Q 2D AIThe SynGAP1 E197Q variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and therefore reports a likely benign outcome. AlphaMissense‑Optimized also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | -6.347 | Likely Benign | 0.723 | Likely Pathogenic | Likely Benign | 0.145 | Likely Benign | -1.75 | Neutral | 0.396 | Benign | 0.067 | Benign | 4.05 | Benign | 0.02 | Affected | 0.0867 | 0.5228 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.58C>G | P20A 2D AIThe SynGAP1 missense variant P20A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Foldetta results are not available, so they do not influence the assessment. Overall, the majority of evidence points to a benign effect for P20A, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.442804 | Uncertain | 0.448 | 0.899 | 0.500 | -3.120 | Likely Benign | 0.128 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.31 | Neutral | 0.805 | Possibly Damaging | 0.539 | Possibly Damaging | 4.31 | Benign | 0.00 | Affected | 0.3753 | 0.5520 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.590A>C | E197A 2D AIThe SynGAP1 E197A missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. Two tools give uncertain results: ESM1b and AlphaMissense‑Optimized. High‑accuracy assessment shows that the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenicity, while AlphaMissense‑Optimized remains uncertain and Foldetta (which would combine FoldX‑MD and Rosetta outputs) has no available result. Overall, the balance of evidence—including the pathogenic majority in the high‑accuracy consensus—suggests that the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | -7.956 | In-Between | 0.787 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -3.56 | Deleterious | 0.055 | Benign | 0.016 | Benign | 4.06 | Benign | 0.02 | Affected | 0.3134 | 0.5290 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||||||||
| c.590A>G | E197G 2D AIThe SynGAP1 missense variant E197G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on benign impact include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict pathogenicity are PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Pathogenic, while AlphaMissense‑Optimized remains benign; Foldetta results are unavailable. Overall, the majority of individual predictors lean toward pathogenicity, and the SGM‑Consensus supports this view. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | -8.480 | Likely Pathogenic | 0.749 | Likely Pathogenic | Likely Benign | 0.165 | Likely Benign | -3.13 | Deleterious | 0.118 | Benign | 0.037 | Benign | 4.03 | Benign | 0.01 | Affected | 0.2638 | 0.4615 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.590A>T | E197V 2D AIThe SynGAP1 missense variant E197V is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split opinion: benign calls come from REVEL, polyPhen‑2 (HumDiv and HumVar) and FATHMM, while pathogenic calls arise from PROVEAN, SIFT, ESM1b and AlphaMissense‑Default. When predictions are grouped by consensus, four tools predict benign and four predict pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability predictor, has no available result for this residue. Taken together, the preponderance of evidence from both general and high‑accuracy predictors indicates that E197V is most likely pathogenic, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | -9.023 | Likely Pathogenic | 0.932 | Likely Pathogenic | Ambiguous | 0.247 | Likely Benign | -4.51 | Deleterious | 0.396 | Benign | 0.099 | Benign | 4.01 | Benign | 0.00 | Affected | 0.0490 | 0.6024 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.597C>G | N199K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N199K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Tools that predict a pathogenic effect are ESM1b and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a benign impact. Overall, the majority of evidence (10 benign vs. 2 pathogenic) supports a benign classification. This consensus does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -8.198 | Likely Pathogenic | 0.686 | Likely Pathogenic | Likely Benign | -0.19 | Likely Benign | 0.1 | 0.03 | Likely Benign | -0.08 | Likely Benign | 0.33 | Likely Benign | 0.024 | Likely Benign | -1.48 | Neutral | 0.276 | Benign | 0.083 | Benign | 4.27 | Benign | 0.13 | Tolerated | 3.47 | 9 | 0.1399 | 0.5218 | 1 | 0 | -0.4 | 14.07 | 207.8 | 21.5 | -0.1 | 1.5 | 0.1 | 0.0 | X | Uncertain | Asn199, located in the N-terminal loop before the first anti-parallel β sheet strand (res. Ile205-Pro208), is replaced by a positively charged lysine. On the protein surface, both the carboxamide group of Asn199 and the amino group of Lys199 side chains can form hydrogen bonds with the backbone carbonyl groups of residues (e.g., Ala249) at the end of an α helix (res. Ala236-Lys251). However, since the model ends abruptly at the N-terminus, no definite conclusions can be drawn from the simulations. | |||||||||||||||||||
| c.570C>A | S190R 2D AIThe SynGAP1 missense variant S190R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority of the four high‑accuracy tools) also predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.422041 | Structured | 0.428613 | Uncertain | 0.338 | 0.615 | 0.250 | -10.137 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.146 | Likely Benign | -2.92 | Deleterious | 0.917 | Possibly Damaging | 0.446 | Benign | 4.07 | Benign | 0.04 | Affected | 0.0809 | 0.4059 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.543C>G | H181Q 2D AIThe SynGAP1 H181Q missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic vs two benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools therefore give a benign prediction from AlphaMissense‑Optimized, an inconclusive SGM Consensus, and an unavailable Foldetta result. Overall, the majority of predictions (six benign vs three pathogenic) indicate that the variant is most likely benign, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.505461 | Disordered | 0.439530 | Uncertain | 0.294 | 0.616 | 0.500 | -9.577 | Likely Pathogenic | 0.692 | Likely Pathogenic | Likely Benign | 0.125 | Likely Benign | -1.45 | Neutral | 0.940 | Possibly Damaging | 0.360 | Benign | 4.19 | Benign | 0.09 | Tolerated | 0.1241 | 0.2835 | 3 | 0 | -0.3 | -9.01 | ||||||||||||||||||||||||||||||||||||||||
| c.550G>C | E184Q 2D AIThe SynGAP1 missense variant E184Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available, so it does not influence the overall assessment. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.431514 | Uncertain | 0.348 | 0.622 | 0.625 | -11.927 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.210 | Likely Benign | -2.55 | Deleterious | 0.990 | Probably Damaging | 0.815 | Possibly Damaging | 3.47 | Benign | 0.00 | Affected | 0.1688 | 0.7914 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.551A>C | E184A 2D AIThe SynGAP1 missense variant E184A has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic impact are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as Likely Pathogenic; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of computational evidence supports a pathogenic effect for E184A. This prediction is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.431514 | Uncertain | 0.348 | 0.622 | 0.625 | -11.486 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.338 | Likely Benign | -4.98 | Deleterious | 0.868 | Possibly Damaging | 0.344 | Benign | 3.48 | Benign | 0.00 | Affected | 0.4419 | 0.7381 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.551A>G | E184G 2D AIThe SynGAP1 missense variant E184G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that E184G is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.431514 | Uncertain | 0.348 | 0.622 | 0.625 | -10.725 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.353 | Likely Benign | -5.52 | Deleterious | 0.970 | Probably Damaging | 0.607 | Possibly Damaging | 3.45 | Benign | 0.00 | Affected | 0.3281 | 0.6534 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.551A>T | E184V 2D AIThe SynGAP1 missense variant E184V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while benign calls are made only by REVEL and FATHMM. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized scores the variant as pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability analysis is available for this variant. Overall, the preponderance of evidence indicates that E184V is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.431514 | Uncertain | 0.348 | 0.622 | 0.625 | -13.119 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.371 | Likely Benign | -5.72 | Deleterious | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 3.44 | Benign | 0.00 | Affected | 0.0995 | 0.8511 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.552G>C | E184D 2D AIThe SynGAP1 E184D missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence (six pathogenic vs. three benign predictions) indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.557691 | Disordered | 0.431514 | Uncertain | 0.348 | 0.622 | 0.625 | -6.753 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.195 | Likely Benign | -2.55 | Deleterious | 0.930 | Possibly Damaging | 0.584 | Possibly Damaging | 3.52 | Benign | 0.00 | Affected | 0.2249 | 0.5394 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.552G>T | E184D 2D AIThe SynGAP1 E184D missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of evidence (six pathogenic vs. three benign predictions) indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.557691 | Disordered | 0.431514 | Uncertain | 0.348 | 0.622 | 0.625 | -6.753 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.195 | Likely Benign | -2.55 | Deleterious | 0.930 | Possibly Damaging | 0.584 | Possibly Damaging | 3.52 | Benign | 0.00 | Affected | 0.2249 | 0.5394 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.553T>A | S185T 2D AIThe SynGAP1 missense variant S185T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, while Foldetta (combining FoldX‑MD and Rosetta outputs) is not available for this variant. Overall, the majority of evidence points to a pathogenic impact. The variant is most likely pathogenic, and this assessment does not contradict any existing ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.545602 | Disordered | 0.430485 | Uncertain | 0.365 | 0.623 | 0.500 | -10.943 | Likely Pathogenic | 0.903 | Likely Pathogenic | Ambiguous | 0.186 | Likely Benign | -2.54 | Deleterious | 0.596 | Possibly Damaging | 0.142 | Benign | 3.60 | Benign | 0.00 | Affected | 0.1484 | 0.6236 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.553T>C | S185P 2D AIThe SynGAP1 missense variant S185P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect are REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default—predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the S185P variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.545602 | Disordered | 0.430485 | Uncertain | 0.365 | 0.623 | 0.500 | -15.129 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.285 | Likely Benign | -4.02 | Deleterious | 0.940 | Possibly Damaging | 0.462 | Possibly Damaging | 3.56 | Benign | 0.00 | Affected | 0.2156 | 0.5242 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.553T>G | S185A 2D AIThe SynGAP1 missense variant S185A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome. AlphaMissense‑Optimized is uncertain, and no Foldetta stability result is available. High‑accuracy assessments therefore show a likely pathogenic consensus from SGM‑Consensus, with no contradictory evidence from ClinVar or population databases. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any existing ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.545602 | Disordered | 0.430485 | Uncertain | 0.365 | 0.623 | 0.500 | -11.839 | Likely Pathogenic | 0.868 | Likely Pathogenic | Ambiguous | 0.203 | Likely Benign | -2.57 | Deleterious | 0.231 | Benign | 0.107 | Benign | 3.65 | Benign | 0.00 | Affected | 0.5147 | 0.4472 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.554C>A | S185Y 2D AIThe SynGAP1 missense variant S185Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts Pathogenic, and the SGM‑Consensus also indicates Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.545602 | Disordered | 0.430485 | Uncertain | 0.365 | 0.623 | 0.500 | -13.633 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.316 | Likely Benign | -4.93 | Deleterious | 0.718 | Possibly Damaging | 0.178 | Benign | 3.56 | Benign | 0.00 | Affected | 0.0736 | 0.5439 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||||
