
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain and Structure information: based on WT protein | Annotated databases | Deep learning-based pathogenicity predictions | Folding stability-based pathogenicity predictions | Sequence/structure-based pathogenicity predictions | Phase Separation | Evolutionary/physical properties | Molecular Dynamics-based analysis | DOI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | IUPred2 | ANCHOR2 | AlphaFold | MobiDB | PhosphoSitePlus | ClinVar | gnomAD | ESM1b | AlphaMissense | FoldX | Rosetta | Foldetta | PremPS | REVEL | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PSMutPred | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | |||||||||||||||||||||||||||||||||||||||||||||
| Score | Prediction | Score | Prediction | pLDDT | disorder | disorder | LTP | HTP | KL | PTM | Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | IP RF | SP RF | Prediction | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.1000A>C | K334Q 2D 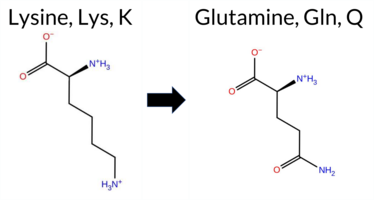 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K334Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, Foldetta, and premPS. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. With seven tools favoring pathogenicity versus five favoring benign, the overall prediction leans toward pathogenic. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.377384 | Structured | 0.325972 | Uncertain | 0.544 | 0.414 | 0.500 | -8.185 | Likely Pathogenic | 0.810 | Likely Pathogenic | Ambiguous | 0.08 | Likely Benign | 0.0 | 0.10 | Likely Benign | 0.09 | Likely Benign | 0.46 | Likely Benign | 0.357 | Likely Benign | -3.67 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.74 | Pathogenic | 0.03 | Affected | 0.4519 | 0.1062 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1000A>G | K334E 2D 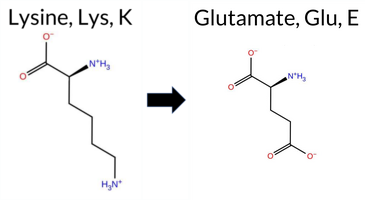 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K334E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and premPS. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.377384 | Structured | 0.325972 | Uncertain | 0.544 | 0.414 | 0.500 | -12.770 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.30 | Likely Benign | 0.2 | -0.07 | Likely Benign | 0.12 | Likely Benign | 0.42 | Likely Benign | 0.372 | Likely Benign | -3.67 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.74 | Pathogenic | 0.02 | Affected | 0.3929 | 0.0882 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1001A>C | K334T 2D 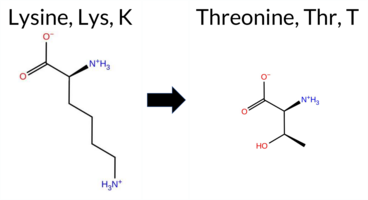 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K334T is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, and premPS. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive or uncertain are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts a likely pathogenic outcome, AlphaMissense‑Optimized is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also uncertain. Overall, the majority of evidence points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.377384 | Structured | 0.325972 | Uncertain | 0.544 | 0.414 | 0.500 | -8.313 | Likely Pathogenic | 0.943 | Likely Pathogenic | Ambiguous | 0.78 | Ambiguous | 0.3 | 0.21 | Likely Benign | 0.50 | Ambiguous | 0.17 | Likely Benign | 0.320 | Likely Benign | -5.51 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.78 | Pathogenic | 0.02 | Affected | 0.2062 | 0.2978 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1001A>G | K334R 2D 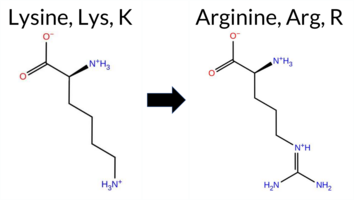 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K334R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.377384 | Structured | 0.325972 | Uncertain | 0.544 | 0.414 | 0.500 | -5.384 | Likely Benign | 0.162 | Likely Benign | Likely Benign | -0.37 | Likely Benign | 0.1 | -0.53 | Ambiguous | -0.45 | Likely Benign | 0.39 | Likely Benign | 0.247 | Likely Benign | -2.62 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.76 | Pathogenic | 0.07 | Tolerated | 0.4647 | 0.0976 | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||
| c.1001A>T | K334M 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K334M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, FoldX, and premPS. In contrast, the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as deleterious. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. Foldetta and Rosetta provide uncertain results. Focusing on high‑accuracy methods, AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus confirms a Likely Pathogenic status, and Foldetta remains inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for K334M, and this assessment does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.377384 | Structured | 0.325972 | Uncertain | 0.544 | 0.414 | 0.500 | -10.530 | Likely Pathogenic | 0.973 | Likely Pathogenic | Likely Pathogenic | 0.44 | Likely Benign | 0.0 | 0.56 | Ambiguous | 0.50 | Ambiguous | 0.14 | Likely Benign | 0.323 | Likely Benign | -5.51 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.77 | Pathogenic | 0.01 | Affected | 0.1027 | 0.3690 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1002G>C | K334N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K334N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and premPS. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic.” Separately, the high‑accuracy AlphaMissense‑Optimized tool predicts pathogenicity, the SGM‑Consensus also predicts pathogenicity, while the Foldetta stability assessment predicts a benign effect. Overall, the majority of predictions (8 pathogenic vs. 5 benign) and the high‑accuracy consensus suggest that K334N is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.377384 | Structured | 0.325972 | Uncertain | 0.544 | 0.414 | 0.500 | -9.581 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.15 | Likely Benign | 0.1 | -0.25 | Likely Benign | -0.05 | Likely Benign | 0.18 | Likely Benign | 0.249 | Likely Benign | -4.59 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.77 | Pathogenic | 0.02 | Affected | 0.3716 | 0.0958 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1002G>T | K334N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K334N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and premPS. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic.” Separately, the high‑accuracy AlphaMissense‑Optimized tool predicts pathogenicity, the SGM‑Consensus also predicts pathogenicity, while the Foldetta stability assessment predicts a benign effect. Overall, the majority of predictions (8 pathogenic vs. 5 benign) and the high‑accuracy consensus suggest that K334N is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.377384 | Structured | 0.325972 | Uncertain | 0.544 | 0.414 | 0.500 | -9.581 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.15 | Likely Benign | 0.1 | -0.25 | Likely Benign | -0.05 | Likely Benign | 0.18 | Likely Benign | 0.249 | Likely Benign | -4.59 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.77 | Pathogenic | 0.02 | Affected | 0.3716 | 0.0958 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1003C>A | R335S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R335S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, premPS, and SIFT; pathogenic predictions from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. No evidence from FoldX or Rosetta alone is available. Based on the preponderance of pathogenic predictions and the high‑accuracy tools, the variant is most likely pathogenic, which is consistent with the absence of ClinVar reporting and gnomAD data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.305330 | Structured | 0.331028 | Uncertain | 0.483 | 0.428 | 0.500 | -9.286 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.68 | Ambiguous | 0.1 | 0.72 | Ambiguous | 0.70 | Ambiguous | 0.14 | Likely Benign | 0.184 | Likely Benign | -3.30 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.89 | Pathogenic | 0.11 | Tolerated | 0.2598 | 0.4005 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||
| c.1003C>G | R335G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R335G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, and SIFT, whereas those that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, more tools predict pathogenicity than benignity, and the high‑accuracy consensus also leans pathogenic. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.305330 | Structured | 0.331028 | Uncertain | 0.483 | 0.428 | 0.500 | -11.860 | Likely Pathogenic | 0.880 | Likely Pathogenic | Ambiguous | 1.01 | Ambiguous | 0.1 | 0.44 | Likely Benign | 0.73 | Ambiguous | 0.20 | Likely Benign | 0.194 | Likely Benign | -4.77 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.01 | Pathogenic | 0.10 | Tolerated | 0.3000 | 0.3554 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1004G>C | R335P 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant R335P has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, and SIFT, whereas pathogenic predictions are reported by FoldX, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments give a more focused view: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a pathogenic effect. Taken together, the majority of evidence points to a pathogenic impact for R335P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.305330 | Structured | 0.331028 | Uncertain | 0.483 | 0.428 | 0.500 | -13.952 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 3.23 | Destabilizing | 0.8 | 5.73 | Destabilizing | 4.48 | Destabilizing | 0.43 | Likely Benign | 0.250 | Likely Benign | -4.22 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.69 | Pathogenic | 0.09 | Tolerated | 0.1866 | 0.4497 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.1004G>T | R335L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R335L is not listed in ClinVar (ClinVar ID None) and has no reported allele in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, and premPS. Tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized and FoldX are uncertain and are treated as unavailable for pathogenicity inference. High‑accuracy assessments: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign. Overall, the majority of predictions (8 pathogenic vs. 4 benign) indicate that the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as no ClinVar assertion is present. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.305330 | Structured | 0.331028 | Uncertain | 0.483 | 0.428 | 0.500 | -13.226 | Likely Pathogenic | 0.938 | Likely Pathogenic | Ambiguous | 0.51 | Ambiguous | 0.0 | -0.19 | Likely Benign | 0.16 | Likely Benign | 0.40 | Likely Benign | 0.196 | Likely Benign | -4.77 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.73 | Pathogenic | 0.04 | Affected | 0.1382 | 0.4753 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.1006A>C | K336Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 K336Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as benign. Because the predictions are evenly split and the high‑accuracy methods give conflicting results, the variant is best classified as of uncertain significance. This assessment does not contradict any ClinVar annotation, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.318242 | Structured | 0.338219 | Uncertain | 0.396 | 0.428 | 0.500 | -12.876 | Likely Pathogenic | 0.829 | Likely Pathogenic | Ambiguous | 0.02 | Likely Benign | 0.0 | -0.17 | Likely Benign | -0.08 | Likely Benign | 0.18 | Likely Benign | 0.211 | Likely Benign | -3.30 | Deleterious | 0.801 | Possibly Damaging | 0.252 | Benign | 1.58 | Pathogenic | 0.02 | Affected | 0.4698 | 0.1514 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1006A>G | K336E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K336E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points toward a pathogenic impact for K336E, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.318242 | Structured | 0.338219 | Uncertain | 0.396 | 0.428 | 0.500 | -16.091 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | -0.28 | Likely Benign | 0.0 | 0.19 | Likely Benign | -0.05 | Likely Benign | 0.35 | Likely Benign | 0.236 | Likely Benign | -3.43 | Deleterious | 0.625 | Possibly Damaging | 0.192 | Benign | 1.60 | Pathogenic | 0.01 | Affected | 0.4003 | 0.1082 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1007A>C | K336T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K336T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy assessment shows AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points toward a pathogenic impact for K336T, and this conclusion does not contradict any ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.318242 | Structured | 0.338219 | Uncertain | 0.396 | 0.428 | 0.500 | -13.468 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.33 | Likely Benign | 0.1 | -0.08 | Likely Benign | 0.13 | Likely Benign | 0.15 | Likely Benign | 0.212 | Likely Benign | -4.94 | Deleterious | 0.891 | Possibly Damaging | 0.315 | Benign | 1.58 | Pathogenic | 0.01 | Affected | 0.2014 | 0.4048 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1007A>T | K336M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K336M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions (REVEL, FoldX, premPS) and pathogenic predictions (SIFT, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized). The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled “Likely Pathogenic.” Stability‑based assessments are inconclusive: Foldetta is uncertain, and Rosetta is also uncertain. High‑accuracy tools specifically indicate pathogenicity: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus confirms pathogenic, while Foldetta remains uncertain. Based on the overall pattern of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.318242 | Structured | 0.338219 | Uncertain | 0.396 | 0.428 | 0.500 | -15.395 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.34 | Likely Benign | 0.1 | 0.82 | Ambiguous | 0.58 | Ambiguous | -0.23 | Likely Benign | 0.301 | Likely Benign | -5.07 | Deleterious | 0.989 | Probably Damaging | 0.832 | Possibly Damaging | 1.53 | Pathogenic | 0.00 | Affected | 0.1185 | 0.4760 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1008G>C | K336N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K336N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show discordant results: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, and polyPhen‑2 HumVar, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome. High‑accuracy assessments further highlight this split: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus also predicts likely pathogenic, while Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts benign. No prediction or stability result is missing. Overall, the majority of tools and the high‑accuracy methods lean toward pathogenicity, which is consistent with the lack of ClinVar annotation and gnomAD absence. Thus, the variant is most likely pathogenic, and this prediction does not contradict ClinVar status because ClinVar has no entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.318242 | Structured | 0.338219 | Uncertain | 0.396 | 0.428 | 0.500 | -13.307 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.20 | Likely Benign | 0.1 | -0.02 | Likely Benign | 0.09 | Likely Benign | 0.20 | Likely Benign | 0.186 | Likely Benign | -4.09 | Deleterious | 0.801 | Possibly Damaging | 0.315 | Benign | 1.59 | Pathogenic | 0.01 | Affected | 0.3710 | 0.1599 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1008G>T | K336N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K336N is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points toward a pathogenic impact for K336N, and this conclusion does not contradict any ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.318242 | Structured | 0.338219 | Uncertain | 0.396 | 0.428 | 0.500 | -13.307 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.20 | Likely Benign | 0.1 | -0.02 | Likely Benign | 0.09 | Likely Benign | 0.20 | Likely Benign | 0.186 | Likely Benign | -4.09 | Deleterious | 0.801 | Possibly Damaging | 0.315 | Benign | 1.59 | Pathogenic | 0.01 | Affected | 0.3710 | 0.1599 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1009A>C | K337Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K337Q missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, and Foldetta. Those that predict a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as benign. Overall, the majority of predictions (8 pathogenic vs. 4 benign) indicate that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.348540 | Uncertain | 0.449 | 0.438 | 0.500 | -9.944 | Likely Pathogenic | 0.934 | Likely Pathogenic | Ambiguous | 0.00 | Likely Benign | 0.0 | 0.88 | Ambiguous | 0.44 | Likely Benign | 0.43 | Likely Benign | 0.305 | Likely Benign | -3.48 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.70 | Pathogenic | 0.01 | Affected | 0.3672 | 0.1219 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1009A>G | K337E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K337E missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, and Foldetta. Those that predict a pathogenic impact comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). The premPS score is uncertain and does not influence the overall assessment. High‑accuracy analyses show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as benign. Based on the majority of predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.348540 | Uncertain | 0.449 | 0.438 | 0.500 | -13.673 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | -0.05 | Likely Benign | 0.1 | 0.49 | Likely Benign | 0.22 | Likely Benign | 0.52 | Ambiguous | 0.316 | Likely Benign | -3.48 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.76 | Pathogenic | 0.02 | Affected | 0.3102 | 0.1039 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.100T>A | Y34N 2D  AIThe SynGAP1 missense variant Y34N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.435847 | Uncertain | 0.303 | 0.855 | 0.375 | -3.153 | Likely Benign | 0.214 | Likely Benign | Likely Benign | 0.165 | Likely Benign | -1.01 | Neutral | 0.824 | Possibly Damaging | 0.828 | Possibly Damaging | 4.16 | Benign | 0.00 | Affected | 0.2410 | 0.1322 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.100T>C | Y34H 2D  AIThe SynGAP1 missense variant Y34H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Y34H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.435847 | Uncertain | 0.303 | 0.855 | 0.375 | -2.929 | Likely Benign | 0.315 | Likely Benign | Likely Benign | 0.102 | Likely Benign | -0.53 | Neutral | 0.824 | Possibly Damaging | 0.775 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 0.2630 | 0.1062 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||||||||||||
| c.100T>G | Y34D 2D 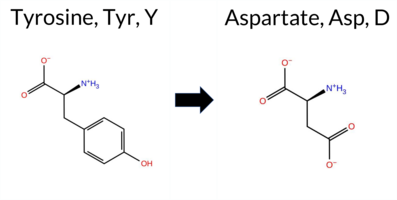 AIThe SynGAP1 missense variant Y34D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized independently predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation; there is no contradictory ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.435847 | Uncertain | 0.303 | 0.855 | 0.375 | -2.653 | Likely Benign | 0.357 | Ambiguous | Likely Benign | 0.199 | Likely Benign | -1.20 | Neutral | 0.824 | Possibly Damaging | 0.828 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 0.4092 | 0.1322 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.1010A>C | K337T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K337T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two consensus groups: benign predictions come from REVEL, FoldX, and premPS, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Three tools report uncertainty: Rosetta, Foldetta, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Pathogenic. In the high‑accuracy subset, AlphaMissense‑Optimized remains uncertain, SGM‑Consensus is Likely Pathogenic, and Foldetta is uncertain. Taken together, the majority of evidence points toward a deleterious effect. Therefore, K337T is most likely pathogenic, and this assessment does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.348540 | Uncertain | 0.449 | 0.438 | 0.500 | -10.896 | Likely Pathogenic | 0.953 | Likely Pathogenic | Ambiguous | 0.45 | Likely Benign | 0.2 | 1.33 | Ambiguous | 0.89 | Ambiguous | 0.25 | Likely Benign | 0.338 | Likely Benign | -5.32 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.70 | Pathogenic | 0.01 | Affected | 0.1741 | 0.3354 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1010A>G | K337R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K337R has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into benign and pathogenic groups: benign predictions come from REVEL, FoldX, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta; pathogenic predictions come from polyPhen2_HumDiv, polyPhen2_HumVar, and FATHMM, while Rosetta remains uncertain. High‑accuracy methods reinforce the benign assessment: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign. Consequently, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.321458 | Structured | 0.348540 | Uncertain | 0.449 | 0.438 | 0.500 | -6.302 | Likely Benign | 0.249 | Likely Benign | Likely Benign | -0.30 | Likely Benign | 0.2 | 1.07 | Ambiguous | 0.39 | Likely Benign | 0.12 | Likely Benign | 0.142 | Likely Benign | -2.36 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.70 | Pathogenic | 0.07 | Tolerated | 0.3913 | 0.1134 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.1010A>T | K337M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K337M missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that classify it as benign include REVEL, FoldX, premPS, and the protein‑folding stability method Foldetta. In contrast, the majority of in‑silico predictors flag it as pathogenic: SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Pathogenic” verdict. For high‑accuracy assessment, AlphaMissense‑Optimized remains pathogenic, the SGM‑Consensus also indicates likely pathogenic, whereas Foldetta predicts benign stability. No prediction is inconclusive; Rosetta is uncertain but not counted as evidence. Overall, the preponderance of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar classification because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.348540 | Uncertain | 0.449 | 0.438 | 0.500 | -13.406 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.28 | Likely Benign | 0.1 | 0.61 | Ambiguous | 0.45 | Likely Benign | -0.24 | Likely Benign | 0.345 | Likely Benign | -5.32 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.66 | Pathogenic | 0.00 | Affected | 0.0862 | 0.3871 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1011G>C | K337N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K337N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, premPS, and SIFT. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions (seven pathogenic vs. five benign) and the high‑accuracy consensus lean toward a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.348540 | Uncertain | 0.449 | 0.438 | 0.500 | -13.095 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.12 | Likely Benign | 0.1 | 0.36 | Likely Benign | 0.24 | Likely Benign | -0.02 | Likely Benign | 0.280 | Likely Benign | -4.38 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.87 | Pathogenic | 0.11 | Tolerated | 0.2945 | 0.1315 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1011G>T | K337N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K337N is not reported in ClinVar and has no entries in gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, Rosetta, premPS, and SIFT, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. With seven tools supporting pathogenicity versus five supporting benign, the overall prediction leans toward pathogenic. No ClinVar entry contradicts this assessment, and the variant is absent from gnomAD, so the pathogenic prediction is not challenged by population data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.348540 | Uncertain | 0.449 | 0.438 | 0.500 | -13.095 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.12 | Likely Benign | 0.1 | 0.36 | Likely Benign | 0.24 | Likely Benign | -0.02 | Likely Benign | 0.280 | Likely Benign | -4.38 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.87 | Pathogenic | 0.11 | Tolerated | 0.2945 | 0.1315 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1012G>A | D338N 2D 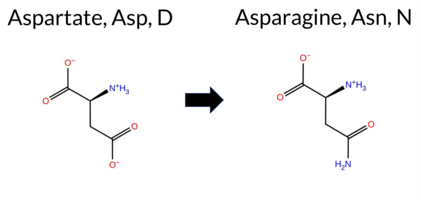 3DClick to see structure in 3D Viewer AIThe SynGAP1 D338N missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, premPS, and polyPhen‑2 HumVar, whereas a majority of tools predict pathogenicity: SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. No evidence from FoldX, Rosetta, or Foldetta supports a benign outcome. Overall, the balance of evidence favors a pathogenic interpretation; this is consistent with the absence of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -9.520 | Likely Pathogenic | 0.809 | Likely Pathogenic | Ambiguous | 0.95 | Ambiguous | 0.4 | 1.34 | Ambiguous | 1.15 | Ambiguous | 0.06 | Likely Benign | 0.442 | Likely Benign | -3.62 | Deleterious | 0.801 | Possibly Damaging | 0.315 | Benign | 1.71 | Pathogenic | 0.02 | Affected | 0.1399 | 0.5970 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1012G>C | D338H 2D 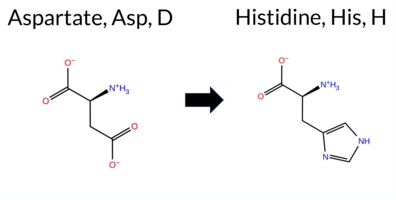 AIThe SynGAP1 missense variant D338H is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to premPS, whereas the remaining 11 tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) all classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, while Foldetta’s stability analysis is inconclusive. Overall, the preponderance of evidence points to a pathogenic impact for D338H, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -12.325 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 1.32 | Ambiguous | 1.2 | 0.76 | Ambiguous | 1.04 | Ambiguous | 0.18 | Likely Benign | 0.515 | Likely Pathogenic | -4.42 | Deleterious | 0.966 | Probably Damaging | 0.770 | Possibly Damaging | 1.71 | Pathogenic | 0.01 | Affected | 0.1671 | 0.6654 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.1012G>T | D338Y 2D 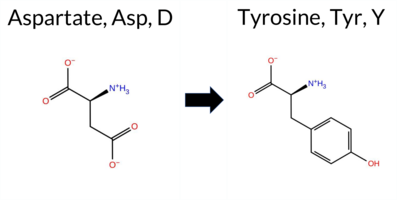 AIThe SynGAP1 D338Y missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools largely agree on a deleterious effect: premPS is the sole predictor labeling it benign, whereas the remaining seven tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—classify it as pathogenic. Predictions from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive. High‑accuracy assessments further support a pathogenic interpretation: the SGM Consensus (derived from a unanimous majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, AlphaMissense‑Optimized remains uncertain, and Foldetta is also uncertain. Overall, the preponderance of evidence indicates that D338Y is most likely pathogenic, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -14.190 | Likely Pathogenic | 0.945 | Likely Pathogenic | Ambiguous | 1.10 | Ambiguous | 1.3 | 0.91 | Ambiguous | 1.01 | Ambiguous | 0.22 | Likely Benign | 0.552 | Likely Pathogenic | -6.49 | Deleterious | 0.989 | Probably Damaging | 0.832 | Possibly Damaging | 1.66 | Pathogenic | 0.00 | Affected | 0.0687 | 0.5609 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1013A>C | D338A 2D  3DClick to see structure in 3D Viewer AISynGAP1 D338A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, polyPhen‑2 HumVar, and SIFT, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. The remaining tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points toward a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -10.639 | Likely Pathogenic | 0.902 | Likely Pathogenic | Ambiguous | 1.22 | Ambiguous | 0.3 | 1.11 | Ambiguous | 1.17 | Ambiguous | 0.16 | Likely Benign | 0.479 | Likely Benign | -5.74 | Deleterious | 0.625 | Possibly Damaging | 0.192 | Benign | 1.73 | Pathogenic | 0.11 | Tolerated | 0.3830 | 0.5988 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.1013A>G | D338G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 D338G missense variant is not reported in ClinVar and has no gnomAD entry. Prediction tools that agree on a benign effect include REVEL, premPS, and polyPhen‑2 HumVar. Those that predict a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Because the majority of available predictors (seven versus three) indicate a deleterious impact, the variant is most likely pathogenic, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -8.875 | Likely Pathogenic | 0.871 | Likely Pathogenic | Ambiguous | 1.33 | Ambiguous | 0.5 | 1.75 | Ambiguous | 1.54 | Ambiguous | 0.15 | Likely Benign | 0.487 | Likely Benign | -5.51 | Deleterious | 0.771 | Possibly Damaging | 0.315 | Benign | 1.69 | Pathogenic | 0.01 | Affected | 0.4014 | 0.5934 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.1013A>T | D338V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 D338V missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only premPS, whereas the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict pathogenicity. High‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain; the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. No prediction or folding‑stability result is missing or inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -11.494 | Likely Pathogenic | 0.927 | Likely Pathogenic | Ambiguous | 1.64 | Ambiguous | 0.2 | 1.08 | Ambiguous | 1.36 | Ambiguous | 0.23 | Likely Benign | 0.553 | Likely Pathogenic | -6.79 | Deleterious | 0.891 | Possibly Damaging | 0.492 | Possibly Damaging | 1.73 | Pathogenic | 0.01 | Affected | 0.0879 | 0.5745 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1014C>A | D338E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D338E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus also indicates a likely benign classification, while Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. No other tools provide conflicting evidence. Based on the preponderance of benign predictions and the lack of pathogenic support from high‑accuracy methods, the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -5.264 | Likely Benign | 0.254 | Likely Benign | Likely Benign | 0.85 | Ambiguous | 0.3 | 0.31 | Likely Benign | 0.58 | Ambiguous | -0.01 | Likely Benign | 0.080 | Likely Benign | -1.95 | Neutral | 0.002 | Benign | 0.005 | Benign | 1.85 | Pathogenic | 0.55 | Tolerated | 0.1584 | 0.5397 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.1014C>G | D338E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D338E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus also indicates a likely benign classification, while Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. No other tools provide conflicting evidence. Based on the preponderance of benign predictions and the lack of pathogenic support from high‑accuracy methods, the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.335645 | Structured | 0.363354 | Uncertain | 0.460 | 0.438 | 0.375 | -5.264 | Likely Benign | 0.254 | Likely Benign | Likely Benign | 0.85 | Ambiguous | 0.3 | 0.31 | Likely Benign | 0.58 | Ambiguous | -0.01 | Likely Benign | 0.080 | Likely Benign | -1.95 | Neutral | 0.002 | Benign | 0.005 | Benign | 1.85 | Pathogenic | 0.55 | Tolerated | 0.1584 | 0.5397 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.1015A>C | K339Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K339Q missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, and Foldetta. Those that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or folding stability result is missing or inconclusive. Overall, the majority of evaluated tools (8 pathogenic vs. 4 benign) indicate a pathogenic effect. This conclusion is not contradicted by ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -10.952 | Likely Pathogenic | 0.863 | Likely Pathogenic | Ambiguous | 0.06 | Likely Benign | 0.0 | -0.50 | Ambiguous | -0.22 | Likely Benign | -0.02 | Likely Benign | 0.458 | Likely Benign | -3.06 | Deleterious | 0.982 | Probably Damaging | 0.824 | Possibly Damaging | 1.90 | Pathogenic | 0.04 | Affected | 0.4041 | 0.1012 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1015A>G | K339E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K339E missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and premPS. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar annotation because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -14.284 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.13 | Likely Benign | 0.1 | -0.03 | Likely Benign | 0.05 | Likely Benign | 0.40 | Likely Benign | 0.482 | Likely Benign | -3.00 | Deleterious | 0.939 | Possibly Damaging | 0.670 | Possibly Damaging | 1.92 | Pathogenic | 0.03 | Affected | 0.3421 | 0.0882 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1016A>C | K339T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K339T missense variant is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools are split: benign calls come from FoldX, Rosetta, Foldetta, premPS, and SIFT, whereas pathogenic calls are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further highlight this discordance: the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect, whereas Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, predicts a benign impact. AlphaMissense‑Optimized returned an uncertain result and is treated as unavailable. Overall, the majority of robust predictors lean toward pathogenicity, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -10.061 | Likely Pathogenic | 0.910 | Likely Pathogenic | Ambiguous | 0.32 | Likely Benign | 0.0 | 0.10 | Likely Benign | 0.21 | Likely Benign | -0.04 | Likely Benign | 0.512 | Likely Pathogenic | -4.73 | Deleterious | 0.991 | Probably Damaging | 0.795 | Possibly Damaging | 1.94 | Pathogenic | 0.10 | Tolerated | 0.1900 | 0.3020 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1016A>G | K339R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K339R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is benign. No predictions or stability results are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -5.773 | Likely Benign | 0.120 | Likely Benign | Likely Benign | -0.15 | Likely Benign | 0.1 | 0.46 | Likely Benign | 0.16 | Likely Benign | 0.08 | Likely Benign | 0.134 | Likely Benign | -2.05 | Neutral | 0.046 | Benign | 0.040 | Benign | 1.94 | Pathogenic | 0.48 | Tolerated | 0.4197 | 0.0976 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.1016A>T | K339M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K339M missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include FoldX and premPS, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools with uncertain or inconclusive results are Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for K339M. This conclusion is not contradicted by ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -13.387 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.23 | Likely Benign | 0.0 | 0.88 | Ambiguous | 0.56 | Ambiguous | -0.37 | Likely Benign | 0.575 | Likely Pathogenic | -4.95 | Deleterious | 0.999 | Probably Damaging | 0.964 | Probably Damaging | 1.92 | Pathogenic | 0.01 | Affected | 0.0967 | 0.3541 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1017G>C | K339N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K339N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and premPS. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the majority of evidence points to a pathogenic impact for K339N, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -11.117 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.20 | Likely Benign | 0.0 | 0.35 | Likely Benign | 0.28 | Likely Benign | 0.00 | Likely Benign | 0.410 | Likely Benign | -3.88 | Deleterious | 0.991 | Probably Damaging | 0.864 | Possibly Damaging | 1.91 | Pathogenic | 0.02 | Affected | 0.3224 | 0.1158 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1017G>T | K339N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K339N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and premPS. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta stability outputs) predicts a benign effect. Overall, the majority of evidence points toward a pathogenic impact for K339N, and this conclusion does not contradict any ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.447574 | Structured | 0.384153 | Uncertain | 0.535 | 0.465 | 0.250 | -11.117 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.20 | Likely Benign | 0.0 | 0.35 | Likely Benign | 0.28 | Likely Benign | 0.00 | Likely Benign | 0.410 | Likely Benign | -3.88 | Deleterious | 0.991 | Probably Damaging | 0.864 | Possibly Damaging | 1.91 | Pathogenic | 0.02 | Affected | 0.3224 | 0.1158 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1018G>C | A340P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A340P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, Foldetta, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The high‑accuracy predictors—AlphaMissense‑Optimized, the SGM‑Consensus, and Foldetta—each report a benign outcome. No prediction or folding‑stability result is missing or inconclusive; all available evidence points to a benign impact. Consequently, the variant is most likely benign based on the aggregate predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | -4.983 | Likely Benign | 0.264 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.5 | -0.58 | Ambiguous | -0.13 | Likely Benign | 0.46 | Likely Benign | 0.279 | Likely Benign | -1.48 | Neutral | 0.891 | Possibly Damaging | 0.575 | Possibly Damaging | 1.91 | Pathogenic | 0.25 | Tolerated | 0.2178 | 0.5468 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1019C>A | A340E 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant A340E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports a likely pathogenic outcome, while the protein‑folding stability method Foldetta is uncertain. AlphaMissense‑Optimized also yields an uncertain result. Overall, the majority of evidence points toward a pathogenic impact, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | -8.225 | Likely Pathogenic | 0.803 | Likely Pathogenic | Ambiguous | 0.63 | Ambiguous | 0.4 | 1.55 | Ambiguous | 1.09 | Ambiguous | 0.06 | Likely Benign | 0.138 | Likely Benign | -0.33 | Neutral | 0.625 | Possibly Damaging | 0.252 | Benign | 1.91 | Pathogenic | 0.39 | Tolerated | 0.1418 | 0.2160 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||
| c.1019C>G | A340G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A340G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive, so they provide no evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of reliable predictors indicate a benign effect, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | -3.763 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.66 | Ambiguous | 0.2 | 1.44 | Ambiguous | 1.05 | Ambiguous | 0.50 | Likely Benign | 0.044 | Likely Benign | -0.34 | Neutral | 0.267 | Benign | 0.127 | Benign | 1.92 | Pathogenic | 0.42 | Tolerated | 0.2355 | 0.4470 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1019C>T | A340V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 A340V variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) is uncertain and therefore unavailable for interpretation. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.390993 | Structured | 0.410781 | Uncertain | 0.558 | 0.485 | 0.250 | -6.427 | Likely Benign | 0.174 | Likely Benign | Likely Benign | 0.69 | Ambiguous | 0.3 | 0.32 | Likely Benign | 0.51 | Ambiguous | 0.40 | Likely Benign | 0.102 | Likely Benign | -1.81 | Neutral | 0.801 | Possibly Damaging | 0.315 | Benign | 2.09 | Pathogenic | 0.57 | Tolerated | 0.1197 | 0.5780 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.101A>C | Y34S 2D  AIThe SynGAP1 missense variant Y34S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Y34S, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.435847 | Uncertain | 0.303 | 0.855 | 0.375 | -0.994 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.178 | Likely Benign | -0.60 | Neutral | 0.824 | Possibly Damaging | 0.775 | Possibly Damaging | 4.19 | Benign | 0.00 | Affected | 0.4760 | 0.2884 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||||||||||||
| c.101A>G | Y34C 2D  AIThe SynGAP1 missense variant Y34C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized reports Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for Y34C, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.435847 | Uncertain | 0.303 | 0.855 | 0.375 | -3.730 | Likely Benign | 0.171 | Likely Benign | Likely Benign | 0.196 | Likely Benign | 0.87 | Neutral | 0.943 | Possibly Damaging | 0.941 | Probably Damaging | 4.32 | Benign | 0.00 | Affected | 0.3010 | 0.2719 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||||||||||||
| c.101A>T | Y34F 2D  AIThe SynGAP1 missense variant Y34F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.435847 | Uncertain | 0.303 | 0.855 | 0.375 | -3.275 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -0.50 | Neutral | 0.458 | Possibly Damaging | 0.481 | Possibly Damaging | 4.20 | Benign | 0.00 | Affected | 0.2604 | 0.3591 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.1021G>A | G341S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G341S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. Predictions that are inconclusive or uncertain are Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta as Uncertain. Overall, the majority of evidence points to a benign impact for G341S, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | -3.206 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.41 | Likely Benign | 0.3 | -1.46 | Ambiguous | -0.53 | Ambiguous | -0.67 | Ambiguous | 0.343 | Likely Benign | 0.73 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 0.37 | Pathogenic | 0.72 | Tolerated | 0.2416 | 0.4215 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1021G>C | G341R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G341R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, polyPhen‑2 HumVar, and Foldetta. Those that predict a pathogenic effect are SGM‑Consensus (Likely Pathogenic), polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show that the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts a likely pathogenic outcome, while Foldetta predicts a benign effect; AlphaMissense‑Optimized remains inconclusive. Overall, the majority of predictions lean toward a benign impact, and this does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign, though the evidence is not definitive. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | -8.263 | Likely Pathogenic | 0.945 | Likely Pathogenic | Ambiguous | 0.28 | Likely Benign | 0.2 | -1.25 | Ambiguous | -0.49 | Likely Benign | 0.18 | Likely Benign | 0.379 | Likely Benign | -1.03 | Neutral | 0.801 | Possibly Damaging | 0.417 | Benign | 0.34 | Pathogenic | 0.17 | Tolerated | 0.0838 | 0.3618 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1021G>T | G341C 2D  AIThe SynGAP1 missense variant G341C has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include FoldX, Foldetta, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (likely benign). Tools that predict a pathogenic effect are REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; Rosetta remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall consensus of the available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | -6.156 | Likely Benign | 0.190 | Likely Benign | Likely Benign | 0.42 | Likely Benign | 1.2 | -0.92 | Ambiguous | -0.25 | Likely Benign | 0.24 | Likely Benign | 0.501 | Likely Pathogenic | -2.40 | Neutral | 0.997 | Probably Damaging | 0.870 | Possibly Damaging | 0.34 | Pathogenic | 0.01 | Affected | 0.1285 | 0.3650 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1022G>C | G341A 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant G341A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenic predictions come only from polyPhen‑2 HumDiv and FATHMM. Uncertain results are reported by Rosetta and Foldetta. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is likely benign; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains inconclusive. Overall, the preponderance of evidence indicates that G341A is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | -3.211 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.16 | Likely Benign | 0.4 | -1.23 | Ambiguous | -0.54 | Ambiguous | -0.03 | Likely Benign | 0.239 | Likely Benign | -1.13 | Neutral | 0.625 | Possibly Damaging | 0.192 | Benign | 0.43 | Pathogenic | 0.15 | Tolerated | 0.3562 | 0.3979 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1022G>T | G341V 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant G341V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, polyPhen‑2 HumVar, and the SGM‑Consensus score (derived from a majority of benign calls among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as uncertain. No evidence from FoldX or premPS is available. Overall, the majority of predictions support a benign classification, and this is consistent with the lack of ClinVar annotation. Therefore, the variant is most likely benign and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | -5.371 | Likely Benign | 0.247 | Likely Benign | Likely Benign | 0.86 | Ambiguous | 0.3 | -2.24 | Stabilizing | -0.69 | Ambiguous | -0.50 | Ambiguous | 0.459 | Likely Benign | -2.29 | Neutral | 0.801 | Possibly Damaging | 0.192 | Benign | 0.42 | Pathogenic | 0.05 | Affected | 0.1019 | 0.3649 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1024T>A | Y342N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y342N has no ClinVar entry (ClinVar status: None) and is not reported in gnomAD (gnomAD ID: None). Prediction tools that assess the variant’s effect are overwhelmingly in agreement that it is deleterious: SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict a pathogenic outcome. No tool predicts a benign effect. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic impact. Taken together, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.366687 | Structured | 0.408200 | Uncertain | 0.866 | 0.487 | 0.250 | -9.685 | Likely Pathogenic | 0.940 | Likely Pathogenic | Ambiguous | 1.76 | Ambiguous | 0.1 | 2.89 | Destabilizing | 2.33 | Destabilizing | 1.02 | Destabilizing | 0.554 | Likely Pathogenic | -6.65 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.71 | Pathogenic | 0.03 | Affected | 0.2189 | 0.0862 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.1024T>G | Y342D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y342D is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include premPS and SIFT, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict it to be pathogenic. FoldX reports an uncertain effect, while Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta is pathogenic. Based on the preponderance of pathogenic predictions and the absence of benign evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.366687 | Structured | 0.408200 | Uncertain | 0.866 | 0.487 | 0.250 | -10.940 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 1.60 | Ambiguous | 0.1 | 2.70 | Destabilizing | 2.15 | Destabilizing | 0.13 | Likely Benign | 0.668 | Likely Pathogenic | -7.19 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.71 | Pathogenic | 0.07 | Tolerated | 0.3925 | 0.0862 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1025A>T | Y342F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y342F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive and therefore treated as unavailable. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.366687 | Structured | 0.408200 | Uncertain | 0.866 | 0.487 | 0.250 | -6.987 | Likely Benign | 0.145 | Likely Benign | Likely Benign | -0.13 | Likely Benign | 0.1 | 0.42 | Likely Benign | 0.15 | Likely Benign | 0.05 | Likely Benign | 0.160 | Likely Benign | -2.67 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.03 | Pathogenic | 0.26 | Tolerated | 0.2626 | 0.3615 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||
| c.1027G>C | V343L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all predict benign or likely benign. Only FATHMM predicts a pathogenic outcome, while FoldX and Rosetta provide uncertain results and are therefore not considered decisive. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates benign. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of ClinVar reporting. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -6.268 | Likely Benign | 0.310 | Likely Benign | Likely Benign | -0.93 | Ambiguous | 0.2 | 0.66 | Ambiguous | -0.14 | Likely Benign | 0.16 | Likely Benign | 0.033 | Likely Benign | -1.09 | Neutral | 0.005 | Benign | 0.013 | Benign | 2.12 | Pathogenic | 0.64 | Tolerated | 0.1361 | 0.5008 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1027G>T | V343F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions are made by REVEL and premPS, whereas pathogenic predictions are made by SIFT, polyPhen‑2 (HumDiv and HumVar), PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain; Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also uncertain. No evidence from FoldX or Rosetta alone is available. Overall, the majority of predictions support a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -10.709 | Likely Pathogenic | 0.799 | Likely Pathogenic | Ambiguous | 1.51 | Ambiguous | 0.4 | 1.28 | Ambiguous | 1.40 | Ambiguous | 0.23 | Likely Benign | 0.324 | Likely Benign | -3.37 | Deleterious | 0.976 | Probably Damaging | 0.759 | Possibly Damaging | 1.61 | Pathogenic | 0.01 | Affected | 0.0915 | 0.4552 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1028T>A | V343D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343D is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, PROVEAN, ESM1b, FATHMM, premPS, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. Rosetta and Foldetta, which evaluate protein‑folding stability, also predict a pathogenic outcome, while FoldX remains uncertain. No tool in the dataset predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta is pathogenic. Consequently, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -15.523 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.57 | Ambiguous | 0.2 | 3.40 | Destabilizing | 2.49 | Destabilizing | 1.73 | Destabilizing | 0.530 | Likely Pathogenic | -5.62 | Deleterious | 0.996 | Probably Damaging | 0.930 | Probably Damaging | 1.59 | Pathogenic | 0.00 | Affected | 0.1781 | 0.2261 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1028T>C | V343A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and AlphaMissense‑Optimized, whereas the remaining tools—SGM‑Consensus, FoldX (uncertain), Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic effect, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenicity. Overall, the preponderance of evidence points to a pathogenic effect for V343A, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -8.088 | Likely Pathogenic | 0.588 | Likely Pathogenic | Likely Benign | 1.66 | Ambiguous | 0.1 | 2.33 | Destabilizing | 2.00 | Destabilizing | 1.69 | Destabilizing | 0.218 | Likely Benign | -3.15 | Deleterious | 0.826 | Possibly Damaging | 0.551 | Possibly Damaging | 1.63 | Pathogenic | 0.01 | Affected | 0.3137 | 0.3301 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.1028T>G | V343G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—classify the variant as pathogenic. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Thus, the preponderance of evidence indicates that V343G is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -11.332 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 2.49 | Destabilizing | 0.1 | 4.40 | Destabilizing | 3.45 | Destabilizing | 1.68 | Destabilizing | 0.421 | Likely Benign | -5.84 | Deleterious | 0.898 | Possibly Damaging | 0.996 | Probably Damaging | 1.60 | Pathogenic | 0.00 | Affected | 0.1982 | 0.3341 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1030G>C | G344R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G344R is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while the only inconclusive result is premPS, which is listed as uncertain. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic status; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports pathogenic. Consequently, the variant is most likely pathogenic, and this assessment is consistent with the absence of a ClinVar entry (no contradiction). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.264545 | Structured | 0.368110 | Uncertain | 0.913 | 0.485 | 0.250 | -12.609 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 14.84 | Destabilizing | 0.8 | 13.66 | Destabilizing | 14.25 | Destabilizing | 0.81 | Ambiguous | 0.801 | Likely Pathogenic | -7.17 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -0.49 | Pathogenic | 0.01 | Affected | 0.0844 | 0.4614 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1030G>T | G344C 2D AIThe SynGAP1 missense variant G344C is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic outcome include SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS is uncertain. High‑accuracy methods specifically show pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No tool predicts a benign effect. Based on the unanimous pathogenic predictions and the absence of any ClinVar or gnomAD evidence, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.264545 | Structured | 0.368110 | Uncertain | 0.913 | 0.485 | 0.250 | -12.880 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 8.52 | Destabilizing | 2.9 | 7.52 | Destabilizing | 8.02 | Destabilizing | 0.59 | Ambiguous | 0.794 | Likely Pathogenic | -8.02 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -0.47 | Pathogenic | 0.01 | Affected | 0.1184 | 0.4788 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1031G>A | G344D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G344D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Based on the unanimous pathogenic predictions and the absence of benign calls, the variant is most likely pathogenic, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.264545 | Structured | 0.368110 | Uncertain | 0.913 | 0.485 | 0.250 | -12.527 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 14.53 | Destabilizing | 1.5 | 11.36 | Destabilizing | 12.95 | Destabilizing | 1.13 | Destabilizing | 0.897 | Likely Pathogenic | -6.30 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -0.49 | Pathogenic | 0.01 | Affected | 0.1572 | 0.1574 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.1031G>C | G344A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G344A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default, while only SIFT predicts a benign outcome. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. Overall, the consensus of the majority of predictors indicates a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.264545 | Structured | 0.368110 | Uncertain | 0.913 | 0.485 | 0.250 | -10.439 | Likely Pathogenic | 0.931 | Likely Pathogenic | Ambiguous | 5.11 | Destabilizing | 0.4 | 4.23 | Destabilizing | 4.67 | Destabilizing | 0.58 | Ambiguous | 0.873 | Likely Pathogenic | -5.38 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -0.32 | Pathogenic | 0.10 | Tolerated | 0.3964 | 0.5685 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1031G>T | G344V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G344V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while the only inconclusive result is premPS, which is listed as uncertain. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic status; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports pathogenic. Consequently, the variant is most likely pathogenic, and this prediction is consistent with the absence of a ClinVar entry (no contradiction). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.264545 | Structured | 0.368110 | Uncertain | 0.913 | 0.485 | 0.250 | -14.913 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 13.33 | Destabilizing | 1.5 | 14.51 | Destabilizing | 13.92 | Destabilizing | 0.53 | Ambiguous | 0.889 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -0.48 | Pathogenic | 0.03 | Affected | 0.1244 | 0.4744 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1033C>A | L345M 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L345M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a benign outcome; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is uncertain. Overall, the majority of evidence points to a benign impact for the L345M variant, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.260850 | Structured | 0.354989 | Uncertain | 0.936 | 0.478 | 0.125 | -3.918 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.75 | Ambiguous | 0.1 | 0.80 | Ambiguous | 0.78 | Ambiguous | 0.54 | Ambiguous | 0.101 | Likely Benign | -0.97 | Neutral | 0.802 | Possibly Damaging | 0.122 | Benign | 1.73 | Pathogenic | 0.07 | Tolerated | 0.0677 | 0.3246 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.1033C>G | L345V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 L345V missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, Rosetta, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also leans toward benign. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.260850 | Structured | 0.354989 | Uncertain | 0.936 | 0.478 | 0.125 | -6.594 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 1.31 | Ambiguous | 0.0 | 0.18 | Likely Benign | 0.75 | Ambiguous | 0.92 | Ambiguous | 0.126 | Likely Benign | -1.96 | Neutral | 0.802 | Possibly Damaging | 0.277 | Benign | 1.77 | Pathogenic | 0.06 | Tolerated | 0.1411 | 0.3455 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1034T>A | L345Q 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant L345Q is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into benign (REVEL, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and pathogenic (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM). FoldX, Rosetta, and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta is unavailable. Overall, the majority of predictions lean toward pathogenicity, while the single high‑accuracy tool that is definitive is benign. Thus, the variant is most likely pathogenic based on the collective evidence, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.260850 | Structured | 0.354989 | Uncertain | 0.936 | 0.478 | 0.125 | -6.034 | Likely Benign | 0.213 | Likely Benign | Likely Benign | 1.32 | Ambiguous | 0.1 | 0.86 | Ambiguous | 1.09 | Ambiguous | 1.26 | Destabilizing | 0.218 | Likely Benign | -3.67 | Deleterious | 0.993 | Probably Damaging | 0.844 | Possibly Damaging | 1.82 | Pathogenic | 0.03 | Affected | 0.0937 | 0.0842 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||
| c.1034T>C | L345P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L345P is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.260850 | Structured | 0.354989 | Uncertain | 0.936 | 0.478 | 0.125 | -10.994 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.33 | Destabilizing | 0.2 | 3.26 | Destabilizing | 3.30 | Destabilizing | 1.27 | Destabilizing | 0.335 | Likely Benign | -4.80 | Deleterious | 0.998 | Probably Damaging | 0.889 | Possibly Damaging | 1.70 | Pathogenic | 0.02 | Affected | 0.3581 | 0.1342 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1034T>G | L345R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L345R is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and AlphaMissense‑Optimized, whereas the majority of tools predict pathogenicity: SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as benign, while the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic prediction. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.260850 | Structured | 0.354989 | Uncertain | 0.936 | 0.478 | 0.125 | -10.325 | Likely Pathogenic | 0.635 | Likely Pathogenic | Likely Benign | 0.96 | Ambiguous | 0.1 | 1.66 | Ambiguous | 1.31 | Ambiguous | 1.28 | Destabilizing | 0.247 | Likely Benign | -3.98 | Deleterious | 0.993 | Probably Damaging | 0.796 | Possibly Damaging | 1.81 | Pathogenic | 0.04 | Affected | 0.1140 | 0.0685 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1036G>A | V346M 2D  3DClick to see structure in 3D Viewer AISynGAP1 variant V346M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL and FoldX, whereas the majority of other in silico methods (PolyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, PROVEAN, AlphaMissense‑Optimized, and the SGM‑Consensus score) indicate pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, remains uncertain. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a pathogenic classification. Thus, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.260850 | Structured | 0.350921 | Uncertain | 0.949 | 0.461 | 0.000 | -10.218 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 0.29 | Likely Benign | 0.1 | 2.52 | Destabilizing | 1.41 | Ambiguous | 0.67 | Ambiguous | 0.367 | Likely Benign | -2.72 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.48 | Pathogenic | 0.05 | Affected | 0.1050 | 0.4749 | 2 | 1 | -2.3 | 32.06 | |||||||||||||||||||||||||||||
| c.1036G>C | V346L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V346L is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, and SIFT, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Four tools (Foldetta, premPS, AlphaMissense‑Optimized, and Rosetta) give uncertain or inconclusive results and are not used as evidence. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of reliable predictors and the SGM‑Consensus support a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.260850 | Structured | 0.350921 | Uncertain | 0.949 | 0.461 | 0.000 | -8.178 | Likely Pathogenic | 0.834 | Likely Pathogenic | Ambiguous | 0.05 | Likely Benign | 0.2 | 1.10 | Ambiguous | 0.58 | Ambiguous | 0.60 | Ambiguous | 0.384 | Likely Benign | -2.68 | Deleterious | 0.994 | Probably Damaging | 0.970 | Probably Damaging | 1.58 | Pathogenic | 0.09 | Tolerated | 0.1237 | 0.5348 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1036G>T | V346L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V346L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, and SIFT, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Four tools (Foldetta, premPS, AlphaMissense‑Optimized, and Rosetta) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points toward a deleterious effect. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.260850 | Structured | 0.350921 | Uncertain | 0.949 | 0.461 | 0.000 | -8.178 | Likely Pathogenic | 0.834 | Likely Pathogenic | Ambiguous | 0.05 | Likely Benign | 0.2 | 1.10 | Ambiguous | 0.58 | Ambiguous | 0.60 | Ambiguous | 0.386 | Likely Benign | -2.68 | Deleterious | 0.994 | Probably Damaging | 0.970 | Probably Damaging | 1.58 | Pathogenic | 0.09 | Tolerated | 0.1237 | 0.5348 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1037T>A | V346E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V346E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). All evaluated in silico predictors classify the change as pathogenic: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts a pathogenic outcome; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic effect; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic impact. **Conclusion:** The variant is most likely pathogenic based on the unanimous computational evidence, and this assessment is not contradicted by the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.260850 | Structured | 0.350921 | Uncertain | 0.949 | 0.461 | 0.000 | -14.004 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.30 | Destabilizing | 0.4 | 4.79 | Destabilizing | 4.05 | Destabilizing | 2.13 | Destabilizing | 0.720 | Likely Pathogenic | -5.52 | Deleterious | 0.999 | Probably Damaging | 0.991 | Probably Damaging | 1.47 | Pathogenic | 0.00 | Affected | 0.1088 | 0.1568 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.1037T>G | V346G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V346G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). All available in‑silico predictors classify it as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a pathogenic verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports pathogenic. With all evidence converging on a deleterious impact and no ClinVar annotation to contradict, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.260850 | Structured | 0.350921 | Uncertain | 0.949 | 0.461 | 0.000 | -12.779 | Likely Pathogenic | 0.969 | Likely Pathogenic | Likely Pathogenic | 4.24 | Destabilizing | 0.2 | 4.62 | Destabilizing | 4.43 | Destabilizing | 2.15 | Destabilizing | 0.667 | Likely Pathogenic | -6.03 | Deleterious | 0.991 | Probably Damaging | 0.999 | Probably Damaging | 1.50 | Pathogenic | 0.00 | Affected | 0.2378 | 0.2522 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1039A>C | T347P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 T347P variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, and FATHMM. Three tools (FoldX, premPS, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.268042 | Structured | 0.349915 | Uncertain | 0.951 | 0.434 | 0.000 | -9.323 | Likely Pathogenic | 0.417 | Ambiguous | Likely Benign | 0.55 | Ambiguous | 0.0 | 0.28 | Likely Benign | 0.42 | Likely Benign | 0.65 | Ambiguous | 0.236 | Likely Benign | -2.41 | Neutral | 0.627 | Possibly Damaging | 0.139 | Benign | 1.63 | Pathogenic | 0.12 | Tolerated | 0.2000 | 0.5350 | 0 | -1 | -0.9 | -3.99 | ||||||||||||||||||||||||||||||
| c.1039A>T | T347S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T347S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive, so they provide no evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of reliable predictors indicate a benign effect, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.268042 | Structured | 0.349915 | Uncertain | 0.951 | 0.434 | 0.000 | -3.342 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.65 | Ambiguous | 0.1 | 0.82 | Ambiguous | 0.74 | Ambiguous | -0.10 | Likely Benign | 0.104 | Likely Benign | 0.63 | Neutral | 0.002 | Benign | 0.001 | Benign | 1.75 | Pathogenic | 0.86 | Tolerated | 0.3205 | 0.4707 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.103G>T | V35F 2D AIThe SynGAP1 missense variant V35F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for V35F, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -4.114 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.133 | Likely Benign | -0.79 | Neutral | 0.923 | Possibly Damaging | 0.865 | Possibly Damaging | 4.14 | Benign | 0.00 | Affected | 0.0765 | 0.3421 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.1040C>G | T347S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T347S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive, so they provide no evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of reliable predictors indicate a benign effect, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.268042 | Structured | 0.349915 | Uncertain | 0.951 | 0.434 | 0.000 | -3.342 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.65 | Ambiguous | 0.1 | 0.82 | Ambiguous | 0.74 | Ambiguous | -0.10 | Likely Benign | 0.054 | Likely Benign | 0.63 | Neutral | 0.002 | Benign | 0.001 | Benign | 1.75 | Pathogenic | 0.86 | Tolerated | 0.3205 | 0.4707 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1040C>T | T347I 2D  3DClick to see structure in 3D Viewer AISynGAP1 T347I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that provide definitive calls cluster into two groups: benign predictions come from REVEL, premPS, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, ESM1b, and FATHMM. Tools with inconclusive outputs (FoldX, Rosetta, Foldetta, AlphaMissense‑Default) are treated as unavailable. High‑accuracy assessments further split the evidence: AlphaMissense‑Optimized predicts benign, while the SGM consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—predicts pathogenic; Foldetta remains uncertain. Consequently, the variant’s pathogenicity is ambiguous, with an equal number of strong benign and pathogenic calls and no consensus from the most reliable methods. The variant is therefore most likely uncertain, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.268042 | Structured | 0.349915 | Uncertain | 0.951 | 0.434 | 0.000 | -10.148 | Likely Pathogenic | 0.387 | Ambiguous | Likely Benign | -0.79 | Ambiguous | 0.1 | -0.94 | Ambiguous | -0.87 | Ambiguous | 0.29 | Likely Benign | 0.079 | Likely Benign | -3.12 | Deleterious | 0.627 | Possibly Damaging | 0.139 | Benign | 1.70 | Pathogenic | 0.08 | Tolerated | 0.0885 | 0.6298 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.1042G>C | V348L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V348L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No other high‑confidence tools provide conflicting evidence. Based on the preponderance of predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.170161 | Structured | 0.346556 | Uncertain | 0.951 | 0.414 | 0.000 | -4.842 | Likely Benign | 0.270 | Likely Benign | Likely Benign | -0.58 | Ambiguous | 0.0 | 0.04 | Likely Benign | -0.27 | Likely Benign | 0.24 | Likely Benign | 0.072 | Likely Benign | -0.75 | Neutral | 0.264 | Benign | 0.030 | Benign | 1.89 | Pathogenic | 0.62 | Tolerated | 0.1088 | 0.4953 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1042G>T | V348L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V348L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No other high‑confidence tools provide conflicting evidence. Based on the preponderance of predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.170161 | Structured | 0.346556 | Uncertain | 0.951 | 0.414 | 0.000 | -4.842 | Likely Benign | 0.270 | Likely Benign | Likely Benign | -0.58 | Ambiguous | 0.0 | 0.04 | Likely Benign | -0.27 | Likely Benign | 0.24 | Likely Benign | 0.072 | Likely Benign | -0.75 | Neutral | 0.264 | Benign | 0.030 | Benign | 1.89 | Pathogenic | 0.62 | Tolerated | 0.1088 | 0.4953 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1043T>A | V348E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V348E is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: the single benign prediction comes from REVEL, while all other evaluated algorithms (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming agreement among the majority of tools, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for V348E. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.170161 | Structured | 0.346556 | Uncertain | 0.951 | 0.414 | 0.000 | -11.135 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 2.41 | Destabilizing | 0.1 | 2.20 | Destabilizing | 2.31 | Destabilizing | 2.14 | Destabilizing | 0.470 | Likely Benign | -5.26 | Deleterious | 0.989 | Probably Damaging | 0.637 | Possibly Damaging | 1.56 | Pathogenic | 0.01 | Affected | 0.1089 | 0.1922 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.1043T>C | V348A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V348A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split opinion: benign predictions come from REVEL, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by SGM‑Consensus, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further highlight the discrepancy: AlphaMissense‑Optimized predicts a benign effect, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) classifies the variant as pathogenic. Taken together, the majority of evidence points toward a pathogenic impact, and this conclusion does not contradict ClinVar status, which currently has no entry for V348A. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.170161 | Structured | 0.346556 | Uncertain | 0.951 | 0.414 | 0.000 | -9.993 | Likely Pathogenic | 0.767 | Likely Pathogenic | Likely Benign | 2.46 | Destabilizing | 0.1 | 3.13 | Destabilizing | 2.80 | Destabilizing | 2.23 | Destabilizing | 0.175 | Likely Benign | -3.42 | Deleterious | 0.622 | Possibly Damaging | 0.152 | Benign | 1.59 | Pathogenic | 0.14 | Tolerated | 0.3223 | 0.2437 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.1043T>G | V348G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V348G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods reinforce this assessment: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates a pathogenic effect. No prediction or stability result is missing or inconclusive. Based on the overwhelming agreement among pathogenic predictions and the lack of a benign consensus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.170161 | Structured | 0.346556 | Uncertain | 0.951 | 0.414 | 0.000 | -12.793 | Likely Pathogenic | 0.964 | Likely Pathogenic | Likely Pathogenic | 3.97 | Destabilizing | 0.2 | 5.70 | Destabilizing | 4.84 | Destabilizing | 2.23 | Destabilizing | 0.419 | Likely Benign | -6.18 | Deleterious | 0.637 | Possibly Damaging | 0.989 | Probably Damaging | 1.56 | Pathogenic | 0.00 | Affected | 0.2229 | 0.2082 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1045C>A | P349T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P349T is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and the SGM‑Consensus score (which is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools with inconclusive results are AlphaMissense‑Default, FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta as uncertain. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for this variant. Thus, the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.167087 | Structured | 0.348607 | Uncertain | 0.947 | 0.396 | 0.000 | -9.736 | Likely Pathogenic | 0.420 | Ambiguous | Likely Benign | 1.37 | Ambiguous | 0.1 | 2.56 | Destabilizing | 1.97 | Ambiguous | 0.76 | Ambiguous | 0.246 | Likely Benign | -6.12 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 1.57 | Pathogenic | 0.07 | Tolerated | 0.1615 | 0.6238 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||
| c.1045C>G | P349A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P349A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM, with the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labeling it likely pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No other stability or pathogenicity scores are available. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.167087 | Structured | 0.348607 | Uncertain | 0.947 | 0.396 | 0.000 | -8.663 | Likely Pathogenic | 0.202 | Likely Benign | Likely Benign | 1.37 | Ambiguous | 0.0 | 1.68 | Ambiguous | 1.53 | Ambiguous | 0.76 | Ambiguous | 0.257 | Likely Benign | -6.01 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.54 | Pathogenic | 0.01 | Affected | 0.3789 | 0.5505 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.1046C>A | P349Q 2D  3DClick to see structure in 3D Viewer AISynGAP1 P349Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. The remaining tools (FoldX, Rosetta, premPS, AlphaMissense‑Default, Foldetta) give uncertain or inconclusive results. High‑accuracy assessments further clarify the variant’s likely effect: AlphaMissense‑Optimized predicts a benign outcome; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, remains uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.167087 | Structured | 0.348607 | Uncertain | 0.947 | 0.396 | 0.000 | -10.545 | Likely Pathogenic | 0.508 | Ambiguous | Likely Benign | 0.98 | Ambiguous | 0.1 | 1.87 | Ambiguous | 1.43 | Ambiguous | 0.91 | Ambiguous | 0.345 | Likely Benign | -6.40 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 1.54 | Pathogenic | 0.06 | Tolerated | 0.1480 | 0.4766 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||
| c.1046C>G | P349R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P349R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and Rosetta. Uncertain or inconclusive results come from FoldX, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show that AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for P349R. This conclusion is not contradicted by ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.167087 | Structured | 0.348607 | Uncertain | 0.947 | 0.396 | 0.000 | -14.001 | Likely Pathogenic | 0.791 | Likely Pathogenic | Ambiguous | 0.99 | Ambiguous | 0.1 | 2.15 | Destabilizing | 1.57 | Ambiguous | 0.93 | Ambiguous | 0.335 | Likely Benign | -7.22 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 1.55 | Pathogenic | 0.01 | Affected | 0.1395 | 0.3009 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.1046C>T | P349L 2D  AIThe SynGAP1 missense variant P349L is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and AlphaMissense‑Optimized, whereas the majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy methods give conflicting results: AlphaMissense‑Optimized reports a benign outcome, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Other stability‑based predictors (FoldX, Rosetta, premPS) are also inconclusive. Overall, the preponderance of evidence from the consensus of multiple in‑silico tools points to a pathogenic effect for P349L. This prediction does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.167087 | Structured | 0.348607 | Uncertain | 0.947 | 0.396 | 0.000 | -11.734 | Likely Pathogenic | 0.650 | Likely Pathogenic | Likely Benign | 0.70 | Ambiguous | 0.6 | 1.17 | Ambiguous | 0.94 | Ambiguous | 0.57 | Ambiguous | 0.326 | Likely Benign | -8.04 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 1.51 | Pathogenic | 0.00 | Affected | 0.2222 | 0.6867 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1048G>A | V350M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V350M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. The remaining tools (FoldX, premPS, ESM1b) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as benign. Taken together, the majority of evidence supports a benign classification. There is no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -7.493 | In-Between | 0.273 | Likely Benign | Likely Benign | -0.65 | Ambiguous | 0.1 | 0.40 | Likely Benign | -0.13 | Likely Benign | 0.69 | Ambiguous | 0.075 | Likely Benign | -1.95 | Neutral | 0.868 | Possibly Damaging | 0.383 | Benign | 1.63 | Pathogenic | 0.06 | Tolerated | 0.1005 | 0.4359 | 2 | 1 | -2.3 | 32.06 | ||||||||||||||||||||||||||||||
| c.1048G>C | V350L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V350L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only FATHMM predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign status. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as uncertain. No other tools provide conflicting evidence. Based on the preponderance of benign predictions and the lack of pathogenic support, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -5.941 | Likely Benign | 0.238 | Likely Benign | Likely Benign | -1.31 | Ambiguous | 0.1 | -0.34 | Likely Benign | -0.83 | Ambiguous | 0.36 | Likely Benign | 0.042 | Likely Benign | -1.63 | Neutral | 0.068 | Benign | 0.022 | Benign | 2.37 | Pathogenic | 0.27 | Tolerated | 0.1173 | 0.4958 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1048G>T | V350L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V350L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only FATHMM predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. No other tools provide conflicting evidence. Based on the preponderance of predictions, V350L is most likely benign, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -5.941 | Likely Benign | 0.238 | Likely Benign | Likely Benign | -1.31 | Ambiguous | 0.1 | -0.34 | Likely Benign | -0.83 | Ambiguous | 0.36 | Likely Benign | 0.042 | Likely Benign | -1.63 | Neutral | 0.068 | Benign | 0.022 | Benign | 2.37 | Pathogenic | 0.27 | Tolerated | 0.1173 | 0.4958 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1049T>A | V350E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V350E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on benign effect include REVEL and SIFT, whereas a majority of tools predict pathogenicity: premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, Foldetta, and Rosetta. Two tools give inconclusive results: FoldX (Uncertain) and AlphaMissense‑Optimized (Uncertain). High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Pathogenic. Overall, the preponderance of evidence points to a pathogenic effect for V350E. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -15.975 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 1.73 | Ambiguous | 0.2 | 2.61 | Destabilizing | 2.17 | Destabilizing | 1.93 | Destabilizing | 0.304 | Likely Benign | -4.67 | Deleterious | 0.976 | Probably Damaging | 0.559 | Possibly Damaging | 1.61 | Pathogenic | 0.08 | Tolerated | 0.1078 | 0.1568 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.1049T>C | V350A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant V350A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), premPS, PROVEAN, ESM1b, and FATHMM. Stability‑based methods FoldX, Rosetta, and Foldetta give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta as unavailable. Overall, the predictions are split, with a slight edge toward pathogenicity from the consensus and high‑accuracy tools. Therefore, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -8.323 | Likely Pathogenic | 0.280 | Likely Benign | Likely Benign | 1.42 | Ambiguous | 0.0 | 1.97 | Ambiguous | 1.70 | Ambiguous | 2.07 | Destabilizing | 0.096 | Likely Benign | -2.73 | Deleterious | 0.435 | Benign | 0.115 | Benign | 1.64 | Pathogenic | 0.55 | Tolerated | 0.3207 | 0.2967 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.1049T>G | V350G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V350G lies in the C2 domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, polyPhen‑2 HumDiv, and AlphaMissense‑Optimized. The remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict it to be pathogenic. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All predictions are available and consistent. Based on the preponderance of pathogenic calls, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -12.267 | Likely Pathogenic | 0.597 | Likely Pathogenic | Likely Benign | 2.57 | Destabilizing | 0.0 | 4.25 | Destabilizing | 3.41 | Destabilizing | 1.93 | Destabilizing | 0.330 | Likely Benign | -5.46 | Deleterious | 0.435 | Benign | 0.920 | Probably Damaging | 1.61 | Pathogenic | 0.02 | Affected | 0.2316 | 0.2522 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.104T>A | V35D 2D AIThe SynGAP1 missense variant V35D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the overall assessment. Overall, the majority of evidence points to the variant being most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -4.232 | Likely Benign | 0.321 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -0.42 | Neutral | 0.824 | Possibly Damaging | 0.828 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.1540 | 0.1547 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.104T>C | V35A 2D AIThe SynGAP1 missense variant V35A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumVar and SIFT, predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -3.552 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.098 | Likely Benign | -0.05 | Neutral | 0.267 | Benign | 0.481 | Possibly Damaging | 4.25 | Benign | 0.00 | Affected | 0.2340 | 0.2144 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||||||||||||
