
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain and Structure information: based on WT protein | Annotated databases | Deep learning-based pathogenicity predictions | Folding stability-based pathogenicity predictions | Sequence/structure-based pathogenicity predictions | Phase Separation | Evolutionary/physical properties | Molecular Dynamics-based analysis | DOI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | IUPred2 | ANCHOR2 | AlphaFold | MobiDB | PhosphoSitePlus | ClinVar | gnomAD | ESM1b | AlphaMissense | FoldX | Rosetta | Foldetta | PremPS | REVEL | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PSMutPred | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | |||||||||||||||||||||||||||||||||||||||||||||
| Score | Prediction | Score | Prediction | pLDDT | disorder | disorder | LTP | HTP | KL | PTM | Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | IP RF | SP RF | Prediction | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.2648T>A | L883Q 2D  AIThe SynGAP1 missense variant L883Q is reported in gnomAD (6‑33443200‑T‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is therefore most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.716283 | Disordered | 0.641952 | Binding | 0.334 | 0.886 | 0.250 | 6-33443200-T-A | 3 | 1.86e-6 | -3.559 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -0.51 | Neutral | 0.934 | Possibly Damaging | 0.637 | Possibly Damaging | 2.66 | Benign | 0.11 | Tolerated | 4.32 | 4 | 0.1119 | 0.1505 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||
| c.2654C>G | P885R 2D  AIThe SynGAP1 missense variant P885R is reported in gnomAD (ID 6‑33443206‑C‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.636133 | Binding | 0.344 | 0.917 | 0.250 | 6-33443206-C-G | 1 | 6.20e-7 | -4.166 | Likely Benign | 0.308 | Likely Benign | Likely Benign | 0.072 | Likely Benign | -1.99 | Neutral | 0.586 | Possibly Damaging | 0.377 | Benign | 2.84 | Benign | 0.00 | Affected | 4.32 | 4 | 0.1311 | 0.3505 | -2 | 0 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||
| c.2665G>A | G889R 2D  AIThe SynGAP1 missense variant G889R is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (ID 6‑33443217‑G‑A). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized; pathogenic predictions come from SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta results are unavailable. Overall, the majority of evidence (six benign vs three pathogenic) supports a benign impact. This conclusion does not contradict ClinVar, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | 6-33443217-G-A | 1 | 6.20e-7 | -3.357 | Likely Benign | 0.685 | Likely Pathogenic | Likely Benign | 0.070 | Likely Benign | -1.96 | Neutral | 0.027 | Benign | 0.009 | Benign | 2.38 | Pathogenic | 0.02 | Affected | 4.32 | 4 | 0.0925 | 0.4165 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.2665G>C | G889R 2D AIThe SynGAP1 missense variant G889R is not reported in ClinVar (ClinVar status: not present) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | -3.357 | Likely Benign | 0.685 | Likely Pathogenic | Likely Benign | 0.072 | Likely Benign | -1.96 | Neutral | 0.027 | Benign | 0.009 | Benign | 2.38 | Pathogenic | 0.02 | Affected | 4.32 | 4 | 0.0925 | 0.4165 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2672T>A | L891H 2D  AIThe SynGAP1 missense variant L891H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.505861 | Binding | 0.305 | 0.923 | 0.750 | -4.604 | Likely Benign | 0.320 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -1.79 | Neutral | 0.122 | Benign | 0.134 | Benign | 2.69 | Benign | 0.00 | Affected | 0.1099 | 0.1189 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2678A>T | Q893L 2D 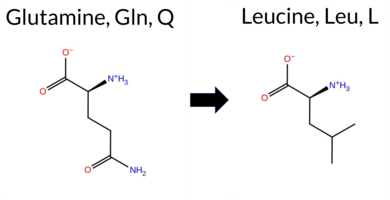 AIThe SynGAP1 missense variant Q893L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this conclusion does not contradict any ClinVar annotation (none present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.447267 | Uncertain | 0.310 | 0.925 | 0.750 | -1.964 | Likely Benign | 0.204 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.92 | Neutral | 0.451 | Benign | 0.209 | Benign | 2.82 | Benign | 1.00 | Tolerated | 0.0904 | 0.5643 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2696T>A | I899N 2D 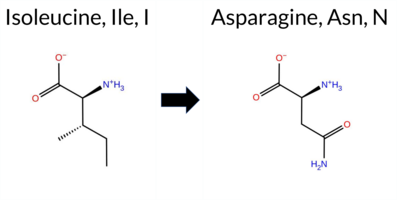 AIThe SynGAP1 missense variant I899N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, SGM‑Consensus is likely benign, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for I899N, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.443727 | Uncertain | 0.292 | 0.928 | 0.375 | -3.328 | Likely Benign | 0.469 | Ambiguous | Likely Benign | 0.058 | Likely Benign | -0.68 | Neutral | 0.611 | Possibly Damaging | 0.140 | Benign | 2.76 | Benign | 0.00 | Affected | 0.0933 | 0.0470 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.269T>A | V90E 2D 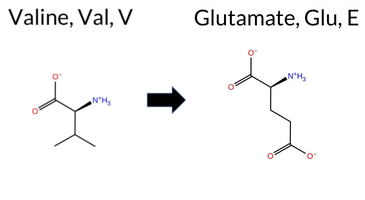 AIThe SynGAP1 missense variant V90E is listed in ClinVar (ID 971665.0) with an uncertain significance status and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all predict benign. Only two tools—SIFT and AlphaMissense‑Default—suggest a pathogenic outcome. When the high‑accuracy consensus is considered, AlphaMissense‑Optimized remains benign, and the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. No Foldetta stability assessment is available for this variant. Overall, the preponderance of evidence points to a benign impact, which does not contradict the ClinVar uncertain classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.570702 | Disordered | 0.542047 | Binding | 0.343 | 0.873 | 0.500 | Uncertain | 1 | -4.079 | Likely Benign | 0.703 | Likely Pathogenic | Likely Benign | 0.108 | Likely Benign | -0.38 | Neutral | 0.001 | Benign | 0.000 | Benign | 4.00 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1323 | 0.2233 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||||||||
| c.26A>T | H9L 2D 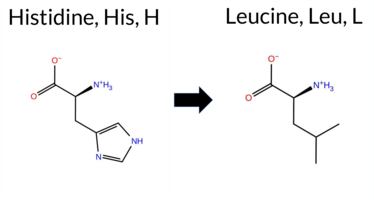 AIThe SynGAP1 missense variant H9L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.486429 | Structured | 0.528099 | Binding | 0.394 | 0.916 | 0.750 | -2.603 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.151 | Likely Benign | 0.48 | Neutral | 0.024 | Benign | 0.002 | Benign | 4.25 | Benign | 0.00 | Affected | 0.1255 | 0.6285 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2714G>T | R905L 2D  AIThe SynGAP1 missense variant R905L is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (allele ID 6‑33443266‑G‑T). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of predictions leans toward a benign interpretation, and this conclusion does not contradict the ClinVar status, which currently has no assertion for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.671169 | Disordered | 0.618085 | Binding | 0.291 | 0.920 | 0.250 | 6-33443266-G-T | -3.284 | Likely Benign | 0.709 | Likely Pathogenic | Likely Benign | 0.242 | Likely Benign | -2.55 | Deleterious | 0.963 | Probably Damaging | 0.753 | Possibly Damaging | 2.61 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1676 | 0.4905 | -2 | -3 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||
| c.2717T>A | L906H 2D AIThe SynGAP1 missense variant L906H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs. two pathogenic votes) and Foldetta results are unavailable. Overall, the majority of standard predictors lean toward pathogenicity, but the most reliable high‑accuracy tool suggests a benign outcome and the consensus is unresolved. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.604312 | Disordered | 0.644316 | Binding | 0.315 | 0.920 | 0.250 | -4.367 | Likely Benign | 0.732 | Likely Pathogenic | Likely Benign | 0.124 | Likely Benign | -1.01 | Neutral | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.18 | Pathogenic | 0.02 | Affected | 0.1125 | 0.1073 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2723A>T | Q908L 2D AIThe SynGAP1 missense variant Q908L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT uniformly predict a pathogenic impact. AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus is Likely Benign, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.678728 | Binding | 0.275 | 0.917 | 0.250 | -3.589 | Likely Benign | 0.420 | Ambiguous | Likely Benign | 0.193 | Likely Benign | -1.40 | Neutral | 0.985 | Probably Damaging | 0.982 | Probably Damaging | 2.53 | Benign | 0.05 | Affected | 0.0706 | 0.5655 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.272A>T | E91V 2D  AIThe SynGAP1 E91V missense variant has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” AlphaMissense‑Optimized yields an uncertain result, and no Foldetta stability assessment is available. Overall, the evidence is mixed, but the consensus of several independent benign predictors and the SGM‑Consensus lean toward a benign interpretation. Thus, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.533667 | Binding | 0.303 | 0.875 | 0.500 | -3.697 | Likely Benign | 0.934 | Likely Pathogenic | Ambiguous | 0.124 | Likely Benign | -2.16 | Neutral | 0.947 | Possibly Damaging | 0.788 | Possibly Damaging | 3.84 | Benign | 0.00 | Affected | 0.0940 | 0.7457 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2732T>A | V911D 2D  AIThe SynGAP1 missense variant V911D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of high‑confidence predictors indicate a benign impact, and this is consistent with the lack of ClinVar evidence. Therefore, the variant is most likely benign and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.685117 | Disordered | 0.724137 | Binding | 0.327 | 0.914 | 0.375 | -4.422 | Likely Benign | 0.624 | Likely Pathogenic | Likely Benign | 0.114 | Likely Benign | -0.41 | Neutral | 0.500 | Possibly Damaging | 0.157 | Benign | 2.71 | Benign | 0.05 | Affected | 0.1416 | 0.1289 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2747T>A | V916D 2D AIThe SynGAP1 missense variant V916D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and SIFT uniformly predict a pathogenic outcome. AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus indicates Likely Benign, and Foldetta (which integrates FoldX‑MD and Rosetta outputs) is not available for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.835395 | Binding | 0.308 | 0.879 | 0.250 | -2.653 | Likely Benign | 0.392 | Ambiguous | Likely Benign | 0.139 | Likely Benign | -1.48 | Neutral | 0.975 | Probably Damaging | 0.767 | Possibly Damaging | 2.69 | Benign | 0.01 | Affected | 0.1580 | 0.1113 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.274G>A | G92R 2D AIThe SynGAP1 missense variant G92R is listed in gnomAD (6-33425882‑G‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain,” SGM‑Consensus as “Likely Benign,” and Foldetta results are unavailable. Taken together, the preponderance of evidence from multiple independent predictors and the consensus score points to a benign classification. This conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.537848 | Binding | 0.337 | 0.874 | 0.625 | 6-33425882-G-A | 1 | 6.20e-7 | -2.909 | Likely Benign | 0.876 | Likely Pathogenic | Ambiguous | 0.139 | Likely Benign | -2.38 | Neutral | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 4.01 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1041 | 0.4605 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||
| c.274G>C | G92R 2D AIThe SynGAP1 missense variant G92R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence tools and the consensus prediction lean toward a benign interpretation, with no conflict with ClinVar status. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.537848 | Binding | 0.337 | 0.874 | 0.625 | -2.909 | Likely Benign | 0.876 | Likely Pathogenic | Ambiguous | 0.139 | Likely Benign | -2.38 | Neutral | 0.999 | Probably Damaging | 0.979 | Probably Damaging | 4.01 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1041 | 0.4605 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.2750C>G | P917R 2D AIThe SynGAP1 missense variant P917R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (gnomAD ID 6‑33443302‑C‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also as benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.863949 | Binding | 0.314 | 0.862 | 0.375 | Uncertain | 1 | 6-33443302-C-G | 5 | 3.10e-6 | -4.475 | Likely Benign | 0.363 | Ambiguous | Likely Benign | 0.142 | Likely Benign | -1.70 | Neutral | 0.642 | Possibly Damaging | 0.316 | Benign | 2.68 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1270 | 0.3012 | -2 | 0 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||
| c.2756A>T | Q919L 2D AIThe SynGAP1 missense variant Q919L is reported in gnomAD (ID 6‑33443308‑A‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.618285 | Disordered | 0.911223 | Binding | 0.299 | 0.841 | 0.250 | 6-33443308-A-T | 1 | 6.20e-7 | -4.492 | Likely Benign | 0.252 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -2.13 | Neutral | 0.891 | Possibly Damaging | 0.596 | Possibly Damaging | 2.40 | Pathogenic | 0.04 | Affected | 4.32 | 4 | 0.0771 | 0.6454 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||
| c.2759A>T | Q920L 2D AIThe SynGAP1 missense variant Q920L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.927260 | Binding | 0.306 | 0.845 | 0.250 | -4.048 | Likely Benign | 0.280 | Likely Benign | Likely Benign | 0.181 | Likely Benign | -2.36 | Neutral | 0.891 | Possibly Damaging | 0.596 | Possibly Damaging | 2.60 | Benign | 0.00 | Affected | 0.0774 | 0.5995 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2762T>A | L921Q 2D AIThe SynGAP1 missense variant L921Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) lean toward a benign interpretation. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.943282 | Binding | 0.311 | 0.845 | 0.375 | -2.389 | Likely Benign | 0.311 | Likely Benign | Likely Benign | 0.132 | Likely Benign | -0.62 | Neutral | 0.994 | Probably Damaging | 0.940 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 0.1097 | 0.1119 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2768T>A | I923N 2D AIThe SynGAP1 missense variant I923N (ClinVar ID 647043.0) is listed as “Uncertain” and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign” because the majority of its constituent tools (three benign, one pathogenic) favor a benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as benign, and the Foldetta stability analysis is unavailable. Overall, the collective predictions point to a benign impact, which does not contradict the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.562014 | Disordered | 0.964857 | Binding | 0.292 | 0.852 | 0.250 | Uncertain | 1 | -0.733 | Likely Benign | 0.712 | Likely Pathogenic | Likely Benign | 0.108 | Likely Benign | -1.16 | Neutral | 0.991 | Probably Damaging | 0.793 | Possibly Damaging | 2.70 | Benign | 0.13 | Tolerated | 3.77 | 5 | 0.1164 | 0.1073 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||
| c.2774T>A | L925H 2D AIThe SynGAP1 missense variant L925H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic.” No Foldetta stability result is available. Overall, the majority of evidence points to a pathogenic effect for L925H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.977963 | Binding | 0.290 | 0.852 | 0.125 | -3.369 | Likely Benign | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.475 | Likely Benign | -5.24 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.28 | Pathogenic | 0.00 | Affected | 0.1219 | 0.1494 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.277C>G | R93G 2D  AIThe SynGAP1 missense variant R93G is listed in ClinVar (ID 2504251.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (derived from the same set of predictors) labels it “Likely Benign”; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that R93G is most likely benign, which does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.549151 | Binding | 0.290 | 0.874 | 0.625 | Uncertain | 1 | -2.674 | Likely Benign | 0.400 | Ambiguous | Likely Benign | 0.093 | Likely Benign | -1.69 | Neutral | 0.103 | Benign | 0.019 | Benign | 3.99 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3809 | 0.3814 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||
| c.2783A>T | Q928L 2D AIThe SynGAP1 missense variant Q928L has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of other in‑silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) and the SGM‑Consensus score (Likely Pathogenic) all indicate a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.497853 | Structured | 0.986260 | Binding | 0.324 | 0.852 | 0.250 | -6.237 | Likely Benign | 0.919 | Likely Pathogenic | Ambiguous | 0.373 | Likely Benign | -4.57 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 1.56 | Pathogenic | 0.00 | Affected | 0.0757 | 0.6091 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.2785A>T | N929Y 2D  AIThe SynGAP1 missense variant N929Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL predicts benign, whereas the remaining eleven predictors—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized—predict pathogenicity. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus result is consistent with a likely pathogenic classification. Foldetta predictions are unavailable. Taken together, the preponderance of evidence indicates that the variant is most likely pathogenic, and this assessment does not conflict with the current ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.986867 | Binding | 0.321 | 0.851 | 0.375 | -10.327 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 0.391 | Likely Benign | -6.02 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.45 | Pathogenic | 0.00 | Affected | 0.0656 | 0.6489 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.2786A>T | N929I 2D  AIThe SynGAP1 missense variant N929I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: REVEL classifies it as benign, whereas the remaining eight tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict pathogenicity. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus result is consistent. Foldetta, a protein‑folding stability method that combines FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not conflict with the current ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.986867 | Binding | 0.321 | 0.851 | 0.375 | -11.799 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.297 | Likely Benign | -6.82 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.45 | Pathogenic | 0.00 | Affected | 0.0780 | 0.6486 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.2792T>A | L931H 2D AIThe SynGAP1 missense variant L931H has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. ESM1b and AlphaMissense‑Optimized are uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.549308 | Disordered | 0.989212 | Binding | 0.335 | 0.856 | 0.375 | -7.495 | In-Between | 0.954 | Likely Pathogenic | Ambiguous | 0.301 | Likely Benign | -3.76 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.38 | Pathogenic | 0.00 | Affected | 0.1198 | 0.1205 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2798A>T | H933L 2D AIThe SynGAP1 H933L missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.666105 | Disordered | 0.987531 | Binding | 0.305 | 0.862 | 0.625 | -0.858 | Likely Benign | 0.470 | Ambiguous | Likely Benign | 0.388 | Likely Benign | -6.26 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.38 | Pathogenic | 0.02 | Affected | 0.1003 | 0.5749 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.2810A>T | D937V 2D  AIThe SynGAP1 missense variant D937V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of predictions (six benign vs. four pathogenic) support a benign classification. This consensus does not contradict ClinVar status, as no ClinVar entry exists for this variant. Thus, based on current computational evidence, the D937V variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.963385 | Binding | 0.348 | 0.883 | 0.625 | -3.418 | Likely Benign | 0.673 | Likely Pathogenic | Likely Benign | 0.141 | Likely Benign | -2.21 | Neutral | 1.000 | Probably Damaging | 0.977 | Probably Damaging | 2.70 | Benign | 0.02 | Affected | 0.1766 | 0.7408 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2816C>G | P939R 2D AIThe SynGAP1 missense variant P939R is reported in gnomAD (ID 6‑33443368‑C‑G) but has no ClinVar entry. Functional prediction tools show mixed results: benign calls come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Grouping by consensus, the benign‑predicted tools outnumbered the pathogenic ones by one, but the overall tally favors pathogenicity (5 pathogenic vs 4 benign). High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is benign, but the SGM Consensus (a 2‑vs‑2 majority among AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields no clear verdict, and Foldetta data are unavailable. Consequently, the variant is most likely pathogenic according to the available predictions, and this assessment does not contradict any ClinVar status because none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.894241 | Disordered | 0.935841 | Binding | 0.397 | 0.897 | 0.625 | 6-33443368-C-G | 1 | 6.20e-7 | -4.863 | Likely Benign | 0.260 | Likely Benign | Likely Benign | 0.199 | Likely Benign | -3.79 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.18 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1360 | 0.3336 | -2 | 0 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||
| c.281C>A | P94H 2D  AIThe SynGAP1 missense variant P94H is reported in gnomAD (variant ID 6‑33425889‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. No Foldetta stability analysis is available, so it does not influence the assessment. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that P94H is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.570978 | Binding | 0.350 | 0.869 | 0.625 | 6-33425889-C-A | 1 | 6.20e-7 | -3.708 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -2.31 | Neutral | 0.637 | Possibly Damaging | 0.102 | Benign | 4.11 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1486 | 0.3913 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||
| c.2827G>C | G943R 2D AIThe SynGAP1 missense variant G943R is reported in gnomAD (variant ID 6-33443379‑G‑C) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity; only ESM1b is uncertain. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions strongly supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.973328 | Disordered | 0.860437 | Binding | 0.372 | 0.910 | 0.750 | 6-33443379-G-C | 1 | 6.20e-7 | -7.159 | In-Between | 0.335 | Likely Benign | Likely Benign | 0.308 | Likely Benign | 1.00 | Neutral | 0.411 | Benign | 0.062 | Benign | 2.86 | Benign | 0.94 | Tolerated | 4.32 | 4 | 0.0976 | 0.4933 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||
| c.2839G>A | G947R 2D AIThe SynGAP1 missense variant G947R is listed in gnomAD (6-33443391-G-A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Optimized) and pathogenic (SIFT). Two tools remain uncertain (ESM1b, AlphaMissense‑Default). High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—does not reach a definitive conclusion (two benign, two uncertain). Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no reported result for this variant. Overall, the preponderance of evidence points to a benign impact, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988695 | Disordered | 0.850554 | Binding | 0.358 | 0.919 | 0.750 | 6-33443391-G-A | 6 | 3.72e-6 | -7.481 | In-Between | 0.343 | Ambiguous | Likely Benign | 0.273 | Likely Benign | -0.60 | Neutral | 0.411 | Benign | 0.140 | Benign | 4.94 | Benign | 0.04 | Affected | 4.32 | 4 | 0.1034 | 0.4733 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.2839G>C | G947R 2D AIThe SynGAP1 missense variant G947R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta stability analysis is unavailable. Overall, the balance of evidence favors a benign classification, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988695 | Disordered | 0.850554 | Binding | 0.358 | 0.919 | 0.750 | -7.481 | In-Between | 0.343 | Ambiguous | Likely Benign | 0.273 | Likely Benign | -0.60 | Neutral | 0.411 | Benign | 0.140 | Benign | 4.94 | Benign | 0.04 | Affected | 4.32 | 4 | 0.1034 | 0.4733 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.2840G>A | G947E 2D  AIThe SynGAP1 missense variant G947E is reported in ClinVar as “Not submitted” and is present in gnomAD (variant ID 6-33443392-G-A). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, creating a single discordant signal. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that G947E is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988695 | Disordered | 0.850554 | Binding | 0.358 | 0.919 | 0.750 | 6-33443392-G-A | 1 | 6.20e-7 | -9.574 | Likely Pathogenic | 0.243 | Likely Benign | Likely Benign | 0.302 | Likely Benign | 0.08 | Neutral | 0.126 | Benign | 0.096 | Benign | 4.92 | Benign | 0.10 | Tolerated | 4.32 | 4 | 0.1555 | 0.4259 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.284A>T | H95L 2D AIThe SynGAP1 missense variant H95L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta results are not available for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.590542 | Binding | 0.335 | 0.875 | 0.625 | -1.967 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -2.31 | Neutral | 0.084 | Benign | 0.007 | Benign | 4.17 | Benign | 0.00 | Affected | 0.1017 | 0.5074 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2852A>T | H951L 2D AIThe SynGAP1 missense variant H951L is not reported in ClinVar and is absent from gnomAD, indicating no documented clinical or population evidence. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification. Foldetta results are not available, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988291 | Disordered | 0.901477 | Binding | 0.415 | 0.925 | 0.750 | -4.822 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.192 | Likely Benign | -0.99 | Neutral | 0.022 | Benign | 0.018 | Benign | 5.47 | Benign | 0.43 | Tolerated | 0.2224 | 0.4526 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2870A>T | H957L 2D AIThe SynGAP1 missense variant H957L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only FATHMM predicts a pathogenic outcome. When predictions are grouped by consensus, the benign group contains eight tools, whereas the pathogenic group contains only FATHMM. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a Likely Benign result. Foldetta, a protein‑folding stability method, has no available output for this variant. Overall, the preponderance of evidence indicates that H957L is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.968874 | Binding | 0.362 | 0.915 | 0.750 | -6.598 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.296 | Likely Benign | -0.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 2.44 | Pathogenic | 0.09 | Tolerated | 0.1525 | 0.5516 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2873A>T | H958L 2D AIThe SynGAP1 missense variant H958L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only two tools, SIFT and ESM1b, predict a pathogenic outcome. When the high‑accuracy consensus is considered, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a likely benign verdict, and AlphaMissense‑Optimized also reports benign. Foldetta predictions are unavailable. Overall, the majority of evidence supports a benign interpretation, and this is consistent with the lack of ClinVar annotation. Thus, the variant is most likely benign and does not contradict existing ClinVar data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.976011 | Binding | 0.371 | 0.913 | 0.750 | -8.422 | Likely Pathogenic | 0.087 | Likely Benign | Likely Benign | 0.181 | Likely Benign | -1.28 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.25 | Benign | 0.03 | Affected | 0.1723 | 0.5338 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2876A>T | H959L 2D AIThe SynGAP1 missense variant H959L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that H959L is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.980566 | Binding | 0.333 | 0.905 | 0.750 | -8.515 | Likely Pathogenic | 0.100 | Likely Benign | Likely Benign | 0.236 | Likely Benign | -1.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.14 | Benign | 0.09 | Tolerated | 0.1698 | 0.5538 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2879A>T | H960L 2D AIThe SynGAP1 missense variant H960L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic prediction comes from ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.987911 | Disordered | 0.983385 | Binding | 0.380 | 0.901 | 0.750 | -8.386 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.130 | Likely Benign | -1.30 | Neutral | 0.174 | Benign | 0.043 | Benign | 4.17 | Benign | 0.33 | Tolerated | 0.1465 | 0.5595 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2882A>T | H961L 2D AIThe SynGAP1 missense variant H961L is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT and ESM1b predict pathogenicity, but these are outliers among the consensus. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, SGM‑Consensus is benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.989835 | Disordered | 0.984562 | Binding | 0.323 | 0.893 | 0.750 | -8.547 | Likely Pathogenic | 0.109 | Likely Benign | Likely Benign | 0.155 | Likely Benign | -1.21 | Neutral | 0.144 | Benign | 0.078 | Benign | 4.13 | Benign | 0.01 | Affected | 0.1465 | 0.5344 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2885A>T | H962L 2D AIThe SynGAP1 missense variant H962L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for H962L, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.984483 | Binding | 0.369 | 0.886 | 0.750 | -8.478 | Likely Pathogenic | 0.108 | Likely Benign | Likely Benign | 0.151 | Likely Benign | -1.49 | Neutral | 0.494 | Possibly Damaging | 0.170 | Benign | 4.15 | Benign | 0.03 | Affected | 0.1738 | 0.5487 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2888A>T | H963L 2D AIThe SynGAP1 missense variant H963L is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates benign, and the SGM‑Consensus likewise suggests a benign effect; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for H963L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.991070 | Disordered | 0.983973 | Binding | 0.325 | 0.886 | 0.750 | -8.110 | Likely Pathogenic | 0.109 | Likely Benign | Likely Benign | 0.172 | Likely Benign | -1.58 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.13 | Benign | 0.43 | Tolerated | 0.1626 | 0.5680 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2891A>T | H964L 2D AIThe SynGAP1 missense variant H964L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. Only SIFT predicts a pathogenic outcome, and ESM1b remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) indicates likely benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.990547 | Disordered | 0.982486 | Binding | 0.364 | 0.886 | 0.750 | -7.568 | In-Between | 0.092 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.20 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.15 | Benign | 0.02 | Affected | 0.1409 | 0.5195 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2894A>T | H965L 2D AIThe SynGAP1 missense variant H965L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect. Benign predictors include REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool in the dataset returned a pathogenic prediction. Consensus predictors such as SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) classify the variant as Likely Benign. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus is Likely Benign; Foldetta results are not available. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | -6.708 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -1.66 | Neutral | 0.033 | Benign | 0.018 | Benign | 4.06 | Benign | 1.00 | Tolerated | 0.1606 | 0.5338 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2897A>T | H966L 2D AIThe SynGAP1 missense variant H966L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. No pathogenic predictions are present among the evaluated algorithms. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) reports likely benign. Foldetta results are not available for this variant. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | -6.119 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -1.74 | Neutral | 0.174 | Benign | 0.062 | Benign | 4.05 | Benign | 0.35 | Tolerated | 0.1514 | 0.5316 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2899C>G | R967G 2D AIThe SynGAP1 missense variant R967G is reported in gnomAD (ID 6‑33443451‑C‑G) and has no ClinVar entry. All evaluated in silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Based on the unanimous benign predictions and absence of a ClinVar pathogenic claim, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.974374 | Disordered | 0.969686 | Binding | 0.340 | 0.888 | 0.750 | 6-33443451-C-G | 1 | 6.20e-7 | -2.214 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.279 | Likely Benign | -0.93 | Neutral | 0.005 | Benign | 0.007 | Benign | 4.17 | Benign | 0.18 | Tolerated | 4.32 | 2 | 0.3187 | 0.4070 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||
| c.2900G>T | R967L 2D AIThe SynGAP1 missense variant R967L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443452‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for R967L, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.974374 | Disordered | 0.969686 | Binding | 0.340 | 0.888 | 0.750 | Uncertain | 1 | 6-33443452-G-T | 1 | 6.20e-7 | -3.496 | Likely Benign | 0.164 | Likely Benign | Likely Benign | 0.123 | Likely Benign | -0.99 | Neutral | 0.959 | Probably Damaging | 0.586 | Possibly Damaging | 4.15 | Benign | 0.75 | Tolerated | 4.32 | 2 | 0.1964 | 0.5093 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||
| c.2909A>T | E970V 2D AIThe SynGAP1 missense variant E970V is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the collective evidence strongly supports a benign interpretation, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.951925 | Disordered | 0.953422 | Binding | 0.342 | 0.902 | 0.750 | -2.791 | Likely Benign | 0.208 | Likely Benign | Likely Benign | 0.245 | Likely Benign | -1.08 | Neutral | 0.002 | Benign | 0.002 | Benign | 4.11 | Benign | 0.08 | Tolerated | 0.1990 | 0.7037 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.290A>T | E97V 2D AIThe SynGAP1 missense variant E97V is listed in gnomAD (ID 6‑33425898‑A‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and Foldetta (FoldX‑MD/Rosetta stability assessment) has no available result. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta data is missing. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.609018 | Binding | 0.340 | 0.867 | 0.625 | 6-33425898-A-T | 1 | 6.20e-7 | -3.743 | Likely Benign | 0.514 | Ambiguous | Likely Benign | 0.124 | Likely Benign | -1.17 | Neutral | 0.947 | Possibly Damaging | 0.788 | Possibly Damaging | 4.07 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1015 | 0.8155 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||
| c.2912C>A | P971H 2D AIThe SynGAP1 missense variant P971H is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443464‑C‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; a Foldetta stability prediction is not available. Overall, the majority of evidence points to a benign impact, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.951523 | Binding | 0.545 | 0.905 | 0.625 | Uncertain | 1 | 6-33443464-C-A | 1 | 6.20e-7 | -5.243 | Likely Benign | 0.086 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -1.11 | Neutral | 0.898 | Possibly Damaging | 0.477 | Possibly Damaging | 3.89 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1584 | 0.4858 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||
| c.2915C>G | P972R 2D AIThe SynGAP1 missense variant P972R is reported in gnomAD (ID 6‑33443467‑C‑G) but has no ClinVar entry. All evaluated in silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Based on the unanimous benign predictions and lack of ClinVar pathogenic annotation, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.954150 | Binding | 0.472 | 0.904 | 0.625 | 6-33443467-C-G | -4.483 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -1.44 | Neutral | 0.290 | Benign | 0.114 | Benign | 4.23 | Benign | 0.12 | Tolerated | 4.32 | 2 | 0.1379 | 0.3931 | -2 | 0 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||
| c.2921A>T | D974V 2D AIThe SynGAP1 missense variant D974V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective predictions strongly suggest that D974V is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.964377 | Binding | 0.389 | 0.897 | 0.625 | -3.629 | Likely Benign | 0.351 | Ambiguous | Likely Benign | 0.211 | Likely Benign | -1.49 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.16 | Benign | 0.01 | Affected | 0.1449 | 0.7286 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2939A>T | H980L 2D AIThe SynGAP1 missense variant H980L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.974598 | Binding | 0.309 | 0.892 | 0.625 | -1.984 | Likely Benign | 0.293 | Likely Benign | Likely Benign | 0.187 | Likely Benign | -1.98 | Neutral | 0.625 | Possibly Damaging | 0.265 | Benign | 4.16 | Benign | 0.00 | Affected | 0.1488 | 0.5538 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.2944T>A | Y982N 2D  AIThe SynGAP1 Y982N variant has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, SGM‑Consensus is benign, and Foldetta results are unavailable. Overall, the majority of consensus‑based and high‑accuracy tools lean toward a benign interpretation. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar status, which currently has no entry for Y982N. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.966717 | Binding | 0.272 | 0.895 | 0.625 | -4.536 | Likely Benign | 0.881 | Likely Pathogenic | Ambiguous | 0.175 | Likely Benign | -1.14 | Neutral | 0.990 | Probably Damaging | 0.900 | Possibly Damaging | 3.88 | Benign | 0.00 | Affected | 0.2090 | 0.0545 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.2957A>T | E986V 2D AIThe SynGAP1 E986V missense variant is not reported in ClinVar and has no gnomAD entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b, whereas pathogenic predictions arise from PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (benign), FATHMM (pathogenic), and PROVEAN (pathogenic)—also indicates pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a pathogenic effect for E986V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.750527 | Disordered | 0.929726 | Binding | 0.349 | 0.902 | 0.750 | -4.511 | Likely Benign | 0.965 | Likely Pathogenic | Likely Pathogenic | 0.220 | Likely Benign | -3.48 | Deleterious | 0.018 | Benign | 0.028 | Benign | 2.10 | Pathogenic | 0.00 | Affected | 0.1146 | 0.7960 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2960A>T | D987V 2D AIThe SynGAP1 missense variant D987V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, while pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that D987V is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.823549 | Disordered | 0.919118 | Binding | 0.299 | 0.903 | 0.750 | -4.647 | Likely Benign | 0.857 | Likely Pathogenic | Ambiguous | 0.389 | Likely Benign | -4.01 | Deleterious | 0.992 | Probably Damaging | 0.913 | Probably Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.0907 | 0.7202 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2963T>A | L988H 2D AIThe SynGAP1 missense variant L988H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2). Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs four benign) lean toward pathogenicity. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.918781 | Binding | 0.360 | 0.913 | 0.750 | -4.779 | Likely Benign | 0.733 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -2.70 | Deleterious | 0.998 | Probably Damaging | 0.947 | Probably Damaging | 2.62 | Benign | 0.00 | Affected | 0.1179 | 0.1414 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.296A>T | E99V 2D AIThe SynGAP1 missense variant E99V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus likewise reports Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign effect for E99V, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.645246 | Binding | 0.325 | 0.874 | 0.500 | -3.628 | Likely Benign | 0.544 | Ambiguous | Likely Benign | 0.208 | Likely Benign | -1.69 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.02 | Benign | 0.00 | Affected | 0.1109 | 0.8175 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.2975T>A | V992D 2D AIThe SynGAP1 missense variant V992D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only SIFT predicts pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates that V992D is most likely benign, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.921728 | Binding | 0.331 | 0.917 | 0.750 | -3.976 | Likely Benign | 0.306 | Likely Benign | Likely Benign | 0.029 | Likely Benign | -1.24 | Neutral | 0.302 | Benign | 0.158 | Benign | 4.21 | Benign | 0.05 | Affected | 0.1645 | 0.1056 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.2987C>G | P996R 2D AIThe SynGAP1 missense variant P996R is listed in ClinVar (ID 2808854.0) as benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates a benign impact. This prediction aligns with the ClinVar benign classification and does not contradict the existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.775545 | Disordered | 0.942262 | Binding | 0.312 | 0.900 | 0.750 | Benign | 1 | -4.457 | Likely Benign | 0.141 | Likely Benign | Likely Benign | 0.040 | Likely Benign | -1.04 | Neutral | 0.144 | Benign | 0.085 | Benign | 4.26 | Benign | 0.01 | Affected | 4.32 | 4 | 0.1297 | 0.3385 | -2 | 0 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||
| c.298T>A | Y100N 2D AIThe SynGAP1 missense variant Y100N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.699094 | Disordered | 0.675421 | Binding | 0.341 | 0.880 | 0.625 | -2.579 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.183 | Likely Benign | -0.66 | Neutral | 0.675 | Possibly Damaging | 0.099 | Benign | 4.23 | Benign | 0.00 | Affected | 0.2487 | 0.0373 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.2999T>A | I1000N 2D AIThe SynGAP1 missense variant I1000N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.745909 | Disordered | 0.957020 | Binding | 0.293 | 0.904 | 0.625 | -5.246 | Likely Benign | 0.677 | Likely Pathogenic | Likely Benign | 0.145 | Likely Benign | -0.82 | Neutral | 0.995 | Probably Damaging | 0.913 | Probably Damaging | 2.72 | Benign | 0.16 | Tolerated | 0.1001 | 0.0900 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3002T>A | L1001H 2D AIThe SynGAP1 missense variant L1001H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.958507 | Binding | 0.269 | 0.902 | 0.375 | -3.119 | Likely Benign | 0.258 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -1.02 | Neutral | 0.997 | Probably Damaging | 0.870 | Possibly Damaging | 2.64 | Benign | 0.00 | Affected | 0.1141 | 0.0958 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3005A>T | H1002L 2D AIThe SynGAP1 H1002L missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and the absence of the variant in population databases, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.953758 | Binding | 0.285 | 0.900 | 0.500 | -6.448 | Likely Benign | 0.556 | Ambiguous | Likely Benign | 0.157 | Likely Benign | -3.12 | Deleterious | 0.801 | Possibly Damaging | 0.602 | Possibly Damaging | 2.79 | Benign | 0.13 | Tolerated | 0.1296 | 0.5088 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3011A>T | H1004L 2D AIThe SynGAP1 missense variant H1004L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two pathogenic vs two benign votes). Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the balance of evidence leans toward a benign interpretation, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.808535 | Disordered | 0.943707 | Binding | 0.271 | 0.901 | 0.750 | -5.179 | Likely Benign | 0.716 | Likely Pathogenic | Likely Benign | 0.220 | Likely Benign | -3.14 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.75 | Benign | 0.55 | Tolerated | 0.1223 | 0.6026 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3016T>A | Y1006N 2D AIThe SynGAP1 missense variant Y1006N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus remains Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.930554 | Binding | 0.264 | 0.896 | 0.750 | -4.238 | Likely Benign | 0.789 | Likely Pathogenic | Ambiguous | 0.118 | Likely Benign | -0.75 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.73 | Benign | 0.75 | Tolerated | 0.2168 | 0.1094 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3023A>T | D1008V 2D AIThe SynGAP1 D1008V variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs. two benign votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, more tools (five) predict pathogenicity than benign (three), and the high‑accuracy assessments do not overturn this trend. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.694846 | Disordered | 0.919416 | Binding | 0.280 | 0.899 | 0.625 | -4.828 | Likely Benign | 0.944 | Likely Pathogenic | Ambiguous | 0.242 | Likely Benign | -3.61 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.64 | Benign | 0.01 | Affected | 0.1447 | 0.6608 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.3026A>T | E1009V 2D AIThe SynGAP1 missense variant E1009V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments give an uncertain result from AlphaMissense‑Optimized, a Likely Pathogenic verdict from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and no available data from Foldetta. Overall, the majority of evidence points to a deleterious effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.728858 | Disordered | 0.914552 | Binding | 0.325 | 0.885 | 0.500 | -3.660 | Likely Benign | 0.815 | Likely Pathogenic | Ambiguous | 0.156 | Likely Benign | -3.81 | Deleterious | 0.998 | Probably Damaging | 0.924 | Probably Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.1111 | 0.7580 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3029T>C | F1010S 2D  AIThe SynGAP1 missense variant F1010S is listed in gnomAD (ID 6‑33443581‑T‑C) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as benign; Foldetta results are unavailable. Overall, the balance of evidence, including the high‑accuracy benign predictions and the consensus benign call, indicates that the variant is most likely benign. This conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.741537 | Disordered | 0.912572 | Binding | 0.286 | 0.881 | 0.625 | 6-33443581-T-C | -1.722 | Likely Benign | 0.744 | Likely Pathogenic | Likely Benign | 0.153 | Likely Benign | -1.97 | Neutral | 0.994 | Probably Damaging | 0.892 | Possibly Damaging | 2.51 | Benign | 0.01 | Affected | 3.77 | 5 | 0.4196 | 0.0775 | -2 | -3 | -3.6 | -60.10 | ||||||||||||||||||||||||||||||||||||
| c.302A>T | H101L 2D AIThe SynGAP1 missense variant H101L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for H101L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.688356 | Binding | 0.370 | 0.884 | 0.625 | -1.376 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.33 | Neutral | 0.824 | Possibly Damaging | 0.840 | Possibly Damaging | 4.19 | Benign | 0.00 | Affected | 0.0924 | 0.4960 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3031G>A | G1011R 2D AIThe SynGAP1 missense variant G1011R is reported in gnomAD (variant ID 6‑33443583‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign, reflecting the majority of benign calls. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect for G1011R, and this conclusion does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.898380 | Binding | 0.332 | 0.869 | 0.625 | 6-33443583-G-A | -4.650 | Likely Benign | 0.609 | Likely Pathogenic | Likely Benign | 0.118 | Likely Benign | -0.79 | Neutral | 0.642 | Possibly Damaging | 0.494 | Possibly Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1041 | 0.4415 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||
| c.3031G>C | G1011R 2D AIThe SynGAP1 missense variant G1011R is not reported in ClinVar and is absent from gnomAD, so no population frequency or clinical assertion data are available. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.898380 | Binding | 0.332 | 0.869 | 0.625 | -4.650 | Likely Benign | 0.609 | Likely Pathogenic | Likely Benign | 0.118 | Likely Benign | -0.79 | Neutral | 0.642 | Possibly Damaging | 0.494 | Possibly Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1041 | 0.4415 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3038C>T | S1013F 2D  AIThe SynGAP1 missense variant S1013F is catalogued in gnomAD (ID 6‑33443590‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen2_HumVar, ESM1b, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen2_HumDiv, and SIFT. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign; the SGM Consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive due to mixed signals, and Foldetta results are unavailable. Overall, the balance of evidence, including the benign call from AlphaMissense‑Optimized, points to a likely benign effect. This conclusion does not conflict with ClinVar, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.823549 | Disordered | 0.899570 | Binding | 0.308 | 0.846 | 0.625 | 6-33443590-C-T | 1 | 6.20e-7 | -5.370 | Likely Benign | 0.353 | Ambiguous | Likely Benign | 0.057 | Likely Benign | -2.54 | Deleterious | 0.453 | Possibly Damaging | 0.272 | Benign | 2.65 | Benign | 0.03 | Affected | 3.77 | 5 | 0.0922 | 0.5803 | -2 | -3 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||
| c.3047A>T | D1016V 2D AIThe SynGAP1 D1016V missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments give an uncertain result from AlphaMissense‑Optimized, a Likely Pathogenic verdict from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and no available data from Foldetta. Overall, the majority of evidence points toward a deleterious effect. Thus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.801317 | Disordered | 0.944705 | Binding | 0.323 | 0.811 | 0.625 | -3.208 | Likely Benign | 0.800 | Likely Pathogenic | Ambiguous | 0.362 | Likely Benign | -3.80 | Deleterious | 0.977 | Probably Damaging | 0.856 | Possibly Damaging | 2.45 | Pathogenic | 0.00 | Affected | 0.1424 | 0.7116 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3056G>T | R1019L 2D AIThe SynGAP1 missense variant R1019L is listed in ClinVar with an “Uncertain” status (ClinVar ID 3364537.0) and is present in gnomAD (gnomAD ID 6‑33443608‑G‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote) remains pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a pathogenic impact, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.856457 | Disordered | 0.966400 | Binding | 0.315 | 0.794 | 0.500 | Uncertain | 1 | 6-33443608-G-T | 2 | 1.24e-6 | -5.194 | Likely Benign | 0.752 | Likely Pathogenic | Likely Benign | 0.110 | Likely Benign | -3.57 | Deleterious | 0.800 | Possibly Damaging | 0.573 | Possibly Damaging | 2.40 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.1850 | 0.4886 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||
| c.305T>G | L102W 2D  AIThe SynGAP1 missense variant L102W is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for L102W, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.696014 | Binding | 0.357 | 0.885 | 0.625 | -5.833 | Likely Benign | 0.202 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.42 | Neutral | 0.996 | Probably Damaging | 0.984 | Probably Damaging | 4.09 | Benign | 0.00 | Affected | 0.0821 | 0.3078 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||||||||||||
| c.3062A>T | Q1021L 2D AIThe SynGAP1 missense variant Q1021L is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta results are unavailable. Overall, the majority of predictions (5 pathogenic vs. 4 benign) indicate that the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as the variant has no existing ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.899122 | Disordered | 0.979641 | Binding | 0.326 | 0.763 | 0.500 | -5.780 | Likely Benign | 0.678 | Likely Pathogenic | Likely Benign | 0.226 | Likely Benign | -3.36 | Deleterious | 0.985 | Probably Damaging | 0.982 | Probably Damaging | 2.58 | Benign | 0.01 | Affected | 0.0688 | 0.5039 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3065T>A | L1022H 2D AIThe SynGAP1 missense variant L1022H is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie (2 pathogenic, 2 benign) and is therefore inconclusive. Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the majority of conventional predictors lean toward pathogenicity, whereas the single high‑accuracy tool predicts benign and the consensus is unresolved. Thus, the variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.859585 | Disordered | 0.986981 | Binding | 0.339 | 0.752 | 0.500 | -2.473 | Likely Benign | 0.589 | Likely Pathogenic | Likely Benign | 0.140 | Likely Benign | -2.21 | Neutral | 0.999 | Probably Damaging | 0.944 | Probably Damaging | 2.49 | Pathogenic | 0.00 | Affected | 0.1165 | 0.1313 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3071T>A | L1024H 2D AIThe SynGAP1 missense variant L1024H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized returns an Uncertain result, SGM‑Consensus indicates Likely Pathogenic, and Foldetta data are unavailable. Overall, the majority of evidence points toward a deleterious effect, suggesting the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.862302 | Disordered | 0.992699 | Binding | 0.327 | 0.753 | 0.500 | -3.271 | Likely Benign | 0.868 | Likely Pathogenic | Ambiguous | 0.123 | Likely Benign | -3.09 | Deleterious | 1.000 | Probably Damaging | 0.981 | Probably Damaging | 2.38 | Pathogenic | 0.01 | Affected | 0.1198 | 0.1483 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3074A>T | Q1025L 2D AIThe SynGAP1 missense variant Q1025L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.879233 | Disordered | 0.993410 | Binding | 0.363 | 0.746 | 0.500 | -6.460 | Likely Benign | 0.463 | Ambiguous | Likely Benign | 0.117 | Likely Benign | -2.48 | Neutral | 0.901 | Possibly Damaging | 0.534 | Possibly Damaging | 2.70 | Benign | 0.05 | Affected | 0.0798 | 0.5497 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3077A>T | D1026V 2D AIThe SynGAP1 missense variant D1026V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Tools that predict a pathogenic effect are PROVEAN, SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and therefore also indicates a likely pathogenic outcome. AlphaMissense‑Optimized is uncertain, and no Foldetta stability result is available, so it does not contribute evidence. Overall, the majority of reliable predictors classify the variant as pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.894241 | Disordered | 0.993931 | Binding | 0.324 | 0.739 | 0.500 | -5.871 | Likely Benign | 0.900 | Likely Pathogenic | Ambiguous | 0.144 | Likely Benign | -3.13 | Deleterious | 0.004 | Benign | 0.004 | Benign | 2.48 | Pathogenic | 0.00 | Affected | 0.0957 | 0.5236 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3079A>T | N1027Y 2D AIThe SynGAP1 missense variant N1027Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -5.799 | Likely Benign | 0.626 | Likely Pathogenic | Likely Benign | 0.074 | Likely Benign | -2.15 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.70 | Benign | 0.03 | Affected | 0.0611 | 0.6133 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3080A>T | N1027I 2D AIThe SynGAP1 missense variant N1027I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.831250 | Disordered | 0.994357 | Binding | 0.347 | 0.745 | 0.500 | -5.847 | Likely Benign | 0.751 | Likely Pathogenic | Likely Benign | 0.065 | Likely Benign | -2.36 | Neutral | 0.970 | Probably Damaging | 0.726 | Possibly Damaging | 2.71 | Benign | 0.02 | Affected | 0.0677 | 0.5724 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3083T>A | L1028Q 2D AIThe SynGAP1 missense variant L1028Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.995137 | Binding | 0.364 | 0.730 | 0.500 | -2.718 | Likely Benign | 0.612 | Likely Pathogenic | Likely Benign | 0.179 | Likely Benign | -0.19 | Neutral | 0.986 | Probably Damaging | 0.825 | Possibly Damaging | 2.80 | Benign | 0.38 | Tolerated | 0.1110 | 0.1403 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3086A>T | Q1029L 2D AIThe SynGAP1 missense variant Q1029L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a benign outcome (2 benign vs. 1 pathogenic, with one uncertain). AlphaMissense‑Default remains uncertain, and Foldetta results are unavailable. High‑accuracy predictions therefore point to a benign impact: AlphaMissense‑Optimized is benign, SGM Consensus is benign, and no Foldetta data are available. Overall, the computational evidence indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.995643 | Binding | 0.375 | 0.734 | 0.500 | -3.984 | Likely Benign | 0.364 | Ambiguous | Likely Benign | 0.067 | Likely Benign | -2.65 | Deleterious | 0.891 | Possibly Damaging | 0.587 | Possibly Damaging | 2.70 | Benign | 0.16 | Tolerated | 0.0685 | 0.5866 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3089A>T | H1030L 2D AIThe SynGAP1 missense variant H1030L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that H1030L is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.849326 | Disordered | 0.995856 | Binding | 0.375 | 0.735 | 0.500 | -3.513 | Likely Benign | 0.270 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -2.35 | Neutral | 0.224 | Benign | 0.120 | Benign | 2.76 | Benign | 0.02 | Affected | 0.0968 | 0.5710 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3095T>A | L1032Q 2D AIThe SynGAP1 missense variant L1032Q is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar) and SIFT. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the aggregate evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.865454 | Disordered | 0.995318 | Binding | 0.365 | 0.735 | 0.500 | -2.992 | Likely Benign | 0.467 | Ambiguous | Likely Benign | 0.134 | Likely Benign | -0.78 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 2.66 | Benign | 0.03 | Affected | 0.1217 | 0.1677 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3098C>A | S1033Y 2D  AIThe SynGAP1 missense variant S1033Y is reported in gnomAD (ID 6‑33443650‑C‑A) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.993473 | Binding | 0.294 | 0.737 | 0.625 | 6-33443650-C-A | 1 | 6.19e-7 | -4.857 | Likely Benign | 0.564 | Ambiguous | Likely Benign | 0.034 | Likely Benign | -1.01 | Neutral | 0.021 | Benign | 0.008 | Benign | 2.69 | Benign | 0.04 | Affected | 3.77 | 5 | 0.0708 | 0.5262 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||
| c.3107A>T | Q1036L 2D AIThe SynGAP1 missense variant Q1036L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors a benign outcome, with two benign votes versus one pathogenic and one uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.948786 | Disordered | 0.987955 | Binding | 0.275 | 0.765 | 0.625 | -4.389 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.092 | Likely Benign | -2.92 | Deleterious | 0.152 | Benign | 0.045 | Benign | 2.52 | Benign | 0.01 | Affected | 0.1069 | 0.5996 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3110T>A | I1037N 2D AIThe SynGAP1 missense variant I1037N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a Likely Benign classification, while AlphaMissense‑Optimized remains uncertain. Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.939629 | Disordered | 0.986140 | Binding | 0.309 | 0.774 | 0.625 | -3.955 | Likely Benign | 0.832 | Likely Pathogenic | Ambiguous | 0.131 | Likely Benign | 1.56 | Neutral | 0.666 | Possibly Damaging | 0.211 | Benign | 2.81 | Benign | 0.34 | Tolerated | 0.1022 | 0.1140 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3116T>A | I1039N 2D AIThe SynGAP1 missense variant I1039N has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status (none is present). Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.979204 | Binding | 0.292 | 0.806 | 0.625 | -3.469 | Likely Benign | 0.951 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.24 | Neutral | 0.011 | Benign | 0.010 | Benign | 2.67 | Benign | 0.02 | Affected | 0.1151 | 0.1311 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.311G>T | R104L 2D AIThe SynGAP1 missense variant R104L is listed in ClinVar (ID 2746314.0) as Benign and is present in gnomAD (6‑33432176‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is benign, and the SGM‑Consensus (majority vote) is also benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the ClinVar benign classification and does not contradict the existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.678998 | Binding | 0.339 | 0.869 | 0.625 | Benign | 1 | 6-33432176-G-T | 1 | 6.20e-7 | -3.563 | Likely Benign | 0.578 | Likely Pathogenic | Likely Benign | 0.170 | Likely Benign | -1.38 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.05 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1681 | 0.4894 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||
| c.3125A>T | Q1042L 2D AIThe SynGAP1 missense variant Q1042L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.969315 | Disordered | 0.959333 | Binding | 0.310 | 0.846 | 0.625 | -3.796 | Likely Benign | 0.203 | Likely Benign | Likely Benign | 0.338 | Likely Benign | -2.47 | Neutral | 0.369 | Benign | 0.120 | Benign | 5.47 | Benign | 0.05 | Affected | 0.1469 | 0.6276 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3140C>T | S1047L 2D  AIThe SynGAP1 missense variant S1047L is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443692‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta stability analysis is not available for this variant. Overall, the preponderance of computational evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.963420 | Disordered | 0.933764 | Binding | 0.409 | 0.909 | 0.750 | Uncertain | 1 | 6-33443692-C-T | 1 | 6.20e-7 | -4.062 | Likely Benign | 0.132 | Likely Benign | Likely Benign | 0.032 | Likely Benign | -0.63 | Neutral | 0.144 | Benign | 0.058 | Benign | 2.60 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1575 | 0.5555 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||
| c.3142G>A | G1048R 2D AIThe SynGAP1 missense variant G1048R is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split: pathogenic calls come from REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar, whereas benign calls are made by PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a benign majority, and AlphaMissense‑Optimized itself predicts benign. The SGM‑Consensus, which aggregates these four predictors, reports a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar assertion; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | -4.305 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.503 | Likely Pathogenic | -0.54 | Neutral | 0.919 | Possibly Damaging | 0.728 | Possibly Damaging | 2.54 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0956 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3142G>C | G1048R 2D AIThe SynGAP1 missense variant G1048R is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are REVEL, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM‑Consensus also indicates a likely benign outcome; the Foldetta protein‑folding stability assessment is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.977651 | Disordered | 0.923876 | Binding | 0.346 | 0.916 | 0.750 | Uncertain | 1 | -4.305 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.503 | Likely Pathogenic | -0.54 | Neutral | 0.919 | Possibly Damaging | 0.728 | Possibly Damaging | 2.54 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0956 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.3148G>A | G1050R 2D AIThe SynGAP1 missense variant G1050R is catalogued in gnomAD (ID 6‑33443700‑G‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign verdict, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | 6-33443700-G-A | 3 | 1.86e-6 | -6.637 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.045 | Likely Benign | -0.68 | Neutral | 0.009 | Benign | 0.008 | Benign | 2.48 | Pathogenic | 0.06 | Tolerated | 3.77 | 5 | 0.0939 | 0.4532 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.3148G>C | G1050R 2D AIThe SynGAP1 missense variant G1050R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. Only FATHMM predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority vote. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact; this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.906802 | Binding | 0.370 | 0.928 | 0.875 | -6.637 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.045 | Likely Benign | -0.68 | Neutral | 0.009 | Benign | 0.008 | Benign | 2.48 | Pathogenic | 0.06 | Tolerated | 3.77 | 5 | 0.0939 | 0.4532 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3154G>A | G1052R 2D AISynGAP1 missense variant G1052R is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores the variant as benign, and the SGM consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar uncertain status but provides additional support toward a likely benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | Uncertain | 1 | -9.050 | Likely Pathogenic | 0.383 | Ambiguous | Likely Benign | 0.497 | Likely Benign | -0.41 | Neutral | 0.990 | Probably Damaging | 0.798 | Possibly Damaging | 3.90 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0976 | 0.4142 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||
| c.3154G>C | G1052R 2D AIThe SynGAP1 missense variant G1052R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and ESM1b predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.984420 | Disordered | 0.892068 | Binding | 0.367 | 0.938 | 0.875 | -9.050 | Likely Pathogenic | 0.383 | Ambiguous | Likely Benign | 0.497 | Likely Benign | -0.41 | Neutral | 0.990 | Probably Damaging | 0.798 | Possibly Damaging | 3.90 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.0976 | 0.4142 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||||
| c.3164G>A | G1055E 2D AIThe SynGAP1 missense variant G1055E is catalogued in gnomAD (variant ID 6‑33443716‑G‑A) but has no ClinVar entry. In silico prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a benign effect. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.982235 | Disordered | 0.872113 | Binding | 0.379 | 0.935 | 0.875 | 6-33443716-G-A | 1 | 6.21e-7 | -10.951 | Likely Pathogenic | 0.290 | Likely Benign | Likely Benign | 0.320 | Likely Benign | -0.03 | Neutral | 0.901 | Possibly Damaging | 0.456 | Possibly Damaging | 3.30 | Benign | 0.10 | Tolerated | 3.77 | 5 | 0.1525 | 0.4459 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.316A>G | R106G 2D AIThe SynGAP1 missense variant R106G is listed in gnomAD (ID 6‑33432181‑A‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Those that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar classification to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.663409 | Binding | 0.345 | 0.862 | 0.875 | 6-33432181-A-G | 1 | 6.20e-7 | -2.617 | Likely Benign | 0.835 | Likely Pathogenic | Ambiguous | 0.161 | Likely Benign | -2.21 | Neutral | 0.421 | Benign | 0.050 | Benign | 3.65 | Benign | 0.00 | Affected | 4.05 | 3 | 0.3680 | 0.3980 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||