| c.554C>G | S185C 2D AIThe SynGAP1 missense variant S185C has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, while the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (derived from the same set of predictors) also reports likely pathogenic. Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.545602 | Disordered | 0.430485 | Uncertain | 0.365 | 0.623 | 0.500 | -10.612 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.266 | Likely Benign | -3.96 | Deleterious | 0.983 | Probably Damaging | 0.635 | Possibly Damaging | 3.54 | Benign | 0.00 | Affected | 0.0989 | 0.6000 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.554C>T | S185F 2D AIThe SynGAP1 missense variant S185F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect are REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic outcome. The SGM‑Consensus, a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for S185F. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.545602 | Disordered | 0.430485 | Uncertain | 0.365 | 0.623 | 0.500 | -13.327 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.356 | Likely Benign | -5.03 | Deleterious | 0.838 | Possibly Damaging | 0.466 | Possibly Damaging | 3.55 | Benign | 0.00 | Affected | 0.0628 | 0.5563 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.556T>A | L186M 2D AIThe SynGAP1 missense variant L186M is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool rates the variant as uncertain, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (5 pathogenic vs. 3 benign) suggest a pathogenic impact. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.458154 | Structured | 0.428613 | Uncertain | 0.397 | 0.617 | 0.500 | -11.783 | Likely Pathogenic | 0.933 | Likely Pathogenic | Ambiguous | 0.146 | Likely Benign | -1.58 | Neutral | 0.952 | Possibly Damaging | 0.694 | Possibly Damaging | 3.50 | Benign | 0.00 | Affected | 0.0671 | 0.3396 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||||||
| c.550G>A | E184K 2D AIThe SynGAP1 missense variant E184K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence indicates that E184K is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.431514 | Uncertain | 0.348 | 0.622 | 0.625 | -14.037 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.289 | Likely Benign | -3.31 | Deleterious | 0.970 | Probably Damaging | 0.681 | Possibly Damaging | 3.50 | Benign | 0.00 | Affected | 0.2890 | 0.8227 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.549T>G | H183Q 2D AIThe SynGAP1 missense variant H183Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.476583 | Structured | 0.432952 | Uncertain | 0.421 | 0.622 | 0.500 | -10.383 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.223 | Likely Benign | -5.43 | Deleterious | 0.838 | Possibly Damaging | 0.276 | Benign | 3.88 | Benign | 0.01 | Affected | 0.1470 | 0.3623 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.544T>A | S182T 2D AIThe SynGAP1 missense variant S182T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, the majority of available predictions (five benign vs three pathogenic) suggest a benign impact. This consensus does not contradict any ClinVar annotation, as no ClinVar entry exists for this variant. Thus, based on current computational evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.501700 | Disordered | 0.436016 | Uncertain | 0.368 | 0.619 | 0.625 | -11.247 | Likely Pathogenic | 0.857 | Likely Pathogenic | Ambiguous | 0.128 | Likely Benign | -2.29 | Neutral | 0.231 | Benign | 0.081 | Benign | 3.68 | Benign | 0.00 | Affected | 0.1624 | 0.5657 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.544T>C | S182P 2D AIThe SynGAP1 missense variant S182P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority of the four high‑accuracy tools) also indicates pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple independent predictors points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.436016 | Uncertain | 0.368 | 0.619 | 0.625 | -13.362 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.279 | Likely Benign | -3.71 | Deleterious | 0.838 | Possibly Damaging | 0.367 | Benign | 3.67 | Benign | 0.00 | Affected | 0.2438 | 0.5030 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.544T>G | S182A 2D AIThe SynGAP1 missense variant S182A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, the majority of standard predictors lean toward a benign impact, and no ClinVar evidence contradicts this assessment. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.501700 | Disordered | 0.436016 | Uncertain | 0.368 | 0.619 | 0.625 | -9.417 | Likely Pathogenic | 0.838 | Likely Pathogenic | Ambiguous | 0.113 | Likely Benign | -2.26 | Neutral | 0.001 | Benign | 0.005 | Benign | 3.70 | Benign | 0.00 | Affected | 0.5466 | 0.4660 | Weaken | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.545C>A | S182Y 2D AIThe SynGAP1 missense variant S182Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while benign calls are made only by REVEL and FATHMM. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized indicates a pathogenic effect, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic. A protein‑folding stability analysis with Foldetta is unavailable, so it does not influence the overall assessment. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.436016 | Uncertain | 0.368 | 0.619 | 0.625 | -15.951 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.274 | Likely Benign | -4.40 | Deleterious | 0.940 | Possibly Damaging | 0.564 | Possibly Damaging | 3.64 | Benign | 0.00 | Affected | 0.0656 | 0.5158 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||||
| c.545C>G | S182C 2D AIThe SynGAP1 missense variant S182C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all classify the variant as damaging. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, while the SGM‑Consensus (majority vote) remains pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple in silico predictors indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.436016 | Uncertain | 0.368 | 0.619 | 0.625 | -12.707 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 0.216 | Likely Benign | -3.14 | Deleterious | 0.983 | Probably Damaging | 0.635 | Possibly Damaging | 3.63 | Benign | 0.00 | Affected | 0.1179 | 0.5800 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.545C>T | S182F 2D AIThe SynGAP1 missense variant S182F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic, and the Foldetta stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the corroborating high‑accuracy tools, the variant is most likely pathogenic, with no conflict with ClinVar status (which is absent). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.436016 | Uncertain | 0.368 | 0.619 | 0.625 | -12.027 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.227 | Likely Benign | -4.30 | Deleterious | 0.838 | Possibly Damaging | 0.466 | Possibly Damaging | 3.64 | Benign | 0.00 | Affected | 0.0597 | 0.5482 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.547C>A | H183N 2D AIThe SynGAP1 H183N missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. In contrast, tools that predict a pathogenic impact are PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (majority vote) also indicates Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the majority of predictions, including the high‑accuracy tools, point to a pathogenic effect, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.476583 | Structured | 0.432952 | Uncertain | 0.421 | 0.622 | 0.500 | -12.028 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.188 | Likely Benign | -4.97 | Deleterious | 0.012 | Benign | 0.006 | Benign | 3.81 | Benign | 0.01 | Affected | 0.1714 | 0.2589 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.547C>G | H183D 2D AIThe SynGAP1 missense variant H183D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Pathogenic, and AlphaMissense‑Optimized independently predicts Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the majority of predictions (seven pathogenic vs. four benign) indicate a pathogenic impact. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for H183D. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.476583 | Structured | 0.432952 | Uncertain | 0.421 | 0.622 | 0.500 | -18.626 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.311 | Likely Benign | -6.55 | Deleterious | 0.421 | Benign | 0.107 | Benign | 3.81 | Benign | 0.01 | Affected | 0.2620 | 0.1817 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||||||||||||
| c.547C>T | H183Y 2D AIThe SynGAP1 H183Y missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of computational evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.476583 | Structured | 0.432952 | Uncertain | 0.421 | 0.622 | 0.500 | -9.481 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.250 | Likely Benign | -4.48 | Deleterious | 0.818 | Possibly Damaging | 0.255 | Benign | 3.77 | Benign | 0.00 | Affected | 0.0831 | 0.4406 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.548A>C | H183P 2D AIThe SynGAP1 missense variant H183P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized also predicts Pathogenic, while the Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic impact, and this conclusion does not contradict any existing ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.476583 | Structured | 0.432952 | Uncertain | 0.421 | 0.622 | 0.500 | -18.527 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 0.410 | Likely Benign | -7.18 | Deleterious | 0.838 | Possibly Damaging | 0.276 | Benign | 3.79 | Benign | 0.01 | Affected | 0.2143 | 0.3712 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.548A>G | H183R 2D AIThe SynGAP1 H183R missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus (majority vote) also pathogenic; Foldetta stability analysis is unavailable. Based on the majority of computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.476583 | Structured | 0.432952 | Uncertain | 0.421 | 0.622 | 0.500 | -10.937 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.368 | Likely Benign | -5.43 | Deleterious | 0.596 | Possibly Damaging | 0.142 | Benign | 3.90 | Benign | 0.13 | Tolerated | 0.1915 | 0.2249 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||||||||||||