| c.104T>G | V35G 2D AIThe SynGAP1 missense variant V35G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus also predicts benign; Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because the variant is not currently catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -3.614 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.143 | Likely Benign | 0.66 | Neutral | 0.693 | Possibly Damaging | 0.824 | Possibly Damaging | 4.41 | Benign | 0.00 | Affected | 0.1514 | 0.2618 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.1051G>A | A351T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 A351T missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. Uncertain predictions come from Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta as inconclusive. Overall, the majority of evidence supports a benign impact for A351T, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.216401 | Structured | 0.362025 | Uncertain | 0.925 | 0.342 | 0.000 | -6.863 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.0 | 0.80 | Ambiguous | 0.51 | Ambiguous | 0.49 | Likely Benign | 0.040 | Likely Benign | -2.42 | Neutral | 0.524 | Possibly Damaging | 0.138 | Benign | 1.71 | Pathogenic | 0.06 | Tolerated | 0.1337 | 0.6713 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.1051G>C | A351P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A351P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are Foldetta, PROVEAN, SIFT, FATHMM, and Rosetta. FoldX and premPS give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of predictions lean toward a benign impact, and this is consistent with the lack of ClinVar reporting; thus the variant is most likely benign rather than pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.216401 | Structured | 0.362025 | Uncertain | 0.925 | 0.342 | 0.000 | -6.559 | Likely Benign | 0.147 | Likely Benign | Likely Benign | -0.89 | Ambiguous | 0.2 | 6.19 | Destabilizing | 2.65 | Destabilizing | 0.62 | Ambiguous | 0.051 | Likely Benign | -3.08 | Deleterious | 0.016 | Benign | 0.007 | Benign | 1.67 | Pathogenic | 0.03 | Affected | 0.1862 | 0.5472 | 1 | -1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||
| c.1051G>T | A351S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A351S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only FATHMM predicts a pathogenic outcome. High‑accuracy methods—AlphaMissense‑Optimized, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (integrating FoldX‑MD and Rosetta)—all report benign or likely benign. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely benign; this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.216401 | Structured | 0.362025 | Uncertain | 0.925 | 0.342 | 0.000 | -4.823 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.24 | Likely Benign | 0.0 | 0.25 | Likely Benign | 0.25 | Likely Benign | 0.27 | Likely Benign | 0.016 | Likely Benign | -1.42 | Neutral | 0.080 | Benign | 0.023 | Benign | 1.88 | Pathogenic | 0.13 | Tolerated | 0.2558 | 0.5334 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1052C>A | A351D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 A351D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and FATHMM; AlphaMissense‑Default and ESM1b are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leaning toward pathogenic due to two pathogenic and two uncertain calls. Overall, the majority of tools predict a benign outcome, and this does not contradict the lack of ClinVar annotation. Thus, based on the available predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.216401 | Structured | 0.362025 | Uncertain | 0.925 | 0.342 | 0.000 | -7.917 | In-Between | 0.543 | Ambiguous | Likely Benign | -0.02 | Likely Benign | 0.0 | -0.12 | Likely Benign | -0.07 | Likely Benign | 0.49 | Likely Benign | 0.114 | Likely Benign | -3.61 | Deleterious | 0.842 | Possibly Damaging | 0.321 | Benign | 1.77 | Pathogenic | 0.11 | Tolerated | 0.1535 | 0.1386 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||
| c.1052C>G | A351G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A351G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL, FoldX, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. Predictions that are uncertain or inconclusive are Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs two pathogenic), and Foldetta is uncertain. Taken together, the majority of available predictions lean toward a benign impact, and this assessment does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.216401 | Structured | 0.362025 | Uncertain | 0.925 | 0.342 | 0.000 | -4.983 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.39 | Likely Benign | 0.0 | 1.04 | Ambiguous | 0.72 | Ambiguous | 0.55 | Ambiguous | 0.041 | Likely Benign | -2.53 | Deleterious | 0.688 | Possibly Damaging | 0.138 | Benign | 1.77 | Pathogenic | 0.04 | Affected | 0.2298 | 0.4737 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||
| c.1052C>T | A351V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A351V is not reported in ClinVar and is absent from gnomAD. Computational predictions cluster into two groups: benign (REVEL, FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen2_HumVar) and pathogenic (PROVEAN, polyPhen2_HumDiv, SIFT, ESM1b, FATHMM, SGM‑Consensus). High‑accuracy tools give a mixed signal: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the balance of evidence leans toward a benign effect, but the presence of several pathogenic predictions introduces uncertainty. The variant is most likely benign based on the current computational data, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.362025 | Uncertain | 0.925 | 0.342 | 0.000 | -9.002 | Likely Pathogenic | 0.124 | Likely Benign | Likely Benign | 0.09 | Likely Benign | 0.0 | 0.19 | Likely Benign | 0.14 | Likely Benign | 0.29 | Likely Benign | 0.052 | Likely Benign | -2.84 | Deleterious | 0.915 | Possibly Damaging | 0.321 | Benign | 1.66 | Pathogenic | 0.03 | Affected | 0.1140 | 0.6565 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.1054A>C | T352P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T352P has no ClinVar entry and is not reported in gnomAD. Prediction tools that converge on a benign outcome include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, FATHMM, and Rosetta. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Overall, the balance of evidence favors a benign interpretation; this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.137348 | Structured | 0.367886 | Uncertain | 0.926 | 0.329 | 0.000 | -3.562 | Likely Benign | 0.138 | Likely Benign | Likely Benign | 1.04 | Ambiguous | 0.1 | 2.57 | Destabilizing | 1.81 | Ambiguous | 0.51 | Ambiguous | 0.159 | Likely Benign | -2.31 | Neutral | 0.627 | Possibly Damaging | 0.196 | Benign | 1.72 | Pathogenic | 0.20 | Tolerated | 0.2244 | 0.5968 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1054A>G | T352A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T352A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus, REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only FATHMM predicts pathogenic, while Rosetta remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign verdict; and Foldetta, integrating FoldX‑MD (benign) and Rosetta (uncertain), reports benign stability. Overall, the preponderance of evidence indicates that T352A is most likely benign, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.137348 | Structured | 0.367886 | Uncertain | 0.926 | 0.329 | 0.000 | -1.952 | Likely Benign | 0.055 | Likely Benign | Likely Benign | 0.12 | Likely Benign | 0.0 | 0.50 | Ambiguous | 0.31 | Likely Benign | 0.45 | Likely Benign | 0.032 | Likely Benign | -1.09 | Neutral | 0.031 | Benign | 0.024 | Benign | 1.73 | Pathogenic | 0.29 | Tolerated | 0.4341 | 0.4938 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1054A>T | T352S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T352S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only FATHMM predicts a pathogenic outcome. The high‑accuracy consensus methods reinforce the benign assessment: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a benign effect. No predictions or stability results are missing or inconclusive. Based on the overwhelming agreement among the majority of tools and the high‑accuracy methods, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.137348 | Structured | 0.367886 | Uncertain | 0.926 | 0.329 | 0.000 | -1.670 | Likely Benign | 0.062 | Likely Benign | Likely Benign | -0.09 | Likely Benign | 0.0 | 0.26 | Likely Benign | 0.09 | Likely Benign | -0.01 | Likely Benign | 0.098 | Likely Benign | 0.57 | Neutral | 0.002 | Benign | 0.002 | Benign | 1.81 | Pathogenic | 1.00 | Tolerated | 0.3583 | 0.4880 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1055C>G | T352S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T352S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only FATHMM predicts a pathogenic outcome. The high‑accuracy consensus methods reinforce the benign assessment: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a benign effect. No predictions or stability results are missing or inconclusive. Based on the overwhelming agreement among the majority of tools and the high‑accuracy methods, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.137348 | Structured | 0.367886 | Uncertain | 0.926 | 0.329 | 0.000 | -1.670 | Likely Benign | 0.062 | Likely Benign | Likely Benign | -0.09 | Likely Benign | 0.0 | 0.26 | Likely Benign | 0.09 | Likely Benign | -0.01 | Likely Benign | 0.104 | Likely Benign | 0.57 | Neutral | 0.002 | Benign | 0.002 | Benign | 1.81 | Pathogenic | 1.00 | Tolerated | 0.3583 | 0.4880 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1055C>T | T352I 2D AISynGAP1 T352I is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, Rosetta, premPS, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Those that predict pathogenicity are SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus majority vote (AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. With seven benign versus five pathogenic predictions and two high‑accuracy benign versus one pathogenic, the evidence leans toward a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.137348 | Structured | 0.367886 | Uncertain | 0.926 | 0.329 | 0.000 | -8.023 | Likely Pathogenic | 0.321 | Likely Benign | Likely Benign | -0.54 | Ambiguous | 0.7 | 0.43 | Likely Benign | -0.06 | Likely Benign | 0.09 | Likely Benign | 0.099 | Likely Benign | -3.02 | Deleterious | 0.627 | Possibly Damaging | 0.196 | Benign | 1.67 | Pathogenic | 0.14 | Tolerated | 0.1009 | 0.6484 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.1057C>A | L353M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L353M has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, FoldX, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. Uncertain results come from Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta as Uncertain. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar classification; there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.137348 | Structured | 0.373584 | Uncertain | 0.926 | 0.315 | 0.000 | -6.943 | Likely Benign | 0.206 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 1.28 | Ambiguous | 0.69 | Ambiguous | 0.60 | Ambiguous | 0.117 | Likely Benign | -0.47 | Neutral | 0.744 | Possibly Damaging | 0.289 | Benign | 1.33 | Pathogenic | 0.03 | Affected | 0.0847 | 0.4080 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.1057C>G | L353V 2D AISynGAP1 missense variant L353V is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools predicting pathogenicity are FoldX and FATHMM, while Rosetta gives an uncertain result. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the balance of evidence favors a benign effect; this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.137348 | Structured | 0.373584 | Uncertain | 0.926 | 0.315 | 0.000 | -2.100 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 3.08 | Destabilizing | 0.9 | 1.48 | Ambiguous | 2.28 | Destabilizing | -0.45 | Likely Benign | 0.074 | Likely Benign | 1.54 | Neutral | 0.002 | Benign | 0.002 | Benign | 1.46 | Pathogenic | 0.89 | Tolerated | 0.1567 | 0.3829 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1058T>A | L353Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L353Q missense variant has no ClinVar entry and is absent from gnomAD. Among in‑silico predictors, only REVEL classifies it as benign, whereas the majority—FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—label it pathogenic. Predictions marked uncertain include Rosetta, Foldetta, ESM1b, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of pathogenic predictions outweighs the single benign call, and no ClinVar record contradicts this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.137348 | Structured | 0.373584 | Uncertain | 0.926 | 0.315 | 0.000 | -7.074 | In-Between | 0.884 | Likely Pathogenic | Ambiguous | 2.38 | Destabilizing | 0.2 | 1.41 | Ambiguous | 1.90 | Ambiguous | 2.00 | Destabilizing | 0.388 | Likely Benign | -3.56 | Deleterious | 0.947 | Possibly Damaging | 0.556 | Possibly Damaging | 1.30 | Pathogenic | 0.01 | Affected | 0.1098 | 0.1303 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1058T>G | L353R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L353R is not reported in ClinVar and is absent from gnomAD. In silico predictors largely agree on a deleterious effect: benign predictions are limited to REVEL, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all classify the variant as pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is uncertain, SGM‑Consensus predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction contradicts the ClinVar status, which is currently unreported. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.137348 | Structured | 0.373584 | Uncertain | 0.926 | 0.315 | 0.000 | -11.213 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 3.60 | Destabilizing | 0.5 | 2.48 | Destabilizing | 3.04 | Destabilizing | 1.91 | Destabilizing | 0.444 | Likely Benign | -4.15 | Deleterious | 0.947 | Possibly Damaging | 0.454 | Possibly Damaging | 1.33 | Pathogenic | 0.01 | Affected | 0.1304 | 0.1145 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1060G>A | A354T 2D AIThe SynGAP1 missense variant A354T is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only FATHMM predicts a pathogenic effect. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a benign result. No predictions or folding‑stability analyses are missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.381329 | Uncertain | 0.882 | 0.335 | 0.125 | -2.812 | Likely Benign | 0.070 | Likely Benign | Likely Benign | 0.37 | Likely Benign | 0.7 | 0.11 | Likely Benign | 0.24 | Likely Benign | -0.52 | Ambiguous | 0.097 | Likely Benign | 1.72 | Neutral | 0.002 | Benign | 0.001 | Benign | 1.75 | Pathogenic | 0.45 | Tolerated | 0.1731 | 0.6496 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.1060G>C | A354P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A354P is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are FoldX, SIFT, and FATHMM. Predictions that are inconclusive are Foldetta, premPS, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta as pathogenic. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.381329 | Uncertain | 0.882 | 0.335 | 0.125 | Uncertain | 1 | -5.971 | Likely Benign | 0.239 | Likely Benign | Likely Benign | 2.66 | Destabilizing | 0.2 | 1.55 | Ambiguous | 2.11 | Destabilizing | 0.57 | Ambiguous | 0.241 | Likely Benign | -1.66 | Neutral | 0.001 | Benign | 0.002 | Benign | 1.76 | Pathogenic | 0.04 | Affected | 0.2259 | 0.5884 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||
| c.1060G>T | A354S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A354S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a benign effect. Taken together, the overwhelming majority of evidence indicates that A354S is most likely benign, and this conclusion is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.381329 | Uncertain | 0.882 | 0.335 | 0.125 | -3.005 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.02 | Likely Benign | 0.1 | 0.27 | Likely Benign | 0.15 | Likely Benign | -0.10 | Likely Benign | 0.071 | Likely Benign | 0.28 | Neutral | 0.005 | Benign | 0.001 | Benign | 1.76 | Pathogenic | 0.21 | Tolerated | 0.2829 | 0.5117 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1061C>A | A354D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A354D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. Those that predict a pathogenic effect are FATHMM and AlphaMissense‑Default. Four tools (Foldetta, premPS, ESM1b, and Rosetta) return uncertain results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors a pathogenic interpretation. Foldetta’s stability prediction is uncertain. Overall, the majority of consensus tools lean toward a benign effect, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.203355 | Structured | 0.381329 | Uncertain | 0.882 | 0.335 | 0.125 | -7.914 | In-Between | 0.725 | Likely Pathogenic | Likely Benign | 0.22 | Likely Benign | 0.1 | 1.10 | Ambiguous | 0.66 | Ambiguous | 0.57 | Ambiguous | 0.087 | Likely Benign | -1.49 | Neutral | 0.255 | Benign | 0.053 | Benign | 1.75 | Pathogenic | 0.07 | Tolerated | 0.1759 | 0.1345 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||
| c.1061C>G | A354G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A354G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. Predictions marked uncertain (Rosetta, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, while Foldetta’s stability analysis is inconclusive. Overall, the majority of evidence points to a benign impact for A354G, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.381329 | Uncertain | 0.882 | 0.335 | 0.125 | -4.285 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.41 | Likely Benign | 0.0 | 1.24 | Ambiguous | 0.83 | Ambiguous | 0.65 | Ambiguous | 0.037 | Likely Benign | -1.87 | Neutral | 0.146 | Benign | 0.038 | Benign | 1.75 | Pathogenic | 0.05 | Affected | 0.2563 | 0.4777 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1063G>A | G355R 2D AIThe SynGAP1 missense variant G355R is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, and Foldetta. Tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are uncertain or inconclusive are AlphaMissense‑Optimized, FoldX, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evaluated tools predict a pathogenic impact, whereas a minority predict benign. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.388832 | Uncertain | 0.810 | 0.354 | 0.125 | -11.022 | Likely Pathogenic | 0.922 | Likely Pathogenic | Ambiguous | 0.61 | Ambiguous | 1.4 | 0.10 | Likely Benign | 0.36 | Likely Benign | 0.60 | Ambiguous | 0.340 | Likely Benign | -6.74 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.80 | Pathogenic | 0.02 | Affected | 0.1041 | 0.4567 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1063G>C | G355R 2D AIThe SynGAP1 missense variant G355R is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, and Foldetta. Tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are uncertain or inconclusive are AlphaMissense‑Optimized, FoldX, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evaluated tools predict a pathogenic impact, whereas a minority predict benign. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.388832 | Uncertain | 0.810 | 0.354 | 0.125 | -11.022 | Likely Pathogenic | 0.922 | Likely Pathogenic | Ambiguous | 0.61 | Ambiguous | 1.4 | 0.10 | Likely Benign | 0.36 | Likely Benign | 0.60 | Ambiguous | 0.340 | Likely Benign | -6.74 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.80 | Pathogenic | 0.02 | Affected | 0.1041 | 0.4567 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1063G>T | G355W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G355W is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL and premPS, and pathogenic predictions from SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results are reported by FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as inconclusive, SGM‑Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.388832 | Uncertain | 0.810 | 0.354 | 0.125 | -12.316 | Likely Pathogenic | 0.938 | Likely Pathogenic | Ambiguous | 1.16 | Ambiguous | 0.5 | 0.88 | Ambiguous | 1.02 | Ambiguous | 0.18 | Likely Benign | 0.455 | Likely Benign | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.73 | Pathogenic | 0.00 | Affected | 0.0796 | 0.4360 | -7 | -2 | -0.5 | 129.16 | |||||||||||||||||||||||||||||
| c.1064G>C | G355A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G355A missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. FoldX and Foldetta are uncertain and therefore not counted as evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta is uncertain. Consequently, the overall prediction leans toward a benign effect, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.155435 | Structured | 0.388832 | Uncertain | 0.810 | 0.354 | 0.125 | -5.431 | Likely Benign | 0.245 | Likely Benign | Likely Benign | 0.88 | Ambiguous | 0.6 | 0.46 | Likely Benign | 0.67 | Ambiguous | 0.49 | Likely Benign | 0.286 | Likely Benign | -4.80 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.80 | Pathogenic | 0.03 | Affected | 0.4111 | 0.5045 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||
| c.1064G>T | G355V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G355V is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain or unavailable results are reported for FoldX, premPS, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, whereas the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect. Foldetta’s stability prediction is unavailable. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.388832 | Uncertain | 0.810 | 0.354 | 0.125 | -10.111 | Likely Pathogenic | 0.675 | Likely Pathogenic | Likely Benign | 1.78 | Ambiguous | 0.6 | 0.14 | Likely Benign | 0.96 | Ambiguous | 0.53 | Ambiguous | 0.346 | Likely Benign | -7.59 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.78 | Pathogenic | 0.04 | Affected | 0.1221 | 0.4685 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1066C>A | R356S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R356S is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL and SIFT, whereas the remaining tools—premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized—predict it to be pathogenic. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support this: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is inconclusive. Taken together, the preponderance of evidence points to a pathogenic effect for R356S, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.219301 | Structured | 0.395028 | Uncertain | 0.802 | 0.373 | 0.250 | -13.059 | Likely Pathogenic | 0.965 | Likely Pathogenic | Likely Pathogenic | 1.00 | Ambiguous | 0.1 | 0.79 | Ambiguous | 0.90 | Ambiguous | 1.04 | Destabilizing | 0.292 | Likely Benign | -5.28 | Deleterious | 0.993 | Probably Damaging | 0.982 | Probably Damaging | 1.84 | Pathogenic | 0.07 | Tolerated | 0.2891 | 0.4491 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||
| c.1066C>G | R356G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R356G missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and SIFT, while the majority of tools predict a pathogenic outcome: SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are uncertain and therefore treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. Overall, the preponderance of pathogenic predictions indicates that the variant is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which contains no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.219301 | Structured | 0.395028 | Uncertain | 0.802 | 0.373 | 0.250 | -12.305 | Likely Pathogenic | 0.883 | Likely Pathogenic | Ambiguous | 1.02 | Ambiguous | 0.0 | 1.80 | Ambiguous | 1.41 | Ambiguous | 1.06 | Destabilizing | 0.271 | Likely Benign | -6.20 | Deleterious | 0.993 | Probably Damaging | 0.982 | Probably Damaging | 1.95 | Pathogenic | 0.08 | Tolerated | 0.3505 | 0.3793 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1067G>C | R356P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R356P missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FoldX, whereas a majority of tools (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. Predictions that are inconclusive are AlphaMissense‑Optimized, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for R356P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.219301 | Structured | 0.395028 | Uncertain | 0.802 | 0.373 | 0.250 | -12.956 | Likely Pathogenic | 0.919 | Likely Pathogenic | Ambiguous | 0.48 | Likely Benign | 0.1 | 0.72 | Ambiguous | 0.60 | Ambiguous | 1.05 | Destabilizing | 0.451 | Likely Benign | -6.23 | Deleterious | 0.998 | Probably Damaging | 0.992 | Probably Damaging | 1.72 | Pathogenic | 0.02 | Affected | 0.2213 | 0.5033 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.1069C>A | H357N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 H357N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Taken together, the overwhelming majority of evidence points to a benign impact. There is no ClinVar annotation to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.399052 | Uncertain | 0.861 | 0.413 | 0.250 | -7.617 | In-Between | 0.103 | Likely Benign | Likely Benign | 0.14 | Likely Benign | 0.2 | 0.31 | Likely Benign | 0.23 | Likely Benign | 0.31 | Likely Benign | 0.126 | Likely Benign | -1.47 | Neutral | 0.495 | Possibly Damaging | 0.169 | Benign | 4.25 | Benign | 0.15 | Tolerated | 0.2052 | 0.3266 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||
| c.1069C>G | H357D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 H357D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, and Foldetta also predicts a benign outcome. No prediction or folding‑stability result is missing or inconclusive. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.203355 | Structured | 0.399052 | Uncertain | 0.861 | 0.413 | 0.250 | -12.013 | Likely Pathogenic | 0.559 | Ambiguous | Likely Benign | -0.23 | Likely Benign | 0.4 | -0.27 | Likely Benign | -0.25 | Likely Benign | 0.48 | Likely Benign | 0.208 | Likely Benign | -1.90 | Neutral | 0.495 | Possibly Damaging | 0.169 | Benign | 4.22 | Benign | 0.08 | Tolerated | 0.2782 | 0.1976 | 1 | -1 | -0.3 | -22.05 | ||||||||||||||||||||||||||||||
| c.106C>A | H36N 2D AIThe SynGAP1 missense variant H36N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -3.614 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.027 | Likely Benign | -0.83 | Neutral | 0.019 | Benign | 0.021 | Benign | 4.21 | Benign | 0.00 | Affected | 0.2353 | 0.4258 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.106C>G | H36D 2D AIThe SynGAP1 missense variant H36D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta results are not available for this variant. Overall, the majority of computational evidence indicates that H36D is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -2.859 | Likely Benign | 0.225 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -1.06 | Neutral | 0.043 | Benign | 0.033 | Benign | 4.21 | Benign | 0.00 | Affected | 0.2805 | 0.3496 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||||||||||||
| c.1070A>C | H357P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 H357P missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are Rosetta, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Two tools give uncertain results: Foldetta (protein‑folding stability) and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign, while Foldetta remains uncertain. Overall, the majority of evidence points to a benign effect. Thus, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.203355 | Structured | 0.399052 | Uncertain | 0.861 | 0.413 | 0.250 | -7.953 | In-Between | 0.267 | Likely Benign | Likely Benign | 0.17 | Likely Benign | 0.9 | 3.52 | Destabilizing | 1.85 | Ambiguous | -0.06 | Likely Benign | 0.197 | Likely Benign | -2.70 | Deleterious | 0.936 | Possibly Damaging | 0.469 | Possibly Damaging | 4.18 | Benign | 0.17 | Tolerated | 0.2433 | 0.4468 | 0 | -2 | 1.6 | -40.02 | ||||||||||||||||||||||||||||||
| c.1070A>G | H357R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 H357R missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. Two tools remain uncertain: Rosetta and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority; and Foldetta also predicts benign. No prediction or stability result is missing or inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.203355 | Structured | 0.399052 | Uncertain | 0.861 | 0.413 | 0.250 | -9.212 | Likely Pathogenic | 0.348 | Ambiguous | Likely Benign | -0.10 | Likely Benign | 0.3 | 1.07 | Ambiguous | 0.49 | Likely Benign | 0.32 | Likely Benign | 0.107 | Likely Benign | -1.03 | Neutral | 0.495 | Possibly Damaging | 0.095 | Benign | 4.22 | Benign | 0.59 | Tolerated | 0.2147 | 0.2818 | 2 | 0 | -1.3 | 19.05 | ||||||||||||||||||||||||||||||
| c.1070A>T | H357L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 H357L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv, while polyPhen‑2 HumVar and ESM1b are benign or uncertain, respectively. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign (3 benign vs 1 pathogenic), and Foldetta also predicts benign. No predictions are missing or inconclusive. Overall, the variant is most likely benign based on the majority of computational evidence, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.203355 | Structured | 0.399052 | Uncertain | 0.861 | 0.413 | 0.250 | -7.281 | In-Between | 0.140 | Likely Benign | Likely Benign | -0.18 | Likely Benign | 0.1 | 0.14 | Likely Benign | -0.02 | Likely Benign | 0.10 | Likely Benign | 0.203 | Likely Benign | -3.39 | Deleterious | 0.704 | Possibly Damaging | 0.169 | Benign | 4.20 | Benign | 0.25 | Tolerated | 0.1084 | 0.5971 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||
| c.1071C>A | H357Q 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H357Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess sequence conservation, structural impact, or functional effect all converge on a benign outcome: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a benign effect. Based on the uniform predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.399052 | Uncertain | 0.861 | 0.413 | 0.250 | -4.370 | Likely Benign | 0.163 | Likely Benign | Likely Benign | 0.06 | Likely Benign | 0.2 | -0.03 | Likely Benign | 0.02 | Likely Benign | -0.35 | Likely Benign | 0.025 | Likely Benign | 1.42 | Neutral | 0.013 | Benign | 0.007 | Benign | 4.49 | Benign | 1.00 | Tolerated | 0.1716 | 0.3850 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.1071C>G | H357Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H357Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess sequence conservation, structural impact, or functional effect all converge on a benign outcome: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a benign effect. Based on the uniform predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.203355 | Structured | 0.399052 | Uncertain | 0.861 | 0.413 | 0.250 | -4.370 | Likely Benign | 0.163 | Likely Benign | Likely Benign | 0.06 | Likely Benign | 0.2 | -0.03 | Likely Benign | 0.02 | Likely Benign | -0.35 | Likely Benign | 0.025 | Likely Benign | 1.42 | Neutral | 0.013 | Benign | 0.007 | Benign | 4.49 | Benign | 1.00 | Tolerated | 0.1716 | 0.3850 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.1072T>A | F358I 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant F358I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, SIFT, and FATHMM; pathogenic predictions from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and ESM1b. Five tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports an uncertain impact. Overall, the preponderance of evidence points to a pathogenic effect for F358I, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.222385 | Structured | 0.407113 | Uncertain | 0.912 | 0.441 | 0.250 | -10.636 | Likely Pathogenic | 0.884 | Likely Pathogenic | Ambiguous | 0.93 | Ambiguous | 0.2 | 1.66 | Ambiguous | 1.30 | Ambiguous | 0.95 | Ambiguous | 0.393 | Likely Benign | -4.45 | Deleterious | 0.993 | Probably Damaging | 0.977 | Probably Damaging | 4.07 | Benign | 0.13 | Tolerated | 0.2331 | 0.2821 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1072T>C | F358L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 F358L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, SIFT, and FATHMM, whereas those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain or inconclusive results are reported for Rosetta, Foldetta, premPS, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, while Foldetta’s stability prediction is unavailable due to inconclusiveness. Overall, the majority of available evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.222385 | Structured | 0.407113 | Uncertain | 0.912 | 0.441 | 0.250 | -7.865 | In-Between | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.18 | Likely Benign | 0.1 | 1.62 | Ambiguous | 0.90 | Ambiguous | 0.97 | Ambiguous | 0.290 | Likely Benign | -4.21 | Deleterious | 0.982 | Probably Damaging | 0.952 | Probably Damaging | 4.12 | Benign | 0.22 | Tolerated | 0.2555 | 0.3602 | 2 | 0 | 1.0 | -34.02 | ||||||||||||||||||||||||||||||
| c.1072T>G | F358V 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant F358V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, SIFT, and FATHMM; pathogenic predictions from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and ESM1b. Five tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is also uncertain. Overall, the preponderance of evidence points to a pathogenic effect for F358V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.222385 | Structured | 0.407113 | Uncertain | 0.912 | 0.441 | 0.250 | -9.021 | Likely Pathogenic | 0.847 | Likely Pathogenic | Ambiguous | 1.42 | Ambiguous | 0.2 | 1.68 | Ambiguous | 1.55 | Ambiguous | 0.93 | Ambiguous | 0.408 | Likely Benign | -5.32 | Deleterious | 0.993 | Probably Damaging | 0.968 | Probably Damaging | 4.09 | Benign | 0.18 | Tolerated | 0.2222 | 0.2978 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1073T>A | F358Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F358Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, ESM1b, FATHMM, PROVEAN, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The high‑accuracy consensus methods reinforce the benign assessment: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a Likely Benign result (3 benign vs. 1 pathogenic), and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a benign effect. premPS is uncertain and is treated as unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.222385 | Structured | 0.407113 | Uncertain | 0.912 | 0.441 | 0.250 | -6.458 | Likely Benign | 0.606 | Likely Pathogenic | Likely Benign | 0.42 | Likely Benign | 0.1 | 0.21 | Likely Benign | 0.32 | Likely Benign | 0.84 | Ambiguous | 0.231 | Likely Benign | -2.17 | Neutral | 0.993 | Probably Damaging | 0.952 | Probably Damaging | 4.05 | Benign | 0.36 | Tolerated | 0.1467 | 0.2697 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1073T>C | F358S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 F358S variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of algorithms predict a pathogenic outcome: FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Rosetta’s assessment is uncertain and is not taken as evidence. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the preponderance of pathogenic predictions and the agreement of the high‑accuracy tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.222385 | Structured | 0.407113 | Uncertain | 0.912 | 0.441 | 0.250 | -9.316 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 2.32 | Destabilizing | 0.2 | 1.97 | Ambiguous | 2.15 | Destabilizing | 1.14 | Destabilizing | 0.493 | Likely Benign | -6.48 | Deleterious | 0.998 | Probably Damaging | 0.986 | Probably Damaging | 4.07 | Benign | 0.20 | Tolerated | 0.3891 | 0.1333 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1073T>G | F358C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F358C is not reported in ClinVar and is absent from gnomAD. Consensus from standard in‑silico predictors shows a split: benign calls come from REVEL, SIFT, and FATHMM, whereas pathogenic calls arise from Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default. High‑accuracy assessments are less definitive: AlphaMissense‑Optimized is inconclusive, Foldetta is inconclusive, and the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans toward pathogenic. Because the majority of available predictions favor a damaging effect, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.222385 | Structured | 0.407113 | Uncertain | 0.912 | 0.441 | 0.250 | -7.966 | In-Between | 0.927 | Likely Pathogenic | Ambiguous | 1.68 | Ambiguous | 0.1 | 2.19 | Destabilizing | 1.94 | Ambiguous | 1.18 | Destabilizing | 0.460 | Likely Benign | -6.36 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 4.02 | Benign | 0.06 | Tolerated | 0.2364 | 0.1800 | -4 | -2 | -0.3 | -44.04 | ||||||||||||||||||||||||||||||
| c.1074C>A | F358L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 F358L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, SIFT, and FATHMM, whereas those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain or inconclusive results are reported for Rosetta, Foldetta, premPS, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, while Foldetta’s stability prediction is unavailable due to inconclusiveness. Overall, the majority of available evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.222385 | Structured | 0.407113 | Uncertain | 0.912 | 0.441 | 0.250 | -7.865 | In-Between | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.18 | Likely Benign | 0.1 | 1.62 | Ambiguous | 0.90 | Ambiguous | 0.97 | Ambiguous | 0.215 | Likely Benign | -4.21 | Deleterious | 0.982 | Probably Damaging | 0.952 | Probably Damaging | 4.12 | Benign | 0.22 | Tolerated | 0.2555 | 0.3602 | 2 | 0 | 1.0 | -34.02 | ||||||||||||||||||||||||||||||
| c.1074C>G | F358L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 F358L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, SIFT, and FATHMM, whereas those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain or inconclusive results come from Rosetta, Foldetta, premPS, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, while Foldetta’s stability prediction is unavailable. Overall, the majority of reliable tools predict a pathogenic impact for F358L. This conclusion does not contradict ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.222385 | Structured | 0.407113 | Uncertain | 0.912 | 0.441 | 0.250 | -7.865 | In-Between | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.18 | Likely Benign | 0.1 | 1.62 | Ambiguous | 0.90 | Ambiguous | 0.97 | Ambiguous | 0.215 | Likely Benign | -4.21 | Deleterious | 0.982 | Probably Damaging | 0.952 | Probably Damaging | 4.12 | Benign | 0.22 | Tolerated | 0.2555 | 0.3602 | 2 | 0 | 1.0 | -34.02 | ||||||||||||||||||||||||||||||
| c.1075A>C | T359P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T359P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.281712 | Structured | 0.414952 | Uncertain | 0.939 | 0.480 | 0.250 | -6.884 | Likely Benign | 0.319 | Likely Benign | Likely Benign | 0.97 | Ambiguous | 0.1 | 1.27 | Ambiguous | 1.12 | Ambiguous | 0.68 | Ambiguous | 0.248 | Likely Benign | -2.36 | Neutral | 0.627 | Possibly Damaging | 0.091 | Benign | 1.78 | Pathogenic | 0.14 | Tolerated | 0.2352 | 0.5611 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1075A>G | T359A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T359A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only FATHMM predicts a pathogenic outcome, while Rosetta remains uncertain. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.281712 | Structured | 0.414952 | Uncertain | 0.939 | 0.480 | 0.250 | -2.948 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.09 | Likely Benign | 0.0 | 0.73 | Ambiguous | 0.41 | Likely Benign | 0.45 | Likely Benign | 0.103 | Likely Benign | -0.73 | Neutral | 0.000 | Benign | 0.001 | Benign | 1.79 | Pathogenic | 0.34 | Tolerated | 0.4215 | 0.4713 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1075A>T | T359S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T359S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score. Only FATHMM predicts a pathogenic outcome, while FoldX, Rosetta, Foldetta, and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also favors benign. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is uncertain. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.281712 | Structured | 0.414952 | Uncertain | 0.939 | 0.480 | 0.250 | -4.624 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.50 | Ambiguous | 0.0 | 0.73 | Ambiguous | 0.62 | Ambiguous | 0.66 | Ambiguous | 0.100 | Likely Benign | -1.87 | Neutral | 0.146 | Benign | 0.024 | Benign | 1.76 | Pathogenic | 0.27 | Tolerated | 0.3637 | 0.4996 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1076C>A | T359K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 T359K missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and FATHMM. The remaining tools—FoldX, Rosetta, premPS, and AlphaMissense‑Default—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of predictions (seven benign vs. two pathogenic) support a benign classification, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.281712 | Structured | 0.414952 | Uncertain | 0.939 | 0.480 | 0.250 | -10.822 | Likely Pathogenic | 0.517 | Ambiguous | Likely Benign | -0.65 | Ambiguous | 0.1 | 0.66 | Ambiguous | 0.01 | Likely Benign | 0.75 | Ambiguous | 0.163 | Likely Benign | -2.23 | Neutral | 0.255 | Benign | 0.045 | Benign | 1.79 | Pathogenic | 0.24 | Tolerated | 0.1618 | 0.3483 | 0 | -1 | -3.2 | 27.07 | ||||||||||||||||||||||||||||||
| c.1076C>G | T359R 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant T359R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on benign effects include REVEL, Rosetta, Foldetta, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and FATHMM. Uncertain results come from FoldX, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, more tools favor a benign outcome, but a significant minority predict pathogenicity, leaving the variant’s clinical significance unresolved. The variant is most likely benign based on the prevailing predictions, and this does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.281712 | Structured | 0.414952 | Uncertain | 0.939 | 0.480 | 0.250 | -8.675 | Likely Pathogenic | 0.402 | Ambiguous | Likely Benign | -0.74 | Ambiguous | 0.1 | -0.20 | Likely Benign | -0.47 | Likely Benign | 0.54 | Ambiguous | 0.157 | Likely Benign | -2.86 | Deleterious | 0.627 | Possibly Damaging | 0.091 | Benign | 1.75 | Pathogenic | 0.23 | Tolerated | 0.1323 | 0.3748 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.1078G>C | E360Q 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E360Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and premPS, while a majority of tools predict a pathogenic outcome: SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E360Q, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.421183 | Uncertain | 0.955 | 0.498 | 0.250 | -11.012 | Likely Pathogenic | 0.925 | Likely Pathogenic | Ambiguous | 0.55 | Ambiguous | 0.1 | 1.38 | Ambiguous | 0.97 | Ambiguous | -0.02 | Likely Benign | 0.343 | Likely Benign | -2.76 | Deleterious | 0.997 | Probably Damaging | 0.986 | Probably Damaging | 1.61 | Pathogenic | 0.03 | Affected | 0.1824 | 0.8282 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1079A>C | E360A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E360A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only premPS. All other evaluated tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict a pathogenic impact. High‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Because the majority of consensus tools predict pathogenicity and no ClinVar entry contradicts this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.421183 | Uncertain | 0.955 | 0.498 | 0.250 | -13.229 | Likely Pathogenic | 0.939 | Likely Pathogenic | Ambiguous | 1.37 | Ambiguous | 0.1 | 1.72 | Ambiguous | 1.55 | Ambiguous | 0.39 | Likely Benign | 0.545 | Likely Pathogenic | -5.52 | Deleterious | 0.997 | Probably Damaging | 0.980 | Probably Damaging | 1.63 | Pathogenic | 0.01 | Affected | 0.4295 | 0.8243 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1079A>G | E360G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E360G is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: premPS is the only tool that predicts a benign outcome, whereas all other evaluated algorithms—including REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the substitution as pathogenic. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized returns a pathogenic prediction; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a pathogenic effect. Taken together, the overwhelming majority of evidence points to a pathogenic impact for E360G, and this conclusion is consistent with the absence of a ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.421183 | Uncertain | 0.955 | 0.498 | 0.250 | -13.972 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 2.55 | Destabilizing | 0.1 | 2.99 | Destabilizing | 2.77 | Destabilizing | 0.31 | Likely Benign | 0.569 | Likely Pathogenic | -6.43 | Deleterious | 0.999 | Probably Damaging | 0.986 | Probably Damaging | 1.68 | Pathogenic | 0.04 | Affected | 0.2993 | 0.6935 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1079A>T | E360V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E360V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only premPS, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) uniformly predict a pathogenic impact. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability results and are therefore not considered evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Taken together, the overwhelming majority of reliable predictors classify E360V as pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Thus, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.421183 | Uncertain | 0.955 | 0.498 | 0.250 | -14.388 | Likely Pathogenic | 0.973 | Likely Pathogenic | Likely Pathogenic | 1.00 | Ambiguous | 0.1 | 1.11 | Ambiguous | 1.06 | Ambiguous | 0.03 | Likely Benign | 0.627 | Likely Pathogenic | -6.43 | Deleterious | 0.999 | Probably Damaging | 0.991 | Probably Damaging | 1.57 | Pathogenic | 0.00 | Affected | 0.1143 | 0.8670 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.107A>C | H36P 2D AIThe SynGAP1 missense variant H36P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -2.285 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 0.171 | Likely Benign | -1.23 | Neutral | 0.182 | Benign | 0.046 | Benign | 4.16 | Benign | 0.00 | Affected | 0.2097 | 0.4723 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.107A>G | H36R 2D AIThe SynGAP1 missense variant H36R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -2.513 | Likely Benign | 0.162 | Likely Benign | Likely Benign | 0.034 | Likely Benign | -0.70 | Neutral | 0.084 | Benign | 0.033 | Benign | 4.25 | Benign | 0.00 | Affected | 0.2417 | 0.3590 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||||||||||||||
| c.107A>T | H36L 2D AIThe SynGAP1 missense variant H36L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign classification. AlphaMissense‑Optimized also reports benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of evidence points to a benign effect. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -2.403 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -1.73 | Neutral | 0.010 | Benign | 0.011 | Benign | 4.19 | Benign | 0.00 | Affected | 0.1310 | 0.6111 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.1080G>C | E360D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E360D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, and SIFT. In contrast, a majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta give uncertain results and are therefore not considered evidence for either side. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus remains Likely Pathogenic, while Foldetta is uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E360D. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.421183 | Uncertain | 0.955 | 0.498 | 0.250 | -9.383 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 1.00 | Ambiguous | 0.0 | 0.63 | Ambiguous | 0.82 | Ambiguous | 0.11 | Likely Benign | 0.290 | Likely Benign | -2.76 | Deleterious | 0.994 | Probably Damaging | 0.970 | Probably Damaging | 1.77 | Pathogenic | 0.07 | Tolerated | 0.2339 | 0.5735 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1080G>T | E360D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E360D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, premPS, and SIFT, whereas the majority of tools predict it to be pathogenic: SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Based on the overall consensus of the available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.421183 | Uncertain | 0.955 | 0.498 | 0.250 | -9.383 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 1.00 | Ambiguous | 0.0 | 0.63 | Ambiguous | 0.82 | Ambiguous | 0.11 | Likely Benign | 0.290 | Likely Benign | -2.76 | Deleterious | 0.994 | Probably Damaging | 0.970 | Probably Damaging | 1.77 | Pathogenic | 0.07 | Tolerated | 0.2339 | 0.5735 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1081C>A | Q361K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q361K is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, FoldX, Rosetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as tolerated, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also classifies it as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic outcome. High‑accuracy tools that integrate structural and evolutionary information—AlphaMissense‑Optimized, the SGM‑Consensus, and Foldetta (combining FoldX‑MD and Rosetta outputs)—all return benign predictions. No prediction or folding‑stability result is missing or inconclusive. Based on the aggregate evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.305330 | Structured | 0.427593 | Uncertain | 0.945 | 0.534 | 0.250 | -5.147 | Likely Benign | 0.286 | Likely Benign | Likely Benign | 0.17 | Likely Benign | 0.0 | 0.36 | Likely Benign | 0.27 | Likely Benign | -0.44 | Likely Benign | 0.217 | Likely Benign | 1.29 | Neutral | 0.969 | Probably Damaging | 0.930 | Probably Damaging | 1.73 | Pathogenic | 1.00 | Tolerated | 0.1739 | 0.4701 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||
| c.1081C>G | Q361E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q361E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Uncertain predictions come from Rosetta, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, while Foldetta remains uncertain. Taken together, the majority of evidence supports a benign impact for Q361E, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.305330 | Structured | 0.427593 | Uncertain | 0.945 | 0.534 | 0.250 | -7.544 | In-Between | 0.289 | Likely Benign | Likely Benign | 0.29 | Likely Benign | 0.0 | 1.02 | Ambiguous | 0.66 | Ambiguous | 0.21 | Likely Benign | 0.153 | Likely Benign | -0.89 | Neutral | 0.969 | Probably Damaging | 0.930 | Probably Damaging | 1.70 | Pathogenic | 0.26 | Tolerated | 0.1217 | 0.3018 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.1082A>G | Q361R 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q361R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, SIFT, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Rosetta; pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus itself is benign; and Foldetta, which integrates FoldX‑MD (Uncertain) and Rosetta (Benign), yields a benign prediction. Overall, the consensus of available computational evidence indicates that the variant is most likely benign, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.305330 | Structured | 0.427593 | Uncertain | 0.945 | 0.534 | 0.250 | -6.898 | Likely Benign | 0.289 | Likely Benign | Likely Benign | -0.88 | Ambiguous | 0.0 | -0.07 | Likely Benign | -0.48 | Likely Benign | -0.20 | Likely Benign | 0.296 | Likely Benign | -0.56 | Neutral | 0.987 | Probably Damaging | 0.953 | Probably Damaging | 1.71 | Pathogenic | 0.41 | Tolerated | 0.1321 | 0.2955 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||
| c.1082A>T | Q361L 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q361L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools (8 benign vs 5 pathogenic) favor a benign effect, and this consensus does not contradict the absence of a ClinVar classification. Thus, the variant is most likely benign based on current predictions, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.305330 | Structured | 0.427593 | Uncertain | 0.945 | 0.534 | 0.250 | -10.678 | Likely Pathogenic | 0.238 | Likely Benign | Likely Benign | 0.12 | Likely Benign | 0.1 | 0.46 | Likely Benign | 0.29 | Likely Benign | 0.21 | Likely Benign | 0.406 | Likely Benign | -3.95 | Deleterious | 0.987 | Probably Damaging | 0.953 | Probably Damaging | 1.71 | Pathogenic | 0.06 | Tolerated | 0.0720 | 0.5213 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||
| c.1083G>C | Q361H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q361H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The remaining tools—FoldX, Rosetta, ESM1b, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also unavailable. Overall, the majority of conventional predictors lean toward pathogenicity, while the single high‑accuracy tool suggests benign. No ClinVar annotation exists to contradict these findings. Thus, based on the collective evidence, the variant is most likely pathogenic, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.305330 | Structured | 0.427593 | Uncertain | 0.945 | 0.534 | 0.250 | -7.269 | In-Between | 0.410 | Ambiguous | Likely Benign | 0.68 | Ambiguous | 0.1 | 0.52 | Ambiguous | 0.60 | Ambiguous | 0.32 | Likely Benign | 0.281 | Likely Benign | -2.73 | Deleterious | 0.996 | Probably Damaging | 0.986 | Probably Damaging | 1.79 | Pathogenic | 0.04 | Affected | 0.1352 | 0.4151 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||
| c.1083G>T | Q361H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q361H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The remaining tools—FoldX, Rosetta, ESM1b, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also unavailable. Overall, the majority of conventional predictors (five pathogenic vs. three benign) lean toward a pathogenic interpretation, while the single high‑accuracy tool suggests benign. No ClinVar entry exists, so there is no contradiction with clinical database status. Based on the collective evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.305330 | Structured | 0.427593 | Uncertain | 0.945 | 0.534 | 0.250 | -7.269 | In-Between | 0.410 | Ambiguous | Likely Benign | 0.68 | Ambiguous | 0.1 | 0.52 | Ambiguous | 0.60 | Ambiguous | 0.32 | Likely Benign | 0.281 | Likely Benign | -2.73 | Deleterious | 0.996 | Probably Damaging | 0.986 | Probably Damaging | 1.79 | Pathogenic | 0.04 | Affected | 0.1352 | 0.4151 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||
| c.1084T>G | W362G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W362G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD, so no population frequency data are available. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while the single uncertain call (premPS) does not alter the overall consensus. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Thus, based on the collective predictions, the variant is most likely pathogenic, and this conclusion is consistent with the absence of any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.430310 | Uncertain | 0.957 | 0.552 | 0.125 | -14.242 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 4.22 | Destabilizing | 0.1 | 5.28 | Destabilizing | 4.75 | Destabilizing | 0.95 | Ambiguous | 0.707 | Likely Pathogenic | -11.95 | Deleterious | 0.997 | Probably Damaging | 0.986 | Probably Damaging | 1.25 | Pathogenic | 0.00 | Affected | 0.4751 | 0.1281 | -7 | -2 | 0.5 | -129.16 | |||||||||||||||||||||||||||||
| c.1085G>C | W362S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W362S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as pathogenic, while premPS remains uncertain. High‑accuracy assessments corroborate this trend: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports likely pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta outputs) also indicates pathogenic. No tool predicts a benign outcome. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.430310 | Uncertain | 0.957 | 0.552 | 0.125 | -13.228 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 4.08 | Destabilizing | 0.0 | 4.55 | Destabilizing | 4.32 | Destabilizing | 0.95 | Ambiguous | 0.625 | Likely Pathogenic | -12.87 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.31 | Pathogenic | 0.00 | Affected | 0.5106 | 0.1215 | Weaken | -2 | -3 | 0.1 | -99.14 | ||||||||||||||||||||||||||||
| c.1085G>T | W362L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W362L is not reported in ClinVar and is absent from gnomAD. All tools except premPS predict pathogenic, leaving no benign predictions. Functional prediction tools uniformly indicate a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while premPS remains uncertain. High‑accuracy methods corroborate this assessment: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Consequently, the variant is most likely pathogenic, and this prediction is consistent with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.430310 | Uncertain | 0.957 | 0.552 | 0.125 | -12.748 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 2.16 | Destabilizing | 0.1 | 2.60 | Destabilizing | 2.38 | Destabilizing | 0.71 | Ambiguous | 0.523 | Likely Pathogenic | -11.95 | Deleterious | 0.997 | Probably Damaging | 0.986 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.2701 | 0.2452 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||
| c.1086G>C | W362C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W362C is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as pathogenic: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect, so the benign group is empty. High‑accuracy methods reinforce this assessment: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Consequently, the variant is most likely pathogenic based on the consensus of all predictions, and this conclusion does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.430310 | Uncertain | 0.957 | 0.552 | 0.125 | -11.200 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.68 | Destabilizing | 0.1 | 4.06 | Destabilizing | 3.87 | Destabilizing | 1.00 | Destabilizing | 0.618 | Likely Pathogenic | -11.95 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 1.24 | Pathogenic | 0.00 | Affected | 0.4158 | 0.1622 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||||
| c.1086G>T | W362C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W362C is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that assess pathogenicity uniformly classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic impact on protein stability. Based on the unanimous pathogenic predictions and the absence of any benign calls, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which simply lacks an entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.430310 | Uncertain | 0.957 | 0.552 | 0.125 | -11.200 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.68 | Destabilizing | 0.1 | 4.06 | Destabilizing | 3.87 | Destabilizing | 1.00 | Destabilizing | 0.618 | Likely Pathogenic | -11.95 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 1.24 | Pathogenic | 0.00 | Affected | 0.4158 | 0.1622 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||||
| c.1087T>A | Y363N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y363N is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify Y363N as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote) predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the overwhelming agreement among these predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.435392 | Uncertain | 0.954 | 0.586 | 0.125 | -12.121 | Likely Pathogenic | 0.958 | Likely Pathogenic | Likely Pathogenic | 2.06 | Destabilizing | 0.1 | 2.85 | Destabilizing | 2.46 | Destabilizing | 1.45 | Destabilizing | 0.477 | Likely Benign | -8.04 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.55 | Pathogenic | 0.01 | Affected | 0.3117 | 0.2021 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.1087T>C | Y363H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y363H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, while the majority of other in silico predictors (Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, Foldetta) indicate a pathogenic impact; FoldX and ESM1b are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the preponderance of evidence points to a pathogenic effect for Y363H, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.435392 | Uncertain | 0.954 | 0.586 | 0.125 | -7.003 | In-Between | 0.747 | Likely Pathogenic | Likely Benign | 1.75 | Ambiguous | 0.1 | 2.40 | Destabilizing | 2.08 | Destabilizing | 1.04 | Destabilizing | 0.419 | Likely Benign | -4.38 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.56 | Pathogenic | 0.01 | Affected | 0.3267 | 0.2021 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||
| c.1087T>G | Y363D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y363D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are none; all evaluated algorithms predict a deleterious impact. Pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions or stability results are missing or inconclusive. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.435392 | Uncertain | 0.954 | 0.586 | 0.125 | -13.840 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.86 | Destabilizing | 0.2 | 3.03 | Destabilizing | 2.95 | Destabilizing | 1.73 | Destabilizing | 0.635 | Likely Pathogenic | -8.94 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.54 | Pathogenic | 0.00 | Affected | 0.4720 | 0.1853 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1088A>C | Y363S 2D AIThe SynGAP1 missense variant Y363S is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Pathogenic”; AlphaMissense‑Optimized is classified as “Uncertain”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a pathogenic effect. Taken together, the overwhelming majority of evidence points to a pathogenic effect for Y363S, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.321458 | Structured | 0.435392 | Uncertain | 0.954 | 0.586 | 0.125 | -11.578 | Likely Pathogenic | 0.952 | Likely Pathogenic | Ambiguous | 3.09 | Destabilizing | 0.6 | 3.92 | Destabilizing | 3.51 | Destabilizing | 1.45 | Destabilizing | 0.420 | Likely Benign | -8.04 | Deleterious | 0.999 | Probably Damaging | 0.991 | Probably Damaging | 1.56 | Pathogenic | 0.01 | Affected | 0.5750 | 0.4208 | Weaken | -3 | -2 | 0.5 | -76.10 | ||||||||||||||||||||||||||||
| c.1088A>T | Y363F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y363F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, seven tools predict benign while four predict pathogenic, with no evidence of pathogenicity from the most accurate methods. Therefore, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.321458 | Structured | 0.435392 | Uncertain | 0.954 | 0.586 | 0.125 | -6.621 | Likely Benign | 0.114 | Likely Benign | Likely Benign | -0.23 | Likely Benign | 0.0 | 0.42 | Likely Benign | 0.10 | Likely Benign | 0.58 | Ambiguous | 0.293 | Likely Benign | -3.42 | Deleterious | 0.997 | Probably Damaging | 0.970 | Probably Damaging | 1.74 | Pathogenic | 0.18 | Tolerated | 0.2781 | 0.3583 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||
| c.108T>A | H36Q 2D AIThe SynGAP1 missense variant H36Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -3.642 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.16 | Neutral | 0.182 | Benign | 0.046 | Benign | 4.34 | Benign | 0.00 | Affected | 0.2042 | 0.4572 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.108T>G | H36Q 2D AIThe SynGAP1 missense variant H36Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.433974 | Uncertain | 0.334 | 0.834 | 0.375 | -3.642 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.044 | Likely Benign | -0.16 | Neutral | 0.182 | Benign | 0.046 | Benign | 4.34 | Benign | 0.00 | Affected | 0.2042 | 0.4572 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||||
| c.1090C>A | P364T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P364T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 (HumDiv and HumVar), and FATHMM. Four tools (FoldX, Foldetta, premPS, ESM1b) return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evaluated tools (five benign versus four pathogenic) lean toward a benign classification, and this assessment does not contradict any ClinVar annotation because no ClinVar claim exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.390993 | Structured | 0.439474 | Uncertain | 0.942 | 0.590 | 0.250 | -7.951 | In-Between | 0.230 | Likely Benign | Likely Benign | 1.18 | Ambiguous | 0.6 | 0.47 | Likely Benign | 0.83 | Ambiguous | 0.53 | Ambiguous | 0.342 | Likely Benign | -5.60 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 1.60 | Pathogenic | 0.09 | Tolerated | 0.1660 | 0.5726 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||
| c.1090C>G | P364A 2D 3DClick to see structure in 3D Viewer AISynGAP1 P364A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Stability‑based methods (FoldX, Rosetta, premPS) give uncertain outcomes, and Foldetta is unavailable. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, does not provide a definitive result. Overall, the predictions are discordant; the majority of tools lean toward pathogenicity, but a substantial subset suggests benign. Based on the predictions, the variant is most likely pathogenic, and this does not contradict ClinVar status because there is no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.390993 | Structured | 0.439474 | Uncertain | 0.942 | 0.590 | 0.250 | -8.077 | Likely Pathogenic | 0.135 | Likely Benign | Likely Benign | 0.79 | Ambiguous | 0.0 | 0.69 | Ambiguous | 0.74 | Ambiguous | 0.60 | Ambiguous | 0.320 | Likely Benign | -6.10 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.56 | Pathogenic | 0.06 | Tolerated | 0.3496 | 0.5186 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.1091C>G | P364R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P364R missense variant is not reported in ClinVar (status: None) and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments: AlphaMissense‑Optimized predicts benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic; Foldetta’s stability analysis is uncertain. Overall, the majority of evaluated tools (seven pathogenic vs. three benign) indicate that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.390993 | Structured | 0.439474 | Uncertain | 0.942 | 0.590 | 0.250 | -12.652 | Likely Pathogenic | 0.715 | Likely Pathogenic | Likely Benign | 0.95 | Ambiguous | 0.7 | 0.88 | Ambiguous | 0.92 | Ambiguous | 0.73 | Ambiguous | 0.416 | Likely Benign | -6.76 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 1.57 | Pathogenic | 0.12 | Tolerated | 0.1520 | 0.3977 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.1094T>A | V365E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V365E is not reported in ClinVar and is absent from gnomAD. Prediction tools uniformly indicate a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. No tool predicts a benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts pathogenic. With all available evidence pointing to a damaging effect and no ClinVar annotation to contradict, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.441505 | Uncertain | 0.923 | 0.608 | 0.250 | -16.263 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.99 | Destabilizing | 0.3 | 3.81 | Destabilizing | 3.90 | Destabilizing | 2.20 | Destabilizing | 0.613 | Likely Pathogenic | -5.02 | Deleterious | 0.989 | Probably Damaging | 0.688 | Possibly Damaging | 1.66 | Pathogenic | 0.00 | Affected | 0.0849 | 0.2083 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.1094T>C | V365A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V365A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions are made by REVEL and polyPhen‑2 HumVar, whereas the remaining tools (SGM‑Consensus, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) all predict a pathogenic effect. High‑accuracy assessments are consistent with a deleterious outcome: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a pathogenic effect. Taken together, the majority of evidence supports a pathogenic classification for V365A, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.441505 | Uncertain | 0.923 | 0.608 | 0.250 | -8.954 | Likely Pathogenic | 0.867 | Likely Pathogenic | Ambiguous | 2.61 | Destabilizing | 0.1 | 2.62 | Destabilizing | 2.62 | Destabilizing | 2.10 | Destabilizing | 0.297 | Likely Benign | -3.18 | Deleterious | 0.622 | Possibly Damaging | 0.235 | Benign | 1.67 | Pathogenic | 0.01 | Affected | 0.2682 | 0.2962 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.1094T>G | V365G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V365G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity unanimously classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign effect. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. All available evidence points to a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.414856 | Structured | 0.441505 | Uncertain | 0.923 | 0.608 | 0.250 | -13.020 | Likely Pathogenic | 0.944 | Likely Pathogenic | Ambiguous | 4.18 | Destabilizing | 0.2 | 4.83 | Destabilizing | 4.51 | Destabilizing | 2.18 | Destabilizing | 0.576 | Likely Pathogenic | -5.88 | Deleterious | 0.688 | Possibly Damaging | 0.989 | Probably Damaging | 1.66 | Pathogenic | 0.00 | Affected | 0.1835 | 0.2717 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1096A>C | T366P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T366P is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors leans toward a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign) all classify the substitution as tolerated. In contrast, polyPhen‑2 HumDiv, FATHMM, Rosetta, and the Foldetta stability analysis predict a damaging or pathogenic outcome. FoldX reports an uncertain effect and is therefore not considered evidence. High‑accuracy tools give mixed results: AlphaMissense‑Optimized and the SGM‑Consensus both indicate benign, whereas Foldetta predicts pathogenic. Overall, the majority of predictors (8 benign vs. 4 pathogenic) support a benign classification, and this is consistent with the lack of ClinVar evidence. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.441902 | Uncertain | 0.897 | 0.642 | 0.250 | -6.483 | Likely Benign | 0.226 | Likely Benign | Likely Benign | 1.75 | Ambiguous | 0.5 | 3.10 | Destabilizing | 2.43 | Destabilizing | 0.47 | Likely Benign | 0.150 | Likely Benign | -2.49 | Neutral | 0.627 | Possibly Damaging | 0.139 | Benign | 1.70 | Pathogenic | 0.24 | Tolerated | 0.2250 | 0.6251 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1096A>G | T366A 2D AIThe SynGAP1 missense variant T366A is not reported in ClinVar and is absent from gnomAD. Consensus among most in silico predictors indicates a benign effect: REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as tolerated. Only FATHMM predicts a pathogenic outcome, while Foldetta, premPS, and Rosetta are inconclusive and are treated as unavailable evidence. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign, and Foldetta provides no definitive stability change. Overall, the computational evidence overwhelmingly favors a benign classification, and this is consistent with the absence of any ClinVar assertion. Therefore, the variant is most likely benign, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.441902 | Uncertain | 0.897 | 0.642 | 0.250 | -3.983 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.5 | 0.86 | Ambiguous | 0.54 | Ambiguous | 0.51 | Ambiguous | 0.046 | Likely Benign | -0.94 | Neutral | 0.031 | Benign | 0.016 | Benign | 1.73 | Pathogenic | 0.45 | Tolerated | 0.4141 | 0.4785 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1096A>T | T366S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T366S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while Rosetta and Foldetta are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign effect. There is no conflict with ClinVar status, as the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.441902 | Uncertain | 0.897 | 0.642 | 0.250 | -3.273 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.13 | Likely Benign | 0.1 | 0.92 | Ambiguous | 0.53 | Ambiguous | 0.27 | Likely Benign | 0.058 | Likely Benign | 0.00 | Neutral | 0.005 | Benign | 0.001 | Benign | 1.73 | Pathogenic | 0.78 | Tolerated | 0.3549 | 0.4878 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1097C>A | T366N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change T366N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate a benign or likely benign outcome. Only two tools—polyPhen‑2 HumDiv and FATHMM—suggest a pathogenic effect, while premPS remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) classifies the variant as benign. Taken together, the majority of evidence supports a benign impact and is consistent with the absence of any ClinVar pathogenic annotation. Therefore, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.441902 | Uncertain | 0.897 | 0.642 | 0.250 | -5.694 | Likely Benign | 0.176 | Likely Benign | Likely Benign | 0.06 | Likely Benign | 0.0 | 0.36 | Likely Benign | 0.21 | Likely Benign | 0.92 | Ambiguous | 0.038 | Likely Benign | -1.72 | Neutral | 0.454 | Possibly Damaging | 0.038 | Benign | 1.72 | Pathogenic | 0.34 | Tolerated | 0.1595 | 0.4934 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.1097C>G | T366S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T366S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while Rosetta and Foldetta are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign effect. There is no conflict with ClinVar status, as the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.441902 | Uncertain | 0.897 | 0.642 | 0.250 | -3.273 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.13 | Likely Benign | 0.1 | 0.92 | Ambiguous | 0.53 | Ambiguous | 0.27 | Likely Benign | 0.051 | Likely Benign | 0.00 | Neutral | 0.005 | Benign | 0.001 | Benign | 1.73 | Pathogenic | 0.78 | Tolerated | 0.3549 | 0.4878 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1099C>A | L367M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L367M variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and FATHMM. The remaining predictions are uncertain: Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also likely benign; Foldetta remains inconclusive. Overall, the majority of evidence supports a benign classification, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.370445 | Structured | 0.441805 | Uncertain | 0.790 | 0.657 | 0.250 | -4.968 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.22 | Likely Benign | 0.1 | 0.92 | Ambiguous | 0.57 | Ambiguous | -0.04 | Likely Benign | 0.078 | Likely Benign | 0.12 | Neutral | 0.947 | Possibly Damaging | 0.360 | Benign | 1.63 | Pathogenic | 0.13 | Tolerated | 0.1402 | 0.3947 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.1099C>G | L367V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L367V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive, so they provide no evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the preponderance of evidence points to a benign effect for L367V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.370445 | Structured | 0.441805 | Uncertain | 0.790 | 0.657 | 0.250 | -2.383 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 1.48 | Ambiguous | 0.2 | 1.72 | Ambiguous | 1.60 | Ambiguous | 0.15 | Likely Benign | 0.040 | Likely Benign | 0.19 | Neutral | 0.410 | Benign | 0.104 | Benign | 1.67 | Pathogenic | 0.13 | Tolerated | 0.2321 | 0.3285 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.109T>A | S37T 2D  AIThe SynGAP1 missense variant S37T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumVar and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.433492 | Uncertain | 0.317 | 0.806 | 0.500 | -3.854 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -0.60 | Neutral | 0.140 | Benign | 0.481 | Possibly Damaging | 3.95 | Benign | 0.00 | Affected | 0.2093 | 0.5974 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.109T>C | S37P 2D  AIThe SynGAP1 missense variant S37P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for S37P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.433492 | Uncertain | 0.317 | 0.806 | 0.500 | -3.788 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.156 | Likely Benign | -1.29 | Neutral | 0.676 | Possibly Damaging | 0.693 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 0.2657 | 0.5327 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.109T>G | S37A 2D  AIThe SynGAP1 missense variant S37A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.433492 | Uncertain | 0.317 | 0.806 | 0.500 | -4.052 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.86 | Neutral | 0.140 | Benign | 0.355 | Benign | 3.98 | Benign | 0.00 | Affected | 0.5089 | 0.4970 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.10T>A | S4T 2D AIThe SynGAP1 missense variant S4T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, all of which are benign, and therefore SGM‑Consensus also predicts benign. AlphaMissense‑Optimized independently predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | -4.598 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.01 | Neutral | 0.140 | Benign | 0.153 | Benign | 4.18 | Benign | 0.00 | Affected | 0.1383 | 0.6572 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.10T>C | S4P 2D AIThe SynGAP1 missense variant S4P is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools largely support a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. In contrast, two tools—polyPhen‑2 HumDiv and SIFT—predict a pathogenic impact. High‑accuracy methods give a consistent benign signal: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | -4.131 | Likely Benign | 0.114 | Likely Benign | Likely Benign | 0.153 | Likely Benign | -0.33 | Neutral | 0.676 | Possibly Damaging | 0.307 | Benign | 4.12 | Benign | 0.00 | Affected | 0.2043 | 0.6112 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||||||
| c.10T>G | S4A 2D AIThe SynGAP1 missense variant S4A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S4A, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | -4.245 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.050 | Likely Benign | 0.02 | Neutral | 0.140 | Benign | 0.097 | Benign | 4.22 | Benign | 0.00 | Affected | 0.4871 | 0.5755 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.1100T>A | L367Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L367Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus also as benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.370445 | Structured | 0.441805 | Uncertain | 0.790 | 0.657 | 0.250 | -3.432 | Likely Benign | 0.150 | Likely Benign | Likely Benign | 1.09 | Ambiguous | 0.3 | 1.63 | Ambiguous | 1.36 | Ambiguous | 0.31 | Likely Benign | 0.061 | Likely Benign | 0.38 | Neutral | 0.002 | Benign | 0.002 | Benign | 1.65 | Pathogenic | 0.02 | Affected | 0.1615 | 0.0973 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1100T>C | L367P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L367P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools that agree on benign impact include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict pathogenicity are FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, SIFT, and FATHMM; premPS remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic effect. Overall, the majority of predictions lean toward a benign effect, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.370445 | Structured | 0.441805 | Uncertain | 0.790 | 0.657 | 0.250 | -2.418 | Likely Benign | 0.160 | Likely Benign | Likely Benign | 2.13 | Destabilizing | 0.4 | 4.05 | Destabilizing | 3.09 | Destabilizing | 0.72 | Ambiguous | 0.212 | Likely Benign | -0.50 | Neutral | 0.627 | Possibly Damaging | 0.196 | Benign | 1.72 | Pathogenic | 0.02 | Affected | 0.3946 | 0.1874 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1100T>G | L367R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L367R missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Those that predict a pathogenic effect are SIFT, FATHMM, and Rosetta. Tools with uncertain or inconclusive results are FoldX, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of evidence points to a benign impact, with a minority of pathogenic predictions. The variant’s status is not contradicted by ClinVar, as it is not yet classified there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.370445 | Structured | 0.441805 | Uncertain | 0.790 | 0.657 | 0.250 | -6.527 | Likely Benign | 0.515 | Ambiguous | Likely Benign | 0.68 | Ambiguous | 0.4 | 6.63 | Destabilizing | 3.66 | Destabilizing | 0.70 | Ambiguous | 0.196 | Likely Benign | -0.39 | Neutral | 0.146 | Benign | 0.057 | Benign | 1.66 | Pathogenic | 0.02 | Affected | 0.1608 | 0.1173 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||
| c.1102C>A | P368T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P368T missense variant is not listed in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. FoldX, Rosetta, and Foldetta report uncertain or inconclusive stability changes and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a tie (2 benign, 2 pathogenic) and thus inconclusive, and Foldetta remains uncertain. Overall, the predictions are evenly split between benign and pathogenic, providing no definitive classification. The variant’s status does not contradict ClinVar, which has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.363090 | Structured | 0.439989 | Uncertain | 0.580 | 0.677 | 0.250 | -5.308 | Likely Benign | 0.284 | Likely Benign | Likely Benign | 1.95 | Ambiguous | 0.6 | 1.61 | Ambiguous | 1.78 | Ambiguous | 0.45 | Likely Benign | 0.188 | Likely Benign | -5.43 | Deleterious | 0.941 | Possibly Damaging | 0.527 | Possibly Damaging | 1.72 | Pathogenic | 0.01 | Affected | 0.1983 | 0.6155 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||