| c.3176G>A | G1059E 2D AIThe SynGAP1 missense variant G1059E is reported in gnomAD (variant ID 6‑33443728‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only two tools—SIFT and ESM1b—predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.983019 | Disordered | 0.898939 | Binding | 0.399 | 0.926 | 0.875 | 6-33443728-G-A | 1 | 6.22e-7 | -10.459 | Likely Pathogenic | 0.297 | Likely Benign | Likely Benign | 0.390 | Likely Benign | -0.71 | Neutral | 0.126 | Benign | 0.066 | Benign | 2.53 | Benign | 0.00 | Affected | 4.32 | 2 | 0.1464 | 0.4069 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.3191A>T | Q1064L 2D AIThe SynGAP1 missense variant Q1064L is not reported in ClinVar and is absent from gnomAD, indicating no documented allele frequency data. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a benign effect. No tool in the dataset indicates pathogenicity. High‑accuracy assessments corroborate this benign prediction: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the consensus of all available predictions strongly supports a benign classification, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.953106 | Binding | 0.378 | 0.914 | 0.875 | -3.492 | Likely Benign | 0.099 | Likely Benign | Likely Benign | 0.133 | Likely Benign | -1.16 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.20 | Benign | 0.13 | Tolerated | 0.1817 | 0.5485 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.31G>A | G11R 2D AIThe SynGAP1 missense variant G11R is catalogued in gnomAD (ID 6‑33420295‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | 6-33420295-G-A | -3.418 | Likely Benign | 0.428 | Ambiguous | Likely Benign | 0.102 | Likely Benign | -0.47 | Neutral | 0.498 | Possibly Damaging | 0.026 | Benign | 3.92 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1022 | 0.4596 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||
| c.31G>C | G11R 2D AIThe SynGAP1 missense variant G11R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | -3.418 | Likely Benign | 0.428 | Ambiguous | Likely Benign | 0.109 | Likely Benign | -0.47 | Neutral | 0.498 | Possibly Damaging | 0.026 | Benign | 3.92 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1022 | 0.4596 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.31G>T | G11W 2D  AIThe SynGAP1 missense variant G11W is catalogued in gnomAD (ID 6‑33420295‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | 6-33420295-G-T | -5.819 | Likely Benign | 0.403 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -0.67 | Neutral | 0.959 | Probably Damaging | 0.318 | Benign | 3.87 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0747 | 0.4731 | -2 | -7 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||||||||
| c.3203T>G | L1068W 2D AIThe SynGAP1 missense variant L1068W is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized remains benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) resolves as pathogenic, and Foldetta’s protein‑folding stability analysis is unavailable. Overall, the majority of evidence points toward a pathogenic impact for L1068W. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.947281 | Disordered | 0.981041 | Binding | 0.362 | 0.907 | 0.875 | -8.574 | Likely Pathogenic | 0.342 | Ambiguous | Likely Benign | 0.151 | Likely Benign | -1.96 | Neutral | 0.994 | Probably Damaging | 0.884 | Possibly Damaging | 2.46 | Pathogenic | 0.00 | Affected | 0.0742 | 0.3763 | -2 | -2 | -4.7 | 73.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3206A>T | Q1069L 2D AIThe SynGAP1 missense variant Q1069L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.943310 | Disordered | 0.981477 | Binding | 0.333 | 0.906 | 0.875 | -4.278 | Likely Benign | 0.141 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -0.96 | Neutral | 0.003 | Benign | 0.008 | Benign | 2.83 | Benign | 0.10 | Tolerated | 0.0936 | 0.6624 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3221A>T | Q1074L 2D AIThe SynGAP1 missense variant Q1074L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.930790 | Disordered | 0.987006 | Binding | 0.339 | 0.897 | 0.750 | -3.561 | Likely Benign | 0.259 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -1.29 | Neutral | 0.625 | Possibly Damaging | 0.266 | Benign | 2.68 | Benign | 1.00 | Tolerated | 0.0840 | 0.6293 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3224A>T | Q1075L 2D AIThe SynGAP1 missense variant Q1075L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy methods confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence supports a benign impact for Q1075L, and this conclusion is consistent with the absence of a ClinVar assertion. The variant is most likely benign based on predictions, and there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.922952 | Disordered | 0.988305 | Binding | 0.354 | 0.894 | 0.750 | -3.976 | Likely Benign | 0.209 | Likely Benign | Likely Benign | 0.128 | Likely Benign | -2.10 | Neutral | 0.985 | Probably Damaging | 0.973 | Probably Damaging | 2.72 | Benign | 0.15 | Tolerated | 0.0888 | 0.6454 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3227T>G | L1076W 2D AIThe SynGAP1 missense variant L1076W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated predictors (five pathogenic vs. three benign) lean toward a pathogenic interpretation. This prediction is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.989617 | Binding | 0.301 | 0.892 | 0.750 | -6.377 | Likely Benign | 0.822 | Likely Pathogenic | Ambiguous | 0.183 | Likely Benign | -1.40 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.38 | Pathogenic | 0.02 | Affected | 0.0749 | 0.3740 | -2 | -2 | -4.7 | 73.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3245A>T | Q1082L 2D AIThe SynGAP1 missense variant Q1082L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Overall, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.979325 | Binding | 0.347 | 0.896 | 0.875 | -3.284 | Likely Benign | 0.171 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -1.30 | Neutral | 0.224 | Benign | 0.058 | Benign | 4.12 | Benign | 1.00 | Tolerated | 0.0979 | 0.6824 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3248A>T | K1083I 2D  AIThe SynGAP1 missense variant K1083I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of a ClinVar entry, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.837511 | Disordered | 0.978906 | Binding | 0.302 | 0.893 | 1.000 | -3.207 | Likely Benign | 0.836 | Likely Pathogenic | Ambiguous | 0.239 | Likely Benign | -1.65 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 4.02 | Benign | 0.30 | Tolerated | 0.1394 | 0.3978 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||||||||||||
| c.3251C>A | P1084H 2D AIThe SynGAP1 missense variant P1084H is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443803‑C‑A). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which reports “Likely Benign”). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar designation of uncertainty rather than a definitive pathogenic claim. Thus, the variant is most likely benign, and its prediction profile does not contradict the current ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | Uncertain | 1 | 6-33443803-C-A | 1 | 6.31e-7 | -4.125 | Likely Benign | 0.323 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -3.16 | Deleterious | 0.997 | Probably Damaging | 0.840 | Possibly Damaging | 3.96 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1751 | 0.5523 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||
| c.3251C>G | P1084R 2D AIThe SynGAP1 missense variant P1084R is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443803‑C‑G). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome (2 benign vs 1 pathogenic vote). Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for P1084R, and this conclusion does not contradict the ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.842060 | Disordered | 0.979020 | Binding | 0.348 | 0.889 | 1.000 | 6-33443803-C-G | 4 | 2.52e-6 | -4.171 | Likely Benign | 0.562 | Ambiguous | Likely Benign | 0.153 | Likely Benign | -2.87 | Deleterious | 0.970 | Probably Damaging | 0.637 | Possibly Damaging | 3.99 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1370 | 0.4153 | -2 | 0 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||
| c.3253C>G | R1085G 2D AIThe SynGAP1 missense variant R1085G is reported in gnomAD (ID 6‑33443805‑C‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions from REVEL, ESM1b, and FATHMM; pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. A high‑accuracy consensus (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a 2‑to‑2 split, leaving the consensus inconclusive. No Foldetta stability assessment is available. Overall, the majority of evidence (five pathogenic versus three benign) points to a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.978838 | Binding | 0.270 | 0.888 | 1.000 | 6-33443805-C-G | -4.225 | Likely Benign | 0.846 | Likely Pathogenic | Ambiguous | 0.179 | Likely Benign | -2.79 | Deleterious | 0.997 | Probably Damaging | 0.993 | Probably Damaging | 2.72 | Benign | 0.01 | Affected | 3.77 | 5 | 0.3310 | 0.3693 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||||
| c.3260C>A | S1087Y 2D AIThe SynGAP1 missense variant S1087Y is catalogued in gnomAD (ID 6‑33443812‑C‑A) and has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as benign, and no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | 6-33443812-C-A | -4.194 | Likely Benign | 0.587 | Likely Pathogenic | Likely Benign | 0.107 | Likely Benign | -2.41 | Neutral | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.56 | Benign | 0.02 | Affected | 3.77 | 5 | 0.0671 | 0.5845 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||
| c.3260C>T | S1087F 2D AISynGAP1 missense variant S1087F is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of reliable predictors and the two high‑accuracy tools suggest a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.974805 | Binding | 0.357 | 0.891 | 1.000 | Uncertain | 1 | -3.843 | Likely Benign | 0.497 | Ambiguous | Likely Benign | 0.105 | Likely Benign | -2.75 | Deleterious | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.56 | Benign | 0.03 | Affected | 3.77 | 5 | 0.0645 | 0.6029 | -2 | -3 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||||||
| c.3263G>T | S1088I 2D  AIThe SynGAP1 missense variant S1088I is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33443815‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. AlphaMissense‑Optimized is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the balance of evidence, the variant is most likely benign; this assessment does not contradict ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.910643 | Disordered | 0.975261 | Binding | 0.336 | 0.889 | 1.000 | 6-33443815-G-T | -4.893 | Likely Benign | 0.891 | Likely Pathogenic | Ambiguous | 0.288 | Likely Benign | -2.05 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.62 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1322 | 0.5712 | -2 | -1 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||
| c.3265G>A | G1089R 2D AIThe SynGAP1 missense variant G1089R is catalogued in gnomAD (ID 6‑33443817‑G‑A) but has no ClinVar entry. Functional prediction tools split in a 6‑to‑3 ratio: benign calls come from REVEL, polyPhen‑2 HumVar, and ESM1b, while pathogenic calls come from PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. AlphaMissense‑Optimized yields an Uncertain result, and no Foldetta stability assessment is available. Overall, the majority of high‑confidence predictors lean toward pathogenicity, and this assessment does not conflict with ClinVar status, which is currently unreported. Thus, the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | 6-33443817-G-A | 1 | 6.35e-7 | -4.757 | Likely Benign | 0.897 | Likely Pathogenic | Ambiguous | 0.222 | Likely Benign | -3.13 | Deleterious | 0.896 | Possibly Damaging | 0.325 | Benign | 2.42 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.0934 | 0.4415 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||
| c.3265G>C | G1089R 2D AIThe SynGAP1 missense variant G1089R is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 HumVar, and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, and AlphaMissense‑Default. Grouping by consensus, seven tools predict pathogenicity and three predict benign, giving a net pathogenic signal. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic. Foldetta stability analysis is unavailable. Overall, the evidence points to the variant being most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.891961 | Disordered | 0.976771 | Binding | 0.366 | 0.890 | 1.000 | -4.757 | Likely Benign | 0.897 | Likely Pathogenic | Ambiguous | 0.228 | Likely Benign | -3.13 | Deleterious | 0.896 | Possibly Damaging | 0.325 | Benign | 2.42 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.0934 | 0.4415 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3268A>T | N1090Y 2D AIThe SynGAP1 missense variant N1090Y is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign, reflecting the majority of benign calls. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect for the variant, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -4.744 | Likely Benign | 0.651 | Likely Pathogenic | Likely Benign | 0.139 | Likely Benign | -2.26 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.66 | Benign | 0.05 | Affected | 0.0661 | 0.5998 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3269A>T | N1090I 2D AIThe SynGAP1 missense variant N1090I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the balance of evidence from both general and high‑accuracy predictors points to a benign classification, and this conclusion does not contradict the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.979886 | Binding | 0.341 | 0.887 | 1.000 | -4.356 | Likely Benign | 0.765 | Likely Pathogenic | Likely Benign | 0.173 | Likely Benign | -2.14 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.67 | Benign | 0.02 | Affected | 0.0781 | 0.6279 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3272T>A | L1091Q 2D AIThe SynGAP1 missense variant L1091Q is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments are less definitive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs 2 benign), and Foldetta results are unavailable. Overall, the majority of available predictions (five pathogenic vs three benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.924947 | Disordered | 0.984454 | Binding | 0.376 | 0.889 | 1.000 | -4.381 | Likely Benign | 0.854 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.32 | Neutral | 0.997 | Probably Damaging | 0.939 | Probably Damaging | 2.47 | Pathogenic | 0.02 | Affected | 0.1192 | 0.1514 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3275T>G | L1092W 2D AIThe SynGAP1 missense variant L1092W is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, while those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. Two tools give uncertain results: ESM1b and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta is unavailable. Overall, the majority of conventional tools predict pathogenicity, but the high‑accuracy consensus leans toward a benign interpretation. Thus, the variant is most likely benign based on the most reliable predictions, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.924947 | Disordered | 0.985431 | Binding | 0.385 | 0.890 | 1.000 | -7.014 | In-Between | 0.804 | Likely Pathogenic | Ambiguous | 0.120 | Likely Benign | -2.00 | Neutral | 0.999 | Probably Damaging | 0.968 | Probably Damaging | 2.58 | Benign | 0.03 | Affected | 0.0787 | 0.4033 | -2 | -2 | -4.7 | 73.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3278A>T | Q1093L 2D AIThe SynGAP1 missense variant Q1093L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.983312 | Binding | 0.351 | 0.886 | 1.000 | -3.242 | Likely Benign | 0.165 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -1.10 | Neutral | 0.224 | Benign | 0.091 | Benign | 2.72 | Benign | 0.03 | Affected | 0.0899 | 0.6837 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3281C>A | S1094Y 2D AIThe SynGAP1 missense variant S1094Y is not reported in ClinVar (status: none) but is present in gnomAD (ID 6‑33443833‑C‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic). Foldetta, a protein‑folding‑stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions therefore show a benign call from AlphaMissense‑Optimized, an inconclusive SGM Consensus, and no Foldetta data. Overall, the balance of evidence leans toward a pathogenic interpretation (five pathogenic versus four benign calls), and this does not contradict the ClinVar status, which currently has no assertion for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.938133 | Disordered | 0.981352 | Binding | 0.358 | 0.877 | 1.000 | 6-33443833-C-A | -5.609 | Likely Benign | 0.631 | Likely Pathogenic | Likely Benign | 0.155 | Likely Benign | -2.07 | Neutral | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 2.45 | Pathogenic | 0.02 | Affected | 3.77 | 5 | 0.1007 | 0.5877 | -2 | -3 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||||||
| c.3287A>T | E1096V 2D AIThe SynGAP1 missense variant E1096V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.926919 | Disordered | 0.976475 | Binding | 0.308 | 0.858 | 1.000 | -3.588 | Likely Benign | 0.650 | Likely Pathogenic | Likely Benign | 0.138 | Likely Benign | -1.06 | Neutral | 0.043 | Benign | 0.017 | Benign | 2.81 | Benign | 0.03 | Affected | 0.1082 | 0.7596 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3293G>T | S1098I 2D AIThe SynGAP1 missense variant S1098I is catalogued in gnomAD (ID 6‑33443845‑G‑T) and has no ClinVar entry. All evaluated in‑silico predictors classify it as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect. Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.973030 | Binding | 0.337 | 0.855 | 1.000 | 6-33443845-G-T | -4.497 | Likely Benign | 0.182 | Likely Benign | Likely Benign | 0.139 | Likely Benign | -0.92 | Neutral | 0.259 | Benign | 0.066 | Benign | 2.67 | Benign | 0.15 | Tolerated | 3.77 | 5 | 0.1219 | 0.5601 | -2 | -1 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||
| c.3295T>A | Y1099N 2D AIThe SynGAP1 missense variant Y1099N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only polyPhen‑2 HumDiv indicates a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the consensus of the available predictions points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | -4.329 | Likely Benign | 0.269 | Likely Benign | Likely Benign | 0.143 | Likely Benign | -1.01 | Neutral | 0.818 | Possibly Damaging | 0.360 | Benign | 2.83 | Benign | 0.16 | Tolerated | 0.2121 | 0.0935 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3296A>G | Y1099C 2D  AIThe SynGAP1 missense variant Y1099C is reported in gnomAD (variant ID 6‑33443848‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.974267 | Binding | 0.400 | 0.862 | 1.000 | 6-33443848-A-G | 3 | 1.95e-6 | -5.670 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -1.13 | Neutral | 0.997 | Probably Damaging | 0.840 | Possibly Damaging | 2.73 | Benign | 0.14 | Tolerated | 3.77 | 5 | 0.3008 | 0.2382 | -2 | 0 | 3.8 | -60.04 | ||||||||||||||||||||||||||||||||||
| c.329T>A | V110D 2D AIThe SynGAP1 missense variant V110D is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic vs. two benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evaluated tools (seven pathogenic vs. three benign) indicate a pathogenic effect. This prediction is consistent with the lack of ClinVar reporting and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.622677 | Disordered | 0.665934 | Binding | 0.347 | 0.860 | 0.750 | -4.536 | Likely Benign | 0.970 | Likely Pathogenic | Likely Pathogenic | 0.189 | Likely Benign | -2.84 | Deleterious | 0.978 | Probably Damaging | 0.500 | Possibly Damaging | 4.04 | Benign | 0.00 | Affected | 0.1849 | 0.1115 | -2 | -3 | -7.7 | 15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.32G>A | G11E 2D AIThe SynGAP1 missense variant G11E is reported in gnomAD (variant ID 6-33420296‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus itself is likely benign. Foldetta results are not available, so they do not influence the assessment. Overall, the preponderance of evidence points to a benign variant, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.444081 | Structured | 0.501027 | Binding | 0.348 | 0.915 | 0.375 | 6-33420296-G-A | 5 | 3.24e-6 | -4.206 | Likely Benign | 0.219 | Likely Benign | Likely Benign | 0.109 | Likely Benign | 0.09 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.95 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1444 | 0.4628 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.3314G>T | R1105L 2D AIThe SynGAP1 missense variant R1105L is not reported in ClinVar and is present in gnomAD (ID 6‑33443866‑G‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized classifies the variant as benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels it pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not provide a result for this residue, so its stability impact is unavailable. Overall, the balance of evidence leans toward a benign effect, with no conflict with ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.901269 | Disordered | 0.954396 | Binding | 0.330 | 0.863 | 0.875 | 6-33443866-G-T | -4.031 | Likely Benign | 0.459 | Ambiguous | Likely Benign | 0.125 | Likely Benign | -3.51 | Deleterious | 0.677 | Possibly Damaging | 0.168 | Benign | 2.46 | Pathogenic | 0.19 | Tolerated | 3.77 | 5 | 0.1734 | 0.5373 | -2 | -3 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||
| c.3317A>T | Q1106L 2D AIThe SynGAP1 missense variant Q1106L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs. four benign) lean toward a pathogenic impact. This assessment does not contradict ClinVar status, as the variant is currently unreported in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.885302 | Disordered | 0.952043 | Binding | 0.382 | 0.870 | 0.875 | -4.219 | Likely Benign | 0.171 | Likely Benign | Likely Benign | 0.169 | Likely Benign | -4.46 | Deleterious | 0.985 | Probably Damaging | 0.973 | Probably Damaging | 1.77 | Pathogenic | 0.05 | Affected | 0.0833 | 0.6282 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3320A>T | Q1107L 2D AIThe SynGAP1 missense variant Q1107L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.951017 | Binding | 0.393 | 0.880 | 0.875 | -3.785 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.119 | Likely Benign | -3.27 | Deleterious | 0.006 | Benign | 0.004 | Benign | 2.53 | Benign | 0.01 | Affected | 0.0820 | 0.6447 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3323G>T | S1108I 2D AIThe SynGAP1 missense variant S1108I is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33443875‑G‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no reported result for this variant. Overall, the balance of evidence (five benign versus four pathogenic predictions) suggests the variant is most likely benign, and this conclusion does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.949221 | Binding | 0.324 | 0.886 | 0.875 | Uncertain | 1 | 6-33443875-G-T | -3.666 | Likely Benign | 0.292 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -3.73 | Deleterious | 0.971 | Probably Damaging | 0.604 | Possibly Damaging | 2.44 | Pathogenic | 0.10 | Tolerated | 3.77 | 5 | 0.0949 | 0.4602 | -2 | -1 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||
| c.3326T>A | L1109H 2D AIThe SynGAP1 missense variant L1109H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.856457 | Disordered | 0.948334 | Binding | 0.343 | 0.893 | 0.875 | -4.353 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.134 | Likely Benign | -0.56 | Neutral | 0.832 | Possibly Damaging | 0.499 | Possibly Damaging | 2.70 | Benign | 0.04 | Affected | 0.1250 | 0.1845 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3329G>T | S1110I 2D AIThe SynGAP1 missense variant S1110I is catalogued in gnomAD (ID 6‑33443881‑G‑T) but has no ClinVar submission. Functional prediction tools show a split: six algorithms (REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a benign effect, whereas three (PROVEAN, SIFT, FATHMM) predict pathogenicity. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized classifies the variant as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign, two pathogenic); Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no available result for this residue. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.912647 | Disordered | 0.934156 | Binding | 0.346 | 0.892 | 0.875 | 6-33443881-G-T | -6.124 | Likely Benign | 0.198 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -2.99 | Deleterious | 0.007 | Benign | 0.003 | Benign | 2.17 | Pathogenic | 0.01 | Affected | 4.32 | 2 | 0.1203 | 0.4971 | -2 | -1 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||
| c.3335A>G | E1112G 2D  AIThe SynGAP1 missense variant E1112G is reported in gnomAD (ID 6‑33443887‑A‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN and SIFT. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yielding a “Likely Benign” classification. AlphaMissense‑Optimized independently predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is assigned). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.909381 | Binding | 0.335 | 0.902 | 0.875 | 6-33443887-A-G | 1 | 6.73e-7 | -3.459 | Likely Benign | 0.277 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -2.58 | Deleterious | 0.058 | Benign | 0.015 | Benign | 2.70 | Benign | 0.01 | Affected | 4.32 | 2 | 0.2795 | 0.6220 | -2 | 0 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||
| c.3335A>T | E1112V 2D AIThe SynGAP1 missense variant E1112V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) also as benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign impact for E1112V, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.894241 | Disordered | 0.909381 | Binding | 0.335 | 0.902 | 0.875 | -3.971 | Likely Benign | 0.579 | Likely Pathogenic | Likely Benign | 0.139 | Likely Benign | -2.28 | Neutral | 0.440 | Benign | 0.140 | Benign | 2.70 | Benign | 0.00 | Affected | 0.1481 | 0.7567 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3341G>T | S1114I 2D AIThe SynGAP1 missense variant S1114I is reported in gnomAD (ID 6‑33443893‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.895196 | Binding | 0.295 | 0.908 | 0.875 | 6-33443893-G-T | -6.718 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.023 | Likely Benign | -1.86 | Neutral | 0.570 | Possibly Damaging | 0.292 | Benign | 2.65 | Benign | 0.02 | Affected | 4.32 | 2 | 0.1159 | 0.5276 | -2 | -1 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||
| c.3344T>A | I1115N 2D AIThe SynGAP1 missense variant I1115N is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | -4.018 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.095 | Likely Benign | -0.60 | Neutral | 0.009 | Benign | 0.011 | Benign | 2.77 | Benign | 0.08 | Tolerated | 0.1299 | 0.1270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3344T>G | I1115S 2D  AIThe SynGAP1 missense variant I1115S is catalogued in gnomAD (ID 6‑33443896‑T‑G) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the variant as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are unavailable. Overall, the consensus of all available predictions is benign, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.892339 | Binding | 0.308 | 0.912 | 0.750 | 6-33443896-T-G | -0.579 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.128 | Likely Benign | 0.67 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.88 | Benign | 0.14 | Tolerated | 4.32 | 2 | 0.2986 | 0.1453 | -2 | -1 | -5.3 | -26.08 | ||||||||||||||||||||||||||||||||||||
| c.3346G>T | G1116W 2D AIThe SynGAP1 missense variant G1116W is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443898‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta’s protein‑folding stability result is unavailable. Taken together, the majority of reliable predictors and the consensus high‑accuracy tools indicate a benign effect. This conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | 6-33443898-G-T | -9.448 | Likely Pathogenic | 0.356 | Ambiguous | Likely Benign | 0.405 | Likely Benign | -1.27 | Neutral | 0.996 | Probably Damaging | 0.946 | Probably Damaging | 4.04 | Benign | 0.01 | Affected | 4.32 | 2 | 0.0776 | 0.4046 | -2 | -7 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||||
| c.3347G>A | G1116E 2D AIThe SynGAP1 missense variant G1116E is reported in gnomAD (variant ID 6-33443899‑G‑A) but has no ClinVar entry. Functional prediction tools uniformly favor a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while AlphaMissense‑Default remains uncertain. No tool predicts pathogenicity. The high‑accuracy consensus (SGM‑Consensus) also indicates a likely benign outcome, and AlphaMissense‑Optimized corroborates this. Foldetta, a protein‑folding stability predictor, was not available for this variant. Overall, the collective evidence strongly supports a benign impact, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.873279 | Binding | 0.320 | 0.909 | 0.750 | 6-33443899-G-A | -6.375 | Likely Benign | 0.375 | Ambiguous | Likely Benign | 0.345 | Likely Benign | -0.33 | Neutral | 0.043 | Benign | 0.022 | Benign | 4.06 | Benign | 0.07 | Tolerated | 4.32 | 2 | 0.1467 | 0.4069 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||||
| c.3355G>T | G1119W 2D AIThe SynGAP1 missense variant G1119W is reported in gnomAD (ID 6‑33443907‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | 6-33443907-G-T | -10.904 | Likely Pathogenic | 0.336 | Likely Benign | Likely Benign | 0.386 | Likely Benign | -1.19 | Neutral | 0.997 | Probably Damaging | 0.949 | Probably Damaging | 3.90 | Benign | 0.02 | Affected | 4.32 | 2 | 0.0948 | 0.4059 | -2 | -7 | -0.5 | 129.16 | ||||||||||||||||||||||||||||||||||||
| c.3356G>A | G1119E 2D AIThe SynGAP1 missense variant G1119E is reported in gnomAD (variant ID 6-33443908‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that G1119E is most likely benign, and this conclusion is not contradicted by any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.818538 | Binding | 0.339 | 0.928 | 0.875 | 6-33443908-G-A | 1 | 6.61e-7 | -9.151 | Likely Pathogenic | 0.338 | Likely Benign | Likely Benign | 0.284 | Likely Benign | -0.41 | Neutral | 0.005 | Benign | 0.013 | Benign | 3.93 | Benign | 0.12 | Tolerated | 4.32 | 2 | 0.1654 | 0.4259 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.3373G>A | G1125R 2D AIThe SynGAP1 missense variant G1125R is catalogued in gnomAD (ID 6‑33443925‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. AlphaMissense‑Default remains uncertain. A high‑accuracy consensus (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a benign majority vote. AlphaMissense‑Optimized also reports benign. No Foldetta stability assessment is available. Collectively, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443925-G-A | -8.463 | Likely Pathogenic | 0.542 | Ambiguous | Likely Benign | 0.291 | Likely Benign | -0.54 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.56 | Benign | 0.04 | Affected | 3.77 | 5 | 0.0980 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||||
| c.3373G>C | G1125R 2D AIThe SynGAP1 missense variant G1125R is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443925‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta’s protein‑folding stability result is unavailable. Overall, the balance of evidence, particularly from the high‑accuracy tools, indicates that the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443925-G-C | 1 | 6.68e-7 | -8.463 | Likely Pathogenic | 0.542 | Ambiguous | Likely Benign | 0.290 | Likely Benign | -0.54 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.56 | Benign | 0.04 | Affected | 3.77 | 5 | 0.0980 | 0.4332 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||||||||||
| c.3374G>A | G1125E 2D AIThe SynGAP1 missense variant G1125E is not reported in ClinVar (ClinVar status: not reported) but is present in gnomAD (gnomAD ID 6‑33443926‑G‑A). Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, AlphaMissense‑Optimized, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; AlphaMissense‑Default remains uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a benign outcome (2 benign vs. 1 pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM Consensus as benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.834292 | Disordered | 0.835839 | Binding | 0.339 | 0.923 | 0.875 | 6-33443926-G-A | 11 | 7.34e-6 | -9.849 | Likely Pathogenic | 0.353 | Ambiguous | Likely Benign | 0.264 | Likely Benign | -0.32 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 4.58 | Benign | 0.09 | Tolerated | 3.77 | 5 | 0.1430 | 0.4063 | -2 | 0 | -3.1 | 72.06 | |||||||||||||||||||||||||||||||||||
| c.3379G>A | G1127R 2D AIThe SynGAP1 missense variant G1127R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443931‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Uncertain | 1 | 6-33443931-G-A | 2 | 1.34e-6 | -5.949 | Likely Benign | 0.629 | Likely Pathogenic | Likely Benign | 0.341 | Likely Benign | -0.87 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.86 | Benign | 0.12 | Tolerated | 4.32 | 4 | 0.0929 | 0.4532 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||
| c.3379G>C | G1127R 2D AIThe SynGAP1 missense variant G1127R is listed in ClinVar (ID 2967461.0) with an “Uncertain” clinical significance and is present in gnomAD (6‑33443931‑G‑C). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that G1127R is most likely benign, which does not contradict the ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | Conflicting | 2 | 6-33443931-G-C | 16 | 1.07e-5 | -5.949 | Likely Benign | 0.629 | Likely Pathogenic | Likely Benign | 0.341 | Likely Benign | -0.87 | Neutral | 0.001 | Benign | 0.001 | Benign | 4.86 | Benign | 0.12 | Tolerated | 4.32 | 4 | 0.0929 | 0.4532 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||
| c.3379G>T | G1127W 2D AIThe SynGAP1 missense variant G1127W is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443931‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta’s protein‑folding stability result is unavailable. Overall, the balance of evidence, particularly from the high‑accuracy tools, indicates that the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.899122 | Disordered | 0.852422 | Binding | 0.344 | 0.915 | 0.875 | 6-33443931-G-T | 3 | 2.01e-6 | -9.850 | Likely Pathogenic | 0.418 | Ambiguous | Likely Benign | 0.394 | Likely Benign | -1.36 | Neutral | 0.983 | Probably Damaging | 0.665 | Possibly Damaging | 4.79 | Benign | 0.03 | Affected | 4.32 | 4 | 0.0782 | 0.4032 | -2 | -7 | -0.5 | 129.16 | |||||||||||||||||||||||||||||||||||
| c.3383G>A | G1128E 2D AIThe SynGAP1 missense variant G1128E is present in gnomAD (6‑33443935‑G‑A) and has no ClinVar entry. In silico predictors that agree on a benign effect include REVEL, PROVEAN, PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as likely benign. High‑accuracy tools reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus itself is a high‑accuracy majority vote. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its stability impact remains unknown. Overall, the computational evidence overwhelmingly supports a benign classification, and this is consistent with the absence of a pathogenic ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.865136 | Binding | 0.309 | 0.911 | 0.875 | 6-33443935-G-A | 1 | 6.70e-7 | -5.580 | Likely Benign | 0.452 | Ambiguous | Likely Benign | 0.235 | Likely Benign | -0.24 | Neutral | 0.002 | Benign | 0.008 | Benign | 4.43 | Benign | 1.00 | Tolerated | 4.32 | 4 | 0.1610 | 0.4497 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.3386T>A | L1129Q 2D AIThe SynGAP1 missense variant L1129Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.876543 | Binding | 0.339 | 0.909 | 0.875 | -3.684 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.453 | Likely Benign | -1.63 | Neutral | 0.846 | Possibly Damaging | 0.525 | Possibly Damaging | 5.49 | Benign | 0.00 | Affected | 0.1278 | 0.1436 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.338G>A | G113E 2D AIThe SynGAP1 missense variant G113E is not reported in ClinVar (ClinVar ID: None) but is present in gnomAD (ID 6‑33432203‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) also as benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.639486 | Binding | 0.350 | 0.870 | 0.750 | 6-33432203-G-A | 1 | 6.20e-7 | -3.169 | Likely Benign | 0.669 | Likely Pathogenic | Likely Benign | 0.092 | Likely Benign | -0.34 | Neutral | 0.028 | Benign | 0.006 | Benign | 4.21 | Benign | 0.05 | Affected | 3.61 | 5 | 0.1464 | 0.4035 | -2 | 0 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||||||
| c.3398T>A | I1133N 2D AIThe SynGAP1 missense variant I1133N is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Pathogenicity is suggested only by polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates Likely Benign; Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign effect for I1133N, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.832785 | Binding | 0.316 | 0.892 | 0.750 | -5.887 | Likely Benign | 0.520 | Ambiguous | Likely Benign | 0.290 | Likely Benign | -1.07 | Neutral | 0.453 | Possibly Damaging | 0.162 | Benign | 5.51 | Benign | 0.02 | Affected | 0.0961 | 0.1140 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3407A>T | Q1136L 2D AIThe SynGAP1 missense variant Q1136L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of all available predictions is benign, and this is consistent with the lack of ClinVar evidence; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.775584 | Binding | 0.321 | 0.884 | 0.875 | -6.020 | Likely Benign | 0.203 | Likely Benign | Likely Benign | 0.242 | Likely Benign | -2.42 | Neutral | 0.005 | Benign | 0.026 | Benign | 5.44 | Benign | 0.23 | Tolerated | 0.0861 | 0.6395 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3410A>C | H1137P 2D  AIThe SynGAP1 missense variant H1137P is listed in ClinVar as a benign alteration (ClinVar ID 3685596.0) and is present in the gnomAD database (gnomAD ID 6‑33444445‑A‑C). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (derived from the same four high‑accuracy predictors) also indicates benign. No Foldetta (FoldX‑MD/Rosetta stability) result is available for this variant. Overall, the majority of predictions, including the most reliable tools, support a benign classification, which is consistent with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.756488 | Binding | 0.314 | 0.879 | 0.875 | Benign | 1 | 6-33444445-A-C | 12 | 7.44e-6 | -2.098 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.419 | Likely Benign | -1.93 | Neutral | 0.925 | Possibly Damaging | 0.703 | Possibly Damaging | 5.29 | Benign | 0.00 | Affected | 4.32 | 4 | 0.2045 | 0.4501 | -2 | 0 | 1.6 | -40.02 | ||||||||||||||||||||||||||||||||
| c.3410A>T | H1137L 2D AIThe SynGAP1 missense variant H1137L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the preponderance of benign predictions and the consensus from high‑accuracy methods, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.903857 | Disordered | 0.756488 | Binding | 0.314 | 0.879 | 0.875 | -2.215 | Likely Benign | 0.080 | Likely Benign | Likely Benign | 0.359 | Likely Benign | -2.77 | Deleterious | 0.802 | Possibly Damaging | 0.534 | Possibly Damaging | 5.30 | Benign | 0.00 | Affected | 0.1199 | 0.6082 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.3413C>A | S1138Y 2D AISynGAP1 missense variant S1138Y is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33444448‑C‑A). Prediction tools that agree on benign impact include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. AlphaMissense‑Default remains uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is ambiguous but leans toward benign (2 benign vs 1 pathogenic, 1 uncertain). High‑accuracy methods further support a benign interpretation: AlphaMissense‑Optimized predicts benign, while Foldetta results are unavailable. Overall, the balance of evidence, especially from high‑accuracy tools, suggests the variant is most likely benign, which does not contradict the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.891961 | Disordered | 0.738250 | Binding | 0.346 | 0.869 | 1.000 | Uncertain | 2 | 6-33444448-C-A | 3 | 1.86e-6 | -6.610 | Likely Benign | 0.449 | Ambiguous | Likely Benign | 0.391 | Likely Benign | -2.51 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 5.41 | Benign | 0.05 | Affected | 4.32 | 4 | 0.1034 | 0.5449 | -2 | -3 | -0.5 | 76.10 | |||||||||||||||||||||||||||||||||
| c.3413C>T | S1138F 2D AIThe SynGAP1 missense variant S1138F is not reported in ClinVar and is present in gnomAD (ID 6‑33444448‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. The high‑accuracy AlphaMissense‑Optimized score is benign. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default (uncertain), ESM1b (benign), FATHMM (benign), and PROVEAN (pathogenic), resolves to benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of available predictions indicates a benign effect. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.891961 | Disordered | 0.738250 | Binding | 0.346 | 0.869 | 1.000 | 6-33444448-C-T | 1 | 6.20e-7 | -6.298 | Likely Benign | 0.379 | Ambiguous | Likely Benign | 0.407 | Likely Benign | -2.88 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 5.42 | Benign | 0.03 | Affected | 4.32 | 4 | 0.0991 | 0.5623 | -2 | -3 | 3.6 | 60.10 | |||||||||||||||||||||||||||||||||||
| c.3416A>T | Q1139L 2D AIThe SynGAP1 missense variant Q1139L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.908098 | Disordered | 0.721191 | Binding | 0.313 | 0.866 | 1.000 | -1.689 | Likely Benign | 0.153 | Likely Benign | Likely Benign | 0.472 | Likely Benign | -3.75 | Deleterious | 0.224 | Benign | 0.237 | Benign | 5.30 | Benign | 0.00 | Affected | 0.0745 | 0.5599 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3431T>G | L1144W 2D AIThe SynGAP1 missense variant L1144W is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33444466‑T‑G). Prediction tools that agree on a benign effect include ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split; Foldetta results are unavailable. Overall, the majority of predictions (six pathogenic vs. three benign) indicate a pathogenic impact. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.950334 | Disordered | 0.726803 | Binding | 0.277 | 0.840 | 1.000 | 6-33444466-T-G | 1 | 6.20e-7 | -5.745 | Likely Benign | 0.707 | Likely Pathogenic | Likely Benign | 0.575 | Likely Pathogenic | -2.87 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 5.36 | Benign | 0.01 | Affected | 4.32 | 4 | 0.0703 | 0.3166 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||||||||
| c.3433A>T | N1145Y 2D AIThe SynGAP1 missense variant N1145Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT uniformly predict a pathogenic impact. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields benign, while Foldetta results are unavailable. Overall, the majority of conventional tools predict pathogenicity, but the high‑accuracy consensus leans benign. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.722723 | Binding | 0.284 | 0.850 | 1.000 | -2.945 | Likely Benign | 0.451 | Ambiguous | Likely Benign | 0.543 | Likely Pathogenic | -4.07 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 5.40 | Benign | 0.01 | Affected | 0.0622 | 0.6337 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||||||||||||
| c.3434A>T | N1145I 2D AIThe SynGAP1 missense variant N1145I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools cluster into two groups: pathogenic predictions come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; benign predictions come from ESM1b, FATHMM, and AlphaMissense‑Optimized. AlphaMissense‑Default is uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, while Foldetta results are unavailable. Overall, the majority of conventional predictors indicate pathogenicity, whereas the high‑accuracy subset leans benign. Based on the aggregate evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.922952 | Disordered | 0.722723 | Binding | 0.284 | 0.850 | 1.000 | -3.172 | Likely Benign | 0.378 | Ambiguous | Likely Benign | 0.504 | Likely Pathogenic | -4.19 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 5.41 | Benign | 0.00 | Affected | 0.0720 | 0.6145 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.344A>T | Q115L 2D AIThe SynGAP1 missense variant Q115L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools, polyPhen‑2 HumDiv and HumVar, predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect for Q115L, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.657256 | Binding | 0.327 | 0.878 | 0.750 | -3.281 | Likely Benign | 0.179 | Likely Benign | Likely Benign | 0.123 | Likely Benign | -1.25 | Neutral | 0.967 | Probably Damaging | 0.901 | Possibly Damaging | 4.10 | Benign | 0.10 | Tolerated | 0.0710 | 0.5452 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3455A>T | E1152V 2D AIThe SynGAP1 missense variant E1152V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.741537 | Disordered | 0.811118 | Binding | 0.395 | 0.846 | 0.500 | -3.304 | Likely Benign | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.408 | Likely Benign | -4.65 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.1247 | 0.6384 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3464T>A | V1155E 2D AIThe SynGAP1 missense variant V1155E is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments highlight a discrepancy: AlphaMissense‑Optimized predicts pathogenic, whereas the SGM‑Consensus indicates likely benign; Foldetta stability analysis is unavailable. Consequently, the evidence is evenly split between benign and pathogenic interpretations, and no ClinVar entry contradicts these findings. The variant’s clinical significance remains uncertain, with no definitive leaning toward benign or pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.750527 | Disordered | 0.855718 | Binding | 0.335 | 0.857 | 0.500 | -3.175 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.204 | Likely Benign | -2.01 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.58 | Benign | 0.02 | Affected | 0.0973 | 0.1727 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||||||||||||
| c.346T>A | Y116N 2D AIThe SynGAP1 missense variant Y116N is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.670235 | Binding | 0.381 | 0.878 | 0.625 | -1.706 | Likely Benign | 0.260 | Likely Benign | Likely Benign | 0.108 | Likely Benign | 0.21 | Neutral | 0.137 | Benign | 0.021 | Benign | 4.27 | Benign | 1.00 | Tolerated | 0.2592 | 0.0545 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3470G>C | W1157S 2D  AIThe SynGAP1 missense variant W1157S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence indicates that W1157S is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.694846 | Disordered | 0.877471 | Binding | 0.364 | 0.861 | 0.375 | -1.158 | Likely Benign | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.394 | Likely Benign | -9.96 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.06 | Pathogenic | 0.00 | Affected | 0.4772 | 0.1227 | -2 | -3 | 0.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3470G>T | W1157L 2D  AIThe SynGAP1 missense variant W1157L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—classify the change as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic. Foldetta results are not available. Overall, the majority of evidence points to a pathogenic impact for W1157L, and this conclusion is consistent with the absence of a ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.694846 | Disordered | 0.877471 | Binding | 0.364 | 0.861 | 0.375 | -1.336 | Likely Benign | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.306 | Likely Benign | -9.46 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 1.08 | Pathogenic | 0.00 | Affected | 0.2400 | 0.2719 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||||||||||||
| c.3473T>A | V1158D 2D AIThe SynGAP1 missense variant V1158D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the majority of computational evidence indicates that V1158D is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.877504 | Binding | 0.369 | 0.847 | 0.250 | -4.076 | Likely Benign | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.486 | Likely Benign | -4.56 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.29 | Pathogenic | 0.00 | Affected | 0.1502 | 0.1820 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3478A>T | N1160Y 2D AIThe SynGAP1 missense variant N1160Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic.” No Foldetta stability prediction is available. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.585406 | Disordered | 0.861611 | Binding | 0.361 | 0.836 | 0.375 | -4.074 | Likely Benign | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.421 | Likely Benign | -4.90 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.79 | Pathogenic | 0.01 | Affected | 0.0591 | 0.5681 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3479A>T | N1160I 2D AIThe SynGAP1 missense variant N1160I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from multiple in silico tools indicates that the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.585406 | Disordered | 0.861611 | Binding | 0.361 | 0.836 | 0.375 | -4.996 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.440 | Likely Benign | -5.35 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.79 | Pathogenic | 0.01 | Affected | 0.0618 | 0.5903 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3488A>T | H1163L 2D AIThe SynGAP1 missense variant H1163L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign) and Foldetta results are unavailable. Overall, the majority of predictions (5 pathogenic vs. 4 benign) lean toward a pathogenic impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.858469 | Binding | 0.328 | 0.825 | 0.375 | -2.401 | Likely Benign | 0.707 | Likely Pathogenic | Likely Benign | 0.568 | Likely Pathogenic | -3.15 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 5.55 | Benign | 0.10 | Tolerated | 0.0938 | 0.5356 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3491T>A | L1164Q 2D AIThe SynGAP1 missense variant L1164Q has no ClinVar record and is not present in gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, whereas the SGM‑Consensus remains benign; Foldetta results are unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not conflict with ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.853935 | Binding | 0.325 | 0.815 | 0.375 | -5.374 | Likely Benign | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.497 | Likely Benign | -0.95 | Neutral | 0.999 | Probably Damaging | 0.999 | Probably Damaging | 5.32 | Benign | 0.18 | Tolerated | 0.1171 | 0.1181 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3494C>G | S1165W 2D  AIThe SynGAP1 missense variant S1165W has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign); pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome; Foldetta stability analysis is unavailable. Overall, the evidence is split, with an equal number of tools supporting benign versus pathogenic effects. Consequently, the variant is most likely pathogenic based on the current computational predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.835017 | Binding | 0.308 | 0.807 | 0.375 | -6.419 | Likely Benign | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.195 | Likely Benign | -2.29 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.54 | Benign | 0.02 | Affected | 0.0671 | 0.4807 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3500A>T | D1167V 2D AIThe SynGAP1 missense variant D1167V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Based on the preponderance of pathogenic predictions and the high‑accuracy tool results, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.783999 | Binding | 0.336 | 0.798 | 0.500 | -2.750 | Likely Benign | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.288 | Likely Benign | -3.34 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.26 | Pathogenic | 0.00 | Affected | 0.0946 | 0.7515 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3503T>A | I1168N 2D AIThe SynGAP1 missense variant I1168N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote) which labels the variant as Likely Benign. In contrast, tools that predict a pathogenic effect are PolyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further show AlphaMissense‑Optimized as Pathogenic, while the SGM‑Consensus remains Likely Benign; Foldetta results are unavailable. Overall, the evidence is split evenly between benign and pathogenic predictions, with no consensus from the most accurate methods. Consequently, the variant’s pathogenicity is uncertain and does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.763262 | Binding | 0.423 | 0.796 | 0.500 | -4.269 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.455 | Likely Benign | -1.79 | Neutral | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 5.53 | Benign | 0.01 | Affected | 0.0943 | 0.0969 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.3506A>T | E1169V 2D AIThe SynGAP1 E1169V missense variant is not reported in ClinVar and is absent from gnomAD. Consensus from standard prediction algorithms shows a split: benign predictions come from REVEL, ESM1b, and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a pathogenic signal: AlphaMissense‑Optimized is pathogenic, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—remains inconclusive (2 pathogenic vs 2 benign), and Foldetta stability analysis is unavailable. Overall, the preponderance of evidence (seven pathogenic vs three benign predictions) indicates that E1169V is most likely pathogenic. This conclusion is consistent with the absence of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.699094 | Disordered | 0.732455 | Binding | 0.400 | 0.781 | 0.625 | -2.482 | Likely Benign | 0.975 | Likely Pathogenic | Likely Pathogenic | 0.227 | Likely Benign | -2.94 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.51 | Benign | 0.00 | Affected | 0.0581 | 0.7028 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3511_3512delinsTG | A1171C 2D  AIThe SynGAP1 missense variant A1171C (ClinVar ID 1723483.0) is listed as “Uncertain” in ClinVar and is not present in gnomAD. Prediction tools that converge on a benign outcome include PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, all of which report a benign or neutral effect. In contrast, PolyPhen‑2 (HumDiv and HumVar) and SIFT uniformly predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign,” reinforcing the benign signal. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote) as benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.599170 | Disordered | 0.702689 | Binding | 0.472 | 0.775 | 0.500 | Uncertain | 1 | -5.363 | Likely Benign | 0.496 | Ambiguous | Likely Benign | -1.16 | Neutral | 0.978 | Probably Damaging | 0.825 | Possibly Damaging | 5.32 | Benign | 0.02 | Affected | 4.32 | 4 | 0.1102 | 0.4921 | -2 | 0 | 0.7 | 32.06 | ||||||||||||||||||||||||||||||||||||
| c.3515A>T | H1172L 2D AIThe SynGAP1 missense variant H1172L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign effect for H1172L, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.626927 | Disordered | 0.673805 | Binding | 0.465 | 0.758 | 0.625 | -0.545 | Likely Benign | 0.446 | Ambiguous | Likely Benign | 0.426 | Likely Benign | -2.30 | Neutral | 0.451 | Benign | 0.265 | Benign | 5.47 | Benign | 0.01 | Affected | 0.0815 | 0.5285 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||
| c.3518T>A | I1173N 2D AIThe SynGAP1 missense variant I1173N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also leans benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Taken together, the preponderance of evidence points to a benign impact for I1173N, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.501700 | Disordered | 0.653145 | Binding | 0.521 | 0.756 | 0.375 | -4.130 | Likely Benign | 0.691 | Likely Pathogenic | Likely Benign | 0.443 | Likely Benign | -1.46 | Neutral | 0.925 | Possibly Damaging | 0.611 | Possibly Damaging | 5.34 | Benign | 0.02 | Affected | 0.0920 | 0.0142 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3521A>T | E1174V 2D AIThe SynGAP1 missense variant E1174V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split opinion: benign calls come from PROVEAN, ESM1b, and FATHMM, while pathogenic calls are made by REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. Grouping by consensus, the majority of tools (four out of six) predict a benign effect, whereas the remaining four predict pathogenicity. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized returns an uncertain result; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. No Foldetta stability analysis is available for this residue. Overall, the preponderance of evidence leans toward a benign impact for E1174V, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.483068 | Structured | 0.618958 | Binding | 0.523 | 0.734 | 0.375 | -4.814 | Likely Benign | 0.877 | Likely Pathogenic | Ambiguous | 0.515 | Likely Pathogenic | -2.41 | Neutral | 0.965 | Probably Damaging | 0.703 | Possibly Damaging | 5.41 | Benign | 0.01 | Affected | 0.0539 | 0.6623 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3527A>T | E1176V 2D AISynGAP1 missense variant E1176V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy consensus methods give a mixed signal: AlphaMissense‑Optimized predicts pathogenic, whereas the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta, a protein‑folding stability approach, has no available result for this variant. Overall, the balance of evidence favors a benign classification, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.538167 | Disordered | 0.572075 | Binding | 0.525 | 0.715 | 0.250 | -3.238 | Likely Benign | 0.974 | Likely Pathogenic | Likely Pathogenic | 0.490 | Likely Benign | -2.41 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 5.69 | Benign | 0.13 | Tolerated | 0.0452 | 0.6423 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3530A>T | E1177V 2D AIThe SynGAP1 E1177V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta results are unavailable. Overall, more tools predict pathogenicity (5) than benignity (3), and no ClinVar evidence contradicts this assessment. Thus, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.541878 | Disordered | 0.566503 | Binding | 0.542 | 0.705 | 0.250 | -3.091 | Likely Benign | 0.892 | Likely Pathogenic | Ambiguous | 0.481 | Likely Benign | -2.90 | Deleterious | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 5.66 | Benign | 0.01 | Affected | 0.0463 | 0.4520 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3532T>A | Y1178N 2D AIThe SynGAP1 missense variant Y1178N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools are divided: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.541878 | Disordered | 0.568433 | Binding | 0.554 | 0.688 | 0.250 | -2.125 | Likely Benign | 0.619 | Likely Pathogenic | Likely Benign | 0.410 | Likely Benign | -1.18 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 5.50 | Benign | 0.09 | Tolerated | 0.2456 | 0.0341 | -2 | -2 | -2.2 | -49.07 | ||||||||||||||||||||||||||||||||||||||
| c.3539T>A | L1180H 2D AIThe SynGAP1 missense variant L1180H is not reported in ClinVar and has no gnomAD allele, so its population frequency is unknown. Functional prediction tools show a split: benign calls come from REVEL, PROVEAN, ESM1b, and FATHMM, whereas pathogenic calls come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments highlight AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus leans benign; Foldetta results are not available. Overall, five of nine individual predictors favor pathogenicity, four favor benign, and the consensus tool suggests benign. Thus, the variant is most likely pathogenic based on the preponderance of high‑confidence predictions, and this assessment is not contradicted by ClinVar, which contains no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.626927 | Disordered | 0.559845 | Binding | 0.591 | 0.672 | 0.250 | -5.621 | Likely Benign | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.213 | Likely Benign | -0.22 | Neutral | 0.987 | Probably Damaging | 0.865 | Possibly Damaging | 2.65 | Benign | 0.00 | Affected | 0.0991 | 0.0860 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||||||||||
| c.3545A>T | E1182V 2D AIThe SynGAP1 missense variant E1182V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the preponderance of evidence (seven pathogenic vs. three benign predictions) indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.566480 | Disordered | 0.530232 | Binding | 0.597 | 0.651 | 0.375 | -4.966 | Likely Benign | 0.966 | Likely Pathogenic | Likely Pathogenic | 0.124 | Likely Benign | -3.21 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.59 | Benign | 0.00 | Affected | 0.0447 | 0.6364 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3547T>A | Y1183N 2D AIThe SynGAP1 missense variant Y1183N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and the SGM‑Consensus score (Likely Benign). In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default all predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized tool returns an uncertain result, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome; Foldetta predictions are unavailable. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.566480 | Disordered | 0.527818 | Binding | 0.523 | 0.652 | 0.500 | -3.413 | Likely Benign | 0.890 | Likely Pathogenic | Ambiguous | 0.083 | Likely Benign | -1.44 | Neutral | 0.905 | Possibly Damaging | 0.543 | Possibly Damaging | 2.88 | Benign | 0.35 | Tolerated | 0.2368 | 0.0243 | -2 | -2 | -2.2 | -49.07 | ||||||||||||||||||||||||||||||||||||||
| c.3557C>G | S1186W 2D AIThe SynGAP1 missense variant S1186W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b remains uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (pathogenic)—also favors pathogenic. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that S1186W is most likely pathogenic, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.562014 | Disordered | 0.506433 | Binding | 0.634 | 0.636 | 0.625 | -7.814 | In-Between | 0.979 | Likely Pathogenic | Likely Pathogenic | 0.214 | Likely Benign | -3.43 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.62 | Benign | 0.01 | Affected | 0.0556 | 0.4158 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3563A>T | D1188V 2D AIThe SynGAP1 D1188V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the balance of evidence (seven pathogenic versus three benign predictions) indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.476583 | Structured | 0.484322 | Uncertain | 0.687 | 0.626 | 0.625 | -4.482 | Likely Benign | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.479 | Likely Benign | -4.13 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 5.49 | Benign | 0.00 | Affected | 0.0592 | 0.4651 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3566A>T | E1189V 2D AIThe SynGAP1 E1189V missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (2 vs 2). Foldetta stability predictions are not available. Overall, more tools (five) predict pathogenicity than benign (three), and the high‑accuracy methods do not overturn this trend. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.562014 | Disordered | 0.466885 | Uncertain | 0.704 | 0.623 | 0.625 | -5.048 | Likely Benign | 0.950 | Likely Pathogenic | Ambiguous | 0.492 | Likely Benign | -3.50 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 5.26 | Benign | 0.02 | Affected | 0.0467 | 0.4252 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.356A>T | E119V 2D AISynGAP1 missense variant E119V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that classify the variant as benign include REVEL, ESM1b, FATHMM, and polyPhen‑2 HumVar, while pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. High‑accuracy methods are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a tie and is therefore unavailable, and Foldetta results are not provided. Consequently, the evidence does not strongly support either outcome. The variant is most likely inconclusive; it does not clearly favor benign or pathogenic status, and this lack of consensus does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.661946 | Binding | 0.346 | 0.881 | 0.750 | -5.696 | Likely Benign | 0.842 | Likely Pathogenic | Ambiguous | 0.151 | Likely Benign | -2.78 | Deleterious | 0.596 | Possibly Damaging | 0.189 | Benign | 3.79 | Benign | 0.00 | Affected | 0.1152 | 0.7753 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3575T>A | L1192Q 2D AIThe SynGAP1 missense variant L1192Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (both HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign; Foldetta results are unavailable. Overall, the preponderance of evidence from multiple prediction algorithms and consensus methods points to a benign impact for this variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.575842 | Disordered | 0.441757 | Uncertain | 0.762 | 0.609 | 0.625 | -3.804 | Likely Benign | 0.535 | Ambiguous | Likely Benign | 0.224 | Likely Benign | -1.09 | Neutral | 0.992 | Probably Damaging | 0.940 | Probably Damaging | 2.70 | Benign | 0.10 | Tolerated | 0.1032 | 0.0558 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3578A>T | D1193V 2D AIThe SynGAP1 D1193V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default, while only FATHMM predicts a benign outcome. ESM1b and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (two pathogenic vs. one benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.585406 | Disordered | 0.433390 | Uncertain | 0.807 | 0.600 | 0.375 | -7.297 | In-Between | 0.855 | Likely Pathogenic | Ambiguous | 0.526 | Likely Pathogenic | -2.92 | Deleterious | 0.977 | Probably Damaging | 0.856 | Possibly Damaging | 5.51 | Benign | 0.00 | Affected | 0.0752 | 0.4174 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3584T>A | V1195E 2D AIThe SynGAP1 missense variant V1195E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. AlphaMissense‑Optimized yields an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. Overall, the balance of evidence leans toward a benign impact, with no conflict with ClinVar status (which has no entry). Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.604312 | Disordered | 0.434133 | Uncertain | 0.842 | 0.603 | 0.250 | -1.722 | Likely Benign | 0.946 | Likely Pathogenic | Ambiguous | 0.499 | Likely Benign | -2.19 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 5.64 | Benign | 0.02 | Affected | 0.0870 | 0.1268 | -2 | -2 | -7.7 | 29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3590A>T | E1197V 2D AIThe SynGAP1 missense variant E1197V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is a 2‑vs‑2 tie and therefore unavailable; Foldetta, which would combine FoldX‑MD and Rosetta outputs, has no reported result. Overall, the balance of evidence (five pathogenic versus three benign predictions, with one uncertain) indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.613573 | Disordered | 0.437361 | Uncertain | 0.827 | 0.599 | 0.250 | -6.298 | Likely Benign | 0.923 | Likely Pathogenic | Ambiguous | 0.472 | Likely Benign | -3.28 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 5.40 | Benign | 0.03 | Affected | 0.0440 | 0.5320 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3592T>A | Y1198N 2D AIThe SynGAP1 missense variant Y1198N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and SIFT, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is “Likely Pathogenic,” and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign evidence, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.626927 | Disordered | 0.439379 | Uncertain | 0.853 | 0.593 | 0.250 | -13.134 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.282 | Likely Benign | -5.92 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.44 | Pathogenic | 0.26 | Tolerated | 0.2093 | 0.0373 | -2 | -2 | -2.2 | -49.07 | ||||||||||||||||||||||||||||||||||||||