| c.548A>T | H183L 2D AIThe SynGAP1 missense variant H183L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. In contrast, tools that predict a pathogenic effect are PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Pathogenic” verdict (3 pathogenic vs. 1 benign votes). AlphaMissense‑Optimized alone also predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.476583 | Structured | 0.432952 | Uncertain | 0.421 | 0.622 | 0.500 | -12.898 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 0.325 | Likely Benign | -8.12 | Deleterious | 0.421 | Benign | 0.058 | Benign | 3.79 | Benign | 0.01 | Affected | 0.0911 | 0.5304 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.549T>A | H183Q 2D AIThe SynGAP1 missense variant H183Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.476583 | Structured | 0.432952 | Uncertain | 0.421 | 0.622 | 0.500 | -10.383 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.223 | Likely Benign | -5.43 | Deleterious | 0.838 | Possibly Damaging | 0.276 | Benign | 3.88 | Benign | 0.01 | Affected | 0.1470 | 0.3623 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.556T>G | L186V 2D AIThe SynGAP1 missense variant L186V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, and FATHMM. Tools that agree on a pathogenic effect include polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments are mixed: AlphaMissense‑Optimized predicts pathogenic, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields no clear majority and is therefore unavailable; Foldetta results are also unavailable. Overall, the majority of available predictions (five pathogenic vs. four benign) indicate a pathogenic effect. There is no ClinVar entry to contradict this assessment, so the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.458154 | Structured | 0.428613 | Uncertain | 0.397 | 0.617 | 0.500 | -9.385 | Likely Pathogenic | 0.958 | Likely Pathogenic | Likely Pathogenic | 0.090 | Likely Benign | -2.27 | Neutral | 0.734 | Possibly Damaging | 0.185 | Benign | 3.60 | Benign | 0.00 | Affected | 0.1419 | 0.3665 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.557T>C | L186S 2D AIThe SynGAP1 missense variant L186S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts a deleterious effect, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.458154 | Structured | 0.428613 | Uncertain | 0.397 | 0.617 | 0.500 | -13.906 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.250 | Likely Benign | -4.73 | Deleterious | 0.952 | Possibly Damaging | 0.601 | Possibly Damaging | 3.51 | Benign | 0.00 | Affected | 0.3214 | 0.0808 | -3 | -2 | -4.6 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.557T>G | L186W 2D AIThe SynGAP1 missense variant L186W has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.458154 | Structured | 0.428613 | Uncertain | 0.397 | 0.617 | 0.500 | -16.177 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.274 | Likely Benign | -4.78 | Deleterious | 0.996 | Probably Damaging | 0.819 | Possibly Damaging | 3.46 | Benign | 0.00 | Affected | 0.0539 | 0.3138 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||||||||||||
| c.564T>G | S188R 2D AIThe SynGAP1 missense variant S188R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect are REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default—predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. Foldetta, a protein‑folding stability method, did not provide a result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.490133 | Structured | 0.428502 | Uncertain | 0.298 | 0.603 | 0.500 | -11.567 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.263 | Likely Benign | -3.82 | Deleterious | 0.985 | Probably Damaging | 0.767 | Possibly Damaging | 3.92 | Benign | 0.00 | Affected | 0.0916 | 0.4236 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.565C>A | P189T 2D AISynGAP1 missense variant P189T is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect comprise PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority‑vote method) reports it as likely pathogenic; Foldetta stability analysis is unavailable. Overall, the majority of computational evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.497853 | Structured | 0.428590 | Uncertain | 0.331 | 0.602 | 0.250 | -8.622 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.265 | Likely Benign | -5.26 | Deleterious | 0.384 | Benign | 0.177 | Benign | 4.05 | Benign | 0.05 | Affected | 0.1603 | 0.6392 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.565C>G | P189A 2D AIThe SynGAP1 missense variant P189A is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two pathogenic, two benign), and Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs. four benign) and the pathogenic call from AlphaMissense‑Optimized suggest that the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.497853 | Structured | 0.428590 | Uncertain | 0.331 | 0.602 | 0.250 | -4.998 | Likely Benign | 0.974 | Likely Pathogenic | Likely Pathogenic | 0.211 | Likely Benign | -5.46 | Deleterious | 0.941 | Possibly Damaging | 0.607 | Possibly Damaging | 4.07 | Benign | 0.07 | Tolerated | 0.3719 | 0.6209 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||||||||
| c.565C>T | P189S 2D AIThe SynGAP1 missense variant P189S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized; ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors pathogenicity (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of computational evidence indicates a pathogenic effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.497853 | Structured | 0.428590 | Uncertain | 0.331 | 0.602 | 0.250 | -7.191 | In-Between | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.211 | Likely Benign | -5.23 | Deleterious | 0.941 | Possibly Damaging | 0.531 | Possibly Damaging | 4.09 | Benign | 0.15 | Tolerated | 0.3626 | 0.6460 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||||||||
| c.566C>A | P189H 2D AIThe SynGAP1 missense variant P189H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence indicates that P189H is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.497853 | Structured | 0.428590 | Uncertain | 0.331 | 0.602 | 0.250 | -9.633 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.298 | Likely Benign | -6.49 | Deleterious | 0.999 | Probably Damaging | 0.936 | Probably Damaging | 4.00 | Benign | 0.00 | Affected | 0.1620 | 0.5327 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.566C>G | P189R 2D AIThe SynGAP1 missense variant P189R has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL and FATHMM, whereas the remaining eight tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—indicate pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus confirms a likely pathogenic status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect for P189R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.497853 | Structured | 0.428590 | Uncertain | 0.331 | 0.602 | 0.250 | -13.503 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.331 | Likely Benign | -6.45 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 4.05 | Benign | 0.01 | Affected | 0.1363 | 0.3894 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.566C>T | P189L 2D AIThe SynGAP1 missense variant P189L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. In contrast, a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all indicate likely pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. Foldetta results are unavailable for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a pathogenic classification, with no ClinVar record to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.497853 | Structured | 0.428590 | Uncertain | 0.331 | 0.602 | 0.250 | -10.132 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.290 | Likely Benign | -7.28 | Deleterious | 0.991 | Probably Damaging | 0.781 | Possibly Damaging | 4.04 | Benign | 0.17 | Tolerated | 0.2283 | 0.7378 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.568A>C | S190R 2D AIThe SynGAP1 missense variant S190R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority of the four high‑accuracy tools) also predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.422041 | Structured | 0.428613 | Uncertain | 0.338 | 0.615 | 0.250 | -10.137 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.156 | Likely Benign | -2.92 | Deleterious | 0.917 | Possibly Damaging | 0.446 | Benign | 4.07 | Benign | 0.04 | Affected | 0.0809 | 0.4059 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.568A>G | S190G 2D AIThe SynGAP1 missense variant S190G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the consensus of all available predictions is that the variant is most likely benign, and this conclusion is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.428613 | Uncertain | 0.338 | 0.615 | 0.250 | -5.440 | Likely Benign | 0.321 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -1.26 | Neutral | 0.243 | Benign | 0.079 | Benign | 4.11 | Benign | 0.19 | Tolerated | 0.2780 | 0.4765 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.568A>T | S190C 2D AIThe SynGAP1 missense variant S190C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.428613 | Uncertain | 0.338 | 0.615 | 0.250 | -6.696 | Likely Benign | 0.617 | Likely Pathogenic | Likely Benign | 0.107 | Likely Benign | -2.04 | Neutral | 0.992 | Probably Damaging | 0.707 | Possibly Damaging | 4.01 | Benign | 0.23 | Tolerated | 0.0937 | 0.6667 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.569G>A | S190N 2D AIThe SynGAP1 missense variant S190N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, and FATHMM, while polyPhen‑2 HumDiv and AlphaMissense‑Default predict a pathogenic outcome. The remaining tools, ESM1b and AlphaMissense‑Optimized, are uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized is uncertain; the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) resolves to benign (two benign votes versus one pathogenic and one uncertain); and Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.422041 | Structured | 0.428613 | Uncertain | 0.338 | 0.615 | 0.250 | -7.497 | In-Between | 0.838 | Likely Pathogenic | Ambiguous | 0.160 | Likely Benign | -1.73 | Neutral | 0.759 | Possibly Damaging | 0.202 | Benign | 4.06 | Benign | 0.08 | Tolerated | 0.1119 | 0.5285 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||||||||||