| c.1102C>G | P368A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P368A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta (combining FoldX‑MD and Rosetta outputs) is also inconclusive. Overall, the balance of evidence leans toward a benign impact for P368A. This conclusion does not contradict any ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.363090 | Structured | 0.439989 | Uncertain | 0.580 | 0.677 | 0.250 | -4.608 | Likely Benign | 0.174 | Likely Benign | Likely Benign | 1.49 | Ambiguous | 0.3 | 1.47 | Ambiguous | 1.48 | Ambiguous | 0.47 | Likely Benign | 0.144 | Likely Benign | -5.42 | Deleterious | 0.767 | Possibly Damaging | 0.344 | Benign | 1.74 | Pathogenic | 0.02 | Affected | 0.3861 | 0.5635 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||
| c.1102C>T | P368S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P368S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and FATHMM. The remaining methods (FoldX, Rosetta, Foldetta, premPS) yield uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta is uncertain, so these do not alter the overall interpretation. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.363090 | Structured | 0.439989 | Uncertain | 0.580 | 0.677 | 0.250 | -4.790 | Likely Benign | 0.247 | Likely Benign | Likely Benign | 1.68 | Ambiguous | 0.4 | 1.60 | Ambiguous | 1.64 | Ambiguous | 0.52 | Ambiguous | 0.090 | Likely Benign | -5.12 | Deleterious | 0.384 | Benign | 0.113 | Benign | 1.80 | Pathogenic | 0.10 | Tolerated | 0.3700 | 0.5635 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||
| c.1103C>A | P368Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P368Q missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Remaining tools (AlphaMissense‑Default, FoldX, Rosetta, Foldetta, premPS) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of definitive predictions (five pathogenic vs. three benign) indicate that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.363090 | Structured | 0.439989 | Uncertain | 0.580 | 0.677 | 0.250 | -6.019 | Likely Benign | 0.413 | Ambiguous | Likely Benign | 1.56 | Ambiguous | 0.8 | 1.71 | Ambiguous | 1.64 | Ambiguous | 0.71 | Ambiguous | 0.205 | Likely Benign | -5.10 | Deleterious | 0.991 | Probably Damaging | 0.881 | Possibly Damaging | 1.71 | Pathogenic | 0.04 | Affected | 0.1694 | 0.5283 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||
| c.1103C>G | P368R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P368R missense variant is not reported in ClinVar (status: None) and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts benign, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is uncertain. Overall, the majority of evidence—including the SGM‑Consensus and several individual high‑accuracy tools—points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.363090 | Structured | 0.439989 | Uncertain | 0.580 | 0.677 | 0.250 | -9.564 | Likely Pathogenic | 0.736 | Likely Pathogenic | Likely Benign | 1.57 | Ambiguous | 1.0 | 1.54 | Ambiguous | 1.56 | Ambiguous | 0.58 | Ambiguous | 0.263 | Likely Benign | -6.07 | Deleterious | 0.991 | Probably Damaging | 0.881 | Possibly Damaging | 1.78 | Pathogenic | 0.00 | Affected | 0.1439 | 0.3922 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.1105A>C | T369P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T369P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only FATHMM predicts a pathogenic outcome, while FoldX and Foldetta are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact for T369P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.468512 | Structured | 0.437011 | Uncertain | 0.417 | 0.707 | 0.500 | -2.743 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 1.20 | Ambiguous | 2.1 | 0.18 | Likely Benign | 0.69 | Ambiguous | 0.17 | Likely Benign | 0.138 | Likely Benign | -2.09 | Neutral | 0.396 | Benign | 0.142 | Benign | 1.83 | Pathogenic | 0.16 | Tolerated | 0.2589 | 0.6046 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1105A>G | T369A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T369A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while Rosetta and Foldetta are inconclusive. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, SGM‑Consensus is Likely Benign, and Foldetta remains uncertain. Overall, the preponderance of evidence indicates that T369A is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.468512 | Structured | 0.437011 | Uncertain | 0.417 | 0.707 | 0.500 | -1.957 | Likely Benign | 0.056 | Likely Benign | Likely Benign | 0.09 | Likely Benign | 0.1 | 1.18 | Ambiguous | 0.64 | Ambiguous | 0.26 | Likely Benign | 0.090 | Likely Benign | -1.93 | Neutral | 0.012 | Benign | 0.016 | Benign | 1.72 | Pathogenic | 0.30 | Tolerated | 0.4538 | 0.5053 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1105A>T | T369S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T369S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. No predictions or stability results are missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.468512 | Structured | 0.437011 | Uncertain | 0.417 | 0.707 | 0.500 | -2.018 | Likely Benign | 0.071 | Likely Benign | Likely Benign | -0.07 | Likely Benign | 0.1 | 0.34 | Likely Benign | 0.14 | Likely Benign | 0.18 | Likely Benign | 0.097 | Likely Benign | -0.81 | Neutral | 0.001 | Benign | 0.001 | Benign | 1.78 | Pathogenic | 0.39 | Tolerated | 0.3746 | 0.4994 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1106C>A | T369K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T369K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FATHMM and AlphaMissense‑Default. Foldetta and Rosetta give uncertain results, which are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs two pathogenic). Foldetta’s stability prediction is uncertain. Overall, the majority of evidence (nine benign vs two pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.468512 | Structured | 0.437011 | Uncertain | 0.417 | 0.707 | 0.500 | -5.884 | Likely Benign | 0.616 | Likely Pathogenic | Likely Benign | -0.10 | Likely Benign | 0.1 | 1.13 | Ambiguous | 0.52 | Ambiguous | 0.27 | Likely Benign | 0.102 | Likely Benign | -1.87 | Neutral | 0.118 | Benign | 0.054 | Benign | 1.84 | Pathogenic | 0.34 | Tolerated | 0.1387 | 0.3855 | 0 | -1 | -3.2 | 27.07 | ||||||||||||||||||||||||||||||
| c.1106C>T | T369I 2D AIThe SynGAP1 missense variant T369I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive, providing no definitive evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact; there is no conflict with ClinVar status, which contains no entry for this variant. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.468512 | Structured | 0.437011 | Uncertain | 0.417 | 0.707 | 0.500 | -6.759 | Likely Benign | 0.289 | Likely Benign | Likely Benign | 0.60 | Ambiguous | 0.8 | 1.41 | Ambiguous | 1.01 | Ambiguous | -0.08 | Likely Benign | 0.078 | Likely Benign | -2.37 | Neutral | 0.396 | Benign | 0.142 | Benign | 1.72 | Pathogenic | 0.13 | Tolerated | 0.1106 | 0.7207 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.1108G>C | G370R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are SGM‑Consensus, FoldX, ESM1b, FATHMM, AlphaMissense‑Default, and Foldetta; Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Overall, the balance of evidence leans toward pathogenicity, with two of the three high‑accuracy tools supporting this view. The variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -8.375 | Likely Pathogenic | 0.731 | Likely Pathogenic | Likely Benign | 3.62 | Destabilizing | 3.7 | 1.72 | Ambiguous | 2.67 | Destabilizing | 0.22 | Likely Benign | 0.373 | Likely Benign | -0.80 | Neutral | 0.016 | Benign | 0.002 | Benign | 1.32 | Pathogenic | 0.55 | Tolerated | 0.0978 | 0.4313 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1108G>T | G370C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370C has no ClinVar entry and is not reported in gnomAD. Functional prediction tools fall into two groups: benign predictions come from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from REVEL, FoldX, Rosetta, Foldetta, and FATHMM. ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. No prediction or stability result is missing. Overall, the majority of tools predict a benign effect, and the high‑accuracy consensus also leans benign, while only one high‑accuracy method (Foldetta) suggests pathogenicity. Thus, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -7.071 | In-Between | 0.119 | Likely Benign | Likely Benign | 3.01 | Destabilizing | 2.1 | 2.03 | Destabilizing | 2.52 | Destabilizing | 0.29 | Likely Benign | 0.511 | Likely Pathogenic | -1.00 | Neutral | 0.353 | Benign | 0.010 | Benign | 1.32 | Pathogenic | 0.06 | Tolerated | 0.1245 | 0.4412 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1109G>A | G370D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, FATHMM, and AlphaMissense‑Default. The high‑accuracy methods give mixed results: AlphaMissense‑Optimized predicts benign, Foldetta (a folding‑stability method that integrates FoldX‑MD and Rosetta outputs) predicts pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive because it yields an equal split of benign and pathogenic calls. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -5.332 | Likely Benign | 0.597 | Likely Pathogenic | Likely Benign | 3.64 | Destabilizing | 3.8 | 0.83 | Ambiguous | 2.24 | Destabilizing | 0.30 | Likely Benign | 0.372 | Likely Benign | -0.44 | Neutral | 0.007 | Benign | 0.001 | Benign | 1.32 | Pathogenic | 0.64 | Tolerated | 0.1632 | 0.1494 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||
| c.1109G>C | G370A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions come from FoldX, Foldetta, and FATHMM, while Rosetta remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus also indicating a likely benign outcome, and Foldetta suggesting a pathogenic impact via combined FoldX‑MD and Rosetta stability analysis. Overall, the majority of evidence points to a benign effect, with only a minority of tools predicting pathogenicity. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -3.334 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 2.44 | Destabilizing | 1.3 | 1.62 | Ambiguous | 2.03 | Destabilizing | -0.14 | Likely Benign | 0.304 | Likely Benign | 0.54 | Neutral | 0.000 | Benign | 0.000 | Benign | 1.33 | Pathogenic | 0.79 | Tolerated | 0.3883 | 0.5247 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1109G>T | G370V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster into two groups: benign (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic (FoldX, Rosetta, Foldetta, and FATHMM). High‑accuracy assessments further refine this picture: AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic outcome. Overall, the majority of evidence points toward a benign impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -5.328 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 4.98 | Destabilizing | 3.8 | 5.61 | Destabilizing | 5.30 | Destabilizing | -0.43 | Likely Benign | 0.427 | Likely Benign | 0.03 | Neutral | 0.000 | Benign | 0.000 | Benign | 1.32 | Pathogenic | 0.29 | Tolerated | 0.1306 | 0.4198 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.110C>G | S37C 2D  AIThe SynGAP1 missense variant S37C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.433492 | Uncertain | 0.317 | 0.806 | 0.500 | -4.304 | Likely Benign | 0.141 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -1.18 | Neutral | 0.880 | Possibly Damaging | 0.923 | Probably Damaging | 3.89 | Benign | 0.00 | Affected | 0.1623 | 0.5775 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.110C>T | S37F 2D  AIThe SynGAP1 missense variant S37F is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT). AlphaMissense‑Default is uncertain, while the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Taken together, the preponderance of evidence from both general and high‑accuracy predictors points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.433492 | Uncertain | 0.317 | 0.806 | 0.500 | -4.258 | Likely Benign | 0.412 | Ambiguous | Likely Benign | 0.131 | Likely Benign | -1.77 | Neutral | 0.676 | Possibly Damaging | 0.828 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 0.1060 | 0.5552 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.1111A>G | S371G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S371G is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, every tool examined—REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized—classifies the substitution as benign. No pathogenic predictions are present. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | -2.073 | Likely Benign | 0.053 | Likely Benign | Likely Benign | 0.47 | Likely Benign | 0.3 | 0.49 | Likely Benign | 0.48 | Likely Benign | 0.35 | Likely Benign | 0.164 | Likely Benign | -1.32 | Neutral | 0.213 | Benign | 0.067 | Benign | 4.63 | Benign | 0.17 | Tolerated | 0.3192 | 0.5295 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||
| c.1111A>T | S371C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S371C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall consensus of the majority of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | -6.330 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.19 | Likely Benign | 0.2 | -0.34 | Likely Benign | -0.08 | Likely Benign | 0.23 | Likely Benign | 0.450 | Likely Benign | -1.41 | Neutral | 0.875 | Possibly Damaging | 0.359 | Benign | 4.61 | Benign | 0.02 | Affected | 0.1786 | 0.6580 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.1112G>A | S371N 2D  AIThe SynGAP1 missense variant S371N is not reported in ClinVar and is absent from gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while Rosetta remains uncertain. High‑accuracy assessments are consistent: AlphaMissense‑Optimized classifies the variant as benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a benign effect. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | -5.950 | Likely Benign | 0.160 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.8 | -0.59 | Ambiguous | -0.19 | Likely Benign | 0.25 | Likely Benign | 0.208 | Likely Benign | -0.31 | Neutral | 0.666 | Possibly Damaging | 0.067 | Benign | 4.64 | Benign | 0.27 | Tolerated | 0.2068 | 0.5100 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||
| c.1112G>C | S371T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S371T is not reported in ClinVar and is absent from gnomAD. All available in silico predictors classify the substitution as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). No tool predicts pathogenicity. High‑accuracy assessments concur: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | -4.512 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.38 | Likely Benign | 0.1 | -0.27 | Likely Benign | 0.06 | Likely Benign | 0.05 | Likely Benign | 0.173 | Likely Benign | -0.65 | Neutral | 0.213 | Benign | 0.067 | Benign | 4.64 | Benign | 0.22 | Tolerated | 0.2245 | 0.6462 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1112G>T | S371I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S371I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from FoldX and Rosetta, which are treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a benign effect. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | -6.888 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.50 | Ambiguous | 0.4 | -0.50 | Ambiguous | 0.00 | Likely Benign | -0.11 | Likely Benign | 0.433 | Likely Benign | -1.06 | Neutral | 0.028 | Benign | 0.016 | Benign | 4.62 | Benign | 0.07 | Tolerated | 0.1423 | 0.5924 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.1114G>A | G372R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G372R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are REVEL, Rosetta, Foldetta, SIFT, FATHMM, and AlphaMissense‑Default. Two tools, FoldX and ESM1b, returned uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicting pathogenic, and Foldetta predicting pathogenic. Overall, the majority of predictions (seven pathogenic vs. five benign) and the consensus of high‑accuracy methods indicate a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.433034 | Structured | 0.430335 | Uncertain | 0.322 | 0.774 | 0.375 | -7.344 | In-Between | 0.617 | Likely Pathogenic | Likely Benign | 1.49 | Ambiguous | 0.3 | 2.87 | Destabilizing | 2.18 | Destabilizing | 0.20 | Likely Benign | 0.572 | Likely Pathogenic | -0.61 | Neutral | 0.001 | Benign | 0.001 | Benign | -0.74 | Pathogenic | 0.02 | Affected | 0.1323 | 0.4313 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1114G>C | G372R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G372R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, Rosetta, Foldetta, SIFT, FATHMM, and AlphaMissense‑Default. Two tools (FoldX and ESM1b) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as pathogenic. Overall, the majority of predictions (seven pathogenic vs five benign) indicate that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.433034 | Structured | 0.430335 | Uncertain | 0.322 | 0.774 | 0.375 | -7.344 | In-Between | 0.617 | Likely Pathogenic | Likely Benign | 1.49 | Ambiguous | 0.3 | 2.87 | Destabilizing | 2.18 | Destabilizing | 0.20 | Likely Benign | 0.572 | Likely Pathogenic | -0.61 | Neutral | 0.001 | Benign | 0.001 | Benign | -0.74 | Pathogenic | 0.02 | Affected | 0.1323 | 0.4313 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1114G>T | G372W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G372W has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include premPS, PROVEAN, polyPhen‑2 HumVar, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are REVEL, FoldX, polyPhen‑2 HumDiv, SIFT, ESM1b, and FATHMM. The remaining tools (Rosetta, Foldetta, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.433034 | Structured | 0.430335 | Uncertain | 0.322 | 0.774 | 0.375 | -8.262 | Likely Pathogenic | 0.478 | Ambiguous | Likely Benign | 2.28 | Destabilizing | 0.5 | 1.14 | Ambiguous | 1.71 | Ambiguous | 0.21 | Likely Benign | 0.649 | Likely Pathogenic | -1.25 | Neutral | 0.657 | Possibly Damaging | 0.075 | Benign | -0.74 | Pathogenic | 0.00 | Affected | 0.1057 | 0.4368 | -7 | -2 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||
| c.1115G>A | G372E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G372E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are REVEL, Rosetta, Foldetta, FATHMM, and AlphaMissense‑Default; FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while Foldetta predicts pathogenic. The SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split and is treated as unavailable. Overall, the majority of evidence (seven benign vs. five pathogenic) supports a benign classification. This conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.433034 | Structured | 0.430335 | Uncertain | 0.322 | 0.774 | 0.375 | -6.682 | Likely Benign | 0.604 | Likely Pathogenic | Likely Benign | 1.58 | Ambiguous | 0.4 | 2.91 | Destabilizing | 2.25 | Destabilizing | 0.34 | Likely Benign | 0.566 | Likely Pathogenic | -0.81 | Neutral | 0.001 | Benign | 0.001 | Benign | -0.74 | Pathogenic | 0.08 | Tolerated | 0.1606 | 0.4103 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||
| c.1115G>C | G372A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G372A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. FoldX and Rosetta individually also return uncertain results. Overall, the majority of evidence points to a benign impact for G372A, and this conclusion does not contradict any ClinVar status, as the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.433034 | Structured | 0.430335 | Uncertain | 0.322 | 0.774 | 0.375 | -5.163 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 1.52 | Ambiguous | 0.2 | 1.58 | Ambiguous | 1.55 | Ambiguous | -0.12 | Likely Benign | 0.465 | Likely Benign | -0.32 | Neutral | 0.000 | Benign | 0.000 | Benign | -0.74 | Pathogenic | 0.03 | Affected | 0.3956 | 0.5247 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1118G>A | G373E 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G373E is listed in ClinVar with an Uncertain significance and is not present in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are FoldX, Foldetta, SIFT, and AlphaMissense‑Default. Predictions from Rosetta and ESM1b are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as pathogenic. Overall, the majority of evidence points to a benign impact, which does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.529623 | Disordered | 0.429267 | Uncertain | 0.295 | 0.799 | 0.625 | Uncertain | 1 | -7.281 | In-Between | 0.569 | Likely Pathogenic | Likely Benign | 4.13 | Destabilizing | 3.2 | 0.52 | Ambiguous | 2.33 | Destabilizing | -0.02 | Likely Benign | 0.420 | Likely Benign | -0.69 | Neutral | 0.001 | Benign | 0.000 | Benign | 3.90 | Benign | 0.01 | Affected | 0.1572 | 0.4309 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||
| c.1118G>C | G373A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G373A is not reported in ClinVar and is absent from gnomAD. Functional prediction consensus shows a predominance of benign calls: REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all predict a benign effect. Pathogenic predictions are limited to SIFT and FoldX, while Rosetta and Foldetta yield uncertain results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta remains inconclusive. Overall, the majority of evidence indicates that G373A is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.529623 | Disordered | 0.429267 | Uncertain | 0.295 | 0.799 | 0.625 | -5.181 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 2.44 | Destabilizing | 0.8 | 0.69 | Ambiguous | 1.57 | Ambiguous | -0.01 | Likely Benign | 0.227 | Likely Benign | -0.47 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.93 | Benign | 0.01 | Affected | 0.4172 | 0.5053 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1120T>A | S374T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S374T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on benign impact include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain but does not indicate destabilization. Overall, the evidence strongly favors a benign effect for S374T, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.642678 | Disordered | 0.428948 | Uncertain | 0.333 | 0.812 | 0.625 | -5.415 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 0.39 | Likely Benign | 0.1 | 0.80 | Ambiguous | 0.60 | Ambiguous | -0.02 | Likely Benign | 0.176 | Likely Benign | -0.47 | Neutral | 0.118 | Benign | 0.049 | Benign | 5.32 | Benign | 0.12 | Tolerated | 0.2212 | 0.6466 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1121C>T | S374F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 S374F missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions come from polyPhen‑2 HumDiv and SIFT. Uncertain or inconclusive results are reported for FoldX, Rosetta, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus also indicates a likely benign outcome, while Foldetta’s stability analysis remains uncertain. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.642678 | Disordered | 0.428948 | Uncertain | 0.333 | 0.812 | 0.625 | -7.907 | In-Between | 0.268 | Likely Benign | Likely Benign | 0.55 | Ambiguous | 0.8 | 0.60 | Ambiguous | 0.58 | Ambiguous | -0.19 | Likely Benign | 0.202 | Likely Benign | -1.19 | Neutral | 0.875 | Possibly Damaging | 0.271 | Benign | 6.29 | Benign | 0.00 | Affected | 0.1045 | 0.6427 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||
| c.1123G>A | G375R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G375R is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized, whereas tools that predict pathogenicity are FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of predictions (7 pathogenic vs. 5 benign) indicate a likely pathogenic effect, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.604312 | Disordered | 0.428340 | Uncertain | 0.301 | 0.836 | 0.625 | -8.955 | Likely Pathogenic | 0.609 | Likely Pathogenic | Likely Benign | 2.97 | Destabilizing | 1.3 | 12.66 | Destabilizing | 7.82 | Destabilizing | 0.36 | Likely Benign | 0.497 | Likely Benign | -1.15 | Neutral | 0.845 | Possibly Damaging | 0.523 | Possibly Damaging | 1.32 | Pathogenic | 0.11 | Tolerated | 0.1335 | 0.4513 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1123G>C | G375R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G375R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized; pathogenic predictions come from FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts benign, whereas Foldetta indicates a destabilizing, pathogenic change, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.604312 | Disordered | 0.428340 | Uncertain | 0.301 | 0.836 | 0.625 | -8.955 | Likely Pathogenic | 0.609 | Likely Pathogenic | Likely Benign | 2.97 | Destabilizing | 1.3 | 12.66 | Destabilizing | 7.82 | Destabilizing | 0.36 | Likely Benign | 0.497 | Likely Benign | -1.15 | Neutral | 0.845 | Possibly Damaging | 0.523 | Possibly Damaging | 1.32 | Pathogenic | 0.11 | Tolerated | 0.1335 | 0.4513 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1124G>A | G375E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G375E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, FATHMM, and AlphaMissense‑Default; ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) favors pathogenic, and Foldetta also predicts pathogenic. Overall, the majority of tools, including the high‑accuracy ones, indicate a pathogenic impact. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.604312 | Disordered | 0.428340 | Uncertain | 0.301 | 0.836 | 0.625 | -7.780 | In-Between | 0.600 | Likely Pathogenic | Likely Benign | 2.89 | Destabilizing | 1.4 | 9.47 | Destabilizing | 6.18 | Destabilizing | 0.45 | Likely Benign | 0.545 | Likely Pathogenic | -1.07 | Neutral | 0.845 | Possibly Damaging | 0.244 | Benign | 1.32 | Pathogenic | 0.09 | Tolerated | 0.1619 | 0.4299 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||
| c.1124G>C | G375A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G375A is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic impact. Overall, the majority of evidence points to a benign effect; this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.604312 | Disordered | 0.428340 | Uncertain | 0.301 | 0.836 | 0.625 | -5.986 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 2.52 | Destabilizing | 1.0 | 3.16 | Destabilizing | 2.84 | Destabilizing | -0.09 | Likely Benign | 0.419 | Likely Benign | -0.61 | Neutral | 0.020 | Benign | 0.008 | Benign | 1.33 | Pathogenic | 0.27 | Tolerated | 0.3991 | 0.5242 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1124G>T | G375V 2D AIThe SynGAP1 missense variant G375V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions (premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic predictions (REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, and FATHMM). High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also benign, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a pathogenic effect. No prediction is missing or inconclusive. Overall, the majority of tools and the consensus score suggest a benign effect, but the Foldetta result introduces uncertainty. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.604312 | Disordered | 0.428340 | Uncertain | 0.301 | 0.836 | 0.625 | -6.149 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 3.93 | Destabilizing | 3.5 | 7.55 | Destabilizing | 5.74 | Destabilizing | 0.02 | Likely Benign | 0.547 | Likely Pathogenic | -0.92 | Neutral | 0.845 | Possibly Damaging | 0.186 | Benign | 1.32 | Pathogenic | 0.06 | Tolerated | 0.1653 | 0.4193 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1126G>C | G376R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G376R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include Rosetta, premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are SGM‑Consensus, REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Foldetta reports an uncertain outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of tools (8 pathogenic vs. 5 benign) and the consensus from high‑accuracy methods lean toward a pathogenic interpretation. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | -8.500 | Likely Pathogenic | 0.658 | Likely Pathogenic | Likely Benign | 3.48 | Destabilizing | 1.3 | -0.46 | Likely Benign | 1.51 | Ambiguous | 0.30 | Likely Benign | 0.589 | Likely Pathogenic | -1.21 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.32 | Pathogenic | 0.09 | Tolerated | 0.1316 | 0.4027 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1126G>T | G376C 2D AISynGAP1 missense variant G376C is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from Rosetta, premPS, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls come from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Two tools report uncertainty: Foldetta and ESM1b. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also yields a benign verdict; Foldetta remains uncertain. Overall, the majority of conventional predictors lean toward pathogenicity, whereas the most accurate methods favor a benign effect. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | Uncertain | 1 | -7.686 | In-Between | 0.125 | Likely Benign | Likely Benign | 2.56 | Destabilizing | 0.5 | 0.22 | Likely Benign | 1.39 | Ambiguous | 0.16 | Likely Benign | 0.560 | Likely Pathogenic | -1.15 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1476 | 0.3929 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||
| c.1127G>A | G376D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G376D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. The remaining tools—Rosetta, Foldetta, premPS, and ESM1b—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicting pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) yielding an uncertain stability change. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | -7.125 | In-Between | 0.569 | Likely Pathogenic | Likely Benign | 3.10 | Destabilizing | 1.1 | -1.08 | Ambiguous | 1.01 | Ambiguous | 0.52 | Ambiguous | 0.572 | Likely Pathogenic | -1.05 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.32 | Pathogenic | 0.09 | Tolerated | 0.1938 | 0.1235 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||
| c.1127G>C | G376A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G376A missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. FoldX and Rosetta give uncertain results and are not considered evidence for either side. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign; and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts benign. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | -6.016 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 1.74 | Ambiguous | 0.3 | -0.84 | Ambiguous | 0.45 | Likely Benign | 0.00 | Likely Benign | 0.392 | Likely Benign | -0.44 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 1.33 | Pathogenic | 0.03 | Affected | 0.3868 | 0.4465 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1127G>T | G376V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G376V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions arise from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The consensus score from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, matching the majority of individual benign calls. High‑accuracy methods give mixed results: AlphaMissense‑Optimized predicts benign, SGM Consensus predicts benign, whereas Foldetta (integrating FoldX‑MD and Rosetta) predicts pathogenic. No prediction is missing or inconclusive. Overall, the balance of evidence leans toward a benign effect; this is consistent with the lack of ClinVar annotation and gnomAD presence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | -6.242 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 4.84 | Destabilizing | 0.8 | -0.81 | Ambiguous | 2.02 | Destabilizing | -0.18 | Likely Benign | 0.541 | Likely Pathogenic | -0.66 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1594 | 0.3525 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.112C>A | P38T 2D AIThe SynGAP1 missense variant P38T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.433285 | Uncertain | 0.344 | 0.791 | 0.375 | -3.248 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -1.91 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.06 | Benign | 0.00 | Affected | 0.1993 | 0.6717 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.112C>G | P38A 2D AIThe SynGAP1 missense variant P38A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P38A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.433285 | Uncertain | 0.344 | 0.791 | 0.375 | -3.179 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -2.03 | Neutral | 0.805 | Possibly Damaging | 0.857 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 0.3888 | 0.5977 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.112C>T | P38S 2D AIThe SynGAP1 missense variant P38S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.433285 | Uncertain | 0.344 | 0.791 | 0.375 | -2.727 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -2.13 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.3837 | 0.6111 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||||
| c.1130T>A | M377K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M377K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are Rosetta and Foldetta. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome, while premPS remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the absence of a ClinVar classification. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.675549 | Disordered | 0.431183 | Uncertain | 0.324 | 0.884 | 0.625 | -3.718 | Likely Benign | 0.226 | Likely Benign | Likely Benign | 0.20 | Likely Benign | 0.2 | 4.44 | Destabilizing | 2.32 | Destabilizing | 0.62 | Ambiguous | 0.440 | Likely Benign | -0.26 | Neutral | 0.002 | Benign | 0.003 | Benign | 5.46 | Benign | 0.07 | Tolerated | 0.2423 | 0.1671 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||
| c.1132G>A | G378S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G378S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as pathogenic, giving a mixed outcome. Because the predictions are evenly split and the high‑accuracy methods are contradictory, the variant’s functional impact is uncertain. Based on the available evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.767246 | Disordered | 0.432858 | Uncertain | 0.341 | 0.915 | 0.625 | -5.331 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 6.75 | Destabilizing | 2.2 | 7.81 | Destabilizing | 7.28 | Destabilizing | 0.12 | Likely Benign | 0.565 | Likely Pathogenic | -0.63 | Neutral | 1.000 | Probably Damaging | 0.986 | Probably Damaging | 1.33 | Pathogenic | 0.08 | Tolerated | 0.3013 | 0.4724 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1132G>C | G378R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G378R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from premPS, PROVEAN, and SIFT, while pathogenic predictions arise from REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts benign, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also indicates pathogenicity. Overall, the preponderance of evidence points to a pathogenic impact for G378R. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.767246 | Disordered | 0.432858 | Uncertain | 0.341 | 0.915 | 0.625 | -8.863 | Likely Pathogenic | 0.745 | Likely Pathogenic | Likely Benign | 12.27 | Destabilizing | 6.3 | 13.17 | Destabilizing | 12.72 | Destabilizing | 0.12 | Likely Benign | 0.653 | Likely Pathogenic | -0.96 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 1.32 | Pathogenic | 0.06 | Tolerated | 0.1343 | 0.4356 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1132G>T | G378C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G378C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of tools (8/12) predict pathogenicity, and the high‑accuracy consensus leans toward benign only for AlphaMissense‑Optimized and SGM Consensus, while Foldetta indicates instability. Thus, the variant is most likely pathogenic based on the preponderance of evidence, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.767246 | Disordered | 0.432858 | Uncertain | 0.341 | 0.915 | 0.625 | -7.981 | In-Between | 0.203 | Likely Benign | Likely Benign | 7.63 | Destabilizing | 2.7 | 12.14 | Destabilizing | 9.89 | Destabilizing | 0.26 | Likely Benign | 0.635 | Likely Pathogenic | -1.18 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1595 | 0.4240 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1133G>C | G378A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G378A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as “Likely Benign.” High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts benign, SGM‑Consensus confirms a benign likelihood, while Foldetta—combining FoldX‑MD and Rosetta outputs—predicts a pathogenic effect on protein folding stability. Overall, the majority of evidence points toward a benign impact, and this conclusion is consistent with the absence of ClinVar annotation and gnomAD data. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.767246 | Disordered | 0.432858 | Uncertain | 0.341 | 0.915 | 0.625 | -6.450 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 5.06 | Destabilizing | 1.3 | 6.00 | Destabilizing | 5.53 | Destabilizing | -0.04 | Likely Benign | 0.497 | Likely Benign | -0.55 | Neutral | 0.999 | Probably Damaging | 0.981 | Probably Damaging | 1.33 | Pathogenic | 0.18 | Tolerated | 0.4062 | 0.4576 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1135T>A | S379T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S379T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are Rosetta, polyPhen‑2 HumDiv, and the Foldetta stability method. FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as pathogenic. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.433206 | Uncertain | 0.327 | 0.931 | 0.625 | -5.646 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 1.42 | Ambiguous | 0.6 | 3.96 | Destabilizing | 2.69 | Destabilizing | 0.06 | Likely Benign | 0.230 | Likely Benign | -0.50 | Neutral | 0.462 | Possibly Damaging | 0.084 | Benign | 3.87 | Benign | 0.16 | Tolerated | 0.2293 | 0.6248 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1135T>C | S379P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S379P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, Rosetta, and Foldetta; FoldX is uncertain. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, whereas Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of evidence—including the high‑accuracy benign predictions—suggests that the variant is most likely benign. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.433206 | Uncertain | 0.327 | 0.931 | 0.625 | -5.007 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 1.10 | Ambiguous | 0.8 | 2.92 | Destabilizing | 2.01 | Destabilizing | 0.17 | Likely Benign | 0.430 | Likely Benign | -0.41 | Neutral | 0.808 | Possibly Damaging | 0.212 | Benign | 3.83 | Benign | 0.10 | Tolerated | 0.3035 | 0.6594 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.1138G>A | G380R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G380R missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized, while those that agree on a pathogenic effect are REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, Foldetta predicts a pathogenic outcome, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of evidence (10 pathogenic vs. 4 benign) points to a pathogenic impact. The variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.724957 | Disordered | 0.432982 | Uncertain | 0.316 | 0.939 | 0.750 | -9.205 | Likely Pathogenic | 0.635 | Likely Pathogenic | Likely Benign | 5.94 | Destabilizing | 2.6 | 0.90 | Ambiguous | 3.42 | Destabilizing | 0.11 | Likely Benign | 0.640 | Likely Pathogenic | -0.86 | Neutral | 0.940 | Possibly Damaging | 0.459 | Possibly Damaging | 2.53 | Benign | 0.01 | Affected | 0.1319 | 0.3956 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1138G>C | G380R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G380R missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized, while those that agree on a pathogenic effect are REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, Foldetta predicts pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of tools (10 pathogenic vs 4 benign) and the Foldetta result support a pathogenic classification. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.724957 | Disordered | 0.432982 | Uncertain | 0.316 | 0.939 | 0.750 | -9.205 | Likely Pathogenic | 0.635 | Likely Pathogenic | Likely Benign | 5.94 | Destabilizing | 2.6 | 0.90 | Ambiguous | 3.42 | Destabilizing | 0.11 | Likely Benign | 0.619 | Likely Pathogenic | -0.86 | Neutral | 0.940 | Possibly Damaging | 0.459 | Possibly Damaging | 2.53 | Benign | 0.01 | Affected | 0.1319 | 0.3956 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1139G>A | G380E 2D AIThe SynGAP1 missense variant G380E has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, FoldX, Foldetta, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give mixed results: AlphaMissense‑Optimized classifies the variant as benign, Foldetta predicts a pathogenic impact on protein stability, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of tools (7 benign vs. 6 pathogenic) lean toward a benign interpretation, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Thus, based on current computational predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.724957 | Disordered | 0.432982 | Uncertain | 0.316 | 0.939 | 0.750 | -9.334 | Likely Pathogenic | 0.617 | Likely Pathogenic | Likely Benign | 6.25 | Destabilizing | 5.1 | -0.28 | Likely Benign | 2.99 | Destabilizing | 0.24 | Likely Benign | 0.582 | Likely Pathogenic | -0.86 | Neutral | 0.056 | Benign | 0.010 | Benign | 2.53 | Benign | 0.03 | Affected | 0.1585 | 0.3741 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||
| c.1139G>C | G380A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G380A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FoldX predicts a pathogenic outcome, while Rosetta and Foldetta are benign or uncertain, respectively. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta as uncertain. Overall, the overwhelming majority of evidence points to a benign effect, with no conflict with ClinVar status (which has no entry for this variant). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.724957 | Disordered | 0.432982 | Uncertain | 0.316 | 0.939 | 0.750 | -6.433 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 3.11 | Destabilizing | 1.0 | -0.15 | Likely Benign | 1.48 | Ambiguous | -0.06 | Likely Benign | 0.422 | Likely Benign | -0.53 | Neutral | 0.056 | Benign | 0.010 | Benign | 2.56 | Benign | 0.17 | Tolerated | 0.3915 | 0.4576 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1139G>T | G380V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G380V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions are reported by REVEL, FoldX, polyPhen‑2 HumDiv, and SIFT; Rosetta is uncertain. The SGM‑Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority of the four high‑accuracy tools) is benign, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a pathogenic impact. Overall, the majority of evidence points to a benign effect, and this does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.724957 | Disordered | 0.432982 | Uncertain | 0.316 | 0.939 | 0.750 | -6.234 | Likely Benign | 0.172 | Likely Benign | Likely Benign | 7.60 | Destabilizing | 2.5 | 1.75 | Ambiguous | 4.68 | Destabilizing | -0.17 | Likely Benign | 0.574 | Likely Pathogenic | -0.70 | Neutral | 0.816 | Possibly Damaging | 0.210 | Benign | 2.54 | Benign | 0.02 | Affected | 0.1618 | 0.3708 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.113C>A | P38Q 2D AIThe SynGAP1 missense variant P38Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P38Q, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.433285 | Uncertain | 0.344 | 0.791 | 0.375 | -3.520 | Likely Benign | 0.183 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -2.16 | Neutral | 0.989 | Probably Damaging | 0.975 | Probably Damaging | 4.00 | Benign | 0.00 | Affected | 0.1737 | 0.5521 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||||||||||||
| c.113C>G | P38R 2D AIThe SynGAP1 missense variant P38R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for P38R, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.497853 | Structured | 0.433285 | Uncertain | 0.344 | 0.791 | 0.375 | -1.799 | Likely Benign | 0.314 | Likely Benign | Likely Benign | 0.168 | Likely Benign | -2.29 | Neutral | 0.989 | Probably Damaging | 0.975 | Probably Damaging | 4.00 | Benign | 0.00 | Affected | 0.1620 | 0.4060 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.1142G>A | G381E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G381E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, FoldX, Foldetta, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Two tools give uncertain results: Rosetta and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as pathogenic. With seven pathogenic versus four benign predictions and two high‑accuracy tools supporting pathogenicity, the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.724957 | Disordered | 0.431692 | Uncertain | 0.301 | 0.951 | 0.750 | -9.360 | Likely Pathogenic | 0.540 | Ambiguous | Likely Benign | 5.52 | Destabilizing | 1.1 | 0.53 | Ambiguous | 3.03 | Destabilizing | 0.24 | Likely Benign | 0.588 | Likely Pathogenic | -0.71 | Neutral | 0.985 | Probably Damaging | 0.720 | Possibly Damaging | 1.32 | Pathogenic | 0.11 | Tolerated | 0.1554 | 0.3735 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||
| c.1144G>A | G382R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G382R is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include premPS, PROVEAN, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts a benign change, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta stability outputs) both predict pathogenicity. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.728858 | Disordered | 0.429690 | Uncertain | 0.315 | 0.951 | 0.750 | -8.997 | Likely Pathogenic | 0.654 | Likely Pathogenic | Likely Benign | 4.53 | Destabilizing | 1.6 | 8.03 | Destabilizing | 6.28 | Destabilizing | 0.23 | Likely Benign | 0.595 | Likely Pathogenic | -0.95 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 1.32 | Pathogenic | 0.02 | Affected | 0.1295 | 0.4346 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1144G>C | G382R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G382R is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include premPS, PROVEAN, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts a benign change, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta stability outputs) both predict pathogenicity. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.728858 | Disordered | 0.429690 | Uncertain | 0.315 | 0.951 | 0.750 | -8.997 | Likely Pathogenic | 0.654 | Likely Pathogenic | Likely Benign | 4.53 | Destabilizing | 1.6 | 8.03 | Destabilizing | 6.28 | Destabilizing | 0.23 | Likely Benign | 0.570 | Likely Pathogenic | -0.95 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 1.32 | Pathogenic | 0.02 | Affected | 0.1295 | 0.4346 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1145G>C | G382A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G382A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic impact. Overall, the majority of predictions lean toward a benign effect, and this consensus does not contradict any ClinVar status (none available). Thus, based on the available computational evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429690 | Uncertain | 0.315 | 0.951 | 0.750 | -6.323 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 2.90 | Destabilizing | 0.6 | 3.00 | Destabilizing | 2.95 | Destabilizing | -0.03 | Likely Benign | 0.495 | Likely Benign | -0.59 | Neutral | 0.953 | Possibly Damaging | 0.952 | Probably Damaging | 1.33 | Pathogenic | 0.12 | Tolerated | 0.4048 | 0.4576 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1145G>T | G382V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G382V has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a split opinion: benign predictions come from PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions are returned by REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM, while premPS is uncertain. High‑accuracy assessments give a mixed signal: AlphaMissense‑Optimized predicts benign, SGM‑Consensus indicates likely benign, but Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of tools lean toward pathogenicity, and the high‑accuracy Foldetta result supports this. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429690 | Uncertain | 0.315 | 0.951 | 0.750 | -5.988 | Likely Benign | 0.155 | Likely Benign | Likely Benign | 6.40 | Destabilizing | 1.6 | 9.28 | Destabilizing | 7.84 | Destabilizing | -0.53 | Ambiguous | 0.571 | Likely Pathogenic | -0.54 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 1.32 | Pathogenic | 0.03 | Affected | 0.1631 | 0.3708 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1147G>A | G383R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G383R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that agree on a pathogenic effect are FoldX, Rosetta, Foldetta, polyPhen2_HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, Foldetta predicts a pathogenic outcome, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs. two benign). Overall, the majority of predictions lean toward pathogenicity, and this does not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.728858 | Disordered | 0.429104 | Uncertain | 0.296 | 0.949 | 0.750 | -9.067 | Likely Pathogenic | 0.660 | Likely Pathogenic | Likely Benign | 4.03 | Destabilizing | 2.3 | 3.36 | Destabilizing | 3.70 | Destabilizing | 0.29 | Likely Benign | 0.449 | Likely Benign | -0.84 | Neutral | 0.498 | Possibly Damaging | 0.119 | Benign | 4.10 | Benign | 0.00 | Affected | 0.1295 | 0.3741 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1147G>C | G383R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G383R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that agree on a pathogenic effect are FoldX, Rosetta, Foldetta, polyPhen2_HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, Foldetta predicts a pathogenic outcome, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs. two benign). Overall, the majority of predictions lean toward pathogenicity, and this does not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.728858 | Disordered | 0.429104 | Uncertain | 0.296 | 0.949 | 0.750 | -9.067 | Likely Pathogenic | 0.660 | Likely Pathogenic | Likely Benign | 4.03 | Destabilizing | 2.3 | 3.36 | Destabilizing | 3.70 | Destabilizing | 0.29 | Likely Benign | 0.440 | Likely Benign | -0.84 | Neutral | 0.498 | Possibly Damaging | 0.119 | Benign | 4.10 | Benign | 0.00 | Affected | 0.1295 | 0.3741 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1148G>A | G383E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G383E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and FATHMM. Tools that predict a pathogenic effect are FoldX, Rosetta, SIFT, ESM1b, AlphaMissense‑Default, and the protein‑folding stability method Foldetta. High‑accuracy assessments give a benign result from AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (pathogenic), FATHMM (benign), and PROVEAN (benign), is inconclusive (tie). Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a pathogenic effect. Overall, the majority of predictions (seven pathogenic vs. six benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.728858 | Disordered | 0.429104 | Uncertain | 0.296 | 0.949 | 0.750 | -9.535 | Likely Pathogenic | 0.570 | Likely Pathogenic | Likely Benign | 4.09 | Destabilizing | 2.4 | 2.62 | Destabilizing | 3.36 | Destabilizing | 0.39 | Likely Benign | 0.308 | Likely Benign | -0.78 | Neutral | 0.000 | Benign | 0.002 | Benign | 4.10 | Benign | 0.01 | Affected | 0.1582 | 0.3741 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||
| c.1148G>C | G383A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G383A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as pathogenic. Thus, the majority of evidence points to a benign impact, with only a minority of high‑accuracy tools suggesting pathogenicity. The variant is most likely benign based on the overall predictions, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429104 | Uncertain | 0.296 | 0.949 | 0.750 | -6.205 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 3.17 | Destabilizing | 1.2 | 3.47 | Destabilizing | 3.32 | Destabilizing | 0.04 | Likely Benign | 0.178 | Likely Benign | -0.64 | Neutral | 0.055 | Benign | 0.037 | Benign | 4.20 | Benign | 0.11 | Tolerated | 0.3880 | 0.4361 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1148G>T | G383V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G383V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect are FoldX, Rosetta, polyPhen‑2 HumDiv, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of predictions lean toward a benign impact, and this conclusion does not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429104 | Uncertain | 0.296 | 0.949 | 0.750 | -5.769 | Likely Benign | 0.145 | Likely Benign | Likely Benign | 5.13 | Destabilizing | 2.1 | 4.06 | Destabilizing | 4.60 | Destabilizing | -0.26 | Likely Benign | 0.406 | Likely Benign | -0.72 | Neutral | 0.668 | Possibly Damaging | 0.207 | Benign | 4.12 | Benign | 0.01 | Affected | 0.1597 | 0.3493 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1150G>C | G384R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G384R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, premPS, PROVEAN, and AlphaMissense‑Optimized, whereas pathogenic calls are made by FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts benign, but SGM‑Consensus and Foldetta both predict pathogenic, with Foldetta integrating FoldX‑MD and Rosetta stability outputs. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.728858 | Disordered | 0.427831 | Uncertain | 0.323 | 0.934 | 0.750 | -9.186 | Likely Pathogenic | 0.719 | Likely Pathogenic | Likely Benign | 2.16 | Destabilizing | 0.4 | 5.06 | Destabilizing | 3.61 | Destabilizing | 0.25 | Likely Benign | 0.475 | Likely Benign | -0.96 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 1.32 | Pathogenic | 0.02 | Affected | 0.1250 | 0.4156 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1150G>T | G384C 2D AIThe SynGAP1 missense variant G384C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic; and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split and is treated as unavailable. Overall, the majority of predictions (seven pathogenic vs. five benign) and the pathogenic Foldetta result indicate that the variant is most likely pathogenic, with no contradiction to ClinVar status because the variant is not yet classified there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.728858 | Disordered | 0.427831 | Uncertain | 0.323 | 0.934 | 0.750 | -8.363 | Likely Pathogenic | 0.135 | Likely Benign | Likely Benign | 2.41 | Destabilizing | 2.0 | 5.50 | Destabilizing | 3.96 | Destabilizing | 0.17 | Likely Benign | 0.456 | Likely Benign | -1.07 | Neutral | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1530 | 0.4240 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1151G>C | G384A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G384A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that classify the variant as benign include REVEL, premPS, PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and Rosetta. Predictions from FoldX and Foldetta are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation. Therefore, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.427831 | Uncertain | 0.323 | 0.934 | 0.750 | -6.927 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 1.81 | Ambiguous | 0.2 | 2.16 | Destabilizing | 1.99 | Ambiguous | 0.04 | Likely Benign | 0.307 | Likely Benign | -0.40 | Neutral | 0.953 | Possibly Damaging | 0.952 | Probably Damaging | 1.33 | Pathogenic | 0.05 | Affected | 0.3986 | 0.4576 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1153T>A | S385T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S385T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumVar and SIFT predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments—AlphaMissense‑Optimized, the SGM‑Consensus, and Foldetta (combining FoldX‑MD and Rosetta outputs)—all indicate a benign effect. Consequently, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.733139 | Disordered | 0.425480 | Uncertain | 0.341 | 0.925 | 0.750 | -5.904 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.65 | Ambiguous | 0.2 | 0.05 | Likely Benign | 0.35 | Likely Benign | -0.12 | Likely Benign | 0.214 | Likely Benign | -0.31 | Neutral | 0.140 | Benign | 0.481 | Possibly Damaging | 4.64 | Benign | 0.03 | Affected | 0.2266 | 0.6259 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1156G>A | G386R 2D 3DClick to see structure in 3D Viewer AIClinVar reports no entry for this SynGAP1 G386R variant, and it is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are FoldX, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default, while Rosetta is uncertain. High‑accuracy methods give a benign call from AlphaMissense‑Optimized, a pathogenic result from Foldetta, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Overall, the evidence is mixed; the variant is most likely benign, and this assessment does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.733139 | Disordered | 0.424156 | Uncertain | 0.334 | 0.898 | 0.750 | -9.024 | Likely Pathogenic | 0.709 | Likely Pathogenic | Likely Benign | 3.62 | Destabilizing | 2.9 | 1.07 | Ambiguous | 2.35 | Destabilizing | 0.29 | Likely Benign | 0.453 | Likely Benign | -0.82 | Neutral | 0.753 | Possibly Damaging | 0.220 | Benign | 4.03 | Benign | 0.01 | Affected | 0.1329 | 0.4032 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1156G>C | G386R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G386R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Computational predictors that agree on a benign effect include REVEL, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise FoldX, polyPhen‑2 (HumDiv), SIFT, ESM1b, AlphaMissense‑Default, and Foldetta; Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the balance of evidence favors a pathogenic classification. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.733139 | Disordered | 0.424156 | Uncertain | 0.334 | 0.898 | 0.750 | -9.024 | Likely Pathogenic | 0.709 | Likely Pathogenic | Likely Benign | 3.62 | Destabilizing | 2.9 | 1.07 | Ambiguous | 2.35 | Destabilizing | 0.29 | Likely Benign | 0.453 | Likely Benign | -0.82 | Neutral | 0.753 | Possibly Damaging | 0.220 | Benign | 4.03 | Benign | 0.01 | Affected | 0.1329 | 0.4032 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1156G>T | G386W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G386W is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Computational predictors that classify the change as benign include REVEL, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. High‑accuracy assessments give a benign verdict from AlphaMissense‑Optimized, a benign consensus from the SGM method (majority of the four contributing tools are benign), and a pathogenic result from Foldetta. Uncertain calls from AlphaMissense‑Default and Rosetta are treated as unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not conflict with the lack of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.733139 | Disordered | 0.424156 | Uncertain | 0.334 | 0.898 | 0.750 | -10.389 | Likely Pathogenic | 0.519 | Ambiguous | Likely Benign | 6.07 | Destabilizing | 5.5 | 1.28 | Ambiguous | 3.68 | Destabilizing | -0.23 | Likely Benign | 0.471 | Likely Benign | -0.85 | Neutral | 0.996 | Probably Damaging | 0.920 | Probably Damaging | 3.90 | Benign | 0.00 | Affected | 0.1058 | 0.4676 | -7 | -2 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||
| c.1157G>C | G386A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change G386A has no ClinVar entry and is not reported in gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (SGM‑Consensus, REVEL, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar) and pathogenic (FoldX, polyPhen‑2 HumDiv, SIFT). Two tools report uncertainty: Rosetta and Foldetta. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, while Foldetta’s stability analysis is inconclusive. Overall, the majority of evidence points to a benign effect for G386A. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.733139 | Disordered | 0.424156 | Uncertain | 0.334 | 0.898 | 0.750 | -6.453 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 2.14 | Destabilizing | 0.7 | 1.05 | Ambiguous | 1.60 | Ambiguous | 0.14 | Likely Benign | 0.331 | Likely Benign | -0.55 | Neutral | 0.718 | Possibly Damaging | 0.332 | Benign | 3.93 | Benign | 0.05 | Affected | 0.3815 | 0.4868 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1159G>C | G387R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G387R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include premPS, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Tools that predict pathogenicity are SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No predictions are missing or inconclusive. Overall, the majority of evidence (10 pathogenic vs. 4 benign) indicates the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.642678 | Disordered | 0.422910 | Uncertain | 0.293 | 0.861 | 0.750 | -8.728 | Likely Pathogenic | 0.683 | Likely Pathogenic | Likely Benign | 4.13 | Destabilizing | 2.9 | 2.57 | Destabilizing | 3.35 | Destabilizing | 0.15 | Likely Benign | 0.516 | Likely Pathogenic | -0.54 | Neutral | 0.003 | Benign | 0.004 | Benign | 1.32 | Pathogenic | 0.01 | Affected | 0.1309 | 0.4356 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.115T>A | Y39N 2D AIThe SynGAP1 missense variant Y39N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.432876 | Uncertain | 0.343 | 0.787 | 0.375 | -3.098 | Likely Benign | 0.229 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -1.84 | Neutral | 0.824 | Possibly Damaging | 0.828 | Possibly Damaging | 3.99 | Benign | 0.00 | Affected | 0.2375 | 0.0903 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.115T>C | Y39H 2D AIThe SynGAP1 missense variant Y39H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.432876 | Uncertain | 0.343 | 0.787 | 0.375 | -3.166 | Likely Benign | 0.267 | Likely Benign | Likely Benign | 0.083 | Likely Benign | -1.08 | Neutral | 0.824 | Possibly Damaging | 0.775 | Possibly Damaging | 4.02 | Benign | 0.00 | Affected | 0.2569 | 0.0843 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||||||||||||
| c.115T>G | Y39D 2D AIThe SynGAP1 missense variant Y39D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for Y39D, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.432876 | Uncertain | 0.343 | 0.787 | 0.375 | -2.338 | Likely Benign | 0.333 | Likely Benign | Likely Benign | 0.166 | Likely Benign | -1.86 | Neutral | 0.824 | Possibly Damaging | 0.828 | Possibly Damaging | 3.98 | Benign | 0.00 | Affected | 0.4099 | 0.0903 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||||||||||||
| c.1162G>C | G388R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G388R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, and AlphaMissense‑Optimized, whereas the remaining eleven tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) uniformly predict a pathogenic impact. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized indicates benign, but the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta outputs) both predict pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overall consensus of the majority of tools and the high‑accuracy predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.736850 | Disordered | 0.420316 | Uncertain | 0.319 | 0.827 | 0.750 | -9.142 | Likely Pathogenic | 0.694 | Likely Pathogenic | Likely Benign | 6.54 | Destabilizing | 8.5 | 4.79 | Destabilizing | 5.67 | Destabilizing | 0.31 | Likely Benign | 0.606 | Likely Pathogenic | -0.74 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 1.32 | Pathogenic | 0.02 | Affected | 0.1296 | 0.4417 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1162G>T | G388C 2D 3DClick to see structure in 3D Viewer AISynGAP1 G388C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, and PROVEAN, while pathogenic predictions arise from REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized predicts benign, Foldetta predicts pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Because the majority of evidence points to pathogenicity and there is no ClinVar entry to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.736850 | Disordered | 0.420316 | Uncertain | 0.319 | 0.827 | 0.750 | -8.088 | Likely Pathogenic | 0.121 | Likely Benign | Likely Benign | 4.01 | Destabilizing | 3.7 | 4.95 | Destabilizing | 4.48 | Destabilizing | 0.03 | Likely Benign | 0.603 | Likely Pathogenic | -0.98 | Neutral | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1497 | 0.4112 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1163G>C | G388A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G388A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as Pathogenic. Overall, the predictions are mixed, but the two most reliable tools (AlphaMissense‑Optimized and SGM‑Consensus) favor a benign interpretation, while Foldetta suggests instability. Based on the available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.736850 | Disordered | 0.420316 | Uncertain | 0.319 | 0.827 | 0.750 | -6.577 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 2.55 | Destabilizing | 1.9 | 3.57 | Destabilizing | 3.06 | Destabilizing | -0.04 | Likely Benign | 0.457 | Likely Benign | -0.57 | Neutral | 0.953 | Possibly Damaging | 0.952 | Probably Damaging | 1.33 | Pathogenic | 0.05 | Affected | 0.3969 | 0.4837 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1168G>A | G390R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G390R is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta) both predict pathogenicity. Overall, the majority of tools and the high‑accuracy methods indicate a pathogenic effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.626927 | Disordered | 0.413274 | Uncertain | 0.304 | 0.763 | 0.875 | -9.242 | Likely Pathogenic | 0.686 | Likely Pathogenic | Likely Benign | 2.43 | Destabilizing | 0.9 | 4.85 | Destabilizing | 3.64 | Destabilizing | 0.16 | Likely Benign | 0.605 | Likely Pathogenic | -0.92 | Neutral | 0.480 | Possibly Damaging | 0.163 | Benign | 1.32 | Pathogenic | 0.08 | Tolerated | 0.1190 | 0.4524 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1168G>C | G390R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G390R is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta) both predict pathogenicity. Overall, the majority of tools and the high‑accuracy methods indicate a pathogenic effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.626927 | Disordered | 0.413274 | Uncertain | 0.304 | 0.763 | 0.875 | -9.242 | Likely Pathogenic | 0.686 | Likely Pathogenic | Likely Benign | 2.43 | Destabilizing | 0.9 | 4.85 | Destabilizing | 3.64 | Destabilizing | 0.16 | Likely Benign | 0.605 | Likely Pathogenic | -0.92 | Neutral | 0.480 | Possibly Damaging | 0.163 | Benign | 1.32 | Pathogenic | 0.08 | Tolerated | 0.1190 | 0.4524 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1168G>T | G390W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G390W is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include AlphaMissense‑Optimized, premPS, and PROVEAN, whereas the remaining tools—REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts benign, but the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic verdict, and Foldetta also indicates pathogenic. With the majority of evidence pointing to a deleterious effect, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.626927 | Disordered | 0.413274 | Uncertain | 0.304 | 0.763 | 0.875 | -9.405 | Likely Pathogenic | 0.487 | Ambiguous | Likely Benign | 3.69 | Destabilizing | 1.9 | 5.31 | Destabilizing | 4.50 | Destabilizing | 0.14 | Likely Benign | 0.639 | Likely Pathogenic | -1.11 | Neutral | 0.987 | Probably Damaging | 0.744 | Possibly Damaging | 1.31 | Pathogenic | 0.00 | Affected | 0.0917 | 0.4368 | -7 | -2 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||
| c.1169G>C | G390A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G390A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and Rosetta. The high‑accuracy assessment shows AlphaMissense‑Optimized as benign, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as pathogenic. FoldX alone is inconclusive. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.626927 | Disordered | 0.413274 | Uncertain | 0.304 | 0.763 | 0.875 | -6.768 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 1.96 | Ambiguous | 0.4 | 2.03 | Destabilizing | 2.00 | Destabilizing | 0.02 | Likely Benign | 0.402 | Likely Benign | -0.55 | Neutral | 0.143 | Benign | 0.028 | Benign | 1.33 | Pathogenic | 0.05 | Affected | 0.4130 | 0.5053 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1169G>T | G390V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G390V has no ClinVar entry and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from REVEL, FoldX, Rosetta, Foldetta, SIFT, and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default (benign), ESM1b (uncertain), FATHMM (pathogenic), and PROVEAN (benign), also yields a benign verdict; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, indicates pathogenic. With two high‑accuracy tools supporting benign and one supporting pathogenic, the overall evidence leans toward a benign effect. This conclusion does not contradict ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.626927 | Disordered | 0.413274 | Uncertain | 0.304 | 0.763 | 0.875 | -7.137 | In-Between | 0.146 | Likely Benign | Likely Benign | 3.74 | Destabilizing | 0.6 | 4.88 | Destabilizing | 4.31 | Destabilizing | -0.16 | Likely Benign | 0.509 | Likely Pathogenic | -0.88 | Neutral | 0.002 | Benign | 0.002 | Benign | 1.32 | Pathogenic | 0.01 | Affected | 0.1537 | 0.4004 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||
| c.116A>C | Y39S 2D AIThe SynGAP1 missense variant Y39S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for Y39S, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.432876 | Uncertain | 0.343 | 0.787 | 0.375 | -1.634 | Likely Benign | 0.174 | Likely Benign | Likely Benign | 0.100 | Likely Benign | -1.57 | Neutral | 0.824 | Possibly Damaging | 0.775 | Possibly Damaging | 4.02 | Benign | 0.00 | Affected | 0.4935 | 0.2649 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||||||||||||
| c.116A>G | Y39C 2D AIThe SynGAP1 missense variant Y39C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the overall assessment. Overall, the majority of evidence points to the variant being most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.432876 | Uncertain | 0.343 | 0.787 | 0.375 | -3.109 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.203 | Likely Benign | -2.00 | Neutral | 0.943 | Possibly Damaging | 0.941 | Probably Damaging | 3.96 | Benign | 0.00 | Affected | 0.3007 | 0.2849 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||||||||||||
| c.116A>T | Y39F 2D AIThe SynGAP1 missense variant Y39F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.432876 | Uncertain | 0.343 | 0.787 | 0.375 | -3.410 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.073 | Likely Benign | -0.78 | Neutral | 0.458 | Possibly Damaging | 0.481 | Possibly Damaging | 4.05 | Benign | 0.00 | Affected | 0.2641 | 0.3363 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||||||||||||
| c.1171G>A | G391S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G391S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and FoldX. Rosetta and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus also as likely benign, while Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact for G391S, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | -5.531 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 2.14 | Destabilizing | 0.6 | 1.01 | Ambiguous | 1.58 | Ambiguous | 0.06 | Likely Benign | 0.462 | Likely Benign | -0.54 | Neutral | 0.978 | Probably Damaging | 0.777 | Possibly Damaging | 1.33 | Pathogenic | 0.35 | Tolerated | 0.2820 | 0.5097 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1171G>C | G391R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G391R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that classify the variant as benign include premPS, PROVEAN, and SIFT, whereas those that predict pathogenicity comprise REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further show AlphaMissense‑Optimized labeling the variant as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic effect. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | -9.115 | Likely Pathogenic | 0.709 | Likely Pathogenic | Likely Benign | 2.80 | Destabilizing | 1.3 | 3.86 | Destabilizing | 3.33 | Destabilizing | 0.32 | Likely Benign | 0.628 | Likely Pathogenic | -0.95 | Neutral | 0.999 | Probably Damaging | 0.960 | Probably Damaging | 1.32 | Pathogenic | 0.17 | Tolerated | 0.1313 | 0.4124 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1171G>T | G391C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G391C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, and PROVEAN, whereas the remaining tools—REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—predict it to be pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence points to a pathogenic effect. This prediction does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | -8.596 | Likely Pathogenic | 0.123 | Likely Benign | Likely Benign | 2.65 | Destabilizing | 0.7 | 5.03 | Destabilizing | 3.84 | Destabilizing | 0.11 | Likely Benign | 0.640 | Likely Pathogenic | -1.29 | Neutral | 1.000 | Probably Damaging | 0.970 | Probably Damaging | 1.32 | Pathogenic | 0.03 | Affected | 0.1483 | 0.4225 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1172G>A | G391D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G391D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. Two tools, Rosetta and Foldetta, return uncertain results. High‑accuracy methods give a benign call from AlphaMissense‑Optimized; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta is also inconclusive. Overall, six tools favor pathogenicity while five favor benignity, with two uncertain. Thus, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | -4.651 | Likely Benign | 0.674 | Likely Pathogenic | Likely Benign | 2.59 | Destabilizing | 1.1 | 1.26 | Ambiguous | 1.93 | Ambiguous | 0.22 | Likely Benign | 0.562 | Likely Pathogenic | -0.95 | Neutral | 0.999 | Probably Damaging | 0.960 | Probably Damaging | 1.32 | Pathogenic | 0.29 | Tolerated | 0.1900 | 0.1305 | 1 | -1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||
| c.1172G>C | G391A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G391A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are FoldX, polyPhen‑2 HumDiv, and FATHMM. Predictions that are inconclusive are Rosetta and Foldetta. The high‑accuracy consensus from AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also likely benign, and Foldetta remains uncertain. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | -5.712 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 2.02 | Destabilizing | 0.5 | 1.92 | Ambiguous | 1.97 | Ambiguous | 0.11 | Likely Benign | 0.442 | Likely Benign | -0.76 | Neutral | 0.633 | Possibly Damaging | 0.219 | Benign | 1.33 | Pathogenic | 0.19 | Tolerated | 0.3828 | 0.4870 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1174A>C | K392Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K392Q has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL and polyPhen‑2 HumDiv. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.541878 | Disordered | 0.405672 | Uncertain | 0.319 | 0.702 | 0.750 | -4.243 | Likely Benign | 0.377 | Ambiguous | Likely Benign | 0.13 | Likely Benign | 0.0 | 0.05 | Likely Benign | 0.09 | Likely Benign | 0.23 | Likely Benign | 0.525 | Likely Pathogenic | -2.09 | Neutral | 0.652 | Possibly Damaging | 0.161 | Benign | 4.61 | Benign | 0.06 | Tolerated | 0.5612 | 0.2106 | Weaken | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||
| c.1174A>G | K392E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K392E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and the SGM‑Consensus score. Tools that predict a pathogenic effect are REVEL, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, while the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta) both indicate a benign outcome. Overall, the majority of evidence supports a benign classification, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.541878 | Disordered | 0.405672 | Uncertain | 0.319 | 0.702 | 0.750 | -4.392 | Likely Benign | 0.850 | Likely Pathogenic | Ambiguous | 0.09 | Likely Benign | 0.0 | -0.04 | Likely Benign | 0.03 | Likely Benign | 0.28 | Likely Benign | 0.529 | Likely Pathogenic | -1.92 | Neutral | 0.276 | Benign | 0.083 | Benign | 4.60 | Benign | 0.02 | Affected | 0.4812 | 0.1916 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1175A>C | K392T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K392T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, PROVEAN, and SIFT. Two tools—FoldX and AlphaMissense‑Default—return uncertain results and are treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign; and Foldetta, a protein‑folding stability method, also predicts benign. No ClinVar entry exists to contradict these predictions. Based on the collective evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.541878 | Disordered | 0.405672 | Uncertain | 0.319 | 0.702 | 0.750 | -3.224 | Likely Benign | 0.481 | Ambiguous | Likely Benign | 0.52 | Ambiguous | 0.1 | -0.29 | Likely Benign | 0.12 | Likely Benign | 0.05 | Likely Benign | 0.512 | Likely Pathogenic | -3.14 | Deleterious | 0.276 | Benign | 0.045 | Benign | 4.60 | Benign | 0.02 | Affected | 0.2807 | 0.4053 | 0 | -1 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||
| c.1175A>G | K392R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K392R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). All available in silico predictors classify the change as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a benign effect. The high‑accuracy folding‑stability tool Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a benign impact. No tool predicts pathogenicity. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.541878 | Disordered | 0.405672 | Uncertain | 0.319 | 0.702 | 0.750 | -4.006 | Likely Benign | 0.091 | Likely Benign | Likely Benign | -0.03 | Likely Benign | 0.0 | 0.44 | Likely Benign | 0.21 | Likely Benign | 0.23 | Likely Benign | 0.131 | Likely Benign | -1.44 | Neutral | 0.436 | Benign | 0.112 | Benign | 7.12 | Benign | 0.08 | Tolerated | 0.5686 | 0.1875 | Weaken | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||
| c.1175A>T | K392M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K392M missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, ESM1b, and FATHMM, whereas those that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The remaining tools—FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized—return uncertain or inconclusive results. High‑accuracy methods give no definitive signal: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a tie and thus unavailable, and Foldetta is uncertain. Overall, the majority of available predictions (six pathogenic vs. three benign) lean toward a pathogenic impact. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.541878 | Disordered | 0.405672 | Uncertain | 0.319 | 0.702 | 0.750 | -3.856 | Likely Benign | 0.788 | Likely Pathogenic | Ambiguous | 0.52 | Ambiguous | 0.1 | 0.67 | Ambiguous | 0.60 | Ambiguous | -0.09 | Likely Benign | 0.665 | Likely Pathogenic | -3.24 | Deleterious | 0.952 | Possibly Damaging | 0.496 | Possibly Damaging | 4.59 | Benign | 0.00 | Affected | 0.1799 | 0.4941 | 0 | -1 | 5.8 | 3.02 | ||||||||||||||||||||||||||||||
| c.1176G>C | K392N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K392N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts benign. Overall, the majority of evidence (9 benign vs 3 pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.541878 | Disordered | 0.405672 | Uncertain | 0.319 | 0.702 | 0.750 | -4.136 | Likely Benign | 0.780 | Likely Pathogenic | Likely Benign | 0.24 | Likely Benign | 0.1 | -0.01 | Likely Benign | 0.12 | Likely Benign | 0.20 | Likely Benign | 0.229 | Likely Benign | -2.61 | Deleterious | 0.276 | Benign | 0.083 | Benign | 4.60 | Benign | 0.02 | Affected | 0.4586 | 0.2454 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||
| c.1176G>T | K392N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K392N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split and is therefore treated as unavailable. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the majority of evidence (nine benign vs. three pathogenic predictions) supports a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.541878 | Disordered | 0.405672 | Uncertain | 0.319 | 0.702 | 0.750 | -4.136 | Likely Benign | 0.780 | Likely Pathogenic | Likely Benign | 0.24 | Likely Benign | 0.1 | -0.01 | Likely Benign | 0.12 | Likely Benign | 0.20 | Likely Benign | 0.229 | Likely Benign | -2.61 | Deleterious | 0.276 | Benign | 0.083 | Benign | 4.60 | Benign | 0.02 | Affected | 0.4586 | 0.2454 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||