| c.3596A>T | E1199V 2D AIThe SynGAP1 missense change E1199V is not reported in ClinVar and is absent from gnomAD. Prediction tools that flag the variant as benign include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic effect. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus likewise reports a likely pathogenic outcome. Foldetta results are not available for this variant. Overall, the preponderance of computational evidence points to a pathogenic effect for E1199V, and this conclusion does not conflict with the current ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.538167 | Disordered | 0.444533 | Uncertain | 0.878 | 0.598 | 0.250 | -12.285 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.360 | Likely Benign | -5.14 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.43 | Pathogenic | 0.00 | Affected | 0.0715 | 0.4581 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3599A>T | E1200V 2D AIThe SynGAP1 E1200V missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2) and Foldetta data are unavailable. Overall, the majority of standard predictors lean toward pathogenicity, but the single high‑accuracy benign prediction and the inconclusive consensus leave the variant’s impact uncertain. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.545602 | Disordered | 0.458056 | Uncertain | 0.889 | 0.596 | 0.250 | -4.987 | Likely Benign | 0.784 | Likely Pathogenic | Likely Benign | 0.274 | Likely Benign | -3.52 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.63 | Benign | 0.00 | Affected | 0.0498 | 0.4648 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3602A>T | E1201V 2D AIThe SynGAP1 missense variant E1201V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: REVEL predicts a benign change, whereas all other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic status. No Foldetta stability analysis is available for this variant. Overall, the preponderance of evidence from multiple prediction tools and consensus methods indicates that E1201V is most likely pathogenic, and this conclusion is consistent with the absence of any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.525368 | Disordered | 0.481868 | Uncertain | 0.870 | 0.596 | 0.250 | -10.865 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.402 | Likely Benign | -5.43 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.59 | Pathogenic | 0.00 | Affected | 0.0455 | 0.6308 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3605T>A | I1202N 2D AIThe SynGAP1 missense variant I1202N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Functional prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote) is likely pathogenic. Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, with no ClinVar status to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.529623 | Disordered | 0.510422 | Binding | 0.874 | 0.593 | 0.250 | -10.922 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.303 | Likely Benign | -5.65 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.79 | Pathogenic | 0.00 | Affected | 0.1014 | 0.0270 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3608A>T | H1203L 2D AIThe SynGAP1 missense variant H1203L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. High‑accuracy assessments confirm this trend: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.618285 | Disordered | 0.527023 | Binding | 0.892 | 0.589 | 0.250 | -4.179 | Likely Benign | 0.153 | Likely Benign | Likely Benign | 0.361 | Likely Benign | -2.23 | Neutral | 0.473 | Possibly Damaging | 0.265 | Benign | 5.53 | Benign | 0.21 | Tolerated | 0.0682 | 0.2951 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||
| c.3611C>T | S1204L 2D AIThe SynGAP1 missense variant S1204L is reported in gnomAD (variant ID 6‑33446603‑C‑T) and has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; Foldetta results are not available. Based on the collective predictions, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.595080 | Disordered | 0.541098 | Binding | 0.887 | 0.587 | 0.375 | 6-33446603-C-T | 1 | 6.20e-7 | -4.672 | Likely Benign | 0.325 | Likely Benign | Likely Benign | 0.375 | Likely Benign | -0.86 | Neutral | 0.035 | Benign | 0.028 | Benign | 5.35 | Benign | 0.32 | Tolerated | 3.77 | 5 | 0.1000 | 0.4082 | -2 | -3 | 4.6 | 26.08 | |||||||||||||||||||||||||||||||||
| c.3614T>A | L1205Q 2D AIThe SynGAP1 missense variant L1205Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus agrees. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.595080 | Disordered | 0.552471 | Binding | 0.880 | 0.576 | 0.375 | -14.466 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.453 | Likely Benign | -5.02 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.46 | Pathogenic | 0.00 | Affected | 0.1032 | 0.0558 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3617A>T | K1206I 2D AIThe SynGAP1 missense variant K1206I is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; Foldetta results are not available. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for K1206I. This conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.585406 | Disordered | 0.555819 | Binding | 0.893 | 0.569 | 0.375 | -13.526 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.302 | Likely Benign | -5.65 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.37 | Pathogenic | 0.01 | Affected | 0.0816 | 0.3521 | -2 | -3 | 8.4 | -15.01 | ||||||||||||||||||||||||||||||||||||||
| c.3620A>T | E1207V 2D AIThe SynGAP1 missense change E1207V is not reported in ClinVar (ClinVar ID = None) and has no entries in gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments show the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, while AlphaMissense‑Optimized remains uncertain and Foldetta data are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for E1207V, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.604312 | Disordered | 0.562696 | Binding | 0.912 | 0.571 | 0.375 | -9.580 | Likely Pathogenic | 0.821 | Likely Pathogenic | Ambiguous | 0.342 | Likely Benign | -5.00 | Deleterious | 0.999 | Probably Damaging | 0.958 | Probably Damaging | 2.07 | Pathogenic | 0.00 | Affected | 0.0511 | 0.4439 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3626T>A | L1209Q 2D AIThe SynGAP1 missense variant L1209Q is not listed in ClinVar and has no entry in gnomAD, indicating it is not a common population variant. Functional prediction tools largely agree on a deleterious effect: REVEL predicts a benign outcome, whereas the remaining predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely pathogenic status. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.595080 | Disordered | 0.583711 | Binding | 0.899 | 0.574 | 0.375 | -12.820 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.379 | Likely Benign | -5.09 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.46 | Pathogenic | 0.00 | Affected | 0.1042 | 0.0558 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3629A>T | H1210L 2D AIThe SynGAP1 missense variant H1210L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools indicates that H1210L is most likely benign, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.599170 | Disordered | 0.587579 | Binding | 0.900 | 0.567 | 0.375 | -4.018 | Likely Benign | 0.116 | Likely Benign | Likely Benign | 0.168 | Likely Benign | -2.90 | Deleterious | 0.000 | Benign | 0.002 | Benign | 2.70 | Benign | 0.04 | Affected | 0.0673 | 0.4282 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||
| c.3637A>T | N1213Y 2D AIThe SynGAP1 missense variant N1213Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. AlphaMissense‑Default is uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as pathogenic. AlphaMissense‑Optimized predicts benign, while high‑accuracy folding‑stability predictions from Foldetta are unavailable. Overall, more tools (five) predict pathogenicity than benign (three), and the consensus and high‑accuracy methods lean toward pathogenic. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.580690 | Disordered | 0.521638 | Binding | 0.888 | 0.561 | 0.500 | -8.972 | Likely Pathogenic | 0.483 | Ambiguous | Likely Benign | 0.083 | Likely Benign | -2.67 | Deleterious | 0.920 | Possibly Damaging | 0.657 | Possibly Damaging | 2.68 | Benign | 0.02 | Affected | 0.0439 | 0.3681 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3638A>T | N1213I 2D AIThe SynGAP1 missense variant N1213I is not reported in ClinVar and is absent from gnomAD. Prediction tools show a split opinion: benign calls come from REVEL, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further highlight this discordance: AlphaMissense‑Optimized predicts a benign effect, whereas the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome. No Foldetta stability analysis is available for this residue. Overall, the preponderance of evidence points to a pathogenic effect for N1213I, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.580690 | Disordered | 0.521638 | Binding | 0.888 | 0.561 | 0.500 | -10.798 | Likely Pathogenic | 0.743 | Likely Pathogenic | Likely Benign | 0.093 | Likely Benign | -3.10 | Deleterious | 0.996 | Probably Damaging | 0.930 | Probably Damaging | 2.71 | Benign | 0.03 | Affected | 0.0437 | 0.4407 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3640C>G | R1214G 2D AIThe SynGAP1 missense variant R1214G is listed in gnomAD (6‑33446632‑C‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect are REVEL and AlphaMissense‑Optimized; those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable. Overall, the majority of tools predict a pathogenic impact, and this conclusion is not contradicted by any ClinVar status (none). Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.497853 | Structured | 0.506868 | Binding | 0.903 | 0.566 | 0.375 | 6-33446632-C-G | 1 | 6.20e-7 | -9.697 | Likely Pathogenic | 0.725 | Likely Pathogenic | Likely Benign | 0.131 | Likely Benign | -4.41 | Deleterious | 0.992 | Probably Damaging | 0.828 | Possibly Damaging | 2.48 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0.3229 | 0.2361 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||
| c.3647T>A | L1216Q 2D AIThe SynGAP1 missense variant L1216Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as pathogenic; Foldetta results are not available. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.580690 | Disordered | 0.504713 | Binding | 0.863 | 0.563 | 0.250 | -8.731 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.404 | Likely Benign | -4.12 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.23 | Pathogenic | 0.00 | Affected | 0.0939 | 0.0488 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3650A>G | E1217G 2D AIThe SynGAP1 missense variant E1217G is reported in gnomAD (ID 6‑33446642‑A‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas the remaining tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) all predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for E1217G. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.599170 | Disordered | 0.493043 | Uncertain | 0.877 | 0.563 | 0.250 | 6-33446642-A-G | -10.803 | Likely Pathogenic | 0.620 | Likely Pathogenic | Likely Benign | 0.331 | Likely Benign | -5.38 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.35 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2205 | 0.4927 | -2 | 0 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||
| c.3650A>T | E1217V 2D AIThe SynGAP1 missense variant E1217V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: REVEL scores the variant as benign, whereas PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenicity. Grouping by consensus, the majority of tools (seven) support a pathogenic effect, while only one tool (REVEL) indicates benign. High‑accuracy assessments further reinforce a deleterious interpretation: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic, and AlphaMissense‑Optimized remains uncertain. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the computational evidence overwhelmingly suggests that E1217V is pathogenic, a finding that aligns with its lack of ClinVar annotation and gnomAD presence. Thus, the variant is most likely pathogenic, and this prediction is consistent with its absence from ClinVar and gnomAD. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.599170 | Disordered | 0.493043 | Uncertain | 0.877 | 0.563 | 0.250 | -12.098 | Likely Pathogenic | 0.843 | Likely Pathogenic | Ambiguous | 0.351 | Likely Benign | -5.48 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.33 | Pathogenic | 0.00 | Affected | 0.0579 | 0.5348 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3653A>T | E1218V 2D AISynGAP1 missense variant E1218V is listed in ClinVar with an uncertain significance (ClinVar ID 1015602.0) and is not reported in gnomAD. Functional prediction tools show a split opinion: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. When the high‑accuracy consensus is considered, AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as likely pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this change. Overall, the majority of evidence points toward a pathogenic effect, which is consistent with the ClinVar designation of uncertain significance rather than a benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.595080 | Disordered | 0.483050 | Uncertain | 0.898 | 0.565 | 0.375 | Uncertain | 2 | -5.647 | Likely Benign | 0.936 | Likely Pathogenic | Ambiguous | 0.418 | Likely Benign | -5.68 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.21 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.0491 | 0.4120 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||
| c.3655T>A | Y1219N 2D AIThe SynGAP1 missense variant Y1219N is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess functional impact largely agree on a deleterious effect: SIFT, polyPhen‑2 (HumDiv and HumVar), PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while the only benign prediction comes from REVEL. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Pathogenic.” High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic status. Foldetta results are not available, so they do not influence the overall assessment. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.613573 | Disordered | 0.474748 | Uncertain | 0.855 | 0.557 | 0.375 | -11.679 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.366 | Likely Benign | -6.41 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.15 | Pathogenic | 0.00 | Affected | 0.2478 | 0.0573 | -2 | -2 | -2.2 | -49.07 | ||||||||||||||||||||||||||||||||||||||
| c.3659A>T | E1220V 2D AIThe SynGAP1 missense variant E1220V is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that E1220V is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.703578 | Disordered | 0.444845 | Uncertain | 0.881 | 0.551 | 0.375 | -15.193 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.444 | Likely Benign | -6.05 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.59 | Pathogenic | 0.00 | Affected | 0.0552 | 0.4452 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3668T>A | L1223Q 2D AIThe SynGAP1 missense variant L1223Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict a pathogenic impact, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of any benign consensus, the variant is most likely pathogenic; this conclusion is not contradicted by ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.608892 | Disordered | 0.436267 | Uncertain | 0.868 | 0.540 | 0.375 | -13.700 | Likely Pathogenic | 0.934 | Likely Pathogenic | Ambiguous | 0.380 | Likely Benign | -4.13 | Deleterious | 1.000 | Probably Damaging | 0.986 | Probably Damaging | 1.46 | Pathogenic | 0.01 | Affected | 0.1010 | 0.1119 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3671T>A | L1224Q 2D AIThe SynGAP1 missense variant L1224Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate a benign or likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports likely benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect for L1224Q, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this is not contradictory to ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.525368 | Disordered | 0.441554 | Uncertain | 0.871 | 0.543 | 0.500 | -6.254 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.87 | Neutral | 0.994 | Probably Damaging | 0.900 | Possibly Damaging | 2.40 | Pathogenic | 0.13 | Tolerated | 0.1018 | 0.0558 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3671T>G | L1224R 2D  AIThe SynGAP1 missense variant L1224R is listed in ClinVar with no assertion (ClinVar status: None) and is present in the gnomAD database (ID 6‑33446663‑T‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; ESM1b remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which currently contains no pathogenic assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.525368 | Disordered | 0.441554 | Uncertain | 0.871 | 0.543 | 0.500 | 6-33446663-T-G | 1 | 6.20e-7 | -7.504 | In-Between | 0.220 | Likely Benign | Likely Benign | 0.113 | Likely Benign | -1.85 | Neutral | 0.989 | Probably Damaging | 0.900 | Possibly Damaging | 2.42 | Pathogenic | 0.33 | Tolerated | 3.77 | 5 | 0.1120 | 0.0558 | -2 | -3 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||
| c.3677A>T | Q1226L 2D AIThe SynGAP1 missense variant Q1226L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments further support a deleterious interpretation: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic,” AlphaMissense‑Optimized is classified as “Uncertain,” and Foldetta’s protein‑folding stability analysis is unavailable. Taken together, the preponderance of evidence points to a pathogenic effect for Q1226L. This conclusion is not contradicted by ClinVar status, as the variant is currently unreported in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.529623 | Disordered | 0.432206 | Uncertain | 0.850 | 0.547 | 0.250 | -11.122 | Likely Pathogenic | 0.879 | Likely Pathogenic | Ambiguous | 0.353 | Likely Benign | -5.62 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 1.77 | Pathogenic | 0.00 | Affected | 0.0493 | 0.4282 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3680A>T | E1227V 2D AIThe SynGAP1 missense variant E1227V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess the variant’s effect fall into two groups: the single benign prediction comes from REVEL, whereas all other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—classify it as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus also indicates likely pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from multiple independent prediction tools and high‑accuracy methods indicates that E1227V is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.513880 | Disordered | 0.433399 | Uncertain | 0.860 | 0.544 | 0.500 | -12.852 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.355 | Likely Benign | -5.49 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.0405 | 0.6559 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3683A>T | E1228V 2D AIThe SynGAP1 missense variant E1228V is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and the SGM‑Consensus score, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further separate the evidence: AlphaMissense‑Optimized indicates a benign effect, whereas the SGM‑Consensus, derived from a consensus of four high‑confidence predictors, flags the variant as pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this change. Overall, the preponderance of pathogenic predictions, including the SGM‑Consensus, outweighs the benign calls. Therefore, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.517562 | Disordered | 0.447051 | Uncertain | 0.892 | 0.546 | 0.500 | -8.077 | Likely Pathogenic | 0.440 | Ambiguous | Likely Benign | 0.293 | Likely Benign | -4.55 | Deleterious | 0.980 | Probably Damaging | 0.833 | Possibly Damaging | 2.43 | Pathogenic | 0.00 | Affected | 0.0470 | 0.4252 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3686A>T | Q1229L 2D AIThe SynGAP1 missense variant Q1229L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into three groups: benign predictions come from REVEL, SIFT, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; the remaining tools (ESM1b and AlphaMissense‑Default) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta data are unavailable. Overall, the majority of conventional predictors favor a pathogenic effect, whereas the single high‑accuracy tool suggests benign. Given the lack of ClinVar evidence, the variant is most likely pathogenic according to the collective predictions, with no contradiction to existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.490133 | Structured | 0.466729 | Uncertain | 0.865 | 0.544 | 0.375 | -7.366 | In-Between | 0.496 | Ambiguous | Likely Benign | 0.349 | Likely Benign | -4.60 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 1.77 | Pathogenic | 0.09 | Tolerated | 0.0541 | 0.4253 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3695A>T | K1232I 2D AIThe SynGAP1 K1232I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. The high‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is classified as Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.505461 | Disordered | 0.542907 | Binding | 0.894 | 0.535 | 0.125 | -12.225 | Likely Pathogenic | 0.896 | Likely Pathogenic | Ambiguous | 0.197 | Likely Benign | -5.98 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.08 | Pathogenic | 0.00 | Affected | 0.0778 | 0.3321 | -2 | -3 | 8.4 | -15.01 | ||||||||||||||||||||||||||||||||||||||
| c.3698T>A | I1233N 2D AIThe SynGAP1 I1233N missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and FATHMM. All other evaluated tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic impact. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.525368 | Disordered | 0.564054 | Binding | 0.881 | 0.531 | 0.125 | -9.586 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.193 | Likely Benign | -4.36 | Deleterious | 0.995 | Probably Damaging | 0.913 | Probably Damaging | 2.52 | Benign | 0.00 | Affected | 0.0879 | 0.0340 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3701T>A | L1234Q 2D AIThe SynGAP1 missense variant L1234Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—classify the variant as pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta results are unavailable. Based on the predominance of pathogenic predictions and the lack of supporting benign evidence, the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.599170 | Disordered | 0.575096 | Binding | 0.844 | 0.527 | 0.125 | -12.969 | Likely Pathogenic | 0.858 | Likely Pathogenic | Ambiguous | 0.272 | Likely Benign | -4.34 | Deleterious | 0.997 | Probably Damaging | 0.955 | Probably Damaging | 1.46 | Pathogenic | 0.01 | Affected | 0.0962 | 0.1049 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3707A>T | Q1236L 2D AIThe SynGAP1 missense variant Q1236L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, while Foldetta results are unavailable. Overall, the majority of high‑confidence predictions lean toward a benign impact, and there is no conflict with ClinVar status. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.680603 | Disordered | 0.567914 | Binding | 0.883 | 0.537 | 0.125 | -6.682 | Likely Benign | 0.409 | Ambiguous | Likely Benign | 0.362 | Likely Benign | -4.51 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.66 | Benign | 0.00 | Affected | 0.0507 | 0.3805 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3709T>A | Y1237N 2D AIThe SynGAP1 missense variant Y1237N is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic. Foldetta results are unavailable. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.653063 | Disordered | 0.563444 | Binding | 0.842 | 0.535 | 0.125 | -10.505 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.413 | Likely Benign | -7.18 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.44 | Pathogenic | 0.00 | Affected | 0.2428 | 0.0173 | -2 | -2 | -2.2 | -49.07 | ||||||||||||||||||||||||||||||||||||||
| c.3710A>G | Y1237C 2D AIThe SynGAP1 missense variant Y1237C is listed in gnomAD (variant ID 6‑33446702‑A‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: the single benign prediction comes from REVEL, while all other evaluated algorithms (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic effect. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized reports a pathogenic outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Pathogenic.” No Foldetta stability result is available, so it does not influence the overall assessment. Based on the collective predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.653063 | Disordered | 0.563444 | Binding | 0.842 | 0.535 | 0.125 | 6-33446702-A-G | 1 | 6.20e-7 | -8.600 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 0.429 | Likely Benign | -7.15 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.43 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.3354 | 0.1549 | -2 | 0 | 3.8 | -60.04 | |||||||||||||||||||||||||||||||||
| c.3713A>T | Q1238L 2D AIThe SynGAP1 missense variant Q1238L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—classify the variant as pathogenic. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments show the SGM‑Consensus as “Likely Pathogenic,” while AlphaMissense‑Optimized remains uncertain and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.562014 | Disordered | 0.548882 | Binding | 0.855 | 0.545 | 0.250 | -14.299 | Likely Pathogenic | 0.876 | Likely Pathogenic | Ambiguous | 0.353 | Likely Benign | -4.89 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.31 | Pathogenic | 0.01 | Affected | 0.0543 | 0.3744 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3722T>A | L1241Q 2D AIThe SynGAP1 missense variant L1241Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: the single benign prediction comes from REVEL, while all other evaluated algorithms (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic effect. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic, and this conclusion is consistent with the absence of any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.545602 | Disordered | 0.488880 | Uncertain | 0.828 | 0.541 | 0.375 | -10.429 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.386 | Likely Benign | -4.65 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.61 | Pathogenic | 0.00 | Affected | 0.1217 | 0.0488 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3725A>T | E1242V 2D AIThe E1242V missense change occurs in the coiled‑coil domain of SynGAP1. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect are REVEL and AlphaMissense‑Optimized. Those that predict a pathogenic outcome include PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (which is “Likely Pathogenic”). ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which simply lacks an entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.557691 | Disordered | 0.456349 | Uncertain | 0.870 | 0.549 | 0.500 | -7.456 | In-Between | 0.691 | Likely Pathogenic | Likely Benign | 0.267 | Likely Benign | -5.46 | Deleterious | 0.991 | Probably Damaging | 0.898 | Possibly Damaging | 2.17 | Pathogenic | 0.00 | Affected | 0.0447 | 0.4320 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3728A>T | Q1243L 2D AIThe SynGAP1 missense variant Q1243L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for this variant, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.545602 | Disordered | 0.433693 | Uncertain | 0.887 | 0.551 | 0.500 | -6.092 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.168 | Likely Benign | -4.00 | Deleterious | 0.912 | Possibly Damaging | 0.629 | Possibly Damaging | 2.65 | Benign | 0.02 | Affected | 0.0541 | 0.3364 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3734A>T | E1245V 2D AIThe SynGAP1 missense change E1245V is not reported in ClinVar and is absent from gnomAD. Prediction tools that flag the variant as benign include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic effect. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus likewise reports a likely pathogenic outcome. Foldetta results are not available for this variant. Overall, the preponderance of computational evidence points to a pathogenic effect for E1245V, and this conclusion does not conflict with the current ClinVar status, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.712013 | Disordered | 0.387847 | Uncertain | 0.869 | 0.554 | 0.625 | -12.988 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.319 | Likely Benign | -5.65 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.21 | Pathogenic | 0.00 | Affected | 0.0518 | 0.6856 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3743T>A | L1248Q 2D AIThe SynGAP1 missense variant L1248Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote) confirms this prediction; the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a pathogenic effect for this variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.834292 | Disordered | 0.371716 | Uncertain | 0.880 | 0.562 | 0.625 | -6.471 | Likely Benign | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.364 | Likely Benign | -4.65 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.64 | Pathogenic | 0.00 | Affected | 0.1067 | 0.0688 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3749A>T | Q1250L 2D AIThe SynGAP1 missense variant Q1250L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions (REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus “Likely Benign”) and pathogenic predictions (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates “Likely Benign.” No Foldetta stability analysis is available for this residue. Overall, the majority of evidence points to a benign effect, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, and this assessment does not contradict ClinVar, which contains no pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.759478 | Disordered | 0.360484 | Uncertain | 0.881 | 0.554 | 0.750 | -4.409 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -3.49 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.65 | Benign | 0.02 | Affected | 0.0613 | 0.3946 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3752A>T | Q1251L 2D AIThe SynGAP1 missense variant Q1251L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b) predict a pathogenic impact; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized remains benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—favors pathogenicity. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence points to a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently contains no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | 0.771762 | Disordered | 0.363872 | Uncertain | 0.869 | 0.551 | 0.875 | -10.298 | Likely Pathogenic | 0.412 | Ambiguous | Likely Benign | 0.279 | Likely Benign | -4.71 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.58 | Benign | 0.00 | Affected | 0.0626 | 0.4605 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3755A>T | Q1252L 2D AIThe SynGAP1 missense variant Q1252L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.759478 | Disordered | 0.371411 | Uncertain | 0.850 | 0.544 | 0.875 | -8.110 | Likely Pathogenic | 0.833 | Likely Pathogenic | Ambiguous | 0.295 | Likely Benign | -5.62 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 1.97 | Pathogenic | 0.00 | Affected | 0.0593 | 0.3689 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3761A>T | E1254V 2D AIThe SynGAP1 missense variant E1254V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: REVEL scores the variant as benign, whereas the remaining seven tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict it to be pathogenic. Grouping by consensus, the benign prediction is represented only by REVEL, while the pathogenic predictions are supported by the majority of in silico methods. High‑accuracy assessments further reinforce a pathogenic interpretation: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as likely pathogenic. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic, which is consistent with the lack of ClinVar annotation and gnomAD absence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.657645 | Disordered | 0.403242 | Uncertain | 0.886 | 0.555 | 0.625 | -9.913 | Likely Pathogenic | 0.814 | Likely Pathogenic | Ambiguous | 0.350 | Likely Benign | -5.15 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.31 | Pathogenic | 0.00 | Affected | 0.0481 | 0.5894 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3767A>T | D1256V 2D AIThe SynGAP1 missense variant D1256V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess functional impact largely agree on a deleterious effect: SIFT, polyPhen‑2 (HumDiv and HumVar), PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while the only benign prediction comes from REVEL. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as “Likely Pathogenic.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic status. Foldetta results are not available, so they do not influence the overall assessment. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.549308 | Disordered | 0.445789 | Uncertain | 0.876 | 0.571 | 0.625 | -14.067 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.446 | Likely Benign | -6.89 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.63 | Pathogenic | 0.00 | Affected | 0.0573 | 0.4407 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||
| c.3773A>T | Q1258L 2D AIThe SynGAP1 missense variant Q1258L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: REVEL scores the variant as benign, whereas PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenicity. Grouping by consensus, the majority of tools (seven) predict pathogenic, while only one tool (REVEL) predicts benign. High‑accuracy assessments further support a deleterious effect: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic; AlphaMissense‑Optimized remains uncertain, and Foldetta results are unavailable. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic. This conclusion aligns with the lack of ClinVar annotation and gnomAD absence, indicating no conflicting evidence from population databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.486429 | Structured | 0.525814 | Binding | 0.859 | 0.577 | 0.250 | -10.302 | Likely Pathogenic | 0.895 | Likely Pathogenic | Ambiguous | 0.341 | Likely Benign | -5.55 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 1.97 | Pathogenic | 0.00 | Affected | 0.0448 | 0.4137 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3776T>A | I1259N 2D AIThe SynGAP1 missense variant I1259N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all indicate likely pathogenicity. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus also reports a likely pathogenic classification. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for I1259N. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.494003 | Structured | 0.576405 | Binding | 0.885 | 0.574 | 0.250 | -10.979 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.459 | Likely Benign | -3.55 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.51 | Benign | 0.00 | Affected | 0.0799 | 0.0340 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3785T>A | I1262N 2D AIThe SynGAP1 missense variant I1262N is not reported in ClinVar and is absent from gnomAD, indicating no known population frequency data. Functional prediction tools largely agree on a deleterious effect: REVEL predicts a benign outcome, whereas all other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely pathogenic status. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus confirms a likely pathogenic classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the preponderance of computational evidence indicates that I1262N is most likely pathogenic, and this conclusion is consistent with the absence of any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.497853 | Structured | 0.707863 | Binding | 0.886 | 0.576 | 0.125 | -14.512 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 0.463 | Likely Benign | -5.81 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.79 | Pathogenic | 0.00 | Affected | 0.0940 | 0.0340 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3788T>A | I1263N 2D AIThe SynGAP1 missense variant I1263N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.425610 | Structured | 0.740957 | Binding | 0.867 | 0.574 | 0.000 | -9.158 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.379 | Likely Benign | -5.80 | Deleterious | 0.995 | Probably Damaging | 0.913 | Probably Damaging | 1.79 | Pathogenic | 0.00 | Affected | 0.1041 | 0.0340 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||
| c.3797T>A | L1266Q 2D AIThe SynGAP1 missense variant L1266Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates likely pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence points to a pathogenic effect for L1266Q, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.802655 | Binding | 0.868 | 0.602 | 0.000 | -16.101 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.374 | Likely Benign | -5.02 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.12 | Pathogenic | 0.00 | Affected | 0.0906 | 0.1249 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3803T>A | L1268Q 2D AIThe SynGAP1 missense variant L1268Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two PolyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus also indicates benign. Foldetta results are not available, so they do not influence the assessment. Overall, the preponderance of evidence points to a benign impact for this variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | 0.458154 | Structured | 0.804315 | Binding | 0.859 | 0.629 | 0.000 | -5.707 | Likely Benign | 0.143 | Likely Benign | Likely Benign | 0.062 | Likely Benign | -0.50 | Neutral | 0.990 | Probably Damaging | 0.637 | Possibly Damaging | 2.68 | Benign | 0.08 | Tolerated | 0.0988 | 0.0558 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||
| c.3806T>A | V1269E 2D AIThe SynGAP1 missense change V1269E is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that flag the variant as benign include only REVEL, whereas the remaining predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classify it as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a “Likely Pathogenic” outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as likely pathogenic; the Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a pathogenic effect, which does not contradict the ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.433034 | Structured | 0.787464 | Binding | 0.843 | 0.647 | 0.125 | Uncertain | 1 | -11.418 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.403 | Likely Benign | -5.05 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.09 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.0899 | 0.1557 | -2 | -2 | -7.7 | 29.98 | ||||||||||||||||||||||||||||||||||
| c.3809A>G | E1270G 2D AIThe SynGAP1 missense variant E1270G is listed in gnomAD (ID 6‑33447857‑A‑G) and has no ClinVar entry. Prediction tools cluster into two groups: the single benign predictor REVEL, and a consensus of pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic, while Foldetta’s protein‑folding stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the absence of a benign consensus, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.490133 | Structured | 0.771865 | Binding | 0.805 | 0.659 | 0.250 | 6-33447857-A-G | -12.022 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.385 | Likely Benign | -5.90 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.05 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2415 | 0.5452 | -2 | 0 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||
| c.3809A>T | E1270V 2D AIThe SynGAP1 missense variant E1270V is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus agrees. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that E1270V is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | 0.490133 | Structured | 0.771865 | Binding | 0.805 | 0.659 | 0.250 | -13.293 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.408 | Likely Benign | -5.90 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.0570 | 0.6461 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3812A>G | E1271G 2D AIThe SynGAP1 missense variant E1271G is catalogued in gnomAD (ID 6‑33447860‑A‑G) but has no entry in ClinVar. Functional prediction tools show a split consensus: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts a benign effect, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic (two pathogenic versus one benign vote). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence, including the SGM Consensus, indicates a pathogenic impact. This prediction is not contradicted by ClinVar, as no ClinVar classification exists for E1271G. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.483068 | Structured | 0.767529 | Binding | 0.832 | 0.666 | 0.375 | 6-33447860-A-G | -4.857 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.288 | Likely Benign | -5.37 | Deleterious | 0.905 | Possibly Damaging | 0.538 | Possibly Damaging | 2.05 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2513 | 0.5485 | -2 | 0 | 3.1 | -72.06 | 10.1016/j.ajhg.2020.11.011 | ||||||||||||||||||||||||||||||||||||
| c.3812A>T | E1271V 2D AISynGAP1 missense variant E1271V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. AlphaMissense‑Optimized yields an uncertain result, and no Foldetta stability assessment is available. Considering the majority of high‑confidence tools and the consensus score, the variant is most likely pathogenic. This assessment does not contradict any ClinVar annotation, as no ClinVar entry exists for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.767529 | Binding | 0.832 | 0.666 | 0.375 | -6.961 | Likely Benign | 0.848 | Likely Pathogenic | Ambiguous | 0.303 | Likely Benign | -5.64 | Deleterious | 0.995 | Probably Damaging | 0.846 | Possibly Damaging | 2.02 | Pathogenic | 0.00 | Affected | 0.0620 | 0.6106 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3815A>G | E1272G 2D AIThe SynGAP1 missense variant E1272G is catalogued in gnomAD (ID 6‑33447863‑A‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL and ESM1b, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score, which is labeled Likely Pathogenic. The high‑accuracy AlphaMissense‑Optimized assessment is uncertain, and the Foldetta protein‑folding stability analysis is not available for this residue. Overall, the majority of evidence points toward a deleterious effect, so the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, as no ClinVar classification exists for E1272G. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.766082 | Binding | 0.799 | 0.677 | 0.500 | 6-33447863-A-G | -1.919 | Likely Benign | 0.863 | Likely Pathogenic | Ambiguous | 0.287 | Likely Benign | -5.89 | Deleterious | 0.999 | Probably Damaging | 0.992 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2804 | 0.5072 | -2 | 0 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||||||||
| c.3815A>T | E1272V 2D AIThe SynGAP1 missense variant E1272V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default all indicate pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. AlphaMissense‑Optimized returns an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this variant. Based on the preponderance of pathogenic predictions and the SGM‑Consensus outcome, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.483068 | Structured | 0.766082 | Binding | 0.799 | 0.677 | 0.500 | -3.628 | Likely Benign | 0.934 | Likely Pathogenic | Ambiguous | 0.278 | Likely Benign | -5.90 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.22 | Pathogenic | 0.00 | Affected | 0.0424 | 0.5894 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3818T>A | L1273Q 2D AIThe SynGAP1 missense variant L1273Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic and the SGM‑Consensus is labeled Likely Pathogenic; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.773625 | Binding | 0.747 | 0.677 | 0.500 | -6.813 | Likely Benign | 0.957 | Likely Pathogenic | Likely Pathogenic | 0.423 | Likely Benign | -5.05 | Deleterious | 0.997 | Probably Damaging | 0.950 | Probably Damaging | 2.13 | Pathogenic | 0.00 | Affected | 0.1139 | 0.1119 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3824G>C | R1275P 2D  AIThe SynGAP1 missense variant R1275P is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33447872‑G‑C). Prediction tools that agree on a benign effect are REVEL and FATHMM, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. Uncertain predictions come from ESM1b and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact. This conclusion is consistent with the lack of ClinVar annotation, as there is no contradictory status to report. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.648219 | Disordered | 0.790317 | Binding | 0.723 | 0.697 | 0.500 | 6-33447872-G-C | -7.155 | In-Between | 0.823 | Likely Pathogenic | Ambiguous | 0.145 | Likely Benign | -3.55 | Deleterious | 0.966 | Probably Damaging | 0.651 | Possibly Damaging | 2.53 | Benign | 0.01 | Affected | 3.77 | 5 | 0.2211 | 0.3083 | -2 | 0 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||
| c.3827A>T | D1276V 2D AIThe SynGAP1 D1276V missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is unavailable for this variant. Based on the preponderance of pathogenic predictions and the SGM‑Consensus result, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.666105 | Disordered | 0.802156 | Binding | 0.636 | 0.705 | 0.625 | -0.725 | Likely Benign | 0.851 | Likely Pathogenic | Ambiguous | 0.331 | Likely Benign | -6.66 | Deleterious | 0.984 | Probably Damaging | 0.825 | Possibly Damaging | 1.19 | Pathogenic | 0.00 | Affected | 0.0614 | 0.5207 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.3830A>C | H1277P 2D AIThe SynGAP1 missense variant H1277P is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33447878‑A‑C). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, and this is consistent with the lack of a ClinVar pathogenic annotation. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | 6-33447878-A-C | -3.829 | Likely Benign | 0.324 | Likely Benign | Likely Benign | 0.237 | Likely Benign | -7.14 | Deleterious | 0.586 | Possibly Damaging | 0.287 | Benign | 2.12 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2266 | 0.3393 | -2 | 0 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||
| c.3830A>T | H1277L 2D AIThe SynGAP1 missense variant H1277L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two benign vs. two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and there is no ClinVar annotation to contradict this assessment. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.805725 | Binding | 0.562 | 0.718 | 0.750 | -2.949 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.215 | Likely Benign | -7.93 | Deleterious | 0.224 | Benign | 0.091 | Benign | 2.17 | Pathogenic | 0.00 | Affected | 0.0822 | 0.4158 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3833C>A | P1278H 2D AIThe SynGAP1 missense variant P1278H is reported in gnomAD (variant ID 6‑33447881‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict pathogenic. High‑accuracy assessments confirm the benign prediction: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (derived from the same set of benign predictions) is benign; Foldetta results are unavailable. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic designation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.806955 | Binding | 0.532 | 0.722 | 0.750 | 6-33447881-C-A | 7 | 4.51e-6 | -5.691 | Likely Benign | 0.142 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -1.69 | Neutral | 0.938 | Possibly Damaging | 0.477 | Possibly Damaging | 2.67 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1997 | 0.3155 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||
| c.3836C>A | A1279D 2D  AIThe SynGAP1 missense variant A1279D is catalogued in gnomAD (ID 6‑33447884‑C‑A) but has no ClinVar entry. Across the spectrum of in‑silico predictors, every tool listed—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classifies the substitution as benign. No pathogenic predictions are reported. Grouping by agreement, all available tools fall into the benign category, with no tools indicating pathogenicity. High‑accuracy assessments further support this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no result for this variant and is therefore considered unavailable. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.814139 | Binding | 0.485 | 0.724 | 0.750 | 6-33447884-C-A | -3.914 | Likely Benign | 0.172 | Likely Benign | Likely Benign | 0.087 | Likely Benign | 1.38 | Neutral | 0.000 | Benign | 0.000 | Benign | 2.89 | Benign | 0.35 | Tolerated | 3.77 | 5 | 0.1505 | 0.2062 | -2 | 0 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||||
| c.3842C>A | A1281D 2D AIThe SynGAP1 missense variant A1281D is reported in gnomAD (ID 6‑33447890‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not conflict with ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.821556 | Binding | 0.434 | 0.721 | 0.875 | 6-33447890-C-A | -5.183 | Likely Benign | 0.178 | Likely Benign | Likely Benign | 0.054 | Likely Benign | -0.76 | Neutral | 0.901 | Possibly Damaging | 0.516 | Possibly Damaging | 2.68 | Benign | 0.15 | Tolerated | 4.32 | 4 | 0.2105 | 0.2462 | -2 | 0 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||||
| c.3845A>T | E1282V 2D AIThe SynGAP1 missense variant E1282V is reported in gnomAD (ID 6‑33447893‑A‑T) but has no ClinVar entry. In silico prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote method) is benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.817364 | Binding | 0.465 | 0.725 | 0.875 | 6-33447893-A-T | -2.790 | Likely Benign | 0.190 | Likely Benign | Likely Benign | 0.102 | Likely Benign | -1.71 | Neutral | 0.369 | Benign | 0.078 | Benign | 2.72 | Benign | 0.04 | Affected | 0.0656 | 0.5905 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||
| c.3851T>A | L1284Q 2D AIThe SynGAP1 missense variant L1284Q is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy tools specifically show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta remain unavailable. Overall, the majority of predictions (five pathogenic vs. four benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.812494 | Disordered | 0.824557 | Binding | 0.441 | 0.748 | 0.875 | -4.730 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 0.138 | Likely Benign | -2.80 | Deleterious | 0.990 | Probably Damaging | 0.796 | Possibly Damaging | 2.48 | Pathogenic | 0.01 | Affected | 0.1065 | 0.0488 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3854C>G | P1285R 2D AIThe SynGAP1 missense variant P1285R is reported in gnomAD (variant ID 6‑33447902‑C‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict pathogenicity. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the majority of evidence points to a benign impact. There is no ClinVar classification to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.834292 | Disordered | 0.821643 | Binding | 0.557 | 0.759 | 0.750 | 6-33447902-C-G | -3.417 | Likely Benign | 0.157 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -2.10 | Neutral | 0.977 | Probably Damaging | 0.632 | Possibly Damaging | 4.22 | Benign | 0.14 | Tolerated | 4.32 | 2 | 0.1564 | 0.2194 | -2 | 0 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||
| c.3857A>T | E1286V 2D AIThe SynGAP1 missense variant E1286V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑benign/2‑pathogenic split. High‑accuracy methods show AlphaMissense‑Optimized as benign; Foldetta results are unavailable. Overall, the majority of available predictions (five pathogenic vs. four benign) indicate that the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.817022 | Binding | 0.544 | 0.765 | 0.750 | -4.195 | Likely Benign | 0.275 | Likely Benign | Likely Benign | 0.259 | Likely Benign | -3.90 | Deleterious | 0.960 | Probably Damaging | 0.679 | Possibly Damaging | 2.42 | Pathogenic | 0.00 | Affected | 0.0828 | 0.5614 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3860C>A | P1287H 2D AIThe SynGAP1 missense variant P1287H is recorded in gnomAD (6‑33447908‑C‑A) and has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) is not available for this variant. Based on the aggregate predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.813701 | Binding | 0.538 | 0.777 | 0.750 | 6-33447908-C-A | -3.606 | Likely Benign | 0.135 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -1.36 | Neutral | 0.938 | Possibly Damaging | 0.596 | Possibly Damaging | 2.80 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1306 | 0.3448 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||
| c.386C>G | S129W 2D AIThe SynGAP1 missense variant S129W is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign, and AlphaMissense‑Optimized is uncertain. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.713635 | Binding | 0.311 | 0.880 | 0.625 | -5.008 | Likely Benign | 0.824 | Likely Pathogenic | Ambiguous | 0.108 | Likely Benign | -1.49 | Neutral | 0.888 | Possibly Damaging | 0.367 | Benign | 4.06 | Benign | 0.01 | Affected | 0.0523 | 0.5471 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||||||||||||
| c.386C>T | S129L 2D AIThe SynGAP1 missense variant S129L is catalogued in gnomAD (ID 6‑33432251‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all report benign or likely benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the preponderance of evidence indicates that S129L is most likely benign, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.517562 | Disordered | 0.713635 | Binding | 0.311 | 0.880 | 0.625 | 6-33432251-C-T | 1 | 6.20e-7 | -4.487 | Likely Benign | 0.456 | Ambiguous | Likely Benign | 0.256 | Likely Benign | -0.18 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.14 | Benign | 0.05 | Affected | 3.74 | 4 | 0.0885 | 0.5249 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||||||
| c.3872T>A | L1291Q 2D AISynGAP1 missense variant L1291Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is equivocal (two benign, two pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence is split, with an equal number of benign and pathogenic calls and no decisive high‑accuracy consensus. Consequently, the variant’s likely effect remains uncertain; it is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.863458 | Binding | 0.579 | 0.797 | 0.625 | -4.450 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.290 | Likely Benign | -4.18 | Deleterious | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 2.49 | Pathogenic | 0.01 | Affected | 0.1052 | 0.1049 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3872T>G | L1291R 2D AIThe SynGAP1 missense variant L1291R is not reported in ClinVar and is present in gnomAD (ID 6‑33447920‑T‑G). Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessment shows AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive and Foldetta results are unavailable. Overall, the majority of conventional predictors indicate a pathogenic effect, whereas the single high‑accuracy tool suggests benign. No ClinVar annotation contradicts these findings. Thus, based on the collective evidence, the variant is most likely pathogenic, though the high‑accuracy prediction introduces uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.827927 | Disordered | 0.863458 | Binding | 0.579 | 0.797 | 0.625 | 6-33447920-T-G | 1 | 6.44e-7 | -3.977 | Likely Benign | 0.302 | Likely Benign | Likely Benign | 0.287 | Likely Benign | -4.29 | Deleterious | 0.990 | Probably Damaging | 0.856 | Possibly Damaging | 2.49 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1160 | 0.1049 | -2 | -3 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||
| c.3878A>T | D1293V 2D AIThe SynGAP1 missense variant D1293V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the majority of available predictions (five pathogenic vs. four benign) lean toward a pathogenic impact. This assessment does not contradict ClinVar status, as the variant has not yet been classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.779859 | Disordered | 0.892346 | Binding | 0.569 | 0.801 | 0.625 | -3.817 | Likely Benign | 0.328 | Likely Benign | Likely Benign | 0.343 | Likely Benign | -6.03 | Deleterious | 0.960 | Probably Damaging | 0.679 | Possibly Damaging | 2.16 | Pathogenic | 0.00 | Affected | 0.0896 | 0.3603 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.3881C>A | A1294D 2D AIThe SynGAP1 missense variant A1294D is listed in gnomAD (ID 6‑33447929‑C‑A) but has no ClinVar entry. Functional prediction tools show mixed results: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessment further clarifies the picture: AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans toward pathogenicity. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a pathogenic effect, which is consistent with the lack of a benign ClinVar classification and the presence of the variant in a general population database. Thus, the variant is most likely pathogenic, and this prediction does not contradict any ClinVar status because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.895011 | Binding | 0.565 | 0.806 | 0.625 | 6-33447929-C-A | -4.179 | Likely Benign | 0.369 | Ambiguous | Likely Benign | 0.224 | Likely Benign | -4.17 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 2.14 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.1893 | 0.2262 | -2 | 0 | -5.3 | 44.01 | |||||||||||||||||||||||||||||||||||||
| c.3884A>T | Q1295L 2D AIThe SynGAP1 missense variant Q1295L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic impact for Q1295L, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.852992 | Disordered | 0.892719 | Binding | 0.499 | 0.801 | 0.625 | -2.862 | Likely Benign | 0.348 | Ambiguous | Likely Benign | 0.379 | Likely Benign | -5.63 | Deleterious | 0.925 | Possibly Damaging | 0.932 | Probably Damaging | 2.25 | Pathogenic | 0.00 | Affected | 0.0810 | 0.5672 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.3887A>T | E1296V 2D AIThe SynGAP1 missense variant E1296V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. High‑accuracy predictions therefore indicate a benign outcome: AlphaMissense‑Optimized is benign, the SGM Consensus is benign, and no Foldetta data are available. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.837511 | Disordered | 0.894444 | Binding | 0.530 | 0.809 | 0.625 | -3.384 | Likely Benign | 0.443 | Ambiguous | Likely Benign | 0.229 | Likely Benign | -4.24 | Deleterious | 0.992 | Probably Damaging | 0.902 | Possibly Damaging | 2.62 | Benign | 0.01 | Affected | 0.0678 | 0.6040 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||||
| c.3889A>G | R1297G 2D AIThe SynGAP1 missense variant R1297G is reported in gnomAD (ID 6‑33451763‑A‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus itself is Benign; Foldetta results are unavailable. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that R1297G is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.859585 | Disordered | 0.895222 | Binding | 0.511 | 0.817 | 0.625 | 6-33451763-A-G | 3 | 1.86e-6 | -2.833 | Likely Benign | 0.168 | Likely Benign | Likely Benign | 0.215 | Likely Benign | -3.65 | Deleterious | 0.224 | Benign | 0.171 | Benign | 2.56 | Benign | 0.01 | Affected | 3.77 | 5 | 0.3089 | 0.2400 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||
| c.3893A>T | Q1298L 2D AIThe SynGAP1 missense variant Q1298L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, PROVEAN and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.823549 | Disordered | 0.895297 | Binding | 0.410 | 0.821 | 0.750 | -2.251 | Likely Benign | 0.147 | Likely Benign | Likely Benign | 0.221 | Likely Benign | -3.04 | Deleterious | 0.224 | Benign | 0.078 | Benign | 2.79 | Benign | 0.04 | Affected | 0.0667 | 0.4164 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3896T>A | L1299H 2D AIThe SynGAP1 missense variant L1299H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.771762 | Disordered | 0.896323 | Binding | 0.398 | 0.832 | 0.750 | -4.586 | Likely Benign | 0.300 | Likely Benign | Likely Benign | 0.317 | Likely Benign | -3.83 | Deleterious | 0.992 | Probably Damaging | 0.750 | Possibly Damaging | 2.67 | Benign | 0.00 | Affected | 0.1293 | 0.1273 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.389A>T | Q130L 2D AIThe SynGAP1 missense variant Q130L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for Q130L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.509769 | Disordered | 0.718853 | Binding | 0.306 | 0.885 | 0.375 | -3.643 | Likely Benign | 0.278 | Likely Benign | Likely Benign | 0.167 | Likely Benign | -1.63 | Neutral | 0.967 | Probably Damaging | 0.901 | Possibly Damaging | 4.11 | Benign | 0.02 | Affected | 0.0975 | 0.5286 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3905T>G | L1302W 2D AIThe SynGAP1 missense variant L1302W is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM; ESM1b is uncertain and not counted. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of predictions (five pathogenic vs. three benign) lean toward pathogenicity, and this is consistent with the SGM Consensus result. Therefore, the variant is most likely pathogenic based on current computational evidence, and this does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.784345 | Disordered | 0.889642 | Binding | 0.429 | 0.842 | 0.875 | -7.133 | In-Between | 0.297 | Likely Benign | Likely Benign | 0.290 | Likely Benign | -4.20 | Deleterious | 0.996 | Probably Damaging | 0.946 | Probably Damaging | 1.49 | Pathogenic | 0.00 | Affected | 0.0671 | 0.2530 | -2 | -2 | -4.7 | 73.05 | ||||||||||||||||||||||||||||||||||||||||
| c.3916A>T | N1306Y 2D AIThe SynGAP1 missense variant N1306Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) as benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the preponderance of evidence points to a benign impact for the variant, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.902190 | Binding | 0.367 | 0.888 | 0.875 | -4.193 | Likely Benign | 0.298 | Likely Benign | Likely Benign | 0.273 | Likely Benign | -5.59 | Deleterious | 0.961 | Probably Damaging | 0.825 | Possibly Damaging | 2.54 | Benign | 0.00 | Affected | 0.0732 | 0.6658 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||||||||||||
| c.3917A>T | N1306I 2D AIThe SynGAP1 missense variant N1306I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.795062 | Disordered | 0.902190 | Binding | 0.367 | 0.888 | 0.875 | -3.234 | Likely Benign | 0.296 | Likely Benign | Likely Benign | 0.188 | Likely Benign | -6.15 | Deleterious | 0.890 | Possibly Damaging | 0.761 | Possibly Damaging | 2.56 | Benign | 0.00 | Affected | 0.0837 | 0.6672 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.391G>C | G131R 2D AIThe SynGAP1 missense variant G131R is listed in ClinVar with an “Uncertain” significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and FATHMM, while pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive due to a 2‑to‑2 split and therefore does not contribute evidence. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of high‑confidence tools predict a pathogenic effect, and the variant is most likely pathogenic based on current predictions, which does not contradict the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.429200 | Structured | 0.724779 | Binding | 0.302 | 0.891 | 0.250 | Uncertain | 1 | -6.564 | Likely Benign | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.099 | Likely Benign | -3.82 | Deleterious | 0.983 | Probably Damaging | 0.656 | Possibly Damaging | 3.92 | Benign | 0.00 | Affected | 3.61 | 5 | 0.1070 | 0.4667 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||||||||
| c.3923G>C | R1308P 2D AIThe SynGAP1 missense variant R1308P is not reported in ClinVar but is present in gnomAD (ID 6‑33451797‑G‑C). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is a 2‑vs‑2 split and therefore inconclusive. Foldetta, a protein‑folding stability method that combines FoldX‑MD and Rosetta outputs, has no available result for this variant. Because the available predictions are evenly divided between benign and pathogenic, the overall computational assessment is inconclusive; the variant is neither clearly benign nor clearly pathogenic. This lack of consensus does not contradict ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.741537 | Disordered | 0.930652 | Binding | 0.378 | 0.904 | 0.750 | 6-33451797-G-C | -1.320 | Likely Benign | 0.216 | Likely Benign | Likely Benign | 0.158 | Likely Benign | -4.62 | Deleterious | 0.995 | Probably Damaging | 0.992 | Probably Damaging | 2.32 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0.2070 | 0.5020 | -2 | 0 | 2.9 | -59.07 | |||||||||||||||||||||||||||||||||||||
| c.3926T>A | V1309E 2D AIThe SynGAP1 missense variant V1309E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.712013 | Disordered | 0.948596 | Binding | 0.402 | 0.907 | 0.750 | -3.393 | Likely Benign | 0.333 | Likely Benign | Likely Benign | 0.294 | Likely Benign | -2.40 | Neutral | 0.767 | Possibly Damaging | 0.473 | Possibly Damaging | 2.40 | Pathogenic | 0.00 | Affected | 0.1098 | 0.1727 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||||||||||||
| c.3932T>A | L1311Q 2D AIThe SynGAP1 missense variant L1311Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.816150 | Disordered | 0.968153 | Binding | 0.393 | 0.907 | 0.750 | -4.009 | Likely Benign | 0.100 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -0.03 | Neutral | 0.579 | Possibly Damaging | 0.413 | Benign | 2.74 | Benign | 0.12 | Tolerated | 0.1476 | 0.1677 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3944G>C | W1315S 2D AIThe SynGAP1 missense variant W1315S is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. No pathogenic predictions are present among the evaluated tools. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are unavailable. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.973142 | Binding | 0.403 | 0.889 | 0.750 | 0.896 | Likely Benign | 0.279 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -0.79 | Neutral | 0.115 | Benign | 0.057 | Benign | 4.26 | Benign | 0.13 | Tolerated | 0.3974 | 0.1211 | -2 | -3 | 0.1 | -99.14 | |||||||||||||||||||||||||||||||||||||||