| c.569G>C | S190T 2D AIThe SynGAP1 missense variant S190T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus confirms Likely Benign, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.428613 | Uncertain | 0.338 | 0.615 | 0.250 | -6.870 | Likely Benign | 0.408 | Ambiguous | Likely Benign | 0.114 | Likely Benign | -1.31 | Neutral | 0.608 | Possibly Damaging | 0.108 | Benign | 4.08 | Benign | 0.20 | Tolerated | 0.1294 | 0.6920 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.569G>T | S190I 2D AIThe SynGAP1 missense variant S190I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. AlphaMissense‑Optimized returns an Uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Overall, the majority of evidence points to a pathogenic impact, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.422041 | Structured | 0.428613 | Uncertain | 0.338 | 0.615 | 0.250 | -9.868 | Likely Pathogenic | 0.954 | Likely Pathogenic | Ambiguous | 0.316 | Likely Benign | -3.39 | Deleterious | 0.845 | Possibly Damaging | 0.368 | Benign | 4.03 | Benign | 0.03 | Affected | 0.0769 | 0.5963 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.564T>A | S188R 2D AIThe SynGAP1 missense variant S188R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default—predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. Foldetta, a protein‑folding stability method, did not provide a result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the S188R variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.490133 | Structured | 0.428502 | Uncertain | 0.298 | 0.603 | 0.500 | -11.567 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.264 | Likely Benign | -3.82 | Deleterious | 0.985 | Probably Damaging | 0.767 | Possibly Damaging | 3.92 | Benign | 0.00 | Affected | 0.0916 | 0.4236 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.563G>T | S188I 2D AIThe SynGAP1 missense variant S188I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and FATHMM, while the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic or likely pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus also pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for S188I, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.490133 | Structured | 0.428502 | Uncertain | 0.298 | 0.603 | 0.500 | -12.133 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.205 | Likely Benign | -3.98 | Deleterious | 0.995 | Probably Damaging | 0.880 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 0.0878 | 0.6335 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.558G>C | L186F 2D AIThe SynGAP1 missense variant L186F is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), and FATHMM. In contrast, tools that predict a pathogenic effect are PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (majority vote) also indicates likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a pathogenic effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.458154 | Structured | 0.428613 | Uncertain | 0.397 | 0.617 | 0.500 | Uncertain | 1 | -11.861 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.132 | Likely Benign | -3.03 | Deleterious | 0.009 | Benign | 0.012 | Benign | 3.50 | Benign | 0.00 | Affected | 0.0524 | 0.3177 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||
| c.558G>T | L186F 2D AIThe SynGAP1 missense variant L186F has no ClinVar entry (ClinVar status: not reported) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Pathogenic.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.458154 | Structured | 0.428613 | Uncertain | 0.397 | 0.617 | 0.500 | -11.861 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.132 | Likely Benign | -3.03 | Deleterious | 0.009 | Benign | 0.012 | Benign | 3.50 | Benign | 0.00 | Affected | 0.0524 | 0.3177 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||||||||||||
| c.559C>A | L187M 2D AIThe SynGAP1 missense variant L187M is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM. Those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, which would assess protein‑folding stability, has no available output for this variant. Overall, the majority of predictions (five pathogenic vs. three benign) lean toward pathogenicity, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.465241 | Structured | 0.428046 | Uncertain | 0.367 | 0.625 | 0.375 | -8.814 | Likely Pathogenic | 0.954 | Likely Pathogenic | Ambiguous | 0.136 | Likely Benign | -1.35 | Neutral | 0.971 | Probably Damaging | 0.641 | Possibly Damaging | 3.72 | Benign | 0.02 | Affected | 0.0848 | 0.3613 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||||||||||
| c.559C>G | L187V 2D AIThe SynGAP1 missense variant L187V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic vs. two benign). High‑accuracy AlphaMissense‑Optimized predicts pathogenic, while Foldetta (combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the majority of standard predictors lean toward a benign classification, but the single high‑accuracy tool indicates pathogenicity. Thus, the variant is most likely benign based on the collective evidence, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.465241 | Structured | 0.428046 | Uncertain | 0.367 | 0.625 | 0.375 | -10.543 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 0.136 | Likely Benign | -2.24 | Neutral | 0.437 | Benign | 0.079 | Benign | 3.81 | Benign | 0.02 | Affected | 0.1525 | 0.3509 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.55G>A | A19T 2D AIThe SynGAP1 missense variant A19T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.443393 | Uncertain | 0.378 | 0.906 | 0.500 | -3.422 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.030 | Likely Benign | -0.02 | Neutral | 0.371 | Benign | 0.036 | Benign | 4.14 | Benign | 0.00 | Affected | 0.2357 | 0.7248 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||||||||||||
| c.55G>T | A19S 2D AIThe SynGAP1 missense variant A19S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.443393 | Uncertain | 0.378 | 0.906 | 0.500 | -2.905 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -0.21 | Neutral | 0.225 | Benign | 0.027 | Benign | 4.17 | Benign | 0.00 | Affected | 0.3273 | 0.6260 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||||||||||||
| c.560T>A | L187Q 2D AIThe SynGAP1 missense variant L187Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all indicate likely pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for the SynGAP1 L187Q variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.465241 | Structured | 0.428046 | Uncertain | 0.367 | 0.625 | 0.375 | -13.063 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.322 | Likely Benign | -4.31 | Deleterious | 0.917 | Possibly Damaging | 0.548 | Possibly Damaging | 3.72 | Benign | 0.00 | Affected | 0.1251 | 0.1060 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.560T>G | L187R 2D AIThe SynGAP1 missense variant L187R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts a deleterious effect, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.465241 | Structured | 0.428046 | Uncertain | 0.367 | 0.625 | 0.375 | -14.118 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.332 | Likely Benign | -4.38 | Deleterious | 0.917 | Possibly Damaging | 0.467 | Possibly Damaging | 3.72 | Benign | 0.00 | Affected | 0.1481 | 0.0702 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.562A>C | S188R 2D AIThe SynGAP1 missense variant S188R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect are REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default—predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the S188R variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.490133 | Structured | 0.428502 | Uncertain | 0.298 | 0.603 | 0.500 | -11.567 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.269 | Likely Benign | -3.82 | Deleterious | 0.985 | Probably Damaging | 0.767 | Possibly Damaging | 3.92 | Benign | 0.00 | Affected | 0.0916 | 0.4236 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||||
| c.562A>G | S188G 2D AIThe SynGAP1 missense variant S188G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy consensus (SGM‑Consensus) derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN is “Likely Pathogenic.” AlphaMissense‑Optimized returns an uncertain result, and Foldetta (which would combine FoldX‑MD and Rosetta outputs) has no available data for this variant. Based on the overall distribution of predictions, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.490133 | Structured | 0.428502 | Uncertain | 0.298 | 0.603 | 0.500 | -10.113 | Likely Pathogenic | 0.919 | Likely Pathogenic | Ambiguous | 0.123 | Likely Benign | -3.10 | Deleterious | 0.882 | Possibly Damaging | 0.404 | Benign | 3.91 | Benign | 0.00 | Affected | 0.3045 | 0.5542 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||||||||||
| c.562A>T | S188C 2D AIThe SynGAP1 missense variant S188C is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default, all of which classify the substitution as deleterious. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized yields an uncertain result, while Foldetta data are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus, the variant is most likely pathogenic; this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.490133 | Structured | 0.428502 | Uncertain | 0.298 | 0.603 | 0.500 | -8.670 | Likely Pathogenic | 0.849 | Likely Pathogenic | Ambiguous | 0.111 | Likely Benign | -2.96 | Deleterious | 0.999 | Probably Damaging | 0.956 | Probably Damaging | 3.87 | Benign | 0.00 | Affected | 0.1007 | 0.6737 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.563G>A | S188N 2D AIThe SynGAP1 missense variant S188N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the majority of available predictions (five benign vs three pathogenic) suggest a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.490133 | Structured | 0.428502 | Uncertain | 0.298 | 0.603 | 0.500 | -10.307 | Likely Pathogenic | 0.952 | Likely Pathogenic | Ambiguous | 0.095 | Likely Benign | -2.34 | Neutral | 0.259 | Benign | 0.048 | Benign | 3.92 | Benign | 0.01 | Affected | 0.1420 | 0.5658 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||||||||||
| c.563G>C | S188T 2D AIThe SynGAP1 missense variant S188T is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and FATHMM, while those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Uncertain predictions come from ESM1b and AlphaMissense‑Optimized. High‑accuracy assessment shows that the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans benign (two benign versus one pathogenic vote). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact; this conclusion does not contradict any ClinVar annotation, as none exists for S188T. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.490133 | Structured | 0.428502 | Uncertain | 0.298 | 0.603 | 0.500 | -7.906 | In-Between | 0.801 | Likely Pathogenic | Ambiguous | 0.124 | Likely Benign | -1.97 | Neutral | 0.948 | Possibly Damaging | 0.484 | Possibly Damaging | 3.92 | Benign | 0.13 | Tolerated | 0.1491 | 0.7492 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||||||||||||