| c.1177G>A | G393S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G393S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from FoldX, polyPhen‑2 HumDiv, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta) as uncertain. No other tools provide decisive evidence. Overall, the majority of reliable predictors lean toward a benign effect, and this consensus does not conflict with the absence of ClinVar annotation. Therefore, G393S is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -5.207 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 2.43 | Destabilizing | 0.7 | -0.78 | Ambiguous | 0.83 | Ambiguous | 0.24 | Likely Benign | 0.466 | Likely Benign | -1.76 | Neutral | 0.889 | Possibly Damaging | 0.444 | Benign | 1.33 | Pathogenic | 0.07 | Tolerated | 0.3011 | 0.5232 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1177G>C | G393R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G393R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include Rosetta and premPS, whereas the remaining tools—SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence points to a pathogenic effect. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -9.148 | Likely Pathogenic | 0.815 | Likely Pathogenic | Ambiguous | 3.88 | Destabilizing | 1.4 | -0.38 | Likely Benign | 1.75 | Ambiguous | 0.47 | Likely Benign | 0.596 | Likely Pathogenic | -2.99 | Deleterious | 0.991 | Probably Damaging | 0.881 | Possibly Damaging | 1.32 | Pathogenic | 0.02 | Affected | 0.1353 | 0.4464 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1177G>T | G393C 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G393C is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Rosetta, premPS, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus (likely pathogenic), REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain (no definitive stability change). The majority of evidence points toward a pathogenic effect, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -8.854 | Likely Pathogenic | 0.181 | Likely Benign | Likely Benign | 2.99 | Destabilizing | 0.9 | -0.26 | Likely Benign | 1.37 | Ambiguous | 0.43 | Likely Benign | 0.769 | Likely Pathogenic | -3.05 | Deleterious | 0.999 | Probably Damaging | 0.936 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1593 | 0.4408 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1178G>A | G393D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G393D is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SIFT, ESM1b, and AlphaMissense‑Optimized. In contrast, the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default all indicate pathogenicity. High‑accuracy assessments further support this: AlphaMissense‑Optimized reports a benign outcome, while the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, yields an uncertain result and is treated as unavailable. Overall, the preponderance of evidence points to a pathogenic effect for G393D, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -5.247 | Likely Benign | 0.717 | Likely Pathogenic | Likely Benign | 3.30 | Destabilizing | 1.4 | -1.00 | Ambiguous | 1.15 | Ambiguous | 0.57 | Ambiguous | 0.528 | Likely Pathogenic | -2.60 | Deleterious | 0.991 | Probably Damaging | 0.831 | Possibly Damaging | 1.32 | Pathogenic | 0.19 | Tolerated | 0.2077 | 0.1645 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.1178G>C | G393A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G393A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta) are inconclusive, so they provide no evidence for or against pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of reliable predictions indicate a benign effect, and there is no ClinVar annotation to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -4.507 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 1.93 | Ambiguous | 0.5 | -0.68 | Ambiguous | 0.63 | Ambiguous | 0.22 | Likely Benign | 0.381 | Likely Benign | -1.89 | Neutral | 0.176 | Benign | 0.039 | Benign | 1.33 | Pathogenic | 0.08 | Tolerated | 0.4143 | 0.5193 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1178G>T | G393V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G393V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Rosetta is uncertain and is treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while Foldetta predicts pathogenic. The SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑vs‑2 split. Overall, the majority of evidence (8 pathogenic vs. 4 benign) points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -6.358 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 5.56 | Destabilizing | 2.3 | -0.72 | Ambiguous | 2.42 | Destabilizing | -0.01 | Likely Benign | 0.639 | Likely Pathogenic | -2.69 | Deleterious | 0.982 | Probably Damaging | 0.648 | Possibly Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1712 | 0.4144 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||
| c.1180A>C | K394Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K394Q missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic outcome are SIFT and Rosetta. The remaining tools—Foldetta, premPS, ESM1b, and AlphaMissense‑Default—return uncertain results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also leans toward benign, with two benign votes and two uncertain votes. Foldetta’s stability prediction is uncertain and thus not considered. Overall, the majority of reliable predictions indicate a benign effect, and this conclusion does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.505461 | Disordered | 0.399336 | Uncertain | 0.387 | 0.634 | 0.625 | -7.261 | In-Between | 0.468 | Ambiguous | Likely Benign | 0.15 | Likely Benign | 0.0 | 2.00 | Destabilizing | 1.08 | Ambiguous | 0.64 | Ambiguous | 0.330 | Likely Benign | -2.46 | Neutral | 0.001 | Benign | 0.009 | Benign | 4.61 | Benign | 0.01 | Affected | 0.5365 | 0.2106 | Weaken | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1181A>C | K394T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K394T missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Those that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. Predictions that are uncertain or inconclusive are FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta is also inconclusive. Overall, the balance of evidence (five benign versus three pathogenic predictions) suggests the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.505461 | Disordered | 0.399336 | Uncertain | 0.387 | 0.634 | 0.625 | -6.487 | Likely Benign | 0.599 | Likely Pathogenic | Likely Benign | 0.50 | Ambiguous | 0.1 | 2.46 | Destabilizing | 1.48 | Ambiguous | 0.57 | Ambiguous | 0.482 | Likely Benign | -3.35 | Deleterious | 0.247 | Benign | 0.166 | Benign | 4.61 | Benign | 0.01 | Affected | 0.2727 | 0.4453 | 0 | -1 | 3.2 | -27.07 | ||||||||||||||||||||||||||||||
| c.1181A>G | K394R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K394R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign. Only SIFT predicts a pathogenic outcome, while Rosetta and Foldetta are uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, SGM‑Consensus is Likely Benign, and Foldetta remains uncertain. Overall, the preponderance of evidence supports a benign classification for K394R, and this conclusion does not contradict the absence of a ClinVar report. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.505461 | Disordered | 0.399336 | Uncertain | 0.387 | 0.634 | 0.625 | -4.902 | Likely Benign | 0.097 | Likely Benign | Likely Benign | -0.01 | Likely Benign | 0.1 | 1.19 | Ambiguous | 0.59 | Ambiguous | 0.42 | Likely Benign | 0.335 | Likely Benign | -1.97 | Neutral | 0.141 | Benign | 0.091 | Benign | 5.11 | Benign | 0.03 | Affected | 0.5488 | 0.2075 | Weaken | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||
| c.1181A>T | K394I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 K394I missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include premPS, polyPhen‑2 HumVar, and FATHMM, while a majority (seven) predict pathogenicity: SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) remains Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. No evidence from these tools contradicts the ClinVar status, which is absent. Overall, the preponderance of pathogenic predictions suggests the variant is most likely pathogenic, with no conflict from ClinVar data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.505461 | Disordered | 0.399336 | Uncertain | 0.387 | 0.634 | 0.625 | -9.244 | Likely Pathogenic | 0.876 | Likely Pathogenic | Ambiguous | 0.78 | Ambiguous | 0.2 | 1.10 | Ambiguous | 0.94 | Ambiguous | 0.19 | Likely Benign | 0.519 | Likely Pathogenic | -3.96 | Deleterious | 0.700 | Possibly Damaging | 0.403 | Benign | 4.59 | Benign | 0.00 | Affected | 0.1728 | 0.4123 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1182A>C | K394N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K394N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, FATHMM, and polyPhen‑2 HumVar. Those that agree on a pathogenic effect are Rosetta, PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. Predictions that are uncertain or inconclusive are Foldetta, premPS, ESM1b, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, Foldetta as uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of available predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.505461 | Disordered | 0.399336 | Uncertain | 0.387 | 0.634 | 0.625 | -7.408 | In-Between | 0.861 | Likely Pathogenic | Ambiguous | 0.08 | Likely Benign | 0.1 | 2.02 | Destabilizing | 1.05 | Ambiguous | 0.66 | Ambiguous | 0.299 | Likely Benign | -3.17 | Deleterious | 0.535 | Possibly Damaging | 0.188 | Benign | 4.60 | Benign | 0.01 | Affected | 0.4353 | 0.2654 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||
| c.1182A>T | K394N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K394N missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a mixed signal: benign calls come from REVEL, FoldX, FATHMM, and polyPhen‑2 HumVar, while pathogenic calls come from Rosetta, PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. Four tools (Foldetta, premPS, ESM1b, AlphaMissense‑Optimized) return uncertain results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized remains uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as pathogenic; and Foldetta is uncertain. Taken together, the majority of evidence—including the high‑accuracy consensus—points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.505461 | Disordered | 0.399336 | Uncertain | 0.387 | 0.634 | 0.625 | -7.408 | In-Between | 0.861 | Likely Pathogenic | Ambiguous | 0.08 | Likely Benign | 0.1 | 2.02 | Destabilizing | 1.05 | Ambiguous | 0.66 | Ambiguous | 0.299 | Likely Benign | -3.17 | Deleterious | 0.535 | Possibly Damaging | 0.188 | Benign | 4.60 | Benign | 0.01 | Affected | 0.4353 | 0.2654 | 1 | 0 | 0.4 | -14.07 | ||||||||||||||||||||||||||||||
| c.1183G>A | G395R 2D AIThe SynGAP1 missense variant G395R has no ClinVar record and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from SIFT, ESM1b, and AlphaMissense‑Default. Two tools, FoldX and Foldetta, yield uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta remains uncertain. Overall, the balance of evidence favors a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.513880 | Disordered | 0.396199 | Uncertain | 0.474 | 0.601 | 0.500 | -9.851 | Likely Pathogenic | 0.768 | Likely Pathogenic | Likely Benign | 1.26 | Ambiguous | 2.1 | -0.25 | Likely Benign | 0.51 | Ambiguous | 0.50 | Likely Benign | 0.430 | Likely Benign | -2.01 | Neutral | 0.037 | Benign | 0.027 | Benign | 4.23 | Benign | 0.02 | Affected | 0.0953 | 0.4313 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1183G>C | G395R 2D AIThe SynGAP1 missense variant G395R has no ClinVar record and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from SIFT, ESM1b, and AlphaMissense‑Default. Two tools, FoldX and Foldetta, yield uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta remains uncertain. Overall, the balance of evidence favors a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.513880 | Disordered | 0.396199 | Uncertain | 0.474 | 0.601 | 0.500 | -9.851 | Likely Pathogenic | 0.768 | Likely Pathogenic | Likely Benign | 1.26 | Ambiguous | 2.1 | -0.25 | Likely Benign | 0.51 | Ambiguous | 0.50 | Likely Benign | 0.430 | Likely Benign | -2.01 | Neutral | 0.037 | Benign | 0.027 | Benign | 4.23 | Benign | 0.02 | Affected | 0.0953 | 0.4313 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1184G>A | G395E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G395E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome, while FoldX and Rosetta give uncertain results. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote) predicts benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Taken together, the majority of evidence points to a benign effect. There is no ClinVar entry to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.513880 | Disordered | 0.396199 | Uncertain | 0.474 | 0.601 | 0.500 | -6.350 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 1.60 | Ambiguous | 0.6 | -0.88 | Ambiguous | 0.36 | Likely Benign | 0.30 | Likely Benign | 0.310 | Likely Benign | -1.81 | Neutral | 0.037 | Benign | 0.010 | Benign | 4.25 | Benign | 0.12 | Tolerated | 0.1328 | 0.4089 | 0 | -2 | -3.1 | 72.06 | |||||||||||||||||||||||||||||
| c.1184G>C | G395A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G395A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from FoldX and Rosetta, which are treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Consequently, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.513880 | Disordered | 0.396199 | Uncertain | 0.474 | 0.601 | 0.500 | -3.964 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 1.51 | Ambiguous | 0.3 | -0.59 | Ambiguous | 0.46 | Likely Benign | 0.08 | Likely Benign | 0.160 | Likely Benign | -0.72 | Neutral | 0.001 | Benign | 0.003 | Benign | 4.24 | Benign | 0.15 | Tolerated | 0.3848 | 0.5047 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1184G>T | G395V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G395V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only FoldX and SIFT predict pathogenic. The SGM‑Consensus score, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and the Foldetta stability analysis is uncertain. Taken together, the preponderance of evidence points to a benign impact for G395V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.513880 | Disordered | 0.396199 | Uncertain | 0.474 | 0.601 | 0.500 | -5.617 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 2.83 | Destabilizing | 0.7 | -0.37 | Likely Benign | 1.23 | Ambiguous | 0.08 | Likely Benign | 0.288 | Likely Benign | -1.49 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.21 | Benign | 0.03 | Affected | 0.1290 | 0.4183 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1186G>A | G396S 2D AIThe SynGAP1 missense variant G396S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, the SGM‑Consensus is benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. No tool predicts pathogenicity. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.394626 | Uncertain | 0.640 | 0.584 | 0.500 | -3.934 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 1.76 | Ambiguous | 1.6 | 1.12 | Ambiguous | 1.44 | Ambiguous | 0.10 | Likely Benign | 0.223 | Likely Benign | -0.99 | Neutral | 0.004 | Benign | 0.003 | Benign | 3.93 | Benign | 0.56 | Tolerated | 0.2668 | 0.4894 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1186G>C | G396R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G396R missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on benign impact include REVEL, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Four tools (FoldX, Rosetta, Foldetta, premPS) returned uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the variant is more frequently predicted to be pathogenic (five tools) than benign (five tools), and the high‑accuracy consensus leans toward pathogenicity, though Foldetta does not provide a definitive verdict. Thus, the variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.394626 | Uncertain | 0.640 | 0.584 | 0.500 | -9.310 | Likely Pathogenic | 0.775 | Likely Pathogenic | Likely Benign | 1.68 | Ambiguous | 1.1 | 1.56 | Ambiguous | 1.62 | Ambiguous | 0.66 | Ambiguous | 0.319 | Likely Benign | -2.65 | Deleterious | 0.718 | Possibly Damaging | 0.216 | Benign | 4.42 | Benign | 0.24 | Tolerated | 0.0986 | 0.4007 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1186G>T | G396C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G396C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar; premPS is uncertain. The high‑accuracy consensus methods give a mixed signal: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of individual predictors and the SGM‑Consensus lean toward a benign interpretation, and the two high‑accuracy tools that are available also favor benign over pathogenic. Therefore, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.394626 | Uncertain | 0.640 | 0.584 | 0.500 | -5.459 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 2.15 | Destabilizing | 0.7 | 2.52 | Destabilizing | 2.34 | Destabilizing | 0.59 | Ambiguous | 0.411 | Likely Benign | -3.00 | Deleterious | 0.983 | Probably Damaging | 0.533 | Possibly Damaging | 3.89 | Benign | 0.08 | Tolerated | 0.1181 | 0.4541 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1187G>A | G396D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G396D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the substitution as benign, whereas only AlphaMissense‑Default predicts a pathogenic outcome. Tools that assess protein stability (FoldX, Rosetta, Foldetta) yield uncertain or inconclusive results. High‑accuracy consensus methods reinforce the benign assessment: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports “Likely Benign”; AlphaMissense‑Optimized also predicts benign; Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.394626 | Uncertain | 0.640 | 0.584 | 0.500 | -4.148 | Likely Benign | 0.678 | Likely Pathogenic | Likely Benign | 1.92 | Ambiguous | 0.8 | 1.33 | Ambiguous | 1.63 | Ambiguous | 0.17 | Likely Benign | 0.272 | Likely Benign | -1.49 | Neutral | 0.421 | Benign | 0.080 | Benign | 3.91 | Benign | 0.35 | Tolerated | 0.1702 | 0.1122 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.1187G>C | G396A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G396A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign verdict, while Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. Consequently, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.394626 | Uncertain | 0.640 | 0.584 | 0.500 | -3.655 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 1.74 | Ambiguous | 0.7 | 1.90 | Ambiguous | 1.82 | Ambiguous | 0.41 | Likely Benign | 0.274 | Likely Benign | -1.28 | Neutral | 0.062 | Benign | 0.024 | Benign | 3.93 | Benign | 0.84 | Tolerated | 0.3846 | 0.5255 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1189T>A | C397S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign, while Rosetta remains uncertain. When predictions are grouped, the benign category contains all available evidence and the pathogenic category is empty. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a benign effect. Consequently, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | -2.324 | Likely Benign | 0.178 | Likely Benign | Likely Benign | 0.37 | Likely Benign | 0.1 | 0.59 | Ambiguous | 0.48 | Likely Benign | 0.43 | Likely Benign | 0.174 | Likely Benign | -0.01 | Neutral | 0.276 | Benign | 0.066 | Benign | 4.68 | Benign | 0.53 | Tolerated | 0.5221 | 0.2474 | Weaken | 0 | -1 | -3.3 | -16.06 | ||||||||||||||||||||||||||||
| c.1189T>C | C397R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Rosetta, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and AlphaMissense‑Default. The remaining tools (FoldX, premPS, ESM1b) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign (2 benign vs. 1 pathogenic, 1 uncertain), and Foldetta predicts a benign impact on protein folding stability. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | -7.094 | In-Between | 0.708 | Likely Pathogenic | Likely Benign | 0.55 | Ambiguous | 0.8 | 0.40 | Likely Benign | 0.48 | Likely Benign | 0.96 | Ambiguous | 0.514 | Likely Pathogenic | -1.46 | Neutral | 0.480 | Possibly Damaging | 0.226 | Benign | 4.65 | Benign | 0.11 | Tolerated | 0.1780 | 0.1853 | -4 | -3 | -7.0 | 53.05 | ||||||||||||||||||||||||||||||
| c.1189T>G | C397G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach consensus all indicate a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the variant as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies it as “Likely Benign.” No tool predicts pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. No pathogenic predictions are present. Therefore, based on the available computational evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | 1.244 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.68 | Ambiguous | 0.2 | 1.42 | Ambiguous | 1.05 | Ambiguous | 0.04 | Likely Benign | 0.272 | Likely Benign | 1.51 | Neutral | 0.276 | Benign | 0.066 | Benign | 5.93 | Benign | 0.60 | Tolerated | 0.3678 | 0.3729 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.118G>A | D40N 2D AIThe SynGAP1 missense variant D40N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The predictions do not contradict ClinVar status, as ClinVar contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -3.841 | Likely Benign | 0.210 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -0.81 | Neutral | 0.028 | Benign | 0.032 | Benign | 4.05 | Benign | 0.00 | Affected | 0.2676 | 0.8761 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.118G>C | D40H 2D AIThe SynGAP1 missense variant D40H is reported in ClinVar as “Not submitted” and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic call comes from SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, SGM‑Consensus indicates likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the consensus of available predictions points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -4.108 | Likely Benign | 0.413 | Ambiguous | Likely Benign | 0.147 | Likely Benign | -1.28 | Neutral | 0.172 | Benign | 0.248 | Benign | 3.99 | Benign | 0.00 | Affected | 0.3123 | 0.9007 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.118G>T | D40Y 2D AIThe SynGAP1 D40Y missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus indicates Likely Benign, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for D40Y, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -4.313 | Likely Benign | 0.483 | Ambiguous | Likely Benign | 0.182 | Likely Benign | -1.72 | Neutral | 0.388 | Benign | 0.328 | Benign | 3.98 | Benign | 0.00 | Affected | 0.1173 | 0.7918 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.1190G>A | C397Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are FoldX, Rosetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and the combined Foldetta method. Uncertain results come from AlphaMissense‑Default and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as pathogenic. Overall, the majority of tools and the high‑accuracy consensus lean toward a benign interpretation, and this does not contradict any ClinVar classification (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | -7.213 | In-Between | 0.397 | Ambiguous | Likely Benign | 2.01 | Destabilizing | 2.3 | 8.64 | Destabilizing | 5.33 | Destabilizing | 0.12 | Likely Benign | 0.455 | Likely Benign | -1.82 | Neutral | 0.952 | Possibly Damaging | 0.497 | Possibly Damaging | 4.64 | Benign | 0.07 | Tolerated | 0.1299 | 0.5084 | 0 | -2 | -3.8 | 60.04 | ||||||||||||||||||||||||||||||
| c.1190G>C | C397S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). In silico prediction tools that assess pathogenicity largely agree on a benign outcome: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. No tool predicts pathogenicity; Rosetta’s assessment is uncertain and therefore treated as unavailable. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is benign. Consequently, the variant is most likely benign based on the collective predictions, and this conclusion does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | -2.324 | Likely Benign | 0.178 | Likely Benign | Likely Benign | 0.37 | Likely Benign | 0.1 | 0.59 | Ambiguous | 0.48 | Likely Benign | 0.43 | Likely Benign | 0.253 | Likely Benign | -0.01 | Neutral | 0.276 | Benign | 0.066 | Benign | 4.68 | Benign | 0.53 | Tolerated | 0.5221 | 0.2474 | Weaken | 0 | -1 | -3.3 | -16.06 | ||||||||||||||||||||||||||||
| c.1191C>G | C397W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C397W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split opinion: benign calls come from premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus, whereas pathogenic calls are made by REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, but Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts pathogenic. Overall, the majority of tools lean toward a benign effect, and this is consistent with the lack of ClinVar evidence. Therefore, the variant is most likely benign, and there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.429200 | Structured | 0.395774 | Uncertain | 0.778 | 0.551 | 0.250 | -6.857 | Likely Benign | 0.614 | Likely Pathogenic | Likely Benign | 2.40 | Destabilizing | 2.3 | 4.46 | Destabilizing | 3.43 | Destabilizing | 0.19 | Likely Benign | 0.545 | Likely Pathogenic | -1.87 | Neutral | 0.987 | Probably Damaging | 0.814 | Possibly Damaging | 4.64 | Benign | 0.02 | Affected | 0.1637 | 0.5092 | -8 | -2 | -3.4 | 83.07 | |||||||||||||||||||||||||||||
| c.1192C>A | P398T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant P398T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, and SIFT. The remaining tools (Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain results. High‑accuracy methods give a benign consensus: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign, and Foldetta is uncertain. Overall, the majority of high‑confidence predictions lean toward a benign impact, although several other predictors indicate pathogenicity. There is no conflict with ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.436924 | Structured | 0.401041 | Uncertain | 0.891 | 0.525 | 0.250 | -6.670 | Likely Benign | 0.536 | Ambiguous | Likely Benign | 2.11 | Destabilizing | 0.4 | 1.57 | Ambiguous | 1.84 | Ambiguous | 0.78 | Ambiguous | 0.608 | Likely Pathogenic | -5.70 | Deleterious | 0.816 | Possibly Damaging | 0.307 | Benign | 5.51 | Benign | 0.01 | Affected | 0.1671 | 0.6607 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||
| c.1192C>T | P398S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P398S missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include polyPhen‑2 (HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv), and SIFT. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of definitive predictions lean toward a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.436924 | Structured | 0.401041 | Uncertain | 0.891 | 0.525 | 0.250 | -6.757 | Likely Benign | 0.490 | Ambiguous | Likely Benign | 1.86 | Ambiguous | 0.3 | 1.78 | Ambiguous | 1.82 | Ambiguous | 0.85 | Ambiguous | 0.544 | Likely Pathogenic | -5.58 | Deleterious | 0.478 | Possibly Damaging | 0.130 | Benign | 5.68 | Benign | 0.03 | Affected | 0.3619 | 0.5737 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||
| c.1193C>A | P398Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P398Q has no ClinVar record (ClinVar ID None) and is not reported in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, while the majority of tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default) predict a pathogenic impact. Two tools report uncertainty: ESM1b and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely pathogenic based on the collective evidence, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.436924 | Structured | 0.401041 | Uncertain | 0.891 | 0.525 | 0.250 | -7.402 | In-Between | 0.792 | Likely Pathogenic | Ambiguous | 2.13 | Destabilizing | 0.1 | 2.01 | Destabilizing | 2.07 | Destabilizing | 1.01 | Destabilizing | 0.719 | Likely Pathogenic | -5.57 | Deleterious | 0.996 | Probably Damaging | 0.724 | Possibly Damaging | 5.48 | Benign | 0.00 | Affected | 0.1522 | 0.5176 | 0 | -1 | -1.9 | 31.01 | ||||||||||||||||||||||||||||||
| c.1193C>G | P398R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P398R variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include only FATHMM. All other evaluated predictors—SGM‑Consensus, REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—indicate a pathogenic effect, while Rosetta, premPS, and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.436924 | Structured | 0.401041 | Uncertain | 0.891 | 0.525 | 0.250 | -9.575 | Likely Pathogenic | 0.889 | Likely Pathogenic | Ambiguous | 3.01 | Destabilizing | 0.5 | 1.35 | Ambiguous | 2.18 | Destabilizing | 0.98 | Ambiguous | 0.755 | Likely Pathogenic | -6.55 | Deleterious | 0.988 | Probably Damaging | 0.724 | Possibly Damaging | 5.49 | Benign | 0.00 | Affected | 0.1479 | 0.3472 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.1195G>C | A399P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A399P missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, FATHMM, and polyPhen‑2 HumVar; pathogenic predictions come from FoldX, Rosetta, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta predicts a pathogenic effect. Overall, the majority of evidence points toward a deleterious impact. The variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.394753 | Structured | 0.407674 | Uncertain | 0.939 | 0.490 | 0.125 | -8.809 | Likely Pathogenic | 0.942 | Likely Pathogenic | Ambiguous | 2.29 | Destabilizing | 0.1 | 4.00 | Destabilizing | 3.15 | Destabilizing | 0.74 | Ambiguous | 0.498 | Likely Benign | -1.82 | Neutral | 0.596 | Possibly Damaging | 0.188 | Benign | 5.56 | Benign | 0.10 | Tolerated | 0.1847 | 0.5472 | 1 | -1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||
| c.1195G>T | A399S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A399S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect, while FoldX, Rosetta, and Foldetta are inconclusive. Grouping by agreement, all available predictors fall into the benign category; none predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign, whereas Foldetta’s stability analysis remains uncertain. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.394753 | Structured | 0.407674 | Uncertain | 0.939 | 0.490 | 0.125 | -4.256 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.65 | Ambiguous | 0.1 | 1.13 | Ambiguous | 0.89 | Ambiguous | -0.33 | Likely Benign | 0.161 | Likely Benign | 0.81 | Neutral | 0.001 | Benign | 0.001 | Benign | 5.65 | Benign | 0.86 | Tolerated | 0.2494 | 0.4897 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1196C>A | A399D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A399D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that agree on a pathogenic effect are REVEL, FoldX, SIFT, ESM1b, and AlphaMissense‑Default. The remaining tools—Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized—return uncertain or inconclusive results. High‑accuracy methods give no definitive verdict: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a tie and thus unavailable, and Foldetta is uncertain. Overall, the majority of available predictions (five pathogenic vs. four benign) lean toward pathogenicity. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.394753 | Structured | 0.407674 | Uncertain | 0.939 | 0.490 | 0.125 | -10.441 | Likely Pathogenic | 0.943 | Likely Pathogenic | Ambiguous | 2.45 | Destabilizing | 0.2 | 1.17 | Ambiguous | 1.81 | Ambiguous | 0.91 | Ambiguous | 0.525 | Likely Pathogenic | -1.84 | Neutral | 0.421 | Benign | 0.054 | Benign | 5.38 | Benign | 0.05 | Affected | 0.1533 | 0.1760 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||
| c.1196C>G | A399G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A399G is not reported in ClinVar and is absent from gnomAD. Across a panel of in silico predictors, all available pathogenicity scores classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. Uncertain results from FoldX, Rosetta, Foldetta, and premPS are treated as unavailable. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates benign. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive and therefore not considered evidence. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.394753 | Structured | 0.407674 | Uncertain | 0.939 | 0.490 | 0.125 | -6.513 | Likely Benign | 0.215 | Likely Benign | Likely Benign | 1.03 | Ambiguous | 0.1 | 1.77 | Ambiguous | 1.40 | Ambiguous | 0.74 | Ambiguous | 0.153 | Likely Benign | -1.59 | Neutral | 0.062 | Benign | 0.024 | Benign | 5.39 | Benign | 0.13 | Tolerated | 0.2174 | 0.4532 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1196C>T | A399V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A399V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). No tool predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Overall, the evidence overwhelmingly supports a benign classification, and this is consistent with the lack of ClinVar reporting. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.394753 | Structured | 0.407674 | Uncertain | 0.939 | 0.490 | 0.125 | -4.943 | Likely Benign | 0.170 | Likely Benign | Likely Benign | 0.49 | Likely Benign | 0.2 | 0.56 | Ambiguous | 0.53 | Ambiguous | 0.22 | Likely Benign | 0.305 | Likely Benign | -1.18 | Neutral | 0.421 | Benign | 0.073 | Benign | 5.37 | Benign | 0.24 | Tolerated | 0.1145 | 0.6807 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.1199T>C | V400A 2D 3DClick to see structure in 3D Viewer AISynGAP1 V400A is not reported in ClinVar and is absent from gnomAD. Consensus from standard predictors shows a split: benign calls from REVEL, polyPhen‑2 (HumDiv and HumVar) and FATHMM, while pathogenic calls come from FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT and AlphaMissense‑Default. Two high‑accuracy tools give a clear signal: AlphaMissense‑Optimized is uncertain, but the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta also predicts pathogenic. With most evidence pointing to deleterious effects, the variant is most likely pathogenic, and this assessment does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.398279 | Structured | 0.415488 | Uncertain | 0.951 | 0.451 | 0.000 | -7.564 | In-Between | 0.871 | Likely Pathogenic | Ambiguous | 3.14 | Destabilizing | 0.1 | 3.12 | Destabilizing | 3.13 | Destabilizing | 2.29 | Destabilizing | 0.479 | Likely Benign | -3.12 | Deleterious | 0.435 | Benign | 0.049 | Benign | 5.32 | Benign | 0.01 | Affected | 0.3291 | 0.2767 | 0 | 0 | -2.4 | -28.05 | ||||||||||||||||||||||||||||||
| c.1199T>G | V400G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V400G missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are polyPhen‑2 HumDiv and FATHMM; all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.398279 | Structured | 0.415488 | Uncertain | 0.951 | 0.451 | 0.000 | -11.508 | Likely Pathogenic | 0.969 | Likely Pathogenic | Likely Pathogenic | 4.84 | Destabilizing | 0.1 | 5.40 | Destabilizing | 5.12 | Destabilizing | 2.40 | Destabilizing | 0.819 | Likely Pathogenic | -5.70 | Deleterious | 0.335 | Benign | 0.920 | Probably Damaging | 5.27 | Benign | 0.00 | Affected | 0.2296 | 0.2522 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.119A>C | D40A 2D AIThe SynGAP1 missense variant D40A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Taken together, the majority of evidence supports a benign interpretation, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -3.630 | Likely Benign | 0.339 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -1.22 | Neutral | 0.006 | Benign | 0.023 | Benign | 4.05 | Benign | 0.00 | Affected | 0.4299 | 0.7993 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.119A>G | D40G 2D AIThe SynGAP1 D40G missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -2.777 | Likely Benign | 0.287 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -1.43 | Neutral | 0.012 | Benign | 0.032 | Benign | 4.01 | Benign | 0.00 | Affected | 0.4110 | 0.7949 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.119A>T | D40V 2D AIThe SynGAP1 missense variant D40V is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -3.350 | Likely Benign | 0.431 | Ambiguous | Likely Benign | 0.222 | Likely Benign | -1.16 | Neutral | 0.028 | Benign | 0.088 | Benign | 4.00 | Benign | 0.00 | Affected | 0.1679 | 0.8357 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.11C>G | S4C 2D AIThe SynGAP1 missense variant S4C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for S4C, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | -5.210 | Likely Benign | 0.124 | Likely Benign | Likely Benign | 0.106 | Likely Benign | 0.41 | Neutral | 0.880 | Possibly Damaging | 0.700 | Possibly Damaging | 4.11 | Benign | 0.00 | Affected | 0.0976 | 0.6129 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||||||
| c.11C>T | S4F 2D AIThe SynGAP1 missense variant S4F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for S4F, and this conclusion is consistent with the lack of any ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.547364 | Binding | 0.390 | 0.924 | 0.750 | -4.880 | Likely Benign | 0.317 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.17 | Neutral | 0.676 | Possibly Damaging | 0.485 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.0686 | 0.6270 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||||||
| c.1201C>G | R401G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R401G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM; all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.314870 | Structured | 0.424277 | Uncertain | 0.961 | 0.419 | 0.000 | -13.353 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.06 | Destabilizing | 0.2 | 4.31 | Destabilizing | 3.69 | Destabilizing | 1.01 | Destabilizing | 0.741 | Likely Pathogenic | -6.45 | Deleterious | 0.997 | Probably Damaging | 0.987 | Probably Damaging | 5.45 | Benign | 0.00 | Affected | 0.3549 | 0.2866 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1202G>C | R401P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R401P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while the remaining twelve tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The high‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.314870 | Structured | 0.424277 | Uncertain | 0.961 | 0.419 | 0.000 | -14.090 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 9.69 | Destabilizing | 0.3 | 8.07 | Destabilizing | 8.88 | Destabilizing | 0.77 | Ambiguous | 0.892 | Likely Pathogenic | -6.45 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 5.42 | Benign | 0.00 | Affected | 0.2265 | 0.4064 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.1204C>A | L402M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L402M is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Uncertain results come from Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.243554 | Structured | 0.431978 | Uncertain | 0.961 | 0.383 | 0.000 | -6.991 | Likely Benign | 0.305 | Likely Benign | Likely Benign | 0.12 | Likely Benign | 0.0 | 1.33 | Ambiguous | 0.73 | Ambiguous | 0.80 | Ambiguous | 0.071 | Likely Benign | -0.99 | Neutral | 0.994 | Probably Damaging | 0.938 | Probably Damaging | 3.73 | Benign | 0.02 | Affected | 0.1064 | 0.4095 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.1204C>G | L402V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L402V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign; the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a benign outcome. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result and is therefore not considered evidence for pathogenicity. In summary, all available predictions support a benign classification for the L402V variant, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.243554 | Structured | 0.431978 | Uncertain | 0.961 | 0.383 | 0.000 | -3.467 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 1.91 | Ambiguous | 0.1 | 0.78 | Ambiguous | 1.35 | Ambiguous | -0.11 | Likely Benign | 0.203 | Likely Benign | 0.18 | Neutral | 0.004 | Benign | 0.004 | Benign | 4.01 | Benign | 0.29 | Tolerated | 0.1807 | 0.3657 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1205T>A | L402Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L402Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels it Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely pathogenic based on the collective computational evidence, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.243554 | Structured | 0.431978 | Uncertain | 0.961 | 0.383 | 0.000 | -13.403 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.93 | Destabilizing | 0.0 | 2.55 | Destabilizing | 2.74 | Destabilizing | 2.09 | Destabilizing | 0.523 | Likely Pathogenic | -4.52 | Deleterious | 0.999 | Probably Damaging | 0.957 | Probably Damaging | 3.69 | Benign | 0.00 | Affected | 0.1384 | 0.1173 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1205T>C | L402P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L402P is not reported in ClinVar and is absent from gnomAD. Consensus among in‑silico predictors is overwhelmingly pathogenic: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a deleterious effect, whereas only FATHMM predicts a benign outcome. High‑accuracy tools reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic effect; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Taken together, the evidence strongly supports a pathogenic classification, and this assessment does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.243554 | Structured | 0.431978 | Uncertain | 0.961 | 0.383 | 0.000 | -12.030 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 7.40 | Destabilizing | 0.1 | 5.82 | Destabilizing | 6.61 | Destabilizing | 2.22 | Destabilizing | 0.616 | Likely Pathogenic | -4.80 | Deleterious | 1.000 | Probably Damaging | 0.980 | Probably Damaging | 3.68 | Benign | 0.01 | Affected | 0.3628 | 0.1874 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1207A>C | K403Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K403Q missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and FATHMM. In contrast, a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score. The premPS assessment is uncertain and does not influence the overall consensus. High‑accuracy analyses show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar claim exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.275179 | Structured | 0.424920 | Uncertain | 0.960 | 0.372 | 0.000 | -12.479 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.36 | Likely Benign | 0.0 | 0.27 | Likely Benign | 0.32 | Likely Benign | 0.70 | Ambiguous | 0.376 | Likely Benign | -3.59 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 3.76 | Benign | 0.01 | Affected | 0.4405 | 0.1954 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1207A>G | K403E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K403E missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact for K403E, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.275179 | Structured | 0.424920 | Uncertain | 0.960 | 0.372 | 0.000 | -15.279 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.01 | Ambiguous | 0.1 | 0.79 | Ambiguous | 0.90 | Ambiguous | 0.70 | Ambiguous | 0.445 | Likely Benign | -3.62 | Deleterious | 0.998 | Probably Damaging | 0.981 | Probably Damaging | 3.79 | Benign | 0.04 | Affected | 0.3650 | 0.1715 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1208A>C | K403T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K403T missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to FATHMM, which scores the variant as benign. In contrast, the majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; SGM‑Consensus predicts likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. Other stability predictors (FoldX, Rosetta, premPS) are also uncertain. Overall, the consensus of the majority of tools indicates a pathogenic effect. This conclusion is not contradicted by ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.275179 | Structured | 0.424920 | Uncertain | 0.960 | 0.372 | 0.000 | -13.135 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.41 | Ambiguous | 0.1 | 0.59 | Ambiguous | 1.00 | Ambiguous | 0.67 | Ambiguous | 0.522 | Likely Pathogenic | -5.43 | Deleterious | 0.999 | Probably Damaging | 1.000 | Probably Damaging | 3.73 | Benign | 0.01 | Affected | 0.1861 | 0.4230 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1208A>G | K403R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K403R missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The remaining tools—FoldX, ESM1b, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a tie, and Foldetta also predicts benign. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.275179 | Structured | 0.424920 | Uncertain | 0.960 | 0.372 | 0.000 | -7.362 | In-Between | 0.355 | Ambiguous | Likely Benign | -0.78 | Ambiguous | 0.1 | 0.23 | Likely Benign | -0.28 | Likely Benign | 0.41 | Likely Benign | 0.335 | Likely Benign | -2.56 | Deleterious | 0.983 | Probably Damaging | 0.926 | Probably Damaging | 3.91 | Benign | 0.15 | Tolerated | 0.4834 | 0.1861 | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||
| c.1208A>T | K403I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K403I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, premPS, and FATHMM, while the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions (nine pathogenic vs. five benign) and the high‑accuracy tools lean toward a pathogenic effect. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.275179 | Structured | 0.424920 | Uncertain | 0.960 | 0.372 | 0.000 | -15.239 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.17 | Likely Benign | 0.1 | -0.46 | Likely Benign | -0.15 | Likely Benign | 0.36 | Likely Benign | 0.519 | Likely Pathogenic | -7.27 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.70 | Benign | 0.00 | Affected | 0.1223 | 0.4141 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1209A>C | K403N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K403N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized) predict a pathogenic impact. Predictions from FoldX, Foldetta, and premPS are uncertain or inconclusive and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.275179 | Structured | 0.424920 | Uncertain | 0.960 | 0.372 | 0.000 | -10.913 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.87 | Ambiguous | 0.1 | 0.41 | Likely Benign | 1.14 | Ambiguous | 0.74 | Ambiguous | 0.201 | Likely Benign | -4.51 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.73 | Benign | 0.05 | Affected | 0.3374 | 0.2492 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1209A>T | K403N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K403N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized) predict a pathogenic impact. Predictions from FoldX, Foldetta, and premPS are uncertain or inconclusive and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.275179 | Structured | 0.424920 | Uncertain | 0.960 | 0.372 | 0.000 | -10.913 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.87 | Ambiguous | 0.1 | 0.41 | Likely Benign | 1.14 | Ambiguous | 0.74 | Ambiguous | 0.201 | Likely Benign | -4.51 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.73 | Benign | 0.05 | Affected | 0.3374 | 0.2492 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.120T>A | D40E 2D AIThe SynGAP1 missense variant D40E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -3.520 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.050 | Likely Benign | -0.21 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.20 | Benign | 0.00 | Affected | 0.2764 | 0.8141 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.120T>G | D40E 2D AIThe SynGAP1 missense variant D40E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Thus, the variant is most likely benign, and this assessment does not contradict the ClinVar record, which contains no pathogenic claim. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.384043 | Structured | 0.432002 | Uncertain | 0.319 | 0.769 | 0.375 | -3.520 | Likely Benign | 0.096 | Likely Benign | Likely Benign | 0.050 | Likely Benign | -0.21 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.20 | Benign | 0.00 | Affected | 0.2764 | 0.8141 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1210G>A | A404T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A404T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: SGM‑Consensus (Likely Benign), REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only FoldX and premPS are inconclusive, so they are treated as unavailable. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta) is benign. Overall, the evidence strongly supports a benign classification, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.232838 | Structured | 0.415505 | Uncertain | 0.965 | 0.355 | 0.000 | -6.674 | Likely Benign | 0.279 | Likely Benign | Likely Benign | 0.74 | Ambiguous | 0.1 | -0.01 | Likely Benign | 0.37 | Likely Benign | 0.56 | Ambiguous | 0.060 | Likely Benign | -1.74 | Neutral | 0.049 | Benign | 0.011 | Benign | 4.12 | Benign | 0.09 | Tolerated | 0.1710 | 0.7367 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.1210G>C | A404P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A404P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, Rosetta, both polyPhen‑2 versions, and FATHMM, while pathogenic calls arise from FoldX, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic verdict. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus confirms likely pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.232838 | Structured | 0.415505 | Uncertain | 0.965 | 0.355 | 0.000 | -11.819 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.03 | Destabilizing | 0.1 | 0.24 | Likely Benign | 1.64 | Ambiguous | 1.17 | Destabilizing | 0.227 | Likely Benign | -3.16 | Deleterious | 0.433 | Benign | 0.080 | Benign | 4.02 | Benign | 0.03 | Affected | 0.2271 | 0.6366 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1210G>T | A404S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A404S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a pathogenic outcome. The high‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates a benign effect. No inconclusive or missing predictions are present. Based on the unanimous benign predictions and the lack of ClinVar evidence, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.232838 | Structured | 0.415505 | Uncertain | 0.965 | 0.355 | 0.000 | -1.937 | Likely Benign | 0.194 | Likely Benign | Likely Benign | 0.13 | Likely Benign | 0.0 | 0.83 | Ambiguous | 0.48 | Likely Benign | 0.28 | Likely Benign | 0.078 | Likely Benign | -0.07 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.18 | Benign | 0.22 | Tolerated | 0.2970 | 0.6178 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1211C>A | A404E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A404E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. In contrast, the majority of tools predict a pathogenic impact: FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate pathogenicity, and the SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.232838 | Structured | 0.415505 | Uncertain | 0.965 | 0.355 | 0.000 | -13.639 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 2.16 | Destabilizing | 0.1 | 3.04 | Destabilizing | 2.60 | Destabilizing | 1.51 | Destabilizing | 0.163 | Likely Benign | -2.77 | Deleterious | 0.179 | Benign | 0.033 | Benign | 4.02 | Benign | 0.02 | Affected | 0.1423 | 0.2383 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||
| c.1211C>G | A404G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A404G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are premPS and SIFT. FoldX, Rosetta, and Foldetta give uncertain or inconclusive results and are treated as unavailable. The high‑accuracy consensus (SGM‑Consensus) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains uncertain. Overall, the majority of evidence points to a benign impact for A404G, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.232838 | Structured | 0.415505 | Uncertain | 0.965 | 0.355 | 0.000 | -5.277 | Likely Benign | 0.137 | Likely Benign | Likely Benign | 0.92 | Ambiguous | 0.0 | 1.88 | Ambiguous | 1.40 | Ambiguous | 1.18 | Destabilizing | 0.099 | Likely Benign | -2.34 | Neutral | 0.045 | Benign | 0.013 | Benign | 4.03 | Benign | 0.00 | Affected | 0.2370 | 0.5054 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1213C>A | R405S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R405S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from REVEL and FATHMM, whereas pathogenic predictions are made by FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts Pathogenic; the SGM‑Consensus itself is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts Pathogenic. Taken together, the overwhelming majority of evidence indicates that R405S is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.404888 | Uncertain | 0.949 | 0.315 | 0.000 | -11.326 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 2.66 | Destabilizing | 0.5 | 2.08 | Destabilizing | 2.37 | Destabilizing | 1.22 | Destabilizing | 0.405 | Likely Benign | -5.40 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.67 | Benign | 0.05 | Affected | 0.2829 | 0.4646 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||
| c.1213C>G | R405G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R405G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.404888 | Uncertain | 0.949 | 0.315 | 0.000 | -11.836 | Likely Pathogenic | 0.958 | Likely Pathogenic | Likely Pathogenic | 3.22 | Destabilizing | 0.5 | 3.26 | Destabilizing | 3.24 | Destabilizing | 1.38 | Destabilizing | 0.419 | Likely Benign | -6.35 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.63 | Benign | 0.01 | Affected | 0.3381 | 0.4139 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1214G>T | R405L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R405L missense change is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are Rosetta and FATHMM, while the majority of other in silico predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) indicate a pathogenic or likely pathogenic impact. Uncertain results come from FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labeling it likely pathogenic, and Foldetta providing an inconclusive stability prediction. Overall, the preponderance of evidence points to a pathogenic effect for R405L, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.250310 | Structured | 0.404888 | Uncertain | 0.949 | 0.315 | 0.000 | -11.576 | Likely Pathogenic | 0.956 | Likely Pathogenic | Likely Pathogenic | 1.43 | Ambiguous | 0.7 | 0.40 | Likely Benign | 0.92 | Ambiguous | 0.72 | Ambiguous | 0.512 | Likely Pathogenic | -6.35 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.64 | Benign | 0.01 | Affected | 0.2126 | 0.5555 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.1216T>A | Y406N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y406N is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely pathogenic outcome, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also indicates pathogenicity. Taken together, the preponderance of evidence points to a pathogenic effect for Y406N, and this conclusion is consistent with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.179055 | Structured | 0.393707 | Uncertain | 0.946 | 0.291 | 0.000 | -13.206 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 2.90 | Destabilizing | 0.1 | 2.88 | Destabilizing | 2.89 | Destabilizing | 1.58 | Destabilizing | 0.288 | Likely Benign | -7.11 | Deleterious | 0.999 | Probably Damaging | 0.966 | Probably Damaging | 3.78 | Benign | 0.02 | Affected | 0.2532 | 0.1071 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.1216T>C | Y406H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Y406H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors—FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently classify the substitution as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely pathogenic outcome, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a pathogenic effect. Taken together, the preponderance of evidence indicates that Y406H is most likely pathogenic, and this conclusion does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.179055 | Structured | 0.393707 | Uncertain | 0.946 | 0.291 | 0.000 | -9.015 | Likely Pathogenic | 0.858 | Likely Pathogenic | Ambiguous | 2.13 | Destabilizing | 0.1 | 2.15 | Destabilizing | 2.14 | Destabilizing | 1.03 | Destabilizing | 0.239 | Likely Benign | -4.21 | Deleterious | 0.997 | Probably Damaging | 0.966 | Probably Damaging | 3.80 | Benign | 0.03 | Affected | 0.2666 | 0.0811 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||
| c.1216T>G | Y406D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y406D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and FATHMM. In contrast, the majority of tools predict a pathogenic impact: FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus score is “Likely Pathogenic,” while premPS is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the preponderance of evidence, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status (which is currently unreported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.179055 | Structured | 0.393707 | Uncertain | 0.946 | 0.291 | 0.000 | -14.832 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.82 | Destabilizing | 0.3 | 4.28 | Destabilizing | 4.05 | Destabilizing | 0.98 | Ambiguous | 0.347 | Likely Benign | -7.64 | Deleterious | 0.999 | Probably Damaging | 0.950 | Probably Damaging | 3.77 | Benign | 0.01 | Affected | 0.4439 | 0.0903 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1217A>C | Y406S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y406S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. In contrast, the majority of tools predict a pathogenic impact: AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FoldX, Rosetta, premPS, and Foldetta all indicate pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No predictions are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.179055 | Structured | 0.393707 | Uncertain | 0.946 | 0.291 | 0.000 | -12.014 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 3.51 | Destabilizing | 0.3 | 4.08 | Destabilizing | 3.80 | Destabilizing | 1.40 | Destabilizing | 0.190 | Likely Benign | -6.72 | Deleterious | 0.991 | Probably Damaging | 0.886 | Possibly Damaging | 3.80 | Benign | 0.06 | Tolerated | 0.4719 | 0.2817 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||
| c.1217A>G | Y406C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y406C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome, and the Foldetta stability assessment (combining FoldX‑MD and Rosetta) predicts a destabilizing, pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while both SGM‑Consensus and Foldetta are pathogenic. Overall, the preponderance of evidence points to a pathogenic effect for Y406C, and this conclusion does not contradict the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.179055 | Structured | 0.393707 | Uncertain | 0.946 | 0.291 | 0.000 | -9.954 | Likely Pathogenic | 0.810 | Likely Pathogenic | Ambiguous | 2.47 | Destabilizing | 0.2 | 3.22 | Destabilizing | 2.85 | Destabilizing | 1.27 | Destabilizing | 0.229 | Likely Benign | -6.72 | Deleterious | 0.998 | Probably Damaging | 0.851 | Possibly Damaging | 3.78 | Benign | 0.01 | Affected | 0.3026 | 0.2861 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||
| c.1217A>T | Y406F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y406F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the variant as benign or likely benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.179055 | Structured | 0.393707 | Uncertain | 0.946 | 0.291 | 0.000 | -3.427 | Likely Benign | 0.080 | Likely Benign | Likely Benign | -0.01 | Likely Benign | 0.2 | 0.40 | Likely Benign | 0.20 | Likely Benign | 0.15 | Likely Benign | 0.079 | Likely Benign | -0.93 | Neutral | 0.002 | Benign | 0.002 | Benign | 3.96 | Benign | 0.31 | Tolerated | 0.2673 | 0.3952 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||
| c.1219C>A | Q407K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q407K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Two tools, FoldX and premPS, give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is unavailable because FoldX is uncertain and Rosetta alone is benign. Overall, the majority of evidence points to a pathogenic impact for Q407K. This conclusion is not contradicted by ClinVar status, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.109221 | Structured | 0.382522 | Uncertain | 0.916 | 0.271 | 0.000 | -14.893 | Likely Pathogenic | 0.765 | Likely Pathogenic | Likely Benign | 0.61 | Ambiguous | 0.1 | 0.19 | Likely Benign | 0.40 | Likely Benign | 0.93 | Ambiguous | 0.246 | Likely Benign | -3.42 | Deleterious | 0.863 | Possibly Damaging | 0.773 | Possibly Damaging | 3.96 | Benign | 0.07 | Tolerated | 0.1656 | 0.3504 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||
| c.1219C>G | Q407E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q407E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome. AlphaMissense‑Default, FoldX, Rosetta, and Foldetta are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of tools and the SGM Consensus support a pathogenic classification, while a minority predict benign. No ClinVar entry exists to contradict these predictions. Thus, the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.109221 | Structured | 0.382522 | Uncertain | 0.916 | 0.271 | 0.000 | -12.631 | Likely Pathogenic | 0.466 | Ambiguous | Likely Benign | 0.50 | Ambiguous | 0.1 | 1.68 | Ambiguous | 1.09 | Ambiguous | 1.30 | Destabilizing | 0.243 | Likely Benign | -2.66 | Deleterious | 0.989 | Probably Damaging | 0.930 | Probably Damaging | 3.96 | Benign | 0.03 | Affected | 0.1199 | 0.2000 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.121C>A | R41S 2D AIThe SynGAP1 missense variant R41S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R41S, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.311707 | Structured | 0.431757 | Uncertain | 0.344 | 0.765 | 0.375 | -3.367 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.130 | Likely Benign | -0.22 | Neutral | 0.686 | Possibly Damaging | 0.735 | Possibly Damaging | 4.24 | Benign | 0.00 | Affected | 0.3190 | 0.5013 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||||||||||||
| c.121C>G | R41G 2D AIThe SynGAP1 missense variant R41G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for R41G, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.311707 | Structured | 0.431757 | Uncertain | 0.344 | 0.765 | 0.375 | -3.737 | Likely Benign | 0.227 | Likely Benign | Likely Benign | 0.086 | Likely Benign | -1.12 | Neutral | 0.686 | Possibly Damaging | 0.630 | Possibly Damaging | 4.16 | Benign | 0.00 | Affected | 0.3620 | 0.4306 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.1220A>C | Q407P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q407P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact; premPS is uncertain and is not counted. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the consensus of these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.109221 | Structured | 0.382522 | Uncertain | 0.916 | 0.271 | 0.000 | -13.578 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.04 | Destabilizing | 0.5 | 2.07 | Destabilizing | 2.56 | Destabilizing | 0.88 | Ambiguous | 0.515 | Likely Pathogenic | -5.40 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.88 | Benign | 0.02 | Affected | 0.1786 | 0.4352 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||
| c.1220A>G | Q407R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q407R has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. With two of the three high‑accuracy methods indicating benign and the remaining one pathogenic, the overall evidence leans toward a benign classification. This conclusion does not contradict ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.109221 | Structured | 0.382522 | Uncertain | 0.916 | 0.271 | 0.000 | -13.263 | Likely Pathogenic | 0.693 | Likely Pathogenic | Likely Benign | 0.15 | Likely Benign | 0.2 | 0.09 | Likely Benign | 0.12 | Likely Benign | 0.65 | Ambiguous | 0.340 | Likely Benign | -3.52 | Deleterious | 0.909 | Possibly Damaging | 0.889 | Possibly Damaging | 4.02 | Benign | 0.17 | Tolerated | 0.1347 | 0.1596 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||
| c.1220A>T | Q407L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q407L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, FATHMM, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Four tools are uncertain (AlphaMissense‑Default, FoldX, Rosetta, Foldetta). High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, more tools predict pathogenicity than benign, and the high‑accuracy consensus leans pathogenic. Therefore, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.109221 | Structured | 0.382522 | Uncertain | 0.916 | 0.271 | 0.000 | -12.730 | Likely Pathogenic | 0.558 | Ambiguous | Likely Benign | -0.65 | Ambiguous | 0.2 | -0.69 | Ambiguous | -0.67 | Ambiguous | 0.35 | Likely Benign | 0.359 | Likely Benign | -6.32 | Deleterious | 0.939 | Possibly Damaging | 0.838 | Possibly Damaging | 3.91 | Benign | 0.02 | Affected | 0.0585 | 0.4977 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||
| c.1222A>C | T408P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T408P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. FoldX, premPS, and Foldetta are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a tie and thus unavailable; Foldetta remains uncertain. Overall, the majority of available predictions (six pathogenic vs. four benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.161087 | Structured | 0.370935 | Uncertain | 0.907 | 0.239 | 0.000 | -10.384 | Likely Pathogenic | 0.230 | Likely Benign | Likely Benign | 1.08 | Ambiguous | 0.3 | 2.27 | Destabilizing | 1.68 | Ambiguous | 0.73 | Ambiguous | 0.323 | Likely Benign | -4.19 | Deleterious | 0.998 | Probably Damaging | 0.963 | Probably Damaging | 4.07 | Benign | 0.05 | Affected | 0.1985 | 0.5779 | 0 | -1 | -0.9 | -3.99 | ||||||||||||||||||||||||||||||
| c.1222A>G | T408A 2D AISynGAP1 missense variant T408A is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv), and ESM1b. The high‑accuracy AlphaMissense‑Optimized score is benign, while the SGM consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Foldetta, a protein‑folding stability method, also predicts benign. Overall, the balance of evidence favors a benign impact, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.161087 | Structured | 0.370935 | Uncertain | 0.907 | 0.239 | 0.000 | Uncertain | 1 | -8.304 | Likely Pathogenic | 0.114 | Likely Benign | Likely Benign | 0.37 | Likely Benign | 0.6 | -0.06 | Likely Benign | 0.16 | Likely Benign | 0.72 | Ambiguous | 0.118 | Likely Benign | -3.07 | Deleterious | 0.540 | Possibly Damaging | 0.131 | Benign | 4.16 | Benign | 0.14 | Tolerated | 0.3970 | 0.4674 | 1 | 0 | 2.5 | -30.03 | ||||||||||||||||||||||||||||
| c.1222A>T | T408S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T408S is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, FoldX, Rosetta, premPS, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). No tool predicts pathogenicity. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates benign stability. Consequently, the variant is most likely benign, and this prediction does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.161087 | Structured | 0.370935 | Uncertain | 0.907 | 0.239 | 0.000 | -6.277 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 0.39 | Likely Benign | 0.25 | Likely Benign | 0.18 | Likely Benign | 0.044 | Likely Benign | -1.48 | Neutral | 0.182 | Benign | 0.127 | Benign | 4.24 | Benign | 0.21 | Tolerated | 0.3256 | 0.4767 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1223C>A | T408K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T408K is not listed in ClinVar (ClinVar ID None) and has no reported allele in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, SIFT, FATHMM, polyPhen‑2 HumVar, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default; premPS remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labeling it likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicting a benign effect. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.161087 | Structured | 0.370935 | Uncertain | 0.907 | 0.239 | 0.000 | -11.841 | Likely Pathogenic | 0.756 | Likely Pathogenic | Likely Benign | -0.44 | Likely Benign | 0.2 | 0.14 | Likely Benign | -0.15 | Likely Benign | 0.88 | Ambiguous | 0.131 | Likely Benign | -3.79 | Deleterious | 0.851 | Possibly Damaging | 0.163 | Benign | 4.17 | Benign | 0.15 | Tolerated | 0.1205 | 0.3384 | 0 | -1 | -3.2 | 27.07 | |||||||||||||||||||||||||||||
| c.1223C>G | T408R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T408R is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The premPS score is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. With six benign versus seven pathogenic calls overall, the evidence is mixed, but the presence of two independent high‑accuracy benign predictions and the lack of ClinVar or gnomAD support suggests the variant is most likely benign, and this does not contradict any existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.161087 | Structured | 0.370935 | Uncertain | 0.907 | 0.239 | 0.000 | -12.075 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | -0.48 | Likely Benign | 0.1 | -0.24 | Likely Benign | -0.36 | Likely Benign | 0.79 | Ambiguous | 0.138 | Likely Benign | -4.06 | Deleterious | 0.991 | Probably Damaging | 0.645 | Possibly Damaging | 4.12 | Benign | 0.15 | Tolerated | 0.1029 | 0.3180 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.1225A>C | M409L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M409L is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts pathogenicity. The only inconclusive result is from premPS, which is marked uncertain and does not influence the overall benign consensus. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -6.809 | Likely Benign | 0.286 | Likely Benign | Likely Benign | -0.04 | Likely Benign | 0.2 | -0.08 | Likely Benign | -0.06 | Likely Benign | 0.51 | Ambiguous | 0.199 | Likely Benign | -0.84 | Neutral | 0.206 | Benign | 0.324 | Benign | 4.21 | Benign | 0.57 | Tolerated | 0.1299 | 0.4411 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1225A>G | M409V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M409V is not reported in ClinVar and is absent from gnomAD. Consensus from most in silico predictors indicates a benign effect: REVEL, Rosetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports likely benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote) is likely benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, reports benign. No conflicting evidence is present. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -4.109 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 1.13 | Ambiguous | 0.2 | -0.25 | Likely Benign | 0.44 | Likely Benign | 0.37 | Likely Benign | 0.163 | Likely Benign | -0.76 | Neutral | 0.996 | Probably Damaging | 0.974 | Probably Damaging | 4.26 | Benign | 1.00 | Tolerated | 0.2706 | 0.4286 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||
| c.1225A>T | M409L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M409L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts pathogenicity. The only inconclusive result is premPS, which is listed as uncertain and does not influence the overall assessment. High‑accuracy methods confirm the benign prediction: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also classifies it as benign. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -6.809 | Likely Benign | 0.286 | Likely Benign | Likely Benign | -0.04 | Likely Benign | 0.2 | -0.08 | Likely Benign | -0.06 | Likely Benign | 0.51 | Ambiguous | 0.199 | Likely Benign | -0.84 | Neutral | 0.206 | Benign | 0.324 | Benign | 4.21 | Benign | 0.57 | Tolerated | 0.1299 | 0.4411 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1226T>A | M409K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M409K has no ClinVar record and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, Rosetta, SIFT, and FATHMM, while pathogenic calls arise from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Uncertain results are reported by FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized is inconclusive; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also inconclusive. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -13.618 | Likely Pathogenic | 0.927 | Likely Pathogenic | Ambiguous | 0.93 | Ambiguous | 0.3 | 0.29 | Likely Benign | 0.61 | Ambiguous | 1.45 | Destabilizing | 0.490 | Likely Benign | -4.26 | Deleterious | 0.769 | Possibly Damaging | 0.750 | Possibly Damaging | 4.18 | Benign | 0.40 | Tolerated | 0.1318 | 0.0656 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||
| c.1226T>C | M409T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M409T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Uncertain results are reported by FoldX, Foldetta, and premPS. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized indicates a benign effect, whereas the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) supports a pathogenic outcome; Foldetta remains inconclusive. Overall, the majority of evidence, including the SGM‑Consensus, points to a pathogenic impact for M409T. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -9.194 | Likely Pathogenic | 0.607 | Likely Pathogenic | Likely Benign | 1.41 | Ambiguous | 0.2 | 0.48 | Likely Benign | 0.95 | Ambiguous | 0.96 | Ambiguous | 0.262 | Likely Benign | -3.18 | Deleterious | 0.987 | Probably Damaging | 0.945 | Probably Damaging | 4.17 | Benign | 0.45 | Tolerated | 0.1835 | 0.2391 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.1227G>A | M409I 2D  AISynGAP1 missense variant M409I is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default). High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, reports a benign effect. FoldX and Rosetta individually give uncertain results and are treated as unavailable. Overall, the majority of reliable predictors indicate a benign impact for M409I. Thus, the variant is most likely benign, and this conclusion does not contradict the lack of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -3.735 | Likely Benign | 0.598 | Likely Pathogenic | Likely Benign | 0.59 | Ambiguous | 0.8 | -0.79 | Ambiguous | -0.10 | Likely Benign | 0.36 | Likely Benign | 0.162 | Likely Benign | -0.58 | Neutral | 0.579 | Possibly Damaging | 0.663 | Possibly Damaging | 4.22 | Benign | 0.92 | Tolerated | 0.1105 | 0.3358 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1227G>C | M409I 2D AISynGAP1 missense variant M409I is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default). High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, reports a benign effect. FoldX and Rosetta individually give uncertain results and are treated as unavailable. Overall, the majority of reliable predictors indicate a benign impact for M409I, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -3.735 | Likely Benign | 0.598 | Likely Pathogenic | Likely Benign | 0.59 | Ambiguous | 0.8 | -0.79 | Ambiguous | -0.10 | Likely Benign | 0.36 | Likely Benign | 0.162 | Likely Benign | -0.58 | Neutral | 0.579 | Possibly Damaging | 0.663 | Possibly Damaging | 4.22 | Benign | 0.92 | Tolerated | 0.1105 | 0.3358 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1227G>T | M409I 2D AISynGAP1 missense variant M409I is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster into two groups: benign (REVEL, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default). High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, reports a benign effect. FoldX and Rosetta individually give uncertain results and are treated as unavailable. Overall, the majority of reliable predictors indicate a benign impact for M409I, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -3.735 | Likely Benign | 0.598 | Likely Pathogenic | Likely Benign | 0.59 | Ambiguous | 0.8 | -0.79 | Ambiguous | -0.10 | Likely Benign | 0.36 | Likely Benign | 0.162 | Likely Benign | -0.58 | Neutral | 0.579 | Possibly Damaging | 0.663 | Possibly Damaging | 4.22 | Benign | 0.92 | Tolerated | 0.1105 | 0.3358 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1228A>C | S410R 2D  3DClick to see structure in 3D Viewer AIClinVar contains no record for the SynGAP1 S410R variant, and it is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL, Foldetta, PROVEAN, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. The remaining tools (FoldX, Rosetta, premPS, AlphaMissense‑Optimized) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, Foldetta predicts a benign impact, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Taken together, the majority of evidence points to a benign outcome. This conclusion does not contradict ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -8.203 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | -0.67 | Ambiguous | 0.2 | 0.56 | Ambiguous | -0.06 | Likely Benign | 0.62 | Ambiguous | 0.242 | Likely Benign | -2.47 | Neutral | 0.871 | Possibly Damaging | 0.298 | Benign | 4.20 | Benign | 0.40 | Tolerated | 0.0955 | 0.3927 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||
| c.1228A>G | S410G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S410G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. The remaining tools (FoldX, Rosetta, Foldetta, premPS, ESM1b) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -7.147 | In-Between | 0.137 | Likely Benign | Likely Benign | 0.58 | Ambiguous | 0.1 | 1.33 | Ambiguous | 0.96 | Ambiguous | 0.85 | Ambiguous | 0.117 | Likely Benign | -2.54 | Deleterious | 0.952 | Possibly Damaging | 0.145 | Benign | 4.13 | Benign | 0.19 | Tolerated | 0.2651 | 0.5143 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||||
| c.1228A>T | S410C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S410C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict it as pathogenic are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -7.552 | In-Between | 0.144 | Likely Benign | Likely Benign | -0.24 | Likely Benign | 0.1 | 0.31 | Likely Benign | 0.04 | Likely Benign | 0.22 | Likely Benign | 0.230 | Likely Benign | -3.10 | Deleterious | 0.993 | Probably Damaging | 0.536 | Possibly Damaging | 4.12 | Benign | 0.11 | Tolerated | 0.1041 | 0.6543 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||
| c.1229G>A | S410N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S410N is predicted to be benign by all evaluated in‑silico tools. Consensus predictors (REVEL, SIFT, polyPhen‑2 HumDiv/HumVar, PROVEAN, premPS, FoldX, Rosetta, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly report a benign effect, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies it as “Likely Benign.” High‑accuracy assessments likewise support a benign outcome: AlphaMissense‑Optimized is benign, the SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) indicates a benign effect. ClinVar contains no entry for this variant, and it is not present in gnomAD. Based on the collective predictions, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -2.901 | Likely Benign | 0.111 | Likely Benign | Likely Benign | -0.16 | Likely Benign | 0.1 | -0.16 | Likely Benign | -0.16 | Likely Benign | 0.38 | Likely Benign | 0.049 | Likely Benign | -0.91 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.20 | Benign | 0.18 | Tolerated | 0.1310 | 0.5471 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||
| c.1229G>C | S410T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S410T has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, FoldX, Foldetta, SGM‑Consensus, REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, and premPS. No tool predicts a pathogenic outcome; the only inconclusive result is from Rosetta, which is treated as unavailable. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. **Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available).** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -6.559 | Likely Benign | 0.123 | Likely Benign | Likely Benign | -0.32 | Likely Benign | 0.1 | -0.52 | Ambiguous | -0.42 | Likely Benign | 0.00 | Likely Benign | 0.066 | Likely Benign | -0.78 | Neutral | 0.080 | Benign | 0.026 | Benign | 4.24 | Benign | 0.84 | Tolerated | 0.1371 | 0.7159 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||
| c.1229G>T | S410I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S410I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Predictions that are uncertain or inconclusive are FoldX, Foldetta, and AlphaMissense‑Default. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized classifies the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leans pathogenic, and Foldetta remains uncertain. Overall, the majority of tools suggest a benign impact, but the high‑accuracy consensus indicates potential pathogenicity, leaving the variant’s effect ambiguous. Based on the aggregate predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -9.383 | Likely Pathogenic | 0.432 | Ambiguous | Likely Benign | -1.08 | Ambiguous | 0.2 | -0.36 | Likely Benign | -0.72 | Ambiguous | -0.41 | Likely Benign | 0.114 | Likely Benign | -3.21 | Deleterious | 0.993 | Probably Damaging | 0.589 | Possibly Damaging | 4.15 | Benign | 0.21 | Tolerated | 0.0965 | 0.6579 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||
| c.122G>C | R41P 2D AIThe SynGAP1 missense variant R41P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for R41P, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.311707 | Structured | 0.431757 | Uncertain | 0.344 | 0.765 | 0.375 | -3.863 | Likely Benign | 0.307 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -0.70 | Neutral | 0.841 | Possibly Damaging | 0.809 | Possibly Damaging | 4.14 | Benign | 0.00 | Affected | 0.2244 | 0.5735 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||||
| c.122G>T | R41L 2D AIThe SynGAP1 missense variant R41L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for R41L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.311707 | Structured | 0.431757 | Uncertain | 0.344 | 0.765 | 0.375 | -3.173 | Likely Benign | 0.261 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -0.58 | Neutral | 0.686 | Possibly Damaging | 0.630 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.2134 | 0.5868 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.1230C>A | S410R 2D 3DClick to see structure in 3D Viewer AIClinVar contains no record for the SynGAP1 S410R variant, and it is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL, Foldetta, PROVEAN, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. The remaining tools (FoldX, Rosetta, premPS, AlphaMissense‑Optimized) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, Foldetta predicts a benign impact, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Taken together, the majority of evidence points to a benign outcome. This conclusion does not contradict ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -8.203 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | -0.67 | Ambiguous | 0.2 | 0.56 | Ambiguous | -0.06 | Likely Benign | 0.62 | Ambiguous | 0.148 | Likely Benign | -2.47 | Neutral | 0.871 | Possibly Damaging | 0.298 | Benign | 4.20 | Benign | 0.40 | Tolerated | 0.0955 | 0.3927 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||