| c.3944G>T | W1315L 2D AIThe SynGAP1 missense variant W1315L is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity. The high‑accuracy consensus methods corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign outcome. Foldetta results are unavailable. Overall, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.885302 | Disordered | 0.973142 | Binding | 0.403 | 0.889 | 0.750 | 0.369 | Likely Benign | 0.304 | Likely Benign | Likely Benign | 0.066 | Likely Benign | -0.65 | Neutral | 0.115 | Benign | 0.057 | Benign | 4.29 | Benign | 0.16 | Tolerated | 0.2256 | 0.3338 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||||||||||||
| c.3946A>T | N1316Y 2D AIThe SynGAP1 missense variant N1316Y is reported in gnomAD (variant ID 6‑33451820‑A‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, polyPhen2_HumVar, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions come from polyPhen2_HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is available). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.971970 | Binding | 0.380 | 0.885 | 0.750 | 6-33451820-A-T | -5.579 | Likely Benign | 0.392 | Ambiguous | Likely Benign | 0.101 | Likely Benign | -2.48 | Neutral | 0.939 | Possibly Damaging | 0.396 | Benign | 3.90 | Benign | 0.00 | Affected | 3.77 | 5 | 0.0671 | 0.5365 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||||||||
| c.3947A>T | N1316I 2D AIThe SynGAP1 missense variant N1316I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.899122 | Disordered | 0.971970 | Binding | 0.380 | 0.885 | 0.750 | -4.829 | Likely Benign | 0.635 | Likely Pathogenic | Likely Benign | 0.147 | Likely Benign | -2.86 | Deleterious | 0.009 | Benign | 0.004 | Benign | 3.91 | Benign | 0.00 | Affected | 0.0719 | 0.5354 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.3953T>A | L1318Q 2D AIThe SynGAP1 missense variant L1318Q is reported in gnomAD (variant ID 6‑33451827‑T‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign. No Foldetta stability analysis is available, so it does not influence the conclusion. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.968271 | Binding | 0.399 | 0.865 | 0.750 | 6-33451827-T-A | 1 | 6.32e-7 | -3.445 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -2.06 | Neutral | 0.834 | Possibly Damaging | 0.307 | Benign | 4.04 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1410 | 0.0903 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||
| c.3953T>G | L1318R 2D AIThe SynGAP1 missense variant L1318R is reported in gnomAD (variant ID 6-33451827‑T‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a benign effect, and this conclusion is not contradicted by any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.968271 | Binding | 0.399 | 0.865 | 0.750 | 6-33451827-T-G | -3.381 | Likely Benign | 0.187 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -1.58 | Neutral | 0.588 | Possibly Damaging | 0.212 | Benign | 4.01 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1429 | 0.1303 | -2 | -3 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||||||||
| c.3956C>A | A1319D 2D AIThe SynGAP1 missense variant A1319D is reported in gnomAD (ID 6‑33451830‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority of the same four tools) is benign. Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for A1319D, and this conclusion is not contradicted by any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.842060 | Disordered | 0.960481 | Binding | 0.454 | 0.851 | 0.750 | 6-33451830-C-A | -5.156 | Likely Benign | 0.297 | Likely Benign | Likely Benign | 0.208 | Likely Benign | -0.85 | Neutral | 0.971 | Probably Damaging | 0.813 | Possibly Damaging | 4.08 | Benign | 0.01 | Affected | 3.77 | 5 | 0.2290 | 0.2702 | -2 | 0 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||||
| c.3959C>A | P1320H 2D AIThe SynGAP1 missense variant P1320H is listed in gnomAD (ID 6‑33451833‑C‑A) and has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.899122 | Disordered | 0.946297 | Binding | 0.510 | 0.833 | 0.750 | 6-33451833-C-A | 2 | 1.29e-6 | -6.296 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -1.05 | Neutral | 0.998 | Probably Damaging | 0.990 | Probably Damaging | 4.17 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1955 | 0.5461 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||
| c.3962C>G | P1321R 2D AIThe SynGAP1 missense variant P1321R is reported in gnomAD (ID 6‑33451836‑C‑G) but has no ClinVar entry. Across a broad panel of in silico predictors, every tool examined—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classifies the substitution as benign. No pathogenic predictions are present. Grouping the results, the benign consensus is unanimous; the pathogenic group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports a likely benign status. Foldetta stability analysis is not available for this variant. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.827927 | Disordered | 0.933505 | Binding | 0.463 | 0.828 | 0.875 | 6-33451836-C-G | 1 | 6.58e-7 | -5.310 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 0.034 | Likely Benign | -1.10 | Neutral | 0.389 | Benign | 0.024 | Benign | 4.25 | Benign | 0.09 | Tolerated | 3.77 | 5 | 0.1722 | 0.3635 | -2 | 0 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||
| c.3965C>A | A1322D 2D AIThe SynGAP1 missense variant A1322D is catalogued in gnomAD (ID 6‑33451839‑C‑A) but has no ClinVar entry. Across the spectrum of in‑silico predictors, every tool listed—REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classifies the substitution as benign. No pathogenic predictions are reported. Grouping by consensus, all available tools fall into the benign category, with no tools indicating pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no result for this variant, so its status is unavailable. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.921040 | Binding | 0.466 | 0.825 | 0.875 | 6-33451839-C-A | -5.744 | Likely Benign | 0.284 | Likely Benign | Likely Benign | 0.092 | Likely Benign | 0.70 | Neutral | 0.013 | Benign | 0.004 | Benign | 4.17 | Benign | 0.45 | Tolerated | 3.77 | 5 | 0.2246 | 0.3134 | -2 | 0 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||||
| c.3974C>A | P1325H 2D AIThe SynGAP1 missense variant P1325H is reported in gnomAD (variant ID 6‑33451848‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumDiv and SIFT. The high‑accuracy consensus methods reinforce the benign assessment: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No result is available from the Foldetta stability analysis. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.876521 | Disordered | 0.893621 | Binding | 0.439 | 0.791 | 0.875 | 6-33451848-C-A | -6.970 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.056 | Likely Benign | 0.10 | Neutral | 0.704 | Possibly Damaging | 0.187 | Benign | 4.04 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2103 | 0.4364 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||
| c.3980C>A | P1327H 2D AIThe SynGAP1 missense variant P1327H is reported in gnomAD (ID 6‑33451854‑C‑A) and has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus is Likely Benign, and Foldetta results are unavailable. Based on the preponderance of evidence from both general and high‑accuracy predictors, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.900145 | Binding | 0.369 | 0.777 | 0.875 | 6-33451854-C-A | -5.496 | Likely Benign | 0.263 | Likely Benign | Likely Benign | 0.144 | Likely Benign | -0.49 | Neutral | 0.998 | Probably Damaging | 0.953 | Probably Damaging | 4.09 | Benign | 0.04 | Affected | 3.77 | 5 | 0.1559 | 0.3932 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||
| c.3980C>G | P1327R 2D AIThe SynGAP1 missense variant P1327R is reported in gnomAD (variant ID 6‑33451854‑C‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely benign. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict the variant to be pathogenic. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a likely benign outcome. No Foldetta stability analysis is available, so it does not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.896620 | Disordered | 0.900145 | Binding | 0.369 | 0.777 | 0.875 | 6-33451854-C-G | -4.724 | Likely Benign | 0.273 | Likely Benign | Likely Benign | 0.137 | Likely Benign | -0.95 | Neutral | 0.994 | Probably Damaging | 0.937 | Probably Damaging | 4.12 | Benign | 0.09 | Tolerated | 3.77 | 5 | 0.1442 | 0.2798 | -2 | 0 | -2.9 | 59.07 | ||||||||||||||||||||||||||||||||||||
| c.3983G>T | R1328L 2D AIThe SynGAP1 missense variant R1328L is listed in gnomAD (ID 6‑33451857‑G‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.911775 | Binding | 0.360 | 0.762 | 0.875 | 6-33451857-G-T | -3.233 | Likely Benign | 0.452 | Ambiguous | Likely Benign | 0.038 | Likely Benign | -1.94 | Neutral | 0.784 | Possibly Damaging | 0.145 | Benign | 4.08 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1978 | 0.3555 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||||
| c.3986T>A | L1329Q 2D AIThe SynGAP1 missense variant L1329Q is reported in gnomAD (ID 6‑33451860‑T‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a deleterious effect, indicating that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.930790 | Disordered | 0.924905 | Binding | 0.336 | 0.748 | 0.875 | 6-33451860-T-A | -4.106 | Likely Benign | 0.956 | Likely Pathogenic | Likely Pathogenic | 0.157 | Likely Benign | -3.31 | Deleterious | 0.994 | Probably Damaging | 0.993 | Probably Damaging | 3.05 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1382 | 0.1505 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||
| c.3986T>G | L1329R 2D AIThe SynGAP1 missense variant L1329R is catalogued in gnomAD (ID 6‑33451860‑T‑G) but has no ClinVar submission. Functional prediction tools cluster into two groups: benign predictions from REVEL, ESM1b, and FATHMM; pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized returns an uncertain result. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive due to a 2‑to‑2 split. No Foldetta stability assessment is available. Overall, the majority of high‑confidence predictors (five pathogenic vs three benign) lean toward a pathogenic effect. Because ClinVar contains no classification, there is no conflict with existing clinical annotations. Thus, based on current in silico evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.930790 | Disordered | 0.924905 | Binding | 0.336 | 0.748 | 0.875 | 6-33451860-T-G | -3.636 | Likely Benign | 0.923 | Likely Pathogenic | Ambiguous | 0.141 | Likely Benign | -3.16 | Deleterious | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 3.05 | Benign | 0.00 | Affected | 3.77 | 5 | 0.1594 | 0.1547 | -2 | -3 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||
| c.3989A>T | Q1330L 2D AIThe SynGAP1 missense variant Q1330L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and SIFT. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction. Foldetta results are unavailable. Overall, the majority of evidence (5 benign vs 3 pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.943310 | Disordered | 0.931969 | Binding | 0.369 | 0.752 | 0.875 | -3.780 | Likely Benign | 0.417 | Ambiguous | Likely Benign | 0.110 | Likely Benign | -2.74 | Deleterious | 0.784 | Possibly Damaging | 0.341 | Benign | 3.92 | Benign | 0.02 | Affected | 0.0741 | 0.5480 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.398T>A | L133Q 2D AIThe SynGAP1 missense variant L133Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; the Foldetta protein‑folding stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.422041 | Structured | 0.718429 | Binding | 0.320 | 0.896 | 0.250 | -9.054 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.317 | Likely Benign | -2.65 | Deleterious | 0.535 | Possibly Damaging | 0.259 | Benign | 3.53 | Benign | 0.01 | Affected | 0.1316 | 0.0879 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.3998A>T | E1333V 2D AIThe SynGAP1 E1333V missense change is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are REVEL, ESM1b, and FATHMM. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive (two pathogenic vs two benign votes), and Foldetta results are unavailable. Overall, the majority of evidence (six pathogenic vs three benign) points to a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.930790 | Disordered | 0.953319 | Binding | 0.347 | 0.746 | 0.750 | -4.322 | Likely Benign | 0.974 | Likely Pathogenic | Likely Pathogenic | 0.289 | Likely Benign | -4.30 | Deleterious | 0.994 | Probably Damaging | 0.981 | Probably Damaging | 2.81 | Benign | 0.00 | Affected | 0.0994 | 0.7615 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||||
| c.4000A>T | N1334Y 2D AIThe SynGAP1 missense variant N1334Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive; Foldetta results are unavailable. Overall, the majority of conventional tools predict a pathogenic impact, but the single high‑accuracy benign prediction and the inconclusive consensus suggest uncertainty. Consequently, the variant is most likely pathogenic based on the prevailing evidence, and this assessment does not contradict any ClinVar annotation because the variant has not yet been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.915074 | Disordered | 0.960403 | Binding | 0.406 | 0.734 | 0.875 | -5.916 | Likely Benign | 0.770 | Likely Pathogenic | Likely Benign | 0.249 | Likely Benign | -4.62 | Deleterious | 0.985 | Probably Damaging | 0.852 | Possibly Damaging | 3.49 | Benign | 0.00 | Affected | 0.0716 | 0.5447 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||||||||||||
| c.4001A>T | N1334I 2D AIThe SynGAP1 missense variant N1334I is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the balance of evidence (seven pathogenic vs. three benign predictions) indicates that the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, as the variant has no existing ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.915074 | Disordered | 0.960403 | Binding | 0.406 | 0.734 | 0.875 | -5.880 | Likely Benign | 0.962 | Likely Pathogenic | Likely Pathogenic | 0.193 | Likely Benign | -5.06 | Deleterious | 0.985 | Probably Damaging | 0.721 | Possibly Damaging | 3.50 | Benign | 0.00 | Affected | 0.0861 | 0.5491 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.4007A>T | E1336V 2D AIThe SynGAP1 missense variant E1336V has no ClinVar record (ClinVar status: None) and is not present in gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is reported as uncertain. High‑accuracy assessments are inconclusive: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a tie (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Consequently, the variant’s predicted impact is ambiguous, with an equal split between benign and pathogenic calls and no evidence from ClinVar to contradict this uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.865454 | Disordered | 0.973342 | Binding | 0.336 | 0.717 | 0.750 | -3.367 | Likely Benign | 0.932 | Likely Pathogenic | Ambiguous | 0.221 | Likely Benign | -4.46 | Deleterious | 0.789 | Possibly Damaging | 0.348 | Benign | 3.18 | Benign | 0.00 | Affected | 0.0991 | 0.7425 | -2 | -2 | 7.7 | -29.98 | ||||||||||||||||||||||||||||||||||||||||
| c.4013G>T | R1338L 2D AIThe SynGAP1 missense variant R1338L is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33451887‑G‑T). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.775545 | Disordered | 0.977425 | Binding | 0.393 | 0.697 | 1.000 | 6-33451887-G-T | -3.359 | Likely Benign | 0.587 | Likely Pathogenic | Likely Benign | 0.232 | Likely Benign | -3.65 | Deleterious | 0.001 | Benign | 0.001 | Benign | 3.78 | Benign | 0.01 | Affected | 3.77 | 5 | 0.2066 | 0.5307 | -2 | -3 | 8.3 | -43.03 | |||||||||||||||||||||||||||||||||||||
| c.4015A>T | N1339Y 2D AIThe SynGAP1 missense variant N1339Y is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic) and Foldetta data are unavailable. Overall, the majority of conventional predictors lean toward pathogenicity, but the single high‑accuracy benign prediction and the inconclusive consensus leave the variant’s impact uncertain. Thus, the variant is most likely pathogenic based on the current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.771762 | Disordered | 0.977585 | Binding | 0.396 | 0.687 | 1.000 | -3.808 | Likely Benign | 0.578 | Likely Pathogenic | Likely Benign | 0.319 | Likely Benign | -4.83 | Deleterious | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 2.87 | Benign | 0.00 | Affected | 0.0697 | 0.6573 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||||||||||||
| c.4016A>T | N1339I 2D AIThe SynGAP1 missense variant N1339I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (five pathogenic vs four benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic based on current computational evidence, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.771762 | Disordered | 0.977585 | Binding | 0.396 | 0.687 | 1.000 | -3.104 | Likely Benign | 0.740 | Likely Pathogenic | Likely Benign | 0.306 | Likely Benign | -5.25 | Deleterious | 0.994 | Probably Damaging | 0.987 | Probably Damaging | 2.87 | Benign | 0.00 | Affected | 0.0747 | 0.6271 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.4025A>T | D1342V 2D AIThe SynGAP1 missense variant D1342V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign and the SGM‑Consensus also indicating a likely benign outcome; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.921076 | Disordered | 0.981682 | Binding | 0.316 | 0.678 | 0.875 | -2.890 | Likely Benign | 0.317 | Likely Benign | Likely Benign | 0.089 | Likely Benign | -1.27 | Neutral | 0.588 | Possibly Damaging | 0.212 | Benign | 4.01 | Benign | 0.00 | Affected | 0.1214 | 0.5617 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.407G>T | R136L 2D AIThe SynGAP1 missense variant R136L is catalogued in gnomAD (ID 6‑33432704‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; pathogenic predictions arise from PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic, and the SGM‑Consensus (majority vote) also indicates Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a pathogenic impact, and this assessment does not conflict with ClinVar, which currently has no classification for R136L. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.433034 | Structured | 0.657394 | Binding | 0.351 | 0.894 | 0.250 | 6-33432704-G-T | 1 | 7.05e-7 | -11.512 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.347 | Likely Benign | -4.19 | Deleterious | 0.190 | Benign | 0.037 | Benign | 3.48 | Benign | 0.01 | Affected | 3.61 | 5 | 0.1718 | 0.4475 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||
| c.410T>A | L137Q 2D AIThe SynGAP1 missense variant L137Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and FATHMM. All other evaluated algorithms—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled “Likely Pathogenic.” The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.553315 | Disordered | 0.639549 | Binding | 0.377 | 0.897 | 0.375 | -12.246 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.356 | Likely Benign | -3.43 | Deleterious | 0.998 | Probably Damaging | 0.995 | Probably Damaging | 3.60 | Benign | 0.00 | Affected | 0.1137 | 0.1049 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.413A>T | K138I 2D AIThe SynGAP1 missense variant K138I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.590140 | Disordered | 0.619482 | Binding | 0.349 | 0.901 | 0.375 | -9.366 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.295 | Likely Benign | -4.74 | Deleterious | 0.535 | Possibly Damaging | 0.259 | Benign | 3.53 | Benign | 0.00 | Affected | 0.1003 | 0.3054 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||||||||||||
| c.41C>A | P14H 2D AIThe SynGAP1 missense variant P14H is listed in gnomAD (ID 6‑33420305‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant, so its status is unavailable. Overall, the majority of predictions—including the high‑accuracy tools—indicate that P14H is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.433034 | Structured | 0.471596 | Uncertain | 0.399 | 0.909 | 0.375 | 6-33420305-C-A | -3.747 | Likely Benign | 0.231 | Likely Benign | Likely Benign | 0.171 | Likely Benign | -0.27 | Neutral | 0.742 | Possibly Damaging | 0.091 | Benign | 4.15 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1999 | 0.5176 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||
| c.422T>A | I141N 2D AIThe SynGAP1 missense variant I141N is not reported in ClinVar and is absent from gnomAD. Consensus from standard prediction algorithms shows a split: benign predictions come from REVEL, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (Likely Pathogenic). High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic. Foldetta stability analysis is unavailable. Overall, the majority of evidence points toward a pathogenic impact for I141N. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.465241 | Structured | 0.577021 | Binding | 0.367 | 0.877 | 0.500 | -12.417 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.258 | Likely Benign | -3.90 | Deleterious | 0.799 | Possibly Damaging | 0.424 | Benign | 3.54 | Benign | 0.00 | Affected | 0.1047 | 0.0270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||||||||||||
| c.425A>T | K142I 2D AIThe SynGAP1 missense variant K142I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts Pathogenic, and the SGM‑Consensus also indicates Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.461924 | Structured | 0.558796 | Binding | 0.374 | 0.859 | 0.500 | -14.597 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.296 | Likely Benign | -4.81 | Deleterious | 0.700 | Possibly Damaging | 0.403 | Benign | 3.44 | Benign | 0.00 | Affected | 0.1005 | 0.3173 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||||||||||||
| c.428G>C | R143P 2D AIThe SynGAP1 missense variant R143P is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33432725‑G‑C). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM‑Consensus as Likely Pathogenic; the Foldetta stability analysis is unavailable. Overall, the majority of computational evidence points to a pathogenic impact, and this is consistent with the ClinVar designation of “Uncertain” rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.575842 | Disordered | 0.538584 | Binding | 0.338 | 0.838 | 0.625 | Uncertain | 2 | 6-33432725-G-C | 2 | 1.35e-6 | -14.564 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.292 | Likely Benign | -3.74 | Deleterious | 0.001 | Benign | 0.000 | Benign | 3.49 | Benign | 0.00 | Affected | 3.61 | 5 | 0.2063 | 0.4457 | -2 | 0 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||
| c.428G>T | R143L 2D AIThe SynGAP1 missense variant R143L is listed in ClinVar with no submitted interpretation and is present in gnomAD (ID 6‑33432725‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect comprise PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.575842 | Disordered | 0.538584 | Binding | 0.338 | 0.838 | 0.625 | 6-33432725-G-T | 1 | 6.77e-7 | -14.250 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.316 | Likely Benign | -4.37 | Deleterious | 0.319 | Benign | 0.124 | Benign | 3.52 | Benign | 0.00 | Affected | 3.61 | 5 | 0.1758 | 0.4886 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||||||||||||
| c.440A>T | Q147L 2D AIThe SynGAP1 missense variant Q147L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default; the SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic. Foldetta results are unavailable. Overall, the majority of evidence points toward a pathogenic interpretation, and this is not contradicted by any ClinVar classification because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.541878 | Disordered | 0.503877 | Binding | 0.349 | 0.840 | 0.625 | -9.879 | Likely Pathogenic | 0.740 | Likely Pathogenic | Likely Benign | 0.185 | Likely Benign | -3.51 | Deleterious | 0.079 | Benign | 0.037 | Benign | 3.92 | Benign | 0.03 | Affected | 0.0861 | 0.4927 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.446A>T | K149I 2D AIThe SynGAP1 missense variant K149I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; the Foldetta protein‑folding stability analysis is unavailable. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.562014 | Disordered | 0.501681 | Binding | 0.302 | 0.839 | 0.625 | -14.426 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 0.305 | Likely Benign | -4.72 | Deleterious | 0.535 | Possibly Damaging | 0.403 | Benign | 3.53 | Benign | 0.00 | Affected | 0.1477 | 0.3965 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||||||||||||
| c.449T>A | L150H 2D AIThe SynGAP1 missense variant L150H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic (3 pathogenic vs. 1 benign). AlphaMissense‑Optimized alone also predicts Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple independent predictors indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.494003 | Structured | 0.505752 | Binding | 0.299 | 0.839 | 0.625 | -14.892 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.303 | Likely Benign | -4.09 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 3.64 | Benign | 0.00 | Affected | 0.1011 | 0.0519 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.452A>C | D151A 2D  AIThe SynGAP1 D151A missense variant is listed in gnomAD (ID 6‑33432749‑A‑C) but has no ClinVar entry. Functional prediction tools fall into two groups: benign predictions come from REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.529623 | Disordered | 0.503277 | Binding | 0.342 | 0.841 | 0.625 | 6-33432749-A-C | 1 | 6.21e-7 | -9.693 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 0.326 | Likely Benign | -4.51 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 3.91 | Benign | 0.01 | Affected | 3.61 | 5 | 0.4286 | 0.7424 | -2 | 0 | 5.3 | -44.01 | ||||||||||||||||||||||||||||||||||
| c.452A>T | D151V 2D AIThe SynGAP1 D151V variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Pathogenic” based on a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is likely pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from multiple in silico predictors indicates that D151V is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.529623 | Disordered | 0.503277 | Binding | 0.342 | 0.841 | 0.625 | -11.927 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.380 | Likely Benign | -5.19 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 3.88 | Benign | 0.00 | Affected | 0.0864 | 0.7781 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.473A>T | Q158L 2D AIThe SynGAP1 missense variant Q158L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign impact for Q158L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.480142 | Structured | 0.527565 | Binding | 0.286 | 0.750 | 0.375 | -5.965 | Likely Benign | 0.229 | Likely Benign | Likely Benign | 0.141 | Likely Benign | -1.11 | Neutral | 0.652 | Possibly Damaging | 0.160 | Benign | 4.14 | Benign | 0.03 | Affected | 0.0711 | 0.4832 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.476T>A | I159N 2D AIThe SynGAP1 missense variant I159N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, whereas polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized result is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑vs‑2 split. Foldetta, which would assess protein‑folding stability, has no available output for this variant. Overall, the majority of tools (five out of eight) predict pathogenicity, while three predict benign and one is uncertain. Thus, the variant is most likely pathogenic based on current computational predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.454136 | Structured | 0.529953 | Binding | 0.278 | 0.731 | 0.125 | -14.684 | Likely Pathogenic | 0.891 | Likely Pathogenic | Ambiguous | 0.218 | Likely Benign | -1.93 | Neutral | 0.995 | Probably Damaging | 0.986 | Probably Damaging | 3.82 | Benign | 0.00 | Affected | 0.0919 | 0.0342 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||||||||||||
| c.479T>A | L160Q 2D AIThe SynGAP1 missense variant L160Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.454136 | Structured | 0.526760 | Binding | 0.275 | 0.728 | 0.125 | -16.626 | Likely Pathogenic | 0.973 | Likely Pathogenic | Likely Pathogenic | 0.227 | Likely Benign | -2.83 | Deleterious | 0.700 | Possibly Damaging | 0.483 | Possibly Damaging | 3.87 | Benign | 0.00 | Affected | 0.1248 | 0.1060 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.484C>G | R162G 2D AIThe SynGAP1 missense variant R162G is listed in ClinVar (ID 2703066.0) with an uncertain significance status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign (three benign votes versus one pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of predictions support a benign impact, and this is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.516348 | Binding | 0.315 | 0.692 | 0.250 | Uncertain | 1 | -6.985 | Likely Benign | 0.664 | Likely Pathogenic | Likely Benign | 0.190 | Likely Benign | -0.73 | Neutral | 0.487 | Possibly Damaging | 0.272 | Benign | 4.09 | Benign | 0.78 | Tolerated | 3.74 | 4 | 0.3562 | 0.4308 | -2 | -3 | 4.1 | -99.14 | |||||||||||||||||||||||||||||||||||
| c.491G>C | R164P 2D AIThe SynGAP1 missense variant R164P is reported in gnomAD (ID 6‑33432788‑G‑C) but has no ClinVar entry. Functional prediction tools split in two groups: benign predictions come from REVEL and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. AlphaMissense‑Optimized returns an Uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available output for this variant. Overall, the majority of high‑confidence tools predict pathogenicity, and this assessment does not contradict any ClinVar status (none is available). Therefore, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.414856 | Structured | 0.512396 | Binding | 0.317 | 0.666 | 0.250 | 6-33432788-G-C | 1 | 6.20e-7 | -12.792 | Likely Pathogenic | 0.898 | Likely Pathogenic | Ambiguous | 0.339 | Likely Benign | -3.42 | Deleterious | 0.910 | Possibly Damaging | 0.578 | Possibly Damaging | 3.77 | Benign | 0.00 | Affected | 3.74 | 4 | 0.2408 | 0.4730 | -2 | 0 | 2.9 | -59.07 | ||||||||||||||||||||||||||||||||||
| c.500A>T | D167V 2D AIThe SynGAP1 D167V variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic impact are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.429200 | Structured | 0.502306 | Binding | 0.377 | 0.667 | 0.375 | -14.388 | Likely Pathogenic | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.396 | Likely Benign | -4.03 | Deleterious | 0.535 | Possibly Damaging | 0.247 | Benign | 3.92 | Benign | 0.00 | Affected | 0.0709 | 0.6905 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.503A>T | H168L 2D AIThe SynGAP1 H168L missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.433034 | Structured | 0.502450 | Binding | 0.402 | 0.678 | 0.125 | -7.936 | In-Between | 0.178 | Likely Benign | Likely Benign | 0.250 | Likely Benign | -2.36 | Neutral | 0.037 | Benign | 0.021 | Benign | 4.21 | Benign | 0.01 | Affected | 0.0752 | 0.5602 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.506A>T | D169V 2D AIThe SynGAP1 D169V missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 (HumDiv and HumVar) and FATHMM, while pathogenic calls arise from PROVEAN, SIFT, ESM1b and AlphaMissense‑Default. When predictions are grouped by consensus, four tools predict benign and four predict pathogenic. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized returns an uncertain result, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this residue. Overall, the balance of evidence, particularly the SGM Consensus and the pathogenic calls from multiple independent predictors, indicates that D169V is most likely pathogenic, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.418646 | Structured | 0.497160 | Uncertain | 0.420 | 0.675 | 0.125 | -12.395 | Likely Pathogenic | 0.925 | Likely Pathogenic | Ambiguous | 0.243 | Likely Benign | -3.77 | Deleterious | 0.380 | Benign | 0.193 | Benign | 4.03 | Benign | 0.00 | Affected | 0.0895 | 0.7166 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.50C>A | S17Y 2D AIThe SynGAP1 missense variant S17Y is listed in gnomAD (ID 6‑33420314‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions come from polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign. No Foldetta stability data are available, so it does not influence the conclusion. Overall, the majority of evidence indicates that S17Y is most likely benign, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.483068 | Structured | 0.452228 | Uncertain | 0.341 | 0.910 | 0.375 | 6-33420314-C-A | -4.492 | Likely Benign | 0.551 | Ambiguous | Likely Benign | 0.050 | Likely Benign | -1.06 | Neutral | 0.742 | Possibly Damaging | 0.047 | Benign | 3.99 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0818 | 0.6366 | -2 | -3 | -0.5 | 76.10 | ||||||||||||||||||||||||||||||||||||
| c.50C>T | S17F 2D AIThe SynGAP1 missense variant S17F is listed in ClinVar (ID 3451958) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33420314‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.483068 | Structured | 0.452228 | Uncertain | 0.341 | 0.910 | 0.375 | Conflicting | 2 | 6-33420314-C-T | 10 | 6.49e-6 | -3.888 | Likely Benign | 0.637 | Likely Pathogenic | Likely Benign | 0.048 | Likely Benign | -0.99 | Neutral | 0.486 | Possibly Damaging | 0.032 | Benign | 3.99 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0729 | 0.6468 | -2 | -3 | 3.6 | 60.10 | ||||||||||||||||||||||||||||||||
| c.518T>A | L173Q 2D AIThe SynGAP1 missense variant L173Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Only AlphaMissense‑Default predicts a pathogenic outcome, while AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from multiple in silico predictors and the SGM‑Consensus indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.491566 | Uncertain | 0.390 | 0.631 | 0.375 | -5.605 | Likely Benign | 0.850 | Likely Pathogenic | Ambiguous | 0.133 | Likely Benign | -1.72 | Neutral | 0.022 | Benign | 0.022 | Benign | 3.94 | Benign | 0.08 | Tolerated | 0.1028 | 0.0919 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.524A>T | Q175L 2D AIThe SynGAP1 missense variant Q175L is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, seven tools favor benign while two favor pathogenic, with no ClinVar evidence to contradict this. Thus, the variant is most likely benign based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.653063 | Disordered | 0.474689 | Uncertain | 0.367 | 0.618 | 0.375 | -8.699 | Likely Pathogenic | 0.579 | Likely Pathogenic | Likely Benign | 0.188 | Likely Benign | -2.46 | Neutral | 0.118 | Benign | 0.039 | Benign | 4.10 | Benign | 0.14 | Tolerated | 0.0647 | 0.5109 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||||||||||||
| c.52T>A | Y18N 2D AIThe SynGAP1 missense variant Y18N is listed in gnomAD (ID 6‑33420316‑T‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.483068 | Structured | 0.446314 | Uncertain | 0.345 | 0.908 | 0.375 | 6-33420316-T-A | -3.094 | Likely Benign | 0.492 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -0.73 | Neutral | 0.872 | Possibly Damaging | 0.114 | Benign | 4.07 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2564 | 0.1253 | -2 | -2 | -2.2 | -49.07 | ||||||||||||||||||||||||||||||||||||
| c.536A>T | E179V 2D AIThe SynGAP1 missense variant E179V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM‑Consensus as Likely Pathogenic; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of computational evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.517562 | Disordered | 0.448169 | Uncertain | 0.329 | 0.635 | 0.500 | -10.930 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.190 | Likely Benign | -4.34 | Deleterious | 0.596 | Possibly Damaging | 0.328 | Benign | 3.94 | Benign | 0.01 | Affected | 0.1077 | 0.7864 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.53A>C | Y18S 2D  AIThe SynGAP1 missense variant Y18S is reported in gnomAD (ID 6‑33420317‑A‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact for Y18S. This conclusion is consistent with the absence of a pathogenic ClinVar classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.483068 | Structured | 0.446314 | Uncertain | 0.345 | 0.908 | 0.375 | 6-33420317-A-C | -1.061 | Likely Benign | 0.280 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -0.50 | Neutral | 0.389 | Benign | 0.036 | Benign | 4.09 | Benign | 0.00 | Affected | 4.32 | 1 | 0.4961 | 0.2640 | -2 | -3 | 0.5 | -76.10 | ||||||||||||||||||||||||||||||||||||
| c.542A>T | H181L 2D AIThe SynGAP1 H181L variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leans pathogenic (2 pathogenic vs 1 benign). Foldetta, which would provide a protein‑folding stability estimate, has no available result for this variant. Overall, the majority of standard predictors classify the variant as benign, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.505461 | Disordered | 0.439530 | Uncertain | 0.294 | 0.616 | 0.500 | -11.014 | Likely Pathogenic | 0.561 | Ambiguous | Likely Benign | 0.212 | Likely Benign | -3.51 | Deleterious | 0.267 | Benign | 0.039 | Benign | 4.17 | Benign | 0.04 | Affected | 0.0734 | 0.4395 | -2 | -3 | 7.0 | -23.98 | ||||||||||||||||||||||||||||||||||||||||
| c.548A>T | H183L 2D AIThe SynGAP1 missense variant H183L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. In contrast, tools that predict a pathogenic effect are PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Pathogenic” verdict (3 pathogenic vs. 1 benign votes). AlphaMissense‑Optimized alone also predicts pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic impact, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.476583 | Structured | 0.432952 | Uncertain | 0.421 | 0.622 | 0.500 | -12.898 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 0.325 | Likely Benign | -8.12 | Deleterious | 0.421 | Benign | 0.058 | Benign | 3.79 | Benign | 0.01 | Affected | 0.0911 | 0.5304 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.551A>T | E184V 2D AIThe SynGAP1 missense variant E184V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while benign calls are made only by REVEL and FATHMM. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized scores the variant as pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Pathogenic. No Foldetta stability analysis is available for this variant. Overall, the preponderance of evidence indicates that E184V is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.557691 | Disordered | 0.431514 | Uncertain | 0.348 | 0.622 | 0.625 | -13.119 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.371 | Likely Benign | -5.72 | Deleterious | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 3.44 | Benign | 0.00 | Affected | 0.0995 | 0.8511 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.557T>G | L186W 2D AIThe SynGAP1 missense variant L186W has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.458154 | Structured | 0.428613 | Uncertain | 0.397 | 0.617 | 0.500 | -16.177 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.274 | Likely Benign | -4.78 | Deleterious | 0.996 | Probably Damaging | 0.819 | Possibly Damaging | 3.46 | Benign | 0.00 | Affected | 0.0539 | 0.3138 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||||||||||||
| c.560T>A | L187Q 2D AIThe SynGAP1 missense variant L187Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all indicate likely pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus also reports it as likely pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, was not available for this variant. Overall, the preponderance of evidence from multiple in silico predictors points to a pathogenic effect for the SynGAP1 L187Q variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.465241 | Structured | 0.428046 | Uncertain | 0.367 | 0.625 | 0.375 | -13.063 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.322 | Likely Benign | -4.31 | Deleterious | 0.917 | Possibly Damaging | 0.548 | Possibly Damaging | 3.72 | Benign | 0.00 | Affected | 0.1251 | 0.1060 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.56C>A | A19D 2D AIThe SynGAP1 missense variant A19D is listed in gnomAD (ID 6‑33420320‑C‑A) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.443393 | Uncertain | 0.378 | 0.906 | 0.500 | 6-33420320-C-A | 2 | 1.30e-6 | -3.746 | Likely Benign | 0.573 | Likely Pathogenic | Likely Benign | 0.055 | Likely Benign | -0.08 | Neutral | 0.588 | Possibly Damaging | 0.054 | Benign | 4.07 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2508 | 0.2734 | -2 | 0 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||
| c.581A>T | E194V 2D AIThe SynGAP1 missense variant E194V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. No Foldetta stability analysis is available for this variant. Overall, the preponderance of computational evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.418646 | Structured | 0.430723 | Uncertain | 0.346 | 0.551 | 0.125 | -10.261 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.284 | Likely Benign | -4.67 | Deleterious | 0.580 | Possibly Damaging | 0.254 | Benign | 3.94 | Benign | 0.00 | Affected | 0.0623 | 0.5469 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.587T>G | L196W 2D AIThe SynGAP1 missense variant L196W has no ClinVar entry (ClinVar status: not reported) and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the remaining tools—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for this variant, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.284882 | Structured | 0.429452 | Uncertain | 0.489 | 0.521 | 0.125 | -12.148 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.293 | Likely Benign | -4.91 | Deleterious | 0.992 | Probably Damaging | 0.869 | Possibly Damaging | 3.58 | Benign | 0.00 | Affected | 0.0551 | 0.2718 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||||||||||||
| c.590A>T | E197V 2D AIThe SynGAP1 missense variant E197V is not reported in ClinVar and has no entries in gnomAD, indicating it is not catalogued in these databases. Functional prediction tools show a split opinion: benign calls come from REVEL, polyPhen‑2 (HumDiv and HumVar) and FATHMM, while pathogenic calls arise from PROVEAN, SIFT, ESM1b and AlphaMissense‑Default. When predictions are grouped by consensus, four tools predict benign and four predict pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, a protein‑folding stability predictor, has no available result for this residue. Taken together, the preponderance of evidence from both general and high‑accuracy predictors indicates that E197V is most likely pathogenic, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.349426 | Structured | 0.431896 | Uncertain | 0.452 | 0.492 | 0.125 | -9.023 | Likely Pathogenic | 0.932 | Likely Pathogenic | Ambiguous | 0.247 | Likely Benign | -4.51 | Deleterious | 0.396 | Benign | 0.099 | Benign | 4.01 | Benign | 0.00 | Affected | 0.0490 | 0.6024 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.593T>A | L198H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L198H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict pathogenicity: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for L198H. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.444081 | Structured | 0.431715 | Uncertain | 0.572 | 0.485 | 0.125 | -15.650 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.09 | Ambiguous | 0.2 | 1.04 | Ambiguous | 1.07 | Ambiguous | 0.74 | Ambiguous | 0.419 | Likely Benign | -5.91 | Deleterious | 0.999 | Probably Damaging | 0.936 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.1057 | 0.0519 | -2 | -3 | -7.0 | 23.98 | ||||||||||||||||||||||||||||||
| c.595A>T | N199Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N199Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumVar), FATHMM, and AlphaMissense‑Optimized; pathogenic predictions arise from SIFT, PROVEAN, polyPhen‑2 (HumDiv), and ESM1b. AlphaMissense‑Default remains uncertain. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates benign stability. Overall, the majority of conventional tools (8 benign vs. 4 pathogenic) favor a benign effect, and this interpretation does not conflict with the absence of ClinVar annotation. Therefore, the variant is most likely benign based on the current predictive evidence, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -8.575 | Likely Pathogenic | 0.391 | Ambiguous | Likely Benign | -0.06 | Likely Benign | 0.1 | 0.14 | Likely Benign | 0.04 | Likely Benign | 0.20 | Likely Benign | 0.097 | Likely Benign | -3.12 | Deleterious | 0.952 | Possibly Damaging | 0.395 | Benign | 4.15 | Benign | 0.04 | Affected | 0.0389 | 0.5630 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||
| c.596A>T | N199I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N199I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields an equal split of benign and pathogenic calls. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.390993 | Structured | 0.431347 | Uncertain | 0.571 | 0.473 | 0.125 | -8.299 | Likely Pathogenic | 0.328 | Likely Benign | Likely Benign | 0.27 | Likely Benign | 0.1 | -0.11 | Likely Benign | 0.08 | Likely Benign | 0.20 | Likely Benign | 0.066 | Likely Benign | -3.27 | Deleterious | 0.316 | Benign | 0.045 | Benign | 4.16 | Benign | 0.01 | Affected | 0.0450 | 0.6009 | -2 | -3 | 8.0 | -0.94 | ||||||||||||||||||||||||||||||
| c.599T>G | L200W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L200W is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of tools (SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and Foldetta) predict a pathogenic impact. Two tools (AlphaMissense‑Optimized and Rosetta) provide inconclusive results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as Pathogenic, the latter integrating stability predictions from FoldX‑MD and Rosetta. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.428168 | Uncertain | 0.687 | 0.453 | 0.125 | -11.550 | Likely Pathogenic | 0.824 | Likely Pathogenic | Ambiguous | 2.90 | Destabilizing | 1.0 | 1.95 | Ambiguous | 2.43 | Destabilizing | 1.20 | Destabilizing | 0.200 | Likely Benign | -2.93 | Deleterious | 0.999 | Probably Damaging | 0.970 | Probably Damaging | 3.98 | Benign | 0.02 | Affected | 0.0627 | 0.2893 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||
| c.59C>A | P20H 2D AIThe SynGAP1 missense variant P20H is reported in gnomAD (ID 6‑33420323‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates benign. Foldetta results are unavailable. Overall, the preponderance of evidence from both consensus and high‑accuracy tools points to a benign effect for P20H, and this conclusion does not contradict any ClinVar classification (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.529623 | Disordered | 0.442804 | Uncertain | 0.448 | 0.899 | 0.500 | 6-33420323-C-A | -4.450 | Likely Benign | 0.352 | Ambiguous | Likely Benign | 0.132 | Likely Benign | -0.69 | Neutral | 0.992 | Probably Damaging | 0.893 | Possibly Damaging | 4.23 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1834 | 0.5066 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||||||||||||||
| c.5G>T | S2I 2D AIThe SynGAP1 missense variant S2I is catalogued in gnomAD (6‑33420269‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all report benign or likely benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments confirm the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus, derived from the majority of the high‑confidence predictors, is benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S2I is most likely benign, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.543646 | Binding | 0.382 | 0.922 | 0.750 | 6-33420269-G-T | -4.947 | Likely Benign | 0.439 | Ambiguous | Likely Benign | 0.031 | Likely Benign | -0.59 | Neutral | 0.212 | Benign | 0.020 | Benign | 4.04 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1021 | 0.5980 | -2 | -1 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||
| c.602A>T | D201V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D201V missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM, while those that predict a pathogenic impact are SGM‑Consensus (Likely Pathogenic), Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. With the majority of tools indicating pathogenicity and no ClinVar record to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.366687 | Structured | 0.428570 | Uncertain | 0.698 | 0.447 | 0.125 | -10.283 | Likely Pathogenic | 0.906 | Likely Pathogenic | Ambiguous | 0.87 | Ambiguous | 0.1 | 2.18 | Destabilizing | 1.53 | Ambiguous | 0.31 | Likely Benign | 0.305 | Likely Benign | -5.01 | Deleterious | 0.999 | Probably Damaging | 0.946 | Probably Damaging | 4.04 | Benign | 0.02 | Affected | 0.0572 | 0.5207 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.605A>T | E202V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E202V missense variant has no ClinVar record and is not present in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are SIFT, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Because the majority of tools (8 benign vs. 5 pathogenic) lean toward a benign outcome, the variant is most likely benign, although the SGM Consensus suggests pathogenicity. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.363090 | Structured | 0.429450 | Uncertain | 0.712 | 0.415 | 0.125 | -8.990 | Likely Pathogenic | 0.783 | Likely Pathogenic | Likely Benign | 0.48 | Likely Benign | 0.0 | 0.34 | Likely Benign | 0.41 | Likely Benign | 0.12 | Likely Benign | 0.270 | Likely Benign | -4.81 | Deleterious | 0.649 | Possibly Damaging | 0.259 | Benign | 3.96 | Benign | 0.01 | Affected | 0.0525 | 0.7007 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.608A>T | D203V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D203V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools and the SGM Consensus lean toward a pathogenic interpretation, whereas two high‑accuracy methods (AlphaMissense‑Optimized and Foldetta) suggest benign. Because ClinVar contains no entry for this variant, there is no contradiction between the predictions and existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.314870 | Structured | 0.427620 | Uncertain | 0.740 | 0.407 | 0.125 | -10.660 | Likely Pathogenic | 0.523 | Ambiguous | Likely Benign | 0.32 | Likely Benign | 0.0 | 0.44 | Likely Benign | 0.38 | Likely Benign | 0.05 | Likely Benign | 0.289 | Likely Benign | -4.07 | Deleterious | 0.991 | Probably Damaging | 0.781 | Possibly Damaging | 3.98 | Benign | 0.02 | Affected | 0.0534 | 0.4084 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||
| c.614T>A | I205N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I205N missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and ESM1b. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. No evidence from these tools contradicts the lack of ClinVar annotation. Overall, the majority of predictions (five pathogenic vs. four benign) and the pathogenic SGM Consensus suggest the variant is most likely pathogenic, with no ClinVar status to conflict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.264545 | Structured | 0.409933 | Uncertain | 0.821 | 0.414 | 0.125 | -10.928 | Likely Pathogenic | 0.552 | Ambiguous | Likely Benign | 0.66 | Ambiguous | 0.1 | 1.16 | Ambiguous | 0.91 | Ambiguous | 1.57 | Destabilizing | 0.138 | Likely Benign | -3.56 | Deleterious | 0.940 | Possibly Damaging | 0.641 | Possibly Damaging | 4.06 | Benign | 0.07 | Tolerated | 0.0802 | 0.0212 | -2 | -3 | -8.0 | 0.94 | ||||||||||||||||||||||||||||||
| c.617T>A | I206N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I206N is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL and FATHMM, while the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is pathogenic. No prediction or folding result is missing or inconclusive. Based on the overwhelming majority of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.298791 | Structured | 0.405123 | Uncertain | 0.863 | 0.391 | 0.125 | -15.211 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.98 | Destabilizing | 0.2 | 2.57 | Destabilizing | 2.78 | Destabilizing | 2.04 | Destabilizing | 0.334 | Likely Benign | -5.55 | Deleterious | 0.940 | Possibly Damaging | 0.641 | Possibly Damaging | 3.62 | Benign | 0.00 | Affected | 0.0689 | 0.0340 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.626T>A | V209E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V209E is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL and FATHMM, whereas the remaining 13 tools (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus) all predict a pathogenic outcome. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming majority of pathogenic predictions, V209E is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.247041 | Structured | 0.397624 | Uncertain | 0.874 | 0.331 | 0.125 | -14.366 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.53 | Destabilizing | 1.3 | 2.35 | Destabilizing | 2.94 | Destabilizing | 1.52 | Destabilizing | 0.403 | Likely Benign | -4.54 | Deleterious | 0.995 | Probably Damaging | 0.829 | Possibly Damaging | 3.65 | Benign | 0.00 | Affected | 0.0826 | 0.1652 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.629A>T | H210L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H210L missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools cluster into two groups: benign predictions come from REVEL, Foldetta, premPS, and FATHMM, while pathogenic predictions arise from SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX and Rosetta provide uncertain results. High‑accuracy assessments further highlight the discrepancy: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, whereas Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.144935 | Structured | 0.390904 | Uncertain | 0.872 | 0.298 | 0.125 | -14.516 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | -0.71 | Ambiguous | 0.1 | 1.35 | Ambiguous | 0.32 | Likely Benign | 0.49 | Likely Benign | 0.421 | Likely Benign | -9.41 | Deleterious | 0.895 | Possibly Damaging | 0.614 | Possibly Damaging | 3.09 | Benign | 0.00 | Affected | 0.0617 | 0.4452 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.638T>A | I213N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I213N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining evaluated algorithms—SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact; FoldX is uncertain and is treated as unavailable. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.158265 | Structured | 0.372201 | Uncertain | 0.850 | 0.295 | 0.125 | -15.069 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.71 | Ambiguous | 0.8 | 2.81 | Destabilizing | 2.26 | Destabilizing | 1.85 | Destabilizing | 0.862 | Likely Pathogenic | -5.81 | Deleterious | 0.995 | Probably Damaging | 0.880 | Possibly Damaging | 5.83 | Benign | 0.00 | Affected | 0.0750 | 0.0412 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.641T>A | L214Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change at residue 214 (Leu→Gln) is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, the majority (SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic effect, while only FATHMM predicts a benign outcome. Three tools (FoldX, Rosetta, Foldetta) return uncertain results. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta is uncertain. Taken together, the overwhelming consensus of pathogenic predictions indicates that the variant is most likely pathogenic, with no ClinVar evidence contradicting this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.155435 | Structured | 0.372604 | Uncertain | 0.818 | 0.302 | 0.125 | -10.119 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 1.60 | Ambiguous | 0.6 | 1.71 | Ambiguous | 1.66 | Ambiguous | 1.73 | Destabilizing | 0.912 | Likely Pathogenic | -5.12 | Deleterious | 0.997 | Probably Damaging | 0.936 | Probably Damaging | 5.74 | Benign | 0.00 | Affected | 0.1140 | 0.0930 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.647A>T | Q216L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q216L is not reported in ClinVar (ClinVar ID: None) and has no entry in gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, and FATHMM, while those that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain (treated as unavailable), SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta predicts benign. Overall, the majority of tools (8 pathogenic vs. 5 benign) and the consensus high‑accuracy prediction lean toward pathogenicity. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.206376 | Structured | 0.396100 | Uncertain | 0.804 | 0.274 | 0.000 | -11.303 | Likely Pathogenic | 0.886 | Likely Pathogenic | Ambiguous | -0.17 | Likely Benign | 0.3 | 0.28 | Likely Benign | 0.06 | Likely Benign | 0.30 | Likely Benign | 0.797 | Likely Pathogenic | -5.58 | Deleterious | 0.963 | Probably Damaging | 0.452 | Possibly Damaging | 5.87 | Benign | 0.01 | Affected | 0.1036 | 0.6410 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||
| c.64A>G | R22G 2D AIThe SynGAP1 missense variant R22G is reported in ClinVar as “Not submitted” and is present in gnomAD (ID 6‑33420328‑A‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which has no pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.538167 | Disordered | 0.441505 | Uncertain | 0.377 | 0.891 | 0.500 | 6-33420328-A-G | -3.628 | Likely Benign | 0.322 | Likely Benign | Likely Benign | 0.185 | Likely Benign | -0.42 | Neutral | 0.462 | Possibly Damaging | 0.152 | Benign | 4.19 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3590 | 0.4098 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||
| c.650A>T | E217V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E217V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include Rosetta, FATHMM, and premPS, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). FoldX and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E217V. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.278302 | Structured | 0.404912 | Uncertain | 0.823 | 0.284 | 0.000 | -10.194 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.76 | Ambiguous | 0.6 | 0.39 | Likely Benign | 0.58 | Ambiguous | 0.23 | Likely Benign | 0.723 | Likely Pathogenic | -4.84 | Deleterious | 0.900 | Possibly Damaging | 0.461 | Possibly Damaging | 5.79 | Benign | 0.03 | Affected | 0.0919 | 0.8340 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.653T>G | F218C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F218C is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33435295‑T‑G). Prediction tools that agree on a benign effect include only FATHMM, whereas the majority of tools (REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default) predict a pathogenic impact. Results that are uncertain or unavailable are FoldX, ESM1b, AlphaMissense‑Optimized, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic prediction (2 pathogenic vs. 1 benign votes); and Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for F218C, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.281712 | Structured | 0.408725 | Uncertain | 0.848 | 0.272 | 0.000 | 6-33435295-T-G | 1 | 6.20e-7 | -7.234 | In-Between | 0.948 | Likely Pathogenic | Ambiguous | 1.49 | Ambiguous | 0.1 | 2.20 | Destabilizing | 1.85 | Ambiguous | 1.02 | Destabilizing | 0.744 | Likely Pathogenic | -4.92 | Deleterious | 0.994 | Probably Damaging | 0.667 | Possibly Damaging | 5.78 | Benign | 0.03 | Affected | 3.41 | 13 | 0.2330 | 0.1321 | -2 | -4 | -0.3 | -44.04 | |||||||||||||||||||||||||
| c.65G>T | R22I 2D  AIThe SynGAP1 missense variant R22I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence favors a benign interpretation, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.538167 | Disordered | 0.441505 | Uncertain | 0.377 | 0.891 | 0.500 | -4.849 | Likely Benign | 0.692 | Likely Pathogenic | Likely Benign | 0.118 | Likely Benign | 0.06 | Neutral | 0.676 | Possibly Damaging | 0.308 | Benign | 4.20 | Benign | 0.00 | Affected | 0.2120 | 0.5048 | -2 | -3 | 9.0 | -43.03 | |||||||||||||||||||||||||||||||||||||||
| c.662A>T | E221V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E221V missense variant is reported in ClinVar as Pathogenic (ClinVar ID 2413181.0) and is not found in gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Benign predictions are limited to premPS, polyPhen‑2 HumVar, and FATHMM. Uncertain results are reported by FoldX, Rosetta, and Foldetta. High‑accuracy assessments reinforce the pathogenic interpretation: AlphaMissense‑Optimized predicts Pathogenic, the SGM‑Consensus also indicates Likely Pathogenic, while Foldetta remains Uncertain. Taken together, the preponderance of evidence supports a pathogenic effect for E221V, and this conclusion aligns with the ClinVar classification, showing no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.413334 | Uncertain | 0.891 | 0.283 | 0.000 | Likely Pathogenic | 1 | -14.954 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | -0.66 | Ambiguous | 0.2 | -0.89 | Ambiguous | -0.78 | Ambiguous | 0.49 | Likely Benign | 0.875 | Likely Pathogenic | -5.54 | Deleterious | 0.596 | Possibly Damaging | 0.203 | Benign | 5.86 | Benign | 0.00 | Affected | 3.41 | 13 | 0.0806 | 0.8138 | -2 | -2 | 7.7 | -29.98 | 234.5 | 50.6 | 0.0 | 0.0 | -0.4 | 0.2 | X | Uncertain | The introduced residue Val221 is located on the outer surface of an anti-parallel β sheet strand (res. Cys219-Thr224). Unlike the carboxylate group of Glu221, Val221 cannot form hydrogen bonds with Thr223 or a salt bridge with the amino group of the Lys207 side chain. Despite this, the WT simulations containing Glu221 do not show significant differences compared to the variant simulations. However, since the model ends abruptly at the N-terminus, no definite conclusions can be drawn from the simulations. | ||||||||||||||||
| c.665T>A | V222E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V222E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is consistent with the lack of ClinVar reporting (i.e., no contradictory evidence). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.116183 | Structured | 0.402706 | Uncertain | 0.885 | 0.310 | 0.125 | -18.017 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.04 | Destabilizing | 0.2 | 3.46 | Destabilizing | 3.75 | Destabilizing | 1.64 | Destabilizing | 0.960 | Likely Pathogenic | -5.20 | Deleterious | 0.998 | Probably Damaging | 0.987 | Probably Damaging | 5.20 | Benign | 0.00 | Affected | 0.0833 | 0.1495 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.674C>T | S225L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S225L is reported in gnomAD (6‑33435525‑C‑T) but has no ClinVar entry. In silico predictors that agree on a benign effect include REVEL, FoldX, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only PROVEAN predicts a pathogenic outcome. Predictions that are inconclusive are Rosetta and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as Uncertain. Overall, the majority of evidence points to a benign effect for S225L, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | 0.150080 | Structured | 0.344191 | Uncertain | 0.817 | 0.319 | 0.250 | 6-33435525-C-T | 1 | 6.20e-7 | -6.644 | Likely Benign | 0.103 | Likely Benign | Likely Benign | 0.29 | Likely Benign | 0.4 | 1.18 | Ambiguous | 0.74 | Ambiguous | 0.18 | Likely Benign | 0.320 | Likely Benign | -3.76 | Deleterious | 0.000 | Benign | 0.001 | Benign | 5.70 | Benign | 0.28 | Tolerated | 3.41 | 13 | 0.1039 | 0.6554 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||||
| c.679G>A | G227R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G227R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. In contrast, the majority of other in silico predictors classify the variant as pathogenic: SGM‑Consensus, REVEL, FoldX, Rosetta, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized (premPS is inconclusive). High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a pathogenic effect. Overall, the preponderance of evidence points to a pathogenic classification for G227R, with no conflict from ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.106997 | Structured | 0.329995 | Uncertain | 0.800 | 0.329 | 0.250 | -9.776 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.31 | Destabilizing | 0.3 | 5.29 | Destabilizing | 3.80 | Destabilizing | 0.90 | Ambiguous | 0.765 | Likely Pathogenic | -6.49 | Deleterious | 0.020 | Benign | 0.018 | Benign | 6.02 | Benign | 0.01 | Affected | 3.43 | 12 | 0.0997 | 0.4195 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||||
| c.679G>C | G227R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G227R is not reported in ClinVar (no ClinVar ID) but is present in gnomAD (ID 6‑33435530‑G‑C). Prediction tools that agree on a benign effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that agree on a pathogenic effect comprise REVEL, FoldX, Rosetta, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta. The premPS score is uncertain and does not influence the consensus. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Based on the overwhelming agreement among high‑confidence predictors, the variant is most likely pathogenic; this conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.106997 | Structured | 0.329995 | Uncertain | 0.800 | 0.329 | 0.250 | 6-33435530-G-C | 1 | 6.20e-7 | -9.776 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.31 | Destabilizing | 0.3 | 5.29 | Destabilizing | 3.80 | Destabilizing | 0.90 | Ambiguous | 0.765 | Likely Pathogenic | -6.49 | Deleterious | 0.020 | Benign | 0.018 | Benign | 6.02 | Benign | 0.01 | Affected | 3.43 | 12 | 0.0997 | 0.4195 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||