| c.56C>G | A19G 2D AIThe SynGAP1 missense variant A19G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The prediction is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.443393 | Uncertain | 0.378 | 0.906 | 0.500 | -3.525 | Likely Benign | 0.194 | Likely Benign | Likely Benign | 0.037 | Likely Benign | -0.45 | Neutral | 0.371 | Benign | 0.036 | Benign | 4.10 | Benign | 0.00 | Affected | 0.2324 | 0.5447 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.598T>A | L200M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L200M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign outcome. Only two tools, polyPhen‑2 HumDiv and HumVar, predict pathogenicity. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from the four high‑confidence predictors, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a benign effect. Taken together, the overwhelming majority of evidence supports a benign classification for L200M, and this conclusion is consistent with the absence of a ClinVar entry. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.366687 | Structured | 0.428168 | Uncertain | 0.687 | 0.453 | 0.125 | -4.107 | Likely Benign | 0.215 | Likely Benign | Likely Benign | 0.08 | Likely Benign | 0.1 | 0.41 | Likely Benign | 0.25 | Likely Benign | -0.25 | Likely Benign | 0.139 | Likely Benign | 0.47 | Neutral | 0.997 | Probably Damaging | 0.960 | Probably Damaging | 4.11 | Benign | 0.31 | Tolerated | 0.0751 | 0.3511 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.626T>A | V209E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V209E is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the remaining 13 tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus) all predict a pathogenic outcome. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming majority of pathogenic predictions, V209E is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.247041 | Structured | 0.397624 | Uncertain | 0.874 | 0.331 | 0.125 | -14.366 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.53 | Destabilizing | 1.3 | 2.35 | Destabilizing | 2.94 | Destabilizing | 1.52 | Destabilizing | 0.403 | Likely Benign | -4.54 | Deleterious | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 3.65 | Benign | 0.00 | Affected | 0.0826 | 0.1652 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.632G>C | S211T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S211T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Two tools, FoldX and Rosetta, give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign and Foldetta as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.209395 | Structured | 0.389893 | Uncertain | 0.846 | 0.300 | 0.125 | -10.301 | Likely Pathogenic | 0.678 | Likely Pathogenic | Likely Benign | 0.98 | Ambiguous | 0.5 | -0.78 | Ambiguous | 0.10 | Likely Benign | 0.50 | Likely Benign | 0.157 | Likely Benign | -2.33 | Neutral | 0.948 | Possibly Damaging | 0.484 | Possibly Damaging | 3.95 | Benign | 0.07 | Tolerated | 0.1678 | 0.5960 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||
| c.632G>T | S211I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 S211I missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, and FATHMM; pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive. No evidence from FoldX or Rosetta is available. Overall, the preponderance of evidence indicates the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.209395 | Structured | 0.389893 | Uncertain | 0.846 | 0.300 | 0.125 | -17.090 | Likely Pathogenic | 0.973 | Likely Pathogenic | Likely Pathogenic | 1.06 | Ambiguous | 1.1 | 1.05 | Ambiguous | 1.06 | Ambiguous | 0.12 | Likely Benign | 0.251 | Likely Benign | -4.94 | Deleterious | 0.995 | Probably Damaging | 0.767 | Possibly Damaging | 3.90 | Benign | 0.01 | Affected | 0.0909 | 0.5220 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.633C>A | S211R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S211R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, FATHMM, and Foldetta. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled “Likely Pathogenic.” The premPS score is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions and the two high‑accuracy pathogenic calls suggest that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.209395 | Structured | 0.389893 | Uncertain | 0.846 | 0.300 | 0.125 | -14.126 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.49 | Likely Benign | 1.0 | 0.20 | Likely Benign | 0.35 | Likely Benign | 0.97 | Ambiguous | 0.144 | Likely Benign | -4.00 | Deleterious | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 3.95 | Benign | 0.02 | Affected | 0.1003 | 0.3592 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.633C>G | S211R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S211R is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, FoldX, Rosetta, FATHMM, and the protein‑stability integrator Foldetta, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic, and premPS is uncertain. High‑accuracy assessments give conflicting signals: AlphaMissense‑Optimized predicts Pathogenic, SGM‑Consensus also predicts Likely Pathogenic, but Foldetta, which combines FoldX‑MD and Rosetta outputs, predicts Benign. Overall, the majority of tools and the consensus score point to a pathogenic effect, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.209395 | Structured | 0.389893 | Uncertain | 0.846 | 0.300 | 0.125 | -14.126 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.49 | Likely Benign | 1.0 | 0.20 | Likely Benign | 0.35 | Likely Benign | 0.97 | Ambiguous | 0.144 | Likely Benign | -4.00 | Deleterious | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 3.95 | Benign | 0.02 | Affected | 0.1003 | 0.3592 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.634T>A | S212T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S212T is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include FoldX and FATHMM, whereas the majority of tools predict a pathogenic impact: REVEL, SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. With most evidence pointing to deleterious effects and no conflicting ClinVar annotation, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.381517 | Uncertain | 0.846 | 0.278 | 0.125 | -11.473 | Likely Pathogenic | 0.939 | Likely Pathogenic | Ambiguous | 0.32 | Likely Benign | 0.2 | 2.03 | Destabilizing | 1.18 | Ambiguous | 0.58 | Ambiguous | 0.767 | Likely Pathogenic | -2.58 | Deleterious | 0.956 | Probably Damaging | 0.931 | Probably Damaging | 5.79 | Benign | 0.01 | Affected | 0.1077 | 0.6284 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.634T>C | S212P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S212P is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include FoldX and FATHMM, whereas the remaining tools—REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict it to be pathogenic. High‑accuracy methods give consistent pathogenic results: AlphaMissense‑Optimized scores it as pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenicity; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a destabilizing, pathogenic effect. With the overwhelming majority of tools supporting pathogenicity and no ClinVar entry to contradict, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.381517 | Uncertain | 0.846 | 0.278 | 0.125 | -13.051 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.02 | Likely Benign | 0.9 | 6.30 | Destabilizing | 3.16 | Destabilizing | 0.90 | Ambiguous | 0.809 | Likely Pathogenic | -4.13 | Deleterious | 0.995 | Probably Damaging | 0.979 | Probably Damaging | 5.75 | Benign | 0.01 | Affected | 0.1859 | 0.5679 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.634T>G | S212A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S212A has no ClinVar record and is not listed in gnomAD. Prediction tools cluster into two groups: benign predictions come from FoldX, Rosetta, Foldetta, PROVEAN, and FATHMM, while pathogenic predictions arise from REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta predicts benign stability. Overall, the majority of conventional tools lean toward pathogenicity, whereas the most reliable high‑accuracy methods are either benign or inconclusive. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.158265 | Structured | 0.381517 | Uncertain | 0.846 | 0.278 | 0.125 | -8.890 | Likely Pathogenic | 0.869 | Likely Pathogenic | Ambiguous | -0.13 | Likely Benign | 0.1 | 0.13 | Likely Benign | 0.00 | Likely Benign | 0.75 | Ambiguous | 0.752 | Likely Pathogenic | -2.48 | Neutral | 0.956 | Probably Damaging | 0.931 | Probably Damaging | 5.83 | Benign | 0.01 | Affected | 0.4639 | 0.4509 | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||
| c.635C>A | S212Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S212Y is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include FoldX, FATHMM, and premPS. In contrast, a majority of tools predict a pathogenic impact: REVEL, SGM‑Consensus, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, is inconclusive for this variant. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; Foldetta remains uncertain. Overall, the preponderance of evidence from multiple in silico predictors indicates that S212Y is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.381517 | Uncertain | 0.846 | 0.278 | 0.125 | -14.812 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.43 | Likely Benign | 0.5 | 2.32 | Destabilizing | 1.38 | Ambiguous | 0.22 | Likely Benign | 0.819 | Likely Pathogenic | -5.15 | Deleterious | 0.995 | Probably Damaging | 0.986 | Probably Damaging | 5.76 | Benign | 0.00 | Affected | 0.0555 | 0.6120 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||
| c.635C>G | S212C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S212C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include FoldX, premPS, and FATHMM, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Predictions that are inconclusive are Rosetta and Foldetta. The high‑accuracy consensus methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence from multiple independent predictors indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.381517 | Uncertain | 0.846 | 0.278 | 0.125 | -11.656 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.16 | Likely Benign | 0.1 | 0.98 | Ambiguous | 0.57 | Ambiguous | 0.47 | Likely Benign | 0.840 | Likely Pathogenic | -4.29 | Deleterious | 0.999 | Probably Damaging | 0.990 | Probably Damaging | 5.73 | Benign | 0.00 | Affected | 0.0785 | 0.6270 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.635C>T | S212F 2D AIThe SynGAP1 missense variant S212F is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include FoldX, FATHMM, and premPS, whereas the majority of other in‑silico predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score (Likely Pathogenic) predict a pathogenic outcome. The remaining tools, Foldetta and Rosetta, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Based on the overall consensus of the majority of predictors and the high‑accuracy tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.381517 | Uncertain | 0.846 | 0.278 | 0.125 | -14.029 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.11 | Likely Benign | 1.6 | 0.88 | Ambiguous | 0.50 | Ambiguous | -0.02 | Likely Benign | 0.820 | Likely Pathogenic | -5.15 | Deleterious | 0.995 | Probably Damaging | 0.986 | Probably Damaging | 5.76 | Benign | 0.00 | Affected | 0.0503 | 0.6259 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||