| c.1230C>G | S410R 2D 3DClick to see structure in 3D Viewer AIClinVar contains no record for the SynGAP1 S410R variant, and it is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL, Foldetta, PROVEAN, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. The remaining tools (FoldX, Rosetta, premPS, AlphaMissense‑Optimized) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, Foldetta predicts a benign impact, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Taken together, the majority of evidence points to a benign outcome. This conclusion does not contradict ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -8.203 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | -0.67 | Ambiguous | 0.2 | 0.56 | Ambiguous | -0.06 | Likely Benign | 0.62 | Ambiguous | 0.146 | Likely Benign | -2.47 | Neutral | 0.871 | Possibly Damaging | 0.298 | Benign | 4.20 | Benign | 0.40 | Tolerated | 0.0955 | 0.3927 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||
| c.1231A>C | I411L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I411L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, whereas a majority (premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. Tools with uncertain or mixed outputs are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, yielding no definitive call; and Foldetta also reports an uncertain stability change. Overall, the majority of standard predictors lean toward pathogenicity, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current computational evidence, though high‑accuracy tools remain inconclusive. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.116183 | Structured | 0.339366 | Uncertain | 0.927 | 0.198 | 0.000 | -10.723 | Likely Pathogenic | 0.819 | Likely Pathogenic | Ambiguous | 1.02 | Ambiguous | 0.3 | 1.89 | Ambiguous | 1.46 | Ambiguous | 1.17 | Destabilizing | 0.285 | Likely Benign | -1.84 | Neutral | 0.908 | Possibly Damaging | 0.943 | Probably Damaging | 3.36 | Benign | 0.01 | Affected | 0.0876 | 0.3539 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1231A>T | I411F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I411F is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact; premPS is uncertain. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.339366 | Uncertain | 0.927 | 0.198 | 0.000 | -13.377 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 5.71 | Destabilizing | 0.1 | 4.53 | Destabilizing | 5.12 | Destabilizing | 0.73 | Ambiguous | 0.503 | Likely Pathogenic | -3.69 | Deleterious | 0.998 | Probably Damaging | 0.973 | Probably Damaging | 3.30 | Benign | 0.00 | Affected | 0.0538 | 0.2853 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.1232T>A | I411N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I411N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.339366 | Uncertain | 0.927 | 0.198 | 0.000 | -14.426 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 3.00 | Destabilizing | 0.1 | 3.71 | Destabilizing | 3.36 | Destabilizing | 2.16 | Destabilizing | 0.556 | Likely Pathogenic | -6.42 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 0.0738 | 0.0700 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1232T>C | I411T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 I411T missense variant is not reported in ClinVar (status: None) and has no entries in gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Rosetta’s assessment is uncertain. High‑accuracy methods give consistent pathogenic predictions: AlphaMissense‑Optimized is Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is Pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.339366 | Uncertain | 0.927 | 0.198 | 0.000 | -11.258 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 2.86 | Destabilizing | 0.0 | 1.75 | Ambiguous | 2.31 | Destabilizing | 1.86 | Destabilizing | 0.579 | Likely Pathogenic | -4.54 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.31 | Benign | 0.00 | Affected | 0.0903 | 0.1389 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.1232T>G | I411S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I411S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.339366 | Uncertain | 0.927 | 0.198 | 0.000 | -13.763 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.59 | Destabilizing | 0.2 | 3.83 | Destabilizing | 3.71 | Destabilizing | 1.91 | Destabilizing | 0.603 | Likely Pathogenic | -5.50 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.30 | Benign | 0.00 | Affected | 0.2262 | 0.1487 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1233C>G | I411M 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I411M is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two consensus groups: benign predictions come from REVEL and FATHMM, while pathogenic predictions are supported by SGM‑Consensus, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX and AlphaMissense‑Optimized yield uncertain results. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is inconclusive, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta also predicts pathogenicity. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.339366 | Uncertain | 0.927 | 0.198 | 0.000 | -10.969 | Likely Pathogenic | 0.911 | Likely Pathogenic | Ambiguous | 1.30 | Ambiguous | 0.2 | 2.76 | Destabilizing | 2.03 | Destabilizing | 1.21 | Destabilizing | 0.344 | Likely Benign | -2.73 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.31 | Benign | 0.00 | Affected | 0.0677 | 0.2848 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||
| c.1234T>A | L412M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L412M has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, and FATHMM, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Five tools give uncertain or inconclusive results (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized). High‑accuracy assessments are likewise inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is a 2‑vs‑2 tie; and Foldetta is uncertain. Consequently, the overall evidence leans toward a pathogenic effect, with no ClinVar record to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.122885 | Structured | 0.331108 | Uncertain | 0.937 | 0.196 | 0.000 | -9.342 | Likely Pathogenic | 0.944 | Likely Pathogenic | Ambiguous | 0.75 | Ambiguous | 0.0 | 1.99 | Ambiguous | 1.37 | Ambiguous | 0.86 | Ambiguous | 0.246 | Likely Benign | -1.84 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.25 | Benign | 0.05 | Affected | 0.0746 | 0.3482 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.1234T>G | L412V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L412V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.122885 | Structured | 0.331108 | Uncertain | 0.937 | 0.196 | 0.000 | -11.726 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 3.65 | Destabilizing | 0.0 | 3.52 | Destabilizing | 3.59 | Destabilizing | 1.54 | Destabilizing | 0.231 | Likely Benign | -2.76 | Deleterious | 0.995 | Probably Damaging | 0.970 | Probably Damaging | 3.25 | Benign | 0.01 | Affected | 0.1275 | 0.3378 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1235T>C | L412S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L412S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (both HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all return pathogenic or likely pathogenic scores, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized indicates pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) also predicts pathogenic. No prediction is inconclusive or missing. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.122885 | Structured | 0.331108 | Uncertain | 0.937 | 0.196 | 0.000 | -13.884 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.03 | Destabilizing | 0.2 | 4.20 | Destabilizing | 4.12 | Destabilizing | 2.01 | Destabilizing | 0.557 | Likely Pathogenic | -5.53 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.22 | Benign | 0.00 | Affected | 0.2821 | 0.0505 | -3 | -2 | -4.6 | -26.08 | |||||||||||||||||||||||||||||
| c.1235T>G | L412W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L412W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.122885 | Structured | 0.331108 | Uncertain | 0.937 | 0.196 | 0.000 | -13.436 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 8.44 | Destabilizing | 0.4 | 9.91 | Destabilizing | 9.18 | Destabilizing | 1.65 | Destabilizing | 0.503 | Likely Pathogenic | -5.53 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.19 | Benign | 0.00 | Affected | 0.0586 | 0.2664 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||
| c.1236G>C | L412F 2D  AIThe SynGAP1 missense variant L412F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM, whereas the majority of tools (SGM‑Consensus, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Taken together, the consensus of the majority of evidence points to a pathogenic effect for L412F, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.122885 | Structured | 0.331108 | Uncertain | 0.937 | 0.196 | 0.000 | -8.710 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 7.00 | Destabilizing | 3.5 | 3.61 | Destabilizing | 5.31 | Destabilizing | 0.48 | Likely Benign | 0.286 | Likely Benign | -3.69 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | 3.27 | Benign | 0.04 | Affected | 0.0602 | 0.2891 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1236G>T | L412F 2D AIThe SynGAP1 missense variant L412F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM, whereas the majority of tools (SGM‑Consensus, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Taken together, the consensus of the majority of evidence points to a pathogenic effect for L412F, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.122885 | Structured | 0.331108 | Uncertain | 0.937 | 0.196 | 0.000 | -8.710 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 7.00 | Destabilizing | 3.5 | 3.61 | Destabilizing | 5.31 | Destabilizing | 0.48 | Likely Benign | 0.283 | Likely Benign | -3.69 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | 3.27 | Benign | 0.04 | Affected | 0.0602 | 0.2891 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1237C>A | P413T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P413T missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining tools—SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this view: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a Likely Pathogenic verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. No predictions are inconclusive or missing. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.332472 | Uncertain | 0.927 | 0.201 | 0.000 | -12.851 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.22 | Destabilizing | 0.2 | 1.87 | Ambiguous | 2.55 | Destabilizing | 0.93 | Ambiguous | 0.554 | Likely Pathogenic | -7.37 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.18 | Benign | 0.01 | Affected | 0.1663 | 0.4670 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||
| c.1237C>G | P413A 2D 3DClick to see structure in 3D Viewer AISynGAP1 P413A is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the majority of algorithms—FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Overall, the preponderance of evidence points to a pathogenic effect for P413A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.332472 | Uncertain | 0.927 | 0.201 | 0.000 | -12.686 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.38 | Destabilizing | 0.0 | 0.79 | Ambiguous | 1.59 | Ambiguous | 0.81 | Ambiguous | 0.392 | Likely Benign | -7.37 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.19 | Benign | 0.02 | Affected | 0.3350 | 0.3863 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.1237C>T | P413S 2D 3DClick to see structure in 3D Viewer AISynGAP1 P413S is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include only FATHMM. All other evaluated predictors—SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta—indicate a pathogenic effect. High‑accuracy methods give a consistent pathogenic verdict: AlphaMissense‑Optimized predicts pathogenicity, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic, and Foldetta (which integrates FoldX‑MD and Rosetta outputs) predicts pathogenic; FoldX alone supports pathogenicity while Rosetta remains uncertain. Taken together, the overwhelming majority of predictions support a pathogenic classification, and this is not contradicted by the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.332472 | Uncertain | 0.927 | 0.201 | 0.000 | -12.586 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.06 | Destabilizing | 0.1 | 1.72 | Ambiguous | 2.39 | Destabilizing | 0.93 | Ambiguous | 0.509 | Likely Pathogenic | -7.37 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.19 | Benign | 0.04 | Affected | 0.3454 | 0.3819 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||
| c.1238C>A | P413H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P413H is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, while the remaining tools—SGM‑Consensus, REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic or likely pathogenic impact; Rosetta is uncertain. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among pathogenic predictors and the high‑accuracy tools, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.332472 | Uncertain | 0.927 | 0.201 | 0.000 | -13.376 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.89 | Destabilizing | 1.0 | 1.85 | Ambiguous | 3.37 | Destabilizing | 1.07 | Destabilizing | 0.546 | Likely Pathogenic | -8.29 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.16 | Benign | 0.00 | Affected | 0.1745 | 0.3480 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||
| c.1238C>G | P413R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P413R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The remaining methods, Foldetta and Rosetta, yield uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.332472 | Uncertain | 0.927 | 0.201 | 0.000 | -15.523 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.40 | Destabilizing | 0.2 | 1.34 | Ambiguous | 1.87 | Ambiguous | 1.07 | Destabilizing | 0.562 | Likely Pathogenic | -8.29 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.32 | Benign | 0.01 | Affected | 0.1550 | 0.2199 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.1238C>T | P413L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P413L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM. Those that predict a pathogenic effect comprise SGM‑Consensus, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Rosetta and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.332472 | Uncertain | 0.927 | 0.201 | 0.000 | -12.735 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.61 | Destabilizing | 0.4 | 1.34 | Ambiguous | 1.98 | Ambiguous | 0.30 | Likely Benign | 0.461 | Likely Benign | -9.21 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.19 | Benign | 0.00 | Affected | 0.2056 | 0.5614 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1240A>C | M414L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M414L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, SIFT, and FATHMM, while polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default predict a pathogenic outcome. Uncertain predictions come from premPS and ESM1b. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. Overall, the preponderance of evidence indicates that M414L is most likely benign, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | -7.980 | In-Between | 0.779 | Likely Pathogenic | Likely Benign | 0.26 | Likely Benign | 0.0 | 0.33 | Likely Benign | 0.30 | Likely Benign | 0.64 | Ambiguous | 0.336 | Likely Benign | -2.40 | Neutral | 0.559 | Possibly Damaging | 0.495 | Possibly Damaging | 3.63 | Benign | 0.90 | Tolerated | 0.1323 | 0.4236 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||
| c.1240A>G | M414V 2D AISynGAP1 M414V is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and ESM1b; the remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) are inconclusive. The SGM consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a pathogenic majority. High‑accuracy assessments give AlphaMissense‑Optimized benign, SGM consensus pathogenic, and Foldetta uncertain. Because the high‑accuracy predictions are divided and the overall tool set is evenly split, there is no definitive evidence for pathogenicity or benignity. Thus, the variant is most likely inconclusive, and this lack of consensus does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | Uncertain | 1 | -8.003 | Likely Pathogenic | 0.541 | Ambiguous | Likely Benign | 1.81 | Ambiguous | 0.4 | 1.73 | Ambiguous | 1.77 | Ambiguous | 0.95 | Ambiguous | 0.261 | Likely Benign | -2.95 | Deleterious | 0.999 | Probably Damaging | 0.987 | Probably Damaging | 3.43 | Benign | 0.24 | Tolerated | 0.2585 | 0.3482 | 2 | 1 | 2.3 | -32.06 | ||||||||||||||||||||||||||||
| c.1240A>T | M414L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M414L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, SIFT, and FATHMM, while polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default predict a pathogenic outcome. Uncertain predictions come from premPS and ESM1b. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. Overall, the preponderance of evidence indicates that M414L is most likely benign, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | -7.980 | In-Between | 0.779 | Likely Pathogenic | Likely Benign | 0.26 | Likely Benign | 0.0 | 0.33 | Likely Benign | 0.30 | Likely Benign | 0.64 | Ambiguous | 0.336 | Likely Benign | -2.40 | Neutral | 0.559 | Possibly Damaging | 0.495 | Possibly Damaging | 3.63 | Benign | 0.90 | Tolerated | 0.1323 | 0.4236 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||
| c.1241T>A | M414K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M414K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. FoldX and Foldetta provide uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta is unavailable. Based on the overwhelming agreement among the majority of prediction tools and the high‑accuracy methods, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | -13.537 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.18 | Ambiguous | 0.1 | 2.39 | Destabilizing | 1.79 | Ambiguous | 1.50 | Destabilizing | 0.503 | Likely Pathogenic | -5.03 | Deleterious | 0.942 | Possibly Damaging | 0.860 | Possibly Damaging | 3.40 | Benign | 0.04 | Affected | 0.1299 | 0.0888 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||
| c.1241T>C | M414T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M414T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions arise from SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta; Rosetta remains uncertain. High‑accuracy methods all support a deleterious effect: AlphaMissense‑Optimized scores the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta predicts a destabilizing, pathogenic outcome. Consequently, the consensus of the most reliable predictors classifies M414T as pathogenic, with no conflict with the ClinVar status, which simply lacks an entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | -8.698 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 2.04 | Destabilizing | 0.1 | 1.96 | Ambiguous | 2.00 | Destabilizing | 1.27 | Destabilizing | 0.406 | Likely Benign | -5.00 | Deleterious | 0.997 | Probably Damaging | 0.972 | Probably Damaging | 3.41 | Benign | 0.08 | Tolerated | 0.1771 | 0.1976 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.1241T>G | M414R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M414R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments further support a damaging outcome: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus remains likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. Overall, the preponderance of evidence indicates that M414R is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | -13.404 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.17 | Ambiguous | 0.1 | 2.58 | Destabilizing | 1.88 | Ambiguous | 1.45 | Destabilizing | 0.525 | Likely Pathogenic | -5.20 | Deleterious | 0.972 | Probably Damaging | 0.895 | Possibly Damaging | 3.38 | Benign | 0.03 | Affected | 0.1509 | 0.1037 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||
| c.1242G>A | M414I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 M414I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, and FATHMM. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta is also inconclusive. Overall, more tools (five) predict pathogenicity than benign (four), and no ClinVar evidence contradicts this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | -6.203 | Likely Benign | 0.972 | Likely Pathogenic | Likely Pathogenic | 0.83 | Ambiguous | 0.0 | 0.86 | Ambiguous | 0.85 | Ambiguous | 0.84 | Ambiguous | 0.265 | Likely Benign | -3.00 | Deleterious | 0.870 | Possibly Damaging | 0.801 | Possibly Damaging | 3.44 | Benign | 0.36 | Tolerated | 0.1168 | 0.3161 | 2 | 1 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||
| c.1242G>C | M414I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M414I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2), and Foldetta is uncertain, so these results are treated as unavailable. Overall, the majority of evaluated tools (5 pathogenic vs 4 benign) and the single high‑accuracy pathogenic prediction support a likely pathogenic classification. This assessment does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | -6.203 | Likely Benign | 0.972 | Likely Pathogenic | Likely Pathogenic | 0.83 | Ambiguous | 0.0 | 0.86 | Ambiguous | 0.85 | Ambiguous | 0.84 | Ambiguous | 0.265 | Likely Benign | -3.00 | Deleterious | 0.870 | Possibly Damaging | 0.801 | Possibly Damaging | 3.44 | Benign | 0.36 | Tolerated | 0.1168 | 0.3161 | 2 | 1 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||
| c.1242G>T | M414I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M414I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2), and Foldetta is uncertain, so these results are treated as unavailable. Overall, the majority of evaluated tools (5 pathogenic vs 4 benign) and the single high‑accuracy pathogenic prediction support a likely pathogenic classification. This assessment does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | -6.203 | Likely Benign | 0.972 | Likely Pathogenic | Likely Pathogenic | 0.83 | Ambiguous | 0.0 | 0.86 | Ambiguous | 0.85 | Ambiguous | 0.84 | Ambiguous | 0.265 | Likely Benign | -3.00 | Deleterious | 0.870 | Possibly Damaging | 0.801 | Possibly Damaging | 3.44 | Benign | 0.36 | Tolerated | 0.1168 | 0.3161 | 2 | 1 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||
| c.1243G>A | E415K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E415K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus shows AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall balance of predictions—particularly the two high‑accuracy pathogenic calls versus one benign—the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.330366 | Uncertain | 0.915 | 0.236 | 0.000 | -11.433 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | -0.07 | Likely Benign | 0.3 | -0.13 | Likely Benign | -0.10 | Likely Benign | 0.46 | Likely Benign | 0.326 | Likely Benign | -3.69 | Deleterious | 0.998 | Probably Damaging | 0.975 | Probably Damaging | 3.22 | Benign | 0.03 | Affected | 0.2174 | 0.3860 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1243G>C | E415Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E415Q missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, seven tools predict pathogenicity versus six predicting benignity, and the two most reliable predictors (AlphaMissense‑Optimized and SGM‑Consensus) both favor pathogenicity. Therefore, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.330366 | Uncertain | 0.915 | 0.236 | 0.000 | -9.085 | Likely Pathogenic | 0.970 | Likely Pathogenic | Likely Pathogenic | 0.29 | Likely Benign | 0.2 | 0.22 | Likely Benign | 0.26 | Likely Benign | 0.01 | Likely Benign | 0.236 | Likely Benign | -2.63 | Deleterious | 0.997 | Probably Damaging | 0.973 | Probably Damaging | 3.26 | Benign | 0.08 | Tolerated | 0.1084 | 0.3474 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1244A>C | E415A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E415A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, Foldetta, and FATHMM, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are given by FoldX and premPS. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the majority of tools lean toward pathogenicity, and the high‑accuracy predictions support this view. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.330366 | Uncertain | 0.915 | 0.236 | 0.000 | -11.743 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.61 | Ambiguous | 0.2 | 0.05 | Likely Benign | 0.33 | Likely Benign | 0.53 | Ambiguous | 0.435 | Likely Benign | -5.56 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.19 | Benign | 0.03 | Affected | 0.2909 | 0.3432 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1244A>G | E415G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E415G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Stability‑based methods (FoldX, Rosetta, premPS) and Foldetta are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact for E415G. This prediction is consistent with the lack of ClinVar annotation and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.330366 | Uncertain | 0.915 | 0.236 | 0.000 | -12.244 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 1.16 | Ambiguous | 0.2 | 0.88 | Ambiguous | 1.02 | Ambiguous | 0.65 | Ambiguous | 0.432 | Likely Benign | -6.45 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.20 | Benign | 0.01 | Affected | 0.2518 | 0.3158 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1244A>T | E415V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E415V missense variant is not reported in ClinVar or gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions are reported by SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods give a mixed signal: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) predicts benign. No evidence is missing or inconclusive. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.330366 | Uncertain | 0.915 | 0.236 | 0.000 | -11.182 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.56 | Ambiguous | 0.2 | 0.14 | Likely Benign | 0.35 | Likely Benign | 0.40 | Likely Benign | 0.441 | Likely Benign | -6.52 | Deleterious | 0.998 | Probably Damaging | 0.983 | Probably Damaging | 3.13 | Benign | 0.02 | Affected | 0.0650 | 0.4218 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1245G>C | E415D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E415D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. Predictions that are inconclusive or uncertain are FoldX, ESM1b, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenicity, while Foldetta predicts a benign effect. Overall, the balance of evidence leans toward a pathogenic impact, and this conclusion does not contradict the ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.100716 | Structured | 0.330366 | Uncertain | 0.915 | 0.236 | 0.000 | -7.766 | In-Between | 0.952 | Likely Pathogenic | Ambiguous | 0.75 | Ambiguous | 0.1 | 0.21 | Likely Benign | 0.48 | Likely Benign | 0.49 | Likely Benign | 0.220 | Likely Benign | -2.60 | Deleterious | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.22 | Benign | 0.04 | Affected | 0.1777 | 0.2075 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1245G>T | E415D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E415D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. Predictions that are uncertain or inconclusive are FoldX, ESM1b, and AlphaMissense‑Optimized. High‑accuracy assessments show that AlphaMissense‑Optimized is uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta indicates a benign effect. Overall, the majority of standard tools lean toward pathogenicity, and the high‑accuracy consensus also supports a pathogenic classification. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.100716 | Structured | 0.330366 | Uncertain | 0.915 | 0.236 | 0.000 | -7.766 | In-Between | 0.952 | Likely Pathogenic | Ambiguous | 0.75 | Ambiguous | 0.1 | 0.21 | Likely Benign | 0.48 | Likely Benign | 0.49 | Likely Benign | 0.220 | Likely Benign | -2.60 | Deleterious | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.22 | Benign | 0.04 | Affected | 0.1777 | 0.2075 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1246C>A | L416I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 L416I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, and FATHMM. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Predictions that remain inconclusive are FoldX, Rosetta, AlphaMissense‑Default, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. Overall, the preponderance of evidence points to a benign effect. This conclusion is not contradicted by any ClinVar annotation, as the variant has no existing ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.104810 | Structured | 0.336105 | Uncertain | 0.935 | 0.227 | 0.000 | -8.613 | Likely Pathogenic | 0.396 | Ambiguous | Likely Benign | 0.57 | Ambiguous | 0.0 | 0.69 | Ambiguous | 0.63 | Ambiguous | 0.50 | Likely Benign | 0.127 | Likely Benign | -1.28 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 3.38 | Benign | 0.37 | Tolerated | 0.1073 | 0.3480 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1246C>G | L416V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L416V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact for the variant. This conclusion is not contradicted by any ClinVar annotation, as the variant is not present in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.104810 | Structured | 0.336105 | Uncertain | 0.935 | 0.227 | 0.000 | -7.861 | In-Between | 0.310 | Likely Benign | Likely Benign | 1.46 | Ambiguous | 0.0 | 1.78 | Ambiguous | 1.62 | Ambiguous | 0.45 | Likely Benign | 0.124 | Likely Benign | -1.47 | Neutral | 0.995 | Probably Damaging | 0.970 | Probably Damaging | 3.42 | Benign | 0.53 | Tolerated | 0.1563 | 0.3550 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1247T>A | L416Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L416Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all predict benign. Only two tools, polyPhen‑2 (HumDiv and HumVar), predict a pathogenic outcome. Stability‑based methods (FoldX, Rosetta, Foldetta, premPS) are inconclusive. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, the SGM‑Consensus is benign, and Foldetta remains uncertain. Overall, the evidence strongly supports a benign classification for this variant, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.104810 | Structured | 0.336105 | Uncertain | 0.935 | 0.227 | 0.000 | -6.445 | Likely Benign | 0.234 | Likely Benign | Likely Benign | 1.17 | Ambiguous | 0.1 | 0.54 | Ambiguous | 0.86 | Ambiguous | 0.88 | Ambiguous | 0.187 | Likely Benign | -1.07 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.39 | Benign | 0.46 | Tolerated | 0.1281 | 0.0860 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1247T>C | L416P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L416P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions from REVEL, SIFT, and FATHMM; pathogenic predictions from the remaining 13 tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen2_HumDiv, polyPhen2_HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, and the SGM Consensus). High‑accuracy methods specifically indicate pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No predictions are inconclusive. Based on the overall consensus of the majority of tools and the high‑accuracy methods, the variant is most likely pathogenic, which is consistent with the lack of ClinVar reporting and gnomAD absence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.336105 | Uncertain | 0.935 | 0.227 | 0.000 | -8.859 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 2.59 | Destabilizing | 0.1 | 6.23 | Destabilizing | 4.41 | Destabilizing | 1.27 | Destabilizing | 0.284 | Likely Benign | -3.56 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.35 | Benign | 0.21 | Tolerated | 0.3589 | 0.1353 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1247T>G | L416R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L416R missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also leans toward benign, while Foldetta remains uncertain. Overall, the majority of reliable predictors classify the variant as benign, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.104810 | Structured | 0.336105 | Uncertain | 0.935 | 0.227 | 0.000 | -8.600 | Likely Pathogenic | 0.511 | Ambiguous | Likely Benign | 0.71 | Ambiguous | 0.0 | 0.97 | Ambiguous | 0.84 | Ambiguous | 0.84 | Ambiguous | 0.238 | Likely Benign | -2.05 | Neutral | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.35 | Benign | 0.43 | Tolerated | 0.1445 | 0.0702 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||
| c.1249T>A | Y417N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y417N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, while the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.346865 | Uncertain | 0.958 | 0.250 | 0.000 | -13.912 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.94 | Destabilizing | 0.2 | 3.69 | Destabilizing | 3.82 | Destabilizing | 2.60 | Destabilizing | 0.561 | Likely Pathogenic | -8.59 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.97 | Benign | 0.00 | Affected | 0.2482 | 0.0728 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.1249T>C | Y417H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y417H is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for Y417H. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.346865 | Uncertain | 0.958 | 0.250 | 0.000 | -11.447 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.22 | Destabilizing | 0.1 | 2.80 | Destabilizing | 3.01 | Destabilizing | 1.54 | Destabilizing | 0.513 | Likely Pathogenic | -4.77 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.97 | Benign | 0.00 | Affected | 0.2596 | 0.0668 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||
| c.1249T>G | Y417D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y417D is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.346865 | Uncertain | 0.958 | 0.250 | 0.000 | -14.955 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.13 | Destabilizing | 0.1 | 5.37 | Destabilizing | 5.25 | Destabilizing | 2.43 | Destabilizing | 0.688 | Likely Pathogenic | -9.55 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.96 | Benign | 0.00 | Affected | 0.4388 | 0.0400 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.124C>A | P42T 2D AIThe SynGAP1 missense variant P42T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P42T, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | -3.626 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -0.97 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.29 | Benign | 0.00 | Affected | 0.1834 | 0.4896 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.124C>G | P42A 2D AIThe SynGAP1 missense variant P42A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign effect for P42A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | -3.945 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -1.27 | Neutral | 0.805 | Possibly Damaging | 0.857 | Possibly Damaging | 4.32 | Benign | 0.00 | Affected | 0.3441 | 0.4323 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.1250A>C | Y417S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y417S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are limited to FATHMM, while all other evaluated algorithms—including SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.346865 | Uncertain | 0.958 | 0.250 | 0.000 | -14.339 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 4.36 | Destabilizing | 0.1 | 5.32 | Destabilizing | 4.84 | Destabilizing | 2.17 | Destabilizing | 0.593 | Likely Pathogenic | -8.59 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.98 | Benign | 0.00 | Affected | 0.4408 | 0.1961 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||
| c.1250A>G | Y417C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y417C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy methods further support this: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic effect; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also classifies the variant as pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.346865 | Uncertain | 0.958 | 0.250 | 0.000 | -12.021 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 4.03 | Destabilizing | 0.1 | 3.81 | Destabilizing | 3.92 | Destabilizing | 2.52 | Destabilizing | 0.597 | Likely Pathogenic | -8.59 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.95 | Benign | 0.00 | Affected | 0.2960 | 0.2005 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||
| c.1250A>T | Y417F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 Y417F variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and FATHMM. Those that predict a pathogenic impact are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Uncertain results come from AlphaMissense‑Optimized and premPS. High‑accuracy assessments show SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, Foldetta (combining FoldX‑MD and Rosetta) as Benign, and AlphaMissense‑Optimized as Uncertain. Overall, the majority of tools and the SGM‑Consensus score suggest a pathogenic effect, while Foldetta indicates a benign effect; the variant’s status is not contradicted by ClinVar (no entry). Thus, based on the available predictions, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.346865 | Uncertain | 0.958 | 0.250 | 0.000 | -11.368 | Likely Pathogenic | 0.852 | Likely Pathogenic | Ambiguous | 0.47 | Likely Benign | 0.1 | -0.09 | Likely Benign | 0.19 | Likely Benign | 0.97 | Ambiguous | 0.367 | Likely Benign | -3.82 | Deleterious | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 3.03 | Benign | 0.06 | Tolerated | 0.2617 | 0.3098 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||
| c.1252A>C | K418Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K418Q missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.360134 | Uncertain | 0.948 | 0.263 | 0.000 | -11.404 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.10 | Likely Benign | 0.1 | 0.17 | Likely Benign | 0.14 | Likely Benign | 0.30 | Likely Benign | 0.263 | Likely Benign | -3.19 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.55 | Benign | 0.13 | Tolerated | 0.4105 | 0.0696 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1252A>G | K418E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K418E is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, and FATHMM. Tools that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. Overall, the majority of evaluated tools (seven pathogenic vs four benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for K418E. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.360134 | Uncertain | 0.948 | 0.263 | 0.000 | -12.443 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.63 | Ambiguous | 0.0 | 0.80 | Ambiguous | 0.72 | Ambiguous | 0.47 | Likely Benign | 0.367 | Likely Benign | -3.42 | Deleterious | 0.999 | Probably Damaging | 0.991 | Probably Damaging | 3.53 | Benign | 0.07 | Tolerated | 0.3578 | 0.0471 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1253A>C | K418T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K418T variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, FATHMM, premPS, and Foldetta. Those that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of tools (10 pathogenic vs. 5 benign) support a pathogenic classification. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar record exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.360134 | Uncertain | 0.948 | 0.263 | 0.000 | -11.994 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.39 | Likely Benign | 0.1 | 0.55 | Ambiguous | 0.47 | Likely Benign | 0.41 | Likely Benign | 0.392 | Likely Benign | -5.36 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.37 | Benign | 0.04 | Affected | 0.1975 | 0.1894 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1253A>G | K418R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K418R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, Foldetta, ESM1b, FATHMM, PROVEAN, and AlphaMissense‑Optimized. Tools that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus methods give a benign verdict: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign (3 benign vs. 1 pathogenic); and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is benign. premPS is uncertain and therefore not considered evidence. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.104810 | Structured | 0.360134 | Uncertain | 0.948 | 0.263 | 0.000 | -6.809 | Likely Benign | 0.635 | Likely Pathogenic | Likely Benign | -0.18 | Likely Benign | 0.1 | 0.12 | Likely Benign | -0.03 | Likely Benign | 0.56 | Ambiguous | 0.229 | Likely Benign | -2.46 | Neutral | 0.994 | Probably Damaging | 0.962 | Probably Damaging | 3.37 | Benign | 0.04 | Affected | 0.4384 | 0.1151 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.1253A>T | K418I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K418I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM. In contrast, the majority of tools predict a pathogenic outcome: SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta provide uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as Uncertain. Overall, the preponderance of evidence points to a pathogenic effect for K418I, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.360134 | Uncertain | 0.948 | 0.263 | 0.000 | -14.895 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.51 | Ambiguous | 0.0 | 0.64 | Ambiguous | 0.58 | Ambiguous | 0.27 | Likely Benign | 0.428 | Likely Benign | -7.27 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.32 | Benign | 0.01 | Affected | 0.1297 | 0.2432 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1254A>C | K418N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K418N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM. The majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Stability‑based methods (FoldX, Rosetta, premPS, Foldetta) are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence indicates that K418N is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.360134 | Uncertain | 0.948 | 0.263 | 0.000 | -13.310 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.57 | Ambiguous | 0.0 | 0.77 | Ambiguous | 0.67 | Ambiguous | 0.56 | Ambiguous | 0.132 | Likely Benign | -4.41 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.42 | Benign | 0.04 | Affected | 0.3483 | 0.0606 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1254A>T | K418N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K418N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two consensus groups: benign predictions are provided by REVEL and FATHMM, whereas pathogenic predictions are given by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus score (Likely Pathogenic). Stability‑based methods FoldX, Rosetta, Foldetta, and premPS returned uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, and Foldetta’s stability analysis is inconclusive. Overall, the majority of reliable predictors classify K418N as pathogenic, and this conclusion does not contradict the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.360134 | Uncertain | 0.948 | 0.263 | 0.000 | -13.310 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.57 | Ambiguous | 0.0 | 0.77 | Ambiguous | 0.67 | Ambiguous | 0.56 | Ambiguous | 0.132 | Likely Benign | -4.41 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.42 | Benign | 0.04 | Affected | 0.3483 | 0.0606 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1255G>A | E419K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E419K missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, and FATHMM. Tools that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Foldetta and Rosetta give uncertain results and are not counted in either group. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus predicts likely pathogenic, and Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic impact for E419K. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.371949 | Uncertain | 0.961 | 0.261 | 0.000 | -12.257 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.01 | Likely Benign | 0.1 | 1.30 | Ambiguous | 0.66 | Ambiguous | -0.03 | Likely Benign | 0.399 | Likely Benign | -3.75 | Deleterious | 0.998 | Probably Damaging | 0.975 | Probably Damaging | 3.36 | Benign | 0.07 | Tolerated | 0.2759 | 0.6899 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1255G>C | E419Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E419Q missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM. Those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the predictions are split, with a slight majority leaning toward pathogenicity. The variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.371949 | Uncertain | 0.961 | 0.261 | 0.000 | -9.268 | Likely Pathogenic | 0.923 | Likely Pathogenic | Ambiguous | 0.01 | Likely Benign | 0.1 | 0.36 | Likely Benign | 0.19 | Likely Benign | 0.02 | Likely Benign | 0.280 | Likely Benign | -2.80 | Deleterious | 0.997 | Probably Damaging | 0.973 | Probably Damaging | 3.41 | Benign | 0.04 | Affected | 0.1499 | 0.6938 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1256A>C | E419A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E419A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM, whereas a majority of tools predict a pathogenic impact: SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic classification; this conclusion does not contradict ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.371949 | Uncertain | 0.961 | 0.261 | 0.000 | -10.951 | Likely Pathogenic | 0.944 | Likely Pathogenic | Ambiguous | 0.56 | Ambiguous | 0.1 | 0.94 | Ambiguous | 0.75 | Ambiguous | 0.28 | Likely Benign | 0.398 | Likely Benign | -5.60 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.32 | Benign | 0.03 | Affected | 0.4051 | 0.6705 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1256A>T | E419V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E419V missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, FoldX, FATHMM, and premPS, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic, the SGM Consensus also indicates Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result. No evidence from the high‑accuracy tools contradicts the pathogenic prediction. Overall, the majority of computational evidence points to a pathogenic effect, which is consistent with the lack of ClinVar reporting and gnomAD absence, suggesting the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.371949 | Uncertain | 0.961 | 0.261 | 0.000 | -12.290 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.45 | Likely Benign | 0.0 | 1.41 | Ambiguous | 0.93 | Ambiguous | 0.27 | Likely Benign | 0.494 | Likely Benign | -6.55 | Deleterious | 0.998 | Probably Damaging | 0.983 | Probably Damaging | 3.35 | Benign | 0.01 | Affected | 0.0953 | 0.6834 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1257G>C | E419D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E419D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, and FATHMM. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Uncertain or inconclusive predictions come from Foldetta, AlphaMissense‑Optimized, ESM1b, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain (treated as unavailable), the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, and Foldetta remains uncertain (unavailable). Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.102787 | Structured | 0.371949 | Uncertain | 0.961 | 0.261 | 0.000 | -7.036 | In-Between | 0.869 | Likely Pathogenic | Ambiguous | 0.17 | Likely Benign | 0.1 | 0.87 | Ambiguous | 0.52 | Ambiguous | 0.48 | Likely Benign | 0.170 | Likely Benign | -2.40 | Neutral | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.33 | Benign | 0.10 | Tolerated | 0.1966 | 0.4906 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1257G>T | E419D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E419D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, and FATHMM. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Uncertain or inconclusive predictions come from Foldetta, AlphaMissense‑Optimized, ESM1b, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain (treated as unavailable), the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, and Foldetta remains uncertain (unavailable). Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.102787 | Structured | 0.371949 | Uncertain | 0.961 | 0.261 | 0.000 | -7.036 | In-Between | 0.869 | Likely Pathogenic | Ambiguous | 0.17 | Likely Benign | 0.1 | 0.87 | Ambiguous | 0.52 | Ambiguous | 0.48 | Likely Benign | 0.170 | Likely Benign | -2.40 | Neutral | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.33 | Benign | 0.10 | Tolerated | 0.1966 | 0.4906 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1258T>A | F420I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F420I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign calls come from REVEL, polyPhen‑2 HumVar, and FATHMM, while pathogenic calls are made by FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.384475 | Uncertain | 0.974 | 0.255 | 0.000 | -12.567 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 2.88 | Destabilizing | 0.0 | 3.64 | Destabilizing | 3.26 | Destabilizing | 1.25 | Destabilizing | 0.330 | Likely Benign | -5.46 | Deleterious | 0.575 | Possibly Damaging | 0.059 | Benign | 3.22 | Benign | 0.02 | Affected | 0.1864 | 0.2113 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1258T>G | F420V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F420V is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM. In contrast, the majority of tools predict a pathogenic outcome: FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts Pathogenic; the SGM‑Consensus itself is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts Pathogenic. Based on the preponderance of pathogenic predictions and the high‑accuracy tool consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.384475 | Uncertain | 0.974 | 0.255 | 0.000 | -11.849 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 3.42 | Destabilizing | 0.1 | 3.35 | Destabilizing | 3.39 | Destabilizing | 1.58 | Destabilizing | 0.402 | Likely Benign | -6.41 | Deleterious | 0.495 | Possibly Damaging | 0.191 | Benign | 3.16 | Benign | 0.01 | Affected | 0.2038 | 0.2022 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1259T>A | F420Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F420Y is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show mixed results. Benign predictions come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic predictions are reported by premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. Predictions from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive. High‑accuracy assessments indicate that the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifies the variant as Likely Pathogenic, while AlphaMissense‑Optimized and Foldetta remain uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.384475 | Uncertain | 0.974 | 0.255 | 0.000 | -12.138 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 1.18 | Ambiguous | 0.1 | 0.96 | Ambiguous | 1.07 | Ambiguous | 1.31 | Destabilizing | 0.228 | Likely Benign | -2.80 | Deleterious | 0.306 | Benign | 0.100 | Benign | 3.09 | Benign | 0.03 | Affected | 0.1609 | 0.1498 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1259T>G | F420C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F420C is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that assess pathogenicity largely agree: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. Only FATHMM predicts a benign outcome. When grouped by consensus, the benign prediction is singular (FATHMM), whereas the pathogenic predictions comprise the remaining eleven tools. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.384475 | Uncertain | 0.974 | 0.255 | 0.000 | -11.931 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.31 | Destabilizing | 0.1 | 5.11 | Destabilizing | 4.71 | Destabilizing | 2.49 | Destabilizing | 0.500 | Likely Pathogenic | -7.40 | Deleterious | 0.998 | Probably Damaging | 0.869 | Possibly Damaging | 3.09 | Benign | 0.00 | Affected | 0.2458 | 0.0662 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.125C>A | P42H 2D AIThe SynGAP1 missense variant P42H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | -3.088 | Likely Benign | 0.167 | Likely Benign | Likely Benign | 0.140 | Likely Benign | -1.95 | Neutral | 0.992 | Probably Damaging | 0.977 | Probably Damaging | 4.16 | Benign | 0.00 | Affected | 0.1995 | 0.3705 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.125C>G | P42R 2D AIThe SynGAP1 missense variant P42R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P42R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | -2.498 | Likely Benign | 0.200 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -1.79 | Neutral | 0.972 | Probably Damaging | 0.954 | Probably Damaging | 4.20 | Benign | 0.00 | Affected | 0.1619 | 0.3082 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.125C>T | P42L 2D AIThe SynGAP1 missense variant P42L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | -3.781 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -1.87 | Neutral | 0.909 | Possibly Damaging | 0.927 | Probably Damaging | 4.19 | Benign | 0.00 | Affected | 0.2312 | 0.5560 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.1261G>C | A421P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A421P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from REVEL, FATHMM, and polyPhen‑2 HumVar, whereas pathogenic predictions are made by FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus also indicates Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, reports a pathogenic effect. Taken together, the overwhelming majority of evidence points to a pathogenic impact for A421P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.404927 | Uncertain | 0.965 | 0.257 | 0.000 | -13.126 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.51 | Destabilizing | 0.2 | 8.77 | Destabilizing | 6.64 | Destabilizing | 1.17 | Destabilizing | 0.371 | Likely Benign | -4.31 | Deleterious | 0.855 | Possibly Damaging | 0.420 | Benign | 3.39 | Benign | 0.04 | Affected | 0.1963 | 0.3343 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1261G>T | A421S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A421S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from PROVEAN, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as Likely Pathogenic. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus predicts pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive. Stability calculations from FoldX and Rosetta are uncertain, and premPS is unavailable. Overall, the majority of tools lean toward a pathogenic interpretation, and this aligns with the SGM‑Consensus result; there is no conflict with ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.404927 | Uncertain | 0.965 | 0.257 | 0.000 | -9.220 | Likely Pathogenic | 0.715 | Likely Pathogenic | Likely Benign | 0.66 | Ambiguous | 0.1 | 1.12 | Ambiguous | 0.89 | Ambiguous | 0.70 | Ambiguous | 0.155 | Likely Benign | -2.50 | Deleterious | 0.058 | Benign | 0.072 | Benign | 3.46 | Benign | 0.08 | Tolerated | 0.2247 | 0.3621 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1262C>A | A421E 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A421E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM, whereas pathogenic calls are made by ESM1b, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, Rosetta, premPS, and the SGM‑Consensus score (Likely Pathogenic). Stability‑based methods give mixed results: FoldX is uncertain, Foldetta is uncertain, and Rosetta alone predicts pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta remains uncertain. Overall, the majority of evidence, including the high‑accuracy tools, points to a pathogenic impact for A421E, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.404927 | Uncertain | 0.965 | 0.257 | 0.000 | -11.993 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.63 | Ambiguous | 0.2 | 2.07 | Destabilizing | 1.35 | Ambiguous | 1.30 | Destabilizing | 0.233 | Likely Benign | -4.24 | Deleterious | 0.368 | Benign | 0.144 | Benign | 3.44 | Benign | 0.14 | Tolerated | 0.1288 | 0.1830 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||
| c.1262C>G | A421G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A421G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). FoldX and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the majority of tools and the SGM Consensus favor a pathogenic interpretation, while a minority suggest benign. Because there is no ClinVar entry, the predictions do not contradict existing clinical classification. The variant is most likely pathogenic based on the collective computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.404927 | Uncertain | 0.965 | 0.257 | 0.000 | -9.699 | Likely Pathogenic | 0.757 | Likely Pathogenic | Likely Benign | 1.47 | Ambiguous | 0.1 | 2.13 | Destabilizing | 1.80 | Ambiguous | 1.19 | Destabilizing | 0.137 | Likely Benign | -3.59 | Deleterious | 0.536 | Possibly Damaging | 0.176 | Benign | 3.41 | Benign | 0.05 | Affected | 0.1692 | 0.2499 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1262C>T | A421V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A421V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, FoldX, Foldetta, premPS, polyPhen‑2 HumVar, SIFT, and FATHMM, while pathogenic predictions are reported by PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote) also predicts pathogenic, whereas Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, predicts benign. Rosetta alone is uncertain and is treated as unavailable. Overall, the majority of high‑confidence tools favor a pathogenic effect, so A421V is most likely pathogenic, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.404927 | Uncertain | 0.965 | 0.257 | 0.000 | -8.167 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | -0.05 | Likely Benign | 0.1 | -0.82 | Ambiguous | -0.44 | Likely Benign | -0.06 | Likely Benign | 0.111 | Likely Benign | -3.15 | Deleterious | 0.538 | Possibly Damaging | 0.113 | Benign | 3.50 | Benign | 0.14 | Tolerated | 0.1055 | 0.3738 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.1264G>A | E422K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E422K missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split opinion: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM, while pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus also indicates Likely Pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a benign effect. Overall, the balance of evidence leans toward pathogenicity, and this assessment does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.426709 | Uncertain | 0.965 | 0.255 | 0.000 | -13.042 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.30 | Likely Benign | 0.0 | 0.10 | Likely Benign | 0.20 | Likely Benign | 0.32 | Likely Benign | 0.346 | Likely Benign | -3.52 | Deleterious | 0.998 | Probably Damaging | 0.975 | Probably Damaging | 3.39 | Benign | 0.04 | Affected | 0.1995 | 0.5129 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1264G>C | E422Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E422Q missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, and FATHMM, while those that predict a pathogenic effect are SIFT, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool rates the variant as uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.067594 | Structured | 0.426709 | Uncertain | 0.965 | 0.255 | 0.000 | -9.460 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 0.32 | Likely Benign | 0.0 | 0.21 | Likely Benign | 0.27 | Likely Benign | -0.15 | Likely Benign | 0.208 | Likely Benign | -2.26 | Neutral | 0.997 | Probably Damaging | 0.973 | Probably Damaging | 3.38 | Benign | 0.03 | Affected | 0.1045 | 0.4913 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.1265A>C | E422A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E422A missense variant is not reported in ClinVar and has no gnomAD entry. Prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM; pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized remains uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized is inconclusive; the SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates likely pathogenic; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts benign. Overall, the balance of evidence (seven pathogenic versus six benign predictions) points to a likely pathogenic effect for E422A, and this conclusion is not contradicted by the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.426709 | Uncertain | 0.965 | 0.255 | 0.000 | -12.088 | Likely Pathogenic | 0.952 | Likely Pathogenic | Ambiguous | 0.49 | Likely Benign | 0.0 | 0.25 | Likely Benign | 0.37 | Likely Benign | 0.26 | Likely Benign | 0.371 | Likely Benign | -5.43 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.2949 | 0.4459 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1265A>G | E422G 2D 3DClick to see structure in 3D Viewer AISynGAP1 E422G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, premPS, and FATHMM, while pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Uncertain results are provided by FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy analyses indicate that the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts a likely pathogenic effect, whereas AlphaMissense‑Optimized remains inconclusive and Foldetta shows no definitive stability change. Overall, the majority of evidence points toward a pathogenic impact for E422G, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.426709 | Uncertain | 0.965 | 0.255 | 0.000 | -11.468 | Likely Pathogenic | 0.952 | Likely Pathogenic | Ambiguous | 1.13 | Ambiguous | 0.0 | 1.25 | Ambiguous | 1.19 | Ambiguous | 0.39 | Likely Benign | 0.488 | Likely Benign | -6.38 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.2440 | 0.4184 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1265A>T | E422V 2D 3DClick to see structure in 3D Viewer AISynGAP1 E422V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, Foldetta, premPS, and FATHMM, while pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenicity, SGM‑Consensus confirms a likely pathogenic outcome, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a benign effect. FoldX remains uncertain. Overall, the majority of high‑confidence tools lean toward pathogenicity, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.426709 | Uncertain | 0.965 | 0.255 | 0.000 | -12.371 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.90 | Ambiguous | 0.1 | -0.02 | Likely Benign | 0.44 | Likely Benign | 0.24 | Likely Benign | 0.489 | Likely Benign | -6.38 | Deleterious | 0.998 | Probably Damaging | 0.983 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 0.0609 | 0.5457 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1266G>C | E422D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E422D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b is uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors pathogenic; whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the majority of tools (six benign vs five pathogenic) lean toward a benign effect, but the two most accurate predictors and the consensus vote indicate a pathogenic tendency. Thus, the variant is most likely benign based on the broader tool set, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.067594 | Structured | 0.426709 | Uncertain | 0.965 | 0.255 | 0.000 | -7.092 | In-Between | 0.969 | Likely Pathogenic | Likely Pathogenic | 0.39 | Likely Benign | 0.0 | -0.03 | Likely Benign | 0.18 | Likely Benign | 0.21 | Likely Benign | 0.199 | Likely Benign | -2.76 | Deleterious | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.38 | Benign | 0.07 | Tolerated | 0.1686 | 0.2864 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1266G>T | E422D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E422D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b is uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors pathogenic; whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the majority of tools (six benign vs five pathogenic) lean toward a benign effect, but the two most accurate predictors and the consensus vote indicate a pathogenic tendency. Thus, the variant is most likely benign based on the broader tool set, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.067594 | Structured | 0.426709 | Uncertain | 0.965 | 0.255 | 0.000 | -7.092 | In-Between | 0.969 | Likely Pathogenic | Likely Pathogenic | 0.39 | Likely Benign | 0.0 | -0.03 | Likely Benign | 0.18 | Likely Benign | 0.21 | Likely Benign | 0.199 | Likely Benign | -2.76 | Deleterious | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.38 | Benign | 0.07 | Tolerated | 0.1686 | 0.2864 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1267T>A | Y423N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y423N is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include SIFT and FATHMM, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. Based on the overwhelming majority of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.421885 | Uncertain | 0.975 | 0.242 | 0.000 | -10.937 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 3.97 | Destabilizing | 0.1 | 4.08 | Destabilizing | 4.03 | Destabilizing | 2.07 | Destabilizing | 0.501 | Likely Pathogenic | -7.47 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.40 | Benign | 0.06 | Tolerated | 0.1405 | 0.0412 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.1267T>C | Y423H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 Y423H missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect comprise FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and AlphaMissense‑Default. AlphaMissense‑Optimized is reported as uncertain. High‑accuracy assessments show that AlphaMissense‑Optimized is inconclusive, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie and therefore inconclusive, while Foldetta predicts a pathogenic impact. Overall, the majority of evaluated tools (10 pathogenic vs. 4 benign) support a pathogenic classification. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.088832 | Structured | 0.421885 | Uncertain | 0.975 | 0.242 | 0.000 | -6.638 | Likely Benign | 0.873 | Likely Pathogenic | Ambiguous | 3.09 | Destabilizing | 0.1 | 2.44 | Destabilizing | 2.77 | Destabilizing | 1.66 | Destabilizing | 0.257 | Likely Benign | -3.88 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.39 | Benign | 0.61 | Tolerated | 0.1603 | 0.0352 | 0 | 2 | -1.9 | -26.03 | ||||||||||||||||||||||||||||||
| c.1267T>G | Y423D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y423D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, while the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.421885 | Uncertain | 0.975 | 0.242 | 0.000 | -13.279 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 4.67 | Destabilizing | 0.1 | 4.79 | Destabilizing | 4.73 | Destabilizing | 1.78 | Destabilizing | 0.553 | Likely Pathogenic | -8.28 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.38 | Benign | 0.04 | Affected | 0.3262 | 0.0412 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1268A>C | Y423S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y423S is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, SIFT, and FATHMM, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Taken together, the preponderance of evidence points to a pathogenic effect for Y423S. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.421885 | Uncertain | 0.975 | 0.242 | 0.000 | -10.847 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 4.28 | Destabilizing | 0.1 | 4.78 | Destabilizing | 4.53 | Destabilizing | 1.95 | Destabilizing | 0.345 | Likely Benign | -7.39 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.40 | Benign | 0.09 | Tolerated | 0.3152 | 0.1259 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||
| c.1268A>G | Y423C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y423C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM. The remaining tools—SGM‑Consensus, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain (treated as unavailable), SGM‑Consensus as Likely Pathogenic, and Foldetta as Pathogenic. With the majority of evidence pointing to deleterious effects, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.421885 | Uncertain | 0.975 | 0.242 | 0.000 | -9.003 | Likely Pathogenic | 0.935 | Likely Pathogenic | Ambiguous | 4.01 | Destabilizing | 0.1 | 4.49 | Destabilizing | 4.25 | Destabilizing | 1.84 | Destabilizing | 0.286 | Likely Benign | -7.51 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.38 | Benign | 0.03 | Affected | 0.2381 | 0.1096 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||
| c.1268A>T | Y423F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y423F is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess sequence conservation and functional impact uniformly classify the substitution as benign: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic effect, while premPS is inconclusive. High‑accuracy methods give consistent benign results: AlphaMissense‑Optimized is benign, the SGM‑Consensus is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta) predicts benign stability. Overall, the overwhelming majority of evidence supports a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.088832 | Structured | 0.421885 | Uncertain | 0.975 | 0.242 | 0.000 | -5.533 | Likely Benign | 0.181 | Likely Benign | Likely Benign | 0.03 | Likely Benign | 0.1 | -0.05 | Likely Benign | -0.01 | Likely Benign | 0.76 | Ambiguous | 0.149 | Likely Benign | -2.30 | Neutral | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 3.45 | Benign | 0.60 | Tolerated | 0.1934 | 0.2337 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||
| c.1270G>A | V424I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V424I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign. The only inconclusive results come from FoldX (uncertain) and Foldetta (uncertain). When high‑accuracy methods are considered separately, AlphaMissense‑Optimized predicts benign, the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also predicts benign, while Foldetta remains uncertain. No tool predicts pathogenicity. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -3.814 | Likely Benign | 0.095 | Likely Benign | Likely Benign | -1.04 | Ambiguous | 0.1 | -0.48 | Likely Benign | -0.76 | Ambiguous | 0.02 | Likely Benign | 0.084 | Likely Benign | -0.15 | Neutral | 0.013 | Benign | 0.006 | Benign | 3.50 | Benign | 0.63 | Tolerated | 0.0874 | 0.2609 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||
| c.1270G>C | V424L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V424L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome, while SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports a likely benign classification. Stability‑based methods are inconclusive: FoldX is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -4.941 | Likely Benign | 0.742 | Likely Pathogenic | Likely Benign | -0.90 | Ambiguous | 0.2 | -0.41 | Likely Benign | -0.66 | Ambiguous | 0.43 | Likely Benign | 0.159 | Likely Benign | -1.68 | Neutral | 0.327 | Benign | 0.026 | Benign | 4.50 | Benign | 0.59 | Tolerated | 0.1018 | 0.2680 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1270G>T | V424F 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant V424F is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and FATHMM, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The remaining tools (FoldX, Foldetta, premPS, AlphaMissense‑Optimized) yield uncertain or unavailable results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -10.947 | Likely Pathogenic | 0.939 | Likely Pathogenic | Ambiguous | 1.99 | Ambiguous | 0.3 | -0.37 | Likely Benign | 0.81 | Ambiguous | 0.55 | Ambiguous | 0.275 | Likely Benign | -3.66 | Deleterious | 0.995 | Probably Damaging | 0.775 | Possibly Damaging | 3.40 | Benign | 0.01 | Affected | 0.0816 | 0.2094 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1271T>A | V424D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V424D is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors indicates a pathogenic effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, whereas only FATHMM predicts a benign outcome. High‑accuracy tools reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels it likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. No prediction is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -15.531 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.60 | Destabilizing | 0.1 | 3.50 | Destabilizing | 3.55 | Destabilizing | 2.13 | Destabilizing | 0.615 | Likely Pathogenic | -5.87 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.1536 | 0.0845 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1271T>C | V424A 2D 3DClick to see structure in 3D Viewer AISynGAP1 V424A is not reported in ClinVar (ClinVar status: not reported) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on benign impact are REVEL and FATHMM, whereas the remaining tools—SGM‑Consensus, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as unavailable, SGM‑Consensus as likely pathogenic, and Foldetta as pathogenic. Based on the collective evidence, the variant is most likely pathogenic; this conclusion is not contradicted by the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -9.665 | Likely Pathogenic | 0.904 | Likely Pathogenic | Ambiguous | 2.31 | Destabilizing | 0.1 | 2.54 | Destabilizing | 2.43 | Destabilizing | 2.10 | Destabilizing | 0.245 | Likely Benign | -3.45 | Deleterious | 0.997 | Probably Damaging | 0.961 | Probably Damaging | 3.39 | Benign | 0.01 | Affected | 0.2105 | 0.1355 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.1271T>G | V424G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V424G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM. All other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic consensus (3 pathogenic vs. 1 benign); and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. No predictions are missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -13.697 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 3.90 | Destabilizing | 0.1 | 4.84 | Destabilizing | 4.37 | Destabilizing | 2.44 | Destabilizing | 0.622 | Likely Pathogenic | -6.26 | Deleterious | 0.994 | Probably Damaging | 1.000 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.1396 | 0.2029 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1273A>C | T425P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T425P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Consensus from multiple in silico predictors indicates a pathogenic effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as deleterious, while only FATHMM predicts a benign outcome; premPS is uncertain. High‑accuracy tools reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic effect. Taken together, the overwhelming majority of evidence supports a pathogenic classification, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.401218 | Uncertain | 0.964 | 0.280 | 0.000 | -12.318 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 2.81 | Destabilizing | 0.3 | 6.49 | Destabilizing | 4.65 | Destabilizing | 0.77 | Ambiguous | 0.512 | Likely Pathogenic | -5.16 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.37 | Benign | 0.03 | Affected | 0.1815 | 0.3469 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1273A>G | T425A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T425A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. Two tools (premPS and AlphaMissense‑Optimized) give uncertain results and are treated as unavailable. High‑accuracy assessments show SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign, and AlphaMissense‑Optimized as uncertain. Overall, the balance of evidence leans toward pathogenicity, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.401218 | Uncertain | 0.964 | 0.280 | 0.000 | -8.945 | Likely Pathogenic | 0.799 | Likely Pathogenic | Ambiguous | -0.31 | Likely Benign | 0.1 | -0.05 | Likely Benign | -0.18 | Likely Benign | 0.70 | Ambiguous | 0.372 | Likely Benign | -4.17 | Deleterious | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.42 | Benign | 0.09 | Tolerated | 0.2971 | 0.2572 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1273A>T | T425S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T425S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Four tools (Foldetta, premPS, ESM1b, Rosetta) return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of tools (five benign vs. four pathogenic) lean toward a benign interpretation, and this assessment does not contradict ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.041405 | Structured | 0.401218 | Uncertain | 0.964 | 0.280 | 0.000 | -7.420 | In-Between | 0.676 | Likely Pathogenic | Likely Benign | 0.22 | Likely Benign | 0.1 | 0.77 | Ambiguous | 0.50 | Ambiguous | 0.63 | Ambiguous | 0.275 | Likely Benign | -3.05 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 3.42 | Benign | 0.07 | Tolerated | 0.2511 | 0.2324 | 1 | 1 | -0.1 | -14.03 | ||||||||||||||||||||||||||||||
| c.1274C>A | T425N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T425N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, FoldX, and FATHMM, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Predictions labeled uncertain (Foldetta, premPS, AlphaMissense‑Optimized, Rosetta) are treated as unavailable. High‑accuracy assessments further indicate a likely pathogenic outcome from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and an uncertain result from AlphaMissense‑Optimized and Foldetta. Overall, the majority of definitive predictions support a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.401218 | Uncertain | 0.964 | 0.280 | 0.000 | -10.709 | Likely Pathogenic | 0.925 | Likely Pathogenic | Ambiguous | 0.19 | Likely Benign | 0.1 | 0.81 | Ambiguous | 0.50 | Ambiguous | 0.82 | Ambiguous | 0.185 | Likely Benign | -3.87 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.44 | Benign | 0.04 | Affected | 0.1169 | 0.2843 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.1274C>G | T425S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T425S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Four tools (Foldetta, premPS, ESM1b, Rosetta) return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of tools (five benign vs. four pathogenic) lean toward a benign interpretation, and this assessment does not contradict ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.041405 | Structured | 0.401218 | Uncertain | 0.964 | 0.280 | 0.000 | -7.420 | In-Between | 0.676 | Likely Pathogenic | Likely Benign | 0.22 | Likely Benign | 0.1 | 0.77 | Ambiguous | 0.50 | Ambiguous | 0.63 | Ambiguous | 0.184 | Likely Benign | -3.05 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 3.42 | Benign | 0.07 | Tolerated | 0.2511 | 0.2324 | 1 | 1 | -0.1 | -14.03 | ||||||||||||||||||||||||||||||
| c.1274C>T | T425I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T425I is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic, whereas Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Overall, the balance of evidence slightly favors a pathogenic interpretation, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.401218 | Uncertain | 0.964 | 0.280 | 0.000 | -10.443 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | -0.04 | Likely Benign | 0.2 | 0.07 | Likely Benign | 0.02 | Likely Benign | 0.30 | Likely Benign | 0.268 | Likely Benign | -5.30 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 3.44 | Benign | 0.06 | Tolerated | 0.0827 | 0.4023 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.1276A>C | N426H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N426H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Predictions that are uncertain or inconclusive are FoldX and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign; and Foldetta also predicts a benign outcome. No prediction tool is missing or inconclusive in this set. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.042364 | Structured | 0.394941 | Uncertain | 0.959 | 0.287 | 0.000 | -7.004 | In-Between | 0.248 | Likely Benign | Likely Benign | 0.64 | Ambiguous | 0.0 | -0.14 | Likely Benign | 0.25 | Likely Benign | 0.24 | Likely Benign | 0.237 | Likely Benign | -3.57 | Deleterious | 0.998 | Probably Damaging | 0.985 | Probably Damaging | 3.29 | Benign | 0.14 | Tolerated | 0.1267 | 0.3124 | 2 | 1 | 0.3 | 23.04 | ||||||||||||||||||||||||||||||
| c.1276A>G | N426D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N426D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Three tools (FoldX, premPS, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of predictions (six benign vs. four pathogenic) and the two high‑accuracy benign calls suggest the variant is most likely benign, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.042364 | Structured | 0.394941 | Uncertain | 0.959 | 0.287 | 0.000 | -11.159 | Likely Pathogenic | 0.554 | Ambiguous | Likely Benign | 0.80 | Ambiguous | 0.0 | 0.18 | Likely Benign | 0.49 | Likely Benign | 0.64 | Ambiguous | 0.173 | Likely Benign | -3.09 | Deleterious | 0.998 | Probably Damaging | 0.980 | Probably Damaging | 3.34 | Benign | 0.09 | Tolerated | 0.1730 | 0.1746 | 2 | 1 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.1276A>T | N426Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N426Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two main groups: benign predictions come from REVEL, FoldX, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and ESM1b. Two tools remain uncertain: Rosetta and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on the current predictive landscape. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.042364 | Structured | 0.394941 | Uncertain | 0.959 | 0.287 | 0.000 | -8.510 | Likely Pathogenic | 0.541 | Ambiguous | Likely Benign | 0.47 | Likely Benign | 0.0 | -0.50 | Ambiguous | -0.02 | Likely Benign | 0.23 | Likely Benign | 0.341 | Likely Benign | -5.48 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.30 | Benign | 0.25 | Tolerated | 0.0578 | 0.3338 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||
| c.1277A>C | N426T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change N426T lies in the GAP domain. It is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Predictions that are uncertain or inconclusive are FoldX, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, while Foldetta remains uncertain and is not taken as evidence. Overall, the majority of tools, including the high‑accuracy methods, predict a benign effect. This consensus does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.042364 | Structured | 0.394941 | Uncertain | 0.959 | 0.287 | 0.000 | -7.389 | In-Between | 0.194 | Likely Benign | Likely Benign | 0.84 | Ambiguous | 0.0 | 0.38 | Likely Benign | 0.61 | Ambiguous | 0.12 | Likely Benign | 0.126 | Likely Benign | -3.14 | Deleterious | 0.983 | Probably Damaging | 0.926 | Probably Damaging | 3.47 | Benign | 0.10 | Tolerated | 0.1090 | 0.3002 | 0 | 0 | 2.8 | -13.00 | ||||||||||||||||||||||||||||||