| c.686A>T | K229I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K229I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from FoldX, Foldetta, premPS, and FATHMM, whereas pathogenic predictions are returned by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports benign. No prediction is inconclusive. Overall, the majority of tools, including the high‑accuracy ones, lean toward pathogenicity, and this does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.179055 | Structured | 0.310912 | Uncertain | 0.843 | 0.306 | 0.000 | -15.276 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | -0.03 | Likely Benign | 0.1 | -0.63 | Ambiguous | -0.33 | Likely Benign | -0.19 | Likely Benign | 0.833 | Likely Pathogenic | -6.52 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | 5.92 | Benign | 0.00 | Affected | 0.1202 | 0.3711 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.68A>T | D23V 2D AIThe SynGAP1 D23V missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie and thus unavailable; Foldetta predictions are not provided. Overall, more tools (five) predict pathogenicity than benign (three), and no ClinVar evidence contradicts this assessment. Therefore, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.557691 | Disordered | 0.440341 | Uncertain | 0.369 | 0.892 | 0.375 | -3.244 | Likely Benign | 0.842 | Likely Pathogenic | Ambiguous | 0.137 | Likely Benign | -2.72 | Deleterious | 0.972 | Probably Damaging | 0.804 | Possibly Damaging | 3.48 | Benign | 0.00 | Affected | 0.1515 | 0.8367 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.713A>G | E238G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E238G is reported in gnomAD (variant ID 6‑33435564‑A‑G) but has no entry in ClinVar. Functional prediction tools largely agree on a deleterious effect: the benign‑predicting tool FATHMM is the only one that classifies it as benign, whereas the pathogenic‑predicting tools (REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus “Likely Pathogenic” call) all predict a harmful impact. Predictions of uncertain status (FoldX, premPS) are treated as unavailable. High‑accuracy assessments reinforce the pathogenic view: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the overwhelming agreement among high‑confidence tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.194234 | Structured | 0.332638 | Uncertain | 0.796 | 0.326 | 0.000 | 6-33435564-A-G | 1 | 6.19e-7 | -12.582 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.09 | Ambiguous | 0.2 | 3.42 | Destabilizing | 2.26 | Destabilizing | 0.87 | Ambiguous | 0.889 | Likely Pathogenic | -6.35 | Deleterious | 0.970 | Probably Damaging | 0.607 | Possibly Damaging | 5.39 | Benign | 0.00 | Affected | 3.40 | 14 | 0.3137 | 0.4833 | -2 | 0 | 3.1 | -72.06 | ||||||||||||||||||||||||
| c.713A>T | E238V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E238V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include premPS and FATHMM, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta give uncertain results. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E238V. This conclusion is not contradicted by ClinVar, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.194234 | Structured | 0.332638 | Uncertain | 0.796 | 0.326 | 0.000 | -14.329 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.88 | Ambiguous | 0.4 | 0.75 | Ambiguous | 0.82 | Ambiguous | 0.45 | Likely Benign | 0.890 | Likely Pathogenic | -6.35 | Deleterious | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 5.42 | Benign | 0.00 | Affected | 0.0848 | 0.6093 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.716G>T | R239I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R239I is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, while the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS (uncertain), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. No predictions are missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.170161 | Structured | 0.336504 | Uncertain | 0.854 | 0.319 | 0.000 | -19.414 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.32 | Destabilizing | 0.5 | 2.53 | Destabilizing | 3.43 | Destabilizing | 0.83 | Ambiguous | 0.890 | Likely Pathogenic | -7.16 | Deleterious | 0.985 | Probably Damaging | 0.724 | Possibly Damaging | 5.69 | Benign | 0.00 | Affected | 0.1480 | 0.3985 | -2 | -3 | 9.0 | -43.03 | |||||||||||||||||||||||||||||
| c.719A>T | D240V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D240V missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta, FATHMM, and premPS, whereas the majority of algorithms—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score—classify the change as pathogenic or likely pathogenic. FoldX and Foldetta return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for D240V. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.127496 | Structured | 0.343480 | Uncertain | 0.822 | 0.333 | 0.000 | -15.095 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 1.32 | Ambiguous | 0.1 | 0.28 | Likely Benign | 0.80 | Ambiguous | 0.11 | Likely Benign | 0.894 | Likely Pathogenic | -7.94 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 5.82 | Benign | 0.00 | Affected | 0.0610 | 0.4949 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.71T>A | V24E 2D AIThe SynGAP1 missense variant V24E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, the SGM‑Consensus also indicates Likely Benign, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.438970 | Uncertain | 0.382 | 0.890 | 0.500 | -3.397 | Likely Benign | 0.517 | Ambiguous | Likely Benign | 0.171 | Likely Benign | -1.73 | Neutral | 0.198 | Benign | 0.074 | Benign | 3.77 | Benign | 0.00 | Affected | 0.1133 | 0.2415 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||||||||||||
| c.722A>T | K241I 2D 3DClick to see structure in 3D Viewer AISynGAP1 K241I is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Rosetta, premPS, and FATHMM. Those that predict a damaging effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). FoldX and Foldetta return uncertain results. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized reports pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, while Foldetta remains inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.196879 | Structured | 0.349250 | Uncertain | 0.797 | 0.347 | 0.000 | -14.370 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 1.31 | Ambiguous | 0.5 | 0.28 | Likely Benign | 0.80 | Ambiguous | 0.41 | Likely Benign | 0.852 | Likely Pathogenic | -6.96 | Deleterious | 0.985 | Probably Damaging | 0.704 | Possibly Damaging | 5.71 | Benign | 0.01 | Affected | 0.1176 | 0.3868 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.725G>C | W242S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W242S, located in the PH domain, is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool predicts a benign outcome. High‑accuracy assessments further support a harmful impact: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic,” while Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. Overall, the consensus of available predictions indicates that W242S is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.328603 | Structured | 0.352582 | Uncertain | 0.847 | 0.341 | 0.000 | -14.674 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 1.96 | Ambiguous | 0.2 | 1.97 | Ambiguous | 1.97 | Ambiguous | 0.83 | Ambiguous | 0.797 | Likely Pathogenic | -12.71 | Deleterious | 0.900 | Possibly Damaging | 0.535 | Possibly Damaging | 1.52 | Pathogenic | 0.00 | Affected | 0.4424 | 0.1418 | -2 | -3 | 0.1 | -99.14 | |||||||||||||||||||||||||||||
| c.725G>T | W242L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W242L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions (FoldX, Rosetta, Foldetta, premPS) and pathogenic predictions (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Pathogenic). High‑accuracy assessments further support this dichotomy: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a benign effect. Overall, the majority of evidence points toward a pathogenic impact for W242L. This conclusion is consistent with the absence of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.328603 | Structured | 0.352582 | Uncertain | 0.847 | 0.341 | 0.000 | -12.297 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | -0.29 | Likely Benign | 0.3 | 0.12 | Likely Benign | -0.09 | Likely Benign | 0.44 | Likely Benign | 0.799 | Likely Pathogenic | -11.80 | Deleterious | 0.948 | Possibly Damaging | 0.533 | Possibly Damaging | 1.52 | Pathogenic | 0.01 | Affected | 0.2370 | 0.2986 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||
| c.728T>A | I243N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant I243N is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into three groups: benign (FATHMM), pathogenic (SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized), and uncertain (FoldX, Rosetta, Foldetta). High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.363090 | Structured | 0.344471 | Uncertain | 0.842 | 0.347 | 0.000 | -14.784 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 1.90 | Ambiguous | 0.2 | 1.43 | Ambiguous | 1.67 | Ambiguous | 1.76 | Destabilizing | 0.811 | Likely Pathogenic | -4.46 | Deleterious | 0.995 | Probably Damaging | 0.854 | Possibly Damaging | 5.49 | Benign | 0.00 | Affected | 0.0899 | 0.0212 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.731A>T | E244V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E244V missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. Four tools (FoldX, Rosetta, Foldetta, premPS) returned uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also predicts pathogenicity; Foldetta remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.450668 | Structured | 0.329406 | Uncertain | 0.778 | 0.360 | 0.000 | -12.118 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.04 | Ambiguous | 0.1 | 1.02 | Ambiguous | 1.03 | Ambiguous | 0.56 | Ambiguous | 0.925 | Likely Pathogenic | -5.90 | Deleterious | 0.997 | Probably Damaging | 0.879 | Possibly Damaging | 5.68 | Benign | 0.00 | Affected | 0.0636 | 0.6073 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.733A>T | N245Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N245Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from FoldX, Rosetta, Foldetta, and FATHMM, whereas pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus, SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a pathogenic majority (3/4). AlphaMissense‑Optimized independently predicts pathogenicity, while Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts benign. premPS remains uncertain. Overall, the majority of evidence points toward a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Thus, the variant is most likely pathogenic, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -12.476 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | -0.36 | Likely Benign | 0.1 | 0.11 | Likely Benign | -0.13 | Likely Benign | 0.59 | Ambiguous | 0.855 | Likely Pathogenic | -6.68 | Deleterious | 0.995 | Probably Damaging | 0.880 | Possibly Damaging | 5.86 | Benign | 0.00 | Affected | 0.0706 | 0.6771 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.734A>T | N245I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N245I is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include Rosetta, Foldetta, and FATHMM, while those that predict a pathogenic effect comprise REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, as the variant is currently unreported in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.454136 | Structured | 0.315864 | Uncertain | 0.831 | 0.351 | 0.000 | -14.527 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | -0.56 | Ambiguous | 0.1 | 0.04 | Likely Benign | -0.26 | Likely Benign | 0.60 | Ambiguous | 0.831 | Likely Pathogenic | -7.46 | Deleterious | 0.995 | Probably Damaging | 0.832 | Possibly Damaging | 5.88 | Benign | 0.00 | Affected | 0.0672 | 0.6999 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.737T>A | L246Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L246Q missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining tools—SGM‑Consensus, REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact; Rosetta is uncertain and is not grouped. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Taken together, the overwhelming majority of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.472492 | Structured | 0.302312 | Uncertain | 0.859 | 0.364 | 0.000 | -15.420 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 2.82 | Destabilizing | 0.3 | 1.79 | Ambiguous | 2.31 | Destabilizing | 1.69 | Destabilizing | 0.921 | Likely Pathogenic | -5.43 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 4.67 | Benign | 0.00 | Affected | 0.1092 | 0.0758 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.740A>T | Q247L 2D AIThe SynGAP1 missense variant Q247L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The remaining tools—FoldX, Rosetta, Foldetta, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence (seven pathogenic versus three benign predictions) points to a pathogenic impact, and this conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | 0.490133 | Structured | 0.283012 | Uncertain | 0.822 | 0.339 | 0.250 | -10.554 | Likely Pathogenic | 0.352 | Ambiguous | Likely Benign | -0.86 | Ambiguous | 0.5 | -1.14 | Ambiguous | -1.00 | Ambiguous | 0.38 | Likely Benign | 0.687 | Likely Pathogenic | -3.89 | Deleterious | 0.982 | Probably Damaging | 0.628 | Possibly Damaging | 5.70 | Benign | 0.02 | Affected | 0.0573 | 0.4129 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||
| c.749T>A | V250E 2D 3DClick to see structure in 3D Viewer AISynGAP1 variant V250E is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, whereas the remaining 12 tools (SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify the change as pathogenic. The high‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates pathogenic. No prediction or stability result is inconclusive. Consequently, the variant is most likely pathogenic, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.447574 | Structured | 0.244075 | Uncertain | 0.778 | 0.324 | 0.125 | -15.723 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 1.85 | Ambiguous | 0.1 | 3.34 | Destabilizing | 2.60 | Destabilizing | 2.01 | Destabilizing | 0.906 | Likely Pathogenic | -5.13 | Deleterious | 0.997 | Probably Damaging | 0.916 | Probably Damaging | 5.74 | Benign | 0.00 | Affected | 0.0787 | 0.1564 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.757A>T | N253Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N253Y is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect include FoldX, FATHMM, and premPS, whereas a larger set—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Because the majority of evidence points to a deleterious impact, the variant is most likely pathogenic; this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | 0.513880 | Disordered | 0.201744 | Uncertain | 0.771 | 0.298 | 0.250 | -14.749 | Likely Pathogenic | 0.920 | Likely Pathogenic | Ambiguous | 0.27 | Likely Benign | 0.1 | 1.13 | Ambiguous | 0.70 | Ambiguous | 0.29 | Likely Benign | 0.896 | Likely Pathogenic | -7.01 | Deleterious | 0.998 | Probably Damaging | 0.994 | Probably Damaging | 5.55 | Benign | 0.01 | Affected | 0.0642 | 0.7055 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.764A>T | D255V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D255V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from premPS and FATHMM, while pathogenic predictions are made by SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are reported by FoldX, Rosetta, and Foldetta. High‑accuracy methods reinforce the pathogenic interpretation: AlphaMissense‑Optimized predicts Pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains Uncertain. Overall, the consensus of available predictions points to a pathogenic effect for D255V, and this conclusion is consistent with the lack of ClinVar annotation (no contradiction). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.501700 | Disordered | 0.219132 | Uncertain | 0.801 | 0.273 | 0.250 | -13.575 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.76 | Ambiguous | 0.2 | 1.07 | Ambiguous | 1.42 | Ambiguous | 0.16 | Likely Benign | 0.895 | Likely Pathogenic | -7.77 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 5.75 | Benign | 0.00 | Affected | 0.0684 | 0.5162 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||
| c.766A>T | N256Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that classify the variant as benign include FoldX, Foldetta, premPS, and FATHMM, whereas the majority of tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—label it pathogenic. The high‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the preponderance of evidence from standard predictors and the two high‑accuracy pathogenic calls suggests that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | -10.881 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.17 | Likely Benign | 0.2 | -0.80 | Ambiguous | -0.32 | Likely Benign | 0.30 | Likely Benign | 0.868 | Likely Pathogenic | -6.85 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 5.83 | Benign | 0.00 | Affected | 0.0493 | 0.5881 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.767A>T | N256I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta, premPS, and FATHMM. Those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy methods give a pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.414856 | Structured | 0.234105 | Uncertain | 0.826 | 0.271 | 0.250 | -14.050 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.64 | Ambiguous | 0.4 | 0.45 | Likely Benign | 0.55 | Ambiguous | 0.31 | Likely Benign | 0.849 | Likely Pathogenic | -7.91 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 5.87 | Benign | 0.00 | Affected | 0.0596 | 0.6260 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.773G>T | R258L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R258L is not reported in ClinVar and is present in gnomAD (ID 6‑33437678‑G‑T). Prediction tools that agree on a benign effect include FoldX, Rosetta, FATHMM, and the combined Foldetta stability method. Tools that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Two tools give inconclusive results: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.295083 | Structured | 0.293667 | Uncertain | 0.894 | 0.260 | 0.250 | 6-33437678-G-T | 1 | 6.20e-7 | -13.302 | Likely Pathogenic | 0.905 | Likely Pathogenic | Ambiguous | 0.14 | Likely Benign | 0.2 | 0.10 | Likely Benign | 0.12 | Likely Benign | 0.52 | Ambiguous | 0.908 | Likely Pathogenic | -5.90 | Deleterious | 0.997 | Probably Damaging | 0.987 | Probably Damaging | 5.84 | Benign | 0.01 | Affected | 3.39 | 15 | 0.1606 | 0.4602 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||
| c.779T>A | V260E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V260E is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect include FoldX, Rosetta, Foldetta, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labeling it likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicting a benign impact on protein stability. Overall, the majority of computational evidence points toward a pathogenic interpretation, and this conclusion is not contradicted by the ClinVar status. Thus, the variant is most likely pathogenic, and this conclusion is not contradicted by the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.254060 | Structured | 0.382651 | Uncertain | 0.888 | 0.259 | 0.250 | -9.516 | Likely Pathogenic | 0.667 | Likely Pathogenic | Likely Benign | -0.02 | Likely Benign | 0.1 | 0.32 | Likely Benign | 0.15 | Likely Benign | 1.10 | Destabilizing | 0.848 | Likely Pathogenic | -3.66 | Deleterious | 0.999 | Probably Damaging | 0.991 | Probably Damaging | 5.79 | Benign | 0.09 | Tolerated | 0.0846 | 0.1636 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.782A>T | D261V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D261V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include premPS, SIFT, and FATHMM, while those that predict a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of available predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.284882 | Structured | 0.422514 | Uncertain | 0.883 | 0.264 | 0.125 | -12.138 | Likely Pathogenic | 0.923 | Likely Pathogenic | Ambiguous | 1.72 | Ambiguous | 0.6 | -0.68 | Ambiguous | 0.52 | Ambiguous | -0.01 | Likely Benign | 0.869 | Likely Pathogenic | -5.50 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 5.73 | Benign | 0.08 | Tolerated | 0.0553 | 0.4734 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.784A>T | N262Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N262Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, FATHMM, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The remaining tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Default) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of predictions (seven pathogenic vs. three benign) lean toward a pathogenic impact, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.284882 | Structured | 0.399879 | Uncertain | 0.912 | 0.240 | 0.000 | -12.177 | Likely Pathogenic | 0.490 | Ambiguous | Likely Benign | 1.86 | Ambiguous | 0.5 | 0.52 | Ambiguous | 1.19 | Ambiguous | 0.36 | Likely Benign | 0.858 | Likely Pathogenic | -6.48 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 5.85 | Benign | 0.01 | Affected | 0.0445 | 0.3877 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||
| c.785A>T | N262I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N262I is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include premPS and FATHMM, whereas the majority of tools predict a pathogenic outcome: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. Predictions from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are uncertain and therefore treated as unavailable. High‑accuracy methods give an uncertain result for AlphaMissense‑Optimized, a pathogenic consensus from SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and an uncertain result for Foldetta. Overall, the evidence points to a pathogenic effect for the variant, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.284882 | Structured | 0.399879 | Uncertain | 0.912 | 0.240 | 0.000 | -15.203 | Likely Pathogenic | 0.868 | Likely Pathogenic | Ambiguous | 1.21 | Ambiguous | 0.4 | 0.54 | Ambiguous | 0.88 | Ambiguous | 0.09 | Likely Benign | 0.777 | Likely Pathogenic | -7.79 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 5.88 | Benign | 0.01 | Affected | 0.0449 | 0.4638 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.788T>A | V263E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V263E missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools largely agree on a deleterious effect: FATHMM predicts the variant as benign, while the remaining twelve tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—classify it as pathogenic. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized remains uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic effect. Overall, the preponderance of evidence points to a pathogenic impact for V263E, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.268042 | Structured | 0.356141 | Uncertain | 0.918 | 0.257 | 0.000 | -13.498 | Likely Pathogenic | 0.809 | Likely Pathogenic | Ambiguous | 2.04 | Destabilizing | 0.3 | 2.18 | Destabilizing | 2.11 | Destabilizing | 1.99 | Destabilizing | 0.862 | Likely Pathogenic | -3.84 | Deleterious | 0.999 | Probably Damaging | 0.991 | Probably Damaging | 5.96 | Benign | 0.01 | Affected | 0.0886 | 0.1436 | -2 | -2 | -7.7 | 29.98 | |||||||||||||||||||||||||||||
| c.791T>A | L264Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L264Q is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. No tool in the dataset predicts a benign outcome. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is pathogenic. Based on the unanimous computational evidence, the variant is most likely pathogenic, a conclusion that contradicts the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.185198 | Structured | 0.323473 | Uncertain | 0.939 | 0.264 | 0.000 | Uncertain | 1 | -15.729 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.43 | Destabilizing | 0.1 | 2.41 | Destabilizing | 2.92 | Destabilizing | 2.48 | Destabilizing | 0.678 | Likely Pathogenic | -5.52 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 0.49 | Pathogenic | 0.00 | Affected | 3.38 | 18 | 0.0942 | 0.0558 | -2 | -2 | -7.3 | 14.97 | 254.7 | -7.6 | 0.0 | 0.0 | 0.0 | 0.3 | X | X | X | Potentially Pathogenic | The iso-butyl branched hydrocarbon side chain of Leu264, located at the end of an anti-parallel β sheet strand (res. Arg259-Arg272), packs against multiple hydrophobic residues such as Leu266, Phe314, Leu317, and Leu323 in the WT simulations. In the variant simulations, the hydrophilic carboxamide group of the Gln264 side chain is not suitable for the hydrophobic niche, causing the hydrophobic residues to make room for the swapped residue. Additionally, the carboxamide group of Gln264 forms hydrogen bonds with the backbone amide groups of Arg405 and Lys256 in the β sheet and the carbonyl group of Val350 in an α helical section of a nearby loop (res. Pro359-Phe358). The residue swap disrupts the packing of the C2 domain, which could adversely affect the C2 domain structure during folding. This disruption could potentially weaken the stability of the SynGAP-membrane association. | ||||||||||||||
| c.797T>A | L266Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L266Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect: none. Tools that agree on a pathogenic effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). The only tool with an uncertain outcome is Rosetta. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic. Based on the overwhelming agreement among these predictions, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.232838 | Structured | 0.297157 | Uncertain | 0.948 | 0.264 | 0.000 | -16.672 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.92 | Destabilizing | 0.1 | 1.49 | Ambiguous | 2.21 | Destabilizing | 2.21 | Destabilizing | 0.563 | Likely Pathogenic | -5.25 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.53 | Pathogenic | 0.00 | Affected | 0.0815 | 0.0558 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.7A>G | R3G 2D AIThe SynGAP1 missense variant R3G is reported in gnomAD (ID 6‑33420271‑A‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign; Foldetta results are unavailable. Taken together, the preponderance of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.538167 | Disordered | 0.550331 | Binding | 0.358 | 0.920 | 0.875 | 6-33420271-A-G | -3.093 | Likely Benign | 0.160 | Likely Benign | Likely Benign | 0.099 | Likely Benign | -0.20 | Neutral | 0.115 | Benign | 0.018 | Benign | 3.99 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3559 | 0.4114 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||||||||||||||
| c.800G>C | W267S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W267S is not listed in ClinVar and has no allele in gnomAD. Functional prediction tools largely agree on a deleterious effect: SIFT is the sole benign predictor, whereas the remaining 12 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all report pathogenicity; premPS is uncertain. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.298060 | Uncertain | 0.943 | 0.274 | 0.000 | -12.833 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 3.42 | Destabilizing | 0.3 | 3.20 | Destabilizing | 3.31 | Destabilizing | 0.87 | Ambiguous | 0.593 | Likely Pathogenic | -12.87 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.03 | Pathogenic | 0.09 | Tolerated | 0.3809 | 0.1415 | -2 | -3 | 0.1 | -99.14 | |||||||||||||||||||||||||||||
| c.800G>T | W267L 2D AIThe SynGAP1 missense variant W267L is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. No tool in the dataset predicts a benign effect. Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS, which are treated as unavailable evidence. High‑accuracy assessments further support a damaging effect: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic,” while Foldetta’s stability prediction is uncertain. Overall, the evidence strongly indicates that W267L is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.298060 | Uncertain | 0.943 | 0.274 | 0.000 | -13.670 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.19 | Ambiguous | 0.7 | 1.31 | Ambiguous | 1.25 | Ambiguous | 0.75 | Ambiguous | 0.628 | Likely Pathogenic | -11.95 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.93 | Pathogenic | 0.01 | Affected | 0.2122 | 0.2843 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||
| c.803T>A | I268N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I268N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic. No tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a destabilizing, pathogenic effect. All available predictions are concordant and supportive. Based on these computational assessments, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.314336 | Uncertain | 0.951 | 0.264 | 0.000 | -13.664 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.48 | Destabilizing | 0.1 | 3.17 | Destabilizing | 3.33 | Destabilizing | 2.21 | Destabilizing | 0.812 | Likely Pathogenic | -6.26 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.51 | Pathogenic | 0.00 | Affected | 0.0708 | 0.0142 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.806T>A | I269K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I269K is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools largely agree on a deleterious effect: SIFT is the sole benign predictor, whereas the remaining methods—SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates pathogenicity, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. FoldX and Rosetta individually report uncertain effects, and Foldetta remains unavailable. Overall, the consensus of the majority of evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.343787 | Uncertain | 0.937 | 0.244 | 0.125 | -14.609 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.45 | Ambiguous | 0.1 | 1.86 | Ambiguous | 1.66 | Ambiguous | 1.55 | Destabilizing | 0.763 | Likely Pathogenic | -5.10 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.76 | Pathogenic | 0.06 | Tolerated | 0.0878 | 0.0713 | -2 | -3 | -8.4 | 15.01 | |||||||||||||||||||||||||||||
| c.806T>G | I269R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I269R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: the benign group contains only SIFT, whereas the pathogenic group includes SGM‑Consensus (Likely Pathogenic), REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX and Rosetta give uncertain results, and Foldetta is also uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as unavailable. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.216401 | Structured | 0.343787 | Uncertain | 0.937 | 0.244 | 0.125 | -14.567 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 1.34 | Ambiguous | 0.1 | 1.99 | Ambiguous | 1.67 | Ambiguous | 1.50 | Destabilizing | 0.765 | Likely Pathogenic | -5.23 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.69 | Pathogenic | 0.07 | Tolerated | 0.1108 | 0.0870 | -2 | -3 | -9.0 | 43.03 | |||||||||||||||||||||||||||||
| c.809A>T | E270V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E270V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that assess sequence conservation and structural impact uniformly classify the substitution as pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return pathogenic scores. No tool predicts a benign effect. Uncertain results are reported only by FoldX, Rosetta, Foldetta, and premPS, which are not considered evidence for or against pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Pathogenic”; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is inconclusive. Overall, the consensus of available predictions indicates that E270V is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.144935 | Structured | 0.382573 | Uncertain | 0.938 | 0.231 | 0.125 | -15.969 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.38 | Ambiguous | 0.6 | 1.88 | Ambiguous | 1.63 | Ambiguous | -0.52 | Ambiguous | 0.574 | Likely Pathogenic | -6.43 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.53 | Pathogenic | 0.00 | Affected | 0.0672 | 0.4498 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.818A>T | E273V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E273V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL and premPS. The majority of tools predict a pathogenic impact: Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, SGM‑Consensus (Likely Pathogenic), and Foldetta. Two tools give inconclusive results: AlphaMissense‑Optimized (Uncertain) and FoldX (Uncertain). High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta as Pathogenic. Overall, the consensus of pathogenic‑predicting tools outweighs the benign predictions, indicating that E273V is most likely pathogenic; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.071867 | Structured | 0.398918 | Uncertain | 0.863 | 0.196 | 0.125 | -11.671 | Likely Pathogenic | 0.814 | Likely Pathogenic | Ambiguous | 1.93 | Ambiguous | 0.3 | 3.31 | Destabilizing | 2.62 | Destabilizing | 0.17 | Likely Benign | 0.361 | Likely Benign | -4.66 | Deleterious | 0.984 | Probably Damaging | 0.825 | Possibly Damaging | 1.70 | Pathogenic | 0.01 | Affected | 0.0811 | 0.3813 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.821T>A | L274Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L274Q is reported in ClinVar with an uncertain significance (ClinVar ID 1810279.0) and is not found in gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while Rosetta remains inconclusive. No tool predicts a benign outcome. High‑accuracy assessments corroborate this trend: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. Consequently, the variant is most likely pathogenic, and this assessment does not contradict the current ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.066181 | Structured | 0.377483 | Uncertain | 0.866 | 0.195 | 0.250 | Uncertain | 1 | -15.518 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 2.54 | Destabilizing | 0.3 | 1.74 | Ambiguous | 2.14 | Destabilizing | 1.97 | Destabilizing | 0.774 | Likely Pathogenic | -5.42 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 0.00 | Pathogenic | 0.00 | Affected | 3.38 | 19 | 0.1128 | 0.0688 | -2 | -2 | -7.3 | 14.97 | 245.9 | 1.8 | 0.0 | 0.0 | 0.1 | 0.2 | X | X | X | Potentially Pathogenic | The aliphatic side chain of Leu274, located in a β hairpin loop (res. Glu273-Lys278) connecting two anti-parallel β sheet strands, packs against multiple hydrophobic residues facing the β sheet (e.g., Ala271, Leu327, Tyr280, Val306). The hydrophilic carboxamide group of the Gln274 side chain is not suitable for this hydrophobic niche, causing nearby residues to adjust to make room for the hydrophilic glutamine. Additionally, a new hydrogen bond forms with the backbone carboxyl group of Arg272 in another β strand (res. Glu273-Arg259).As a result, the backbone amide group of Ala399 and the carbonyl group of Arg272, which connect two β strands at the β sheet end, form fewer hydrogen bonds in the variant than in the WT simulations. Although no major secondary structure disruption is observed in the variant simulations, the residue swap could profoundly affect the C2 domain folding, as the hydrophobic packing of Leu274 is crucial for maintaining the loop's contact with the rest of the C2 domain. Lastly, because the Leu274-containing loop faces the membrane surface, the residue swap could also negatively impact the SynGAP-membrane association. | ||||||||||||||
| c.827C>A | P276H 2D 3DClick to see structure in 3D Viewer AISynGAP1 P276H is reported in gnomAD (ID 6‑33437732‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all classify the variant as pathogenic, while only AlphaMissense‑Optimized predicts a benign outcome. Uncertain results are provided by FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.037156 | Structured | 0.338937 | Uncertain | 0.724 | 0.230 | 0.250 | 6-33437732-C-A | 1 | 6.20e-7 | -10.469 | Likely Pathogenic | 0.575 | Likely Pathogenic | Likely Benign | 1.98 | Ambiguous | 0.1 | 1.09 | Ambiguous | 1.54 | Ambiguous | 0.85 | Ambiguous | 0.534 | Likely Pathogenic | -4.80 | Deleterious | 1.000 | Probably Damaging | 0.961 | Probably Damaging | 1.84 | Pathogenic | 0.00 | Affected | 3.38 | 19 | 0.1739 | 0.3439 | -2 | 0 | -1.6 | 40.02 | ||||||||||||||||||||||||
| c.827C>G | P276R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P276R missense variant is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (gnomAD ID 6‑33437732‑C‑G). Prediction tools that agree on a benign effect include REVEL and AlphaMissense‑Optimized, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. Stability‑based predictors (FoldX, Rosetta, premPS, Foldetta) are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic classification; this conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.037156 | Structured | 0.338937 | Uncertain | 0.724 | 0.230 | 0.250 | 6-33437732-C-G | 7 | 4.34e-6 | -10.983 | Likely Pathogenic | 0.714 | Likely Pathogenic | Likely Benign | 1.78 | Ambiguous | 0.2 | 1.02 | Ambiguous | 1.40 | Ambiguous | 0.78 | Ambiguous | 0.498 | Likely Benign | -4.52 | Deleterious | 0.994 | Probably Damaging | 0.892 | Possibly Damaging | 1.89 | Pathogenic | 0.01 | Affected | 3.38 | 19 | 0.1445 | 0.2828 | -2 | 0 | -2.9 | 59.07 | ||||||||||||||||||||||||
| c.836G>T | R279L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R279L is reported in gnomAD (ID 6‑33437741‑G‑T) but has no ClinVar entry. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, and premPS. Those that predict a pathogenic effect comprise SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a unanimous majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a pathogenic impact. This conclusion is not contradicted by ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.309382 | Uncertain | 0.887 | 0.257 | 0.125 | 6-33437741-G-T | 1 | 6.20e-7 | -12.390 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 0.01 | Likely Benign | 0.2 | 0.14 | Likely Benign | 0.08 | Likely Benign | 0.39 | Likely Benign | 0.576 | Likely Pathogenic | -5.37 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.91 | Pathogenic | 0.03 | Affected | 3.39 | 18 | 0.1682 | 0.3266 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||
| c.838T>A | Y280N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y280N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only SIFT, whereas all other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Based on the overwhelming consensus of pathogenic predictions and the absence of any benign consensus, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for Y280N. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.161087 | Structured | 0.321243 | Uncertain | 0.917 | 0.248 | 0.125 | -13.090 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 4.26 | Destabilizing | 0.2 | 4.18 | Destabilizing | 4.22 | Destabilizing | 1.82 | Destabilizing | 0.589 | Likely Pathogenic | -8.27 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.95 | Pathogenic | 0.06 | Tolerated | 0.1902 | 0.0212 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.841T>A | Y281N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y281N is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions are provided only by SIFT, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict pathogenicity. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized remains inconclusive, SGM‑Consensus is labeled Likely Pathogenic, and Foldetta predicts a destabilizing, pathogenic outcome. Overall, the collective evidence indicates that Y281N is most likely pathogenic, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.098513 | Structured | 0.337647 | Uncertain | 0.927 | 0.254 | 0.000 | -11.620 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 3.29 | Destabilizing | 0.2 | 2.49 | Destabilizing | 2.89 | Destabilizing | 1.71 | Destabilizing | 0.713 | Likely Pathogenic | -6.20 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 0.99 | Pathogenic | 0.09 | Tolerated | 0.2494 | 0.1503 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.848A>T | E283V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E283V missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are FoldX and premPS, while the majority of tools predict a pathogenic impact: SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools give inconclusive results: Rosetta (Uncertain) and Foldetta (Uncertain). High‑accuracy methods specifically indicate that AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus (a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as likely pathogenic, whereas Foldetta’s stability assessment is uncertain. Taken together, the preponderance of evidence from both general and high‑accuracy predictors points to a pathogenic effect for E283V. This conclusion is consistent with the absence of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.098513 | Structured | 0.358602 | Uncertain | 0.950 | 0.249 | 0.000 | -13.602 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.44 | Likely Benign | 0.2 | 0.56 | Ambiguous | 0.50 | Ambiguous | 0.36 | Likely Benign | 0.558 | Likely Pathogenic | -6.43 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.83 | Pathogenic | 0.00 | Affected | 0.0874 | 0.6011 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.851T>A | L284H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L284H is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts a pathogenic impact. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.094817 | Structured | 0.371601 | Uncertain | 0.950 | 0.255 | 0.000 | -16.581 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.26 | Destabilizing | 0.1 | 3.06 | Destabilizing | 3.16 | Destabilizing | 2.57 | Destabilizing | 0.772 | Likely Pathogenic | -6.22 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.65 | Pathogenic | 0.00 | Affected | 0.0954 | 0.0726 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.857T>A | L286Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L286Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity are unanimous: SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a deleterious effect. No tool predicts a benign outcome. Two tools, Rosetta and Foldetta, return uncertain results. High‑accuracy methods give the following: AlphaMissense‑Optimized predicts pathogenic; SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which simply lacks an entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.122885 | Structured | 0.385647 | Uncertain | 0.932 | 0.260 | 0.000 | -13.056 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.37 | Destabilizing | 0.3 | 1.42 | Ambiguous | 1.90 | Ambiguous | 2.06 | Destabilizing | 0.852 | Likely Pathogenic | -5.52 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.48 | Pathogenic | 0.00 | Affected | 0.1123 | 0.0958 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.860A>C | D287A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D287A is listed in ClinVar with an Uncertain significance status and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, FoldX, Rosetta, Foldetta, and premPS, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta) as benign. The overall tally favors pathogenicity (8 tools vs 5 benign), but the conflicting high‑accuracy results leave uncertainty. Thus, the variant is most likely pathogenic according to the majority of predictions, which does not contradict its ClinVar Uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.102787 | Structured | 0.389029 | Uncertain | 0.912 | 0.268 | 0.000 | Uncertain | 1 | -14.686 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.30 | Likely Benign | 0.1 | -0.04 | Likely Benign | 0.13 | Likely Benign | 0.40 | Likely Benign | 0.484 | Likely Benign | -7.35 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.58 | Pathogenic | 0.01 | Affected | 3.38 | 23 | 0.4448 | 0.7431 | -2 | 0 | 5.3 | -44.01 | |||||||||||||||||||||||||
| c.860A>T | D287V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D287V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only premPS; all other evaluated algorithms (SGM‑Consensus, REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact, while FoldX is uncertain and thus not counted as evidence. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.102787 | Structured | 0.389029 | Uncertain | 0.912 | 0.268 | 0.000 | -14.418 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.71 | Ambiguous | 0.4 | 3.94 | Destabilizing | 2.83 | Destabilizing | 0.25 | Likely Benign | 0.516 | Likely Pathogenic | -8.27 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.57 | Pathogenic | 0.01 | Affected | 0.0884 | 0.7788 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.863A>T | D288V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D288V missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions are provided by REVEL and premPS, whereas the remaining tools—FoldX (uncertain), Rosetta, Foldetta, SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—all indicate a pathogenic effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports pathogenic. Consequently, the variant is most likely pathogenic based on the aggregate predictions, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.125101 | Structured | 0.395525 | Uncertain | 0.873 | 0.261 | 0.000 | -15.812 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 1.74 | Ambiguous | 0.5 | 5.44 | Destabilizing | 3.59 | Destabilizing | 0.13 | Likely Benign | 0.481 | Likely Benign | -7.14 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.63 | Pathogenic | 0.05 | Affected | 0.0838 | 0.5545 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.869T>A | L290Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L290Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only SIFT, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which itself is “Likely Pathogenic”). Uncertain or inconclusive results come from FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, and Foldetta’s stability prediction is unavailable. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.127496 | Structured | 0.399723 | Uncertain | 0.904 | 0.255 | 0.000 | -12.776 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.12 | Ambiguous | 0.1 | 1.38 | Ambiguous | 1.25 | Ambiguous | 0.68 | Ambiguous | 0.697 | Likely Pathogenic | -5.52 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.97 | Pathogenic | 0.06 | Tolerated | 0.1079 | 0.1014 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.871T>A | Y291N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y291N is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess functional impact uniformly indicate a deleterious effect. Benign predictions are absent; all evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the substitution as pathogenic. High‑accuracy methods corroborate this: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a likely pathogenic verdict, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a pathogenic effect. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.173081 | Structured | 0.383842 | Uncertain | 0.912 | 0.251 | 0.000 | -13.599 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.82 | Destabilizing | 0.6 | 3.37 | Destabilizing | 3.10 | Destabilizing | 2.16 | Destabilizing | 0.533 | Likely Pathogenic | -7.43 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.76 | Pathogenic | 0.01 | Affected | 0.2255 | 0.0499 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.89A>T | H30L 2D AIThe SynGAP1 H30L missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.570702 | Disordered | 0.438063 | Uncertain | 0.373 | 0.883 | 0.250 | -2.073 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.163 | Likely Benign | -2.88 | Deleterious | 0.462 | Possibly Damaging | 0.599 | Possibly Damaging | 3.94 | Benign | 0.00 | Affected | 0.1523 | 0.5793 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.907G>A | G303R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G303R is catalogued in gnomAD (6-33437812-G-A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome; the remaining tools (FoldX, Foldetta, premPS, ESM1b) are inconclusive. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, while Foldetta remains uncertain. Taken together, the majority of evidence points to a benign effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.450668 | Structured | 0.271087 | Uncertain | 0.630 | 0.254 | 0.250 | 6-33437812-G-A | 1 | 6.20e-7 | -7.493 | In-Between | 0.724 | Likely Pathogenic | Likely Benign | 1.44 | Ambiguous | 0.4 | 0.23 | Likely Benign | 0.84 | Ambiguous | 0.73 | Ambiguous | 0.048 | Likely Benign | -1.38 | Neutral | 0.001 | Benign | 0.003 | Benign | 3.99 | Benign | 0.06 | Tolerated | 3.55 | 18 | 0.0839 | 0.4490 | -2 | -3 | -4.1 | 99.14 | |||||||||||||||||||||||||
| c.907G>C | G303R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G303R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome. The remaining tools—FoldX, Foldetta, premPS, and ESM1b—return uncertain or inconclusive results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign, while Foldetta remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.450668 | Structured | 0.271087 | Uncertain | 0.630 | 0.254 | 0.250 | -7.493 | In-Between | 0.724 | Likely Pathogenic | Likely Benign | 1.44 | Ambiguous | 0.4 | 0.23 | Likely Benign | 0.84 | Ambiguous | 0.73 | Ambiguous | 0.048 | Likely Benign | -1.38 | Neutral | 0.001 | Benign | 0.003 | Benign | 3.99 | Benign | 0.06 | Tolerated | 3.55 | 18 | 0.0839 | 0.4490 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||||||
| c.908G>A | G303E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G303E is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33437813‑G‑A). Across the available in‑silico predictors, benign calls are made by REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls are made by SIFT and ESM1b; the remaining tools (FoldX, Foldetta, premPS, AlphaMissense‑Default) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, and Foldetta remains uncertain. Taken together, the majority of evidence points to a benign effect; this conclusion is not contradicted by any ClinVar annotation, as no pathogenic classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.450668 | Structured | 0.271087 | Uncertain | 0.630 | 0.254 | 0.250 | 6-33437813-G-A | 3 | 1.86e-6 | -9.339 | Likely Pathogenic | 0.549 | Ambiguous | Likely Benign | 1.87 | Ambiguous | 0.5 | 0.37 | Likely Benign | 1.12 | Ambiguous | 0.89 | Ambiguous | 0.063 | Likely Benign | -1.56 | Neutral | 0.001 | Benign | 0.005 | Benign | 4.04 | Benign | 0.05 | Affected | 3.55 | 18 | 0.1155 | 0.4172 | -2 | 0 | -3.1 | 72.06 | |||||||||||||||||||||||||
| c.911A>T | D304V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D304V missense variant is not reported in ClinVar (ClinVar status: None) and has no entry in gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include Rosetta and premPS, while the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain or inconclusive results come from FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for D304V. This conclusion is not contradicted by ClinVar, which currently has no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.352862 | Structured | 0.285053 | Uncertain | 0.764 | 0.271 | 0.250 | -8.280 | Likely Pathogenic | 0.803 | Likely Pathogenic | Ambiguous | 1.35 | Ambiguous | 0.3 | 0.14 | Likely Benign | 0.75 | Ambiguous | 0.35 | Likely Benign | 0.541 | Likely Pathogenic | -6.22 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.81 | Pathogenic | 0.00 | Affected | 0.0971 | 0.6999 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.917T>A | V306D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V306D is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All available in‑silico predictors that were evaluated return a pathogenic or likely‑pathogenic assessment: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a benign effect. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Based on the unanimous pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.363090 | Structured | 0.315026 | Uncertain | 0.896 | 0.287 | 0.125 | Uncertain | 1 | -18.289 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 4.40 | Destabilizing | 0.3 | 4.29 | Destabilizing | 4.35 | Destabilizing | 2.44 | Destabilizing | 0.530 | Likely Pathogenic | -5.44 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.74 | Pathogenic | 0.00 | Affected | 3.38 | 19 | 0.1317 | 0.0768 | -2 | -3 | -7.7 | 15.96 | 212.3 | -18.3 | -0.2 | 0.4 | 0.0 | 0.2 | X | X | X | Potentially Pathogenic | The isopropyl group of Val396, located at the beginning of an anti-parallel β sheet strand (res. Thr305-Asn315), packs against multiple hydrophobic residues (e.g., Leu274, Trp308, Ala271) in the WT simulations. However, in the variant simulations, the negatively charged carboxylate group of the Asp306 side chain is not suitable for this hydrophobic niche. Consequently, the side chain moves out to interact with Ser300 in the β strand (res. Met289-Arg299) and the guanidinium group of Arg299 in the β hairpin loop.In the third simulation, the residue swap disrupts the C2 domain secondary structure and tertiary assembly to a large degree when the amino group of the Lys297 side chain rotates to form a salt bridge with Asp306. This drastic effect could potentially reflect the challenge presented by the residue swap during the C2 domain folding. Because the residue swap affects the C2 domain structure, the SynGAP-membrane association could also be impacted. However, this is beyond the scope of the solvent-only simulations to unravel. | ||||||||||||||
| c.923G>C | W308S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W308S is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. No tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates pathogenicity; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic effect. No predictions or stability results are missing or inconclusive. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.333942 | Uncertain | 0.907 | 0.314 | 0.125 | -15.425 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 6.42 | Destabilizing | 0.2 | 5.31 | Destabilizing | 5.87 | Destabilizing | 1.93 | Destabilizing | 0.861 | Likely Pathogenic | -12.87 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 0.48 | Pathogenic | 0.00 | Affected | 0.5089 | 0.1406 | Weaken | -2 | -3 | 0.1 | -99.14 | ||||||||||||||||||||||||||||
| c.923G>T | W308L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W308L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates “Likely Pathogenic.” Only Rosetta and premPS yield uncertain results, which are treated as unavailable evidence. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No tools predict benign effects. **Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available).** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.328603 | Structured | 0.333942 | Uncertain | 0.907 | 0.314 | 0.125 | -13.349 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 3.53 | Destabilizing | 0.5 | 1.52 | Ambiguous | 2.53 | Destabilizing | 0.53 | Ambiguous | 0.811 | Likely Pathogenic | -11.95 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 0.49 | Pathogenic | 0.00 | Affected | 0.2731 | 0.2754 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||
| c.929A>G | E310G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E310G is listed in ClinVar as Pathogenic (ClinVar ID 2732842.0) and is not reported in gnomAD. Prediction tools that assess the variant’s effect largely agree on a deleterious outcome: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while only premPS predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Consequently, the variant is most likely pathogenic, and this prediction is consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.222385 | Structured | 0.346136 | Uncertain | 0.914 | 0.337 | 0.125 | Pathogenic | 1 | -14.132 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 2.38 | Destabilizing | 0.7 | 3.56 | Destabilizing | 2.97 | Destabilizing | 0.36 | Likely Benign | 0.848 | Likely Pathogenic | -6.43 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 1.12 | Pathogenic | 0.00 | Affected | 3.38 | 19 | 0.2736 | 0.6932 | -2 | 0 | 3.1 | -72.06 | |||||||||||||||||||||||||
| c.929A>T | E310V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E310V missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are Rosetta and premPS, while the majority of tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. FoldX and Foldetta are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence indicates that E310V is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.222385 | Structured | 0.346136 | Uncertain | 0.914 | 0.337 | 0.125 | -15.494 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 1.08 | Ambiguous | 0.9 | 0.06 | Likely Benign | 0.57 | Ambiguous | 0.14 | Likely Benign | 0.871 | Likely Pathogenic | -6.43 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.12 | Pathogenic | 0.00 | Affected | 0.0849 | 0.8729 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.932A>T | H311L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H311L missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include AlphaMissense‑Optimized, Foldetta, premPS, and Rosetta. Tools that predict a pathogenic outcome are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labeling it likely pathogenic, and Foldetta predicting a benign effect. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.229226 | Structured | 0.354792 | Uncertain | 0.902 | 0.314 | 0.125 | -10.119 | Likely Pathogenic | 0.575 | Likely Pathogenic | Likely Benign | -0.53 | Ambiguous | 0.0 | -0.04 | Likely Benign | -0.29 | Likely Benign | 0.43 | Likely Benign | 0.663 | Likely Pathogenic | -7.99 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.87 | Pathogenic | 0.02 | Affected | 0.0961 | 0.5292 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.938A>T | E313V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E313V missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, and premPS, whereas a majority of tools (REVEL, SIFT, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic impact. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the preponderance of evidence points to a pathogenic effect for E313V. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.170161 | Structured | 0.366526 | Uncertain | 0.898 | 0.304 | 0.125 | -13.169 | Likely Pathogenic | 0.821 | Likely Pathogenic | Ambiguous | 0.34 | Likely Benign | 0.1 | -0.02 | Likely Benign | 0.16 | Likely Benign | 0.21 | Likely Benign | 0.655 | Likely Pathogenic | -5.73 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.93 | Pathogenic | 0.05 | Affected | 0.1132 | 0.7900 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.943A>T | N315Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N315Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. The high‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the balance of evidence slightly favors a pathogenic interpretation, with six pathogenic‑predicted tools versus five benign‑predicted tools, and the high‑accuracy consensus leaning pathogenic. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -8.303 | Likely Pathogenic | 0.339 | Likely Benign | Likely Benign | -1.03 | Ambiguous | 0.5 | -0.85 | Ambiguous | -0.94 | Ambiguous | -0.10 | Likely Benign | 0.420 | Likely Benign | -3.81 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.93 | Pathogenic | 1.00 | Tolerated | 0.0613 | 0.6732 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.944A>T | N315I 2D 3DClick to see structure in 3D Viewer AISynGAP1 variant N315I is not reported in ClinVar and is absent from gnomAD. In silico predictors that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, and AlphaMissense‑Optimized. Predictors that agree on a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. The high‑accuracy AlphaMissense‑Optimized score is benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; Foldetta, which integrates FoldX‑MD (uncertain) and Rosetta (benign), is considered unavailable. Overall, the balance of evidence leans toward pathogenicity, and this assessment does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.118441 | Structured | 0.379740 | Uncertain | 0.862 | 0.253 | 0.125 | -9.666 | Likely Pathogenic | 0.500 | Ambiguous | Likely Benign | -0.72 | Ambiguous | 0.4 | -0.17 | Likely Benign | -0.45 | Likely Benign | 0.36 | Likely Benign | 0.496 | Likely Benign | -5.19 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.90 | Pathogenic | 0.43 | Tolerated | 0.0763 | 0.7235 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.946A>T | N316Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N316Y is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labeling it likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicting a benign impact. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.118441 | Structured | 0.385187 | Uncertain | 0.817 | 0.246 | 0.125 | -11.226 | Likely Pathogenic | 0.767 | Likely Pathogenic | Likely Benign | 0.53 | Ambiguous | 0.5 | -0.09 | Likely Benign | 0.22 | Likely Benign | 0.50 | Likely Benign | 0.454 | Likely Benign | -5.70 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.81 | Pathogenic | 0.01 | Affected | 0.0574 | 0.6971 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.947A>T | N316I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N316I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and premPS, whereas the majority of tools predict a pathogenic impact: FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. No prediction is missing or inconclusive beyond the uncertain AlphaMissense‑Optimized result. Based on the preponderance of pathogenic predictions and the high‑accuracy tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.118441 | Structured | 0.385187 | Uncertain | 0.817 | 0.246 | 0.125 | -11.164 | Likely Pathogenic | 0.899 | Likely Pathogenic | Ambiguous | 2.74 | Destabilizing | 0.2 | 4.10 | Destabilizing | 3.42 | Destabilizing | 0.18 | Likely Benign | 0.318 | Likely Benign | -6.37 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.00 | Pathogenic | 0.03 | Affected | 0.0734 | 0.7422 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.950T>A | L317Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L317Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on these predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.106997 | Structured | 0.394031 | Uncertain | 0.874 | 0.240 | 0.125 | -13.424 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 2.87 | Destabilizing | 0.2 | 2.47 | Destabilizing | 2.67 | Destabilizing | 1.61 | Destabilizing | 0.607 | Likely Pathogenic | -5.52 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.75 | Pathogenic | 0.00 | Affected | 0.1252 | 0.1251 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.959T>A | V320D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V320D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL and SIFT; pathogenic predictions from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Stability‑based methods FoldX and Rosetta returned uncertain results, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also yielded an uncertain prediction. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points toward a pathogenic effect, which is consistent with the lack of ClinVar annotation and gnomAD absence. Thus, the variant is most likely pathogenic, and this prediction does not contradict ClinVar status because ClinVar has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.185198 | Structured | 0.419626 | Uncertain | 0.905 | 0.266 | 0.125 | -11.269 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.99 | Ambiguous | 1.0 | 1.67 | Ambiguous | 1.83 | Ambiguous | 1.50 | Destabilizing | 0.405 | Likely Benign | -5.58 | Deleterious | 0.999 | Probably Damaging | 0.972 | Probably Damaging | 1.93 | Pathogenic | 0.07 | Tolerated | 0.1101 | 0.0482 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.968T>A | L323Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L323Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify it as pathogenic. Benign predictions are absent; all evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—return pathogenic or likely pathogenic calls. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports pathogenic. Consequently, the variant is most likely pathogenic, with no conflict with ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.268042 | Structured | 0.428564 | Uncertain | 0.956 | 0.369 | 0.000 | -14.487 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 3.19 | Destabilizing | 0.2 | 3.28 | Destabilizing | 3.24 | Destabilizing | 1.85 | Destabilizing | 0.728 | Likely Pathogenic | -4.54 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 0.59 | Pathogenic | 0.00 | Affected | 0.0915 | 0.0758 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.971G>T | R324L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R324L is catalogued in gnomAD (6-33437876‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, and SIFT; pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome, and Foldetta (integrating FoldX‑MD and Rosetta outputs) reports a benign stability change. Overall, the majority of evidence points toward a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.257454 | Structured | 0.426893 | Uncertain | 0.954 | 0.397 | 0.000 | 6-33437876-G-T | 1 | 6.20e-7 | -10.328 | Likely Pathogenic | 0.575 | Likely Pathogenic | Likely Benign | -0.28 | Likely Benign | 0.0 | -0.08 | Likely Benign | -0.18 | Likely Benign | 0.29 | Likely Benign | 0.489 | Likely Benign | -2.20 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.86 | Pathogenic | 0.63 | Tolerated | 3.39 | 22 | 0.2310 | 0.5607 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||
| c.974T>A | L325Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L325Q is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify it as pathogenic. Benign predictions are absent; all evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—return pathogenic or likely pathogenic calls. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports pathogenic. Consequently, the variant is most likely pathogenic, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.352862 | Structured | 0.424577 | Uncertain | 0.955 | 0.436 | 0.000 | -17.005 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.23 | Destabilizing | 0.0 | 2.68 | Destabilizing | 2.96 | Destabilizing | 2.02 | Destabilizing | 0.547 | Likely Pathogenic | -3.97 | Deleterious | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 1.33 | Pathogenic | 0.00 | Affected | 0.1235 | 0.1405 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.977A>T | H326L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 H326L missense variant is not reported in ClinVar (status: None) and has no entry in gnomAD. Prediction tools that agree on a benign effect include SIFT, premPS, Rosetta, and Foldetta. Tools that agree on a pathogenic effect include REVEL, SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic; and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) predicts benign. No evidence is available from FoldX or AlphaMissense‑Optimized to support either outcome. Overall, the majority of predictions (nine pathogenic vs. four benign) indicate that H326L is most likely pathogenic, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.342579 | Structured | 0.418150 | Uncertain | 0.944 | 0.455 | 0.000 | -10.421 | Likely Pathogenic | 0.888 | Likely Pathogenic | Ambiguous | -0.85 | Ambiguous | 0.2 | 0.36 | Likely Benign | -0.25 | Likely Benign | 0.44 | Likely Benign | 0.627 | Likely Pathogenic | -9.64 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 1.95 | Pathogenic | 0.08 | Tolerated | 0.0992 | 0.5799 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||