| c.637A>C | I213L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I213L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM all classify the variant as benign, while AlphaMissense‑Default predicts it as pathogenic and ESM1b remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores the variant benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also reports a benign effect. No prediction or stability result is missing or inconclusive. Overall, the variant is most likely benign based on the collective evidence, and this benign prediction does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.158265 | Structured | 0.372201 | Uncertain | 0.850 | 0.295 | 0.125 | -7.673 | In-Between | 0.577 | Likely Pathogenic | Likely Benign | 0.29 | Likely Benign | 0.4 | 0.20 | Likely Benign | 0.25 | Likely Benign | 0.45 | Likely Benign | 0.489 | Likely Benign | -1.45 | Neutral | 0.447 | Benign | 0.177 | Benign | 5.89 | Benign | 0.18 | Tolerated | 0.0771 | 0.2857 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.637A>G | I213V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I213V missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). No tool predicts a pathogenic outcome. Predictions that are inconclusive are FoldX, Rosetta, Foldetta, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, while Foldetta’s stability analysis is uncertain. Overall, the evidence strongly favors a benign classification, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.158265 | Structured | 0.372201 | Uncertain | 0.850 | 0.295 | 0.125 | -6.133 | Likely Benign | 0.364 | Ambiguous | Likely Benign | 0.67 | Ambiguous | 0.2 | 0.58 | Ambiguous | 0.63 | Ambiguous | 0.47 | Likely Benign | 0.413 | Likely Benign | -0.69 | Neutral | 0.128 | Benign | 0.048 | Benign | 5.82 | Benign | 0.10 | Tolerated | 0.1008 | 0.2959 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||
| c.637A>T | I213F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I213F missense variant is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, while the remaining evaluated algorithms (SGM‑Consensus, REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact; FoldX and premPS are uncertain and are treated as unavailable evidence. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain and thus not considered. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which simply lacks an entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.372201 | Uncertain | 0.850 | 0.295 | 0.125 | -12.212 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.96 | Ambiguous | 0.7 | 2.66 | Destabilizing | 1.81 | Ambiguous | 0.73 | Ambiguous | 0.815 | Likely Pathogenic | -3.30 | Deleterious | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 5.79 | Benign | 0.01 | Affected | 0.0473 | 0.2560 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.632G>A | S211N 2D AIThe SynGAP1 S211N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and FATHMM, whereas those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and premPS. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments are likewise inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta is uncertain. Consequently, the overall prediction leans toward pathogenicity, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.209395 | Structured | 0.389893 | Uncertain | 0.846 | 0.300 | 0.125 | -9.995 | Likely Pathogenic | 0.794 | Likely Pathogenic | Ambiguous | 0.60 | Ambiguous | 1.2 | 1.39 | Ambiguous | 1.00 | Ambiguous | 1.21 | Destabilizing | 0.174 | Likely Benign | -2.22 | Neutral | 0.982 | Probably Damaging | 0.747 | Possibly Damaging | 3.95 | Benign | 0.09 | Tolerated | 0.1534 | 0.4863 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||
| c.631A>T | S211C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S211C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, FATHMM, AlphaMissense‑Optimized, and premPS, whereas a separate group predicts pathogenicity: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Two tools (Rosetta and AlphaMissense‑Default) return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Because the consensus of the most reliable predictors is split (two benign, one pathogenic) and the overall tool distribution is evenly divided, the variant’s impact remains ambiguous. Based on the available predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.209395 | Structured | 0.389893 | Uncertain | 0.846 | 0.300 | 0.125 | -10.567 | Likely Pathogenic | 0.547 | Ambiguous | Likely Benign | 0.28 | Likely Benign | 0.1 | 0.63 | Ambiguous | 0.46 | Likely Benign | 0.11 | Likely Benign | 0.263 | Likely Benign | -4.14 | Deleterious | 0.999 | Probably Damaging | 0.908 | Possibly Damaging | 3.89 | Benign | 0.01 | Affected | 0.1292 | 0.5638 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||
| c.626T>G | V209G 2D AIThe SynGAP1 missense variant V209G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the remaining tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a pathogenic classification; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a destabilizing, pathogenic effect. Taken together, the overwhelming majority of computational evidence indicates that V209G is likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.247041 | Structured | 0.397624 | Uncertain | 0.874 | 0.331 | 0.125 | -13.763 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 3.05 | Destabilizing | 0.5 | 3.99 | Destabilizing | 3.52 | Destabilizing | 1.27 | Destabilizing | 0.390 | Likely Benign | -5.49 | Deleterious | 0.829 | Possibly Damaging | 0.995 | Probably Damaging | 3.65 | Benign | 0.04 | Affected | 0.1600 | 0.2035 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.628C>A | H210N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H210N missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, and FATHMM, while pathogenic predictions are made by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus score (Likely Pathogenic). Two tools give uncertain results: Foldetta (combining FoldX‑MD and Rosetta outputs) and Rosetta alone. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as Likely Pathogenic, and Foldetta as inconclusive. Overall, the majority of evidence points toward a pathogenic effect for H210N. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.144935 | Structured | 0.390904 | Uncertain | 0.872 | 0.298 | 0.125 | -13.699 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.19 | Likely Benign | 0.3 | 1.24 | Ambiguous | 0.72 | Ambiguous | 1.12 | Destabilizing | 0.375 | Likely Benign | -6.01 | Deleterious | 0.895 | Possibly Damaging | 0.533 | Possibly Damaging | 3.11 | Benign | 0.00 | Affected | 0.1166 | 0.1839 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||
| c.628C>G | H210D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H210D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, and FATHMM. In contrast, the majority of tools predict a pathogenic impact: premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain, and Rosetta alone is also uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for H210D, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.144935 | Structured | 0.390904 | Uncertain | 0.872 | 0.298 | 0.125 | -16.440 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.13 | Likely Benign | 0.4 | 1.23 | Ambiguous | 0.68 | Ambiguous | 1.23 | Destabilizing | 0.489 | Likely Benign | -7.73 | Deleterious | 0.895 | Possibly Damaging | 0.533 | Possibly Damaging | 3.18 | Benign | 0.00 | Affected | 0.2025 | 0.1255 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||
| c.628C>T | H210Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H210Y missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; the SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as Benign. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar classification because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.144935 | Structured | 0.390904 | Uncertain | 0.872 | 0.298 | 0.125 | -12.828 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.27 | Likely Benign | 0.8 | 0.38 | Likely Benign | 0.33 | Likely Benign | 0.39 | Likely Benign | 0.427 | Likely Benign | -5.12 | Deleterious | 0.963 | Probably Damaging | 0.628 | Possibly Damaging | 3.07 | Benign | 0.00 | Affected | 0.0497 | 0.3662 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||
| c.629A>C | H210P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H210P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM; all other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.144935 | Structured | 0.390904 | Uncertain | 0.872 | 0.298 | 0.125 | -14.634 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 2.26 | Destabilizing | 0.5 | 2.80 | Destabilizing | 2.53 | Destabilizing | 1.00 | Destabilizing | 0.512 | Likely Pathogenic | -8.55 | Deleterious | 0.989 | Probably Damaging | 0.795 | Possibly Damaging | 3.09 | Benign | 0.00 | Affected | 0.1805 | 0.3175 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||
| c.629A>G | H210R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H210R missense variant is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FATHMM, and FoldX, whereas a majority of tools predict a pathogenic impact: premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). The high‑accuracy AlphaMissense‑Optimized result is pathogenic; the SGM Consensus, based on the same set of predictors, is also pathogenic; Foldetta’s stability assessment is uncertain and therefore not considered evidence. Overall, the balance of evidence points to a pathogenic effect for H210R. This prediction does not contradict the ClinVar “Uncertain” classification, which remains unresolved. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.144935 | Structured | 0.390904 | Uncertain | 0.872 | 0.298 | 0.125 | Uncertain | 1 | -14.254 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.40 | Likely Benign | 0.4 | 3.05 | Destabilizing | 1.73 | Ambiguous | 1.12 | Destabilizing | 0.431 | Likely Benign | -6.74 | Deleterious | 0.808 | Possibly Damaging | 0.452 | Possibly Damaging | 3.09 | Benign | 0.00 | Affected | 0.1427 | 0.1982 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||
| c.629A>T | H210L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H210L missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools cluster into two groups: benign predictions come from REVEL, Foldetta, premPS, and FATHMM, while pathogenic predictions arise from SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX and Rosetta provide uncertain results. High‑accuracy assessments further highlight the discrepancy: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, whereas Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.144935 | Structured | 0.390904 | Uncertain | 0.872 | 0.298 | 0.125 | -14.516 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | -0.71 | Ambiguous | 0.1 | 1.35 | Ambiguous | 0.32 | Likely Benign | 0.49 | Likely Benign | 0.421 | Likely Benign | -9.41 | Deleterious | 0.895 | Possibly Damaging | 0.614 | Possibly Damaging | 3.09 | Benign | 0.00 | Affected | 0.0617 | 0.4452 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.62T>C | F21S 2D AIThe SynGAP1 missense variant F21S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.443284 | Uncertain | 0.369 | 0.897 | 0.500 | -2.396 | Likely Benign | 0.745 | Likely Pathogenic | Likely Benign | 0.182 | Likely Benign | -0.22 | Neutral | 0.462 | Possibly Damaging | 0.307 | Benign | 4.15 | Benign | 0.00 | Affected | 0.4793 | 0.1133 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||||||||||||
| c.62T>G | F21C 2D AIThe SynGAP1 missense variant F21C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for F21C. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.443284 | Uncertain | 0.369 | 0.897 | 0.500 | -3.698 | Likely Benign | 0.686 | Likely Pathogenic | Likely Benign | 0.151 | Likely Benign | -0.31 | Neutral | 0.880 | Possibly Damaging | 0.759 | Possibly Damaging | 4.12 | Benign | 0.00 | Affected | 0.2873 | 0.2171 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||||||||||||