| c.1277A>G | N426S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N426S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The high‑accuracy AlphaMissense‑Optimized score is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and the Foldetta stability assessment is inconclusive. No evidence from FoldX or Rosetta is available. Overall, the majority of predictions support a benign impact, and this is consistent with the absence of a ClinVar assertion. Therefore, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.042364 | Structured | 0.394941 | Uncertain | 0.959 | 0.287 | 0.000 | -5.215 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.81 | Ambiguous | 0.0 | 0.52 | Ambiguous | 0.67 | Ambiguous | 0.03 | Likely Benign | 0.131 | Likely Benign | -2.37 | Neutral | 0.998 | Probably Damaging | 0.921 | Probably Damaging | 3.47 | Benign | 0.33 | Tolerated | 0.2224 | 0.3188 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||
| c.1277A>T | N426I 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant N426I has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and the protein‑folding stability method Foldetta. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicting pathogenicity, and Foldetta indicating a benign folding stability outcome. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.394941 | Uncertain | 0.959 | 0.287 | 0.000 | -10.158 | Likely Pathogenic | 0.570 | Likely Pathogenic | Likely Benign | 0.67 | Ambiguous | 0.0 | 0.14 | Likely Benign | 0.41 | Likely Benign | 0.23 | Likely Benign | 0.216 | Likely Benign | -5.71 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 3.31 | Benign | 0.09 | Tolerated | 0.0656 | 0.3244 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.1278C>A | N426K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N426K resides in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and FATHMM. Those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Two tools give inconclusive results: AlphaMissense‑Optimized and premPS. High‑accuracy assessments show that AlphaMissense‑Optimized is uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a benign effect. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which is currently unclassified. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.394941 | Uncertain | 0.959 | 0.287 | 0.000 | -11.659 | Likely Pathogenic | 0.867 | Likely Pathogenic | Ambiguous | 0.03 | Likely Benign | 0.0 | 0.00 | Likely Benign | 0.02 | Likely Benign | 0.54 | Ambiguous | 0.197 | Likely Benign | -3.86 | Deleterious | 0.998 | Probably Damaging | 0.978 | Probably Damaging | 3.38 | Benign | 0.12 | Tolerated | 0.1906 | 0.2822 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.1278C>G | N426K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N426K is located in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, SIFT, and FATHMM, while those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Two tools report uncertainty: AlphaMissense‑Optimized and premPS. High‑accuracy assessments show that AlphaMissense‑Optimized is uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a benign effect on protein stability. Overall, the majority of predictions lean toward pathogenicity, with a split in the most reliable methods. Therefore, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.394941 | Uncertain | 0.959 | 0.287 | 0.000 | -11.659 | Likely Pathogenic | 0.867 | Likely Pathogenic | Ambiguous | 0.03 | Likely Benign | 0.0 | 0.00 | Likely Benign | 0.02 | Likely Benign | 0.54 | Ambiguous | 0.197 | Likely Benign | -3.86 | Deleterious | 0.998 | Probably Damaging | 0.978 | Probably Damaging | 3.38 | Benign | 0.12 | Tolerated | 0.1906 | 0.2822 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.1279C>A | H427N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H427N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, Rosetta, and AlphaMissense‑Default all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a benign outcome. With all available evidence pointing to a neutral effect and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.081712 | Structured | 0.394261 | Uncertain | 0.962 | 0.287 | 0.000 | 1.162 | Likely Benign | 0.045 | Likely Benign | Likely Benign | -0.11 | Likely Benign | 0.1 | 0.39 | Likely Benign | 0.14 | Likely Benign | -0.48 | Likely Benign | 0.100 | Likely Benign | 1.63 | Neutral | 0.010 | Benign | 0.006 | Benign | 3.62 | Benign | 0.46 | Tolerated | 0.1507 | 0.2418 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||
| c.1279C>G | H427D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant H427D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign or likely benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, SGM‑Consensus is likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. No evidence from the available tools suggests pathogenicity, and the absence of a ClinVar classification means there is no contradiction. Therefore, based on the current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.081712 | Structured | 0.394261 | Uncertain | 0.962 | 0.287 | 0.000 | -3.684 | Likely Benign | 0.500 | Ambiguous | Likely Benign | 0.76 | Ambiguous | 0.1 | 1.26 | Ambiguous | 1.01 | Ambiguous | 0.10 | Likely Benign | 0.163 | Likely Benign | -1.43 | Neutral | 0.677 | Possibly Damaging | 0.236 | Benign | 3.58 | Benign | 0.12 | Tolerated | 0.2241 | 0.1678 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||
| c.1279C>T | H427Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 H427Y missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar, as well as the SGM‑Consensus “Likely Benign” call. Tools that predict a pathogenic outcome are PROVEAN, polyPhen‑2 HumDiv, and SIFT. High‑accuracy assessments—AlphaMissense‑Optimized, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta)—all uniformly indicate a benign effect. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.081712 | Structured | 0.394261 | Uncertain | 0.962 | 0.287 | 0.000 | -6.824 | Likely Benign | 0.328 | Likely Benign | Likely Benign | -0.08 | Likely Benign | 0.0 | -0.37 | Likely Benign | -0.23 | Likely Benign | 0.18 | Likely Benign | 0.180 | Likely Benign | -3.47 | Deleterious | 0.815 | Possibly Damaging | 0.073 | Benign | 3.44 | Benign | 0.01 | Affected | 0.0958 | 0.4029 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||
| c.127G>C | G43R 2D AIThe SynGAP1 missense variant G43R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for G43R, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431462 | Uncertain | 0.396 | 0.762 | 0.375 | -2.104 | Likely Benign | 0.266 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -1.26 | Neutral | 0.870 | Possibly Damaging | 0.500 | Possibly Damaging | 4.22 | Benign | 0.00 | Affected | 0.1025 | 0.3645 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.127G>T | G43C 2D AIThe SynGAP1 missense variant G43C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431462 | Uncertain | 0.396 | 0.762 | 0.375 | -4.599 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.90 | Neutral | 0.987 | Probably Damaging | 0.750 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.1445 | 0.3438 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||||||||||||
| c.1280A>C | H427P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H427P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic impact. Two tools are uncertain: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as inconclusive, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta outputs) both indicate a pathogenic effect. Overall, the preponderance of evidence points to a pathogenic classification for H427P, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.081712 | Structured | 0.394261 | Uncertain | 0.962 | 0.287 | 0.000 | -11.098 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 4.74 | Destabilizing | 0.1 | 7.61 | Destabilizing | 6.18 | Destabilizing | 0.68 | Ambiguous | 0.324 | Likely Benign | -4.22 | Deleterious | 0.993 | Probably Damaging | 0.819 | Possibly Damaging | 3.51 | Benign | 0.01 | Affected | 0.2061 | 0.3442 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||
| c.1280A>T | H427L 2D 3DClick to see structure in 3D Viewer AISynGAP1 H427L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: nine tools (REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Optimized) predict a benign effect, while five tools (SGM‑Consensus, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default) predict pathogenicity. High‑accuracy methods provide a more focused view: AlphaMissense‑Optimized indicates a benign outcome; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, remains pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. Taken together, the majority of evidence supports a benign impact for H427L, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.081712 | Structured | 0.394261 | Uncertain | 0.962 | 0.287 | 0.000 | -9.691 | Likely Pathogenic | 0.755 | Likely Pathogenic | Likely Benign | -0.17 | Likely Benign | 0.0 | 0.05 | Likely Benign | -0.06 | Likely Benign | 0.31 | Likely Benign | 0.272 | Likely Benign | -5.38 | Deleterious | 0.299 | Benign | 0.033 | Benign | 3.42 | Benign | 0.01 | Affected | 0.0942 | 0.4953 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.1281T>A | H427Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 H427Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and SIFT. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign effect for H427Q. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.081712 | Structured | 0.394261 | Uncertain | 0.962 | 0.287 | 0.000 | -5.858 | Likely Benign | 0.367 | Ambiguous | Likely Benign | 0.77 | Ambiguous | 0.1 | 0.62 | Ambiguous | 0.70 | Ambiguous | 0.73 | Ambiguous | 0.142 | Likely Benign | -2.41 | Neutral | 0.864 | Possibly Damaging | 0.088 | Benign | 3.52 | Benign | 0.02 | Affected | 0.1556 | 0.3382 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.1281T>G | H427Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 H427Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and SIFT. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign effect for H427Q. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.081712 | Structured | 0.394261 | Uncertain | 0.962 | 0.287 | 0.000 | -5.858 | Likely Benign | 0.367 | Ambiguous | Likely Benign | 0.77 | Ambiguous | 0.1 | 0.62 | Ambiguous | 0.70 | Ambiguous | 0.73 | Ambiguous | 0.142 | Likely Benign | -2.41 | Neutral | 0.864 | Possibly Damaging | 0.088 | Benign | 3.52 | Benign | 0.02 | Affected | 0.1556 | 0.3382 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.1282T>A | Y428N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y428N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are limited to FATHMM, which classifies the change as benign. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic or likely pathogenic impact. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.389652 | Uncertain | 0.965 | 0.292 | 0.000 | -12.164 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 2.34 | Destabilizing | 0.0 | 3.81 | Destabilizing | 3.08 | Destabilizing | 1.85 | Destabilizing | 0.533 | Likely Pathogenic | -8.59 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.48 | Benign | 0.03 | Affected | 0.2607 | 0.0971 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.1282T>G | Y428D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Y428D is not reported in ClinVar and is absent from gnomAD. Across a broad panel of in silico predictors, 15 tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) uniformly predict a deleterious effect, whereas only FATHMM classifies it as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote) is likely pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic effect. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.389652 | Uncertain | 0.965 | 0.292 | 0.000 | -13.473 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.24 | Destabilizing | 0.0 | 4.19 | Destabilizing | 3.72 | Destabilizing | 1.64 | Destabilizing | 0.554 | Likely Pathogenic | -9.55 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.50 | Benign | 0.01 | Affected | 0.4458 | 0.1003 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1283A>C | Y428S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y428S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM. All other evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact, while AlphaMissense‑Optimized is uncertain. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus indicates “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic effect. Based on the overwhelming majority of predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.389652 | Uncertain | 0.965 | 0.292 | 0.000 | -10.936 | Likely Pathogenic | 0.928 | Likely Pathogenic | Ambiguous | 3.00 | Destabilizing | 0.0 | 3.42 | Destabilizing | 3.21 | Destabilizing | 2.00 | Destabilizing | 0.505 | Likely Pathogenic | -8.59 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.45 | Benign | 0.03 | Affected | 0.4623 | 0.2352 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||
| c.1283A>G | Y428C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y428C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are REVEL and FATHMM. The remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus—consistently predict a pathogenic impact. High‑accuracy assessments show the SGM‑Consensus result as “Likely Pathogenic,” Foldetta predicts a destabilizing, pathogenic effect, while AlphaMissense‑Optimized is uncertain. No prediction or stability result is missing; the only inconclusive outcome is the AlphaMissense‑Optimized score. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar status exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.118441 | Structured | 0.389652 | Uncertain | 0.965 | 0.292 | 0.000 | -11.312 | Likely Pathogenic | 0.852 | Likely Pathogenic | Ambiguous | 2.82 | Destabilizing | 0.0 | 2.99 | Destabilizing | 2.91 | Destabilizing | 2.02 | Destabilizing | 0.454 | Likely Benign | -8.59 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.39 | Benign | 0.01 | Affected | 0.3322 | 0.2220 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||
| c.1283A>T | Y428F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y428F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b) predict a pathogenic impact. Uncertain or inconclusive results are reported for FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as unavailable. Overall, the balance of evidence—five pathogenic versus three benign predictions, with a pathogenic SGM Consensus and a benign AlphaMissense‑Optimized—suggests that the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.118441 | Structured | 0.389652 | Uncertain | 0.965 | 0.292 | 0.000 | -10.464 | Likely Pathogenic | 0.530 | Ambiguous | Likely Benign | 0.82 | Ambiguous | 0.1 | 0.52 | Ambiguous | 0.67 | Ambiguous | 0.89 | Ambiguous | 0.366 | Likely Benign | -3.82 | Deleterious | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 3.41 | Benign | 0.04 | Affected | 0.2719 | 0.3146 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||
| c.1285C>G | R429G 2D 3DClick to see structure in 3D Viewer AISynGAP1 R429G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta’s stability analysis is uncertain. No evidence from FoldX or Rosetta is available. Overall, the predictions are evenly split, with no clear consensus. The variant is most likely of uncertain significance; it is not contradicted by ClinVar status, which has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.390504 | Uncertain | 0.959 | 0.290 | 0.000 | -9.157 | Likely Pathogenic | 0.333 | Likely Benign | Likely Benign | 1.58 | Ambiguous | 0.0 | 1.68 | Ambiguous | 1.63 | Ambiguous | 1.21 | Destabilizing | 0.257 | Likely Benign | -3.37 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.43 | Benign | 0.34 | Tolerated | 0.2888 | 0.2717 | -3 | -2 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||
| c.1286G>C | R429P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R429P is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and ESM1b; AlphaMissense‑Default and FoldX are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of tools and the high‑accuracy methods favor a pathogenic interpretation. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.074921 | Structured | 0.390504 | Uncertain | 0.959 | 0.290 | 0.000 | -9.771 | Likely Pathogenic | 0.556 | Ambiguous | Likely Benign | 1.53 | Ambiguous | 0.2 | 5.13 | Destabilizing | 3.33 | Destabilizing | 1.01 | Destabilizing | 0.265 | Likely Benign | -2.89 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.43 | Benign | 0.25 | Tolerated | 0.1881 | 0.3790 | 0 | -2 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||
| c.1286G>T | R429L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R429L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. No contradictory evidence is present from ClinVar or gnomAD. **Based on the aggregate predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.074921 | Structured | 0.390504 | Uncertain | 0.959 | 0.290 | 0.000 | -6.495 | Likely Benign | 0.408 | Ambiguous | Likely Benign | -0.05 | Likely Benign | 0.1 | 0.06 | Likely Benign | 0.01 | Likely Benign | -0.12 | Likely Benign | 0.241 | Likely Benign | -1.20 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.59 | Benign | 0.37 | Tolerated | 0.1555 | 0.4025 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.1288A>C | M430L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M430L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, FoldX, Rosetta, PolyPhen‑2 (HumDiv and HumVar), SIFT, and REVEL; no tool predicts pathogenicity. The high‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. With all available evidence pointing to a benign effect and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.071867 | Structured | 0.385298 | Uncertain | 0.952 | 0.306 | 0.000 | -5.873 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.33 | Likely Benign | 0.1 | -0.10 | Likely Benign | 0.12 | Likely Benign | 0.72 | Ambiguous | 0.107 | Likely Benign | -1.07 | Neutral | 0.028 | Benign | 0.004 | Benign | 3.69 | Benign | 0.66 | Tolerated | 0.1767 | 0.5067 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1288A>G | M430V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M430V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign or tolerated changes. Only polyPhen‑2 HumDiv flags it as pathogenic, while the SGM‑Consensus score is Likely Benign. Stability‑based methods are inconclusive: FoldX and premPS return Uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports Uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta remains Uncertain. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.071867 | Structured | 0.385298 | Uncertain | 0.952 | 0.306 | 0.000 | -3.555 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 1.48 | Ambiguous | 0.1 | -0.23 | Likely Benign | 0.63 | Ambiguous | 0.87 | Ambiguous | 0.103 | Likely Benign | -1.35 | Neutral | 0.918 | Possibly Damaging | 0.185 | Benign | 3.53 | Benign | 0.51 | Tolerated | 0.3397 | 0.4953 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||
| c.1288A>T | M430L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M430L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, FoldX, Rosetta, PolyPhen‑2 (HumDiv and HumVar), SIFT, and REVEL; no tool predicts pathogenicity. The high‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. With all available evidence pointing to a benign effect and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.071867 | Structured | 0.385298 | Uncertain | 0.952 | 0.306 | 0.000 | -5.873 | Likely Benign | 0.104 | Likely Benign | Likely Benign | 0.33 | Likely Benign | 0.1 | -0.10 | Likely Benign | 0.12 | Likely Benign | 0.72 | Ambiguous | 0.107 | Likely Benign | -1.07 | Neutral | 0.028 | Benign | 0.004 | Benign | 3.69 | Benign | 0.66 | Tolerated | 0.1767 | 0.5067 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1289T>A | M430K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M430K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN and ESM1b. The remaining tools—FoldX, Foldetta, premPS, and AlphaMissense‑Default—return uncertain or inconclusive results. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized classifies the variant as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors a pathogenic outcome; Foldetta remains uncertain. Overall, the majority of individual predictors lean toward a benign interpretation, whereas the SGM Consensus suggests pathogenicity. Given the lack of ClinVar evidence, there is no contradiction with existing clinical annotations. Based on the aggregate predictions, the variant is most likely benign, and this assessment does not conflict with the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.071867 | Structured | 0.385298 | Uncertain | 0.952 | 0.306 | 0.000 | -10.816 | Likely Pathogenic | 0.494 | Ambiguous | Likely Benign | 0.78 | Ambiguous | 0.1 | 0.44 | Likely Benign | 0.61 | Ambiguous | 0.86 | Ambiguous | 0.149 | Likely Benign | -2.66 | Deleterious | 0.134 | Benign | 0.033 | Benign | 3.47 | Benign | 0.32 | Tolerated | 0.1660 | 0.1271 | 0 | -1 | -5.8 | -3.02 | ||||||||||||||||||||||||||||||
| c.1289T>G | M430R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M430R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and ESM1b. The remaining tools—FoldX, Foldetta, premPS, and AlphaMissense‑Default—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of consensus tools lean toward a benign classification, while a subset of high‑accuracy methods suggest pathogenicity, leaving the variant’s impact ambiguous. Based on the aggregate predictions, the variant is most likely benign, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.071867 | Structured | 0.385298 | Uncertain | 0.952 | 0.306 | 0.000 | -10.636 | Likely Pathogenic | 0.558 | Ambiguous | Likely Benign | 0.98 | Ambiguous | 0.1 | 0.47 | Likely Benign | 0.73 | Ambiguous | 0.83 | Ambiguous | 0.153 | Likely Benign | -2.87 | Deleterious | 0.620 | Possibly Damaging | 0.046 | Benign | 3.48 | Benign | 0.38 | Tolerated | 0.1775 | 0.1652 | 0 | -1 | -6.4 | 24.99 | ||||||||||||||||||||||||||||||
| c.128G>A | G43D 2D AIThe SynGAP1 missense variant G43D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of any ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431462 | Uncertain | 0.396 | 0.762 | 0.375 | -5.867 | Likely Benign | 0.204 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.84 | Neutral | 0.870 | Possibly Damaging | 0.500 | Possibly Damaging | 4.21 | Benign | 0.00 | Affected | 0.2018 | 0.2339 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||||||
| c.128G>T | G43V 2D AIThe SynGAP1 missense variant G43V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431462 | Uncertain | 0.396 | 0.762 | 0.375 | -3.745 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -0.79 | Neutral | 0.320 | Benign | 0.140 | Benign | 4.28 | Benign | 0.00 | Affected | 0.1156 | 0.3120 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.1291C>A | L431M 2D AIThe SynGAP1 missense variant L431M (ClinVar ID 4327026) is not found in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, FATHMM, AlphaMissense‑Optimized, and Foldetta, whereas premPS, polyPhen‑2 (HumDiv and HumVar), and SIFT all predict a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. No predictions or stability results are missing or inconclusive. Overall, the majority of computational evidence indicates that the variant is most likely benign, which contradicts the ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.094817 | Structured | 0.374755 | Uncertain | 0.959 | 0.300 | 0.000 | 1 | -7.471 | In-Between | 0.375 | Ambiguous | Likely Benign | 0.18 | Likely Benign | 0.3 | 0.50 | Ambiguous | 0.34 | Likely Benign | 1.07 | Destabilizing | 0.102 | Likely Benign | -1.50 | Neutral | 0.995 | Probably Damaging | 0.849 | Possibly Damaging | 2.94 | Benign | 0.02 | Affected | 0.0748 | 0.3121 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.1291C>G | L431V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L431V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are FoldX, premPS, PROVEAN, and polyPhen‑2 HumDiv. Four tools (Rosetta, Foldetta, ESM1b, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 1‑to‑1 split between benign and pathogenic signals, and Foldetta also yields an uncertain outcome. Overall, the balance of evidence—including the higher number of benign predictions and the benign call from the most accurate tool—suggests that the variant is most likely benign. This conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.094817 | Structured | 0.374755 | Uncertain | 0.959 | 0.300 | 0.000 | -7.949 | In-Between | 0.505 | Ambiguous | Likely Benign | 2.17 | Destabilizing | 0.0 | 1.50 | Ambiguous | 1.84 | Ambiguous | 1.32 | Destabilizing | 0.093 | Likely Benign | -2.58 | Deleterious | 0.861 | Possibly Damaging | 0.332 | Benign | 3.04 | Benign | 0.20 | Tolerated | 0.1377 | 0.3198 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||
| c.1292T>A | L431Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L431Q resides in the GAP domain. ClinVar has no entry for this variant, and it is not present in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM; all other evaluated algorithms—including FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions or stability results are missing or inconclusive. Based on the overwhelming agreement among pathogenic predictions and the high‑accuracy tools, the variant is most likely pathogenic; this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.094817 | Structured | 0.374755 | Uncertain | 0.959 | 0.300 | 0.000 | -12.399 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 2.69 | Destabilizing | 0.0 | 2.37 | Destabilizing | 2.53 | Destabilizing | 1.99 | Destabilizing | 0.493 | Likely Benign | -5.40 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 2.92 | Benign | 0.01 | Affected | 0.1036 | 0.1088 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1292T>G | L431R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L431R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.094817 | Structured | 0.374755 | Uncertain | 0.959 | 0.300 | 0.000 | -14.777 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.45 | Destabilizing | 0.0 | 4.76 | Destabilizing | 4.11 | Destabilizing | 1.93 | Destabilizing | 0.654 | Likely Pathogenic | -5.47 | Deleterious | 0.997 | Probably Damaging | 0.833 | Possibly Damaging | 2.93 | Benign | 0.00 | Affected | 0.1191 | 0.0730 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1294T>A | C432S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C432S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools (SGM Consensus, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show SGM Consensus as likely pathogenic, AlphaMissense‑Optimized as uncertain, and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic classification for C432S, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.362533 | Uncertain | 0.960 | 0.285 | 0.000 | -8.229 | Likely Pathogenic | 0.913 | Likely Pathogenic | Ambiguous | 0.61 | Ambiguous | 0.1 | 0.96 | Ambiguous | 0.79 | Ambiguous | 1.52 | Destabilizing | 0.496 | Likely Benign | -9.55 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.53 | Benign | 0.12 | Tolerated | 0.4262 | 0.1415 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.1294T>C | C432R 2D 3DClick to see structure in 3D Viewer AIClinVar has no entry for this SynGAP1 missense variant, and it is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM. The majority of algorithms predict a pathogenic impact: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). FoldX and Foldetta report uncertain results. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, while Foldetta remains inconclusive. Overall, the consensus of the available predictions indicates that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.362533 | Uncertain | 0.960 | 0.285 | 0.000 | -13.858 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.03 | Ambiguous | 0.2 | 2.05 | Destabilizing | 1.54 | Ambiguous | 1.73 | Destabilizing | 0.690 | Likely Pathogenic | -11.46 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.45 | Benign | 0.01 | Affected | 0.1359 | 0.1766 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.1294T>G | C432G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C432G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM. Those that predict a pathogenic effect comprise SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Predictions that are inconclusive or uncertain are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points to a pathogenic impact. This conclusion is consistent with the absence of a ClinVar classification; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.362533 | Uncertain | 0.960 | 0.285 | 0.000 | -10.247 | Likely Pathogenic | 0.831 | Likely Pathogenic | Ambiguous | 1.37 | Ambiguous | 0.0 | 2.25 | Destabilizing | 1.81 | Ambiguous | 1.60 | Destabilizing | 0.631 | Likely Pathogenic | -11.46 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.45 | Benign | 0.03 | Affected | 0.2796 | 0.2130 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1295G>A | C432Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant C432Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while benign predictions are reported by REVEL, Rosetta, FATHMM, and Foldetta. Tools with uncertain or missing results (FoldX, premPS) are not considered evidence. High‑accuracy methods give mixed signals: AlphaMissense‑Optimized and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predict pathogenicity, whereas Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a benign effect. Overall, the majority of predictions support a pathogenic impact, and this conclusion does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.362533 | Uncertain | 0.960 | 0.285 | 0.000 | -13.720 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.62 | Ambiguous | 0.3 | 0.31 | Likely Benign | 0.47 | Likely Benign | 0.71 | Ambiguous | 0.441 | Likely Benign | -10.50 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.45 | Benign | 0.02 | Affected | 0.0857 | 0.3438 | 0 | -2 | -3.8 | 60.04 | |||||||||||||||||||||||||||||
| c.1295G>C | C432S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C432S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools (SGM Consensus, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show SGM Consensus as likely pathogenic, AlphaMissense‑Optimized as uncertain, and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic classification for C432S, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.362533 | Uncertain | 0.960 | 0.285 | 0.000 | -8.229 | Likely Pathogenic | 0.913 | Likely Pathogenic | Ambiguous | 0.61 | Ambiguous | 0.1 | 0.96 | Ambiguous | 0.79 | Ambiguous | 1.52 | Destabilizing | 0.417 | Likely Benign | -9.55 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.53 | Benign | 0.12 | Tolerated | 0.4262 | 0.1415 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.1295G>T | C432F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C432F is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact; AlphaMissense‑Optimized and premPS are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the balance of evidence favors a pathogenic classification, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.362533 | Uncertain | 0.960 | 0.285 | 0.000 | -12.862 | Likely Pathogenic | 0.943 | Likely Pathogenic | Ambiguous | -0.44 | Likely Benign | 0.2 | -0.34 | Likely Benign | -0.39 | Likely Benign | 0.57 | Ambiguous | 0.434 | Likely Benign | -10.50 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.46 | Benign | 0.01 | Affected | 0.1090 | 0.3956 | -4 | -2 | 0.3 | 44.04 | |||||||||||||||||||||||||||||
| c.1296T>G | C432W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C432W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments reinforce this: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus indicates a likely pathogenic status, and Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result, providing no counter‑evidence. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a pathogenic effect. This conclusion is consistent with the absence of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.362533 | Uncertain | 0.960 | 0.285 | 0.000 | -13.936 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.17 | Ambiguous | 0.4 | 0.72 | Ambiguous | 0.95 | Ambiguous | 0.58 | Ambiguous | 0.433 | Likely Benign | -10.50 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 3.43 | Benign | 0.00 | Affected | 0.1217 | 0.3246 | -8 | -2 | -3.4 | 83.07 | |||||||||||||||||||||||||||||
| c.1297G>A | A433T 2D AIThe SynGAP1 A433T missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only two tools—polyPhen‑2 HumDiv and polyPhen‑2 HumVar—predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.098513 | Structured | 0.352258 | Uncertain | 0.938 | 0.302 | 0.000 | -5.499 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.35 | Likely Benign | 0.5 | -0.02 | Likely Benign | 0.17 | Likely Benign | 0.40 | Likely Benign | 0.088 | Likely Benign | -1.41 | Neutral | 0.964 | Probably Damaging | 0.481 | Possibly Damaging | 3.41 | Benign | 0.08 | Tolerated | 0.0891 | 0.5554 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.1297G>C | A433P 2D 3DClick to see structure in 3D Viewer AISynGAP1 A433P is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, SIFT, and FATHMM. Those that predict pathogenicity comprise FoldX, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and ESM1b. The remaining tools, premPS and AlphaMissense‑Optimized, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of evidence points to a pathogenic effect, which is consistent with the lack of ClinVar annotation and gnomAD absence. Therefore, the variant is most likely pathogenic, and this prediction does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.352258 | Uncertain | 0.938 | 0.302 | 0.000 | -9.887 | Likely Pathogenic | 0.883 | Likely Pathogenic | Ambiguous | 2.48 | Destabilizing | 0.0 | 7.09 | Destabilizing | 4.79 | Destabilizing | 0.55 | Ambiguous | 0.217 | Likely Benign | -2.64 | Deleterious | 0.998 | Probably Damaging | 0.820 | Possibly Damaging | 3.37 | Benign | 0.07 | Tolerated | 0.1471 | 0.4150 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1297G>T | A433S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A433S is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign or likely benign. Only polyPhen‑2 (HumDiv) predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized reports benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a likely benign classification; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Overall, the preponderance of evidence points to a benign impact for A433S, and this conclusion is consistent with the lack of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.098513 | Structured | 0.352258 | Uncertain | 0.938 | 0.302 | 0.000 | -3.861 | Likely Benign | 0.077 | Likely Benign | Likely Benign | -0.04 | Likely Benign | 0.0 | 0.41 | Likely Benign | 0.19 | Likely Benign | -0.21 | Likely Benign | 0.077 | Likely Benign | 0.35 | Neutral | 0.597 | Possibly Damaging | 0.391 | Benign | 3.46 | Benign | 0.28 | Tolerated | 0.1938 | 0.3975 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1298C>A | A433E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A433E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Two tools (premPS and AlphaMissense‑Default) give uncertain results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign prediction; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports benign. No prediction is missing or inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.098513 | Structured | 0.352258 | Uncertain | 0.938 | 0.302 | 0.000 | -8.210 | Likely Pathogenic | 0.506 | Ambiguous | Likely Benign | -0.20 | Likely Benign | 0.0 | 0.15 | Likely Benign | -0.03 | Likely Benign | 0.58 | Ambiguous | 0.148 | Likely Benign | -1.65 | Neutral | 0.998 | Probably Damaging | 0.824 | Possibly Damaging | 3.42 | Benign | 0.18 | Tolerated | 0.0783 | 0.1695 | 0 | -1 | -5.3 | 58.04 | ||||||||||||||||||||||||||||||
| c.1298C>G | A433G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A433G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach a consensus all indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as tolerated. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. The protein‑folding stability method Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result and is therefore treated as unavailable evidence. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.098513 | Structured | 0.352258 | Uncertain | 0.938 | 0.302 | 0.000 | -5.044 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 0.59 | Ambiguous | 0.0 | 1.21 | Ambiguous | 0.90 | Ambiguous | 0.70 | Ambiguous | 0.038 | Likely Benign | -1.54 | Neutral | 0.035 | Benign | 0.014 | Benign | 3.38 | Benign | 0.15 | Tolerated | 0.1629 | 0.2919 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1300G>C | V434L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Two tools, Rosetta and AlphaMissense‑Default, give uncertain results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign consensus; and Foldetta, a protein‑folding stability method, also predicts benign. No prediction or stability result is missing or inconclusive. Overall, the preponderance of evidence indicates that V434L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | -8.697 | Likely Pathogenic | 0.477 | Ambiguous | Likely Benign | -0.05 | Likely Benign | 0.0 | 1.02 | Ambiguous | 0.49 | Likely Benign | 0.20 | Likely Benign | 0.225 | Likely Benign | -2.30 | Neutral | 0.947 | Possibly Damaging | 0.851 | Possibly Damaging | 3.64 | Benign | 0.27 | Tolerated | 0.0895 | 0.3813 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||
| c.1300G>T | V434F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434F is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that indicate a benign effect include REVEL, premPS, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; SGM‑Consensus predicts pathogenic; Foldetta predicts pathogenic. All predictions are available and none are inconclusive. Based on the overall distribution of predictions, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | -12.553 | Likely Pathogenic | 0.672 | Likely Pathogenic | Likely Benign | 3.64 | Destabilizing | 0.1 | 3.27 | Destabilizing | 3.46 | Destabilizing | 0.27 | Likely Benign | 0.417 | Likely Benign | -3.93 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.44 | Benign | 0.04 | Affected | 0.0596 | 0.3391 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1301T>A | V434D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign are SIFT and FATHMM; all other evaluated algorithms—including REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, and ESM1b—predict it to be pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized returns a pathogenic score; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among pathogenic predictors and the corroborating high‑accuracy tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | -14.765 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 3.26 | Destabilizing | 0.0 | 3.33 | Destabilizing | 3.30 | Destabilizing | 1.78 | Destabilizing | 0.592 | Likely Pathogenic | -5.18 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.38 | Benign | 0.13 | Tolerated | 0.1307 | 0.0741 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1301T>C | V434A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V434A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and ESM1b; FoldX and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as pathogenic. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | -8.531 | Likely Pathogenic | 0.441 | Ambiguous | Likely Benign | 1.72 | Ambiguous | 0.0 | 2.51 | Destabilizing | 2.12 | Destabilizing | 1.00 | Destabilizing | 0.238 | Likely Benign | -2.51 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.58 | Benign | 0.75 | Tolerated | 0.2338 | 0.2292 | 0 | 0 | -2.4 | -28.05 | ||||||||||||||||||||||||||||||
| c.1301T>G | V434G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are SIFT and FATHMM, while the remaining tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default) all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as pathogenic. Taken together, the overwhelming majority of evidence indicates a deleterious effect. Therefore, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | -12.519 | Likely Pathogenic | 0.809 | Likely Pathogenic | Ambiguous | 3.08 | Destabilizing | 0.0 | 4.37 | Destabilizing | 3.73 | Destabilizing | 1.91 | Destabilizing | 0.564 | Likely Pathogenic | -5.16 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.39 | Benign | 0.07 | Tolerated | 0.1758 | 0.2408 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1303T>A | L435M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L435M missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions come from premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Uncertain results are reported by Rosetta, Foldetta, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign effect; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.229226 | Structured | 0.333584 | Uncertain | 0.954 | 0.292 | 0.000 | -4.705 | Likely Benign | 0.534 | Ambiguous | Likely Benign | 0.24 | Likely Benign | 0.0 | 1.02 | Ambiguous | 0.63 | Ambiguous | 1.05 | Destabilizing | 0.233 | Likely Benign | -1.74 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.17 | Benign | 0.01 | Affected | 0.0675 | 0.2753 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.1303T>G | L435V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L435V has no ClinVar assertion and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and AlphaMissense‑Default. ESM1b is uncertain. High‑accuracy assessments further support a pathogenic bias: AlphaMissense‑Optimized predicts benign, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic)—favors pathogenicity. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. With the preponderance of pathogenic calls from both general and high‑accuracy tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.229226 | Structured | 0.333584 | Uncertain | 0.954 | 0.292 | 0.000 | -7.134 | In-Between | 0.571 | Likely Pathogenic | Likely Benign | 2.97 | Destabilizing | 0.1 | 2.33 | Destabilizing | 2.65 | Destabilizing | 1.36 | Destabilizing | 0.217 | Likely Benign | -2.80 | Deleterious | 0.995 | Probably Damaging | 0.970 | Probably Damaging | 3.23 | Benign | 0.06 | Tolerated | 0.1216 | 0.2628 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||
| c.1304T>C | L435S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L435S is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity largely agree: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. Only FATHMM predicts a benign outcome. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No predictions are missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.333584 | Uncertain | 0.954 | 0.292 | 0.000 | -12.662 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 4.18 | Destabilizing | 0.0 | 4.09 | Destabilizing | 4.14 | Destabilizing | 1.83 | Destabilizing | 0.596 | Likely Pathogenic | -5.63 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.18 | Benign | 0.01 | Affected | 0.2705 | 0.0505 | -3 | -2 | -4.6 | -26.08 | |||||||||||||||||||||||||||||
| c.1305G>C | L435F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L435F is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote). High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also uncertain, providing no definitive evidence for either outcome. Given the preponderance of pathogenic predictions and the lack of any ClinVar annotation to contradict this assessment, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.333584 | Uncertain | 0.954 | 0.292 | 0.000 | -11.871 | Likely Pathogenic | 0.932 | Likely Pathogenic | Ambiguous | 0.51 | Ambiguous | 0.1 | 0.95 | Ambiguous | 0.73 | Ambiguous | 0.69 | Ambiguous | 0.222 | Likely Benign | -3.75 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | 3.26 | Benign | 0.01 | Affected | 0.0587 | 0.2723 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1305G>T | L435F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L435F is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (Likely Pathogenic). High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also uncertain, providing no definitive evidence for either outcome. Given the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.333584 | Uncertain | 0.954 | 0.292 | 0.000 | -11.871 | Likely Pathogenic | 0.932 | Likely Pathogenic | Ambiguous | 0.51 | Ambiguous | 0.1 | 0.95 | Ambiguous | 0.73 | Ambiguous | 0.69 | Ambiguous | 0.222 | Likely Benign | -3.75 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | 3.26 | Benign | 0.01 | Affected | 0.0587 | 0.2723 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1306G>C | E436Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E436Q is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include premPS and FATHMM, while the majority of tools predict a pathogenic outcome: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Tools with uncertain or inconclusive results are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the consensus of pathogenic predictions outweighs benign ones, and the high‑accuracy tools reinforce a pathogenic classification. Therefore, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.321046 | Uncertain | 0.934 | 0.289 | 0.000 | -12.413 | Likely Pathogenic | 0.952 | Likely Pathogenic | Ambiguous | 0.51 | Ambiguous | 0.1 | 1.58 | Ambiguous | 1.05 | Ambiguous | 0.50 | Likely Benign | 0.607 | Likely Pathogenic | -2.76 | Deleterious | 0.992 | Probably Damaging | 0.946 | Probably Damaging | 4.64 | Benign | 0.01 | Affected | 0.1237 | 0.5809 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1307A>C | E436A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E436A is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include Foldetta, premPS, and FATHMM, whereas the majority of algorithms—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. Two high‑accuracy predictors give concordant results: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a benign effect. No prediction is missing or inconclusive. Overall, the preponderance of evidence points to a pathogenic impact for E436A, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.321046 | Uncertain | 0.934 | 0.289 | 0.000 | -12.678 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 1.03 | Ambiguous | 0.0 | -0.88 | Ambiguous | 0.08 | Likely Benign | 0.23 | Likely Benign | 0.825 | Likely Pathogenic | -5.68 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 4.65 | Benign | 0.05 | Affected | 0.3901 | 0.5116 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1307A>G | E436G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E436G missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, while the majority of algorithms (SGM‑Consensus, REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; FoldX, premPS, and Foldetta are inconclusive. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta’s stability prediction is uncertain. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.321046 | Uncertain | 0.934 | 0.289 | 0.000 | -12.355 | Likely Pathogenic | 0.966 | Likely Pathogenic | Likely Pathogenic | 1.50 | Ambiguous | 0.1 | 2.02 | Destabilizing | 1.76 | Ambiguous | 0.58 | Ambiguous | 0.802 | Likely Pathogenic | -6.63 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 4.61 | Benign | 0.03 | Affected | 0.2910 | 0.4841 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1307A>T | E436V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E436V variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include premPS and FATHMM, while the remaining evaluated algorithms (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) all predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Based on the preponderance of pathogenic predictions and the high‑accuracy tools’ results, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.321046 | Uncertain | 0.934 | 0.289 | 0.000 | -13.364 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.45 | Ambiguous | 0.0 | 1.62 | Ambiguous | 1.54 | Ambiguous | 0.30 | Likely Benign | 0.875 | Likely Pathogenic | -6.63 | Deleterious | 0.995 | Probably Damaging | 0.967 | Probably Damaging | 4.64 | Benign | 0.03 | Affected | 0.0727 | 0.5952 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1308G>C | E436D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E436D missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions are limited to FATHMM, while the remaining nine tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. Four tools (FoldX, Rosetta, Foldetta, premPS) give uncertain results and are treated as unavailable. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized is pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also pathogenic (3 pathogenic vs. 1 benign). Foldetta’s stability prediction is uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.321046 | Uncertain | 0.934 | 0.289 | 0.000 | -10.355 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.55 | Ambiguous | 0.1 | 0.71 | Ambiguous | 0.63 | Ambiguous | 0.73 | Ambiguous | 0.554 | Likely Pathogenic | -2.85 | Deleterious | 0.986 | Probably Damaging | 0.921 | Probably Damaging | 4.61 | Benign | 0.01 | Affected | 0.1938 | 0.3553 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1308G>T | E436D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E436D missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions are limited to FATHMM, while the remaining nine tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity. Four tools (FoldX, Rosetta, Foldetta, premPS) give uncertain results and are treated as unavailable. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized is pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also pathogenic (3 pathogenic vs. 1 benign). Foldetta’s stability prediction is uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.239899 | Structured | 0.321046 | Uncertain | 0.934 | 0.289 | 0.000 | -10.355 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.55 | Ambiguous | 0.1 | 0.71 | Ambiguous | 0.63 | Ambiguous | 0.73 | Ambiguous | 0.554 | Likely Pathogenic | -2.85 | Deleterious | 0.986 | Probably Damaging | 0.921 | Probably Damaging | 4.61 | Benign | 0.01 | Affected | 0.1938 | 0.3553 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1309C>A | P437T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P437T missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, FATHMM, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The remaining tools—FoldX, Foldetta, premPS, and AlphaMissense‑Default—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicting pathogenic, and Foldetta yielding an uncertain stability change. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.306196 | Uncertain | 0.921 | 0.298 | 0.000 | -13.011 | Likely Pathogenic | 0.484 | Ambiguous | Likely Benign | 1.35 | Ambiguous | 0.0 | -3.46 | Stabilizing | -1.06 | Ambiguous | 0.54 | Ambiguous | 0.305 | Likely Benign | -6.67 | Deleterious | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 3.45 | Benign | 0.01 | Affected | 0.1659 | 0.5782 | 0 | -1 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||
| c.1309C>G | P437A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P437A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, Rosetta, FATHMM, AlphaMissense‑Optimized, and premPS. Those that predict pathogenicity are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Tools with uncertain or inconclusive results are AlphaMissense‑Default, FoldX, and Foldetta. High‑accuracy assessments give AlphaMissense‑Optimized a benign prediction. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default (uncertain), ESM1b (pathogenic), FATHMM (benign), and PROVEAN (pathogenic), yields a pathogenic consensus. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. Overall, the balance of evidence leans toward a pathogenic effect for P437A, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.306196 | Uncertain | 0.921 | 0.298 | 0.000 | -12.059 | Likely Pathogenic | 0.392 | Ambiguous | Likely Benign | 1.23 | Ambiguous | 0.0 | -3.14 | Stabilizing | -0.96 | Ambiguous | 0.50 | Likely Benign | 0.266 | Likely Benign | -6.53 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 3.51 | Benign | 0.03 | Affected | 0.3489 | 0.4775 | 1 | -1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||
| c.130T>A | W44R 2D  AIThe SynGAP1 W44R missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta results are unavailable. Overall, the majority of tools (seven pathogenic vs. three benign) indicate a pathogenic impact. Thus, the variant is most likely pathogenic, and this prediction does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -4.850 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.291 | Likely Benign | -5.06 | Deleterious | 0.943 | Possibly Damaging | 0.888 | Possibly Damaging | 3.16 | Benign | 0.00 | Affected | 0.4268 | 0.0749 | 2 | -3 | -3.6 | -30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.130T>C | W44R 2D AISynGAP1 W44R is not reported in ClinVar and is absent from gnomAD. Consensus from standard predictors shows a split: benign calls come from REVEL, ESM1b, and FATHMM, while pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessment focuses on AlphaMissense‑Optimized, which predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive, and Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -4.850 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.291 | Likely Benign | -5.06 | Deleterious | 0.943 | Possibly Damaging | 0.888 | Possibly Damaging | 3.16 | Benign | 0.00 | Affected | 0.4268 | 0.0749 | 2 | -3 | -3.6 | -30.03 | ||||||||||||||||||||||||||||||||||||||||
| c.130T>G | W44G 2D AIThe SynGAP1 missense variant W44G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, more tools predict pathogenicity (5) than benign (3), and no ClinVar evidence contradicts this assessment. Thus, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -4.658 | Likely Benign | 0.850 | Likely Pathogenic | Ambiguous | 0.323 | Likely Benign | -4.80 | Deleterious | 0.659 | Possibly Damaging | 0.693 | Possibly Damaging | 3.16 | Benign | 0.00 | Affected | 0.4198 | 0.2164 | -7 | -2 | 0.5 | -129.16 | ||||||||||||||||||||||||||||||||||||||||
| c.1310C>A | P437H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P437H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, and FATHMM, whereas those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Predictions that are inconclusive or unavailable are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts a likely pathogenic outcome, while AlphaMissense‑Optimized and Foldetta remain uncertain. Overall, the balance of evidence favors a pathogenic classification for P437H, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.306196 | Uncertain | 0.921 | 0.298 | 0.000 | -13.496 | Likely Pathogenic | 0.826 | Likely Pathogenic | Ambiguous | 1.19 | Ambiguous | 0.0 | -3.28 | Stabilizing | -1.05 | Ambiguous | 0.31 | Likely Benign | 0.343 | Likely Benign | -7.75 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 3.44 | Benign | 0.01 | Affected | 0.1819 | 0.4328 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||
| c.1310C>G | P437R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant P437R is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, and FATHMM, whereas pathogenic predictions are made by SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. With the majority of tools indicating pathogenicity and the high‑accuracy consensus leaning toward pathogenic, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.306196 | Uncertain | 0.921 | 0.298 | 0.000 | -13.094 | Likely Pathogenic | 0.879 | Likely Pathogenic | Ambiguous | 0.34 | Likely Benign | 0.1 | -3.52 | Stabilizing | -1.59 | Ambiguous | 0.61 | Ambiguous | 0.461 | Likely Benign | -7.72 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.51 | Benign | 0.03 | Affected | 0.1448 | 0.2846 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.1310C>T | P437L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P437L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. Predictions that are inconclusive or uncertain are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts a likely pathogenic outcome, while AlphaMissense‑Optimized and Foldetta are uncertain. Taken together, the majority of evidence points toward a pathogenic impact for P437L, and this conclusion does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.306196 | Uncertain | 0.921 | 0.298 | 0.000 | -13.554 | Likely Pathogenic | 0.787 | Likely Pathogenic | Ambiguous | 1.10 | Ambiguous | 0.0 | -3.52 | Stabilizing | -1.21 | Ambiguous | 0.35 | Likely Benign | 0.324 | Likely Benign | -8.48 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.67 | Benign | 0.04 | Affected | 0.2228 | 0.6354 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1312G>C | A438P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A438P missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas those that predict a pathogenic impact are FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv), SIFT, and AlphaMissense‑Default; premPS and ESM1b are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as pathogenic. Overall, six tools predict pathogenicity versus four predicting benign, and the high‑accuracy consensus is split, but the majority of evidence points toward a deleterious effect. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.147574 | Structured | 0.290154 | Uncertain | 0.929 | 0.293 | 0.000 | -7.955 | In-Between | 0.721 | Likely Pathogenic | Likely Benign | 2.21 | Destabilizing | 0.1 | 6.36 | Destabilizing | 4.29 | Destabilizing | 0.83 | Ambiguous | 0.158 | Likely Benign | -2.46 | Neutral | 0.815 | Possibly Damaging | 0.137 | Benign | 4.09 | Benign | 0.05 | Affected | 0.1624 | 0.4150 | 1 | -1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||
| c.1312G>T | A438S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A438S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree that the substitution is benign: REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as benign. The only inconclusive results come from Rosetta and premPS, which are listed as uncertain. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports benign. Overall, the evidence strongly supports a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.147574 | Structured | 0.290154 | Uncertain | 0.929 | 0.293 | 0.000 | -6.085 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.30 | Likely Benign | 0.0 | 0.62 | Ambiguous | 0.46 | Likely Benign | 0.68 | Ambiguous | 0.012 | Likely Benign | -1.27 | Neutral | 0.042 | Benign | 0.035 | Benign | 4.14 | Benign | 0.15 | Tolerated | 0.2143 | 0.3995 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1313C>A | A438D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A438D missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, polyPhen‑2 HumVar, and FATHMM; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default; the remaining tools (FoldX, Rosetta, Foldetta, premPS, ESM1b, AlphaMissense‑Optimized) are uncertain. High‑accuracy assessments indicate that AlphaMissense‑Optimized is inconclusive, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta is also inconclusive. Overall, the majority of evidence points toward a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Thus, based on the available predictions, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.147574 | Structured | 0.290154 | Uncertain | 0.929 | 0.293 | 0.000 | -7.410 | In-Between | 0.808 | Likely Pathogenic | Ambiguous | 1.60 | Ambiguous | 0.1 | 0.70 | Ambiguous | 1.15 | Ambiguous | 0.92 | Ambiguous | 0.175 | Likely Benign | -3.07 | Deleterious | 0.859 | Possibly Damaging | 0.124 | Benign | 4.10 | Benign | 0.04 | Affected | 0.1490 | 0.1941 | 0 | -2 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||
| c.1313C>G | A438G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A438G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only PROVEAN predicts a pathogenic outcome. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact for A438G, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.147574 | Structured | 0.290154 | Uncertain | 0.929 | 0.293 | 0.000 | -5.790 | Likely Benign | 0.182 | Likely Benign | Likely Benign | 1.08 | Ambiguous | 0.1 | 1.78 | Ambiguous | 1.43 | Ambiguous | 0.80 | Ambiguous | 0.034 | Likely Benign | -2.51 | Deleterious | 0.247 | Benign | 0.037 | Benign | 4.12 | Benign | 0.11 | Tolerated | 0.1788 | 0.2719 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1315C>G | L439V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L439V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar; premPS is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of predictions (8 benign vs. 5 pathogenic) favor a benign classification, and this consensus does not contradict the absence of ClinVar evidence. Thus, based on current computational predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.222385 | Structured | 0.281542 | Uncertain | 0.942 | 0.265 | 0.000 | -6.340 | Likely Benign | 0.279 | Likely Benign | Likely Benign | 2.34 | Destabilizing | 0.2 | 2.40 | Destabilizing | 2.37 | Destabilizing | 0.91 | Ambiguous | 0.121 | Likely Benign | -1.13 | Neutral | 0.976 | Probably Damaging | 0.941 | Probably Damaging | 3.36 | Benign | 0.12 | Tolerated | 0.1272 | 0.2628 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1316T>A | L439Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L439Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.281542 | Uncertain | 0.942 | 0.265 | 0.000 | -11.162 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | 2.50 | Destabilizing | 0.1 | 2.06 | Destabilizing | 2.28 | Destabilizing | 1.35 | Destabilizing | 0.575 | Likely Pathogenic | -5.15 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.21 | Benign | 0.01 | Affected | 0.1065 | 0.0488 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1316T>C | L439P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L439P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a deleterious effect: pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a benign outcome. High‑accuracy methods reinforce the pathogenic view: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.281542 | Uncertain | 0.942 | 0.265 | 0.000 | -9.929 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 6.30 | Destabilizing | 0.2 | 7.62 | Destabilizing | 6.96 | Destabilizing | 1.28 | Destabilizing | 0.539 | Likely Pathogenic | -5.72 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.21 | Benign | 0.02 | Affected | 0.3426 | 0.1192 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1316T>G | L439R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L439R lies in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining evaluated algorithms (SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic or likely pathogenic impact; FoldX is uncertain and is treated as unavailable. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.281542 | Uncertain | 0.942 | 0.265 | 0.000 | -12.870 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 1.90 | Ambiguous | 0.5 | 2.62 | Destabilizing | 2.26 | Destabilizing | 1.40 | Destabilizing | 0.554 | Likely Pathogenic | -5.25 | Deleterious | 0.996 | Probably Damaging | 0.983 | Probably Damaging | 3.22 | Benign | 0.01 | Affected | 0.1217 | 0.0488 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1318A>C | N440H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N440H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenicity is suggested only by polyPhen‑2 HumDiv and ESM1b, while FoldX, Rosetta, and Foldetta provide uncertain or inconclusive results. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta’s stability prediction is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.191378 | Structured | 0.267204 | Uncertain | 0.929 | 0.245 | 0.000 | -8.064 | Likely Pathogenic | 0.226 | Likely Benign | Likely Benign | 1.12 | Ambiguous | 0.1 | 0.83 | Ambiguous | 0.98 | Ambiguous | 0.00 | Likely Benign | 0.140 | Likely Benign | -2.48 | Neutral | 0.835 | Possibly Damaging | 0.217 | Benign | 3.40 | Benign | 0.19 | Tolerated | 0.0935 | 0.3270 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.1318A>G | N440D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N440D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome, while FoldX, Foldetta, and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.191378 | Structured | 0.267204 | Uncertain | 0.929 | 0.245 | 0.000 | -9.335 | Likely Pathogenic | 0.407 | Ambiguous | Likely Benign | -0.62 | Ambiguous | 0.0 | -0.41 | Likely Benign | -0.52 | Ambiguous | 0.47 | Likely Benign | 0.074 | Likely Benign | -1.71 | Neutral | 0.229 | Benign | 0.045 | Benign | 3.43 | Benign | 0.43 | Tolerated | 0.1544 | 0.2025 | 2 | 1 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.1318A>T | N440Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N440Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen2_HumDiv, AlphaMissense‑Default, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicating pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, yielding an uncertain stability change. Overall, the majority of predictions lean toward a benign interpretation, and this is consistent with the lack of ClinVar annotation. Therefore, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.267204 | Uncertain | 0.929 | 0.245 | 0.000 | -10.586 | Likely Pathogenic | 0.674 | Likely Pathogenic | Likely Benign | 0.81 | Ambiguous | 0.1 | 1.25 | Ambiguous | 1.03 | Ambiguous | 0.20 | Likely Benign | 0.135 | Likely Benign | -3.81 | Deleterious | 0.931 | Possibly Damaging | 0.230 | Benign | 3.43 | Benign | 0.07 | Tolerated | 0.0535 | 0.3624 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.1319A>C | N440T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N440T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly favor a benign effect: AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, REVEL, premPS, Rosetta, Foldetta, and the SGM‑Consensus (majority vote) all predict benign or likely benign. No tool predicts pathogenicity; the only inconclusive result is from FoldX, which is listed as uncertain. High‑accuracy methods corroborate the benign assessment: AlphaMissense‑Optimized is benign, the SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Therefore, based on the available predictions, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.191378 | Structured | 0.267204 | Uncertain | 0.929 | 0.245 | 0.000 | -5.371 | Likely Benign | 0.143 | Likely Benign | Likely Benign | 0.58 | Ambiguous | 0.0 | 0.16 | Likely Benign | 0.37 | Likely Benign | 0.11 | Likely Benign | 0.079 | Likely Benign | -1.27 | Neutral | 0.007 | Benign | 0.005 | Benign | 3.48 | Benign | 0.14 | Tolerated | 0.0969 | 0.3341 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.1319A>G | N440S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N440S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: SGM‑Consensus, REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign or likely benign. Only FoldX and premPS returned uncertain results, which are treated as unavailable. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is benign. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.191378 | Structured | 0.267204 | Uncertain | 0.929 | 0.245 | 0.000 | -1.753 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.52 | Ambiguous | 0.1 | 0.08 | Likely Benign | 0.30 | Likely Benign | -0.50 | Ambiguous | 0.104 | Likely Benign | 1.15 | Neutral | 0.001 | Benign | 0.000 | Benign | 3.53 | Benign | 0.92 | Tolerated | 0.2024 | 0.3556 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||
| c.1319A>T | N440I 2D AISynGAP1 missense variant N440I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. Uncertain results are reported by FoldX, Rosetta, and Foldetta. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as likely pathogenic; Foldetta remains inconclusive. Overall, the balance of evidence favors a pathogenic interpretation, and this conclusion does not contradict the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.267204 | Uncertain | 0.929 | 0.245 | 0.000 | -10.365 | Likely Pathogenic | 0.778 | Likely Pathogenic | Likely Benign | 0.97 | Ambiguous | 0.9 | 1.10 | Ambiguous | 1.04 | Ambiguous | 0.10 | Likely Benign | 0.100 | Likely Benign | -4.07 | Deleterious | 0.322 | Benign | 0.109 | Benign | 3.47 | Benign | 0.03 | Affected | 0.0554 | 0.3772 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.131G>C | W44S 2D AIThe SynGAP1 missense variant W44S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default) predict a pathogenic impact; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, the balance of evidence favors a pathogenic classification, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -5.113 | Likely Benign | 0.921 | Likely Pathogenic | Ambiguous | 0.275 | Likely Benign | -4.68 | Deleterious | 0.824 | Possibly Damaging | 0.775 | Possibly Damaging | 3.16 | Benign | 0.00 | Affected | 0.4139 | 0.2371 | -2 | -3 | 0.1 | -99.14 | ||||||||||||||||||||||||||||||||||||||||
| c.131G>T | W44L 2D AIThe SynGAP1 missense variant W44L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Consensus from standard predictors shows a split: benign calls come from REVEL, ESM1b, and FATHMM, while pathogenic calls come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized returns an uncertain result. High‑accuracy assessment is inconclusive: the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is a 2‑vs‑2 tie, and Foldetta stability analysis is unavailable. Overall, the balance of evidence favors a pathogenic interpretation, with no conflict with ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -5.743 | Likely Benign | 0.869 | Likely Pathogenic | Ambiguous | 0.211 | Likely Benign | -4.37 | Deleterious | 0.659 | Possibly Damaging | 0.693 | Possibly Damaging | 3.20 | Benign | 0.00 | Affected | 0.2584 | 0.3413 | -2 | -2 | 4.7 | -73.05 | ||||||||||||||||||||||||||||||||||||||||
| c.1320T>A | N440K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N440K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Tools that predict a pathogenic effect are ESM1b and AlphaMissense‑Default. The remaining methods—FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized—yield uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments are likewise inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a 2‑vs‑2 tie, and Foldetta is uncertain. Based on the overall distribution of predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.191378 | Structured | 0.267204 | Uncertain | 0.929 | 0.245 | 0.000 | -10.114 | Likely Pathogenic | 0.895 | Likely Pathogenic | Ambiguous | 0.92 | Ambiguous | 0.1 | 1.04 | Ambiguous | 0.98 | Ambiguous | 0.40 | Likely Benign | 0.058 | Likely Benign | -1.97 | Neutral | 0.206 | Benign | 0.021 | Benign | 3.50 | Benign | 0.19 | Tolerated | 0.1660 | 0.2550 | 1 | 0 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||
| c.1320T>G | N440K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N440K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Tools that predict a pathogenic effect are ESM1b and AlphaMissense‑Default. The remaining methods—FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized—yield uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments are likewise inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a 2‑vs‑2 tie, and Foldetta is uncertain. Based on the overall distribution of predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.191378 | Structured | 0.267204 | Uncertain | 0.929 | 0.245 | 0.000 | -10.114 | Likely Pathogenic | 0.895 | Likely Pathogenic | Ambiguous | 0.92 | Ambiguous | 0.1 | 1.04 | Ambiguous | 0.98 | Ambiguous | 0.40 | Likely Benign | 0.057 | Likely Benign | -1.97 | Neutral | 0.206 | Benign | 0.021 | Benign | 3.50 | Benign | 0.19 | Tolerated | 0.1660 | 0.2550 | 1 | 0 | -0.4 | 14.07 | ||||||||||||||||||||||||||||||
| c.1321G>A | V441I 2D AIThe SynGAP1 missense variant V441I is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign, while only ESM1b predicts it as pathogenic. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized reports benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates benign stability. No contradictory evidence is present. Based on the collective predictions, the variant is most likely benign, and this conclusion does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -8.773 | Likely Pathogenic | 0.122 | Likely Benign | Likely Benign | -0.24 | Likely Benign | 0.3 | 0.11 | Likely Benign | -0.07 | Likely Benign | 0.25 | Likely Benign | 0.135 | Likely Benign | -0.82 | Neutral | 0.287 | Benign | 0.038 | Benign | 3.41 | Benign | 0.16 | Tolerated | 0.0694 | 0.3412 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||
| c.1321G>C | V441L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V441L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome, while Rosetta and AlphaMissense‑Default are uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. No prediction or folding result is missing or inconclusive. Overall, the variant is most likely benign based on the preponderance of evidence, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -9.546 | Likely Pathogenic | 0.395 | Ambiguous | Likely Benign | -0.43 | Likely Benign | 0.0 | 0.59 | Ambiguous | 0.08 | Likely Benign | 0.45 | Likely Benign | 0.135 | Likely Benign | -2.27 | Neutral | 0.165 | Benign | 0.028 | Benign | 3.43 | Benign | 0.11 | Tolerated | 0.0961 | 0.3881 | 2 | 1 | -0.4 | 14.03 | ||||||||||||||||||||||||||||||
| c.1321G>T | V441F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V441F missense variant is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools lean toward a benign interpretation, and the high‑accuracy predictions are split but favor benign. Thus, the variant is most likely benign based on current computational evidence, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -13.519 | Likely Pathogenic | 0.653 | Likely Pathogenic | Likely Benign | -0.26 | Likely Benign | 0.0 | 0.32 | Likely Benign | 0.03 | Likely Benign | 0.54 | Ambiguous | 0.355 | Likely Benign | -4.22 | Deleterious | 0.992 | Probably Damaging | 0.658 | Possibly Damaging | 3.37 | Benign | 0.08 | Tolerated | 0.0670 | 0.3269 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1322T>A | V441D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V441D is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and Foldetta, whereas a majority of tools (SGM Consensus, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. FoldX and Rosetta are inconclusive, and AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show that the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenicity, while Foldetta predicts benign stability. Overall, the balance of evidence leans toward pathogenicity, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -15.392 | Likely Pathogenic | 0.934 | Likely Pathogenic | Ambiguous | -0.57 | Ambiguous | 0.1 | 0.56 | Ambiguous | -0.01 | Likely Benign | 1.15 | Destabilizing | 0.308 | Likely Benign | -6.07 | Deleterious | 1.000 | Probably Damaging | 0.959 | Probably Damaging | 3.38 | Benign | 0.10 | Tolerated | 0.1232 | 0.0698 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1322T>G | V441G 2D 3DClick to see structure in 3D Viewer AISynGAP1 variant V441G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into three groups: benign predictions come from REVEL, FoldX, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; the remaining tools (Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized remains benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) favors pathogenic, and Foldetta is inconclusive. Taken together, the majority of evidence points to a benign effect, but the SGM Consensus and several individual pathogenic predictors introduce uncertainty. Therefore, the variant is most likely benign based on the overall prediction landscape, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -12.380 | Likely Pathogenic | 0.478 | Ambiguous | Likely Benign | 0.18 | Likely Benign | 0.0 | 1.25 | Ambiguous | 0.72 | Ambiguous | 0.95 | Ambiguous | 0.273 | Likely Benign | -5.88 | Deleterious | 0.841 | Possibly Damaging | 0.997 | Probably Damaging | 3.41 | Benign | 0.24 | Tolerated | 0.1664 | 0.1715 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1324A>C | K442Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K442Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.170161 | Structured | 0.255766 | Uncertain | 0.912 | 0.225 | 0.000 | -10.410 | Likely Pathogenic | 0.562 | Ambiguous | Likely Benign | 0.05 | Likely Benign | 0.1 | 0.03 | Likely Benign | 0.04 | Likely Benign | 0.25 | Likely Benign | 0.268 | Likely Benign | -3.10 | Deleterious | 0.998 | Probably Damaging | 0.995 | Probably Damaging | 3.39 | Benign | 0.18 | Tolerated | 0.3353 | 0.1014 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||
| c.1324A>G | K442E 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K442E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split consensus: benign predictions come from REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments give mixed results: AlphaMissense‑Optimized remains uncertain; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. No single evidence decisively outweighs the others. Therefore, the variant’s pathogenicity remains uncertain; the predictions do not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.170161 | Structured | 0.255766 | Uncertain | 0.912 | 0.225 | 0.000 | -12.813 | Likely Pathogenic | 0.939 | Likely Pathogenic | Ambiguous | -0.05 | Likely Benign | 0.1 | -0.21 | Likely Benign | -0.13 | Likely Benign | 0.24 | Likely Benign | 0.324 | Likely Benign | -3.34 | Deleterious | 0.997 | Probably Damaging | 0.966 | Probably Damaging | 3.47 | Benign | 0.16 | Tolerated | 0.2943 | 0.0789 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1325A>C | K442T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K442T missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM. Those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) indicates a benign stability change; AlphaMissense‑Optimized remains inconclusive. Overall, the predictions are evenly split, with no single consensus. Thus, the variant is most likely benign based on the majority of evidence, and this assessment does not contradict any ClinVar record (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.170161 | Structured | 0.255766 | Uncertain | 0.912 | 0.225 | 0.000 | -11.273 | Likely Pathogenic | 0.865 | Likely Pathogenic | Ambiguous | 0.30 | Likely Benign | 0.2 | 0.21 | Likely Benign | 0.26 | Likely Benign | 0.22 | Likely Benign | 0.330 | Likely Benign | -5.01 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 3.43 | Benign | 0.07 | Tolerated | 0.1510 | 0.2859 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1325A>T | K442I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K442I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy assessments are: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of predictions, including the two high‑accuracy pathogenic calls, indicate a pathogenic impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.170161 | Structured | 0.255766 | Uncertain | 0.912 | 0.225 | 0.000 | -14.921 | Likely Pathogenic | 0.965 | Likely Pathogenic | Likely Pathogenic | 0.16 | Likely Benign | 0.1 | -0.16 | Likely Benign | 0.00 | Likely Benign | 0.30 | Likely Benign | 0.350 | Likely Benign | -6.91 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.37 | Benign | 0.02 | Affected | 0.0943 | 0.3073 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1326A>C | K442N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K442N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, and the folding‑stability method Foldetta (which integrates FoldX‑MD and Rosetta outputs). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta as benign. Overall, the majority of predictions lean toward pathogenicity, but the presence of several strong benign predictions and a benign folding‑stability result introduces uncertainty. Based on the current computational evidence, the variant is most likely pathogenic, and this does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.170161 | Structured | 0.255766 | Uncertain | 0.912 | 0.225 | 0.000 | -11.039 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.37 | Likely Benign | 0.1 | 0.53 | Ambiguous | 0.45 | Likely Benign | 0.31 | Likely Benign | 0.161 | Likely Benign | -4.05 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.46 | Benign | 0.10 | Tolerated | 0.2686 | 0.1314 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1326A>T | K442N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K442N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, and the folding‑stability method Foldetta (which integrates FoldX‑MD and Rosetta outputs). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta as benign. Overall, the majority of predictions lean toward pathogenicity, but the presence of several strong benign predictions and a benign folding‑stability result introduces uncertainty. Based on the current computational evidence, the variant is most likely pathogenic, and this does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.170161 | Structured | 0.255766 | Uncertain | 0.912 | 0.225 | 0.000 | -11.039 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.37 | Likely Benign | 0.1 | 0.53 | Ambiguous | 0.45 | Likely Benign | 0.31 | Likely Benign | 0.161 | Likely Benign | -4.05 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.46 | Benign | 0.10 | Tolerated | 0.2686 | 0.1314 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1327G>A | G443S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree that the substitution is benign: REVEL, FoldX, PROVEAN, polyPhen‑2 (both HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as benign. Only Rosetta and premPS yield uncertain results, which are treated as unavailable. Grouping by consensus, the benign‑predicting tools outnumber any pathogenic calls (none). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports benign. Therefore, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -1.258 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.25 | Likely Benign | 0.1 | -0.72 | Ambiguous | -0.24 | Likely Benign | -0.65 | Ambiguous | 0.087 | Likely Benign | 0.40 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.51 | Benign | 0.34 | Tolerated | 0.2409 | 0.3416 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1327G>C | G443R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM all classify it as benign. Only two tools predict pathogenicity: polyPhen‑2 HumDiv and AlphaMissense‑Default. Predictions that are inconclusive—FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized—are treated as unavailable. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments are: AlphaMissense‑Optimized (uncertain), SGM Consensus (likely benign), and Foldetta (uncertain). Overall, the preponderance of evidence points to a benign impact for G443R, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -6.954 | Likely Benign | 0.886 | Likely Pathogenic | Ambiguous | -0.88 | Ambiguous | 0.3 | -1.19 | Ambiguous | -1.04 | Ambiguous | 0.28 | Likely Benign | 0.132 | Likely Benign | -1.49 | Neutral | 0.832 | Possibly Damaging | 0.286 | Benign | 3.40 | Benign | 0.21 | Tolerated | 0.0934 | 0.3197 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1327G>T | G443C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The remaining tools are inconclusive: Foldetta is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta remains uncertain. Overall, the consensus of the majority of evidence points to a benign impact for G443C, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -4.308 | Likely Benign | 0.208 | Likely Benign | Likely Benign | -0.06 | Likely Benign | 0.0 | -0.95 | Ambiguous | -0.51 | Ambiguous | -0.10 | Likely Benign | 0.260 | Likely Benign | -2.94 | Deleterious | 0.977 | Probably Damaging | 0.504 | Possibly Damaging | 3.37 | Benign | 0.16 | Tolerated | 0.1258 | 0.2782 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1328G>A | G443D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome. Uncertain results come from Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, and Foldetta as inconclusive. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and gnomAD presence, so there is no contradiction with existing clinical data. The variant is most likely benign based on the current predictive landscape. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -6.818 | Likely Benign | 0.761 | Likely Pathogenic | Likely Benign | -0.19 | Likely Benign | 0.2 | -1.72 | Ambiguous | -0.96 | Ambiguous | 0.32 | Likely Benign | 0.072 | Likely Benign | -0.50 | Neutral | 0.345 | Benign | 0.072 | Benign | 3.63 | Benign | 0.43 | Tolerated | 0.1687 | 0.2090 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.1328G>C | G443A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that reach a consensus all indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as tolerated. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. The protein‑folding stability predictor Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result and is therefore treated as unavailable evidence. Overall, the collective predictions strongly suggest that the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -0.332 | Likely Benign | 0.101 | Likely Benign | Likely Benign | -0.59 | Ambiguous | 0.1 | -1.61 | Ambiguous | -1.10 | Ambiguous | -0.52 | Ambiguous | 0.033 | Likely Benign | -1.04 | Neutral | 0.022 | Benign | 0.011 | Benign | 3.41 | Benign | 0.54 | Tolerated | 0.3539 | 0.3192 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1328G>T | G443V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Overall, the majority of evidence points to a benign impact for G443V, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -4.130 | Likely Benign | 0.274 | Likely Benign | Likely Benign | 0.17 | Likely Benign | 0.2 | -2.19 | Stabilizing | -1.01 | Ambiguous | 0.21 | Likely Benign | 0.099 | Likely Benign | -2.90 | Deleterious | 0.585 | Possibly Damaging | 0.195 | Benign | 3.36 | Benign | 0.12 | Tolerated | 0.1089 | 0.3375 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.132G>C | W44C 2D AIThe SynGAP1 missense variant W44C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the majority of evaluated tools (seven pathogenic vs. three benign) indicate a pathogenic impact. Thus, the variant is most likely pathogenic, and this prediction does not contradict any ClinVar status because the variant has not yet been classified in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -6.216 | Likely Benign | 0.975 | Likely Pathogenic | Likely Pathogenic | 0.321 | Likely Benign | -4.70 | Deleterious | 0.943 | Possibly Damaging | 0.941 | Probably Damaging | 3.14 | Benign | 0.00 | Affected | 0.3542 | 0.2370 | -8 | -2 | 3.4 | -83.07 | ||||||||||||||||||||||||||||||||||||||||