| c.980T>A | L327Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L327Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a pathogenic or likely pathogenic outcome. No tool in the dataset predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is pathogenic. With all available evidence pointing to a harmful impact and no ClinVar entry to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.335645 | Structured | 0.409189 | Uncertain | 0.939 | 0.490 | 0.000 | -14.243 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.03 | Destabilizing | 0.1 | 2.17 | Destabilizing | 2.60 | Destabilizing | 2.11 | Destabilizing | 0.605 | Likely Pathogenic | -5.26 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.52 | Pathogenic | 0.00 | Affected | 0.1131 | 0.1042 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.982T>A | Y328N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y328N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the consensus of all available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.411940 | Structured | 0.392519 | Uncertain | 0.916 | 0.497 | 0.000 | -13.778 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.72 | Destabilizing | 0.1 | 3.88 | Destabilizing | 3.30 | Destabilizing | 2.06 | Destabilizing | 0.548 | Likely Pathogenic | -8.07 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.53 | Pathogenic | 0.01 | Affected | 0.2374 | 0.0703 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.989A>T | D330V 2D 3DClick to see structure in 3D Viewer AISynGAP1 D330V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL and premPS, whereas a majority of tools predict a pathogenic impact: polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, and AlphaMissense‑Default. Uncertain predictions from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are treated as unavailable. High‑accuracy assessments show that the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. AlphaMissense‑Optimized and Foldetta provide inconclusive results and are therefore not considered evidence. Overall, the preponderance of evidence points to a pathogenic effect for D330V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.380708 | Structured | 0.360008 | Uncertain | 0.805 | 0.488 | 0.250 | -13.176 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 2.06 | Destabilizing | 0.5 | 0.57 | Ambiguous | 1.32 | Ambiguous | 0.45 | Likely Benign | 0.428 | Likely Benign | -6.40 | Deleterious | 0.994 | Probably Damaging | 0.892 | Possibly Damaging | 0.89 | Pathogenic | 0.01 | Affected | 0.0862 | 0.4633 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.98A>T | Q33L 2D AIThe SynGAP1 missense variant Q33L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.707965 | Disordered | 0.436712 | Uncertain | 0.342 | 0.860 | 0.375 | 0.253 | Likely Benign | 0.174 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -1.24 | Neutral | 0.084 | Benign | 0.033 | Benign | 4.18 | Benign | 0.00 | Affected | 0.1127 | 0.6214 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||||||||||||
| c.995A>T | D332V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D332V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Foldetta, and premPS, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Uncertain results come from FoldX, Rosetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the balance of evidence favors a pathogenic classification; this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.339168 | Structured | 0.336528 | Uncertain | 0.537 | 0.445 | 0.375 | -13.710 | Likely Pathogenic | 0.933 | Likely Pathogenic | Ambiguous | 1.39 | Ambiguous | 0.1 | -0.52 | Ambiguous | 0.44 | Likely Benign | 0.50 | Likely Benign | 0.484 | Likely Benign | -7.27 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.21 | Pathogenic | 0.03 | Affected | 0.0573 | 0.4132 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.998A>T | K333I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K333I missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX and premPS, whereas the majority of tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; Rosetta and Foldetta give uncertain results. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as Likely Pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.311707 | Structured | 0.330781 | Uncertain | 0.537 | 0.447 | 0.500 | -14.517 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 0.30 | Likely Benign | 0.0 | 0.95 | Ambiguous | 0.63 | Ambiguous | 0.43 | Likely Benign | 0.544 | Likely Pathogenic | -6.49 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.89 | Pathogenic | 0.03 | Affected | 0.0931 | 0.3521 | -2 | -3 | 8.4 | -15.01 | |||||||||||||||||||||||||||||
| c.1022G>A | G341D 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant G341D is not reported in ClinVar (ClinVar ID: None) but is present in gnomAD (ID: 6-33437927‑G‑A). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, FATHMM, and AlphaMissense‑Default. The remaining tools—Foldetta, AlphaMissense‑Optimized, ESM1b, and Rosetta—return uncertain or inconclusive results and are treated as unavailable for pathogenicity inference. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Considering the majority of standard tools lean benign but the high‑accuracy consensus indicates pathogenicity, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | 6-33437927-G-A | 6 | 3.72e-6 | -7.402 | In-Between | 0.871 | Likely Pathogenic | Ambiguous | 0.28 | Likely Benign | 0.1 | -1.32 | Ambiguous | -0.52 | Ambiguous | -0.04 | Likely Benign | 0.295 | Likely Benign | -0.11 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 0.34 | Pathogenic | 0.25 | Tolerated | 3.42 | 13 | 0.1727 | 0.2241 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||
| c.1022G>T | G341V 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant G341V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, polyPhen‑2 HumVar, and the SGM‑Consensus score (derived from a majority of benign calls among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as uncertain. No evidence from FoldX or premPS is available. Overall, the majority of predictions support a benign classification, and this is consistent with the lack of ClinVar annotation. Therefore, the variant is most likely benign and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.356642 | Structured | 0.431164 | Uncertain | 0.745 | 0.479 | 0.250 | -5.371 | Likely Benign | 0.247 | Likely Benign | Likely Benign | 0.86 | Ambiguous | 0.3 | -2.24 | Stabilizing | -0.69 | Ambiguous | -0.50 | Ambiguous | 0.459 | Likely Benign | -2.29 | Neutral | 0.801 | Possibly Damaging | 0.192 | Benign | 0.42 | Pathogenic | 0.05 | Affected | 0.1019 | 0.3649 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1027G>T | V343F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions are made by REVEL and premPS, whereas pathogenic predictions are made by SIFT, polyPhen‑2 (HumDiv and HumVar), PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain; Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also uncertain. No evidence from FoldX or Rosetta alone is available. Overall, the majority of predictions support a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -10.709 | Likely Pathogenic | 0.799 | Likely Pathogenic | Ambiguous | 1.51 | Ambiguous | 0.4 | 1.28 | Ambiguous | 1.40 | Ambiguous | 0.23 | Likely Benign | 0.324 | Likely Benign | -3.37 | Deleterious | 0.976 | Probably Damaging | 0.759 | Possibly Damaging | 1.61 | Pathogenic | 0.01 | Affected | 0.0915 | 0.4552 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1028T>G | V343G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—classify the variant as pathogenic. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Thus, the preponderance of evidence indicates that V343G is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.291804 | Structured | 0.383911 | Uncertain | 0.882 | 0.497 | 0.250 | -11.332 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 2.49 | Destabilizing | 0.1 | 4.40 | Destabilizing | 3.45 | Destabilizing | 1.68 | Destabilizing | 0.421 | Likely Benign | -5.84 | Deleterious | 0.898 | Possibly Damaging | 0.996 | Probably Damaging | 1.60 | Pathogenic | 0.00 | Affected | 0.1982 | 0.3341 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1031G>T | G344V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G344V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while the only inconclusive result is premPS, which is listed as uncertain. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic status; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports pathogenic. Consequently, the variant is most likely pathogenic, and this prediction is consistent with the absence of a ClinVar entry (no contradiction). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.264545 | Structured | 0.368110 | Uncertain | 0.913 | 0.485 | 0.250 | -14.913 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 13.33 | Destabilizing | 1.5 | 14.51 | Destabilizing | 13.92 | Destabilizing | 0.53 | Ambiguous | 0.889 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -0.48 | Pathogenic | 0.03 | Affected | 0.1244 | 0.4744 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1037T>G | V346G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V346G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). All available in‑silico predictors classify it as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a pathogenic verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports pathogenic. With all evidence converging on a deleterious impact and no ClinVar annotation to contradict, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.260850 | Structured | 0.350921 | Uncertain | 0.949 | 0.461 | 0.000 | -12.779 | Likely Pathogenic | 0.969 | Likely Pathogenic | Likely Pathogenic | 4.24 | Destabilizing | 0.2 | 4.62 | Destabilizing | 4.43 | Destabilizing | 2.15 | Destabilizing | 0.667 | Likely Pathogenic | -6.03 | Deleterious | 0.991 | Probably Damaging | 0.999 | Probably Damaging | 1.50 | Pathogenic | 0.00 | Affected | 0.2378 | 0.2522 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.103G>T | V35F 2D AIThe SynGAP1 missense variant V35F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact for V35F, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -4.114 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.133 | Likely Benign | -0.79 | Neutral | 0.923 | Possibly Damaging | 0.865 | Possibly Damaging | 4.14 | Benign | 0.00 | Affected | 0.0765 | 0.3421 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.1043T>G | V348G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V348G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to REVEL, which scores the variant as benign. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods reinforce this assessment: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates a pathogenic effect. No prediction or stability result is missing or inconclusive. Based on the overwhelming agreement among pathogenic predictions and the lack of a benign consensus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.170161 | Structured | 0.346556 | Uncertain | 0.951 | 0.414 | 0.000 | -12.793 | Likely Pathogenic | 0.964 | Likely Pathogenic | Likely Pathogenic | 3.97 | Destabilizing | 0.2 | 5.70 | Destabilizing | 4.84 | Destabilizing | 2.23 | Destabilizing | 0.419 | Likely Benign | -6.18 | Deleterious | 0.637 | Possibly Damaging | 0.989 | Probably Damaging | 1.56 | Pathogenic | 0.00 | Affected | 0.2229 | 0.2082 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1049T>G | V350G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V350G lies in the C2 domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that classify it as benign include REVEL, polyPhen‑2 HumDiv, and AlphaMissense‑Optimized. The remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—predict it to be pathogenic. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All predictions are available and consistent. Based on the preponderance of pathogenic calls, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.179055 | Structured | 0.353227 | Uncertain | 0.931 | 0.371 | 0.000 | -12.267 | Likely Pathogenic | 0.597 | Likely Pathogenic | Likely Benign | 2.57 | Destabilizing | 0.0 | 4.25 | Destabilizing | 3.41 | Destabilizing | 1.93 | Destabilizing | 0.330 | Likely Benign | -5.46 | Deleterious | 0.435 | Benign | 0.920 | Probably Damaging | 1.61 | Pathogenic | 0.02 | Affected | 0.2316 | 0.2522 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.104T>G | V35G 2D AIThe SynGAP1 missense variant V35G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus also predicts benign; Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because the variant is not currently catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.613573 | Disordered | 0.434838 | Uncertain | 0.360 | 0.851 | 0.375 | -3.614 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.143 | Likely Benign | 0.66 | Neutral | 0.693 | Possibly Damaging | 0.824 | Possibly Damaging | 4.41 | Benign | 0.00 | Affected | 0.1514 | 0.2618 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.1064G>T | G355V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G355V is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain or unavailable results are reported for FoldX, premPS, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, whereas the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic effect. Foldetta’s stability prediction is unavailable. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.155435 | Structured | 0.388832 | Uncertain | 0.810 | 0.354 | 0.125 | -10.111 | Likely Pathogenic | 0.675 | Likely Pathogenic | Likely Benign | 1.78 | Ambiguous | 0.6 | 0.14 | Likely Benign | 0.96 | Ambiguous | 0.53 | Ambiguous | 0.346 | Likely Benign | -7.59 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.78 | Pathogenic | 0.04 | Affected | 0.1221 | 0.4685 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1072T>G | F358V 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant F358V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL, SIFT, and FATHMM; pathogenic predictions from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and ESM1b. Five tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is also uncertain. Overall, the preponderance of evidence points to a pathogenic effect for F358V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.222385 | Structured | 0.407113 | Uncertain | 0.912 | 0.441 | 0.250 | -9.021 | Likely Pathogenic | 0.847 | Likely Pathogenic | Ambiguous | 1.42 | Ambiguous | 0.2 | 1.68 | Ambiguous | 1.55 | Ambiguous | 0.93 | Ambiguous | 0.408 | Likely Benign | -5.32 | Deleterious | 0.993 | Probably Damaging | 0.968 | Probably Damaging | 4.09 | Benign | 0.18 | Tolerated | 0.2222 | 0.2978 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1076C>G | T359R 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant T359R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on benign effects include REVEL, Rosetta, Foldetta, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and FATHMM. Uncertain results come from FoldX, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, more tools favor a benign outcome, but a significant minority predict pathogenicity, leaving the variant’s clinical significance unresolved. The variant is most likely benign based on the prevailing predictions, and this does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.281712 | Structured | 0.414952 | Uncertain | 0.939 | 0.480 | 0.250 | -8.675 | Likely Pathogenic | 0.402 | Ambiguous | Likely Benign | -0.74 | Ambiguous | 0.1 | -0.20 | Likely Benign | -0.47 | Likely Benign | 0.54 | Ambiguous | 0.157 | Likely Benign | -2.86 | Deleterious | 0.627 | Possibly Damaging | 0.091 | Benign | 1.75 | Pathogenic | 0.23 | Tolerated | 0.1323 | 0.3748 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.1076C>T | T359I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T359I is reported in gnomAD (variant ID 6‑33437981‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. Uncertain results are reported for FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as Uncertain. Overall, the majority of evidence points to a benign effect. There is no ClinVar status to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.281712 | Structured | 0.414952 | Uncertain | 0.939 | 0.480 | 0.250 | 6-33437981-C-T | 1 | 6.22e-7 | -2.594 | Likely Benign | 0.181 | Likely Benign | Likely Benign | -0.66 | Ambiguous | 0.1 | -0.64 | Ambiguous | -0.65 | Ambiguous | -0.63 | Ambiguous | 0.085 | Likely Benign | 0.77 | Neutral | 0.070 | Benign | 0.006 | Benign | 1.80 | Pathogenic | 0.23 | Tolerated | 3.38 | 24 | 0.1123 | 0.5921 | -1 | 0 | 5.2 | 12.05 | ||||||||||||||||||||||||
| c.1082A>C | Q361P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q361P (ClinVar ID 3235087) is listed as Pathogenic and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL and premPS, whereas the remaining tools—FoldX, Rosetta, Foldetta, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, PROVEAN, polyPhen‑2 (HumDiv and HumVar)—all predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming majority of pathogenic predictions, the variant is most likely pathogenic, and this conclusion aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.305330 | Structured | 0.427593 | Uncertain | 0.945 | 0.534 | 0.250 | Likely Pathogenic | 2 | -13.280 | Likely Pathogenic | 0.956 | Likely Pathogenic | Likely Pathogenic | 3.12 | Destabilizing | 0.0 | 3.45 | Destabilizing | 3.29 | Destabilizing | 0.38 | Likely Benign | 0.482 | Likely Benign | -3.03 | Deleterious | 0.996 | Probably Damaging | 0.979 | Probably Damaging | 1.63 | Pathogenic | 0.05 | Affected | 3.37 | 25 | 0.1986 | 0.5151 | -1 | 0 | 1.9 | -31.01 | |||||||||||||||||||||||||
| c.1090C>T | P364S 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant P364S resides in the C2 domain. It is not reported in ClinVar and is present in gnomAD (ID 6‑33437995‑C‑T). Prediction tools that classify the variant as benign include REVEL, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM; the SGM‑Consensus score is also labeled Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. No prediction or stability result is missing or inconclusive beyond the Uncertain labels. Based on the majority of evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.390993 | Structured | 0.439474 | Uncertain | 0.942 | 0.590 | 0.250 | 6-33437995-C-T | 1 | 6.20e-7 | -8.318 | Likely Pathogenic | 0.214 | Likely Benign | Likely Benign | 1.34 | Ambiguous | 0.3 | 0.68 | Ambiguous | 1.01 | Ambiguous | 0.83 | Ambiguous | 0.307 | Likely Benign | -5.98 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 1.62 | Pathogenic | 0.05 | Affected | 3.39 | 20 | 0.3390 | 0.5512 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||
| c.1094T>G | V365G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V365G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity unanimously classify the variant as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign effect. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. All available evidence points to a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.414856 | Structured | 0.441505 | Uncertain | 0.923 | 0.608 | 0.250 | -13.020 | Likely Pathogenic | 0.944 | Likely Pathogenic | Ambiguous | 4.18 | Destabilizing | 0.2 | 4.83 | Destabilizing | 4.51 | Destabilizing | 2.18 | Destabilizing | 0.576 | Likely Pathogenic | -5.88 | Deleterious | 0.688 | Possibly Damaging | 0.989 | Probably Damaging | 1.66 | Pathogenic | 0.00 | Affected | 0.1835 | 0.2717 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1097C>T | T366I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T366I is reported in gnomAD (6‑33438002‑C‑T) and has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. No other tools provide conclusive evidence for pathogenicity. **Based on the aggregate predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none available).** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.414856 | Structured | 0.441902 | Uncertain | 0.897 | 0.642 | 0.250 | 6-33438002-C-T | 1 | 6.20e-7 | -4.921 | Likely Benign | 0.279 | Likely Benign | Likely Benign | -0.62 | Ambiguous | 0.1 | -0.31 | Likely Benign | -0.47 | Likely Benign | -0.14 | Likely Benign | 0.058 | Likely Benign | -1.22 | Neutral | 0.002 | Benign | 0.001 | Benign | 1.77 | Pathogenic | 0.26 | Tolerated | 3.38 | 23 | 0.1062 | 0.6215 | -1 | 0 | 5.2 | 12.05 | ||||||||||||||||||||||||
| c.1106C>G | T369R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T369R is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33438011‑C‑G). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FATHMM and AlphaMissense‑Default. Rosetta and Foldetta are uncertain, so their results are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta is also uncertain. Overall, the majority of evidence (nine benign vs two pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.468512 | Structured | 0.437011 | Uncertain | 0.417 | 0.707 | 0.500 | 6-33438011-C-G | 3 | 1.93e-6 | -6.772 | Likely Benign | 0.571 | Likely Pathogenic | Likely Benign | -0.27 | Likely Benign | 0.1 | 1.48 | Ambiguous | 0.61 | Ambiguous | 0.29 | Likely Benign | 0.148 | Likely Benign | -2.15 | Neutral | 0.244 | Benign | 0.107 | Benign | 1.72 | Pathogenic | 0.32 | Tolerated | 3.42 | 19 | 0.1217 | 0.3737 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||
| c.1109G>T | G370V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G370V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster into two groups: benign (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic (FoldX, Rosetta, Foldetta, and FATHMM). High‑accuracy assessments further refine this picture: AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic outcome. Overall, the majority of evidence points toward a benign impact, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.461924 | Structured | 0.434325 | Uncertain | 0.359 | 0.720 | 0.500 | -5.328 | Likely Benign | 0.094 | Likely Benign | Likely Benign | 4.98 | Destabilizing | 3.8 | 5.61 | Destabilizing | 5.30 | Destabilizing | -0.43 | Likely Benign | 0.427 | Likely Benign | 0.03 | Neutral | 0.000 | Benign | 0.000 | Benign | 1.32 | Pathogenic | 0.29 | Tolerated | 0.1306 | 0.4198 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1111A>C | S371R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change S371R is catalogued in gnomAD (ID 6‑33438016‑A‑C) but has no ClinVar entry. Functional prediction programs largely agree on a benign effect: REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, Foldetta, and the SGM‑Consensus score (Likely Benign) all report a non‑pathogenic outcome. Only AlphaMissense‑Default predicts a pathogenic effect, while FoldX and premPS are inconclusive. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta) is benign. Taken together, the majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic report. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | 6-33438016-A-C | -6.415 | Likely Benign | 0.762 | Likely Pathogenic | Likely Benign | 0.51 | Ambiguous | 1.2 | -0.25 | Likely Benign | 0.13 | Likely Benign | 0.57 | Ambiguous | 0.295 | Likely Benign | -1.17 | Neutral | 0.396 | Benign | 0.099 | Benign | 5.35 | Benign | 0.26 | Tolerated | 3.52 | 18 | 0.1362 | 0.4131 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||
| c.1112G>T | S371I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S371I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from FoldX and Rosetta, which are treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a benign effect. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | -6.888 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.50 | Ambiguous | 0.4 | -0.50 | Ambiguous | 0.00 | Likely Benign | -0.11 | Likely Benign | 0.433 | Likely Benign | -1.06 | Neutral | 0.028 | Benign | 0.016 | Benign | 4.62 | Benign | 0.07 | Tolerated | 0.1423 | 0.5924 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.1113T>A | S371R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S371R is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus, REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the substitution as benign, while AlphaMissense‑Optimized also predicts benign. Only AlphaMissense‑Default indicates a pathogenic outcome; FoldX and premPS are inconclusive. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Overall, the majority of evidence supports a benign impact, and this is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | -6.415 | Likely Benign | 0.762 | Likely Pathogenic | Likely Benign | 0.51 | Ambiguous | 1.2 | -0.25 | Likely Benign | 0.13 | Likely Benign | 0.57 | Ambiguous | 0.344 | Likely Benign | -1.17 | Neutral | 0.396 | Benign | 0.099 | Benign | 5.35 | Benign | 0.26 | Tolerated | 3.52 | 18 | 0.1362 | 0.4131 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||
| c.1113T>G | S371R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S371R is reported in gnomAD (variant ID 6‑33438018‑T‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only AlphaMissense‑Default predicts a pathogenic outcome. The SGM‑Consensus, which is a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves as “Likely Benign” (3 benign vs. 1 pathogenic). High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM‑Consensus is benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No prediction or stability result is missing or inconclusive. Overall, the evidence strongly favors a benign classification, and this is consistent with the absence of a ClinVar pathogenic report. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.444081 | Structured | 0.432086 | Uncertain | 0.294 | 0.746 | 0.375 | 6-33438018-T-G | 1 | 1.18e-6 | -6.415 | Likely Benign | 0.762 | Likely Pathogenic | Likely Benign | 0.51 | Ambiguous | 1.2 | -0.25 | Likely Benign | 0.13 | Likely Benign | 0.57 | Ambiguous | 0.340 | Likely Benign | -1.17 | Neutral | 0.396 | Benign | 0.099 | Benign | 5.35 | Benign | 0.26 | Tolerated | 3.52 | 18 | 0.1362 | 0.4131 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||
| c.1118G>T | G373V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G373V is listed in ClinVar with an uncertain significance and is present in gnomAD (variant ID 6‑33438023‑G‑T). Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are FoldX, Foldetta, and SIFT, while Rosetta is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as pathogenic. Overall, the majority of predictions support a benign impact, and this consensus does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.529623 | Disordered | 0.429267 | Uncertain | 0.295 | 0.799 | 0.625 | Uncertain | 1 | 6-33438023-G-T | 6 | 5.03e-6 | -6.062 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 5.32 | Destabilizing | 3.2 | 0.82 | Ambiguous | 3.07 | Destabilizing | 0.09 | Likely Benign | 0.428 | Likely Benign | -0.98 | Neutral | 0.007 | Benign | 0.001 | Benign | 3.90 | Benign | 0.00 | Affected | 3.53 | 16 | 0.1424 | 0.4004 | -1 | -3 | 4.6 | 42.08 | 207.6 | -68.1 | 1.9 | 1.1 | -0.6 | 0.1 | Uncertain | Gly373 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364, res. Ala399-Ile411). Because the Ω loop is assumed to directly interact with the membrane, it moves arbitrarily throughout the WT solvent simulations. The Ω loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play major roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are observed in the variant simulations, Val373 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. However, since the effect on the Gly-rich Ω loop dynamics can only be studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1120T>C | S374P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S374P is reported in gnomAD (6‑33438025‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and Rosetta; FoldX and Foldetta are inconclusive. The high‑accuracy consensus (SGM‑Consensus) is “Likely Benign,” derived from the unanimous benign calls of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN. AlphaMissense‑Optimized also predicts benign, while Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact. There is no ClinVar status to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.642678 | Disordered | 0.428948 | Uncertain | 0.333 | 0.812 | 0.625 | 6-33438025-T-C | 1 | 7.85e-7 | -4.849 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.66 | Ambiguous | 0.6 | 2.22 | Destabilizing | 1.44 | Ambiguous | 0.34 | Likely Benign | 0.388 | Likely Benign | -0.89 | Neutral | 0.396 | Benign | 0.099 | Benign | 5.30 | Benign | 0.02 | Affected | 4.32 | 13 | 0.3012 | 0.6813 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||
| c.1121C>G | S374C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S374C is reported in gnomAD (6-33438026-C-G) and has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen2_HumVar all indicate benign. Only two tools (polyPhen2_HumDiv and SIFT) predict pathogenicity, while the consensus score SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM‑Consensus itself is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. No prediction or stability result is missing or inconclusive. Based on the aggregate evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.642678 | Disordered | 0.428948 | Uncertain | 0.333 | 0.812 | 0.625 | 6-33438026-C-G | -6.242 | Likely Benign | 0.106 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 0.79 | Ambiguous | 0.45 | Likely Benign | 0.08 | Likely Benign | 0.317 | Likely Benign | -0.99 | Neutral | 0.875 | Possibly Damaging | 0.430 | Benign | 5.30 | Benign | 0.00 | Affected | 4.32 | 13 | 0.1749 | 0.6584 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||
| c.1124G>T | G375V 2D AIThe SynGAP1 missense variant G375V is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions (premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score of Likely Benign) and pathogenic predictions (REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, and FATHMM). High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also benign, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a pathogenic effect. No prediction is missing or inconclusive. Overall, the majority of tools and the consensus score suggest a benign effect, but the Foldetta result introduces uncertainty. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.604312 | Disordered | 0.428340 | Uncertain | 0.301 | 0.836 | 0.625 | -6.149 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 3.93 | Destabilizing | 3.5 | 7.55 | Destabilizing | 5.74 | Destabilizing | 0.02 | Likely Benign | 0.547 | Likely Pathogenic | -0.92 | Neutral | 0.845 | Possibly Damaging | 0.186 | Benign | 1.32 | Pathogenic | 0.06 | Tolerated | 0.1653 | 0.4193 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1127G>T | G376V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G376V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from premPS, PROVEAN, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions arise from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The consensus score from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, matching the majority of individual benign calls. High‑accuracy methods give mixed results: AlphaMissense‑Optimized predicts benign, SGM Consensus predicts benign, whereas Foldetta (integrating FoldX‑MD and Rosetta) predicts pathogenic. No prediction is missing or inconclusive. Overall, the balance of evidence leans toward a benign effect; this is consistent with the lack of ClinVar annotation and gnomAD presence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.680603 | Disordered | 0.428979 | Uncertain | 0.326 | 0.869 | 0.625 | -6.242 | Likely Benign | 0.120 | Likely Benign | Likely Benign | 4.84 | Destabilizing | 0.8 | -0.81 | Ambiguous | 2.02 | Destabilizing | -0.18 | Likely Benign | 0.541 | Likely Pathogenic | -0.66 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1594 | 0.3525 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1130T>C | M377T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M377T is reported in gnomAD (variant ID 6‑33438035‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. Uncertain results are reported for FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign, and Foldetta as Uncertain. Overall, the majority of evidence points to a benign effect for M377T, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.675549 | Disordered | 0.431183 | Uncertain | 0.324 | 0.884 | 0.625 | 6-33438035-T-C | 5 | 1.17e-5 | -1.881 | Likely Benign | 0.090 | Likely Benign | Likely Benign | 0.90 | Ambiguous | 0.4 | 1.95 | Ambiguous | 1.43 | Ambiguous | 0.59 | Ambiguous | 0.245 | Likely Benign | -0.65 | Neutral | 0.000 | Benign | 0.002 | Benign | 5.47 | Benign | 0.05 | Affected | 4.32 | 12 | 0.3064 | 0.3267 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||
| c.1130T>G | M377R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M377R is present in gnomAD (6‑33438035‑T‑G) and has no ClinVar entry. Prediction tools that report a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are Rosetta and Foldetta. The high‑accuracy consensus (SGM‑Consensus) is “Likely Benign,” derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—all of which are benign. AlphaMissense‑Optimized also predicts benign, whereas Foldetta predicts pathogenic. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict the ClinVar status (which is currently absent). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.675549 | Disordered | 0.431183 | Uncertain | 0.324 | 0.884 | 0.625 | 6-33438035-T-G | -3.150 | Likely Benign | 0.219 | Likely Benign | Likely Benign | 0.49 | Likely Benign | 0.4 | 4.81 | Destabilizing | 2.65 | Destabilizing | 0.69 | Ambiguous | 0.471 | Likely Benign | -0.64 | Neutral | 0.004 | Benign | 0.009 | Benign | 5.46 | Benign | 0.18 | Tolerated | 4.32 | 12 | 0.2369 | 0.1918 | -1 | 0 | -6.4 | 24.99 | ||||||||||||||||||||||||||
| c.1133G>A | G378D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G378D is not reported in ClinVar (ClinVar ID: None) but is present in gnomAD (ID 6‑33438038‑G‑A). Prediction tools that classify the variant as benign include premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized. Tools that predict pathogenicity are REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. ESM1b is uncertain, and no other high‑accuracy predictions are available. Overall, the majority of evidence supports a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.767246 | Disordered | 0.432858 | Uncertain | 0.341 | 0.915 | 0.625 | 6-33438038-G-A | 1 | 6.97e-7 | -7.767 | In-Between | 0.576 | Likely Pathogenic | Likely Benign | 11.41 | Destabilizing | 5.0 | 11.84 | Destabilizing | 11.63 | Destabilizing | 0.50 | Likely Benign | 0.619 | Likely Pathogenic | -0.63 | Neutral | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 1.32 | Pathogenic | 0.08 | Tolerated | 4.32 | 12 | 0.2130 | 0.2035 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||
| c.1139G>T | G380V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G380V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions are reported by REVEL, FoldX, polyPhen‑2 HumDiv, and SIFT; Rosetta is uncertain. The SGM‑Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority of the four high‑accuracy tools) is benign, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a pathogenic impact. Overall, the majority of evidence points to a benign effect, and this does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.724957 | Disordered | 0.432982 | Uncertain | 0.316 | 0.939 | 0.750 | -6.234 | Likely Benign | 0.172 | Likely Benign | Likely Benign | 7.60 | Destabilizing | 2.5 | 1.75 | Ambiguous | 4.68 | Destabilizing | -0.17 | Likely Benign | 0.574 | Likely Pathogenic | -0.70 | Neutral | 0.816 | Possibly Damaging | 0.210 | Benign | 2.54 | Benign | 0.02 | Affected | 0.1618 | 0.3708 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1142G>T | G381V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G381V is listed in ClinVar with an uncertain significance (ClinVar ID 1940172.0) and is present in the gnomAD database (6‑33438047‑G‑T). Functional prediction tools that report a benign effect include premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, FoldX, Rosetta, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a majority‑benign vote and is reported as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of predictions lean toward a benign impact, and this is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.724957 | Disordered | 0.431692 | Uncertain | 0.301 | 0.951 | 0.750 | Uncertain | 1 | 6-33438047-G-T | 2 | 1.25e-6 | -5.967 | Likely Benign | 0.146 | Likely Benign | Likely Benign | 7.16 | Destabilizing | 1.0 | 4.10 | Destabilizing | 5.63 | Destabilizing | -0.32 | Likely Benign | 0.618 | Likely Pathogenic | -0.95 | Neutral | 0.386 | Benign | 0.157 | Benign | 1.32 | Pathogenic | 0.10 | Tolerated | 4.32 | 9 | 0.1621 | 0.3902 | -1 | -3 | 4.6 | 42.08 | 214.6 | -68.8 | 0.3 | 0.7 | -0.5 | 0.3 | Uncertain | Gly381 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364, res. Ala399-Ile411). Because the Ω loop is assumed to directly interact with the membrane, it moves arbitrarily throughout the WT solvent simulations. The Ω loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play major roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are observed in the variant simulations, Val381 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. However, since the effects on Gly-rich Ω loop dynamics can only be well studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1145G>T | G382V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G382V has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a split opinion: benign predictions come from PROVEAN, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Pathogenic predictions are returned by REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM, while premPS is uncertain. High‑accuracy assessments give a mixed signal: AlphaMissense‑Optimized predicts benign, SGM‑Consensus indicates likely benign, but Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of tools lean toward pathogenicity, and the high‑accuracy Foldetta result supports this. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429690 | Uncertain | 0.315 | 0.951 | 0.750 | -5.988 | Likely Benign | 0.155 | Likely Benign | Likely Benign | 6.40 | Destabilizing | 1.6 | 9.28 | Destabilizing | 7.84 | Destabilizing | -0.53 | Ambiguous | 0.571 | Likely Pathogenic | -0.54 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 1.32 | Pathogenic | 0.03 | Affected | 0.1631 | 0.3708 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1148G>T | G383V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G383V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect are FoldX, Rosetta, polyPhen‑2 HumDiv, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of predictions lean toward a benign impact, and this conclusion does not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.728858 | Disordered | 0.429104 | Uncertain | 0.296 | 0.949 | 0.750 | -5.769 | Likely Benign | 0.145 | Likely Benign | Likely Benign | 5.13 | Destabilizing | 2.1 | 4.06 | Destabilizing | 4.60 | Destabilizing | -0.26 | Likely Benign | 0.406 | Likely Benign | -0.72 | Neutral | 0.668 | Possibly Damaging | 0.207 | Benign | 4.12 | Benign | 0.01 | Affected | 0.1597 | 0.3493 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1151G>A | G384D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G384D is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33438056‑G‑A). Prediction tools that classify the variant as benign include REVEL, PROVEAN, and AlphaMissense‑Optimized. Those that predict pathogenicity are FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; premPS is uncertain. Separately, the high‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of tools—including the high‑accuracy methods—indicate a pathogenic effect. This prediction does not contradict any ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.728858 | Disordered | 0.427831 | Uncertain | 0.323 | 0.934 | 0.750 | 6-33438056-G-A | -9.142 | Likely Pathogenic | 0.610 | Likely Pathogenic | Likely Benign | 2.06 | Destabilizing | 0.5 | 2.15 | Destabilizing | 2.11 | Destabilizing | 0.53 | Ambiguous | 0.439 | Likely Benign | -0.93 | Neutral | 0.994 | Probably Damaging | 0.986 | Probably Damaging | 1.32 | Pathogenic | 0.04 | Affected | 4.32 | 2 | 0.2071 | 0.2235 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||||
| c.1160G>A | G387D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G387D is reported in gnomAD (6‑33438065‑G‑A) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, Foldetta, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. With two of the three high‑accuracy methods indicating pathogenicity and a majority of general predictors leaning toward pathogenic, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.642678 | Disordered | 0.422910 | Uncertain | 0.293 | 0.861 | 0.750 | 6-33438065-G-A | 2 | 1.24e-6 | -8.625 | Likely Pathogenic | 0.612 | Likely Pathogenic | Likely Benign | 3.57 | Destabilizing | 2.3 | 3.22 | Destabilizing | 3.40 | Destabilizing | 0.39 | Likely Benign | 0.459 | Likely Benign | -0.37 | Neutral | 0.069 | Benign | 0.041 | Benign | 1.32 | Pathogenic | 0.02 | Affected | 4.32 | 3 | 0.2145 | 0.2035 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||
| c.1160G>T | G387V 2D 3DClick to see structure in 3D Viewer AISynGAP1 G387V is listed in ClinVar with an uncertain significance and is present in gnomAD (variant ID 6-33438065-G-T). Functional prediction tools that report a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, SIFT, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as likely benign, while the Foldetta stability assessment (combining FoldX‑MD and Rosetta) indicates a pathogenic change. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as pathogenic. Overall, the majority of predictions favor a benign impact, and this consensus does not contradict the ClinVar uncertain status; thus the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.642678 | Disordered | 0.422910 | Uncertain | 0.293 | 0.861 | 0.750 | Uncertain | 1 | 6-33438065-G-T | 22 | 1.37e-5 | -6.199 | Likely Benign | 0.153 | Likely Benign | Likely Benign | 5.13 | Destabilizing | 1.8 | 6.44 | Destabilizing | 5.79 | Destabilizing | -0.33 | Likely Benign | 0.390 | Likely Benign | -0.54 | Neutral | 0.069 | Benign | 0.077 | Benign | 1.32 | Pathogenic | 0.01 | Affected | 4.32 | 3 | 0.1586 | 0.3708 | -1 | -3 | 4.6 | 42.08 | 207.7 | -68.4 | -0.7 | 0.8 | -0.5 | 0.1 | Uncertain | Gly387 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364 and res. Ala399-Ile411). The Ω loop is assumed to directly interact with the membrane, and it is observed to move arbitrarily throughout the WT solvent simulations. This loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play significant roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are visualized in the variant’s simulations, Val387 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. Since the effects on the Gly-rich Ω loop dynamics can only be well studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1163G>A | G388D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G388D is reported in gnomAD (variant ID 6-33438068‑G‑A) but has no entry in ClinVar. Functional prediction tools cluster into two groups: benign predictions come from premPS, PROVEAN, and AlphaMissense‑Optimized; pathogenic predictions arise from SGM‑Consensus, REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments reinforce this split: AlphaMissense‑Optimized indicates a benign effect, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as likely pathogenic; Foldetta, integrating FoldX‑MD and Rosetta outputs, also predicts a pathogenic impact on protein stability. Taken together, the preponderance of evidence from both general and high‑accuracy predictors points to a pathogenic effect for G388D, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.736850 | Disordered | 0.420316 | Uncertain | 0.319 | 0.827 | 0.750 | 6-33438068-G-A | -8.416 | Likely Pathogenic | 0.574 | Likely Pathogenic | Likely Benign | 5.56 | Destabilizing | 7.3 | 3.08 | Destabilizing | 4.32 | Destabilizing | 0.42 | Likely Benign | 0.618 | Likely Pathogenic | -0.67 | Neutral | 0.994 | Probably Damaging | 0.986 | Probably Damaging | 1.32 | Pathogenic | 0.04 | Affected | 4.32 | 3 | 0.1923 | 0.1624 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||||
| c.1165T>C | S389P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S389P is listed in gnomAD (variant ID 6‑33438070‑T‑C) and has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.703578 | Disordered | 0.417444 | Uncertain | 0.306 | 0.803 | 0.875 | 6-33438070-T-C | -5.286 | Likely Benign | 0.130 | Likely Benign | Likely Benign | 1.85 | Ambiguous | 2.0 | -0.99 | Ambiguous | 0.43 | Likely Benign | 0.16 | Likely Benign | 0.460 | Likely Benign | -0.44 | Neutral | 0.676 | Possibly Damaging | 0.693 | Possibly Damaging | 5.05 | Benign | 0.01 | Affected | 4.32 | 8 | 0.3098 | 0.7002 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||
| c.1169G>T | G390V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G390V has no ClinVar entry and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from REVEL, FoldX, Rosetta, Foldetta, SIFT, and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default (benign), ESM1b (uncertain), FATHMM (pathogenic), and PROVEAN (benign), also yields a benign verdict; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, indicates pathogenic. With two high‑accuracy tools supporting benign and one supporting pathogenic, the overall evidence leans toward a benign effect. This conclusion does not contradict ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.626927 | Disordered | 0.413274 | Uncertain | 0.304 | 0.763 | 0.875 | -7.137 | In-Between | 0.146 | Likely Benign | Likely Benign | 3.74 | Destabilizing | 0.6 | 4.88 | Destabilizing | 4.31 | Destabilizing | -0.16 | Likely Benign | 0.509 | Likely Pathogenic | -0.88 | Neutral | 0.002 | Benign | 0.002 | Benign | 1.32 | Pathogenic | 0.01 | Affected | 0.1537 | 0.4004 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||
| c.1172G>T | G391V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G391V is listed in ClinVar as Benign (ClinVar ID 1014488.0) and is present in gnomAD (variant ID 6‑33438077‑G‑T). Prediction tools that classify the variant as benign include premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus. Tools that predict pathogenicity are REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. With two high‑accuracy tools supporting benign and one supporting pathogenic, the overall prediction leans toward a benign effect. This conclusion aligns with the ClinVar benign classification, so there is no contradiction with the existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.637480 | Disordered | 0.409509 | Uncertain | 0.279 | 0.741 | 0.750 | Likely Benign | 1 | 6-33438077-G-T | 3 | 1.86e-6 | -6.642 | Likely Benign | 0.133 | Likely Benign | Likely Benign | 4.23 | Destabilizing | 1.3 | 4.81 | Destabilizing | 4.52 | Destabilizing | -0.11 | Likely Benign | 0.595 | Likely Pathogenic | -0.98 | Neutral | 0.994 | Probably Damaging | 0.887 | Possibly Damaging | 1.32 | Pathogenic | 0.10 | Tolerated | 3.69 | 8 | 0.1621 | 0.3821 | -1 | -3 | 4.6 | 42.08 | 228.6 | -69.0 | 0.0 | 0.8 | -0.5 | 0.3 | Uncertain | Gly387 is located in the Gly-rich Ω loop (res. Pro364-Pro398) between two anti-parallel β sheet strands (res. Thr359-Pro364 and res. Ala399-Ile411). The Ω loop is assumed to directly interact with the membrane, and it is observed to move arbitrarily throughout the WT solvent simulations. This loop potentially plays a crucial role in the SynGAP-membrane complex association, stability, and dynamics. However, this aspect cannot be fully addressed through solvent simulations alone.Ω loops are known to play significant roles in protein functions that require flexibility, and thus hydrophobic residues like valine are rarely tolerated. Although no negative structural effects are visualized in the variant’s simulations, Val391 may exert drastic effects on the SynGAP-membrane complex dynamics and stability. Since the effects on the Gly-rich Ω loop dynamics can only be well studied through the SynGAP-membrane complex, no definite conclusions can be drawn. | ||||||||||||||
| c.1178G>T | G393V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 G393V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Rosetta is uncertain and is treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while Foldetta predicts pathogenic. The SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑vs‑2 split. Overall, the majority of evidence (8 pathogenic vs. 4 benign) points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.538167 | Disordered | 0.402365 | Uncertain | 0.333 | 0.670 | 0.625 | -6.358 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 5.56 | Destabilizing | 2.3 | -0.72 | Ambiguous | 2.42 | Destabilizing | -0.01 | Likely Benign | 0.639 | Likely Pathogenic | -2.69 | Deleterious | 0.982 | Probably Damaging | 0.648 | Possibly Damaging | 1.32 | Pathogenic | 0.01 | Affected | 0.1712 | 0.4144 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||
| c.1184G>T | G395V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G395V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only FoldX and SIFT predict pathogenic. The SGM‑Consensus score, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely benign outcome. High‑accuracy assessments further support this view: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and the Foldetta stability analysis is uncertain. Taken together, the preponderance of evidence points to a benign impact for G395V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.513880 | Disordered | 0.396199 | Uncertain | 0.474 | 0.601 | 0.500 | -5.617 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 2.83 | Destabilizing | 0.7 | -0.37 | Likely Benign | 1.23 | Ambiguous | 0.08 | Likely Benign | 0.288 | Likely Benign | -1.49 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.21 | Benign | 0.03 | Affected | 0.1290 | 0.4183 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1192C>G | P398A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P398A is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33438097‑C‑G). Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only PROVEAN predicts a pathogenic outcome. Tools with inconclusive results—FoldX, Rosetta, Foldetta, and premPS—are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign, and Foldetta as Uncertain. Overall, the majority of evidence points to a benign effect for P398A, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | 0.436924 | Structured | 0.401041 | Uncertain | 0.891 | 0.525 | 0.250 | 6-33438097-C-G | 2 | 1.24e-6 | -5.321 | Likely Benign | 0.184 | Likely Benign | Likely Benign | 1.80 | Ambiguous | 0.2 | 1.15 | Ambiguous | 1.48 | Ambiguous | 0.92 | Ambiguous | 0.290 | Likely Benign | -5.17 | Deleterious | 0.008 | Benign | 0.005 | Benign | 5.55 | Benign | 0.12 | Tolerated | 3.40 | 16 | 0.3644 | 0.5487 | -1 | 1 | 3.4 | -26.04 | ||||||||||||||||||||||||
| c.1199T>G | V400G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V400G missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are polyPhen‑2 HumDiv and FATHMM; all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.398279 | Structured | 0.415488 | Uncertain | 0.951 | 0.451 | 0.000 | -11.508 | Likely Pathogenic | 0.969 | Likely Pathogenic | Likely Pathogenic | 4.84 | Destabilizing | 0.1 | 5.40 | Destabilizing | 5.12 | Destabilizing | 2.40 | Destabilizing | 0.819 | Likely Pathogenic | -5.70 | Deleterious | 0.335 | Benign | 0.920 | Probably Damaging | 5.27 | Benign | 0.00 | Affected | 0.2296 | 0.2522 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1223C>G | T408R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T408R is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The premPS score is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. With six benign versus seven pathogenic calls overall, the evidence is mixed, but the presence of two independent high‑accuracy benign predictions and the lack of ClinVar or gnomAD support suggests the variant is most likely benign, and this does not contradict any existing clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.161087 | Structured | 0.370935 | Uncertain | 0.907 | 0.239 | 0.000 | -12.075 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | -0.48 | Likely Benign | 0.1 | -0.24 | Likely Benign | -0.36 | Likely Benign | 0.79 | Ambiguous | 0.138 | Likely Benign | -4.06 | Deleterious | 0.991 | Probably Damaging | 0.645 | Possibly Damaging | 4.12 | Benign | 0.15 | Tolerated | 0.1029 | 0.3180 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.1223C>T | T408I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T408I missense variant is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33438128‑C‑T). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. Tools with uncertain or mixed outputs are AlphaMissense‑Default and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of evidence points toward a benign effect, and this conclusion does not contradict ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | 0.161087 | Structured | 0.370935 | Uncertain | 0.907 | 0.239 | 0.000 | 6-33438128-C-T | 1 | 6.19e-7 | -10.023 | Likely Pathogenic | 0.542 | Ambiguous | Likely Benign | -0.16 | Likely Benign | 0.1 | -0.67 | Ambiguous | -0.42 | Likely Benign | 0.38 | Likely Benign | 0.131 | Likely Benign | -4.53 | Deleterious | 0.976 | Probably Damaging | 0.607 | Possibly Damaging | 4.08 | Benign | 0.05 | Affected | 3.38 | 28 | 0.0905 | 0.6700 | -1 | 0 | 5.2 | 12.05 | |||||||||||||||||||||||||
| c.1226T>C | M409T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M409T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Uncertain results are reported by FoldX, Foldetta, and premPS. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized indicates a benign effect, whereas the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) supports a pathogenic outcome; Foldetta remains inconclusive. Overall, the majority of evidence, including the SGM‑Consensus, points to a pathogenic impact for M409T. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | -9.194 | Likely Pathogenic | 0.607 | Likely Pathogenic | Likely Benign | 1.41 | Ambiguous | 0.2 | 0.48 | Likely Benign | 0.95 | Ambiguous | 0.96 | Ambiguous | 0.262 | Likely Benign | -3.18 | Deleterious | 0.987 | Probably Damaging | 0.945 | Probably Damaging | 4.17 | Benign | 0.45 | Tolerated | 0.1835 | 0.2391 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.1226T>G | M409R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M409R is not reported in ClinVar and is present in gnomAD (ID 6‑33438131‑T‑G). Functional prediction tools cluster into two groups: benign (REVEL, SIFT, FATHMM, Rosetta) and pathogenic (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default). Three tools give uncertain results (FoldX, Foldetta, AlphaMissense‑Optimized). The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Overall, the balance of evidence favors a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | 0.150080 | Structured | 0.360643 | Uncertain | 0.884 | 0.219 | 0.000 | 6-33438131-T-G | -12.795 | Likely Pathogenic | 0.911 | Likely Pathogenic | Ambiguous | 1.47 | Ambiguous | 0.4 | 0.44 | Likely Benign | 0.96 | Ambiguous | 1.30 | Destabilizing | 0.485 | Likely Benign | -4.39 | Deleterious | 0.877 | Possibly Damaging | 0.807 | Possibly Damaging | 4.15 | Benign | 0.27 | Tolerated | 3.38 | 28 | 0.1537 | 0.0957 | -1 | 0 | -6.4 | 24.99 | ||||||||||||||||||||||||||
| c.1229G>T | S410I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S410I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Predictions that are uncertain or inconclusive are FoldX, Foldetta, and AlphaMissense‑Default. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized classifies the variant as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leans pathogenic, and Foldetta remains uncertain. Overall, the majority of tools suggest a benign impact, but the high‑accuracy consensus indicates potential pathogenicity, leaving the variant’s effect ambiguous. Based on the aggregate predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.098513 | Structured | 0.349627 | Uncertain | 0.908 | 0.206 | 0.000 | -9.383 | Likely Pathogenic | 0.432 | Ambiguous | Likely Benign | -1.08 | Ambiguous | 0.2 | -0.36 | Likely Benign | -0.72 | Ambiguous | -0.41 | Likely Benign | 0.114 | Likely Benign | -3.21 | Deleterious | 0.993 | Probably Damaging | 0.589 | Possibly Damaging | 4.15 | Benign | 0.21 | Tolerated | 0.0965 | 0.6579 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||
| c.1232T>G | I411S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I411S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.339366 | Uncertain | 0.927 | 0.198 | 0.000 | -13.763 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.59 | Destabilizing | 0.2 | 3.83 | Destabilizing | 3.71 | Destabilizing | 1.91 | Destabilizing | 0.603 | Likely Pathogenic | -5.50 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.30 | Benign | 0.00 | Affected | 0.2262 | 0.1487 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1241T>C | M414T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M414T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions arise from SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta; Rosetta remains uncertain. High‑accuracy methods all support a deleterious effect: AlphaMissense‑Optimized scores the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta predicts a destabilizing, pathogenic outcome. Consequently, the consensus of the most reliable predictors classifies M414T as pathogenic, with no conflict with the ClinVar status, which simply lacks an entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.081712 | Structured | 0.329108 | Uncertain | 0.914 | 0.217 | 0.000 | -8.698 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 2.04 | Destabilizing | 0.1 | 1.96 | Ambiguous | 2.00 | Destabilizing | 1.27 | Destabilizing | 0.406 | Likely Benign | -5.00 | Deleterious | 0.997 | Probably Damaging | 0.972 | Probably Damaging | 3.41 | Benign | 0.08 | Tolerated | 0.1771 | 0.1976 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.124C>T | P42S 2D AIThe SynGAP1 missense variant P42S is listed in gnomAD (ID 6‑33423533‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions are reported by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for P42S, and this conclusion is not contradicted by ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431487 | Uncertain | 0.420 | 0.771 | 0.375 | 6-33423533-C-T | 1 | 6.20e-7 | -3.472 | Likely Benign | 0.109 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -1.12 | Neutral | 0.909 | Possibly Damaging | 0.901 | Possibly Damaging | 4.24 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3462 | 0.4470 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.1258T>G | F420V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F420V is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM. In contrast, the majority of tools predict a pathogenic outcome: FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts Pathogenic; the SGM‑Consensus itself is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts Pathogenic. Based on the preponderance of pathogenic predictions and the high‑accuracy tool consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.384475 | Uncertain | 0.974 | 0.255 | 0.000 | -11.849 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 3.42 | Destabilizing | 0.1 | 3.35 | Destabilizing | 3.39 | Destabilizing | 1.58 | Destabilizing | 0.402 | Likely Benign | -6.41 | Deleterious | 0.495 | Possibly Damaging | 0.191 | Benign | 3.16 | Benign | 0.01 | Affected | 0.2038 | 0.2022 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1270G>T | V424F 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant V424F is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and FATHMM, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The remaining tools (FoldX, Foldetta, premPS, AlphaMissense‑Optimized) yield uncertain or unavailable results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -10.947 | Likely Pathogenic | 0.939 | Likely Pathogenic | Ambiguous | 1.99 | Ambiguous | 0.3 | -0.37 | Likely Benign | 0.81 | Ambiguous | 0.55 | Ambiguous | 0.275 | Likely Benign | -3.66 | Deleterious | 0.995 | Probably Damaging | 0.775 | Possibly Damaging | 3.40 | Benign | 0.01 | Affected | 0.0816 | 0.2094 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1271T>G | V424G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V424G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM. All other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic consensus (3 pathogenic vs. 1 benign); and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. No predictions are missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.050641 | Structured | 0.411431 | Uncertain | 0.973 | 0.248 | 0.000 | -13.697 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 3.90 | Destabilizing | 0.1 | 4.84 | Destabilizing | 4.37 | Destabilizing | 2.44 | Destabilizing | 0.622 | Likely Pathogenic | -6.26 | Deleterious | 0.994 | Probably Damaging | 1.000 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.1396 | 0.2029 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1289T>C | M430T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M430T is reported in gnomAD (6‑33438194‑T‑C) and has no ClinVar entry. Consensus from multiple in‑silico predictors indicates a benign effect: REVEL, FoldX (uncertain), Rosetta, Foldetta (uncertain), premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify it as benign or likely benign. No tool predicts pathogenicity. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is benign, the SGM Consensus is likely benign, and Foldetta is uncertain. Overall, the computational evidence strongly supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic report. Thus, the variant is most likely benign, and this is not contradictory to ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.071867 | Structured | 0.385298 | Uncertain | 0.952 | 0.306 | 0.000 | 6-33438194-T-C | 1 | 6.19e-7 | -3.430 | Likely Benign | 0.107 | Likely Benign | Likely Benign | 1.89 | Ambiguous | 0.1 | 0.01 | Likely Benign | 0.95 | Ambiguous | 0.43 | Likely Benign | 0.103 | Likely Benign | -1.48 | Neutral | 0.016 | Benign | 0.010 | Benign | 3.48 | Benign | 0.40 | Tolerated | 3.37 | 29 | 0.2002 | 0.3184 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||
| c.128G>T | G43V 2D AIThe SynGAP1 missense variant G43V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.308712 | Structured | 0.431462 | Uncertain | 0.396 | 0.762 | 0.375 | -3.745 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -0.79 | Neutral | 0.320 | Benign | 0.140 | Benign | 4.28 | Benign | 0.00 | Affected | 0.1156 | 0.3120 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.1300G>T | V434F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434F is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that indicate a benign effect include REVEL, premPS, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; SGM‑Consensus predicts pathogenic; Foldetta predicts pathogenic. All predictions are available and none are inconclusive. Based on the overall distribution of predictions, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | -12.553 | Likely Pathogenic | 0.672 | Likely Pathogenic | Likely Benign | 3.64 | Destabilizing | 0.1 | 3.27 | Destabilizing | 3.46 | Destabilizing | 0.27 | Likely Benign | 0.417 | Likely Benign | -3.93 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.44 | Benign | 0.04 | Affected | 0.0596 | 0.3391 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1301T>G | V434G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are SIFT and FATHMM, while the remaining tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default) all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as pathogenic. Taken together, the overwhelming majority of evidence indicates a deleterious effect. Therefore, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.158265 | Structured | 0.342846 | Uncertain | 0.954 | 0.306 | 0.000 | -12.519 | Likely Pathogenic | 0.809 | Likely Pathogenic | Ambiguous | 3.08 | Destabilizing | 0.0 | 4.37 | Destabilizing | 3.73 | Destabilizing | 1.91 | Destabilizing | 0.564 | Likely Pathogenic | -5.16 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.39 | Benign | 0.07 | Tolerated | 0.1758 | 0.2408 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1309C>T | P437S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P437S is not reported in ClinVar (ClinVar ID: None) but is present in gnomAD (ID 6‑33438214‑C‑T). Prediction tools that agree on a benign effect include REVEL, Rosetta, FATHMM, and AlphaMissense‑Optimized, whereas PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all predict a pathogenic outcome; the remaining methods (FoldX, Foldetta, premPS, AlphaMissense‑Default) are uncertain and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic interpretation. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.306196 | Uncertain | 0.921 | 0.298 | 0.000 | 6-33438214-C-T | 2 | 1.24e-6 | -11.964 | Likely Pathogenic | 0.518 | Ambiguous | Likely Benign | 1.14 | Ambiguous | 0.0 | -3.31 | Stabilizing | -1.09 | Ambiguous | 0.60 | Ambiguous | 0.226 | Likely Benign | -6.60 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.49 | Benign | 0.01 | Affected | 3.38 | 26 | 0.3548 | 0.4843 | -1 | 1 | 0.8 | -10.04 | |||||||||||||||||||||||||