| c.630C>A | H210Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant H210Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign calls come from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar) and FATHMM, whereas pathogenic calls are made by premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM and PROVEAN, favors a pathogenic outcome (3/4). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote) also predicts pathogenic. Foldetta, a protein‑folding stability predictor that integrates FoldX‑MD and Rosetta, is inconclusive and therefore not used as evidence. Overall, the majority of reliable predictors indicate a pathogenic effect for H210Q, and this conclusion does not conflict with any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.144935 | Structured | 0.390904 | Uncertain | 0.872 | 0.298 | 0.125 | -12.639 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.26 | Likely Benign | 0.3 | 1.96 | Ambiguous | 1.11 | Ambiguous | 1.20 | Destabilizing | 0.258 | Likely Benign | -6.84 | Deleterious | 0.141 | Benign | 0.064 | Benign | 3.10 | Benign | 0.00 | Affected | 0.1016 | 0.2852 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.630C>G | H210Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H210Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar) and FATHMM, whereas pathogenic calls come from premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default and AlphaMissense‑Optimized. When predictions are grouped, five tools favor a benign effect and six favor a pathogenic effect. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates pathogenic, and the Foldetta stability analysis is inconclusive. No evidence from ClinVar contradicts these findings. Therefore, the variant is most likely pathogenic based on the aggregate computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.144935 | Structured | 0.390904 | Uncertain | 0.872 | 0.298 | 0.125 | -12.639 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.26 | Likely Benign | 0.3 | 1.96 | Ambiguous | 1.11 | Ambiguous | 1.20 | Destabilizing | 0.258 | Likely Benign | -6.84 | Deleterious | 0.141 | Benign | 0.064 | Benign | 3.10 | Benign | 0.00 | Affected | 0.1016 | 0.2852 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.631A>C | S211R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S211R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, and FATHMM, whereas a pathogenic signal is reported by SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as benign. Overall, the majority of tools predict a pathogenic effect, and the high‑accuracy predictions reinforce this conclusion. The variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.209395 | Structured | 0.389893 | Uncertain | 0.846 | 0.300 | 0.125 | -14.126 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.49 | Likely Benign | 1.0 | 0.20 | Likely Benign | 0.35 | Likely Benign | 0.97 | Ambiguous | 0.132 | Likely Benign | -4.00 | Deleterious | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 3.95 | Benign | 0.02 | Affected | 0.1003 | 0.3592 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.631A>G | S211G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S211G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are premPS, PROVEAN, polyPhen‑2 HumDiv, and ESM1b; Rosetta is uncertain. High‑accuracy methods give a benign result for AlphaMissense‑Optimized and for Foldetta. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs two pathogenic). No evidence from ClinVar contradicts these predictions. Overall, the balance of evidence—including the unanimous benign calls from the high‑accuracy tools—suggests that the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.209395 | Structured | 0.389893 | Uncertain | 0.846 | 0.300 | 0.125 | -9.895 | Likely Pathogenic | 0.265 | Likely Benign | Likely Benign | 0.30 | Likely Benign | 0.2 | 0.54 | Ambiguous | 0.42 | Likely Benign | 1.07 | Destabilizing | 0.067 | Likely Benign | -3.17 | Deleterious | 0.787 | Possibly Damaging | 0.404 | Benign | 3.92 | Benign | 0.07 | Tolerated | 0.2560 | 0.4337 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||
| c.638T>A | I213N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I213N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining evaluated algorithms—SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact; FoldX is uncertain and is treated as unavailable. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.372201 | Uncertain | 0.850 | 0.295 | 0.125 | -15.069 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.71 | Ambiguous | 0.8 | 2.81 | Destabilizing | 2.26 | Destabilizing | 1.85 | Destabilizing | 0.862 | Likely Pathogenic | -5.81 | Deleterious | 0.995 | Probably Damaging | 0.880 | Possibly Damaging | 5.83 | Benign | 0.00 | Affected | 0.0750 | 0.0412 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.638T>C | I213T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I213T missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while the remaining evidence—SGM‑Consensus (Likely Pathogenic), REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently indicates pathogenicity. FoldX, Rosetta, and Foldetta provide uncertain results. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence supports a pathogenic classification for I213T, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.372201 | Uncertain | 0.850 | 0.295 | 0.125 | -11.080 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.46 | Ambiguous | 0.8 | 1.32 | Ambiguous | 1.39 | Ambiguous | 1.49 | Destabilizing | 0.882 | Likely Pathogenic | -3.99 | Deleterious | 0.948 | Possibly Damaging | 0.588 | Possibly Damaging | 5.82 | Benign | 0.00 | Affected | 0.0905 | 0.0708 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.638T>G | I213S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I213S missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while all other evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) classify the change as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.372201 | Uncertain | 0.850 | 0.295 | 0.125 | -12.858 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.00 | Destabilizing | 1.1 | 2.95 | Destabilizing | 2.48 | Destabilizing | 1.53 | Destabilizing | 0.879 | Likely Pathogenic | -4.88 | Deleterious | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 5.83 | Benign | 0.00 | Affected | 0.2290 | 0.0800 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.647A>C | Q216P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q216P is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to FATHMM, which scores the variant as benign. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic or likely pathogenic impact. The high‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; SGM‑Consensus indicates likely pathogenic; Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. premPS is inconclusive and is treated as unavailable. Based on the collective predictions, the variant is most likely pathogenic, and this assessment is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.206376 | Structured | 0.396100 | Uncertain | 0.804 | 0.274 | 0.000 | -13.128 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 4.35 | Destabilizing | 0.8 | 4.05 | Destabilizing | 4.20 | Destabilizing | 0.53 | Ambiguous | 0.830 | Likely Pathogenic | -4.69 | Deleterious | 0.989 | Probably Damaging | 0.737 | Possibly Damaging | 5.84 | Benign | 0.01 | Affected | 0.2603 | 0.5736 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||
| c.647A>G | Q216R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q216R is not reported in ClinVar and has no gnomAD entry. Prediction tools that call the variant benign include FoldX, Rosetta, Foldetta, premPS, SIFT, and FATHMM, while pathogenic calls come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta as Benign. Overall, the predictions are split, with a slight edge toward pathogenicity. The variant is therefore most likely pathogenic based on the current computational evidence, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.206376 | Structured | 0.396100 | Uncertain | 0.804 | 0.274 | 0.000 | -10.149 | Likely Pathogenic | 0.821 | Likely Pathogenic | Ambiguous | -0.42 | Likely Benign | 0.0 | 0.09 | Likely Benign | -0.17 | Likely Benign | -0.01 | Likely Benign | 0.513 | Likely Pathogenic | -2.70 | Deleterious | 0.963 | Probably Damaging | 0.549 | Possibly Damaging | 6.00 | Benign | 0.19 | Tolerated | 0.1972 | 0.2748 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||
| c.647A>T | Q216L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q216L is not reported in ClinVar (ClinVar ID: None) and has no entry in gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, and FATHMM, while those that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain (treated as unavailable), SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta predicts benign. Overall, the majority of tools (8 pathogenic vs. 5 benign) and the consensus high‑accuracy prediction lean toward pathogenicity. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.206376 | Structured | 0.396100 | Uncertain | 0.804 | 0.274 | 0.000 | -11.303 | Likely Pathogenic | 0.886 | Likely Pathogenic | Ambiguous | -0.17 | Likely Benign | 0.3 | 0.28 | Likely Benign | 0.06 | Likely Benign | 0.30 | Likely Benign | 0.797 | Likely Pathogenic | -5.58 | Deleterious | 0.963 | Probably Damaging | 0.452 | Possibly Damaging | 5.87 | Benign | 0.01 | Affected | 0.1036 | 0.6410 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||
| c.648G>C | Q216H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q216H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, and FATHMM, whereas those that predict a pathogenic impact are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the balance of evidence favors a pathogenic classification; this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.206376 | Structured | 0.396100 | Uncertain | 0.804 | 0.274 | 0.000 | -10.532 | Likely Pathogenic | 0.951 | Likely Pathogenic | Ambiguous | 0.29 | Likely Benign | 0.0 | 0.30 | Likely Benign | 0.30 | Likely Benign | 0.50 | Likely Benign | 0.638 | Likely Pathogenic | -3.84 | Deleterious | 0.989 | Probably Damaging | 0.809 | Possibly Damaging | 5.85 | Benign | 0.01 | Affected | 0.1909 | 0.4660 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.648G>T | Q216H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q216H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, and FATHMM, whereas a larger group predicts pathogenicity: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.206376 | Structured | 0.396100 | Uncertain | 0.804 | 0.274 | 0.000 | -10.532 | Likely Pathogenic | 0.951 | Likely Pathogenic | Ambiguous | 0.29 | Likely Benign | 0.0 | 0.30 | Likely Benign | 0.30 | Likely Benign | 0.50 | Likely Benign | 0.638 | Likely Pathogenic | -3.84 | Deleterious | 0.989 | Probably Damaging | 0.809 | Possibly Damaging | 5.85 | Benign | 0.01 | Affected | 0.1909 | 0.4660 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.649G>A | E217K 2D 3DClick to see structure in 3D Viewer AISynGAP1 E217K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Computational predictions cluster into two groups: benign calls from premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, and FATHMM; pathogenic calls from REVEL, Rosetta, polyPhen‑2 HumDiv, AlphaMissense‑Default, and AlphaMissense‑Optimized. Three tools give uncertain results: FoldX, Foldetta, and ESM1b. High‑accuracy methods give conflicting outcomes: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields benign; Foldetta remains uncertain. Because the majority of standard tools are split evenly and the high‑accuracy predictions are discordant, the evidence does not decisively support either outcome. The variant is therefore most likely pathogenic based on the preponderance of pathogenic predictions, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.278302 | Structured | 0.404912 | Uncertain | 0.823 | 0.284 | 0.000 | -7.169 | In-Between | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.52 | Ambiguous | 0.5 | 2.14 | Destabilizing | 1.33 | Ambiguous | 0.45 | Likely Benign | 0.563 | Likely Pathogenic | -2.38 | Neutral | 0.900 | Possibly Damaging | 0.307 | Benign | 5.95 | Benign | 0.13 | Tolerated | 0.2742 | 0.8216 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||