| c.132G>T | W44C 2D AIThe SynGAP1 missense variant W44C is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic vs. two benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated tools (seven pathogenic vs. three benign) indicate a pathogenic effect. This prediction is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.301917 | Structured | 0.431379 | Uncertain | 0.377 | 0.748 | 0.375 | -6.216 | Likely Benign | 0.975 | Likely Pathogenic | Likely Pathogenic | 0.321 | Likely Benign | -4.70 | Deleterious | 0.943 | Possibly Damaging | 0.941 | Probably Damaging | 3.14 | Benign | 0.00 | Affected | 0.3542 | 0.2370 | -8 | -2 | 3.4 | -83.07 | ||||||||||||||||||||||||||||||||||||||||
| c.1330A>C | K444Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 K444Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, SIFT, and FATHMM; pathogenic predictions from premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (majority vote) is pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for K444Q, and this conclusion does not conflict with any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.203355 | Structured | 0.262172 | Uncertain | 0.955 | 0.213 | 0.000 | -12.876 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.34 | Ambiguous | 0.0 | 1.36 | Ambiguous | 1.35 | Ambiguous | 1.04 | Destabilizing | 0.382 | Likely Benign | -3.82 | Deleterious | 0.998 | Probably Damaging | 0.997 | Probably Damaging | 3.43 | Benign | 0.07 | Tolerated | 0.4112 | 0.1057 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1330A>G | K444E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K444E missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and FATHMM; all other evaluated predictors—including FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a pathogenic effect. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.203355 | Structured | 0.262172 | Uncertain | 0.955 | 0.213 | 0.000 | -15.571 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.99 | Destabilizing | 0.1 | 3.75 | Destabilizing | 3.37 | Destabilizing | 1.10 | Destabilizing | 0.437 | Likely Benign | -3.82 | Deleterious | 0.997 | Probably Damaging | 0.981 | Probably Damaging | 3.40 | Benign | 0.01 | Affected | 0.3486 | 0.0793 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1331A>C | K444T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K444T is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are REVEL and FATHMM. Those that predict a pathogenic effect include FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools with uncertain or inconclusive results are Foldetta, Rosetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain (treated as unavailable for pathogenicity inference). Overall, the majority of reliable predictions indicate a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.203355 | Structured | 0.262172 | Uncertain | 0.955 | 0.213 | 0.000 | -15.557 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 2.12 | Destabilizing | 0.1 | 1.17 | Ambiguous | 1.65 | Ambiguous | 0.96 | Ambiguous | 0.442 | Likely Benign | -5.73 | Deleterious | 0.999 | Probably Damaging | 1.000 | Probably Damaging | 3.45 | Benign | 0.01 | Affected | 0.1643 | 0.3416 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1331A>G | K444R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K444R is not reported in ClinVar and has no gnomAD allele. Prediction tools cluster into benign (REVEL, FoldX, SIFT, FATHMM, AlphaMissense‑Optimized) and pathogenic (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default). Two tools (Rosetta, premPS) give uncertain results. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta predicts benign. No prediction or folding stability result is missing. Overall, six tools favor pathogenicity while five favor benignity, and the high‑accuracy consensus leans toward benign. Thus the variant is most likely pathogenic based on the broader set of predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.203355 | Structured | 0.262172 | Uncertain | 0.955 | 0.213 | 0.000 | -10.753 | Likely Pathogenic | 0.693 | Likely Pathogenic | Likely Benign | -0.12 | Likely Benign | 0.1 | -0.61 | Ambiguous | -0.37 | Likely Benign | 0.77 | Ambiguous | 0.213 | Likely Benign | -2.86 | Deleterious | 0.972 | Probably Damaging | 0.926 | Probably Damaging | 3.45 | Benign | 0.12 | Tolerated | 0.4597 | 0.0972 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.1331A>T | K444M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K444M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions are returned by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further highlight the discrepancy: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a benign effect. Overall, the majority of tools lean toward a pathogenic interpretation, and this is not contradicted by any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.203355 | Structured | 0.262172 | Uncertain | 0.955 | 0.213 | 0.000 | -14.223 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.39 | Likely Benign | 0.1 | -0.47 | Likely Benign | -0.04 | Likely Benign | 0.33 | Likely Benign | 0.442 | Likely Benign | -5.73 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.41 | Benign | 0.00 | Affected | 0.0934 | 0.3890 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1332G>C | K444N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K444N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the majority of tools (FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict a pathogenic impact. The high‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic; Foldetta’s stability assessment is uncertain and therefore not used as evidence. Overall, the preponderance of evidence points to a pathogenic effect for K444N. This conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.203355 | Structured | 0.262172 | Uncertain | 0.955 | 0.213 | 0.000 | -14.797 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.41 | Destabilizing | 0.0 | 1.52 | Ambiguous | 1.97 | Ambiguous | 1.18 | Destabilizing | 0.286 | Likely Benign | -4.77 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.39 | Benign | 0.01 | Affected | 0.3282 | 0.1213 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1332G>T | K444N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K444N is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are REVEL and FATHMM. Tools that predict a pathogenic effect include FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain predictions come from Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact. There is no ClinVar annotation to contradict this assessment, so the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.203355 | Structured | 0.262172 | Uncertain | 0.955 | 0.213 | 0.000 | -14.797 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.41 | Destabilizing | 0.0 | 1.52 | Ambiguous | 1.97 | Ambiguous | 1.18 | Destabilizing | 0.286 | Likely Benign | -4.77 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.39 | Benign | 0.01 | Affected | 0.3282 | 0.1213 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1333G>A | E445K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E445K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from FoldX, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic calls arise from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. When high‑accuracy methods are considered separately, AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of evidence—including the high‑accuracy tools—supports a pathogenic effect. This conclusion does not conflict with ClinVar, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.270205 | Uncertain | 0.947 | 0.228 | 0.000 | -15.371 | Likely Pathogenic | 0.958 | Likely Pathogenic | Likely Pathogenic | 0.02 | Likely Benign | 0.0 | 0.02 | Likely Benign | 0.02 | Likely Benign | 0.46 | Likely Benign | 0.524 | Likely Pathogenic | -3.82 | Deleterious | 0.991 | Probably Damaging | 0.951 | Probably Damaging | 3.35 | Benign | 0.02 | Affected | 0.1784 | 0.5311 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1334A>C | E445A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E445A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and FATHMM. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as Benign. Overall, the majority of predictions lean toward pathogenicity, and the high‑accuracy consensus supports a likely pathogenic classification. Thus, the variant is most likely pathogenic, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.270205 | Uncertain | 0.947 | 0.228 | 0.000 | -12.659 | Likely Pathogenic | 0.849 | Likely Pathogenic | Ambiguous | 0.09 | Likely Benign | 0.0 | -0.34 | Likely Benign | -0.13 | Likely Benign | 0.56 | Ambiguous | 0.476 | Likely Benign | -5.73 | Deleterious | 0.997 | Probably Damaging | 0.991 | Probably Damaging | 3.34 | Benign | 0.01 | Affected | 0.2846 | 0.4876 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1334A>G | E445G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E445G missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, while the majority of tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic impact. Predictions that are inconclusive or uncertain are FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E445G, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.270205 | Uncertain | 0.947 | 0.228 | 0.000 | -12.294 | Likely Pathogenic | 0.823 | Likely Pathogenic | Ambiguous | 0.80 | Ambiguous | 0.1 | 0.79 | Ambiguous | 0.80 | Ambiguous | 0.69 | Ambiguous | 0.523 | Likely Pathogenic | -6.68 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.39 | Benign | 0.01 | Affected | 0.2446 | 0.4202 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1334A>T | E445V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E445V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Foldetta, premPS, and FATHMM, whereas a larger group predicts pathogenicity: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Two tools give uncertain results: Rosetta and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of evidence points toward a pathogenic effect. The variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.270205 | Uncertain | 0.947 | 0.228 | 0.000 | -14.830 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 0.34 | Likely Benign | 0.1 | -0.67 | Ambiguous | -0.17 | Likely Benign | 0.32 | Likely Benign | 0.572 | Likely Pathogenic | -6.68 | Deleterious | 0.992 | Probably Damaging | 0.967 | Probably Damaging | 3.34 | Benign | 0.01 | Affected | 0.0414 | 0.5233 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1336G>C | E446Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E446Q missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No other folding‑stability predictions are available. Overall, the balance of evidence from the consensus of multiple in silico predictors points to a pathogenic classification for E446Q. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.276479 | Uncertain | 0.940 | 0.216 | 0.000 | -11.107 | Likely Pathogenic | 0.752 | Likely Pathogenic | Likely Benign | 0.92 | Ambiguous | 0.5 | 0.54 | Ambiguous | 0.73 | Ambiguous | 0.84 | Ambiguous | 0.337 | Likely Benign | -2.80 | Deleterious | 0.992 | Probably Damaging | 0.973 | Probably Damaging | 3.24 | Benign | 0.04 | Affected | 0.1049 | 0.6218 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1337A>C | E446A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E446A is not reported in ClinVar and is absent from gnomAD. In silico predictors that classify the variant as benign include REVEL, Rosetta, FATHMM, and AlphaMissense‑Optimized. Predictors that classify it as pathogenic comprise SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, yielded an inconclusive result and is treated as unavailable. Overall, the majority of predictions lean toward pathogenicity, and this conclusion is not contradicted by the absence of a ClinVar entry. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.276479 | Uncertain | 0.940 | 0.216 | 0.000 | -9.868 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | 1.66 | Ambiguous | 0.7 | 0.39 | Likely Benign | 1.03 | Ambiguous | 0.67 | Ambiguous | 0.443 | Likely Benign | -5.60 | Deleterious | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 3.28 | Benign | 0.01 | Affected | 0.3195 | 0.5877 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1337A>G | E446G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E446G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM. The majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and Foldetta. Predictions that are uncertain or inconclusive are AlphaMissense‑Optimized, Rosetta, and premPS. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is Pathogenic. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.276479 | Uncertain | 0.940 | 0.216 | 0.000 | -11.457 | Likely Pathogenic | 0.866 | Likely Pathogenic | Ambiguous | 2.62 | Destabilizing | 0.7 | 1.63 | Ambiguous | 2.13 | Destabilizing | 0.83 | Ambiguous | 0.510 | Likely Pathogenic | -6.42 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.24 | Benign | 0.00 | Affected | 0.2665 | 0.5202 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1337A>T | E446V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E446V missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta, premPS, and FATHMM, while those that agree on a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Predictions that are uncertain or inconclusive are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evaluated tools (8 pathogenic vs. 3 benign) indicate a pathogenic effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.276479 | Uncertain | 0.940 | 0.216 | 0.000 | -12.231 | Likely Pathogenic | 0.884 | Likely Pathogenic | Ambiguous | 1.72 | Ambiguous | 0.7 | 0.34 | Likely Benign | 1.03 | Ambiguous | 0.37 | Likely Benign | 0.513 | Likely Pathogenic | -6.55 | Deleterious | 0.995 | Probably Damaging | 0.983 | Probably Damaging | 3.19 | Benign | 0.00 | Affected | 0.0618 | 0.6433 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1338G>C | E446D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E446D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a damaging effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports it as Likely Benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, yields an uncertain result. Taken together, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.271506 | Structured | 0.276479 | Uncertain | 0.940 | 0.216 | 0.000 | -5.668 | Likely Benign | 0.562 | Ambiguous | Likely Benign | 1.65 | Ambiguous | 0.7 | 0.80 | Ambiguous | 1.23 | Ambiguous | 0.68 | Ambiguous | 0.171 | Likely Benign | -2.40 | Neutral | 0.986 | Probably Damaging | 0.960 | Probably Damaging | 3.30 | Benign | 0.18 | Tolerated | 0.1657 | 0.4275 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1338G>T | E446D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change E446D lies in the GAP domain. ClinVar has no entry for this variant and it is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Two tools report a pathogenic signal: polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The remaining predictors (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also benign, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Taken together, the majority of evidence points to a benign effect. Thus, the variant is most likely benign, and this is consistent with the lack of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.271506 | Structured | 0.276479 | Uncertain | 0.940 | 0.216 | 0.000 | -5.668 | Likely Benign | 0.562 | Ambiguous | Likely Benign | 1.65 | Ambiguous | 0.7 | 0.80 | Ambiguous | 1.23 | Ambiguous | 0.68 | Ambiguous | 0.171 | Likely Benign | -2.40 | Neutral | 0.986 | Probably Damaging | 0.960 | Probably Damaging | 3.30 | Benign | 0.18 | Tolerated | 0.1657 | 0.4275 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1339G>A | V447I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V447I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate a benign or likely benign outcome. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta) predicts benign stability. Thus, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.283801 | Uncertain | 0.970 | 0.243 | 0.000 | -5.067 | Likely Benign | 0.167 | Likely Benign | Likely Benign | -0.33 | Likely Benign | 0.1 | 0.27 | Likely Benign | -0.03 | Likely Benign | -0.44 | Likely Benign | 0.117 | Likely Benign | -0.06 | Neutral | 0.947 | Possibly Damaging | 0.851 | Possibly Damaging | 3.37 | Benign | 0.28 | Tolerated | 0.0625 | 0.3059 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||||
| c.1339G>T | V447F 2D 3DClick to see structure in 3D Viewer AISynGAP1 variant V447F is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools that classify the variant as benign include REVEL, premPS, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports an uncertain effect on protein folding. Overall, the majority of predictions lean toward pathogenicity, suggesting the variant is most likely pathogenic, a conclusion that does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.283801 | Uncertain | 0.970 | 0.243 | 0.000 | Uncertain | 1 | -8.673 | Likely Pathogenic | 0.701 | Likely Pathogenic | Likely Benign | 1.40 | Ambiguous | 0.3 | 0.61 | Ambiguous | 1.01 | Ambiguous | 0.20 | Likely Benign | 0.206 | Likely Benign | -2.62 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.44 | Benign | 0.03 | Affected | 0.0551 | 0.3055 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||
| c.133A>C | N45H 2D AIThe SynGAP1 missense variant N45H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for the variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.401658 | Structured | 0.431853 | Uncertain | 0.498 | 0.741 | 0.375 | -2.620 | Likely Benign | 0.285 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -0.62 | Neutral | 0.943 | Possibly Damaging | 0.924 | Probably Damaging | 4.05 | Benign | 0.00 | Affected | 0.2009 | 0.8046 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||||||||||||
| c.133A>G | N45D 2D AIThe SynGAP1 missense variant N45D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; no Foldetta stability result is available. Overall, the majority of evidence—including the consensus and high‑accuracy predictions—supports a benign classification for this variant, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.401658 | Structured | 0.431853 | Uncertain | 0.498 | 0.741 | 0.375 | -3.340 | Likely Benign | 0.278 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.37 | Neutral | 0.458 | Possibly Damaging | 0.678 | Possibly Damaging | 4.17 | Benign | 0.00 | Affected | 0.2234 | 0.4999 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||||||||||||
| c.133A>T | N45Y 2D AIThe SynGAP1 missense variant N45Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic outcome. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized independently predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence—including the high‑accuracy tools—points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.401658 | Structured | 0.431853 | Uncertain | 0.498 | 0.741 | 0.375 | -5.773 | Likely Benign | 0.502 | Ambiguous | Likely Benign | 0.180 | Likely Benign | -1.18 | Neutral | 0.943 | Possibly Damaging | 0.924 | Probably Damaging | 4.04 | Benign | 0.00 | Affected | 0.0741 | 0.7255 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.1340T>A | V447D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant V447D lies in the GAP domain. ClinVar has no entry for this change, and it is absent from gnomAD. Prediction tools that agree on benign impact are REVEL and FATHMM, whereas the remaining predictors—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classify the variant as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta also reports a pathogenic effect. Overall, the evidence points to a pathogenic effect for V447D, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.283801 | Uncertain | 0.970 | 0.243 | 0.000 | -16.643 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.34 | Destabilizing | 0.1 | 3.59 | Destabilizing | 3.97 | Destabilizing | 2.29 | Destabilizing | 0.491 | Likely Benign | -5.33 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.30 | Benign | 0.01 | Affected | 0.1424 | 0.0541 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1340T>C | V447A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V447A missense variant is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta both predict pathogenicity. No predictions are missing or inconclusive. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.283801 | Uncertain | 0.970 | 0.243 | 0.000 | -9.852 | Likely Pathogenic | 0.692 | Likely Pathogenic | Likely Benign | 2.18 | Destabilizing | 0.0 | 2.72 | Destabilizing | 2.45 | Destabilizing | 1.56 | Destabilizing | 0.266 | Likely Benign | -2.84 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.37 | Benign | 0.15 | Tolerated | 0.2456 | 0.2124 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.1340T>G | V447G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V447G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and FATHMM, while the remaining tools—FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. No prediction or stability result is missing or inconclusive. Overall, the preponderance of evidence indicates that V447G is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.250310 | Structured | 0.283801 | Uncertain | 0.970 | 0.243 | 0.000 | -13.648 | Likely Pathogenic | 0.861 | Likely Pathogenic | Ambiguous | 3.81 | Destabilizing | 0.1 | 4.62 | Destabilizing | 4.22 | Destabilizing | 2.28 | Destabilizing | 0.499 | Likely Benign | -5.43 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.1863 | 0.2240 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1342G>A | A448T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A448T missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact; premPS is uncertain and therefore not counted. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Taken together, the overwhelming majority of evidence indicates that A448T is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.225814 | Structured | 0.292774 | Uncertain | 0.973 | 0.257 | 0.000 | -9.192 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 3.06 | Destabilizing | 0.2 | 2.40 | Destabilizing | 2.73 | Destabilizing | 0.63 | Ambiguous | 0.558 | Likely Pathogenic | -3.95 | Deleterious | 0.996 | Probably Damaging | 0.973 | Probably Damaging | 3.19 | Benign | 0.00 | Affected | 0.1187 | 0.5050 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.1342G>C | A448P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A448P is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that assess pathogenicity largely agree: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect, while only FATHMM predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized returns a pathogenic prediction; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a likely pathogenic result; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. No predictions or stability results are missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.225814 | Structured | 0.292774 | Uncertain | 0.973 | 0.257 | 0.000 | -13.706 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 5.42 | Destabilizing | 0.0 | 8.74 | Destabilizing | 7.08 | Destabilizing | 1.16 | Destabilizing | 0.650 | Likely Pathogenic | -4.94 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.13 | Benign | 0.01 | Affected | 0.1758 | 0.3626 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1342G>T | A448S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A448S missense variant is not reported in ClinVar (status: None) and has no entry in gnomAD. Prediction tools that indicate a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Uncertain or inconclusive results are reported for FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as uncertain. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.225814 | Structured | 0.292774 | Uncertain | 0.973 | 0.257 | 0.000 | -9.213 | Likely Pathogenic | 0.590 | Likely Pathogenic | Likely Benign | 1.18 | Ambiguous | 0.1 | 1.97 | Ambiguous | 1.58 | Ambiguous | 0.55 | Ambiguous | 0.310 | Likely Benign | -2.96 | Deleterious | 0.965 | Probably Damaging | 0.972 | Probably Damaging | 3.27 | Benign | 0.06 | Tolerated | 0.2420 | 0.3471 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1343C>A | A448D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A448D is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy methods further support this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No prediction or folding result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.225814 | Structured | 0.292774 | Uncertain | 0.973 | 0.257 | 0.000 | -17.290 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 8.13 | Destabilizing | 0.2 | 4.35 | Destabilizing | 6.24 | Destabilizing | 1.40 | Destabilizing | 0.662 | Likely Pathogenic | -5.93 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.13 | Benign | 0.00 | Affected | 0.1541 | 0.1741 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.1343C>G | A448G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A448G resides in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized, whereas those that predict pathogenicity are Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default; FoldX is inconclusive. High‑accuracy assessments further clarify the impact: AlphaMissense‑Optimized reports a benign effect, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates pathogenicity, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a pathogenic effect. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.225814 | Structured | 0.292774 | Uncertain | 0.973 | 0.257 | 0.000 | -9.984 | Likely Pathogenic | 0.640 | Likely Pathogenic | Likely Benign | 1.76 | Ambiguous | 0.0 | 2.45 | Destabilizing | 2.11 | Destabilizing | 1.00 | Destabilizing | 0.378 | Likely Benign | -3.95 | Deleterious | 0.998 | Probably Damaging | 0.980 | Probably Damaging | 3.15 | Benign | 0.06 | Tolerated | 0.2135 | 0.2936 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1343C>T | A448V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A448V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict a pathogenic impact: FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). Two tools (Rosetta and premPS) yield uncertain results. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Overall, the preponderance of evidence indicates that A448V is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.225814 | Structured | 0.292774 | Uncertain | 0.973 | 0.257 | 0.000 | -10.372 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 3.31 | Destabilizing | 0.2 | 1.80 | Ambiguous | 2.56 | Destabilizing | 0.66 | Ambiguous | 0.487 | Likely Benign | -3.95 | Deleterious | 0.998 | Probably Damaging | 0.955 | Probably Damaging | 3.26 | Benign | 0.02 | Affected | 0.0950 | 0.4955 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.1345A>C | S449R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S449R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are SGM Consensus, PROVEAN, polyPhen‑2 HumDiv, AlphaMissense‑Default, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classifying it as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) yielding an uncertain stability change. No folding‑stability prediction is definitive. Overall, the majority of tools predict a benign outcome, and this does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.301437 | Uncertain | 0.958 | 0.251 | 0.000 | -8.486 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | -0.69 | Ambiguous | 0.2 | -1.33 | Ambiguous | -1.01 | Ambiguous | 0.50 | Likely Benign | 0.145 | Likely Benign | -3.36 | Deleterious | 0.950 | Possibly Damaging | 0.214 | Benign | 3.40 | Benign | 0.18 | Tolerated | 0.0762 | 0.3250 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1345A>T | S449C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S449C is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, FoldX, Rosetta, premPS (uncertain), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments concur: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. Based on the unanimous benign predictions and lack of ClinVar evidence, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.254060 | Structured | 0.301437 | Uncertain | 0.958 | 0.251 | 0.000 | -2.207 | Likely Benign | 0.050 | Likely Benign | Likely Benign | 0.48 | Likely Benign | 0.0 | -0.31 | Likely Benign | 0.09 | Likely Benign | -0.57 | Ambiguous | 0.067 | Likely Benign | 0.48 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.32 | Benign | 0.21 | Tolerated | 0.0864 | 0.5012 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.1346G>C | S449T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S449T is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts pathogenicity. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus predicts likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on these predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status (which has no entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.254060 | Structured | 0.301437 | Uncertain | 0.958 | 0.251 | 0.000 | -6.262 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.36 | Likely Benign | 0.1 | -0.46 | Likely Benign | -0.05 | Likely Benign | -0.15 | Likely Benign | 0.039 | Likely Benign | -1.48 | Neutral | 0.038 | Benign | 0.008 | Benign | 3.42 | Benign | 0.33 | Tolerated | 0.1219 | 0.5077 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1346G>T | S449I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S449I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen2_HumVar. Those that predict a pathogenic effect are PROVEAN, polyPhen2_HumDiv, and ESM1b. Rosetta and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs. 2 pathogenic). Foldetta also remains uncertain. Overall, the majority of evidence (7 benign vs. 3 pathogenic) points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.254060 | Structured | 0.301437 | Uncertain | 0.958 | 0.251 | 0.000 | -9.549 | Likely Pathogenic | 0.310 | Likely Benign | Likely Benign | 0.04 | Likely Benign | 0.1 | -1.39 | Ambiguous | -0.68 | Ambiguous | 0.13 | Likely Benign | 0.105 | Likely Benign | -3.23 | Deleterious | 0.559 | Possibly Damaging | 0.044 | Benign | 3.40 | Benign | 0.14 | Tolerated | 0.0756 | 0.5006 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||
| c.1347T>A | S449R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S449R is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, FATHMM, and polyPhen‑2 HumVar, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments provide a mixed picture: AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an inconclusive result. FoldX and Rosetta predictions are also uncertain and are treated as unavailable. Overall, the evidence is balanced, with an equal number of benign and pathogenic calls, and the high‑accuracy tools do not converge on a single conclusion. Consequently, the variant is most likely pathogenic based on the preponderance of pathogenic predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.301437 | Uncertain | 0.958 | 0.251 | 0.000 | -8.486 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | -0.69 | Ambiguous | 0.2 | -1.33 | Ambiguous | -1.01 | Ambiguous | 0.50 | Likely Benign | 0.168 | Likely Benign | -3.36 | Deleterious | 0.950 | Possibly Damaging | 0.214 | Benign | 3.40 | Benign | 0.18 | Tolerated | 0.0762 | 0.3250 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1347T>G | S449R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S449R is not reported in ClinVar (ClinVar status: None) and has no entry in gnomAD (gnomAD ID: None). Prediction tools that classify the variant as benign include REVEL, premPS, SIFT, FATHMM, and polyPhen‑2 HumVar. Those that predict pathogenicity are SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicting a benign effect, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; Foldetta’s stability prediction is uncertain and therefore treated as unavailable. Overall, the predictions are split, with an equal number of benign and pathogenic calls and conflicting high‑accuracy results. Consequently, the variant’s impact remains inconclusive, and there is no contradiction with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.301437 | Uncertain | 0.958 | 0.251 | 0.000 | -8.486 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | -0.69 | Ambiguous | 0.2 | -1.33 | Ambiguous | -1.01 | Ambiguous | 0.50 | Likely Benign | 0.167 | Likely Benign | -3.36 | Deleterious | 0.950 | Possibly Damaging | 0.214 | Benign | 3.40 | Benign | 0.18 | Tolerated | 0.0762 | 0.3250 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1348G>A | A450T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A450T missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Because the majority of conventional tools lean toward benign and no ClinVar evidence contradicts this, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.321458 | Structured | 0.306281 | Uncertain | 0.963 | 0.234 | 0.000 | -9.149 | Likely Pathogenic | 0.380 | Ambiguous | Likely Benign | 0.50 | Ambiguous | 0.2 | 0.98 | Ambiguous | 0.74 | Ambiguous | 0.81 | Ambiguous | 0.233 | Likely Benign | -3.35 | Deleterious | 0.996 | Probably Damaging | 0.973 | Probably Damaging | 3.40 | Benign | 0.06 | Tolerated | 0.0943 | 0.5902 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||
| c.1348G>C | A450P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A450P missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, SIFT, and FATHMM, whereas the majority of other in silico predictors (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM Consensus score all classify the change as pathogenic. The high‑accuracy predictors give consistent pathogenic results: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. premPS remains uncertain. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, which is consistent with the absence of a benign ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.321458 | Structured | 0.306281 | Uncertain | 0.963 | 0.234 | 0.000 | -15.378 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.75 | Destabilizing | 0.3 | 8.32 | Destabilizing | 5.54 | Destabilizing | 0.72 | Ambiguous | 0.459 | Likely Benign | -4.67 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.40 | Benign | 0.08 | Tolerated | 0.1494 | 0.4309 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1348G>T | A450S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A450S missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. The remaining methods—FoldX, Rosetta, Foldetta, and premPS—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie and thus unavailable; Foldetta is uncertain. Overall, the balance of evidence (five benign versus four pathogenic predictions, with three uncertain) suggests the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.321458 | Structured | 0.306281 | Uncertain | 0.963 | 0.234 | 0.000 | -9.257 | Likely Pathogenic | 0.274 | Likely Benign | Likely Benign | 0.81 | Ambiguous | 0.0 | 1.35 | Ambiguous | 1.08 | Ambiguous | 0.69 | Ambiguous | 0.268 | Likely Benign | -2.70 | Deleterious | 0.965 | Probably Damaging | 0.972 | Probably Damaging | 3.47 | Benign | 0.10 | Tolerated | 0.2003 | 0.4322 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||
| c.1349C>G | A450G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A450G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. The majority of other in silico predictors (SGM‑Consensus, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) classify the variant as pathogenic; FoldX is inconclusive. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized predicts benign, but the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta) both predict pathogenic. Overall, the preponderance of evidence indicates that A450G is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.321458 | Structured | 0.306281 | Uncertain | 0.963 | 0.234 | 0.000 | -11.090 | Likely Pathogenic | 0.695 | Likely Pathogenic | Likely Benign | 1.79 | Ambiguous | 0.0 | 2.29 | Destabilizing | 2.04 | Destabilizing | 1.23 | Destabilizing | 0.355 | Likely Benign | -3.82 | Deleterious | 0.998 | Probably Damaging | 0.980 | Probably Damaging | 3.40 | Benign | 0.04 | Affected | 0.1670 | 0.3078 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.134A>C | N45T 2D AIThe SynGAP1 missense variant N45T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.401658 | Structured | 0.431853 | Uncertain | 0.498 | 0.741 | 0.375 | -2.425 | Likely Benign | 0.367 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -0.81 | Neutral | 0.659 | Possibly Damaging | 0.775 | Possibly Damaging | 4.08 | Benign | 0.00 | Affected | 0.1642 | 0.8318 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||||||||||||
| c.134A>G | N45S 2D AIThe SynGAP1 missense variant N45S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for the variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.401658 | Structured | 0.431853 | Uncertain | 0.498 | 0.741 | 0.375 | -2.740 | Likely Benign | 0.217 | Likely Benign | Likely Benign | 0.050 | Likely Benign | -0.38 | Neutral | 0.458 | Possibly Damaging | 0.678 | Possibly Damaging | 4.14 | Benign | 0.00 | Affected | 0.3949 | 0.7617 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||||||||||||
| c.134A>T | N45I 2D AIThe SynGAP1 missense variant N45I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.401658 | Structured | 0.431853 | Uncertain | 0.498 | 0.741 | 0.375 | -4.063 | Likely Benign | 0.568 | Likely Pathogenic | Likely Benign | 0.147 | Likely Benign | -1.32 | Neutral | 0.943 | Possibly Damaging | 0.924 | Probably Damaging | 4.04 | Benign | 0.00 | Affected | 0.0861 | 0.7406 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.1351C>A | L451I 2D AIThe SynGAP1 L451I missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Uncertain results come from FoldX, Rosetta, premPS, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta is also inconclusive. Overall, the majority of available predictions (five pathogenic versus four benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict the absence of a ClinVar record. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.281712 | Structured | 0.314017 | Uncertain | 0.978 | 0.232 | 0.000 | -11.046 | Likely Pathogenic | 0.658 | Likely Pathogenic | Likely Benign | 1.43 | Ambiguous | 0.8 | 0.70 | Ambiguous | 1.07 | Ambiguous | 0.94 | Ambiguous | 0.284 | Likely Benign | -1.94 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 2.75 | Benign | 0.02 | Affected | 0.0681 | 0.2958 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1351C>G | L451V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L451V is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. In contrast, the majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Rosetta is uncertain and does not contribute to the consensus. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic; and Foldetta predicts pathogenic. Overall, the preponderance of evidence points to a pathogenic effect for L451V. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.281712 | Structured | 0.314017 | Uncertain | 0.978 | 0.232 | 0.000 | -10.732 | Likely Pathogenic | 0.635 | Likely Pathogenic | Likely Benign | 2.83 | Destabilizing | 0.1 | 1.81 | Ambiguous | 2.32 | Destabilizing | 1.39 | Destabilizing | 0.325 | Likely Benign | -2.90 | Deleterious | 0.995 | Probably Damaging | 0.970 | Probably Damaging | 2.54 | Benign | 0.01 | Affected | 0.1065 | 0.2856 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1352T>A | L451Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L451Q is not reported in ClinVar and is absent from gnomAD. Prediction tools uniformly indicate a deleterious effect: pathogenic predictions are returned by REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the change as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels it likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a pathogenic effect. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.281712 | Structured | 0.314017 | Uncertain | 0.978 | 0.232 | 0.000 | -15.426 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.91 | Destabilizing | 0.0 | 2.48 | Destabilizing | 2.70 | Destabilizing | 2.30 | Destabilizing | 0.708 | Likely Pathogenic | -5.79 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.43 | Pathogenic | 0.00 | Affected | 0.0869 | 0.0558 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1352T>G | L451R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L451R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). All available in‑silico predictors classify the substitution as pathogenic: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a pathogenic effect. Based on the unanimous pathogenic predictions and the absence of benign calls, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.281712 | Structured | 0.314017 | Uncertain | 0.978 | 0.232 | 0.000 | -16.162 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.32 | Destabilizing | 0.1 | 3.76 | Destabilizing | 3.54 | Destabilizing | 2.25 | Destabilizing | 0.726 | Likely Pathogenic | -5.82 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.43 | Pathogenic | 0.00 | Affected | 0.1130 | 0.0558 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1354G>A | V452I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V452I variant is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Rosetta and AlphaMissense‑Default return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.335645 | Structured | 0.315167 | Uncertain | 0.970 | 0.229 | 0.000 | Uncertain | 2 | -8.985 | Likely Pathogenic | 0.361 | Ambiguous | Likely Benign | -0.08 | Likely Benign | 0.1 | 0.51 | Ambiguous | 0.22 | Likely Benign | 0.25 | Likely Benign | 0.218 | Likely Benign | -0.99 | Neutral | 0.947 | Possibly Damaging | 0.851 | Possibly Damaging | 3.26 | Benign | 0.05 | Affected | 0.0630 | 0.3476 | 4 | 3 | 0.3 | 14.03 | ||||||||||||||||||||||||||||
| c.1354G>C | V452L 2D 3DClick to see structure in 3D Viewer AISynGAP1 V452L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM, while those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Because the majority of tools (six) predict benign and the high‑accuracy Foldetta also supports benign, the variant is most likely benign, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.335645 | Structured | 0.315167 | Uncertain | 0.970 | 0.229 | 0.000 | -11.285 | Likely Pathogenic | 0.929 | Likely Pathogenic | Ambiguous | 0.29 | Likely Benign | 0.1 | -0.25 | Likely Benign | 0.02 | Likely Benign | 0.34 | Likely Benign | 0.316 | Likely Benign | -2.96 | Deleterious | 0.947 | Possibly Damaging | 0.851 | Possibly Damaging | 3.54 | Benign | 0.11 | Tolerated | 0.0777 | 0.4061 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1355T>A | V452D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V452D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, while all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy predictors, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.335645 | Structured | 0.315167 | Uncertain | 0.970 | 0.229 | 0.000 | -15.793 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.92 | Destabilizing | 0.1 | 3.37 | Destabilizing | 3.65 | Destabilizing | 2.52 | Destabilizing | 0.630 | Likely Pathogenic | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.16 | Benign | 0.00 | Affected | 0.1320 | 0.0610 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1355T>C | V452A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V452A is not reported in ClinVar and has no entry in gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, yields an uncertain result and is treated as unavailable evidence. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta remains inconclusive. Overall, the consensus of the available tools points to a pathogenic effect for V452A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.335645 | Structured | 0.315167 | Uncertain | 0.970 | 0.229 | 0.000 | -10.423 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 2.21 | Destabilizing | 0.0 | 1.51 | Ambiguous | 1.86 | Ambiguous | 2.18 | Destabilizing | 0.505 | Likely Pathogenic | -3.95 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.21 | Benign | 0.00 | Affected | 0.2642 | 0.2521 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.1355T>G | V452G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V452G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. AlphaMissense‑Optimized remains uncertain. Overall, the preponderance of evidence indicates that V452G is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.335645 | Structured | 0.315167 | Uncertain | 0.970 | 0.229 | 0.000 | -13.410 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 3.70 | Destabilizing | 0.1 | 3.37 | Destabilizing | 3.54 | Destabilizing | 2.46 | Destabilizing | 0.570 | Likely Pathogenic | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.17 | Benign | 0.00 | Affected | 0.1948 | 0.2567 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1357C>A | H453N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant H453N is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. A third set of methods (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) yield uncertain or inconclusive results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also uncertain. Overall, the majority of evidence points toward a pathogenic effect for H453N. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.352862 | Structured | 0.316097 | Uncertain | 0.946 | 0.200 | 0.000 | -8.416 | Likely Pathogenic | 0.789 | Likely Pathogenic | Ambiguous | 0.93 | Ambiguous | 0.1 | 0.97 | Ambiguous | 0.95 | Ambiguous | 0.78 | Ambiguous | 0.347 | Likely Benign | -6.92 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.48 | Benign | 0.09 | Tolerated | 0.1435 | 0.2548 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||
| c.1357C>G | H453D 2D 3DClick to see structure in 3D Viewer AISynGAP1 H453D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions are made by FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus methods reinforce a pathogenic interpretation: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates pathogenicity. premPS remains uncertain. Overall, the majority of evidence points to a pathogenic effect for H453D, and this conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.352862 | Structured | 0.316097 | Uncertain | 0.946 | 0.200 | 0.000 | -15.256 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 3.33 | Destabilizing | 0.0 | 2.68 | Destabilizing | 3.01 | Destabilizing | 0.97 | Ambiguous | 0.443 | Likely Benign | -8.89 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.48 | Benign | 0.09 | Tolerated | 0.2225 | 0.1815 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||
| c.1357C>T | H453Y 2D AIThe SynGAP1 missense variant H453Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign calls come from REVEL, premPS, FATHMM, and AlphaMissense‑Optimized; pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus. Predictions from FoldX, Rosetta, and Foldetta are uncertain. High‑accuracy methods provide a mixed picture: AlphaMissense‑Optimized predicts benign, SGM‑Consensus predicts likely pathogenic, and Foldetta is inconclusive. Overall, the majority of evidence points toward a pathogenic effect, aligning with the SGM‑Consensus but opposing the benign predictions from several tools. Because ClinVar contains no entry for this variant, there is no conflict with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.352862 | Structured | 0.316097 | Uncertain | 0.946 | 0.200 | 0.000 | -12.060 | Likely Pathogenic | 0.753 | Likely Pathogenic | Likely Benign | -0.52 | Ambiguous | 0.7 | -0.68 | Ambiguous | -0.60 | Ambiguous | 0.09 | Likely Benign | 0.320 | Likely Benign | -5.93 | Deleterious | 0.995 | Probably Damaging | 0.961 | Probably Damaging | 3.41 | Benign | 0.04 | Affected | 0.0815 | 0.3843 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||
| c.1358A>C | H453P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H453P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM. The majority of tools predict a pathogenic impact: FoldX, Rosetta, Foldetta, premPS (uncertain), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No predictions are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.352862 | Structured | 0.316097 | Uncertain | 0.946 | 0.200 | 0.000 | -15.704 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 3.29 | Destabilizing | 0.4 | 7.04 | Destabilizing | 5.17 | Destabilizing | 0.84 | Ambiguous | 0.488 | Likely Benign | -9.88 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.40 | Benign | 0.04 | Affected | 0.2115 | 0.3938 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||
| c.1358A>G | H453R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant H453R is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools predicting a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Uncertain results come from FoldX and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of standard predictors are split, but the two most reliable methods (AlphaMissense‑Optimized and Foldetta) favor a benign outcome, while the SGM Consensus suggests pathogenicity. Thus, the variant is most likely benign based on the available predictions, and this does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.352862 | Structured | 0.316097 | Uncertain | 0.946 | 0.200 | 0.000 | Uncertain | 1 | -9.239 | Likely Pathogenic | 0.573 | Likely Pathogenic | Likely Benign | -0.52 | Ambiguous | 0.1 | 0.37 | Likely Benign | -0.08 | Likely Benign | 0.56 | Ambiguous | 0.396 | Likely Benign | -7.91 | Deleterious | 0.993 | Probably Damaging | 0.957 | Probably Damaging | 3.53 | Benign | 0.39 | Tolerated | 0.1646 | 0.2031 | 2 | 0 | -1.3 | 19.05 | |||||||||||||||||||||||||||
| c.1358A>T | H453L 2D 3DClick to see structure in 3D Viewer AISynGAP1 H453L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, premPS, SIFT, FATHMM, and the protein‑folding stability method Foldetta; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments indicate that AlphaMissense‑Optimized is uncertain, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta predicts benign. Overall, the predictions are split, with a slight edge toward pathogenicity from the consensus and high‑accuracy tools, but the presence of several benign calls and the uncertainty of AlphaMissense‑Optimized temper this conclusion. Thus, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.352862 | Structured | 0.316097 | Uncertain | 0.946 | 0.200 | 0.000 | -10.438 | Likely Pathogenic | 0.834 | Likely Pathogenic | Ambiguous | -0.59 | Ambiguous | 0.1 | 0.04 | Likely Benign | -0.28 | Likely Benign | 0.31 | Likely Benign | 0.413 | Likely Benign | -10.87 | Deleterious | 0.998 | Probably Damaging | 0.973 | Probably Damaging | 3.45 | Benign | 0.11 | Tolerated | 0.0841 | 0.4964 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.1359C>A | H453Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H453Q is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic outcome: SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Tools with uncertain or inconclusive results—FoldX, Rosetta, premPS, AlphaMissense‑Optimized, and Foldetta—are treated as unavailable for pathogenicity assessment. High‑accuracy methods specifically show SGM‑Consensus as Likely Pathogenic, AlphaMissense‑Optimized as uncertain, and Foldetta as uncertain. Overall, the preponderance of evidence from consensus and high‑accuracy predictors indicates that H453Q is most likely pathogenic, and this assessment does not contradict the current ClinVar status, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.352862 | Structured | 0.316097 | Uncertain | 0.946 | 0.200 | 0.000 | -10.894 | Likely Pathogenic | 0.893 | Likely Pathogenic | Ambiguous | 0.90 | Ambiguous | 0.1 | 1.55 | Ambiguous | 1.23 | Ambiguous | 0.92 | Ambiguous | 0.259 | Likely Benign | -7.91 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.45 | Benign | 0.09 | Tolerated | 0.1223 | 0.3112 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.1359C>G | H453Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant H453Q is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic outcome: SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Tools with uncertain or inconclusive results—FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized—are treated as unavailable for pathogenicity assessment. High‑accuracy methods specifically show SGM‑Consensus as Likely Pathogenic, AlphaMissense‑Optimized as Uncertain, and Foldetta as Uncertain. Overall, the preponderance of evidence points to a pathogenic effect for H453Q. This prediction does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.352862 | Structured | 0.316097 | Uncertain | 0.946 | 0.200 | 0.000 | -10.894 | Likely Pathogenic | 0.893 | Likely Pathogenic | Ambiguous | 0.90 | Ambiguous | 0.1 | 1.55 | Ambiguous | 1.23 | Ambiguous | 0.92 | Ambiguous | 0.258 | Likely Benign | -7.91 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.45 | Benign | 0.09 | Tolerated | 0.1223 | 0.3112 | 3 | 0 | -0.3 | -9.01 | |||||||||||||||||||||||||||||
| c.135C>A | N45K 2D AISynGAP1 missense variant N45K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a benign outcome; Foldetta results are unavailable. Overall, the balance of evidence—five benign versus four pathogenic predictions, with two high‑accuracy tools supporting benign—suggests that the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.401658 | Structured | 0.431853 | Uncertain | 0.498 | 0.741 | 0.375 | -1.711 | Likely Benign | 0.697 | Likely Pathogenic | Likely Benign | 0.082 | Likely Benign | -0.58 | Neutral | 0.659 | Possibly Damaging | 0.775 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.2409 | 0.6724 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||||
| c.135C>G | N45K 2D AISynGAP1 missense variant N45K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a benign outcome; Foldetta results are unavailable. Overall, the balance of evidence—five benign versus four pathogenic predictions, with two high‑accuracy tools supporting benign—suggests that the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.401658 | Structured | 0.431853 | Uncertain | 0.498 | 0.741 | 0.375 | -1.711 | Likely Benign | 0.697 | Likely Pathogenic | Likely Benign | 0.082 | Likely Benign | -0.58 | Neutral | 0.659 | Possibly Damaging | 0.775 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.2409 | 0.6724 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||||||||||||
| c.1360A>C | I454L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I454L is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and FATHMM, while polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default predict a pathogenic impact. The remaining tools—FoldX, Rosetta, Foldetta, premPS, ESM1b, and AlphaMissense‑Optimized—yield uncertain or inconclusive results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leaning toward benign, and Foldetta also uncertain. Overall, the majority of reliable predictors and the SGM Consensus favor a benign classification. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.254060 | Structured | 0.312811 | Uncertain | 0.965 | 0.182 | 0.000 | -7.852 | In-Between | 0.786 | Likely Pathogenic | Ambiguous | 0.57 | Ambiguous | 0.1 | 1.36 | Ambiguous | 0.97 | Ambiguous | 0.80 | Ambiguous | 0.246 | Likely Benign | -1.98 | Neutral | 0.908 | Possibly Damaging | 0.943 | Probably Damaging | 3.51 | Benign | 0.26 | Tolerated | 0.0653 | 0.2857 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1360A>G | I454V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I454V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as likely benign, and Foldetta as uncertain. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.254060 | Structured | 0.312811 | Uncertain | 0.965 | 0.182 | 0.000 | -4.719 | Likely Benign | 0.657 | Likely Pathogenic | Likely Benign | 1.47 | Ambiguous | 0.0 | 1.36 | Ambiguous | 1.42 | Ambiguous | 0.69 | Ambiguous | 0.132 | Likely Benign | -0.79 | Neutral | 0.935 | Possibly Damaging | 0.858 | Possibly Damaging | 3.40 | Benign | 0.18 | Tolerated | 0.0833 | 0.2959 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||
| c.1360A>T | I454F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I454F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and FATHMM. All other evaluated algorithms—FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. premPS is uncertain and does not influence the overall assessment. Based on the preponderance of pathogenic predictions and the absence of benign evidence, the variant is most likely pathogenic, with no ClinVar status to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.312811 | Uncertain | 0.965 | 0.182 | 0.000 | -12.468 | Likely Pathogenic | 0.960 | Likely Pathogenic | Likely Pathogenic | 4.15 | Destabilizing | 0.7 | 2.96 | Destabilizing | 3.56 | Destabilizing | 0.70 | Ambiguous | 0.464 | Likely Benign | -3.95 | Deleterious | 0.998 | Probably Damaging | 0.973 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.0422 | 0.2560 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.1361T>A | I454N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I454N resides in the GAP domain. ClinVar has no entry for this variant, and it is absent from gnomAD. Prediction tools that agree on benign impact are REVEL and FATHMM, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict pathogenicity. High‑accuracy methods further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic, and Foldetta predicts a destabilizing, pathogenic change. Overall, the evidence strongly favors a pathogenic classification, and this assessment does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.312811 | Uncertain | 0.965 | 0.182 | 0.000 | -14.246 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.24 | Destabilizing | 0.1 | 3.56 | Destabilizing | 3.90 | Destabilizing | 2.44 | Destabilizing | 0.497 | Likely Benign | -6.82 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.25 | Benign | 0.00 | Affected | 0.0750 | 0.0412 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1361T>C | I454T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I454T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.312811 | Uncertain | 0.965 | 0.182 | 0.000 | -9.045 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.20 | Destabilizing | 0.0 | 2.83 | Destabilizing | 3.02 | Destabilizing | 2.02 | Destabilizing | 0.502 | Likely Pathogenic | -4.74 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.26 | Benign | 0.00 | Affected | 0.0877 | 0.0708 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.1361T>G | I454S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I454S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy predictors, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.312811 | Uncertain | 0.965 | 0.182 | 0.000 | -13.025 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.50 | Destabilizing | 0.1 | 4.45 | Destabilizing | 4.48 | Destabilizing | 2.20 | Destabilizing | 0.574 | Likely Pathogenic | -5.83 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.26 | Benign | 0.00 | Affected | 0.2116 | 0.0800 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1362C>G | I454M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I454M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools (SGM‑Consensus, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. Tools with inconclusive results are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for I454M. This conclusion is not contradicted by ClinVar, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.312811 | Uncertain | 0.965 | 0.182 | 0.000 | -8.437 | Likely Pathogenic | 0.871 | Likely Pathogenic | Ambiguous | 1.06 | Ambiguous | 0.1 | 2.32 | Destabilizing | 1.69 | Ambiguous | 1.08 | Destabilizing | 0.292 | Likely Benign | -2.86 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.27 | Benign | 0.01 | Affected | 0.0566 | 0.2524 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||
| c.1363C>A | L455M 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L455M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a tie, and Foldetta is uncertain. No folding‑stability method provides a definitive result. Consequently, the computational evidence does not favor either benign or pathogenic classification. The variant’s status is therefore inconclusive, and this lack of consensus does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.188120 | Structured | 0.310377 | Uncertain | 0.963 | 0.168 | 0.000 | -10.086 | Likely Pathogenic | 0.802 | Likely Pathogenic | Ambiguous | 0.88 | Ambiguous | 0.0 | 1.27 | Ambiguous | 1.08 | Ambiguous | 0.59 | Ambiguous | 0.147 | Likely Benign | -1.64 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.25 | Benign | 0.11 | Tolerated | 0.0606 | 0.3362 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.1363C>G | L455V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L455V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from REVEL and FATHMM, whereas the remaining 11 tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict pathogenicity. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Taken together, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is consistent with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.310377 | Uncertain | 0.963 | 0.168 | 0.000 | -12.773 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 2.89 | Destabilizing | 0.1 | 2.38 | Destabilizing | 2.64 | Destabilizing | 1.41 | Destabilizing | 0.276 | Likely Benign | -2.83 | Deleterious | 0.995 | Probably Damaging | 0.970 | Probably Damaging | 3.29 | Benign | 0.02 | Affected | 0.1149 | 0.3056 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1364T>A | L455Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L455Q is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.310377 | Uncertain | 0.963 | 0.168 | 0.000 | -13.075 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.84 | Destabilizing | 0.0 | 3.42 | Destabilizing | 3.13 | Destabilizing | 2.24 | Destabilizing | 0.655 | Likely Pathogenic | -5.66 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.23 | Benign | 0.00 | Affected | 0.0871 | 0.0958 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1364T>C | L455P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L455P is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.310377 | Uncertain | 0.963 | 0.168 | 0.000 | -14.150 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 7.45 | Destabilizing | 0.2 | 11.99 | Destabilizing | 9.72 | Destabilizing | 2.19 | Destabilizing | 0.695 | Likely Pathogenic | -6.72 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.23 | Benign | 0.00 | Affected | 0.3211 | 0.1221 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1364T>G | L455R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L455R resides in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM; all other evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.188120 | Structured | 0.310377 | Uncertain | 0.963 | 0.168 | 0.000 | -16.347 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 6.36 | Destabilizing | 0.2 | 4.96 | Destabilizing | 5.66 | Destabilizing | 2.08 | Destabilizing | 0.648 | Likely Pathogenic | -5.73 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 3.24 | Benign | 0.00 | Affected | 0.1126 | 0.0600 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1366C>A | Q456K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q456K is not reported in ClinVar and has no gnomAD entry. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and FATHMM, whereas those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default; premPS and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of tools (six pathogenic vs five benign) and the SGM‑Consensus result point toward a pathogenic interpretation, while Foldetta suggests stability‑preserving benignity. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.170161 | Structured | 0.302348 | Uncertain | 0.939 | 0.164 | 0.000 | -13.768 | Likely Pathogenic | 0.806 | Likely Pathogenic | Ambiguous | 0.21 | Likely Benign | 0.1 | 0.23 | Likely Benign | 0.22 | Likely Benign | 0.78 | Ambiguous | 0.391 | Likely Benign | -3.75 | Deleterious | 0.969 | Probably Damaging | 0.875 | Possibly Damaging | 3.36 | Benign | 0.13 | Tolerated | 0.1321 | 0.2547 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||
| c.1366C>G | Q456E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q456E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) yield uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicting pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) remaining uncertain. Overall, the predictions are split, but the high‑accuracy consensus leans toward pathogenicity. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.170161 | Structured | 0.302348 | Uncertain | 0.939 | 0.164 | 0.000 | -12.982 | Likely Pathogenic | 0.503 | Ambiguous | Likely Benign | 0.71 | Ambiguous | 0.1 | 0.87 | Ambiguous | 0.79 | Ambiguous | 0.65 | Ambiguous | 0.233 | Likely Benign | -2.76 | Deleterious | 0.998 | Probably Damaging | 0.964 | Probably Damaging | 3.41 | Benign | 0.15 | Tolerated | 0.1191 | 0.1276 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.1367A>G | Q456R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q456R has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, and FATHMM. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain; the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, whereas Foldetta (a folding‑stability method combining FoldX‑MD and Rosetta outputs) predicts benign. No evidence from the data contradicts ClinVar status, which is currently unclassified. Based on the available predictions, the variant is most likely benign, though the evidence is conflicting and does not conflict with the lack of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.170161 | Structured | 0.302348 | Uncertain | 0.939 | 0.164 | 0.000 | -11.326 | Likely Pathogenic | 0.801 | Likely Pathogenic | Ambiguous | 0.21 | Likely Benign | 0.1 | 0.12 | Likely Benign | 0.17 | Likely Benign | 0.33 | Likely Benign | 0.295 | Likely Benign | -3.69 | Deleterious | 0.980 | Probably Damaging | 0.943 | Probably Damaging | 3.37 | Benign | 0.20 | Tolerated | 0.1241 | 0.1124 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||
| c.1367A>T | Q456L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q456L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Tools with uncertain or inconclusive results are FoldX, Foldetta, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation, with no evidence of contradiction with the ClinVar status. Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.170161 | Structured | 0.302348 | Uncertain | 0.939 | 0.164 | 0.000 | -10.272 | Likely Pathogenic | 0.492 | Ambiguous | Likely Benign | -0.88 | Ambiguous | 0.1 | -0.30 | Likely Benign | -0.59 | Ambiguous | 0.28 | Likely Benign | 0.347 | Likely Benign | -6.65 | Deleterious | 0.987 | Probably Damaging | 0.914 | Probably Damaging | 3.49 | Benign | 0.36 | Tolerated | 0.0554 | 0.4317 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||
| c.1368G>C | Q456H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q456H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Tools with uncertain or inconclusive results are FoldX, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of predictions lean toward a benign impact, and this does not contradict the lack of ClinVar annotation. Thus, based on the available computational evidence, the variant is most likely benign, though a high‑accuracy consensus suggests a possible pathogenic effect that warrants further investigation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.170161 | Structured | 0.302348 | Uncertain | 0.939 | 0.164 | 0.000 | -7.993 | In-Between | 0.598 | Likely Pathogenic | Likely Benign | 0.74 | Ambiguous | 0.1 | 0.39 | Likely Benign | 0.57 | Ambiguous | 0.32 | Likely Benign | 0.214 | Likely Benign | -4.07 | Deleterious | 0.996 | Probably Damaging | 0.974 | Probably Damaging | 3.37 | Benign | 0.19 | Tolerated | 0.0987 | 0.2358 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||
| c.1368G>T | Q456H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q456H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Tools with uncertain or inconclusive results are FoldX, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of predictions lean toward a benign impact, and this does not contradict the lack of ClinVar annotation. Thus, based on the available computational evidence, the variant is most likely benign, though a high‑accuracy consensus suggests a possible pathogenic effect that warrants further investigation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.170161 | Structured | 0.302348 | Uncertain | 0.939 | 0.164 | 0.000 | -7.993 | In-Between | 0.598 | Likely Pathogenic | Likely Benign | 0.74 | Ambiguous | 0.1 | 0.39 | Likely Benign | 0.57 | Ambiguous | 0.32 | Likely Benign | 0.214 | Likely Benign | -4.07 | Deleterious | 0.996 | Probably Damaging | 0.974 | Probably Damaging | 3.37 | Benign | 0.19 | Tolerated | 0.0987 | 0.2358 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||
| c.1369A>C | S457R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S457R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split opinion: benign calls come from REVEL, Rosetta, and FATHMM, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX, Foldetta, and premPS are inconclusive. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates pathogenic, whereas Foldetta’s stability analysis is uncertain. Overall, the majority of evidence points toward a pathogenic impact. This conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.164327 | Structured | 0.297330 | Uncertain | 0.909 | 0.159 | 0.000 | -10.882 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | -1.04 | Ambiguous | 0.0 | -0.06 | Likely Benign | -0.55 | Ambiguous | 0.74 | Ambiguous | 0.468 | Likely Benign | -4.47 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 3.30 | Benign | 0.05 | Affected | 0.0781 | 0.3426 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1369A>G | S457G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S457G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for S457G. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.164327 | Structured | 0.297330 | Uncertain | 0.909 | 0.159 | 0.000 | -9.154 | Likely Pathogenic | 0.811 | Likely Pathogenic | Ambiguous | 0.86 | Ambiguous | 0.0 | 0.87 | Ambiguous | 0.87 | Ambiguous | 0.65 | Ambiguous | 0.382 | Likely Benign | -3.82 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 3.35 | Benign | 0.13 | Tolerated | 0.2735 | 0.4074 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||
| c.1369A>T | S457C 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S457C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, FoldX, Foldetta, and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy methods give mixed results: AlphaMissense‑Optimized is inconclusive and therefore not used as evidence; the SGM‑Consensus majority vote (AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a benign effect. Overall, the majority of individual predictors lean toward pathogenicity, while the high‑accuracy Foldetta result suggests benign stability. Given the predominance of pathogenic calls and the lack of ClinVar evidence, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.164327 | Structured | 0.297330 | Uncertain | 0.909 | 0.159 | 0.000 | -8.152 | Likely Pathogenic | 0.815 | Likely Pathogenic | Ambiguous | -0.12 | Likely Benign | 0.0 | -0.79 | Ambiguous | -0.46 | Likely Benign | 0.56 | Ambiguous | 0.497 | Likely Benign | -4.81 | Deleterious | 0.999 | Probably Damaging | 0.987 | Probably Damaging | 3.30 | Benign | 0.01 | Affected | 0.0865 | 0.6284 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.136C>A | P46T 2D AIThe SynGAP1 missense variant P46T is reported in ClinVar as “Not submitted” and is not present in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, all of which classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT uniformly predict a pathogenic impact. AlphaMissense‑Default remains uncertain, and no Foldetta stability assessment is available. High‑accuracy methods give a consistent benign signal: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Benign,” and Foldetta data are missing. Overall, the majority of reliable predictors and the consensus analysis indicate that P46T is most likely benign, and this conclusion does not contradict the ClinVar status, which contains no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.390993 | Structured | 0.433588 | Uncertain | 0.549 | 0.741 | 0.375 | -4.329 | Likely Benign | 0.383 | Ambiguous | Likely Benign | 0.092 | Likely Benign | -0.68 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.2194 | 0.6400 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.136C>G | P46A 2D AIThe SynGAP1 missense variant P46A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for P46A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.390993 | Structured | 0.433588 | Uncertain | 0.549 | 0.741 | 0.375 | -4.811 | Likely Benign | 0.270 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.68 | Neutral | 0.805 | Possibly Damaging | 0.857 | Possibly Damaging | 4.16 | Benign | 0.00 | Affected | 0.3987 | 0.5696 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||||||
| c.136C>T | P46S 2D AIThe SynGAP1 missense variant P46S is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence—including high‑accuracy tools—points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.390993 | Structured | 0.433588 | Uncertain | 0.549 | 0.741 | 0.375 | Uncertain | 1 | -3.338 | Likely Benign | 0.302 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.60 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 0.3904 | 0.5771 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||||||
| c.1370G>A | S457N 2D AIThe SynGAP1 missense variant S457N is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, FoldX, Rosetta, SIFT, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The high‑accuracy consensus methods give a mixed picture: AlphaMissense‑Optimized is inconclusive, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) favors pathogenicity, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a benign effect. Overall, the majority of individual predictors lean toward pathogenicity, but the high‑accuracy Foldetta result suggests a benign impact. Thus, the variant is most likely pathogenic based on the preponderance of predictions, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.164327 | Structured | 0.297330 | Uncertain | 0.909 | 0.159 | 0.000 | Uncertain | 1 | -10.221 | Likely Pathogenic | 0.949 | Likely Pathogenic | Ambiguous | 0.19 | Likely Benign | 0.0 | -0.22 | Likely Benign | -0.02 | Likely Benign | 0.67 | Ambiguous | 0.241 | Likely Benign | -2.76 | Deleterious | 0.940 | Possibly Damaging | 0.843 | Possibly Damaging | 3.28 | Benign | 0.06 | Tolerated | 0.1015 | 0.4980 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||