| c.1321G>T | V441F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V441F missense variant is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools lean toward a benign interpretation, and the high‑accuracy predictions are split but favor benign. Thus, the variant is most likely benign based on current computational evidence, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -13.519 | Likely Pathogenic | 0.653 | Likely Pathogenic | Likely Benign | -0.26 | Likely Benign | 0.0 | 0.32 | Likely Benign | 0.03 | Likely Benign | 0.54 | Ambiguous | 0.355 | Likely Benign | -4.22 | Deleterious | 0.992 | Probably Damaging | 0.658 | Possibly Damaging | 3.37 | Benign | 0.08 | Tolerated | 0.0670 | 0.3269 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1322T>G | V441G 2D 3DClick to see structure in 3D Viewer AISynGAP1 variant V441G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into three groups: benign predictions come from REVEL, FoldX, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; the remaining tools (Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments further refine the picture: AlphaMissense‑Optimized remains benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) favors pathogenic, and Foldetta is inconclusive. Taken together, the majority of evidence points to a benign effect, but the SGM Consensus and several individual pathogenic predictors introduce uncertainty. Therefore, the variant is most likely benign based on the overall prediction landscape, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.161087 | Structured | 0.259875 | Uncertain | 0.918 | 0.249 | 0.000 | -12.380 | Likely Pathogenic | 0.478 | Ambiguous | Likely Benign | 0.18 | Likely Benign | 0.0 | 1.25 | Ambiguous | 0.72 | Ambiguous | 0.95 | Ambiguous | 0.273 | Likely Benign | -5.88 | Deleterious | 0.841 | Possibly Damaging | 0.997 | Probably Damaging | 3.41 | Benign | 0.24 | Tolerated | 0.1664 | 0.1715 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1328G>T | G443V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G443V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Overall, the majority of evidence points to a benign impact for G443V, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.250310 | Structured | 0.258623 | Uncertain | 0.935 | 0.206 | 0.000 | -4.130 | Likely Benign | 0.274 | Likely Benign | Likely Benign | 0.17 | Likely Benign | 0.2 | -2.19 | Stabilizing | -1.01 | Ambiguous | 0.21 | Likely Benign | 0.099 | Likely Benign | -2.90 | Deleterious | 0.585 | Possibly Damaging | 0.195 | Benign | 3.36 | Benign | 0.12 | Tolerated | 0.1089 | 0.3375 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1339G>T | V447F 2D 3DClick to see structure in 3D Viewer AISynGAP1 variant V447F is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools that classify the variant as benign include REVEL, premPS, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports an uncertain effect on protein folding. Overall, the majority of predictions lean toward pathogenicity, suggesting the variant is most likely pathogenic, a conclusion that does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.250310 | Structured | 0.283801 | Uncertain | 0.970 | 0.243 | 0.000 | Uncertain | 1 | -8.673 | Likely Pathogenic | 0.701 | Likely Pathogenic | Likely Benign | 1.40 | Ambiguous | 0.3 | 0.61 | Ambiguous | 1.01 | Ambiguous | 0.20 | Likely Benign | 0.206 | Likely Benign | -2.62 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.44 | Benign | 0.03 | Affected | 0.0551 | 0.3055 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||
| c.1340T>G | V447G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V447G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL and FATHMM, while the remaining tools—FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. No prediction or stability result is missing or inconclusive. Overall, the preponderance of evidence indicates that V447G is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.250310 | Structured | 0.283801 | Uncertain | 0.970 | 0.243 | 0.000 | -13.648 | Likely Pathogenic | 0.861 | Likely Pathogenic | Ambiguous | 3.81 | Destabilizing | 0.1 | 4.62 | Destabilizing | 4.22 | Destabilizing | 2.28 | Destabilizing | 0.499 | Likely Benign | -5.43 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.1863 | 0.2240 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1346G>T | S449I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S449I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen2_HumVar. Those that predict a pathogenic effect are PROVEAN, polyPhen2_HumDiv, and ESM1b. Rosetta and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs. 2 pathogenic). Foldetta also remains uncertain. Overall, the majority of evidence (7 benign vs. 3 pathogenic) points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.254060 | Structured | 0.301437 | Uncertain | 0.958 | 0.251 | 0.000 | -9.549 | Likely Pathogenic | 0.310 | Likely Benign | Likely Benign | 0.04 | Likely Benign | 0.1 | -1.39 | Ambiguous | -0.68 | Ambiguous | 0.13 | Likely Benign | 0.105 | Likely Benign | -3.23 | Deleterious | 0.559 | Possibly Damaging | 0.044 | Benign | 3.40 | Benign | 0.14 | Tolerated | 0.0756 | 0.5006 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||
| c.1354G>T | V452F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V452F variant is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect are Rosetta and FATHMM, whereas the remaining tools (REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) all predict a pathogenic impact. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Based on the preponderance of evidence, the variant is most likely pathogenic, a conclusion that contradicts its current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.335645 | Structured | 0.315167 | Uncertain | 0.970 | 0.229 | 0.000 | Uncertain | 1 | -14.769 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 9.21 | Destabilizing | 0.1 | 0.37 | Likely Benign | 4.79 | Destabilizing | 0.61 | Ambiguous | 0.511 | Likely Pathogenic | -4.94 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 3.37 | 34 | 0.0564 | 0.3451 | -1 | -1 | -1.4 | 48.04 | 249.4 | -35.7 | 0.0 | 0.0 | 0.4 | 0.1 | X | Potentially Pathogenic | The iso-propyl side chain of Val452, located in the middle of an α helix (res. Val441-Ser457), packs against hydrophobic residues in the inter-helix space at the intersection of three α helices (e.g., Leu500, His453, Leu465). In the variant simulations, the larger side chain of Phe452 cannot pack against the opposing α helix (res. Leu489-Glu519) as efficiently as valine. Due to space restrictions, the phenol ring adjusts to make room by rotating slightly sideways in the inter-helix space. Besides this small and local shift, no large-scale effects on the protein structure are seen based on the simulations. However, the size difference between the swapped residues could affect the protein folding process. | ||||||||||||||||
| c.1355T>G | V452G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V452G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. AlphaMissense‑Optimized is uncertain, providing no definitive direction. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. AlphaMissense‑Optimized remains uncertain. Overall, the preponderance of evidence indicates that V452G is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.335645 | Structured | 0.315167 | Uncertain | 0.970 | 0.229 | 0.000 | -13.410 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 3.70 | Destabilizing | 0.1 | 3.37 | Destabilizing | 3.54 | Destabilizing | 2.46 | Destabilizing | 0.570 | Likely Pathogenic | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.17 | Benign | 0.00 | Affected | 0.1948 | 0.2567 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1361T>G | I454S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I454S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy predictors, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.312811 | Uncertain | 0.965 | 0.182 | 0.000 | -13.025 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.50 | Destabilizing | 0.1 | 4.45 | Destabilizing | 4.48 | Destabilizing | 2.20 | Destabilizing | 0.574 | Likely Pathogenic | -5.83 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.26 | Benign | 0.00 | Affected | 0.2116 | 0.0800 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1367A>C | Q456P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q456P is listed in ClinVar with an uncertain significance (ClinVar ID 2697090.0) and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions are made by FoldX, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta. High‑accuracy methods specifically report pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are inconclusive. Overall, the preponderance of evidence indicates the variant is most likely pathogenic, which contradicts the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.170161 | Structured | 0.302348 | Uncertain | 0.939 | 0.164 | 0.000 | Uncertain | 1 | -15.250 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 3.68 | Destabilizing | 0.2 | 8.43 | Destabilizing | 6.06 | Destabilizing | 0.82 | Ambiguous | 0.469 | Likely Benign | -5.66 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.34 | Benign | 0.07 | Tolerated | 3.37 | 34 | 0.2256 | 0.3631 | -1 | 0 | 1.9 | -31.01 | |||||||||||||||||||||||||
| c.1370G>T | S457I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S457I lies within the GAP domain. ClinVar has no entry for this change, and it is absent from gnomAD. Prediction tools that report a benign effect include FoldX and FATHMM, whereas the majority of other in silico predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default and AlphaMissense‑Optimized—classify it as pathogenic. Uncertain results come from Rosetta, Foldetta and premPS. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM and PROVEAN) is Likely Pathogenic, and Foldetta remains inconclusive. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.164327 | Structured | 0.297330 | Uncertain | 0.909 | 0.159 | 0.000 | -13.170 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | -0.26 | Likely Benign | 0.3 | -0.96 | Ambiguous | -0.61 | Ambiguous | 0.65 | Ambiguous | 0.557 | Likely Pathogenic | -5.73 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.34 | Benign | 0.02 | Affected | 0.0779 | 0.6095 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.1373C>G | T458R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T458R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, SIFT, and FATHMM, whereas a larger set—SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict pathogenicity. Uncertain results come from FoldX and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.294848 | Uncertain | 0.915 | 0.144 | 0.000 | -12.984 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | -0.81 | Ambiguous | 0.1 | -0.11 | Likely Benign | -0.46 | Likely Benign | 0.61 | Ambiguous | 0.390 | Likely Benign | -5.26 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.52 | Benign | 0.20 | Tolerated | 0.1016 | 0.3013 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.1376G>T | G459V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G459V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, while the remaining evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact; premPS is inconclusive and therefore not counted. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the consensus of these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for G459V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.185198 | Structured | 0.289888 | Uncertain | 0.903 | 0.150 | 0.125 | -13.606 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.79 | Destabilizing | 0.1 | 5.01 | Destabilizing | 4.40 | Destabilizing | 0.71 | Ambiguous | 0.605 | Likely Pathogenic | -8.46 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.99 | Benign | 0.00 | Affected | 0.0997 | 0.4240 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1390T>G | F464V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 F464V variant is listed in ClinVar with an “Uncertain” status (ClinVar ID 1716596.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM; all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the collective predictions, the variant is most likely pathogenic, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.313424 | Uncertain | 0.961 | 0.178 | 0.000 | Uncertain | 1 | -12.254 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.61 | Destabilizing | 0.1 | 2.89 | Destabilizing | 3.25 | Destabilizing | 1.40 | Destabilizing | 0.592 | Likely Pathogenic | -6.96 | Deleterious | 0.998 | Probably Damaging | 0.996 | Probably Damaging | 3.36 | Benign | 0.04 | Affected | 3.37 | 34 | 0.1587 | 0.2219 | -1 | -1 | 1.4 | -48.04 | 210.1 | 40.5 | -0.1 | 0.0 | -0.9 | 0.3 | X | Potentially Pathogenic | The phenyl ring of Phe464, located in the middle of an α helix (res. Ala461–Phe476), packs against hydrophobic residues (e.g., Met468, Leu451, Leu455, and Tyr428) in the inter-helix space formed with two other α helices (res. Asn440-Lys460 and res. Pro413-Glu436). The iso-propyl side chain of Val464 is similarly hydrophobic but considerably smaller than the original phenyl ring of Phe464. To compensate for the size difference, neighboring residues need to fill in the gap in the variant simulations.The phenolic side chain of Tyr428, located at the middle bend of an α helix (res. Glu436-Pro413), assumes a new position in the inter-helix space or rotates inward next to the third α helix (res. Asn440-Lys460) when the stable H-bond between Tyr428 and Asp467 seen in the WT simulations breaks. The residue swap also leads to the loss of the methionine-aromatic interaction between the Met468 and Phe464 side chains, which could weaken the integrity of the parent α helix (res. Ala461-Phe476). Although the simulations likely underestimate the full adverse effect of the introduced mutation during folding, the two opposing α helices (res. Ala461–Phe476 and res. Glu436-Pro413) move substantially closer to each other in the variant simulations. | ||||||||||||||||
| c.1403T>C | M468T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M468T is listed in ClinVar with an “Uncertain” status and is present in the gnomAD database. Prediction tools that are available all converge on a pathogenic interpretation: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool reports a benign outcome. High‑accuracy assessments are consistent: AlphaMissense‑Optimized is “Uncertain,” SGM Consensus is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. **Based on the aggregate predictions, the variant is most likely pathogenic, which does not contradict the ClinVar “Uncertain” classification.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.284882 | Structured | 0.339253 | Uncertain | 0.932 | 0.257 | 0.000 | Uncertain | 2 | 6-33438435-T-C | 1 | 6.20e-7 | -12.399 | Likely Pathogenic | 0.862 | Likely Pathogenic | Ambiguous | 3.47 | Destabilizing | 0.1 | 3.10 | Destabilizing | 3.29 | Destabilizing | 1.84 | Destabilizing | 0.801 | Likely Pathogenic | -3.85 | Deleterious | 0.994 | Probably Damaging | 0.985 | Probably Damaging | -1.31 | Pathogenic | 0.01 | Affected | 3.37 | 31 | 0.2094 | 0.1950 | -1 | -1 | -2.6 | -30.09 | 214.6 | 47.1 | 0.0 | 0.0 | 0.1 | 0.0 | X | Potentially Pathogenic | The thioether group of Met468, located in the middle of an α helix (res. Ala461–Phe476), interacts with hydrophobic residues (e.g., Phe464, Leu465, Leu489) in an inter-helix space formed by two other α helices (res. Ala461–Phe476, res. Thr488–Gly502). In the variant simulations, the hydrophilic side chain of Thr468 does not pack favorably in the hydrophobic niche, and the methionine-aromatic stacking is lost. Although the hydroxyl group of Thr468 forms an H-bond with the backbone carbonyl group of Phe464, the integrity of the α helix is not affected in the simulations. No large-scale structural changes are observed during the variant simulations; however, due to the importance of hydrophobic packing, the effects could be more pronounced during protein folding. | |||||||||||||
| c.1409T>C | M470T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M470T is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include only SIFT, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all predict a pathogenic impact. High‑accuracy methods further support this: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding‑stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, which is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.298791 | Structured | 0.351497 | Uncertain | 0.908 | 0.272 | 0.000 | Uncertain | 1 | -8.104 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 3.19 | Destabilizing | 0.1 | 2.68 | Destabilizing | 2.94 | Destabilizing | 1.49 | Destabilizing | 0.763 | Likely Pathogenic | -5.30 | Deleterious | 0.996 | Probably Damaging | 0.985 | Probably Damaging | -1.08 | Pathogenic | 0.24 | Tolerated | 3.37 | 34 | 0.1782 | 0.1950 | -1 | -1 | -2.6 | -30.09 | 213.8 | 46.5 | 0.0 | 0.0 | -0.2 | 0.2 | X | X | Potentially Pathogenic | The thioether group of Met470, located in the middle of an α helix (res. Ala461–Phe476), interacts with hydrophobic residues in the inter-helix space (e.g., Val473, Leu558, Cys576, Trp572) formed by two other α helices (res. Ser604–Arg581, res. Pro562–Arg579). In the WT simulations, the Met470 side chain also packs against the positively charged guanidinium groups of Arg575, Arg429, and Arg579, which form salt bridges with the negatively charged carboxylate groups of the Asp474 and Asp467 side chains at the protein surface. In the variant simulations, the hydroxyl group of the Thr470 side chain forms an H-bond with the backbone carbonyl group of Ser466 in the α helix, potentially lowering its structural integrity. Importantly, the hydroxyl group of Thr470 also forms an H-bond with the guanidinium group of Arg575, which helps it form a more permanent salt bridge with Asp467. | |||||||||||||||
| c.1412C>G | S471C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S471C is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33438444‑C‑G). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of predictions (10 benign vs. 3 pathogenic) indicate that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no reported pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.305330 | Structured | 0.355411 | Uncertain | 0.888 | 0.261 | 0.000 | 6-33438444-C-G | 1 | 6.20e-7 | -3.454 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.36 | Likely Benign | 0.0 | -0.05 | Likely Benign | 0.16 | Likely Benign | 0.07 | Likely Benign | 0.273 | Likely Benign | -2.90 | Deleterious | 0.000 | Benign | 0.001 | Benign | -1.32 | Pathogenic | 0.01 | Affected | 3.37 | 34 | 0.1048 | 0.4913 | -1 | 0 | 3.3 | 16.06 | |||||||||||||||||||||||||
| c.1418T>G | V473G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V473G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, while the remaining eleven tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) uniformly predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) votes pathogenic, and Foldetta also predicts pathogenic. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic based on the collective evidence, and this conclusion does not contradict any ClinVar status, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.191378 | Structured | 0.362529 | Uncertain | 0.884 | 0.239 | 0.000 | -14.782 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 3.45 | Destabilizing | 0.0 | 3.40 | Destabilizing | 3.43 | Destabilizing | 2.69 | Destabilizing | 0.683 | Likely Pathogenic | -6.91 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.13 | Benign | 0.00 | Affected | 0.1885 | 0.2567 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1421A>G | D474G 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant D474G is not reported in ClinVar and is present in gnomAD (ID 6‑33438453‑A‑G). Functional prediction tools show a split: benign calls come from FoldX, Foldetta, premPS, and SIFT, while pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; Rosetta remains uncertain. High‑accuracy assessments give a pathogenic verdict from AlphaMissense‑Optimized and a pathogenic consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). In contrast, Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts benign stability. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.373433 | Uncertain | 0.864 | 0.255 | 0.000 | 6-33438453-A-G | 1 | 6.20e-7 | -11.215 | Likely Pathogenic | 0.959 | Likely Pathogenic | Likely Pathogenic | -0.38 | Likely Benign | 0.0 | 0.82 | Ambiguous | 0.22 | Likely Benign | 0.44 | Likely Benign | 0.823 | Likely Pathogenic | -6.13 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.28 | Pathogenic | 0.07 | Tolerated | 3.37 | 34 | 0.3245 | 0.4933 | -1 | 1 | 3.1 | -58.04 | ||||||||||||||||||||||||
| c.1426T>G | F476V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 F476V variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and FATHMM. Those that agree on a pathogenic effect are FoldX, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and Foldetta. Tools with uncertain or mixed outputs are Rosetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta predicts a pathogenic impact. Overall, the majority of evidence points toward a pathogenic effect. The variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.257454 | Structured | 0.397815 | Uncertain | 0.821 | 0.250 | 0.000 | -12.329 | Likely Pathogenic | 0.918 | Likely Pathogenic | Ambiguous | 3.01 | Destabilizing | 0.2 | 1.90 | Ambiguous | 2.46 | Destabilizing | 0.64 | Ambiguous | 0.353 | Likely Benign | -1.63 | Neutral | 0.996 | Probably Damaging | 0.993 | Probably Damaging | 3.49 | Benign | 0.53 | Tolerated | 0.1478 | 0.2251 | -1 | -1 | 1.4 | -48.04 | ||||||||||||||||||||||||||||||
| c.142T>G | F48V 2D AIThe SynGAP1 missense variant F48V is not reported in ClinVar and is absent from gnomAD. Consensus from routine in silico predictors shows six tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Optimized) classifying the change as benign, while two (SIFT, AlphaMissense‑Default) predict pathogenicity; ESM1b remains uncertain. High‑accuracy assessment further supports a benign outcome: AlphaMissense‑Optimized is benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority vote, and Foldetta data are unavailable. Consequently, the overall evidence points to a benign effect for F48V, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.298791 | Structured | 0.440452 | Uncertain | 0.558 | 0.707 | 0.125 | -7.591 | In-Between | 0.756 | Likely Pathogenic | Likely Benign | 0.144 | Likely Benign | -1.87 | Neutral | 0.092 | Benign | 0.037 | Benign | 4.00 | Benign | 0.00 | Affected | 0.2533 | 0.2300 | -1 | -1 | 1.4 | -48.04 | ||||||||||||||||||||||||||||||||||||||||
| c.1430T>C | M477T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M477T has no ClinVar entry and is present in gnomAD (ID 6‑33438462‑T‑C). Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. The remaining tools (FoldX, Foldetta, premPS, AlphaMissense‑Default) return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign (2 benign vs. 1 pathogenic votes); Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.268042 | Structured | 0.408680 | Uncertain | 0.761 | 0.250 | 0.000 | 6-33438462-T-C | 2 | 1.24e-6 | -2.509 | Likely Benign | 0.373 | Ambiguous | Likely Benign | 1.62 | Ambiguous | 0.2 | 0.16 | Likely Benign | 0.89 | Ambiguous | 0.51 | Ambiguous | 0.273 | Likely Benign | -1.33 | Neutral | 0.765 | Possibly Damaging | 0.363 | Benign | -1.10 | Pathogenic | 0.40 | Tolerated | 3.37 | 34 | 0.2177 | 0.1950 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||
| c.1450T>G | F484V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F484V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming agreement among the majority of tools and the high‑accuracy methods, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.182256 | Structured | 0.403079 | Uncertain | 0.798 | 0.245 | 0.125 | -15.492 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 6.08 | Destabilizing | 0.1 | 6.07 | Destabilizing | 6.08 | Destabilizing | 1.28 | Destabilizing | 0.455 | Likely Benign | -6.70 | Deleterious | 0.859 | Possibly Damaging | 0.526 | Possibly Damaging | 2.75 | Benign | 0.00 | Affected | 0.1696 | 0.1902 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1463C>G | T488R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T488R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only FATHMM, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) all predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.332663 | Uncertain | 0.928 | 0.233 | 0.125 | -14.353 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 1.29 | Ambiguous | 0.3 | 1.55 | Ambiguous | 1.42 | Ambiguous | 0.85 | Ambiguous | 0.726 | Likely Pathogenic | -5.70 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.22 | Benign | 0.00 | Affected | 0.0711 | 0.2048 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.1463C>T | T488M 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant T488M is listed in ClinVar with an uncertain significance (ClinVar ID 2824521.0) and is present in gnomAD (ID 6‑33438495‑C‑T). Prediction tools that indicate a benign effect include premPS and FATHMM, whereas the majority of algorithms predict a pathogenic outcome: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as inconclusive. No other tools provide definitive evidence. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.332663 | Uncertain | 0.928 | 0.233 | 0.125 | Uncertain | 1 | 6-33438495-C-T | 2 | 1.24e-6 | -12.459 | Likely Pathogenic | 0.973 | Likely Pathogenic | Likely Pathogenic | 0.66 | Ambiguous | 0.3 | 1.62 | Ambiguous | 1.14 | Ambiguous | 0.46 | Likely Benign | 0.746 | Likely Pathogenic | -5.70 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.21 | Benign | 0.00 | Affected | 3.37 | 35 | 0.1027 | 0.4857 | -1 | -1 | 2.6 | 30.09 | ||||||||||||||||||||||
| c.1468G>C | A490P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A490P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Among the available in‑silico predictors, 10 tools (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) uniformly predict a pathogenic effect, whereas only Foldetta predicts a benign outcome; FoldX, Rosetta, and AlphaMissense‑Optimized are inconclusive. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) is benign. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.120615 | Structured | 0.322979 | Uncertain | 0.938 | 0.210 | 0.125 | Uncertain | 1 | -12.905 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | -1.27 | Ambiguous | 0.1 | 1.31 | Ambiguous | 0.02 | Likely Benign | 1.07 | Destabilizing | 0.878 | Likely Pathogenic | -4.81 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.42 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.1755 | 0.3071 | -1 | 1 | -3.4 | 26.04 | |||||||||||||||||||||||||
| c.1493T>C | M498T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M498T is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: all but one (polyPhen‑2 HumVar) predict pathogenicity, while polyPhen‑2 HumVar alone predicts benign. Uncertain predictions (ESM1b and AlphaMissense‑Optimized) are treated as unavailable. High‑accuracy methods reinforce the pathogenic view: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports the variant as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic effect. AlphaMissense‑Optimized remains inconclusive. Overall, the preponderance of evidence indicates that M498T is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.092881 | Structured | 0.399612 | Uncertain | 0.932 | 0.158 | 0.000 | -7.477 | In-Between | 0.869 | Likely Pathogenic | Ambiguous | 2.46 | Destabilizing | 0.1 | 2.62 | Destabilizing | 2.54 | Destabilizing | 1.41 | Destabilizing | 0.672 | Likely Pathogenic | -3.80 | Deleterious | 0.803 | Possibly Damaging | 0.343 | Benign | -1.19 | Pathogenic | 0.02 | Affected | 0.1925 | 0.1630 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.1496G>C | R499T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R499T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumVar and ESM1b, whereas a majority of tools (REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, SIFT, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates Likely Pathogenic, AlphaMissense‑Optimized is inconclusive, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. No evidence from ClinVar contradicts these predictions. Overall, the preponderance of computational evidence points to a pathogenic effect for R499T. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.071867 | Structured | 0.386723 | Uncertain | 0.899 | 0.146 | 0.000 | -6.797 | Likely Benign | 0.855 | Likely Pathogenic | Ambiguous | 2.47 | Destabilizing | 0.1 | 1.43 | Ambiguous | 1.95 | Ambiguous | 0.73 | Ambiguous | 0.664 | Likely Pathogenic | -3.51 | Deleterious | 0.843 | Possibly Damaging | 0.403 | Benign | -1.44 | Pathogenic | 0.02 | Affected | 0.1732 | 0.2074 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||
| c.1497A>C | R499S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R499S is catalogued in gnomAD (ID 6‑33438529‑A‑C) but has no ClinVar entry. Functional prediction tools largely converge on a deleterious effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate pathogenicity. No tool reports a benign outcome. High‑accuracy assessments are mixed: AlphaMissense‑Optimized and Foldetta are uncertain, whereas the SGM‑Consensus remains likely pathogenic. Protein‑stability predictions are inconclusive (FoldX uncertain, Rosetta pathogenic, Foldetta uncertain). Taken together, the overwhelming majority of evidence supports a pathogenic classification for R499S. This conclusion is consistent with the absence of a ClinVar status, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.071867 | Structured | 0.386723 | Uncertain | 0.899 | 0.146 | 0.000 | 6-33438529-A-C | 1 | 6.20e-7 | -9.559 | Likely Pathogenic | 0.935 | Likely Pathogenic | Ambiguous | 1.03 | Ambiguous | 0.0 | 2.19 | Destabilizing | 1.61 | Ambiguous | 1.40 | Destabilizing | 0.632 | Likely Pathogenic | -2.69 | Deleterious | 0.958 | Probably Damaging | 0.702 | Possibly Damaging | -1.43 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.2443 | 0.1649 | -1 | 0 | 3.7 | -69.11 | ||||||||||||||||||||||||
| c.1497A>T | R499S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R499S is not reported in ClinVar and has no entry in gnomAD. Consensus from multiple in silico predictors indicates a pathogenic effect: SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and Rosetta all predict pathogenicity, while FoldX, AlphaMissense‑Optimized, and Foldetta are uncertain. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports likely pathogenic, and Foldetta likewise yields an uncertain result. Taken together, the overwhelming majority of reliable tools predict a pathogenic effect, and there is no ClinVar annotation to contradict this assessment. Therefore, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.071867 | Structured | 0.386723 | Uncertain | 0.899 | 0.146 | 0.000 | -9.559 | Likely Pathogenic | 0.935 | Likely Pathogenic | Ambiguous | 1.03 | Ambiguous | 0.0 | 2.19 | Destabilizing | 1.61 | Ambiguous | 1.40 | Destabilizing | 0.632 | Likely Pathogenic | -2.69 | Deleterious | 0.958 | Probably Damaging | 0.702 | Possibly Damaging | -1.43 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.2443 | 0.1649 | -1 | 0 | 3.7 | -69.11 | |||||||||||||||||||||||||||
| c.149T>G | I50S 2D AIThe SynGAP1 I50S missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM all classify it as benign. In contrast, SIFT and AlphaMissense‑Default predict a pathogenic impact. AlphaMissense‑Optimized yields an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this residue. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments therefore point to a benign outcome: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus is likely benign, and Foldetta data are missing. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.295083 | Structured | 0.449965 | Uncertain | 0.545 | 0.708 | 0.000 | -4.257 | Likely Benign | 0.946 | Likely Pathogenic | Ambiguous | 0.209 | Likely Benign | -2.03 | Neutral | 0.334 | Benign | 0.099 | Benign | 3.74 | Benign | 0.00 | Affected | 0.2626 | 0.0800 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.1502T>G | I501S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I501S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are FATHMM and AlphaMissense‑Optimized; all other evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized indicates a benign change, whereas the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta outputs) both predict pathogenicity. Overall, the preponderance of evidence points to a pathogenic effect for I501S. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.079919 | Structured | 0.366596 | Uncertain | 0.886 | 0.153 | 0.000 | -9.914 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | 3.23 | Destabilizing | 0.2 | 2.40 | Destabilizing | 2.82 | Destabilizing | 1.79 | Destabilizing | 0.511 | Likely Pathogenic | -5.14 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.43 | Benign | 0.04 | Affected | 0.2370 | 0.0858 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1505G>T | G502V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G502V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: all evaluated algorithms except premPS (which predicts benign) classify the substitution as pathogenic or likely pathogenic. The consensus of high‑accuracy predictors is consistent: AlphaMissense‑Optimized reports a pathogenic effect; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a destabilizing, pathogenic outcome. In contrast, premPS is the sole tool suggesting a benign impact. Overall, the overwhelming majority of evidence points to a pathogenic effect for G502V, and this conclusion does not conflict with the absence of a ClinVar classification. Thus, the variant is most likely pathogenic, and this assessment is not contradicted by ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.083462 | Structured | 0.340113 | Uncertain | 0.882 | 0.152 | 0.000 | -15.278 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 3.56 | Destabilizing | 1.0 | 5.50 | Destabilizing | 4.53 | Destabilizing | 0.43 | Likely Benign | 0.917 | Likely Pathogenic | -8.65 | Deleterious | 0.999 | Probably Damaging | 0.944 | Probably Damaging | -1.67 | Pathogenic | 0.00 | Affected | 0.1406 | 0.3387 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1529T>G | I510S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I510S is listed in ClinVar as Pathogenic (ClinVar ID 449946.0) and is not reported in gnomAD. Prediction tools that assess the variant’s effect all converge on a deleterious outcome: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate pathogenicity. No tool predicts a benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized is uncertain, SGM‑Consensus is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this prediction aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.025762 | Structured | 0.250630 | Uncertain | 0.945 | 0.273 | 0.000 | Likely Pathogenic | 1 | -11.661 | Likely Pathogenic | 0.955 | Likely Pathogenic | Ambiguous | 4.00 | Destabilizing | 0.1 | 3.78 | Destabilizing | 3.89 | Destabilizing | 2.34 | Destabilizing | 0.926 | Likely Pathogenic | -4.63 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.44 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.2395 | 0.0858 | -1 | -2 | -5.3 | -26.08 | 201.4 | 45.9 | -0.4 | 0.2 | 0.0 | 0.3 | X | Potentially Pathogenic | Ile510 is located in the middle of an α-helix (res. Gly502-Tyr518) within the inter-helix space of three helices (res. Gly502-Tyr518, Ala533-Val560, and res. Glu582-Met603). In the WT simulations, the sec-butyl side chain of Ile510 hydrophobically packs with other residues in the inter-helix space (e.g., Leu506, Leu610, Ile514, Ile602, Leu598). In the variant simulations, the hydroxyl group of Ser510 forms a hydrogen bond with the backbone atoms of Leu506 and Gly511 in the same α-helix, which could further weaken the α-helix integrity. This α-helix already shows weakness in the WT simulations due to Gly511. Although the simulations do not show large-scale effects, the residue swap could have a substantial impact due to the fundamental role of hydrophobic packing during protein folding. | ||||||||||||||||
| c.152T>G | I51S 2D AIThe SynGAP1 missense variant I51S is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The remaining tools, ESM1b and AlphaMissense‑Optimized, are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign (2 benign vs. 1 pathogenic vote). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.291804 | Structured | 0.454181 | Uncertain | 0.606 | 0.710 | 0.000 | -7.603 | In-Between | 0.879 | Likely Pathogenic | Ambiguous | 0.220 | Likely Benign | -1.39 | Neutral | 0.182 | Benign | 0.099 | Benign | 4.15 | Benign | 0.00 | Affected | 0.3142 | 0.0957 | -1 | -2 | -5.3 | -26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.1532G>T | G511V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G511V is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, which currently has no classification for G511V. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.244404 | Uncertain | 0.924 | 0.287 | 0.000 | -11.738 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 3.47 | Destabilizing | 0.2 | 0.79 | Ambiguous | 2.13 | Destabilizing | 0.65 | Ambiguous | 0.540 | Likely Pathogenic | -8.71 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.16 | Benign | 0.01 | Affected | 0.1557 | 0.3019 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1537T>G | F513V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F513V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only SIFT, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the overwhelming agreement among these predictions, the variant is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.250651 | Uncertain | 0.949 | 0.269 | 0.000 | -10.675 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 3.54 | Destabilizing | 0.4 | 2.68 | Destabilizing | 3.11 | Destabilizing | 1.13 | Destabilizing | 0.799 | Likely Pathogenic | -6.70 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.21 | Pathogenic | 0.44 | Tolerated | 0.1656 | 0.1583 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1541T>G | I514S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I514S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.049374 | Structured | 0.221408 | Uncertain | 0.948 | 0.266 | 0.000 | -12.512 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 4.03 | Destabilizing | 0.2 | 3.70 | Destabilizing | 3.87 | Destabilizing | 2.11 | Destabilizing | 0.625 | Likely Pathogenic | -5.86 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.82 | Benign | 0.00 | Affected | 0.2296 | 0.0530 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1570T>A | C524S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C524S is listed in gnomAD (variant ID 6‑33438813‑T‑A) but has no ClinVar entry. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report a pathogenic outcome, while FoldX, Rosetta, and Foldetta are uncertain and therefore not counted as evidence. Grouping by agreement yields a benign‑prediction set that is empty and a pathogenic‑prediction set that contains the eleven tools above. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; Foldetta remains uncertain. Consequently, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.134866 | Structured | 0.024729 | Uncertain | 0.916 | 0.385 | 0.125 | 6-33438813-T-A | 1 | 6.20e-7 | -11.174 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.80 | Ambiguous | 0.1 | 1.55 | Ambiguous | 1.18 | Ambiguous | 1.58 | Destabilizing | 0.915 | Likely Pathogenic | -9.94 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.38 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.5362 | 0.1848 | Weaken | -1 | 0 | -3.3 | -16.06 | |||||||||||||||||||||||
| c.1571G>C | C524S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 C524S variant has no ClinVar entry and is not present in gnomAD. Prediction tools that agree on a benign effect are none; those that predict pathogenicity include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX, Rosetta, and Foldetta returned inconclusive results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.134866 | Structured | 0.024729 | Uncertain | 0.916 | 0.385 | 0.125 | -11.174 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.80 | Ambiguous | 0.1 | 1.55 | Ambiguous | 1.18 | Ambiguous | 1.58 | Destabilizing | 0.904 | Likely Pathogenic | -9.94 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.38 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.5362 | 0.1848 | Weaken | -1 | 0 | -3.3 | -16.06 | ||||||||||||||||||||||||||
| c.1577T>G | V526G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V526G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on pathogenicity include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; no tool predicts it benign. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as pathogenic. All available evidence points to a deleterious effect. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.139895 | Structured | 0.023118 | Uncertain | 0.943 | 0.403 | 0.000 | -15.811 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 3.21 | Destabilizing | 0.3 | 4.79 | Destabilizing | 4.00 | Destabilizing | 2.19 | Destabilizing | 0.935 | Likely Pathogenic | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.49 | Pathogenic | 0.00 | Affected | 0.1636 | 0.1837 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1586T>G | I529S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I529S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate benign or likely benign. In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM predict pathogenicity, while Rosetta remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Overall, the majority of evidence supports a benign classification and is consistent with the absence of a ClinVar assertion. Therefore, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.318242 | Structured | 0.019545 | Uncertain | 0.901 | 0.403 | 0.000 | -0.171 | Likely Benign | 0.197 | Likely Benign | Likely Benign | 0.13 | Likely Benign | 0.3 | -0.53 | Ambiguous | -0.20 | Likely Benign | 0.00 | Likely Benign | 0.383 | Likely Benign | 1.62 | Neutral | 0.979 | Probably Damaging | 0.820 | Possibly Damaging | -1.14 | Pathogenic | 0.40 | Tolerated | 0.2402 | 0.0887 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.158G>T | G53V 2D AIThe SynGAP1 missense variant G53V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM. Tools that agree on a pathogenic effect include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign). Foldetta results are unavailable. Overall, the majority of predictions (six pathogenic vs. three benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.268042 | Structured | 0.460894 | Uncertain | 0.386 | 0.666 | 0.000 | -8.308 | Likely Pathogenic | 0.959 | Likely Pathogenic | Likely Pathogenic | 0.238 | Likely Benign | -1.71 | Neutral | 0.994 | Probably Damaging | 0.990 | Probably Damaging | 4.11 | Benign | 0.00 | Affected | 0.1193 | 0.4833 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.1595C>G | T532R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T532R missense variant is not listed in ClinVar and has no reported allele in gnomAD. Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, FATHMM, and AlphaMissense‑Default. FoldX and Rosetta provide uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labeling it likely pathogenic, and Foldetta predicting a benign effect. Overall, the balance of evidence (seven benign versus five pathogenic predictions) indicates that the variant is most likely benign, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.021478 | Uncertain | 0.889 | 0.385 | 0.000 | -6.564 | Likely Benign | 0.608 | Likely Pathogenic | Likely Benign | -0.57 | Ambiguous | 0.2 | 0.68 | Ambiguous | 0.06 | Likely Benign | 0.48 | Likely Benign | 0.495 | Likely Benign | -2.62 | Deleterious | 0.694 | Possibly Damaging | 0.230 | Benign | -1.20 | Pathogenic | 0.06 | Tolerated | 0.0841 | 0.1755 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.1600T>C | S534P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S534P is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33438843‑T‑C). Functional prediction tools that report a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The high‑accuracy assessments are consistent with a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Based on the aggregate predictions, the variant is most likely benign, which does not contradict the ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.167087 | Structured | 0.032173 | Uncertain | 0.860 | 0.362 | 0.000 | Uncertain | 1 | 6-33438843-T-C | 3 | 1.86e-6 | -5.056 | Likely Benign | 0.265 | Likely Benign | Likely Benign | -0.40 | Likely Benign | 0.2 | 0.35 | Likely Benign | -0.03 | Likely Benign | 0.47 | Likely Benign | 0.203 | Likely Benign | -3.81 | Deleterious | 0.993 | Probably Damaging | 0.993 | Probably Damaging | 3.32 | Benign | 0.05 | Affected | 3.37 | 35 | 0.2071 | 0.4650 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||
| c.1603A>C | S535R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S535R is not reported in ClinVar and is present in gnomAD (ID 6‑33438846‑A‑C). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results come from AlphaMissense‑Optimized, Foldetta, Rosetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain. Overall, the balance of evidence, especially the SGM Consensus, points to a pathogenic interpretation. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.041365 | Uncertain | 0.918 | 0.343 | 0.000 | 6-33438846-A-C | 3 | 1.86e-6 | -9.363 | Likely Pathogenic | 0.913 | Likely Pathogenic | Ambiguous | -0.37 | Likely Benign | 0.0 | -0.97 | Ambiguous | -0.67 | Ambiguous | 0.64 | Ambiguous | 0.390 | Likely Benign | -1.99 | Neutral | 0.830 | Possibly Damaging | 0.274 | Benign | -1.23 | Pathogenic | 0.19 | Tolerated | 3.37 | 35 | 0.1086 | 0.3743 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||
| c.1604G>T | S535I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S535I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and FATHMM. The high‑accuracy AlphaMissense‑Optimized score is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a benign outcome. Overall, the preponderance of evidence points to a benign impact for S535I, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.236433 | Structured | 0.041365 | Uncertain | 0.918 | 0.343 | 0.000 | -8.073 | Likely Pathogenic | 0.330 | Likely Benign | Likely Benign | 0.50 | Ambiguous | 0.0 | -0.42 | Likely Benign | 0.04 | Likely Benign | 0.14 | Likely Benign | 0.246 | Likely Benign | -2.49 | Neutral | 0.004 | Benign | 0.004 | Benign | -1.25 | Pathogenic | 0.07 | Tolerated | 0.1123 | 0.5571 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||
| c.1605T>A | S535R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S535R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, FoldX, PROVEAN, polyPhen‑2 HumVar, and SIFT, while pathogenic calls come from polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results are reported by Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments indicate that AlphaMissense‑Optimized is inconclusive, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta is also inconclusive. Overall, the balance of evidence, particularly the SGM Consensus, points to a pathogenic effect. This conclusion does not conflict with ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.041365 | Uncertain | 0.918 | 0.343 | 0.000 | -9.363 | Likely Pathogenic | 0.913 | Likely Pathogenic | Ambiguous | -0.37 | Likely Benign | 0.0 | -0.97 | Ambiguous | -0.67 | Ambiguous | 0.64 | Ambiguous | 0.432 | Likely Benign | -1.99 | Neutral | 0.830 | Possibly Damaging | 0.274 | Benign | -1.23 | Pathogenic | 0.19 | Tolerated | 3.37 | 35 | 0.1086 | 0.3743 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||
| c.1605T>G | S535R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S535R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, FoldX, PROVEAN, polyPhen‑2 HumVar, and SIFT, while pathogenic calls come from polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results are reported by Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments indicate that AlphaMissense‑Optimized is inconclusive, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta is also inconclusive. Overall, the balance of evidence, particularly the SGM Consensus, points to a pathogenic effect. This conclusion does not conflict with ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.041365 | Uncertain | 0.918 | 0.343 | 0.000 | -9.363 | Likely Pathogenic | 0.913 | Likely Pathogenic | Ambiguous | -0.37 | Likely Benign | 0.0 | -0.97 | Ambiguous | -0.67 | Ambiguous | 0.64 | Ambiguous | 0.432 | Likely Benign | -1.99 | Neutral | 0.830 | Possibly Damaging | 0.274 | Benign | -1.23 | Pathogenic | 0.19 | Tolerated | 3.37 | 35 | 0.1086 | 0.3743 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||
| c.1634T>C | M545T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M545T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only SIFT, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) uniformly predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta’s protein‑folding stability analysis is inconclusive. Taken together, the preponderance of evidence points to a pathogenic effect for M545T. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.025762 | Structured | 0.012875 | Uncertain | 0.955 | 0.311 | 0.000 | -8.070 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.94 | Ambiguous | 0.2 | 0.78 | Ambiguous | 0.86 | Ambiguous | 0.97 | Ambiguous | 0.722 | Likely Pathogenic | -5.03 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | -1.24 | Pathogenic | 0.36 | Tolerated | 0.1727 | 0.1847 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.1636T>A | C546S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C546S is reported in gnomAD (ID 6‑33438879‑T‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions from FoldX and SIFT; pathogenic predictions from REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools give uncertain results: Rosetta and Foldetta. High‑accuracy assessments reinforce a pathogenic signal: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains inconclusive. Overall, the preponderance of evidence, including the high‑accuracy tools, indicates that C546S is most likely pathogenic, and this assessment does not conflict with any ClinVar classification because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.027463 | Structured | 0.009041 | Uncertain | 0.960 | 0.288 | 0.000 | 6-33438879-T-A | 1 | 6.20e-7 | -8.079 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.44 | Likely Benign | 0.1 | 1.39 | Ambiguous | 0.92 | Ambiguous | 1.65 | Destabilizing | 0.836 | Likely Pathogenic | -8.04 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.21 | Pathogenic | 0.17 | Tolerated | 3.37 | 35 | 0.4343 | 0.1950 | -1 | 0 | -3.3 | -16.06 | ||||||||||||||||||||||||
| c.1637G>C | C546S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C546S is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX and SIFT, whereas the majority of tools predict a pathogenic impact: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Rosetta and Foldetta are uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for C546S, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.027463 | Structured | 0.009041 | Uncertain | 0.960 | 0.288 | 0.000 | -8.079 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.44 | Likely Benign | 0.1 | 1.39 | Ambiguous | 0.92 | Ambiguous | 1.65 | Destabilizing | 0.788 | Likely Pathogenic | -8.04 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.21 | Pathogenic | 0.17 | Tolerated | 3.37 | 35 | 0.4343 | 0.1950 | -1 | 0 | -3.3 | -16.06 | |||||||||||||||||||||||||||
| c.164A>C | Q55P 2D AIThe SynGAP1 missense variant Q55P is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33423573‑A‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, whereas those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs two benign), and Foldetta results are unavailable. Overall, more tools predict pathogenicity than benignity, and no ClinVar entry contradicts this assessment. Therefore, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.332115 | Structured | 0.470108 | Uncertain | 0.461 | 0.657 | 0.000 | 6-33423573-A-C | 1 | 6.20e-7 | -13.163 | Likely Pathogenic | 0.897 | Likely Pathogenic | Ambiguous | 0.260 | Likely Benign | -2.06 | Neutral | 0.462 | Possibly Damaging | 0.480 | Possibly Damaging | 3.83 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2557 | 0.5508 | -1 | 0 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||
| c.1661T>G | V554G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V554G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, while the remaining eleven tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as pathogenic. No predictions are missing or inconclusive. Overall, the variant is most likely pathogenic based on the collective evidence, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.020522 | Structured | 0.007349 | Uncertain | 0.955 | 0.226 | 0.000 | -13.879 | Likely Pathogenic | 0.942 | Likely Pathogenic | Ambiguous | 3.31 | Destabilizing | 0.1 | 4.13 | Destabilizing | 3.72 | Destabilizing | 2.40 | Destabilizing | 0.594 | Likely Pathogenic | -6.96 | Deleterious | 0.997 | Probably Damaging | 1.000 | Probably Damaging | 3.21 | Benign | 0.00 | Affected | 0.1619 | 0.1548 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1663G>T | V555F 2D 3DClick to see structure in 3D Viewer AISynGAP1 V555F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or stability result is missing or inconclusive. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation because none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.013265 | Structured | 0.008218 | Uncertain | 0.943 | 0.225 | 0.000 | -10.965 | Likely Pathogenic | 0.599 | Likely Pathogenic | Likely Benign | 0.17 | Likely Benign | 0.3 | -2.01 | Stabilizing | -0.92 | Ambiguous | 0.16 | Likely Benign | 0.440 | Likely Benign | -2.85 | Deleterious | 0.011 | Benign | 0.013 | Benign | -1.20 | Pathogenic | 0.16 | Tolerated | 0.0727 | 0.2366 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1664T>G | V555G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V555G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on pathogenicity include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; no tool predicts it benign. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as pathogenic. No predictions are inconclusive or missing. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.013265 | Structured | 0.008218 | Uncertain | 0.943 | 0.225 | 0.000 | -13.327 | Likely Pathogenic | 0.899 | Likely Pathogenic | Ambiguous | 2.07 | Destabilizing | 0.1 | 2.22 | Destabilizing | 2.15 | Destabilizing | 1.07 | Destabilizing | 0.798 | Likely Pathogenic | -6.35 | Deleterious | 0.984 | Probably Damaging | 1.000 | Probably Damaging | -1.39 | Pathogenic | 0.01 | Affected | 0.1994 | 0.1677 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1679T>G | V560G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V560G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only SIFT, whereas the majority of tools (REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. Uncertain results come from FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, AlphaMissense‑Optimized is uncertain, and Foldetta is uncertain. Overall, the preponderance of evidence indicates that V560G is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.021381 | Structured | 0.013872 | Uncertain | 0.853 | 0.204 | 0.000 | -12.485 | Likely Pathogenic | 0.799 | Likely Pathogenic | Ambiguous | 0.66 | Ambiguous | 0.1 | 2.12 | Destabilizing | 1.39 | Ambiguous | 1.80 | Destabilizing | 0.753 | Likely Pathogenic | -5.87 | Deleterious | 0.981 | Probably Damaging | 1.000 | Probably Damaging | -1.25 | Pathogenic | 0.19 | Tolerated | 0.1738 | 0.2036 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1681T>G | F561V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F561V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Pathogenic. All available evidence points to a pathogenic impact. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.024826 | Structured | 0.018013 | Uncertain | 0.903 | 0.196 | 0.000 | -11.371 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 3.11 | Destabilizing | 0.1 | 2.58 | Destabilizing | 2.85 | Destabilizing | 1.48 | Destabilizing | 0.827 | Likely Pathogenic | -6.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.25 | Pathogenic | 0.04 | Affected | 0.1985 | 0.1575 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1685C>A | P562Q 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P562Q is recorded in gnomAD (variant ID 6‑33440737‑C‑A) but has no ClinVar entry. All available in silico predictors classify the substitution as pathogenic: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a benign outcome, so the benign group is empty. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Consequently, the variant is most likely pathogenic based on the consensus of predictive tools, and this assessment is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.022306 | Structured | 0.023606 | Uncertain | 0.893 | 0.200 | 0.000 | 6-33440737-C-A | -15.705 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.18 | Destabilizing | 0.4 | 1.22 | Ambiguous | 2.70 | Destabilizing | 1.24 | Destabilizing | 0.778 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 0.57 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.1634 | 0.3184 | -1 | 0 | -1.9 | 31.01 | ||||||||||||||||||||||||||
| c.1688G>C | R563T 2D 3DClick to see structure in 3D Viewer AISynGAP1 R563T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, and FATHMM, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The remaining tools—FoldX, Rosetta, ESM1b, AlphaMissense‑Optimized, and Foldetta—give uncertain or inconclusive results. High‑accuracy methods provide the following: AlphaMissense‑Optimized is unavailable; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic; Foldetta is unavailable. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.039760 | Structured | 0.031987 | Uncertain | 0.876 | 0.209 | 0.000 | -7.088 | In-Between | 0.913 | Likely Pathogenic | Ambiguous | 1.13 | Ambiguous | 0.1 | 0.77 | Ambiguous | 0.95 | Ambiguous | 0.18 | Likely Benign | 0.307 | Likely Benign | -4.77 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.49 | Benign | 0.23 | Tolerated | 0.2115 | 0.3148 | -1 | -1 | 3.8 | -55.08 | ||||||||||||||||||||||||||||||
| c.1688G>T | R563M 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant R563M is reported in gnomAD (ID 6‑33440740‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, FATHMM, premPS, and Foldetta; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (integrating FoldX‑MD and Rosetta outputs) as benign. No prediction or stability result is missing; all available data are considered. Overall, the balance of evidence leans toward a pathogenic effect, with a single high‑accuracy tool (Foldetta) suggesting benign stability. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.039760 | Structured | 0.031987 | Uncertain | 0.876 | 0.209 | 0.000 | 6-33440740-G-T | -8.910 | Likely Pathogenic | 0.934 | Likely Pathogenic | Ambiguous | -0.18 | Likely Benign | 0.1 | 0.70 | Ambiguous | 0.26 | Likely Benign | 0.17 | Likely Benign | 0.311 | Likely Benign | -4.91 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.43 | Benign | 0.04 | Affected | 3.37 | 35 | 0.1636 | 0.2230 | -1 | 0 | 6.4 | -24.99 | ||||||||||||||||||||||||||
| c.1705T>G | F569V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F569V is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.024393 | Structured | 0.054289 | Uncertain | 0.941 | 0.242 | 0.000 | -14.248 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 4.74 | Destabilizing | 0.2 | 5.40 | Destabilizing | 5.07 | Destabilizing | 1.73 | Destabilizing | 0.874 | Likely Pathogenic | -6.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.29 | Pathogenic | 0.00 | Affected | 0.2054 | 0.1422 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1739G>T | G580V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G580V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess functional impact uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. The only inconclusive result is premPS, which is listed as uncertain. No tool predicts a benign effect. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenic. Based on the unanimous pathogenic predictions and the absence of any benign calls, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.025952 | Uncertain | 0.853 | 0.236 | 0.000 | -13.705 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 4.10 | Destabilizing | 0.1 | 3.89 | Destabilizing | 4.00 | Destabilizing | 0.79 | Ambiguous | 0.750 | Likely Pathogenic | -8.66 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.18 | Pathogenic | 0.04 | Affected | 0.1270 | 0.2909 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.173T>C | M58T 2D AIThe SynGAP1 missense variant M58T is listed in gnomAD (ID 6‑33423582‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | 6-33423582-T-C | 1 | 6.20e-7 | -4.308 | Likely Benign | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.159 | Likely Benign | -1.58 | Neutral | 0.018 | Benign | 0.184 | Benign | 4.09 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1999 | 0.2357 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.1751T>G | I584S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I584S missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on pathogenicity include SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign effect. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as pathogenic. No contradictory evidence is present. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not conflict with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.059222 | Structured | 0.046673 | Uncertain | 0.846 | 0.244 | 0.000 | -13.379 | Likely Pathogenic | 0.945 | Likely Pathogenic | Ambiguous | 3.15 | Destabilizing | 0.1 | 2.53 | Destabilizing | 2.84 | Destabilizing | 1.70 | Destabilizing | 0.710 | Likely Pathogenic | -5.54 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | -1.18 | Pathogenic | 0.03 | Affected | 0.2391 | 0.0858 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1760G>C | R587T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R587T is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include SIFT and AlphaMissense‑Optimized, whereas a majority of tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic outcome. Uncertain predictions from FoldX, Rosetta, Foldetta, and premPS are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for R587T, which does not contradict the ClinVar “Uncertain” classification but suggests that the variant is more likely pathogenic rather than benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | Uncertain | 1 | -9.697 | Likely Pathogenic | 0.784 | Likely Pathogenic | Likely Benign | 1.14 | Ambiguous | 0.2 | 0.74 | Ambiguous | 0.94 | Ambiguous | 0.98 | Ambiguous | 0.603 | Likely Pathogenic | -4.71 | Deleterious | 0.998 | Probably Damaging | 0.847 | Possibly Damaging | -1.19 | Pathogenic | 0.08 | Tolerated | 3.37 | 35 | 0.1958 | 0.4578 | -1 | -1 | 3.8 | -55.08 | 227.2 | 87.4 | 0.0 | 0.0 | 0.5 | 0.1 | X | Potentially Pathogenic | The guanidinium group of Arg587, located on an α helix (res. Glu582-Met603), is constantly rotating and breaking/forming multiple hydrogen bonds and/or salt bridges at the surface intersection of α helices in the WT simulations. The positively charged Arg587 side chain can form a salt bridge with either the carboxylate group of Asp583 or Asp586 in the same helix, or with Glu480 on the opposing short helical loop structure (res. Glu480-Leu482).Importantly, the Arg587 side chain also hydrogen bonds with the backbone carbonyl groups of Ala634 and Asn635, as well as the carboxamide group of Asn635 at the end of another α helix (res. Asp616-Phe636). However, in the variant simulations, the neutral hydroxyl group of the Thr587 side chain is unable to form these salt bridges. Due to its smaller size, it also does not form the hydrogen bonds that the Arg587 side chain could. Instead, the hydroxyl group of Thr587 hydrogen bonds with the backbone carbonyl group of Asp583, which could weaken the integrity of the α helix, although this is not observed in the simulations.Overall, the residue swap could weaken the tertiary structure assembly and negatively affect the overall protein folding process. | ||||||||||||||||
| c.1761G>C | R587S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R587S is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: the single benign prediction comes from SIFT, while the remaining tools—REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus—indicate pathogenicity. High‑accuracy assessments further support a deleterious effect: the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; AlphaMissense‑Optimized is “Uncertain”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is also “Uncertain.” Taken together, the preponderance of evidence points to a pathogenic impact for R587S. This conclusion is not contradicted by ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | -12.264 | Likely Pathogenic | 0.830 | Likely Pathogenic | Ambiguous | 0.84 | Ambiguous | 0.1 | 1.79 | Ambiguous | 1.32 | Ambiguous | 1.17 | Destabilizing | 0.508 | Likely Pathogenic | -4.84 | Deleterious | 0.990 | Probably Damaging | 0.779 | Possibly Damaging | -1.20 | Pathogenic | 0.09 | Tolerated | 3.37 | 35 | 0.2852 | 0.4165 | -1 | 0 | 3.7 | -69.11 | |||||||||||||||||||||||||||
| c.1761G>T | R587S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R587S missense variant is catalogued in gnomAD (ID 6‑33440813‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default, while only SIFT predicts a benign outcome. Uncertain results are reported by FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized remains uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as pathogenic, and Foldetta likewise yields an uncertain stability change. Overall, the preponderance of evidence indicates that R587S is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | 6-33440813-G-T | 4 | 2.48e-6 | -12.264 | Likely Pathogenic | 0.830 | Likely Pathogenic | Ambiguous | 0.84 | Ambiguous | 0.1 | 1.79 | Ambiguous | 1.32 | Ambiguous | 1.17 | Destabilizing | 0.508 | Likely Pathogenic | -4.84 | Deleterious | 0.990 | Probably Damaging | 0.779 | Possibly Damaging | -1.20 | Pathogenic | 0.09 | Tolerated | 3.37 | 35 | 0.2852 | 0.4165 | -1 | 0 | 3.7 | -69.11 | ||||||||||||||||||||||||