| c.650A>C | E217A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E217A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, polyPhen‑2 HumVar, SIFT, and FATHMM, whereas those that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, AlphaMissense‑Default, and AlphaMissense‑Optimized. Four tools (FoldX, Rosetta, Foldetta, and ESM1b) returned uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; Foldetta’s stability prediction is uncertain. Overall, the majority of available predictions support a pathogenic impact for E217A. This conclusion is not contradicted by ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.278302 | Structured | 0.404912 | Uncertain | 0.823 | 0.284 | 0.000 | -7.294 | In-Between | 0.960 | Likely Pathogenic | Likely Pathogenic | 0.67 | Ambiguous | 0.5 | 0.61 | Ambiguous | 0.64 | Ambiguous | 0.50 | Likely Benign | 0.619 | Likely Pathogenic | -3.88 | Deleterious | 0.900 | Possibly Damaging | 0.307 | Benign | 5.81 | Benign | 0.12 | Tolerated | 0.4442 | 0.8090 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||
| c.650A>G | E217G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E217G missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumVar, SIFT, and FATHMM, while those that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). Predictions that are uncertain or inconclusive are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized predicts a pathogenic impact, the SGM‑Consensus also indicates a likely pathogenic outcome, and Foldetta’s stability analysis is inconclusive. Overall, the majority of evidence points to a pathogenic effect for E217G, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.278302 | Structured | 0.404912 | Uncertain | 0.823 | 0.284 | 0.000 | -8.076 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 1.13 | Ambiguous | 0.4 | 1.87 | Ambiguous | 1.50 | Ambiguous | 0.59 | Ambiguous | 0.684 | Likely Pathogenic | -3.93 | Deleterious | 0.816 | Possibly Damaging | 0.307 | Benign | 5.82 | Benign | 0.06 | Tolerated | 0.3278 | 0.6941 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.650A>T | E217V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E217V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include Rosetta, FATHMM, and premPS, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). FoldX and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E217V. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.278302 | Structured | 0.404912 | Uncertain | 0.823 | 0.284 | 0.000 | -10.194 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.76 | Ambiguous | 0.6 | 0.39 | Likely Benign | 0.58 | Ambiguous | 0.23 | Likely Benign | 0.723 | Likely Pathogenic | -4.84 | Deleterious | 0.900 | Possibly Damaging | 0.461 | Possibly Damaging | 5.79 | Benign | 0.03 | Affected | 0.0919 | 0.8340 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.651G>C | E217D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E217D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the change as benign. The only inconclusive result comes from AlphaMissense‑Default, which is listed as uncertain. High‑accuracy assessments corroborate this benign profile: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a benign outcome. No pathogenic predictions are present. Therefore, based on the available computational evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.278302 | Structured | 0.404912 | Uncertain | 0.823 | 0.284 | 0.000 | -2.268 | Likely Benign | 0.453 | Ambiguous | Likely Benign | 0.24 | Likely Benign | 0.3 | -0.07 | Likely Benign | 0.09 | Likely Benign | -0.34 | Likely Benign | 0.276 | Likely Benign | 0.66 | Neutral | 0.014 | Benign | 0.007 | Benign | 5.85 | Benign | 0.85 | Tolerated | 3.41 | 13 | 0.2066 | 0.5976 | 2 | 3 | 0.0 | -14.03 | |||||||||||||||||||||||||||
| c.652T>C | F218L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F218L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict a pathogenic effect comprise REVEL, Rosetta, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). FoldX and Foldetta provide uncertain results and are therefore considered unavailable for interpretation. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact for F218L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.281712 | Structured | 0.408725 | Uncertain | 0.848 | 0.272 | 0.000 | -8.861 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.96 | Ambiguous | 0.4 | 2.36 | Destabilizing | 1.66 | Ambiguous | 1.08 | Destabilizing | 0.546 | Likely Pathogenic | -3.80 | Deleterious | 0.158 | Benign | 0.025 | Benign | 5.90 | Benign | 0.15 | Tolerated | 0.2331 | 0.3241 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.652T>G | F218V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F218V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; pathogenic predictions arise from REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled “Likely Pathogenic.” High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus agrees on a likely pathogenic outcome, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates pathogenicity. No predictions or stability results are missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion aligns with the absence of a ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.281712 | Structured | 0.408725 | Uncertain | 0.848 | 0.272 | 0.000 | -10.081 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 4.18 | Destabilizing | 0.2 | 6.35 | Destabilizing | 5.27 | Destabilizing | 1.15 | Destabilizing | 0.691 | Likely Pathogenic | -4.14 | Deleterious | 0.300 | Benign | 0.066 | Benign | 5.81 | Benign | 0.08 | Tolerated | 0.2173 | 0.2258 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.653T>A | F218Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F218Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). All available in silico predictors classify the substitution as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign effect. The high‑accuracy folding‑stability tool Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a benign impact. No tool predicts pathogenicity, and no contradictory evidence exists in ClinVar. **Based on the consensus of all predictions, the variant is most likely benign, and this assessment does not conflict with ClinVar status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.281712 | Structured | 0.408725 | Uncertain | 0.848 | 0.272 | 0.000 | -3.596 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.26 | Likely Benign | 0.1 | -0.40 | Likely Benign | -0.07 | Likely Benign | -0.44 | Likely Benign | 0.229 | Likely Benign | 0.63 | Neutral | 0.001 | Benign | 0.002 | Benign | 5.95 | Benign | 0.52 | Tolerated | 0.1532 | 0.2725 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.646C>G | Q216E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q216E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as benign. No predictions or stability results are missing or inconclusive. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.206376 | Structured | 0.396100 | Uncertain | 0.804 | 0.274 | 0.000 | -9.215 | Likely Pathogenic | 0.550 | Ambiguous | Likely Benign | 0.49 | Likely Benign | 0.3 | 0.30 | Likely Benign | 0.40 | Likely Benign | 0.35 | Likely Benign | 0.474 | Likely Benign | -1.89 | Neutral | 0.779 | Possibly Damaging | 0.351 | Benign | 5.85 | Benign | 0.13 | Tolerated | 0.1872 | 0.3117 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.646C>A | Q216K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q216K is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or folding stability result is missing or inconclusive. Overall, the majority of tools lean toward a benign classification, but the presence of several pathogenic predictions and a high‑accuracy consensus that is pathogenic introduces uncertainty. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.206376 | Structured | 0.396100 | Uncertain | 0.804 | 0.274 | 0.000 | -10.908 | Likely Pathogenic | 0.826 | Likely Pathogenic | Ambiguous | -0.35 | Likely Benign | 0.1 | 0.38 | Likely Benign | 0.02 | Likely Benign | 0.17 | Likely Benign | 0.617 | Likely Pathogenic | -2.71 | Deleterious | 0.779 | Possibly Damaging | 0.351 | Benign | 5.92 | Benign | 0.12 | Tolerated | 0.2431 | 0.4591 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||
| c.639C>G | I213M 2D AIThe SynGAP1 missense variant I213M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are PROVEAN and FATHMM, while a larger group—REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) return uncertain or inconclusive results and are therefore treated as unavailable. High‑accuracy assessments are likewise inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a tie, and Foldetta is uncertain. Overall, the majority of available predictions lean toward pathogenicity, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.158265 | Structured | 0.372201 | Uncertain | 0.850 | 0.295 | 0.125 | -10.777 | Likely Pathogenic | 0.906 | Likely Pathogenic | Ambiguous | 0.66 | Ambiguous | 0.5 | 1.58 | Ambiguous | 1.12 | Ambiguous | 0.85 | Ambiguous | 0.680 | Likely Pathogenic | -2.31 | Neutral | 0.995 | Probably Damaging | 0.880 | Possibly Damaging | 5.85 | Benign | 0.01 | Affected | 0.0611 | 0.2524 | 2 | 1 | -2.6 | 18.03 | ||||||||||||||||||||||||||||||
| c.63C>G | F21L 2D AIThe SynGAP1 missense variant F21L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. The high‑accuracy consensus (SGM‑Consensus) classifies the variant as Likely Benign, while AlphaMissense‑Optimized remains uncertain and Foldetta is unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict the ClinVar status, which is currently unreported. Thus, based on the available computational predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.443284 | Uncertain | 0.369 | 0.897 | 0.500 | -2.480 | Likely Benign | 0.940 | Likely Pathogenic | Ambiguous | 0.104 | Likely Benign | 0.15 | Neutral | 0.140 | Benign | 0.153 | Benign | 4.34 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2680 | 0.3250 | 0 | 2 | 1.0 | -34.02 | |||||||||||||||||||||||||||||||||||||
Found 8751 rows. Show 800 rows per page. Page 3/11 « Previous | Next »