| c.1370G>C | S457T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S457T has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, Foldetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. FoldX and Rosetta provide uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicting likely pathogenic, and Foldetta predicting benign. Overall, the majority of high‑confidence tools lean toward a benign effect, and there is no conflict with the ClinVar status, which is currently unreported. Thus, the variant is most likely benign based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.164327 | Structured | 0.297330 | Uncertain | 0.909 | 0.159 | 0.000 | -8.772 | Likely Pathogenic | 0.723 | Likely Pathogenic | Likely Benign | 0.82 | Ambiguous | 0.1 | -0.57 | Ambiguous | 0.13 | Likely Benign | 0.38 | Likely Benign | 0.273 | Likely Benign | -2.83 | Deleterious | 0.866 | Possibly Damaging | 0.780 | Possibly Damaging | 3.34 | Benign | 0.09 | Tolerated | 0.1154 | 0.6487 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1370G>T | S457I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S457I lies within the GAP domain. ClinVar has no entry for this change, and it is absent from gnomAD. Prediction tools that report a benign effect include FoldX and FATHMM, whereas the majority of other in silico predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default and AlphaMissense‑Optimized—classify it as pathogenic. Uncertain results come from Rosetta, Foldetta and premPS. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM and PROVEAN) is Likely Pathogenic, and Foldetta remains inconclusive. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.164327 | Structured | 0.297330 | Uncertain | 0.909 | 0.159 | 0.000 | -13.170 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | -0.26 | Likely Benign | 0.3 | -0.96 | Ambiguous | -0.61 | Ambiguous | 0.65 | Ambiguous | 0.557 | Likely Pathogenic | -5.73 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.34 | Benign | 0.02 | Affected | 0.0779 | 0.6095 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.1371T>A | S457R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S457R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, SGM‑Consensus confirms a likely pathogenic status, and Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result. No other high‑confidence stability predictions are available. Overall, the preponderance of evidence from multiple independent predictors indicates that S457R is most likely pathogenic, and this assessment does not contradict any existing ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.164327 | Structured | 0.297330 | Uncertain | 0.909 | 0.159 | 0.000 | -10.882 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | -1.04 | Ambiguous | 0.0 | -0.06 | Likely Benign | -0.55 | Ambiguous | 0.74 | Ambiguous | 0.343 | Likely Benign | -4.47 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 3.30 | Benign | 0.05 | Affected | 0.0781 | 0.3426 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1371T>G | S457R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S457R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, SGM‑Consensus confirms a likely pathogenic status, and Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result. No other high‑confidence stability predictions are available. Overall, the preponderance of evidence from multiple independent predictors indicates that S457R is most likely pathogenic, and this assessment does not contradict any existing ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.164327 | Structured | 0.297330 | Uncertain | 0.909 | 0.159 | 0.000 | -10.882 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | -1.04 | Ambiguous | 0.0 | -0.06 | Likely Benign | -0.55 | Ambiguous | 0.74 | Ambiguous | 0.343 | Likely Benign | -4.47 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 3.30 | Benign | 0.05 | Affected | 0.0781 | 0.3426 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1372A>C | T458P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T458P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are SIFT and FATHMM, while the remaining tools (REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, ESM1b, and the SGM‑Consensus) all predict a pathogenic impact; premPS is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a pathogenic effect. No prediction or folding stability result is missing or inconclusive. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.294848 | Uncertain | 0.915 | 0.144 | 0.000 | -13.547 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 3.05 | Destabilizing | 0.2 | 5.36 | Destabilizing | 4.21 | Destabilizing | 0.78 | Ambiguous | 0.557 | Likely Pathogenic | -5.33 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.35 | Benign | 0.06 | Tolerated | 0.2098 | 0.5395 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1372A>G | T458A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T458A missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and FATHMM. Those that agree on a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Two tools give uncertain results: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show that AlphaMissense‑Optimized is uncertain, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign. Overall, the majority of reliable predictors (six pathogenic vs. five benign) indicate a pathogenic effect. This conclusion does not contradict ClinVar status, as the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.294848 | Uncertain | 0.915 | 0.144 | 0.000 | -10.734 | Likely Pathogenic | 0.924 | Likely Pathogenic | Ambiguous | 0.36 | Likely Benign | 0.0 | 0.31 | Likely Benign | 0.34 | Likely Benign | 0.56 | Ambiguous | 0.358 | Likely Benign | -4.27 | Deleterious | 0.995 | Probably Damaging | 0.960 | Probably Damaging | 3.40 | Benign | 0.15 | Tolerated | 0.4137 | 0.4257 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1372A>T | T458S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T458S missense variant has no ClinVar entry and is not present in gnomAD. Functional prediction tools cluster into two groups: benign calls from REVEL, FoldX, Rosetta, SIFT, and FATHMM; pathogenic calls from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments further split the signal: AlphaMissense‑Optimized remains uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Because the majority of standard predictors are evenly divided and the high‑accuracy methods disagree, the variant’s effect cannot be confidently classified as benign or pathogenic. Thus, the variant is of uncertain significance, and this uncertainty does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.294848 | Uncertain | 0.915 | 0.144 | 0.000 | -8.465 | Likely Pathogenic | 0.915 | Likely Pathogenic | Ambiguous | 0.44 | Likely Benign | 0.1 | 0.35 | Likely Benign | 0.40 | Likely Benign | 0.55 | Ambiguous | 0.260 | Likely Benign | -3.49 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 3.48 | Benign | 0.13 | Tolerated | 0.3525 | 0.4349 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1373C>A | T458K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T458K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, Rosetta, SIFT, and FATHMM. Tools that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results from FoldX and premPS are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicting pathogenicity, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicting pathogenicity, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicting benign stability. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.294848 | Uncertain | 0.915 | 0.144 | 0.000 | -13.734 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | -0.59 | Ambiguous | 0.1 | -0.26 | Likely Benign | -0.43 | Likely Benign | 0.80 | Ambiguous | 0.373 | Likely Benign | -5.23 | Deleterious | 0.999 | Probably Damaging | 0.973 | Probably Damaging | 3.40 | Benign | 0.14 | Tolerated | 0.1153 | 0.3326 | 0 | -1 | -3.2 | 27.07 | |||||||||||||||||||||||||||||
| c.1373C>G | T458R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T458R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, SIFT, and FATHMM, whereas a larger set—SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict pathogenicity. Uncertain results come from FoldX and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.294848 | Uncertain | 0.915 | 0.144 | 0.000 | -12.984 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | -0.81 | Ambiguous | 0.1 | -0.11 | Likely Benign | -0.46 | Likely Benign | 0.61 | Ambiguous | 0.390 | Likely Benign | -5.26 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.52 | Benign | 0.20 | Tolerated | 0.1016 | 0.3013 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.1373C>T | T458I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T458I missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM. Those that predict a pathogenic impact are SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools (seven versus six) and the two high‑accuracy pathogenic predictions suggest the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as no ClinVar classification is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.294848 | Uncertain | 0.915 | 0.144 | 0.000 | -9.436 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | -0.40 | Likely Benign | 0.1 | 0.29 | Likely Benign | -0.06 | Likely Benign | 0.50 | Likely Benign | 0.337 | Likely Benign | -4.76 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 3.40 | Benign | 0.09 | Tolerated | 0.0724 | 0.6128 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.1375G>A | G459S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G459S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign calls come from REVEL and FATHMM, while pathogenic predictions are made by FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). Uncertain results are reported by Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as inconclusive, SGM‑Consensus as Likely Pathogenic, and Foldetta as inconclusive. Overall, the majority of evidence points to a pathogenic impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.289888 | Uncertain | 0.903 | 0.150 | 0.125 | -10.979 | Likely Pathogenic | 0.873 | Likely Pathogenic | Ambiguous | 2.34 | Destabilizing | 0.1 | 0.77 | Ambiguous | 1.56 | Ambiguous | 0.60 | Ambiguous | 0.414 | Likely Benign | -5.46 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.09 | Benign | 0.05 | Affected | 0.2340 | 0.5197 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1375G>C | G459R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G459R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM Consensus score (Likely Pathogenic) all indicate a pathogenic impact. Stability‑based methods (FoldX, Rosetta, premPS) are inconclusive, and Foldetta likewise reports an uncertain effect. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as Likely Pathogenic, and Foldetta as unavailable. Taken together, the consensus of the majority of tools points to a pathogenic effect for G459R. This prediction is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.289888 | Uncertain | 0.903 | 0.150 | 0.125 | -12.958 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.84 | Ambiguous | 0.2 | 0.79 | Ambiguous | 1.32 | Ambiguous | 0.79 | Ambiguous | 0.486 | Likely Benign | -7.41 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.09 | Benign | 0.00 | Affected | 0.0903 | 0.4248 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1375G>T | G459C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G459C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the majority of algorithms (SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. Uncertain or inconclusive results come from Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Taken together, the preponderance of evidence points to a pathogenic effect for G459C, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.289888 | Uncertain | 0.903 | 0.150 | 0.125 | -12.109 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 2.46 | Destabilizing | 0.2 | 1.49 | Ambiguous | 1.98 | Ambiguous | 0.65 | Ambiguous | 0.586 | Likely Pathogenic | -8.46 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.00 | Benign | 0.00 | Affected | 0.0960 | 0.4103 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1376G>A | G459D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G459D is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate benign effects include REVEL and FATHMM, while the majority of tools predict pathogenicity: FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain predictions come from Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as inconclusive. Overall, the evidence points to a pathogenic effect for G459D, and this conclusion does not conflict with the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.289888 | Uncertain | 0.903 | 0.150 | 0.125 | -11.258 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 2.21 | Destabilizing | 0.1 | 0.56 | Ambiguous | 1.39 | Ambiguous | 0.93 | Ambiguous | 0.454 | Likely Benign | -6.18 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.07 | Benign | 0.05 | Affected | 0.1466 | 0.1905 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.1376G>C | G459A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G459A missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two consensus groups: benign predictions come from REVEL and FATHMM, while pathogenic predictions are made by FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Predictions labeled Uncertain (Rosetta, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is Uncertain and therefore not considered evidence. Overall, the majority of reliable tools indicate a pathogenic impact, and this conclusion does not contradict any ClinVar annotation (none present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.289888 | Uncertain | 0.903 | 0.150 | 0.125 | -11.684 | Likely Pathogenic | 0.961 | Likely Pathogenic | Likely Pathogenic | 2.44 | Destabilizing | 0.1 | 0.90 | Ambiguous | 1.67 | Ambiguous | 0.72 | Ambiguous | 0.469 | Likely Benign | -5.63 | Deleterious | 0.999 | Probably Damaging | 0.991 | Probably Damaging | 3.06 | Benign | 0.01 | Affected | 0.3529 | 0.5161 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1376G>T | G459V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G459V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact; premPS is inconclusive and therefore not counted. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the consensus of these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for G459V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.289888 | Uncertain | 0.903 | 0.150 | 0.125 | -13.606 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.79 | Destabilizing | 0.1 | 5.01 | Destabilizing | 4.40 | Destabilizing | 0.71 | Ambiguous | 0.605 | Likely Pathogenic | -8.46 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.99 | Benign | 0.00 | Affected | 0.0997 | 0.4240 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1378A>C | K460Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K460Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, SIFT, and FATHMM, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and ESM1b. Predictions that are inconclusive include FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments give a pathogenic consensus from the SGM method (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) and an uncertain result from AlphaMissense‑Optimized; Foldetta likewise reports no definitive stability change. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.289547 | Uncertain | 0.938 | 0.150 | 0.125 | -9.404 | Likely Pathogenic | 0.793 | Likely Pathogenic | Ambiguous | 0.71 | Ambiguous | 0.0 | 0.86 | Ambiguous | 0.79 | Ambiguous | 0.86 | Ambiguous | 0.312 | Likely Benign | -3.15 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 3.35 | Benign | 0.14 | Tolerated | 0.4523 | 0.1454 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1378A>G | K460E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K460E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. In contrast, a majority of tools predict a pathogenic impact: AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, PROVEAN, polyPhen‑2 (HumDiv and HumVar), Rosetta, and premPS all indicate pathogenicity, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No prediction or folding‑stability result is missing or inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for K460E, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.289547 | Uncertain | 0.938 | 0.150 | 0.125 | -13.304 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 1.68 | Ambiguous | 0.0 | 2.02 | Destabilizing | 1.85 | Ambiguous | 1.05 | Destabilizing | 0.398 | Likely Benign | -3.40 | Deleterious | 0.999 | Probably Damaging | 0.991 | Probably Damaging | 3.34 | Benign | 0.09 | Tolerated | 0.3966 | 0.1022 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1379A>C | K460T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K460T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two consensus groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Stability‑based methods (FoldX, Rosetta, premPS, Foldetta) yield uncertain or inconclusive results. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus majority vote (AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, while Foldetta’s combined FoldX‑MD/Rosetta analysis remains inconclusive. Overall, the majority of evidence points to a pathogenic impact for K460T, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.289547 | Uncertain | 0.938 | 0.150 | 0.125 | -11.187 | Likely Pathogenic | 0.967 | Likely Pathogenic | Likely Pathogenic | 1.23 | Ambiguous | 0.1 | 1.16 | Ambiguous | 1.20 | Ambiguous | 0.77 | Ambiguous | 0.208 | Likely Benign | -5.02 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.35 | Benign | 0.07 | Tolerated | 0.1858 | 0.3848 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1379A>G | K460R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K460R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Foldetta, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign or likely benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity, while Rosetta and premPS are inconclusive. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Taken together, the overwhelming majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.155435 | Structured | 0.289547 | Uncertain | 0.938 | 0.150 | 0.125 | -5.349 | Likely Benign | 0.175 | Likely Benign | Likely Benign | -0.41 | Likely Benign | 0.0 | 0.63 | Ambiguous | 0.11 | Likely Benign | 0.55 | Ambiguous | 0.103 | Likely Benign | -1.52 | Neutral | 0.991 | Probably Damaging | 0.962 | Probably Damaging | 3.46 | Benign | 0.63 | Tolerated | 0.5093 | 0.1169 | Weaken | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||
| c.1379A>T | K460M 2D 3DClick to see structure in 3D Viewer AISynGAP1 K460M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, Rosetta, Foldetta, premPS, and FATHMM, while pathogenic calls arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Two tools give uncertain results: FoldX and AlphaMissense‑Optimized. When high‑accuracy methods are considered separately, AlphaMissense‑Optimized remains inconclusive, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of predictions lean toward pathogenicity, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.289547 | Uncertain | 0.938 | 0.150 | 0.125 | -10.351 | Likely Pathogenic | 0.943 | Likely Pathogenic | Ambiguous | 0.61 | Ambiguous | 0.0 | 0.18 | Likely Benign | 0.40 | Likely Benign | 0.20 | Likely Benign | 0.252 | Likely Benign | -4.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.29 | Benign | 0.02 | Affected | 0.1148 | 0.4522 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.137C>A | P46H 2D AIThe SynGAP1 missense variant P46H is evaluated by multiple in silico tools. Consensus from SGM‑Consensus indicates a likely benign effect, and the variant is not reported in ClinVar or gnomAD. Functional predictors that agree on a benign outcome include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Predictors that flag a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports likely benign, while Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact for P46H, and this conclusion is not contradicted by any ClinVar annotation. The variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.390993 | Structured | 0.433588 | Uncertain | 0.549 | 0.741 | 0.375 | -4.022 | Likely Benign | 0.428 | Ambiguous | Likely Benign | 0.097 | Likely Benign | -0.43 | Neutral | 0.992 | Probably Damaging | 0.977 | Probably Damaging | 4.10 | Benign | 0.00 | Affected | 0.2360 | 0.5229 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.137C>T | P46L 2D AIThe SynGAP1 missense variant P46L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for P46L, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.390993 | Structured | 0.433588 | Uncertain | 0.549 | 0.741 | 0.375 | -5.135 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 0.076 | Likely Benign | -1.14 | Neutral | 0.909 | Possibly Damaging | 0.927 | Probably Damaging | 4.12 | Benign | 0.00 | Affected | 0.2469 | 0.6633 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.1380G>C | K460N 2D AIThe SynGAP1 missense variant K460N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, SIFT, and FATHMM, whereas those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a pathogenic signal: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic; and the protein‑folding stability method Foldetta, which integrates FoldX‑MD and Rosetta outputs, indicates a benign effect. Overall, the balance of evidence—six pathogenic versus five benign predictions, a pathogenic SGM Consensus, and a pathogenic AlphaMissense‑Optimized—suggests that K460N is most likely pathogenic. This conclusion does not contradict ClinVar status, as the variant is not currently catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.289547 | Uncertain | 0.938 | 0.150 | 0.125 | -11.988 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.25 | Likely Benign | 0.5 | 0.45 | Likely Benign | 0.35 | Likely Benign | 0.89 | Ambiguous | 0.202 | Likely Benign | -4.13 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.29 | Benign | 0.06 | Tolerated | 0.3669 | 0.1599 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1380G>T | K460N 2D AIThe SynGAP1 K460N missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus methods further support a pathogenic interpretation: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also pathogenic; whereas Foldetta, which integrates FoldX‑MD and Rosetta outputs, indicates a benign effect. Overall, the majority of evidence—including the high‑accuracy tools—points to a pathogenic impact for K460N. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.289547 | Uncertain | 0.938 | 0.150 | 0.125 | -11.988 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.25 | Likely Benign | 0.5 | 0.45 | Likely Benign | 0.35 | Likely Benign | 0.89 | Ambiguous | 0.202 | Likely Benign | -4.13 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.29 | Benign | 0.06 | Tolerated | 0.3669 | 0.1599 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1381G>A | A461T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A461T missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and ESM1b. Four tools (FoldX, Rosetta, Foldetta, premPS) returned uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta is also inconclusive. Overall, the balance of evidence favors a benign classification, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.179055 | Structured | 0.292531 | Uncertain | 0.936 | 0.151 | 0.125 | -10.885 | Likely Pathogenic | 0.323 | Likely Benign | Likely Benign | 1.21 | Ambiguous | 0.2 | 0.51 | Ambiguous | 0.86 | Ambiguous | 0.65 | Ambiguous | 0.214 | Likely Benign | -3.27 | Deleterious | 0.508 | Possibly Damaging | 0.042 | Benign | 3.40 | Benign | 0.13 | Tolerated | 0.1083 | 0.5930 | 1 | 0 | -2.5 | 30.03 | ||||||||||||||||||||||||||||||
| c.1381G>C | A461P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A461P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, and FATHMM, whereas the majority of tools predict a pathogenic outcome: SGM‑Consensus (Likely Pathogenic), Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; premPS remains uncertain. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is likely pathogenic, and Foldetta is pathogenic. Based on the overall consensus of the available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.292531 | Uncertain | 0.936 | 0.151 | 0.125 | -13.869 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | -0.35 | Likely Benign | 0.1 | 5.09 | Destabilizing | 2.37 | Destabilizing | 0.84 | Ambiguous | 0.451 | Likely Benign | -4.52 | Deleterious | 0.999 | Probably Damaging | 0.849 | Possibly Damaging | 3.32 | Benign | 0.03 | Affected | 0.1748 | 0.3949 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1381G>T | A461S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A461S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 (HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv), SIFT, and ESM1b. Four tools (FoldX, Rosetta, Foldetta, premPS) give uncertain results and are not considered evidence for either side. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta’s stability analysis is also unavailable. Overall, the balance of evidence leans toward a benign effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.179055 | Structured | 0.292531 | Uncertain | 0.936 | 0.151 | 0.125 | -10.663 | Likely Pathogenic | 0.309 | Likely Benign | Likely Benign | 0.87 | Ambiguous | 0.0 | 1.18 | Ambiguous | 1.03 | Ambiguous | 0.63 | Ambiguous | 0.236 | Likely Benign | -2.74 | Deleterious | 0.600 | Possibly Damaging | 0.289 | Benign | 3.36 | Benign | 0.02 | Affected | 0.2401 | 0.4551 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||
| c.1382C>A | A461D 2D AIThe SynGAP1 missense variant A461D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and FATHMM. The majority of tools predict a pathogenic impact: premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). FoldX and Foldetta are inconclusive, providing no definitive evidence. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence indicates that A461D is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.292531 | Uncertain | 0.936 | 0.151 | 0.125 | -14.918 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.89 | Ambiguous | 1.1 | 2.18 | Destabilizing | 1.54 | Ambiguous | 1.09 | Destabilizing | 0.477 | Likely Benign | -5.47 | Deleterious | 0.997 | Probably Damaging | 0.792 | Possibly Damaging | 3.32 | Benign | 0.01 | Affected | 0.1533 | 0.2016 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.1382C>G | A461G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A461G has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into three groups: benign predictions come from REVEL, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions arise from Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b; the remaining tools (FoldX, premPS, AlphaMissense‑Default, Foldetta) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points toward a pathogenic effect, with no ClinVar record to contradict this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.179055 | Structured | 0.292531 | Uncertain | 0.936 | 0.151 | 0.125 | -11.360 | Likely Pathogenic | 0.556 | Ambiguous | Likely Benign | 1.46 | Ambiguous | 0.1 | 2.06 | Destabilizing | 1.76 | Ambiguous | 0.83 | Ambiguous | 0.276 | Likely Benign | -3.79 | Deleterious | 0.991 | Probably Damaging | 0.628 | Possibly Damaging | 3.32 | Benign | 0.00 | Affected | 0.1988 | 0.3317 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||
| c.1382C>T | A461V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A461V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that predict a pathogenic effect are PROVEAN, polyPhen2_HumDiv, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Because the majority of standard predictors (nine benign vs. three pathogenic) favor a benign outcome, the variant is most likely benign, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.179055 | Structured | 0.292531 | Uncertain | 0.936 | 0.151 | 0.125 | -9.968 | Likely Pathogenic | 0.436 | Ambiguous | Likely Benign | -0.08 | Likely Benign | 0.3 | 0.34 | Likely Benign | 0.13 | Likely Benign | 0.02 | Likely Benign | 0.141 | Likely Benign | -3.05 | Deleterious | 0.983 | Probably Damaging | 0.273 | Benign | 3.43 | Benign | 0.66 | Tolerated | 0.0819 | 0.5078 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||
| c.1384A>C | K462Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K462Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL, FoldX, Rosetta, premPS, SIFT, and FATHMM, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. Overall, the majority of tools and the protein‑stability analysis favor a benign effect, while the consensus pathogenic score introduces uncertainty. Thus, the variant is most likely benign; this assessment does not contradict ClinVar status, which has no entry for K462Q. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.297737 | Uncertain | 0.921 | 0.159 | 0.125 | -12.144 | Likely Pathogenic | 0.809 | Likely Pathogenic | Ambiguous | 0.12 | Likely Benign | 0.1 | 0.34 | Likely Benign | 0.23 | Likely Benign | 0.48 | Likely Benign | 0.384 | Likely Benign | -3.85 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 3.40 | Benign | 0.15 | Tolerated | 0.4639 | 0.1286 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1384A>G | K462E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K462E is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, and FATHMM. Tools that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Rosetta and Foldetta are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta is uncertain. Overall, the majority of predictions (7 pathogenic vs. 5 benign) and the high‑accuracy tools support a pathogenic classification. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.297737 | Uncertain | 0.921 | 0.159 | 0.125 | -14.696 | Likely Pathogenic | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.34 | Likely Benign | 0.0 | 1.56 | Ambiguous | 0.95 | Ambiguous | 0.33 | Likely Benign | 0.433 | Likely Benign | -3.88 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 3.49 | Benign | 0.15 | Tolerated | 0.4145 | 0.1022 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1385A>C | K462T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K462T missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, and FATHMM, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. Overall, the majority of evidence points toward a pathogenic effect. This conclusion is not contradicted by ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.297737 | Uncertain | 0.921 | 0.159 | 0.125 | -11.586 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 0.55 | Ambiguous | 0.0 | 1.08 | Ambiguous | 0.82 | Ambiguous | 0.30 | Likely Benign | 0.414 | Likely Benign | -5.82 | Deleterious | 0.999 | Probably Damaging | 1.000 | Probably Damaging | 3.47 | Benign | 0.08 | Tolerated | 0.2095 | 0.3257 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1385A>T | K462M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K462M missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, FATHMM, premPS, and the protein‑folding stability method Foldetta; pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score. Rosetta’s output is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also pathogenic, while Foldetta indicates a benign effect on protein stability. No evidence from ClinVar contradicts these findings. Overall, the majority of predictive tools and the consensus score support a pathogenic classification, suggesting that K462M is most likely pathogenic rather than benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.297737 | Uncertain | 0.921 | 0.159 | 0.125 | -12.837 | Likely Pathogenic | 0.970 | Likely Pathogenic | Likely Pathogenic | -0.23 | Likely Benign | 0.1 | 0.86 | Ambiguous | 0.32 | Likely Benign | 0.17 | Likely Benign | 0.475 | Likely Benign | -5.82 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.40 | Benign | 0.02 | Affected | 0.1282 | 0.3932 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1387G>A | D463N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D463N missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, and FATHMM. Those that predict a pathogenic impact are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. Predictions that are uncertain (Foldetta, AlphaMissense‑Optimized, Rosetta) are treated as unavailable and do not influence the overall assessment. High‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; Foldetta is uncertain. Overall, the majority of available predictions lean toward a benign effect, and this conclusion does not contradict any ClinVar status (none reported). Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | -11.428 | Likely Pathogenic | 0.812 | Likely Pathogenic | Ambiguous | 0.10 | Likely Benign | 0.1 | 1.19 | Ambiguous | 0.65 | Ambiguous | 0.40 | Likely Benign | 0.217 | Likely Benign | -4.37 | Deleterious | 0.880 | Possibly Damaging | 0.430 | Benign | 3.33 | Benign | 0.10 | Tolerated | 0.1152 | 0.5531 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1387G>C | D463H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant D463H is not reported in ClinVar and is absent from gnomAD. Consensus from standard prediction algorithms shows a split: benign predictions come from REVEL, FoldX, SIFT, and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic, SGM Consensus confirms Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains Uncertain. No evidence from ClinVar contradicts these findings. Overall, the preponderance of computational evidence indicates that D463H is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | -13.151 | Likely Pathogenic | 0.970 | Likely Pathogenic | Likely Pathogenic | 0.20 | Likely Benign | 0.1 | 0.85 | Ambiguous | 0.53 | Ambiguous | 0.57 | Ambiguous | 0.356 | Likely Benign | -5.96 | Deleterious | 0.996 | Probably Damaging | 0.852 | Possibly Damaging | 3.35 | Benign | 0.11 | Tolerated | 0.1341 | 0.6156 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.1387G>T | D463Y 2D 3DClick to see structure in 3D Viewer AIClinVar reports no entry for this SynGAP1 D463Y variant, and it is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, and FATHMM. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Uncertain results come from AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of conventional tools lean toward pathogenicity, while the high‑accuracy Foldetta suggests benign. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | -14.387 | Likely Pathogenic | 0.904 | Likely Pathogenic | Ambiguous | -0.14 | Likely Benign | 0.1 | 0.77 | Ambiguous | 0.32 | Likely Benign | 0.07 | Likely Benign | 0.399 | Likely Benign | -7.95 | Deleterious | 0.998 | Probably Damaging | 0.904 | Possibly Damaging | 3.35 | Benign | 0.14 | Tolerated | 0.0540 | 0.6213 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1388A>C | D463A 2D 3DClick to see structure in 3D Viewer AISynGAP1 D463A is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, and FATHMM, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Tools with inconclusive results are AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the predictions are split, with an equal number of benign and pathogenic calls, and the high‑accuracy methods provide conflicting outcomes. Therefore, the variant is most likely of uncertain significance, which does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | Uncertain | 1 | -8.607 | Likely Pathogenic | 0.894 | Likely Pathogenic | Ambiguous | -0.04 | Likely Benign | 0.1 | 0.96 | Ambiguous | 0.46 | Likely Benign | 0.43 | Likely Benign | 0.425 | Likely Benign | -6.96 | Deleterious | 0.978 | Probably Damaging | 0.602 | Possibly Damaging | 3.33 | Benign | 0.29 | Tolerated | 0.3591 | 0.5169 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||
| c.1388A>G | D463G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D463G missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and ESM1b. Five tools favor pathogenicity versus three favor benign, with the remaining five (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) yielding uncertain results. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is inconclusive; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, also remains inconclusive. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | -10.713 | Likely Pathogenic | 0.921 | Likely Pathogenic | Ambiguous | 0.66 | Ambiguous | 0.1 | 1.93 | Ambiguous | 1.30 | Ambiguous | 0.54 | Ambiguous | 0.422 | Likely Benign | -6.36 | Deleterious | 0.994 | Probably Damaging | 0.824 | Possibly Damaging | 3.32 | Benign | 0.09 | Tolerated | 0.3751 | 0.4898 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.1388A>T | D463V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D463V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, premPS, SIFT, and FATHMM, whereas those that agree on a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Uncertain predictions come from Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points toward a pathogenic impact for D463V, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | -12.374 | Likely Pathogenic | 0.880 | Likely Pathogenic | Ambiguous | 0.23 | Likely Benign | 0.1 | 0.98 | Ambiguous | 0.61 | Ambiguous | 0.33 | Likely Benign | 0.521 | Likely Pathogenic | -7.95 | Deleterious | 0.973 | Probably Damaging | 0.658 | Possibly Damaging | 3.31 | Benign | 0.09 | Tolerated | 0.0713 | 0.5526 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1389C>A | D463E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D463E missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and premPS. Only PROVEAN predicts a pathogenic outcome, while Rosetta and AlphaMissense‑Default are uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. No prediction or stability result is missing or inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | -5.170 | Likely Benign | 0.527 | Ambiguous | Likely Benign | -0.42 | Likely Benign | 0.1 | 0.50 | Ambiguous | 0.04 | Likely Benign | 0.47 | Likely Benign | 0.162 | Likely Benign | -2.58 | Deleterious | 0.012 | Benign | 0.003 | Benign | 3.47 | Benign | 0.29 | Tolerated | 0.1330 | 0.5177 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||
| c.1389C>G | D463E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D463E missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and premPS. Only PROVEAN predicts a pathogenic outcome, while Rosetta and AlphaMissense‑Default are uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) resolves to benign, and Foldetta also indicates benign stability. No prediction or stability result is missing or inconclusive. Overall, the variant is most likely benign based on the collective evidence, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.260850 | Structured | 0.305622 | Uncertain | 0.940 | 0.176 | 0.000 | -5.170 | Likely Benign | 0.527 | Ambiguous | Likely Benign | -0.42 | Likely Benign | 0.1 | 0.50 | Ambiguous | 0.04 | Likely Benign | 0.47 | Likely Benign | 0.162 | Likely Benign | -2.58 | Deleterious | 0.012 | Benign | 0.003 | Benign | 3.47 | Benign | 0.29 | Tolerated | 0.1330 | 0.5177 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||
| c.1390T>A | F464I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F464I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are limited to FATHMM, while all other evaluated algorithms—including SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.313424 | Uncertain | 0.961 | 0.178 | 0.000 | -11.210 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 4.77 | Destabilizing | 0.2 | 4.35 | Destabilizing | 4.56 | Destabilizing | 1.23 | Destabilizing | 0.539 | Likely Pathogenic | -5.97 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 3.44 | Benign | 0.03 | Affected | 0.1439 | 0.2002 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1390T>C | F464L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F464L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions are returned by SGM‑Consensus (Likely Pathogenic), premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability assessments. High‑accuracy methods specifically indicate pathogenicity: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, while Foldetta’s combined FoldX‑MD and Rosetta output is uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this assessment does not conflict with any ClinVar annotation because the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.313424 | Uncertain | 0.961 | 0.178 | 0.000 | -10.482 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.63 | Ambiguous | 0.3 | 0.95 | Ambiguous | 1.29 | Ambiguous | 1.12 | Destabilizing | 0.435 | Likely Benign | -5.97 | Deleterious | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 3.93 | Benign | 0.22 | Tolerated | 0.1664 | 0.2883 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1391T>A | F464Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F464Y is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM, whereas a majority of tools (SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show that the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome, while AlphaMissense‑Optimized and Foldetta provide uncertain results and are treated as unavailable evidence. Overall, the balance of evidence points to a pathogenic effect for F464Y, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.313424 | Uncertain | 0.961 | 0.178 | 0.000 | -10.056 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 1.35 | Ambiguous | 0.1 | 0.24 | Likely Benign | 0.80 | Ambiguous | 1.31 | Destabilizing | 0.387 | Likely Benign | -2.98 | Deleterious | 0.979 | Probably Damaging | 0.953 | Probably Damaging | 3.28 | Benign | 0.01 | Affected | 0.1081 | 0.1523 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1391T>C | F464S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F464S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.313424 | Uncertain | 0.961 | 0.178 | 0.000 | -13.361 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 4.84 | Destabilizing | 0.0 | 4.90 | Destabilizing | 4.87 | Destabilizing | 2.52 | Destabilizing | 0.748 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.27 | Benign | 0.00 | Affected | 0.3700 | 0.0358 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1391T>G | F464C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F464C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.313424 | Uncertain | 0.961 | 0.178 | 0.000 | -13.011 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.18 | Destabilizing | 0.0 | 4.31 | Destabilizing | 4.25 | Destabilizing | 2.20 | Destabilizing | 0.740 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.26 | Benign | 0.00 | Affected | 0.2298 | 0.1219 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.1392C>A | F464L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F464L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions are returned by SGM‑Consensus (Likely Pathogenic), premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability assessments. High‑accuracy methods specifically indicate pathogenicity: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, while Foldetta’s combined FoldX‑MD and Rosetta output is uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this assessment does not conflict with any ClinVar annotation because the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.313424 | Uncertain | 0.961 | 0.178 | 0.000 | -10.482 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.63 | Ambiguous | 0.3 | 0.95 | Ambiguous | 1.29 | Ambiguous | 1.12 | Destabilizing | 0.279 | Likely Benign | -5.97 | Deleterious | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 3.93 | Benign | 0.22 | Tolerated | 0.1664 | 0.2883 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1392C>G | F464L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F464L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions are returned by SGM‑Consensus (Likely Pathogenic), premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability assessments. High‑accuracy methods specifically indicate pathogenicity: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, while Foldetta’s combined FoldX‑MD and Rosetta output is uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this assessment does not conflict with any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.313424 | Uncertain | 0.961 | 0.178 | 0.000 | -10.482 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.63 | Ambiguous | 0.3 | 0.95 | Ambiguous | 1.29 | Ambiguous | 1.12 | Destabilizing | 0.279 | Likely Benign | -5.97 | Deleterious | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 3.93 | Benign | 0.22 | Tolerated | 0.1664 | 0.2883 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1393C>A | L465I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L465I missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score classifies the variant as benign, whereas the Foldetta stability assessment is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive due to a 2‑to‑2 split. Overall, the evidence is mixed; the balance of predictions leans toward a benign interpretation, and this does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.346032 | Structured | 0.319240 | Uncertain | 0.956 | 0.202 | 0.000 | -9.672 | Likely Pathogenic | 0.770 | Likely Pathogenic | Likely Benign | 1.21 | Ambiguous | 0.1 | 1.27 | Ambiguous | 1.24 | Ambiguous | 0.78 | Ambiguous | 0.258 | Likely Benign | -1.99 | Neutral | 0.998 | Probably Damaging | 0.997 | Probably Damaging | 2.54 | Benign | 0.08 | Tolerated | 0.0967 | 0.3539 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1393C>T | L465F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L465F is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL and Rosetta, whereas the majority of tools predict a pathogenic impact: FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Two tools (Foldetta and premPS) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus also pathogenic, and Foldetta remains uncertain. Overall, the consensus of high‑confidence predictors points to a pathogenic effect for L465F. This conclusion is not contradicted by ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.346032 | Structured | 0.319240 | Uncertain | 0.956 | 0.202 | 0.000 | -12.626 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | 3.11 | Destabilizing | 0.7 | 0.05 | Likely Benign | 1.58 | Ambiguous | 0.52 | Ambiguous | 0.432 | Likely Benign | -3.98 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.44 | Pathogenic | 0.01 | Affected | 0.0771 | 0.2759 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1394T>A | L465H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L465H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic. Based on the consensus of all available predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.346032 | Structured | 0.319240 | Uncertain | 0.956 | 0.202 | 0.000 | -16.751 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.15 | Destabilizing | 0.4 | 2.29 | Destabilizing | 2.72 | Destabilizing | 2.54 | Destabilizing | 0.764 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.1289 | 0.1089 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1394T>G | L465R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L465R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. No inconclusive or missing results are present. Based on the consensus of all available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.346032 | Structured | 0.319240 | Uncertain | 0.956 | 0.202 | 0.000 | -17.976 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.52 | Destabilizing | 0.7 | 4.44 | Destabilizing | 4.48 | Destabilizing | 2.41 | Destabilizing | 0.761 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.1597 | 0.0963 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1396T>A | S466T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S466T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Uncertain results come from Rosetta and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta as Benign. Taken together, the majority of evidence points to a benign impact. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.311707 | Structured | 0.322353 | Uncertain | 0.933 | 0.227 | 0.000 | -5.488 | Likely Benign | 0.314 | Likely Benign | Likely Benign | 0.30 | Likely Benign | 0.2 | -1.02 | Ambiguous | -0.36 | Likely Benign | -0.65 | Ambiguous | 0.414 | Likely Benign | 1.05 | Neutral | 0.740 | Possibly Damaging | 0.872 | Possibly Damaging | -1.51 | Pathogenic | 1.00 | Tolerated | 0.0928 | 0.5152 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1396T>C | S466P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S466P is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect; the only inconclusive result is from premPS (Uncertain). High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts Pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.311707 | Structured | 0.322353 | Uncertain | 0.933 | 0.227 | 0.000 | -13.107 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.67 | Destabilizing | 0.2 | 2.83 | Destabilizing | 2.75 | Destabilizing | 0.95 | Ambiguous | 0.811 | Likely Pathogenic | -3.02 | Deleterious | 0.995 | Probably Damaging | 0.986 | Probably Damaging | -1.55 | Pathogenic | 0.04 | Affected | 0.1617 | 0.4915 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.1396T>G | S466A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S466A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: pathogenic scores are given by REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM, whereas benign predictions come from SIFT, PROVEAN, premPS, ESM1b, Rosetta, Foldetta, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, the SGM‑Consensus itself is Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports Benign. FoldX alone is Uncertain, but this does not alter the overall consensus. Taken together, the majority of evidence indicates the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.311707 | Structured | 0.322353 | Uncertain | 0.933 | 0.227 | 0.000 | -6.928 | Likely Benign | 0.228 | Likely Benign | Likely Benign | -0.50 | Ambiguous | 0.1 | 0.06 | Likely Benign | -0.22 | Likely Benign | 0.37 | Likely Benign | 0.537 | Likely Pathogenic | -1.46 | Neutral | 0.909 | Possibly Damaging | 0.987 | Probably Damaging | -1.51 | Pathogenic | 0.10 | Tolerated | 0.4245 | 0.3996 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.1397C>T | S466L 2D  AIThe SynGAP1 missense variant S466L is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include FoldX, while the majority of tools predict a pathogenic impact: REVEL, SIFT, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. Tools with uncertain or inconclusive results are AlphaMissense‑Optimized, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for S466L. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.311707 | Structured | 0.322353 | Uncertain | 0.933 | 0.227 | 0.000 | -9.417 | Likely Pathogenic | 0.806 | Likely Pathogenic | Ambiguous | -0.36 | Likely Benign | 1.6 | -1.60 | Ambiguous | -0.98 | Ambiguous | -0.52 | Ambiguous | 0.768 | Likely Pathogenic | -3.05 | Deleterious | 0.995 | Probably Damaging | 0.991 | Probably Damaging | -1.56 | Pathogenic | 0.03 | Affected | 0.0666 | 0.4884 | -3 | -2 | 4.6 | 26.08 | |||||||||||||||||||||||||||||
| c.1399G>A | D467N 2D 3DClick to see structure in 3D Viewer AISynGAP1 D467N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a predominance of pathogenic calls: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of the four high‑accuracy predictors) all predict a deleterious effect. Benign predictions come from FoldX, premPS, and SIFT. Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive. High‑accuracy methods specifically give an uncertain result for AlphaMissense‑Optimized, a pathogenic verdict for the SGM‑Consensus, and an uncertain outcome for Foldetta. Overall, the balance of evidence favors a pathogenic impact for D467N, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.329932 | Uncertain | 0.940 | 0.246 | 0.000 | -11.881 | Likely Pathogenic | 0.913 | Likely Pathogenic | Ambiguous | 0.43 | Likely Benign | 0.1 | 1.63 | Ambiguous | 1.03 | Ambiguous | 0.38 | Likely Benign | 0.673 | Likely Pathogenic | -4.82 | Deleterious | 0.987 | Probably Damaging | 0.990 | Probably Damaging | -1.22 | Pathogenic | 0.06 | Tolerated | 0.0936 | 0.4879 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1399G>C | D467H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D467H is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only premPS, whereas the remaining evaluated algorithms uniformly predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta yield uncertain results and are treated as unavailable. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also pathogenic; Foldetta remains uncertain. Overall, the variant is most likely pathogenic based on the consensus of the available predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.329932 | Uncertain | 0.940 | 0.246 | 0.000 | -13.348 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.05 | Ambiguous | 0.1 | 0.59 | Ambiguous | 0.82 | Ambiguous | 0.32 | Likely Benign | 0.851 | Likely Pathogenic | -6.71 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.31 | Pathogenic | 0.02 | Affected | 0.1074 | 0.5564 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.1399G>T | D467Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D467Y is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from FoldX, Foldetta, premPS, and SIFT, whereas pathogenic calls are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy methods give a clearer picture: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. No evidence from Rosetta is available due to its uncertain status. Overall, the majority of high‑confidence predictions lean toward pathogenicity, and this does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.329932 | Uncertain | 0.940 | 0.246 | 0.000 | -14.373 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.36 | Likely Benign | 0.1 | -0.60 | Ambiguous | -0.12 | Likely Benign | 0.07 | Likely Benign | 0.846 | Likely Pathogenic | -8.70 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.28 | Pathogenic | 0.07 | Tolerated | 0.0517 | 0.5521 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.139C>G | R47G 2D AIThe SynGAP1 missense variant R47G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence points to a benign impact for R47G, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.318242 | Structured | 0.436559 | Uncertain | 0.520 | 0.719 | 0.125 | -6.670 | Likely Benign | 0.616 | Likely Pathogenic | Likely Benign | 0.169 | Likely Benign | -1.79 | Neutral | 0.686 | Possibly Damaging | 0.630 | Possibly Damaging | 4.04 | Benign | 0.00 | Affected | 0.3471 | 0.3758 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.1400A>C | D467A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D467A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS and SIFT, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools with uncertain or inconclusive results—FoldX, Rosetta, and Foldetta—do not provide decisive evidence. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.329932 | Uncertain | 0.940 | 0.246 | 0.000 | -12.499 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.83 | Ambiguous | 0.1 | 0.79 | Ambiguous | 0.81 | Ambiguous | 0.23 | Likely Benign | 0.790 | Likely Pathogenic | -7.71 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.19 | Pathogenic | 0.07 | Tolerated | 0.3134 | 0.5117 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.1400A>G | D467G 2D AISynGAP1 missense variant D467G is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: the single benign prediction comes from premPS, while all other evaluated algorithms—including SGM‑Consensus, REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—label the change as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports pathogenic. No prediction is inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.329932 | Uncertain | 0.940 | 0.246 | 0.000 | -12.973 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 1.87 | Ambiguous | 0.8 | 2.55 | Destabilizing | 2.21 | Destabilizing | 0.23 | Likely Benign | 0.910 | Likely Pathogenic | -6.81 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | -1.32 | Pathogenic | 0.03 | Affected | 0.3455 | 0.4908 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.1400A>T | D467V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D467V missense variant is not reported in ClinVar or gnomAD. Prediction tools cluster into two groups: benign predictions come from Rosetta and premPS, while the majority—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—label the change as pathogenic. Two tools, FoldX and Foldetta, give uncertain results. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus indicates likely pathogenic, and Foldetta remains uncertain. Overall, the consensus of high‑confidence predictors points to a pathogenic effect, and this conclusion is consistent with the absence of any ClinVar annotation or gnomAD observation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.329932 | Uncertain | 0.940 | 0.246 | 0.000 | -15.041 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 1.24 | Ambiguous | 0.1 | 0.08 | Likely Benign | 0.66 | Ambiguous | 0.02 | Likely Benign | 0.893 | Likely Pathogenic | -8.70 | Deleterious | 0.997 | Probably Damaging | 0.997 | Probably Damaging | -1.28 | Pathogenic | 0.02 | Affected | 0.0644 | 0.5274 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1402A>C | M468L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M468L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumVar and FATHMM. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments—all available—show benign predictions: AlphaMissense‑Optimized (Benign), SGM‑Consensus (Likely Benign), and Foldetta (Benign). Based on the overall consensus of the majority of tools and the high‑accuracy predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.284882 | Structured | 0.339253 | Uncertain | 0.932 | 0.257 | 0.000 | -6.886 | Likely Benign | 0.290 | Likely Benign | Likely Benign | 0.43 | Likely Benign | 0.1 | -0.38 | Likely Benign | 0.03 | Likely Benign | -0.35 | Likely Benign | 0.412 | Likely Benign | 0.19 | Neutral | 0.359 | Benign | 0.654 | Possibly Damaging | -0.73 | Pathogenic | 1.00 | Tolerated | 0.1564 | 0.4408 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1402A>T | M468L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M468L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumVar and FATHMM. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments—AlphaMissense‑Optimized, the SGM‑Consensus, and Foldetta (combining FoldX‑MD and Rosetta outputs)—all indicate a benign outcome. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.284882 | Structured | 0.339253 | Uncertain | 0.932 | 0.257 | 0.000 | -6.886 | Likely Benign | 0.290 | Likely Benign | Likely Benign | 0.43 | Likely Benign | 0.1 | -0.38 | Likely Benign | 0.03 | Likely Benign | -0.35 | Likely Benign | 0.412 | Likely Benign | 0.19 | Neutral | 0.359 | Benign | 0.654 | Possibly Damaging | -0.73 | Pathogenic | 1.00 | Tolerated | 0.1564 | 0.4408 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1403T>G | M468R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M468R is not reported in ClinVar and is absent from gnomAD. All evaluated in‑silico predictors classify the substitution as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Consequently, the variant is most likely pathogenic based on the consensus of predictive tools, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.284882 | Structured | 0.339253 | Uncertain | 0.932 | 0.257 | 0.000 | -16.180 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 2.66 | Destabilizing | 0.2 | 2.23 | Destabilizing | 2.45 | Destabilizing | 2.43 | Destabilizing | 0.837 | Likely Pathogenic | -4.64 | Deleterious | 0.939 | Possibly Damaging | 0.943 | Probably Damaging | -1.34 | Pathogenic | 0.00 | Affected | 0.1717 | 0.0637 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||
| c.1405G>A | A469T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A469T is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized; pathogenic predictions arise from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and the Foldetta stability assessment (combining FoldX‑MD and Rosetta). The high‑accuracy subset shows AlphaMissense‑Optimized as benign, whereas SGM Consensus and Foldetta both predict pathogenic. Overall, the majority of evidence supports a pathogenic effect, and this conclusion does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.343926 | Uncertain | 0.910 | 0.276 | 0.000 | Uncertain | 1 | -9.540 | Likely Pathogenic | 0.723 | Likely Pathogenic | Likely Benign | 2.26 | Destabilizing | 0.1 | 1.90 | Ambiguous | 2.08 | Destabilizing | 0.34 | Likely Benign | 0.527 | Likely Pathogenic | -1.46 | Neutral | 0.994 | Probably Damaging | 0.986 | Probably Damaging | -1.21 | Pathogenic | 0.42 | Tolerated | 0.1005 | 0.5884 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||
| c.1405G>C | A469P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A469P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only SIFT, whereas all other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Based on the overwhelming consensus of these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.343926 | Uncertain | 0.910 | 0.276 | 0.000 | -16.072 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 5.06 | Destabilizing | 0.3 | 8.83 | Destabilizing | 6.95 | Destabilizing | 1.02 | Destabilizing | 0.774 | Likely Pathogenic | -2.69 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | -1.33 | Pathogenic | 0.21 | Tolerated | 0.1606 | 0.4092 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1405G>T | A469S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A469S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include PROVEAN, SIFT, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are SGM‑Consensus, REVEL, Rosetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. FoldX, Foldetta, and premPS give uncertain or inconclusive results. High‑accuracy methods give the following: AlphaMissense‑Optimized predicts a benign change; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is uncertain. Overall, the majority of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.343926 | Uncertain | 0.910 | 0.276 | 0.000 | -9.051 | Likely Pathogenic | 0.627 | Likely Pathogenic | Likely Benign | 0.86 | Ambiguous | 0.0 | 2.10 | Destabilizing | 1.48 | Ambiguous | 0.81 | Ambiguous | 0.558 | Likely Pathogenic | -1.53 | Neutral | 0.953 | Possibly Damaging | 0.985 | Probably Damaging | -1.30 | Pathogenic | 0.41 | Tolerated | 0.2113 | 0.4505 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1406C>G | A469G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A469G is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD. Prediction tools that agree on a benign effect include PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise REVEL, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default; FoldX is uncertain. High‑accuracy methods give mixed results: AlphaMissense‑Optimized indicates benign, Foldetta indicates pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of tools (12 vs. 4) predict pathogenicity, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.278302 | Structured | 0.343926 | Uncertain | 0.910 | 0.276 | 0.000 | -4.438 | Likely Benign | 0.733 | Likely Pathogenic | Likely Benign | 1.99 | Ambiguous | 0.1 | 2.22 | Destabilizing | 2.11 | Destabilizing | 1.01 | Destabilizing | 0.569 | Likely Pathogenic | -2.42 | Neutral | 0.997 | Probably Damaging | 0.990 | Probably Damaging | -1.32 | Pathogenic | 0.37 | Tolerated | 0.1797 | 0.3078 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||
| c.1406C>T | A469V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A469V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect comprise FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and Foldetta; premPS and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta predicts pathogenic. With seven tools supporting pathogenicity versus four supporting benignity, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.278302 | Structured | 0.343926 | Uncertain | 0.910 | 0.276 | 0.000 | -8.858 | Likely Pathogenic | 0.459 | Ambiguous | Likely Benign | 2.30 | Destabilizing | 0.4 | 2.66 | Destabilizing | 2.48 | Destabilizing | -0.77 | Ambiguous | 0.426 | Likely Benign | 0.26 | Neutral | 0.997 | Probably Damaging | 0.976 | Probably Damaging | -1.20 | Pathogenic | 0.60 | Tolerated | 0.0827 | 0.5421 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||
| c.1409T>A | M470K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M470K is not reported in ClinVar (status: None) and is absent from gnomAD. Prediction tools cluster into two groups: the single benign call comes from SIFT, while all other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—label the change as pathogenic or likely pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports pathogenic. Thus, the variant is most likely pathogenic based on the consensus of available predictions, and this assessment does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.351497 | Uncertain | 0.908 | 0.272 | 0.000 | -14.327 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.77 | Destabilizing | 0.1 | 2.67 | Destabilizing | 2.72 | Destabilizing | 1.55 | Destabilizing | 0.823 | Likely Pathogenic | -5.42 | Deleterious | 0.923 | Possibly Damaging | 0.922 | Probably Damaging | -1.06 | Pathogenic | 0.20 | Tolerated | 0.1157 | 0.0656 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||
| c.1409T>G | M470R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M470R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only SIFT, whereas all other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.351497 | Uncertain | 0.908 | 0.272 | 0.000 | -13.161 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 2.75 | Destabilizing | 0.1 | 2.75 | Destabilizing | 2.75 | Destabilizing | 1.57 | Destabilizing | 0.823 | Likely Pathogenic | -5.47 | Deleterious | 0.963 | Probably Damaging | 0.943 | Probably Damaging | -1.19 | Pathogenic | 0.17 | Tolerated | 0.1399 | 0.0757 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||
| c.140G>C | R47P 2D AIThe SynGAP1 missense variant R47P is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic, two benign) and therefore unavailable as evidence. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant and is likewise unavailable. High‑accuracy tools specifically indicate a benign prediction from AlphaMissense‑Optimized, while the other high‑accuracy methods (SGM Consensus, Foldetta) provide no definitive assessment. Overall, the balance of evidence (five pathogenic vs. four benign predictions) suggests the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.318242 | Structured | 0.436559 | Uncertain | 0.520 | 0.719 | 0.125 | -10.316 | Likely Pathogenic | 0.746 | Likely Pathogenic | Likely Benign | 0.195 | Likely Benign | -1.63 | Neutral | 0.841 | Possibly Damaging | 0.809 | Possibly Damaging | 4.02 | Benign | 0.00 | Affected | 0.2050 | 0.4956 | 0 | -2 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||||||||||
| c.140G>T | R47L 2D AIThe SynGAP1 missense variant R47L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for R47L. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.318242 | Structured | 0.436559 | Uncertain | 0.520 | 0.719 | 0.125 | -5.758 | Likely Benign | 0.664 | Likely Pathogenic | Likely Benign | 0.130 | Likely Benign | -1.76 | Neutral | 0.686 | Possibly Damaging | 0.630 | Possibly Damaging | 4.04 | Benign | 0.00 | Affected | 0.1904 | 0.5206 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.1410G>A | M470I 2D AIThe SynGAP1 missense variant M470I is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include only SIFT, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) all predict a pathogenic impact. Predictions marked as uncertain (AlphaMissense‑Optimized, FoldX, Rosetta, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments show the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, while AlphaMissense‑Optimized and Foldetta are uncertain. Overall, the preponderance of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.351497 | Uncertain | 0.908 | 0.272 | 0.000 | -9.474 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 1.53 | Ambiguous | 0.7 | 1.34 | Ambiguous | 1.44 | Ambiguous | 0.84 | Ambiguous | 0.747 | Likely Pathogenic | -3.55 | Deleterious | 0.833 | Possibly Damaging | 0.886 | Possibly Damaging | -1.26 | Pathogenic | 0.07 | Tolerated | 0.1061 | 0.2827 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1410G>C | M470I 2D AIThe SynGAP1 missense variant M470I is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include only SIFT, whereas the remaining evidence—REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—predict pathogenicity. Results from high‑accuracy methods are mixed: AlphaMissense‑Optimized is uncertain, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta is uncertain. Overall, the preponderance of predictions supports a pathogenic effect for M470I, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.351497 | Uncertain | 0.908 | 0.272 | 0.000 | -9.474 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 1.53 | Ambiguous | 0.7 | 1.34 | Ambiguous | 1.44 | Ambiguous | 0.84 | Ambiguous | 0.747 | Likely Pathogenic | -3.55 | Deleterious | 0.833 | Possibly Damaging | 0.886 | Possibly Damaging | -1.26 | Pathogenic | 0.07 | Tolerated | 0.1061 | 0.2827 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1410G>T | M470I 2D AIThe SynGAP1 missense variant M470I is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include only SIFT, whereas the remaining evidence—REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—predict pathogenicity. Results from high‑accuracy methods are mixed: AlphaMissense‑Optimized is uncertain, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta is uncertain. Overall, the preponderance of predictions supports a pathogenic effect for M470I, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.351497 | Uncertain | 0.908 | 0.272 | 0.000 | -9.474 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 1.53 | Ambiguous | 0.7 | 1.34 | Ambiguous | 1.44 | Ambiguous | 0.84 | Ambiguous | 0.747 | Likely Pathogenic | -3.55 | Deleterious | 0.833 | Possibly Damaging | 0.886 | Possibly Damaging | -1.26 | Pathogenic | 0.07 | Tolerated | 0.1061 | 0.2827 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1411T>A | S471T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S471T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only FATHMM predicts pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign; the SGM‑Consensus (3 benign vs. 1 pathogenic) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is benign. No prediction or stability result is inconclusive. Overall, the evidence overwhelmingly supports a benign classification, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.305330 | Structured | 0.355411 | Uncertain | 0.888 | 0.261 | 0.000 | -5.780 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.59 | Ambiguous | 0.2 | -0.47 | Likely Benign | 0.06 | Likely Benign | 0.20 | Likely Benign | 0.257 | Likely Benign | -1.96 | Neutral | 0.000 | Benign | 0.000 | Benign | -1.18 | Pathogenic | 0.09 | Tolerated | 0.1307 | 0.5046 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1411T>C | S471P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S471P is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only polyPhen‑2 HumVar, whereas the remaining evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) all predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among the majority of tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.305330 | Structured | 0.355411 | Uncertain | 0.888 | 0.261 | 0.000 | -12.379 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 3.65 | Destabilizing | 0.2 | 9.24 | Destabilizing | 6.45 | Destabilizing | 0.84 | Ambiguous | 0.530 | Likely Pathogenic | -4.03 | Deleterious | 0.552 | Possibly Damaging | 0.141 | Benign | -1.31 | Pathogenic | 0.05 | Affected | 0.2007 | 0.4769 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.1411T>G | S471A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S471A is not reported in ClinVar and is absent from gnomAD. Across the spectrum of in‑silico predictors, the majority (REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) classify the change as benign, whereas only FATHMM predicts it as pathogenic; Rosetta is inconclusive. High‑accuracy assessments reinforce the benign interpretation: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. No evidence suggests pathogenicity, and the predictions do not contradict the absence of ClinVar annotation. Therefore, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.305330 | Structured | 0.355411 | Uncertain | 0.888 | 0.261 | 0.000 | -5.516 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.11 | Likely Benign | 0.1 | -0.61 | Ambiguous | -0.25 | Likely Benign | 0.23 | Likely Benign | 0.252 | Likely Benign | -1.93 | Neutral | 0.010 | Benign | 0.037 | Benign | -1.22 | Pathogenic | 0.29 | Tolerated | 0.4439 | 0.3691 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.1412C>A | S471Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S471Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, FoldX, premPS, and AlphaMissense‑Optimized; pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The high‑accuracy AlphaMissense‑Optimized scores the variant as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels it as likely pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, returns an uncertain stability change. No evidence from ClinVar contradicts these computational assessments. Overall, the preponderance of pathogenic predictions and the SGM Consensus suggest the variant is most likely pathogenic, consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.305330 | Structured | 0.355411 | Uncertain | 0.888 | 0.261 | 0.000 | -11.257 | Likely Pathogenic | 0.493 | Ambiguous | Likely Benign | -0.33 | Likely Benign | 0.1 | -1.31 | Ambiguous | -0.82 | Ambiguous | 0.19 | Likely Benign | 0.446 | Likely Benign | -4.79 | Deleterious | 0.980 | Probably Damaging | 0.584 | Possibly Damaging | -1.27 | Pathogenic | 0.03 | Affected | 0.0607 | 0.4417 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||
| c.1412C>T | S471F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S471F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, and AlphaMissense‑Optimized, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. The remaining tools—AlphaMissense‑Default, FoldX, Rosetta, and Foldetta—return uncertain or inconclusive results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts a benign effect; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Taken together, the majority of evidence points toward a pathogenic impact for S471F, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.305330 | Structured | 0.355411 | Uncertain | 0.888 | 0.261 | 0.000 | -10.777 | Likely Pathogenic | 0.492 | Ambiguous | Likely Benign | -0.54 | Ambiguous | 0.1 | -1.51 | Ambiguous | -1.03 | Ambiguous | 0.12 | Likely Benign | 0.423 | Likely Benign | -4.75 | Deleterious | 0.942 | Possibly Damaging | 0.487 | Possibly Damaging | -1.26 | Pathogenic | 0.02 | Affected | 0.0553 | 0.4691 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||
| c.1414G>A | E472K 2D AIThe SynGAP1 missense variant E472K is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect; the only inconclusive result is from premPS, which is listed as uncertain. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a destabilizing, pathogenic effect. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -15.214 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.01 | Destabilizing | 1.2 | 3.23 | Destabilizing | 2.62 | Destabilizing | 0.78 | Ambiguous | 0.670 | Likely Pathogenic | -3.95 | Deleterious | 0.996 | Probably Damaging | 0.987 | Probably Damaging | 2.33 | Pathogenic | 0.03 | Affected | 0.3077 | 0.5651 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1414G>C | E472Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E472Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only Foldetta, which classifies the variant as benign. Tools that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Uncertain or inconclusive predictions come from FoldX, Rosetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic impact for E472Q, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -13.760 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 1.34 | Ambiguous | 0.3 | -0.67 | Ambiguous | 0.34 | Likely Benign | 0.89 | Ambiguous | 0.555 | Likely Pathogenic | -2.95 | Deleterious | 0.994 | Probably Damaging | 0.986 | Probably Damaging | 2.39 | Pathogenic | 0.01 | Affected | 0.1388 | 0.5726 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1415A>C | E472A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E472A is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; all these methods uniformly classify the change as deleterious. Tools that are inconclusive or uncertain for this variant are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts a pathogenic outcome, the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates pathogenicity, while Foldetta’s stability analysis remains uncertain. Taken together, the overwhelming majority of evidence points to a pathogenic effect for E472A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -15.356 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 1.81 | Ambiguous | 0.3 | 0.67 | Ambiguous | 1.24 | Ambiguous | 0.69 | Ambiguous | 0.732 | Likely Pathogenic | -5.90 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.32 | Pathogenic | 0.01 | Affected | 0.4639 | 0.6315 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1415A>G | E472G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E472G is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that assess the variant’s effect largely agree on a deleterious outcome: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic or likely pathogenic. Only premPS predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -15.239 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 3.06 | Destabilizing | 0.2 | 2.86 | Destabilizing | 2.96 | Destabilizing | 0.24 | Likely Benign | 0.744 | Likely Pathogenic | -6.83 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.30 | Pathogenic | 0.00 | Affected | 0.3343 | 0.4950 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1415A>T | E472V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E472V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Foldetta and premPS, whereas the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as benign. No prediction or folding‑stability result is missing or inconclusive; uncertain outputs from FoldX and Rosetta are treated as unavailable. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -14.957 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 1.05 | Ambiguous | 0.3 | -0.64 | Ambiguous | 0.21 | Likely Benign | 0.37 | Likely Benign | 0.733 | Likely Pathogenic | -6.90 | Deleterious | 0.996 | Probably Damaging | 0.991 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.0881 | 0.6347 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1416G>C | E472D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E472D is not reported in ClinVar and has no gnomAD entry. Consensus from multiple in‑silico predictors shows a split: benign calls come from REVEL and SIFT, whereas the majority of tools—SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict pathogenicity. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability estimates. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (derived from the four high‑confidence predictors) is pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E472D, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -9.798 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.27 | Ambiguous | 0.3 | 1.99 | Ambiguous | 1.63 | Ambiguous | 1.00 | Destabilizing | 0.304 | Likely Benign | -2.75 | Deleterious | 0.989 | Probably Damaging | 0.979 | Probably Damaging | 2.43 | Pathogenic | 0.06 | Tolerated | 0.1975 | 0.3918 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1416G>T | E472D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E472D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL and SIFT, whereas a majority of tools predict a pathogenic impact: polyPhen‑2 (HumDiv and HumVar), FATHMM, ESM1b, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Stability‑based methods (FoldX, Rosetta, Foldetta) yield uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence from multiple independent predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.264545 | Structured | 0.359300 | Uncertain | 0.878 | 0.231 | 0.000 | -9.798 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.27 | Ambiguous | 0.3 | 1.99 | Ambiguous | 1.63 | Ambiguous | 1.00 | Destabilizing | 0.303 | Likely Benign | -2.75 | Deleterious | 0.989 | Probably Damaging | 0.979 | Probably Damaging | 2.43 | Pathogenic | 0.06 | Tolerated | 0.1975 | 0.3918 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1417G>C | V473L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V473L is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, and Foldetta, while those that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as benign. Overall, the majority of tools (seven pathogenic vs. six benign) indicate a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion is not contradicted by the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.362529 | Uncertain | 0.884 | 0.239 | 0.000 | -9.073 | Likely Pathogenic | 0.966 | Likely Pathogenic | Likely Pathogenic | -0.29 | Likely Benign | 0.1 | -0.52 | Ambiguous | -0.41 | Likely Benign | 0.35 | Likely Benign | 0.336 | Likely Benign | -2.85 | Deleterious | 0.929 | Possibly Damaging | 0.917 | Probably Damaging | 3.43 | Benign | 0.21 | Tolerated | 0.0708 | 0.3921 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1417G>T | V473L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V473L is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, and Foldetta, while those that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as benign. Overall, the majority of tools (seven pathogenic vs. six benign) indicate a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion is not contradicted by the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.362529 | Uncertain | 0.884 | 0.239 | 0.000 | -9.073 | Likely Pathogenic | 0.966 | Likely Pathogenic | Likely Pathogenic | -0.29 | Likely Benign | 0.1 | -0.52 | Ambiguous | -0.41 | Likely Benign | 0.35 | Likely Benign | 0.335 | Likely Benign | -2.85 | Deleterious | 0.929 | Possibly Damaging | 0.917 | Probably Damaging | 3.43 | Benign | 0.21 | Tolerated | 0.0708 | 0.3921 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1418T>A | V473E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V473E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools largely agree on a deleterious effect: all evaluated algorithms except FATHMM predict pathogenicity, while FATHMM alone predicts benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also reports a pathogenic effect. No predictions or stability results are missing or inconclusive. Based on the consensus of the majority of tools and the high‑accuracy methods, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.362529 | Uncertain | 0.884 | 0.239 | 0.000 | -15.296 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 3.20 | Destabilizing | 0.2 | 2.24 | Destabilizing | 2.72 | Destabilizing | 2.22 | Destabilizing | 0.664 | Likely Pathogenic | -5.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.13 | Benign | 0.00 | Affected | 0.0903 | 0.1396 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.1418T>C | V473A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V473A missense variant is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM. Tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are all uncertain or unavailable, providing no decisive evidence. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the majority of available predictions support a pathogenic impact. This conclusion is consistent with the lack of ClinVar annotation (no contradiction). Thus, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.191378 | Structured | 0.362529 | Uncertain | 0.884 | 0.239 | 0.000 | -10.867 | Likely Pathogenic | 0.925 | Likely Pathogenic | Ambiguous | 1.88 | Ambiguous | 0.0 | 1.76 | Ambiguous | 1.82 | Ambiguous | 2.17 | Destabilizing | 0.485 | Likely Benign | -3.95 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.18 | Benign | 0.00 | Affected | 0.2529 | 0.2521 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.1418T>G | V473G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V473G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, while the remaining eleven tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) uniformly predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) votes pathogenic, and Foldetta also predicts pathogenic. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic based on the collective evidence, and this conclusion does not contradict any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.191378 | Structured | 0.362529 | Uncertain | 0.884 | 0.239 | 0.000 | -14.782 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 3.45 | Destabilizing | 0.0 | 3.40 | Destabilizing | 3.43 | Destabilizing | 2.69 | Destabilizing | 0.683 | Likely Pathogenic | -6.91 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.13 | Benign | 0.00 | Affected | 0.1885 | 0.2567 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
Found 8751 rows. Show 800 rows per page. Page 1/11 | Next »