| c.1766T>G | I589S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I589S is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a pathogenic or likely pathogenic outcome. No tool in the dataset predicts a benign effect. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also labels it pathogenic. Based on the uniform predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.018415 | Structured | 0.084536 | Uncertain | 0.927 | 0.214 | 0.000 | -15.223 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.62 | Destabilizing | 0.1 | 4.12 | Destabilizing | 3.87 | Destabilizing | 2.21 | Destabilizing | 0.954 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.99 | Pathogenic | 0.00 | Affected | 0.2786 | 0.1130 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1769G>T | S590I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S590I is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include FoldX, premPS, and FATHMM, whereas the majority of tools predict it to be pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools give uncertain results: Rosetta and Foldetta. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence from multiple independent predictors indicates that S590I is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.022667 | Structured | 0.088943 | Uncertain | 0.918 | 0.199 | 0.000 | -17.956 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | -0.04 | Likely Benign | 0.5 | 1.15 | Ambiguous | 0.56 | Ambiguous | 0.31 | Likely Benign | 0.686 | Likely Pathogenic | -5.78 | Deleterious | 0.998 | Probably Damaging | 0.948 | Probably Damaging | 3.05 | Benign | 0.02 | Affected | 0.0975 | 0.5025 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.1780T>G | F594V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F594V lies within the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that assess pathogenicity unanimously favor a deleterious effect: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity; only Rosetta is uncertain. No tool predicts a benign outcome. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which simply lacks an entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009187 | Structured | 0.120166 | Uncertain | 0.946 | 0.147 | 0.000 | -12.648 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 2.91 | Destabilizing | 0.2 | 1.90 | Ambiguous | 2.41 | Destabilizing | 1.80 | Destabilizing | 0.931 | Likely Pathogenic | -6.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.91 | Pathogenic | 0.01 | Affected | 0.1813 | 0.1575 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1789T>G | F597V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F597V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All available evidence points to a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion is consistent with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | -13.883 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 3.75 | Destabilizing | 0.7 | 2.02 | Destabilizing | 2.89 | Destabilizing | 1.60 | Destabilizing | 0.939 | Likely Pathogenic | -6.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -2.16 | Pathogenic | 0.01 | Affected | 0.2237 | 0.1583 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1805T>G | I602S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I602S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. No conflicting benign evidence is available. Therefore, based on the consensus of all available predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010221 | Structured | 0.186541 | Uncertain | 0.963 | 0.171 | 0.000 | -15.558 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 3.50 | Destabilizing | 0.1 | 3.79 | Destabilizing | 3.65 | Destabilizing | 2.04 | Destabilizing | 0.935 | Likely Pathogenic | -5.89 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -2.01 | Pathogenic | 0.00 | Affected | 0.2213 | 0.1087 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1808T>C | M603T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M603T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that provide a clear verdict all classify the substitution as pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool in the dataset reports a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus indicates likely pathogenic, while the Foldetta stability analysis is inconclusive and therefore not considered evidence. No contradictory evidence is present in ClinVar. Consequently, the variant is most likely pathogenic based on the available predictions, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -13.152 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 1.71 | Ambiguous | 0.2 | 1.71 | Ambiguous | 1.71 | Ambiguous | 0.66 | Ambiguous | 0.903 | Likely Pathogenic | -5.48 | Deleterious | 0.996 | Probably Damaging | 0.985 | Probably Damaging | -1.29 | Pathogenic | 0.00 | Affected | 0.1883 | 0.1495 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.1817G>T | S606I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S606I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, Rosetta, Foldetta, premPS, and FATHMM, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points toward a pathogenic impact for S606I. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.191720 | Uncertain | 0.875 | 0.247 | 0.000 | -14.552 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | -0.60 | Ambiguous | 0.1 | -0.08 | Likely Benign | -0.34 | Likely Benign | 0.41 | Likely Benign | 0.288 | Likely Benign | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.0891 | 0.4162 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.1822T>G | F608V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F608V is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; all of these return a pathogenic or likely pathogenic label. No tool in the dataset predicts a benign outcome. Uncertain results are reported only for Rosetta and Foldetta, which are treated as unavailable evidence. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta remains uncertain. Taken together, the overwhelming majority of predictions indicate that F608V is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.106997 | Structured | 0.197190 | Uncertain | 0.891 | 0.247 | 0.000 | -16.084 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 2.21 | Destabilizing | 0.1 | 1.39 | Ambiguous | 1.80 | Ambiguous | 1.47 | Destabilizing | 0.917 | Likely Pathogenic | -6.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.59 | Pathogenic | 0.01 | Affected | 0.2193 | 0.2199 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1826G>T | G609V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G609V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from premPS and AlphaMissense‑Optimized, whereas the remaining 10 tools (SGM Consensus, REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM) all predict pathogenicity. High‑accuracy assessments further highlight the discordance: AlphaMissense‑Optimized reports a benign effect, whereas the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta outputs) both indicate pathogenicity. With the majority of evidence pointing to deleterious impact, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.203786 | Uncertain | 0.851 | 0.252 | 0.000 | -11.049 | Likely Pathogenic | 0.374 | Ambiguous | Likely Benign | 4.17 | Destabilizing | 0.3 | 3.77 | Destabilizing | 3.97 | Destabilizing | 0.34 | Likely Benign | 0.734 | Likely Pathogenic | -4.47 | Deleterious | 0.974 | Probably Damaging | 0.818 | Possibly Damaging | -1.48 | Pathogenic | 0.02 | Affected | 0.1269 | 0.3317 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1832T>C | M611T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M611T is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33440884‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. Four tools (FoldX, Rosetta, Foldetta, premPS) return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | Uncertain | 1 | 6-33440884-T-C | 1 | 6.19e-7 | -5.696 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 1.98 | Ambiguous | 0.2 | 0.94 | Ambiguous | 1.46 | Ambiguous | 0.87 | Ambiguous | 0.240 | Likely Benign | -2.40 | Neutral | 0.034 | Benign | 0.038 | Benign | -1.19 | Pathogenic | 0.29 | Tolerated | 3.37 | 35 | 0.1635 | 0.1415 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||
| c.1877T>G | I626S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I626S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.040732 | Uncertain | 0.970 | 0.223 | 0.000 | -14.449 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 3.80 | Destabilizing | 0.0 | 4.54 | Destabilizing | 4.17 | Destabilizing | 2.16 | Destabilizing | 0.706 | Likely Pathogenic | -5.41 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.04 | Benign | 0.00 | Affected | 0.2308 | 0.0858 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1885G>T | V629F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V629F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact; premPS is uncertain and is not counted as evidence. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta is pathogenic. No prediction is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions and the absence of any ClinVar annotation, the variant is most likely pathogenic, with no contradiction from ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -13.484 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 3.41 | Destabilizing | 0.5 | 4.40 | Destabilizing | 3.91 | Destabilizing | 0.64 | Ambiguous | 0.642 | Likely Pathogenic | -4.68 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.07 | Benign | 0.00 | Affected | 0.0560 | 0.2915 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1886T>G | V629G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 V629G missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining eleven tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) uniformly predict a pathogenic outcome; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. Taken together, the overwhelming majority of evidence indicates that V629G is likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -13.150 | Likely Pathogenic | 0.871 | Likely Pathogenic | Ambiguous | 3.81 | Destabilizing | 0.0 | 4.52 | Destabilizing | 4.17 | Destabilizing | 1.94 | Destabilizing | 0.678 | Likely Pathogenic | -6.47 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.07 | Benign | 0.00 | Affected | 0.1903 | 0.2240 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1889T>G | I630S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I630S is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as pathogenic. No contradictory evidence is present. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not conflict with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.036106 | Uncertain | 0.966 | 0.236 | 0.000 | -12.527 | Likely Pathogenic | 0.952 | Likely Pathogenic | Ambiguous | 3.61 | Destabilizing | 0.1 | 3.74 | Destabilizing | 3.68 | Destabilizing | 2.25 | Destabilizing | 0.851 | Likely Pathogenic | -3.86 | Deleterious | 1.000 | Probably Damaging | 0.981 | Probably Damaging | -1.44 | Pathogenic | 0.00 | Affected | 0.2363 | 0.0858 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1906T>G | F636V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F636V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | -14.603 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 3.06 | Destabilizing | 0.1 | 5.13 | Destabilizing | 4.10 | Destabilizing | 1.07 | Destabilizing | 0.533 | Likely Pathogenic | -6.74 | Deleterious | 0.991 | Probably Damaging | 0.985 | Probably Damaging | 3.44 | Benign | 0.04 | Affected | 0.1841 | 0.2030 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1915T>G | F639V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F639V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while the remaining 12 tools (SGM‑Consensus, REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity; Rosetta is uncertain and is therefore not counted. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.117665 | Uncertain | 0.942 | 0.284 | 0.000 | -12.910 | Likely Pathogenic | 0.965 | Likely Pathogenic | Likely Pathogenic | 4.17 | Destabilizing | 0.1 | 1.18 | Ambiguous | 2.68 | Destabilizing | 1.47 | Destabilizing | 0.560 | Likely Pathogenic | -6.97 | Deleterious | 0.992 | Probably Damaging | 0.756 | Possibly Damaging | 3.08 | Benign | 0.02 | Affected | 0.2074 | 0.1583 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.191T>C | I64T 2D  AIThe SynGAP1 missense variant I64T is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33425799‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Those that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Benign, and the Foldetta protein‑folding stability analysis is unavailable. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | 6-33425799-T-C | 1 | 6.20e-7 | -3.183 | Likely Benign | 0.943 | Likely Pathogenic | Ambiguous | 0.075 | Likely Benign | -0.51 | Neutral | 0.092 | Benign | 0.007 | Benign | 4.08 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0978 | 0.0851 | -1 | 0 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||||
| c.1933T>G | F645V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F645V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that agree on a pathogenic effect are premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy assessments give an uncertain result from AlphaMissense‑Optimized, a pathogenic outcome from the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and an uncertain outcome from Foldetta (combining FoldX‑MD and Rosetta). No definitive folding‑stability change is reported. Overall, the majority of evidence points to a pathogenic impact for F645V, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -12.175 | Likely Pathogenic | 0.810 | Likely Pathogenic | Ambiguous | 1.98 | Ambiguous | 0.1 | 1.70 | Ambiguous | 1.84 | Ambiguous | 1.02 | Destabilizing | 0.316 | Likely Benign | -4.41 | Deleterious | 0.036 | Benign | 0.018 | Benign | 3.40 | Benign | 0.02 | Affected | 0.2163 | 0.2919 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1940G>A | G647D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G647D is reported in ClinVar as having no entry and is present in gnomAD (6‑33441199‑G‑A). Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized all predict benign. Only AlphaMissense‑Default predicts pathogenic, while FoldX, Foldetta, premPS, and ESM1b are uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields benign; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. Overall, the consensus of most tools and the high‑accuracy predictions indicate that G647D is most likely benign, and this is consistent with the absence of a pathogenic ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | 6-33441199-G-A | 1 | 6.20e-7 | -7.643 | In-Between | 0.582 | Likely Pathogenic | Likely Benign | 0.68 | Ambiguous | 0.1 | 0.33 | Likely Benign | 0.51 | Ambiguous | 0.56 | Ambiguous | 0.098 | Likely Benign | -1.63 | Neutral | 0.282 | Benign | 0.128 | Benign | 3.45 | Benign | 0.24 | Tolerated | 3.37 | 30 | 0.1630 | 0.1372 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||
| c.1940G>T | G647V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G647V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that assess sequence conservation and structural impact uniformly classify the substitution as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a tolerated change. No tool predicts pathogenicity; only FoldX and Rosetta report uncertain stability effects, which are treated as unavailable evidence. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a benign effect. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -4.713 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.85 | Ambiguous | 0.1 | -0.59 | Ambiguous | 0.13 | Likely Benign | -0.13 | Likely Benign | 0.115 | Likely Benign | -1.77 | Neutral | 0.178 | Benign | 0.073 | Benign | 3.52 | Benign | 0.12 | Tolerated | 0.1318 | 0.3946 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1942T>G | F648V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F648V is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include SIFT and FATHMM, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and the SGM‑Consensus—predict it to be pathogenic. High‑accuracy methods give consistent results: AlphaMissense‑Optimized indicates pathogenicity; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a destabilizing, pathogenic effect. No prediction is missing or inconclusive. Overall, the evidence overwhelmingly supports a pathogenic classification, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.346782 | Uncertain | 0.943 | 0.339 | 0.000 | -11.574 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 3.11 | Destabilizing | 0.1 | 2.80 | Destabilizing | 2.96 | Destabilizing | 1.14 | Destabilizing | 0.514 | Likely Pathogenic | -6.98 | Deleterious | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 3.49 | Benign | 0.06 | Tolerated | 0.1848 | 0.1983 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1946T>C | M649T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M649T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.360413 | Uncertain | 0.962 | 0.345 | 0.000 | -11.916 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 3.19 | Destabilizing | 0.2 | 3.45 | Destabilizing | 3.32 | Destabilizing | 1.92 | Destabilizing | 0.531 | Likely Pathogenic | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.779 | Possibly Damaging | 3.35 | Benign | 0.00 | Affected | 0.1732 | 0.1615 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.1954T>G | F652V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F652V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, whereas only FATHMM predicts a benign outcome. The high‑accuracy subset confirms this trend: AlphaMissense‑Optimized is pathogenic; SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.356594 | Uncertain | 0.966 | 0.338 | 0.000 | -11.576 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 2.66 | Destabilizing | 0.1 | 3.21 | Destabilizing | 2.94 | Destabilizing | 1.49 | Destabilizing | 0.595 | Likely Pathogenic | -6.71 | Deleterious | 0.995 | Probably Damaging | 0.885 | Possibly Damaging | 3.05 | Benign | 0.00 | Affected | 0.1656 | 0.1783 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1973G>T | G658V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G658V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv; Rosetta’s output is uncertain and therefore not used as evidence. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta) as Benign. Taken together, the majority of reliable predictors indicate a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.029376 | Structured | 0.180299 | Uncertain | 0.942 | 0.251 | 0.000 | -6.815 | Likely Benign | 0.166 | Likely Benign | Likely Benign | -0.07 | Likely Benign | 0.0 | -0.71 | Ambiguous | -0.39 | Likely Benign | 0.34 | Likely Benign | 0.117 | Likely Benign | -3.23 | Deleterious | 0.841 | Possibly Damaging | 0.264 | Benign | 3.38 | Benign | 0.13 | Tolerated | 0.1337 | 0.3330 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1979T>C | M660T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M660T is catalogued in gnomAD (ID 6‑33441238‑T‑C) but has no ClinVar entry, so its clinical status is currently unreported. In silico prediction tools largely agree that the substitution is deleterious: pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a benign effect. High‑accuracy assessments reinforce the pathogenic view: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | 6-33441238-T-C | 1 | 6.20e-7 | -9.791 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 3.62 | Destabilizing | 0.1 | 2.05 | Destabilizing | 2.84 | Destabilizing | 1.91 | Destabilizing | 0.561 | Likely Pathogenic | -5.99 | Deleterious | 0.967 | Probably Damaging | 0.633 | Possibly Damaging | 3.36 | Benign | 0.02 | Affected | 3.38 | 28 | 0.1811 | 0.1630 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||
| c.1987T>G | F663V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F663V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all indicate pathogenicity, while only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the change as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic effect. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.056825 | Structured | 0.093963 | Uncertain | 0.944 | 0.355 | 0.000 | -14.196 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 3.04 | Destabilizing | 0.1 | 3.31 | Destabilizing | 3.18 | Destabilizing | 1.75 | Destabilizing | 0.655 | Likely Pathogenic | -6.98 | Deleterious | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 3.02 | Benign | 0.00 | Affected | 0.1844 | 0.1983 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.19T>C | S7P 2D AIThe SynGAP1 missense variant S7P is reported in gnomAD (ID 6‑33420283‑T‑C) but has no ClinVar entry. In silico prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign status. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, points to a benign effect. This prediction is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | 6-33420283-T-C | -3.798 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.210 | Likely Benign | -0.23 | Neutral | 0.267 | Benign | 0.029 | Benign | 4.07 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2474 | 0.4925 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||||
| c.2000T>G | I667S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I667S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.083597 | Uncertain | 0.927 | 0.379 | 0.000 | -14.328 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 3.69 | Destabilizing | 0.1 | 3.97 | Destabilizing | 3.83 | Destabilizing | 2.48 | Destabilizing | 0.690 | Likely Pathogenic | -5.90 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.96 | Benign | 0.00 | Affected | 0.2462 | 0.0858 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.2015C>G | T672R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T672R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, Foldetta, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Uncertain results from Rosetta and premPS are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, while the SGM Consensus indicates a likely pathogenic outcome. Overall, the majority of individual predictors lean toward pathogenicity, but the high‑accuracy tools provide conflicting benign signals. Therefore, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.102069 | Uncertain | 0.586 | 0.362 | 0.000 | -12.454 | Likely Pathogenic | 0.626 | Likely Pathogenic | Likely Benign | -0.48 | Likely Benign | 0.4 | 1.19 | Ambiguous | 0.36 | Likely Benign | 0.60 | Ambiguous | 0.084 | Likely Benign | -4.47 | Deleterious | 0.886 | Possibly Damaging | 0.377 | Benign | 3.43 | Benign | 0.02 | Affected | 0.0996 | 0.2919 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.2015C>T | T672M 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T672M is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33441274‑C‑T). Prediction tools that classify the variant as benign include REVEL, FoldX, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. Rosetta and Foldetta report uncertain results, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while SGM Consensus and Foldetta remain unavailable. Overall, the balance of evidence favors a benign effect, and this conclusion does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.116183 | Structured | 0.102069 | Uncertain | 0.586 | 0.362 | 0.000 | Conflicting | 3 | 6-33441274-C-T | 19 | 1.18e-5 | -9.472 | Likely Pathogenic | 0.174 | Likely Benign | Likely Benign | 0.31 | Likely Benign | 0.4 | 1.52 | Ambiguous | 0.92 | Ambiguous | 0.41 | Likely Benign | 0.127 | Likely Benign | -4.34 | Deleterious | 0.993 | Probably Damaging | 0.520 | Possibly Damaging | 3.39 | Benign | 0.00 | Affected | 3.40 | 25 | 0.1332 | 0.6677 | -1 | -1 | 2.6 | 30.09 | 231.9 | -52.9 | 1.1 | 0.1 | 0.5 | 0.0 | X | X | Potentially Pathogenic | The hydroxyl group of Thr672, located in an entangled α-α loop connecting the two α-helices (res. Ser641-Glu666 and res. Leu685-Val699), is involved in a highly coordinated hydrogen-bonding network between residues from two α-helices (res. Ser641-Glu666 and res. Arg563-Glu578) and from the α-α loop itself, such as Lys566, Glu666, and Asn669. Met672 can only form a hydrogen bond with the amino group of the Lys566 side chain via its backbone carbonyl group. Nevertheless, the Lys566-Glu666 salt bridge forms intermittently. This is possible because Asn669 keeps the carboxylate group of Glu666 in the vicinity through hydrogen bonding, and the hydrophobic side chain of Met stays mostly rotated away from the salt bridge. Consequently, no drastic disruption of the hydrogen-bond network that keeps the loop close to the helices occurs in the variant simulations. | |||||||||||||
| c.2021C>T | T674I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T674I is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (ID 6‑33441280‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and ESM1b (polyPhen‑2 HumVar is benign). AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | 6-33441280-C-T | 2 | 1.24e-6 | -8.951 | Likely Pathogenic | 0.522 | Ambiguous | Likely Benign | -0.05 | Likely Benign | 0.1 | 0.28 | Likely Benign | 0.12 | Likely Benign | -0.04 | Likely Benign | 0.289 | Likely Benign | -3.18 | Deleterious | 0.981 | Probably Damaging | 0.444 | Benign | 3.46 | Benign | 0.11 | Tolerated | 3.40 | 24 | 0.0898 | 0.6945 | -1 | 0 | 5.2 | 12.05 | |||||||||||||||||||||||||
| c.2027G>T | S676I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and ESM1b. FoldX, Rosetta, Foldetta, and AlphaMissense‑Default are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, while Foldetta remains uncertain. Overall, the balance of evidence (five benign versus three pathogenic predictions) indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -10.165 | Likely Pathogenic | 0.383 | Ambiguous | Likely Benign | 1.25 | Ambiguous | 0.3 | 1.12 | Ambiguous | 1.19 | Ambiguous | 0.14 | Likely Benign | 0.170 | Likely Benign | -2.46 | Neutral | 0.801 | Possibly Damaging | 0.063 | Benign | 3.38 | Benign | 0.02 | Affected | 0.0879 | 0.5720 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||
| c.2029A>T | S677C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677C is reported in ClinVar as Benign (ClinVar ID 2825814.0) and is not present in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 HumDiv, SIFT, and ESM1b predict a pathogenic impact. High‑accuracy predictors all support a benign outcome: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No prediction or folding‑stability result is missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this assessment aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | Benign | 1 | -8.496 | Likely Pathogenic | 0.076 | Likely Benign | Likely Benign | -0.51 | Ambiguous | 0.3 | -0.30 | Likely Benign | -0.41 | Likely Benign | 0.15 | Likely Benign | 0.153 | Likely Benign | -2.41 | Neutral | 0.932 | Possibly Damaging | 0.222 | Benign | 3.25 | Benign | 0.04 | Affected | 3.41 | 23 | 0.1375 | 0.6697 | -1 | 0 | 3.3 | 16.06 | |||||||||||||||||||||||||
| c.2030G>T | S677I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenicity is suggested only by PROVEAN and SIFT, while Foldetta and ESM1b give uncertain results. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign, and Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | -7.942 | In-Between | 0.156 | Likely Benign | Likely Benign | -0.09 | Likely Benign | 0.0 | -2.25 | Stabilizing | -1.17 | Ambiguous | -0.01 | Likely Benign | 0.097 | Likely Benign | -2.81 | Deleterious | 0.002 | Benign | 0.002 | Benign | 3.34 | Benign | 0.05 | Affected | 0.1040 | 0.6463 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||
| c.2033G>T | S678I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of evidence (10 benign vs. 3 pathogenic) supports a benign classification. This consensus does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | -9.735 | Likely Pathogenic | 0.253 | Likely Benign | Likely Benign | 0.36 | Likely Benign | 0.3 | -0.01 | Likely Benign | 0.18 | Likely Benign | -0.11 | Likely Benign | 0.119 | Likely Benign | -3.99 | Deleterious | 0.294 | Benign | 0.057 | Benign | 3.45 | Benign | 0.01 | Affected | 0.0941 | 0.5954 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||
| c.2035T>G | F679V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F679V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic, while AlphaMissense‑Optimized and Foldetta provide inconclusive results and are treated as unavailable. Overall, the balance of evidence favors a pathogenic classification, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.129316 | Uncertain | 0.700 | 0.320 | 0.000 | -11.868 | Likely Pathogenic | 0.900 | Likely Pathogenic | Ambiguous | 1.92 | Ambiguous | 0.5 | 0.29 | Likely Benign | 1.11 | Ambiguous | 0.89 | Ambiguous | 0.497 | Likely Benign | -6.86 | Deleterious | 0.993 | Probably Damaging | 0.968 | Probably Damaging | 3.50 | Benign | 0.01 | Affected | 0.1830 | 0.2349 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.2042G>T | G681V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G681V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the majority of algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—classify the change as pathogenic. High‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain; SGM‑Consensus predicts a likely pathogenic effect; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic outcome. No other high‑confidence predictions are available. Taken together, the consensus of pathogenic predictions outweighs the single benign call, indicating that G681V is most likely pathogenic. This assessment is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | -14.043 | Likely Pathogenic | 0.953 | Likely Pathogenic | Ambiguous | 3.21 | Destabilizing | 2.0 | 6.12 | Destabilizing | 4.67 | Destabilizing | 0.64 | Ambiguous | 0.572 | Likely Pathogenic | -8.98 | Deleterious | 0.999 | Probably Damaging | 0.928 | Probably Damaging | 3.33 | Benign | 0.01 | Affected | 0.1350 | 0.3840 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2048T>G | I683S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I683S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to FATHMM, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is Pathogenic. No prediction or folding stability result is missing or inconclusive; all available evidence points to a deleterious effect. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.143268 | Uncertain | 0.848 | 0.314 | 0.000 | -11.443 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 2.53 | Destabilizing | 0.2 | 1.94 | Ambiguous | 2.24 | Destabilizing | 1.35 | Destabilizing | 0.552 | Likely Pathogenic | -5.88 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 3.29 | Benign | 0.05 | Affected | 0.1936 | 0.1320 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.2050G>C | D684H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684H is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM; all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact, and the SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are inconclusive or missing. Based on the overwhelming majority of pathogenic predictions, the variant is most likely pathogenic, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | Uncertain | 1 | -14.194 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.36 | Destabilizing | 1.0 | 2.95 | Destabilizing | 3.16 | Destabilizing | 0.55 | Ambiguous | 0.613 | Likely Pathogenic | -6.98 | Deleterious | 1.000 | Probably Damaging | 0.972 | Probably Damaging | 3.36 | Benign | 0.00 | Affected | 3.42 | 17 | 0.1344 | 0.6618 | -1 | 1 | 0.3 | 22.05 | |||||||||||||||||||||||||
| c.2057G>T | G686V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G686V has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on benign impact are Rosetta and FATHMM, while the majority of other in silico predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score (Likely Pathogenic) indicate a pathogenic effect. Uncertain results come from FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for G686V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.177104 | Uncertain | 0.919 | 0.268 | 0.000 | -13.751 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.89 | Ambiguous | 0.5 | 0.21 | Likely Benign | 0.55 | Ambiguous | 0.74 | Ambiguous | 0.570 | Likely Pathogenic | -8.08 | Deleterious | 1.000 | Probably Damaging | 0.979 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.1059 | 0.3646 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.206T>C | I69T 2D AIThe SynGAP1 missense variant I69T is listed in gnomAD (ID 6‑33425814‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as “Likely Benign” (three benign votes versus one pathogenic). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of evidence points to a benign effect for I69T, and this conclusion does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | 6-33425814-T-C | 1 | 6.20e-7 | -2.978 | Likely Benign | 0.755 | Likely Pathogenic | Likely Benign | 0.116 | Likely Benign | -0.79 | Neutral | 0.824 | Possibly Damaging | 0.507 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1022 | 0.0891 | -1 | 0 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||||
| c.206T>G | I69S 2D AIThe SynGAP1 I69S missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign based on current predictive data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.466129 | Uncertain | 0.437 | 0.786 | 0.375 | -1.880 | Likely Benign | 0.634 | Likely Pathogenic | Likely Benign | 0.152 | Likely Benign | -0.78 | Neutral | 0.824 | Possibly Damaging | 0.507 | Possibly Damaging | 4.14 | Benign | 0.00 | Affected | 0.2672 | 0.0782 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2072C>G | T691R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T691R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as Benign, SGM Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Overall, the preponderance of evidence from standard and high‑accuracy predictors points to a pathogenic effect. Based on the aggregate predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which has no entry for it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.271308 | Uncertain | 0.941 | 0.232 | 0.000 | -12.228 | Likely Pathogenic | 0.621 | Likely Pathogenic | Likely Benign | -1.27 | Ambiguous | 0.3 | -0.94 | Ambiguous | -1.11 | Ambiguous | 1.35 | Destabilizing | 0.180 | Likely Benign | -3.66 | Deleterious | 0.993 | Probably Damaging | 0.566 | Possibly Damaging | 3.48 | Benign | 0.02 | Affected | 0.0729 | 0.2478 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.2072C>T | T691I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T691I is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (gnomAD ID: 6‑33441331‑C‑T). Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from FoldX, Foldetta, and premPS. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, while Foldetta remains uncertain. Overall, the evidence overwhelmingly indicates that T691I is most likely benign, and this conclusion does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.271308 | Uncertain | 0.941 | 0.232 | 0.000 | 6-33441331-C-T | 1 | 6.20e-7 | -5.857 | Likely Benign | 0.202 | Likely Benign | Likely Benign | -1.08 | Ambiguous | 0.1 | -2.12 | Stabilizing | -1.60 | Ambiguous | -0.61 | Ambiguous | 0.052 | Likely Benign | -1.19 | Neutral | 0.040 | Benign | 0.003 | Benign | 3.49 | Benign | 0.34 | Tolerated | 3.43 | 14 | 0.0644 | 0.5397 | -1 | 0 | 5.2 | 12.05 | ||||||||||||||||||||||||
| c.2096T>G | V699G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V699G has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include FATHMM and AlphaMissense‑Optimized, while the remaining tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b) uniformly predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic verdict; and Foldetta also predicts pathogenic. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.069024 | Structured | 0.432975 | Uncertain | 0.935 | 0.315 | 0.000 | -11.912 | Likely Pathogenic | 0.481 | Ambiguous | Likely Benign | 2.22 | Destabilizing | 0.0 | 3.25 | Destabilizing | 2.74 | Destabilizing | 1.77 | Destabilizing | 0.514 | Likely Pathogenic | -5.11 | Deleterious | 0.972 | Probably Damaging | 0.999 | Probably Damaging | 3.37 | Benign | 0.01 | Affected | 0.1830 | 0.2240 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.2101C>T | P701S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P701S (ClinVar ID 2995856.0) is listed as “Uncertain” in ClinVar and is present in gnomAD (ID 6‑33441360‑C‑T). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). No tool in the dataset predicts a pathogenic outcome; all remaining predictions are either benign or uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.074921 | Structured | 0.404318 | Uncertain | 0.918 | 0.345 | 0.000 | Uncertain | 1 | 6-33441360-C-T | 3 | 1.86e-6 | -4.375 | Likely Benign | 0.221 | Likely Benign | Likely Benign | 1.33 | Ambiguous | 0.0 | 0.12 | Likely Benign | 0.73 | Ambiguous | -0.36 | Likely Benign | 0.132 | Likely Benign | 0.78 | Neutral | 0.044 | Benign | 0.025 | Benign | 3.48 | Benign | 1.00 | Tolerated | 3.47 | 10 | 0.3149 | 0.3782 | -1 | 1 | 0.8 | -10.04 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||
| c.2111G>T | S704I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S704I lies in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM, whereas the majority of other in silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus) report a pathogenic or likely pathogenic outcome. FoldX, Rosetta, and Foldetta provide uncertain results. High‑accuracy methods specifically give AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, and Foldetta as uncertain. Based on the overall consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.096677 | Structured | 0.383620 | Uncertain | 0.928 | 0.363 | 0.000 | -14.222 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 1.63 | Ambiguous | 0.1 | 1.32 | Ambiguous | 1.48 | Ambiguous | 0.29 | Likely Benign | 0.232 | Likely Benign | -4.05 | Deleterious | 0.997 | Probably Damaging | 0.758 | Possibly Damaging | 3.49 | Benign | 0.02 | Affected | 0.0727 | 0.4798 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.212A>G | D71G 2D AIThe SynGAP1 missense variant D71G is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33425820‑A‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.456046 | Uncertain | 0.350 | 0.799 | 0.375 | 6-33425820-A-G | 1 | 6.20e-7 | -3.136 | Likely Benign | 0.439 | Ambiguous | Likely Benign | 0.111 | Likely Benign | -1.82 | Neutral | 0.171 | Benign | 0.021 | Benign | 4.03 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3802 | 0.5704 | -1 | 1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||||||
| c.2135G>A | G712D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G712D is catalogued in gnomAD (ID 6‑33441600‑G‑A) but has no ClinVar entry. In silico predictors show mixed results: benign calls come from REVEL, SIFT, and FATHMM, while pathogenic calls are made by FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Uncertain predictions are reported by premPS and AlphaMissense‑Optimized. The high‑accuracy consensus tools give a pathogenic verdict: AlphaMissense‑Optimized is inconclusive, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Pathogenic. Overall, the majority of reliable predictors and the consensus methods lean toward a pathogenic effect, and this assessment does not conflict with ClinVar, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | 6-33441600-G-A | 1 | 6.20e-7 | -8.211 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 2.87 | Destabilizing | 0.1 | 5.37 | Destabilizing | 4.12 | Destabilizing | 0.90 | Ambiguous | 0.320 | Likely Benign | -5.69 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.67 | Benign | 0.12 | Tolerated | 3.50 | 9 | 0.2146 | 0.2533 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||
| c.2135G>T | G712V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G712V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, while the majority of tools (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic impact. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.278302 | Structured | 0.384858 | Uncertain | 0.947 | 0.365 | 0.000 | -10.466 | Likely Pathogenic | 0.877 | Likely Pathogenic | Ambiguous | 3.79 | Destabilizing | 0.0 | 6.18 | Destabilizing | 4.99 | Destabilizing | 0.79 | Ambiguous | 0.410 | Likely Benign | -7.55 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.32 | Benign | 0.00 | Affected | 0.1395 | 0.3712 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2162T>G | I721S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I721S is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that assess the variant’s effect fall into two groups: the single benign prediction comes from REVEL, while all other evaluated algorithms (FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic outcome. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, which does not contradict its current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.394753 | Structured | 0.454550 | Uncertain | 0.957 | 0.437 | 0.125 | Uncertain | 1 | -14.032 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 3.91 | Destabilizing | 0.1 | 3.96 | Destabilizing | 3.94 | Destabilizing | 2.28 | Destabilizing | 0.466 | Likely Benign | -5.26 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.21 | Pathogenic | 0.00 | Affected | 3.50 | 9 | 0.2606 | 0.1110 | -1 | -2 | -5.3 | -26.08 | 203.3 | 49.3 | -0.1 | 0.0 | -1.1 | 0.0 | X | Uncertain | The sec-butyl side chain of Ile721, located on an α-helix (res. Leu714-Arg726), engages in hydrophobic packing with other residues in the hydrophobic inter-helix space, such as Phe420, Tyr417, His693, and Leu717. In the variant simulations, the hydroxyl side chain of Ser721 forms hydrogen bonds with nearby residues, such as Leu717 and His693. Although no major structural changes are observed during the variant simulations, the hydrophilic residue Ser721 could disrupt the hydrophobic packing during folding. However, because the model ends abruptly at the C-terminus, no definite conclusions can be drawn based on the simulations. | ||||||||||||||||
| c.2165G>T | S722I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S722I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, premPS, SIFT, and the folding‑stability method Foldetta, whereas pathogenic predictions are reported by SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results are provided by AlphaMissense‑Optimized, FoldX, and Rosetta. High‑accuracy analyses further clarify the picture: AlphaMissense‑Optimized remains inconclusive; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect; Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a benign impact on protein stability. Overall, the majority of evidence leans toward a pathogenic interpretation, and this conclusion does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.468512 | Structured | 0.457186 | Uncertain | 0.950 | 0.431 | 0.375 | -11.165 | Likely Pathogenic | 0.867 | Likely Pathogenic | Ambiguous | 0.69 | Ambiguous | 0.1 | -0.65 | Ambiguous | 0.02 | Likely Benign | 0.18 | Likely Benign | 0.232 | Likely Benign | -3.88 | Deleterious | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.48 | Pathogenic | 0.07 | Tolerated | 0.0776 | 0.4187 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.2168C>G | T723R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T723R has no ClinVar entry and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify it as benign. Two tools (polyPhen‑2 HumDiv and HumVar) predict it to be pathogenic, while FoldX, Foldetta, and AlphaMissense‑Default are uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM Consensus as likely benign, and Foldetta as uncertain. Overall, the preponderance of evidence points to a benign impact for T723R, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.476583 | Structured | 0.458243 | Uncertain | 0.945 | 0.447 | 0.375 | -3.654 | Likely Benign | 0.408 | Ambiguous | Likely Benign | -0.90 | Ambiguous | 0.1 | -0.13 | Likely Benign | -0.52 | Ambiguous | 0.44 | Likely Benign | 0.146 | Likely Benign | -1.50 | Neutral | 0.931 | Possibly Damaging | 0.458 | Possibly Damaging | 3.51 | Benign | 0.29 | Tolerated | 0.0794 | 0.2437 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.2177G>C | R726T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R726T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default; FoldX is uncertain. High‑accuracy methods give a consistent benign signal: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the aggregate predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.521092 | Disordered | 0.449098 | Uncertain | 0.888 | 0.513 | 0.625 | -5.249 | Likely Benign | 0.661 | Likely Pathogenic | Likely Benign | 0.82 | Ambiguous | 0.0 | -0.11 | Likely Benign | 0.36 | Likely Benign | -0.01 | Likely Benign | 0.200 | Likely Benign | -0.90 | Neutral | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.74 | Benign | 1.00 | Tolerated | 0.1685 | 0.4569 | -1 | -1 | 3.8 | -55.08 | ||||||||||||||||||||||||||||||
| c.2182C>T | P728S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant P728S is not reported in ClinVar and is present in gnomAD (ID 6‑33441647‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, whereas the majority of tools predict pathogenicity: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results from FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized are treated as unavailable. High‑accuracy consensus methods give a Likely Pathogenic verdict from the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and an Uncertain outcome from AlphaMissense‑Optimized; Foldetta also reports Uncertain. Overall, the preponderance of evidence points to a pathogenic effect for P728S, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.632174 | Disordered | 0.434760 | Uncertain | 0.725 | 0.567 | 0.625 | 6-33441647-C-T | 1 | 6.20e-7 | -9.047 | Likely Pathogenic | 0.897 | Likely Pathogenic | Ambiguous | 0.89 | Ambiguous | 0.0 | 0.98 | Ambiguous | 0.94 | Ambiguous | 0.54 | Ambiguous | 0.280 | Likely Benign | -6.38 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 0.68 | Pathogenic | 0.00 | Affected | 3.59 | 7 | 0.3571 | 0.3571 | -1 | 1 | 0.8 | -10.04 | |||||||||||||||||||||||||
| c.2189T>G | I730S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I730S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar; AlphaMissense‑Default remains uncertain. High‑accuracy assessments—AlphaMissense‑Optimized, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta outputs)—all uniformly predict a benign impact. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.420109 | Uncertain | 0.591 | 0.619 | 0.750 | -6.220 | Likely Benign | 0.349 | Ambiguous | Likely Benign | 0.25 | Likely Benign | 0.2 | -0.12 | Likely Benign | 0.07 | Likely Benign | 0.47 | Likely Benign | 0.123 | Likely Benign | -0.51 | Neutral | 1.000 | Probably Damaging | 0.967 | Probably Damaging | 3.60 | Benign | 0.34 | Tolerated | 0.2646 | 0.0903 | -1 | -2 | -5.3 | -26.08 | ||||||||||||||||||||||||||||||
| c.218G>C | R73T 2D AIThe SynGAP1 missense variant R73T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are unavailable and therefore not considered. Overall, the preponderance of evidence from multiple prediction algorithms and consensus methods indicates that R73T is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.501700 | Disordered | 0.453164 | Uncertain | 0.332 | 0.826 | 0.375 | -4.061 | Likely Benign | 0.343 | Ambiguous | Likely Benign | 0.070 | Likely Benign | -1.05 | Neutral | 0.115 | Benign | 0.012 | Benign | 4.05 | Benign | 0.00 | Affected | 0.1980 | 0.4439 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||||||||||||
| c.2195G>C | R732T 2D AISynGAP1 missense variant R732T is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign (REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Optimized) and pathogenic (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b). AlphaMissense‑Default remains uncertain. The high‑accuracy AlphaMissense‑Optimized predicts a benign effect, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also favors a benign outcome. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors points to a benign impact, which does not contradict the current ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.846163 | Disordered | 0.412403 | Uncertain | 0.427 | 0.673 | 0.750 | Uncertain | 1 | -8.545 | Likely Pathogenic | 0.434 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -1.96 | Neutral | 0.999 | Probably Damaging | 0.892 | Possibly Damaging | 2.59 | Benign | 0.12 | Tolerated | 3.59 | 7 | 0.1915 | 0.3153 | -1 | -1 | 3.8 | -55.08 | ||||||||||||||||||||||||||||||||||||
| c.2204G>T | S735I 2D AIThe SynGAP1 missense variant S735I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S735I, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.916840 | Disordered | 0.412174 | Uncertain | 0.290 | 0.752 | 0.875 | -5.669 | Likely Benign | 0.167 | Likely Benign | Likely Benign | 0.117 | Likely Benign | -1.71 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.64 | Benign | 0.09 | Tolerated | 0.0933 | 0.5069 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.220A>C | S74R 2D AIThe SynGAP1 missense variant S74R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority of the high‑accuracy tools) also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | -3.271 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.065 | Likely Benign | -1.34 | Neutral | 0.361 | Benign | 0.019 | Benign | 4.08 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0943 | 0.3562 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2210A>C | Q737P 2D AIThe SynGAP1 missense variant Q737P is listed in ClinVar (ID 2580571.0) with an uncertain significance designation and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while only SIFT indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta stability analysis is unavailable. Taken together, the preponderance of evidence supports a benign classification for Q737P, which is consistent with its ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.425743 | Uncertain | 0.323 | 0.803 | 0.875 | Uncertain | 1 | -2.407 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.154 | Likely Benign | -1.22 | Neutral | 0.005 | Benign | 0.013 | Benign | 2.78 | Benign | 0.04 | Affected | 4.07 | 3 | 0.2366 | 0.4981 | -1 | 0 | 1.9 | -31.01 | |||||||||||||||||||||||||||||||||||
| c.2212A>T | S738C 2D AIThe SynGAP1 missense variant S738C is reported in ClinVar as not yet classified and is present in gnomAD (variant ID 6‑33441677‑A‑T). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict pathogenic. High‑accuracy methods give a benign result from AlphaMissense‑Optimized and a likely benign consensus from SGM‑Consensus; Foldetta predictions are unavailable. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | 6-33441677-A-T | 4 | 2.48e-6 | -6.373 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -2.09 | Neutral | 0.975 | Probably Damaging | 0.815 | Possibly Damaging | 2.63 | Benign | 0.01 | Affected | 4.32 | 2 | 0.1112 | 0.3982 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||||
| c.2213G>T | S738I 2D AIThe SynGAP1 missense variant S738I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.812494 | Disordered | 0.441162 | Uncertain | 0.284 | 0.827 | 0.875 | -4.312 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -1.78 | Neutral | 0.642 | Possibly Damaging | 0.393 | Benign | 2.66 | Benign | 0.01 | Affected | 0.0847 | 0.3636 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.221G>T | S74I 2D AIThe SynGAP1 missense variant S74I is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports the variant as likely benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is benign; Foldetta results are not available. Overall, the consensus of available predictions indicates that S74I is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | -4.668 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.036 | Likely Benign | -1.78 | Neutral | 0.099 | Benign | 0.007 | Benign | 4.06 | Benign | 0.00 | Affected | 0.0886 | 0.4680 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2227C>T | P743S 2D AIThe SynGAP1 missense variant P743S is listed in gnomAD (ID 6‑33441692‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the change as benign. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is “Likely Benign.” No Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence indicates that P743S is most likely benign, and this conclusion is not contradicted by any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.852992 | Disordered | 0.526809 | Binding | 0.317 | 0.862 | 0.875 | 6-33441692-C-T | 1 | 6.19e-7 | -4.286 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.085 | Likely Benign | -0.33 | Neutral | 0.021 | Benign | 0.015 | Benign | 2.93 | Benign | 0.00 | Affected | 4.32 | 2 | 0.3087 | 0.3948 | -1 | 1 | 0.8 | -10.04 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||||||||||
| c.222C>A | S74R 2D AIThe SynGAP1 missense variant S74R is catalogued in gnomAD (ID 6‑33425830‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all report benign or likely benign. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments confirm the benign consensus: AlphaMissense‑Optimized is benign, and the SGM‑Consensus, derived from the majority of the high‑confidence predictors, is benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that S74R is most likely benign, and this assessment does not contradict any ClinVar status, as none is reported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | 6-33425830-C-A | 1 | 6.20e-7 | -3.271 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.070 | Likely Benign | -1.34 | Neutral | 0.361 | Benign | 0.019 | Benign | 4.08 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0943 | 0.3562 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||
| c.222C>G | S74R 2D AIThe SynGAP1 missense variant S74R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority of the high‑accuracy tools) is also benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.505461 | Disordered | 0.450156 | Uncertain | 0.294 | 0.831 | 0.500 | -3.271 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.070 | Likely Benign | -1.34 | Neutral | 0.361 | Benign | 0.019 | Benign | 4.08 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0943 | 0.3562 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2233C>T | P745S 2D AIThe SynGAP1 missense variant P745S is reported in gnomAD (variant ID 6-33441698-C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence from multiple prediction algorithms and consensus methods points to a benign effect for P745S, and this conclusion is not contradicted by any ClinVar classification (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.791621 | Disordered | 0.558331 | Binding | 0.341 | 0.860 | 0.875 | 6-33441698-C-T | 1 | 6.19e-7 | -4.300 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.160 | Likely Benign | -2.32 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.54 | Benign | 0.05 | Affected | 4.32 | 2 | 0.3397 | 0.3882 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.2237T>G | V746G 2D AIThe SynGAP1 missense variant V746G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized scores benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective predictions, V746G is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.808535 | Disordered | 0.576597 | Binding | 0.336 | 0.867 | 0.875 | -2.971 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -0.89 | Neutral | 0.136 | Benign | 0.270 | Benign | 2.74 | Benign | 0.02 | Affected | 0.1897 | 0.2550 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2240T>G | V747G 2D AIThe SynGAP1 missense variant V747G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as benign, and Foldetta results are unavailable. Overall, the consensus of the available predictions indicates that V747G is most likely benign, and this conclusion does not contradict any ClinVar status because the variant has not been reported there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.816150 | Disordered | 0.594069 | Binding | 0.343 | 0.873 | 0.750 | -3.883 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -1.22 | Neutral | 0.589 | Possibly Damaging | 0.917 | Probably Damaging | 2.56 | Benign | 0.00 | Affected | 0.1960 | 0.2374 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2249G>T | G750V 2D AIThe SynGAP1 missense variant G750V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 benign vs 2 pathogenic votes). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions therefore show AlphaMissense‑Optimized as benign, while the remaining high‑confidence tools (PROVEAN, polyPhen‑2, SIFT, FATHMM) all indicate pathogenicity. Overall, the majority of evidence (five pathogenic vs four benign) points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.618285 | Disordered | 0.646832 | Binding | 0.348 | 0.866 | 0.625 | -4.512 | Likely Benign | 0.134 | Likely Benign | Likely Benign | 0.145 | Likely Benign | -2.54 | Deleterious | 0.984 | Probably Damaging | 0.850 | Possibly Damaging | 2.45 | Pathogenic | 0.01 | Affected | 0.1270 | 0.4212 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2264T>C | M755T 2D AIThe SynGAP1 missense variant M755T is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the change as benign, and AlphaMissense‑Optimized also predicts benign. No tool predicts pathogenicity; AlphaMissense‑Default is uncertain. The SGM‑Consensus, which aggregates majority votes from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta data are not available. Overall, the computational evidence overwhelmingly suggests the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.783855 | Binding | 0.336 | 0.873 | 0.375 | -4.116 | Likely Benign | 0.358 | Ambiguous | Likely Benign | 0.040 | Likely Benign | -1.23 | Neutral | 0.159 | Benign | 0.053 | Benign | 2.65 | Benign | 0.19 | Tolerated | 0.1944 | 0.1767 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2270G>T | G757V 2D AIThe SynGAP1 missense variant G757V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.436924 | Structured | 0.830995 | Binding | 0.310 | 0.869 | 0.375 | -4.840 | Likely Benign | 0.149 | Likely Benign | Likely Benign | 0.087 | Likely Benign | -1.47 | Neutral | 0.801 | Possibly Damaging | 0.494 | Possibly Damaging | 2.68 | Benign | 0.07 | Tolerated | 0.1089 | 0.3512 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2276T>C | M759T 2D AIThe SynGAP1 missense variant M759T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also reports a benign prediction. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence—including the SGM consensus and AlphaMissense‑Optimized—supports a benign classification, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.879389 | Binding | 0.299 | 0.864 | 0.375 | -4.202 | Likely Benign | 0.380 | Ambiguous | Likely Benign | 0.197 | Likely Benign | -1.90 | Neutral | 0.891 | Possibly Damaging | 0.315 | Benign | 2.58 | Benign | 0.08 | Tolerated | 0.2063 | 0.1534 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2279T>C | M760T 2D AIThe SynGAP1 missense variant M760T is reported in gnomAD (ID 6‑33441744‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.541878 | Disordered | 0.893402 | Binding | 0.346 | 0.865 | 0.375 | 6-33441744-T-C | 1 | 6.20e-7 | -2.365 | Likely Benign | 0.519 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -1.56 | Neutral | 0.425 | Benign | 0.252 | Benign | 2.71 | Benign | 0.41 | Tolerated | 3.99 | 5 | 0.2515 | 0.2875 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.2293A>C | S765R 2D AIThe SynGAP1 missense variant S765R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus (majority vote) as Benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this conclusion, so the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | -5.422 | Likely Benign | 0.791 | Likely Pathogenic | Ambiguous | 0.157 | Likely Benign | -1.57 | Neutral | 0.996 | Probably Damaging | 0.985 | Probably Damaging | 4.16 | Benign | 0.07 | Tolerated | 3.64 | 6 | 0.0741 | 0.3859 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2294G>T | S765I 2D AIThe SynGAP1 missense variant S765I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also resolves to benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | -6.891 | Likely Benign | 0.699 | Likely Pathogenic | Likely Benign | 0.187 | Likely Benign | -1.24 | Neutral | 0.996 | Probably Damaging | 0.985 | Probably Damaging | 4.09 | Benign | 0.69 | Tolerated | 0.0768 | 0.5577 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2295C>A | S765R 2D AIThe SynGAP1 missense variant S765R is reported in gnomAD (ID 6‑33442453‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM. Those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Taken together, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status because none is assigned. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | 6-33442453-C-A | -5.422 | Likely Benign | 0.791 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.57 | Neutral | 0.996 | Probably Damaging | 0.985 | Probably Damaging | 4.16 | Benign | 0.07 | Tolerated | 3.64 | 6 | 0.0741 | 0.3859 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||||
| c.2295C>G | S765R 2D AIThe SynGAP1 missense variant S765R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the balance of evidence points to a benign impact for S765R, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.370445 | Structured | 0.922652 | Binding | 0.335 | 0.865 | 0.250 | -5.422 | Likely Benign | 0.791 | Likely Pathogenic | Ambiguous | 0.155 | Likely Benign | -1.57 | Neutral | 0.996 | Probably Damaging | 0.985 | Probably Damaging | 4.16 | Benign | 0.07 | Tolerated | 3.64 | 6 | 0.0741 | 0.3859 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2296T>C | S766P 2D AIThe SynGAP1 missense variant S766P is reported in gnomAD (ID 6‑33442454‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while SIFT uniquely predicts it as pathogenic. The remaining tools, ESM1b and AlphaMissense‑Default, are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta stability analysis is unavailable. Overall, the preponderance of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.405110 | Structured | 0.923125 | Binding | 0.338 | 0.874 | 0.250 | 6-33442454-T-C | 1 | 1.28e-6 | -7.343 | In-Between | 0.374 | Ambiguous | Likely Benign | 0.193 | Likely Benign | -0.91 | Neutral | 0.006 | Benign | 0.013 | Benign | 4.07 | Benign | 0.01 | Affected | 3.64 | 6 | 0.2216 | 0.5862 | -1 | 1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||||||||
| c.2300T>G | I767S 2D AIThe SynGAP1 missense variant I767S is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate benign. Only polyPhen‑2 HumDiv flags it as pathogenic, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Taken together, the preponderance of evidence points to a benign classification for I767S, and this assessment does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.321458 | Structured | 0.927771 | Binding | 0.369 | 0.872 | 0.125 | -3.030 | Likely Benign | 0.388 | Ambiguous | Likely Benign | 0.126 | Likely Benign | -0.64 | Neutral | 0.925 | Possibly Damaging | 0.329 | Benign | 4.13 | Benign | 0.25 | Tolerated | 0.3242 | 0.1782 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.230G>T | S77I 2D AIThe SynGAP1 missense variant S77I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.446124 | Uncertain | 0.310 | 0.855 | 0.375 | -3.918 | Likely Benign | 0.140 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -1.41 | Neutral | 0.604 | Possibly Damaging | 0.029 | Benign | 4.07 | Benign | 0.00 | Affected | 0.0758 | 0.5165 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2314T>G | F772V 2D AIThe SynGAP1 missense variant F772V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are not available for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.450668 | Structured | 0.922662 | Binding | 0.329 | 0.884 | 0.250 | -2.886 | Likely Benign | 0.191 | Likely Benign | Likely Benign | 0.161 | Likely Benign | -0.43 | Neutral | 0.845 | Possibly Damaging | 0.899 | Possibly Damaging | 4.27 | Benign | 0.37 | Tolerated | 0.1687 | 0.2419 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2318T>C | M773T 2D AIThe SynGAP1 missense variant M773T is listed in gnomAD (ID 6‑33442476‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only polyPhen‑2 HumVar predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | 6-33442476-T-C | 1 | 1.28e-6 | -4.225 | Likely Benign | 0.377 | Ambiguous | Likely Benign | 0.112 | Likely Benign | -1.63 | Neutral | 0.106 | Benign | 0.471 | Possibly Damaging | 4.22 | Benign | 0.06 | Tolerated | 3.64 | 6 | 0.2196 | 0.1586 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.2318T>G | M773R 2D AIThe SynGAP1 missense variant M773R is listed in gnomAD (ID 6‑33442476‑T‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates benign. No Foldetta (FoldX‑MD/Rosetta stability) result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.408655 | Structured | 0.916222 | Binding | 0.325 | 0.893 | 0.250 | 6-33442476-T-G | 1 | 1.28e-6 | -4.340 | Likely Benign | 0.597 | Likely Pathogenic | Likely Benign | 0.183 | Likely Benign | -1.87 | Neutral | 0.220 | Benign | 0.471 | Possibly Damaging | 4.18 | Benign | 0.02 | Affected | 3.64 | 6 | 0.1778 | 0.0800 | -1 | 0 | -6.4 | 24.99 | ||||||||||||||||||||||||||||||||||
| c.2327G>A | G776D 2D AIThe SynGAP1 missense variant G776D is catalogued in gnomAD (ID 6‑33442485‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions come from AlphaMissense‑Default, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus (majority of the four high‑accuracy tools) also indicates benign. Foldetta stability analysis is unavailable, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for G776D, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.377384 | Structured | 0.886983 | Binding | 0.296 | 0.888 | 0.250 | 6-33442485-G-A | 0.388 | Likely Benign | 0.566 | Likely Pathogenic | Likely Benign | 0.188 | Likely Benign | 0.16 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 4.30 | Benign | 0.10 | Tolerated | 3.64 | 6 | 0.1835 | 0.2971 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||||
| c.2336G>T | S779I 2D AIThe SynGAP1 missense variant S779I is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Default. High‑accuracy methods give a benign call from AlphaMissense‑Optimized; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta results are unavailable. Overall, the majority of tools (five pathogenic vs. four benign) suggest a pathogenic impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.509769 | Disordered | 0.834974 | Binding | 0.321 | 0.890 | 0.375 | -6.261 | Likely Benign | 0.578 | Likely Pathogenic | Likely Benign | 0.198 | Likely Benign | -2.10 | Neutral | 0.918 | Possibly Damaging | 0.827 | Possibly Damaging | 2.28 | Pathogenic | 0.05 | Affected | 0.1046 | 0.6248 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2338T>C | S780P 2D AIThe SynGAP1 missense variant S780P is reported in gnomAD (ID 6‑33442890‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.812415 | Binding | 0.283 | 0.883 | 0.500 | 6-33442890-T-C | 1 | 6.22e-7 | -6.055 | Likely Benign | 0.234 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -1.11 | Neutral | 0.995 | Probably Damaging | 0.892 | Possibly Damaging | 2.64 | Benign | 0.30 | Tolerated | 3.64 | 6 | 0.2086 | 0.6171 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||
| c.2339C>G | S780C 2D AIThe SynGAP1 missense variant S780C is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33442891‑C‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). No tool in the dataset predicts a pathogenic outcome; ESM1b is inconclusive and therefore treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while Foldetta results are not reported and thus unavailable. Based on the collective predictions, the variant is most likely benign, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.812415 | Binding | 0.283 | 0.883 | 0.500 | Uncertain | 4 | 6-33442891-C-G | 16 | 9.94e-6 | -7.603 | In-Between | 0.278 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.41 | Neutral | 0.065 | Benign | 0.043 | Benign | 2.59 | Benign | 0.10 | Tolerated | 3.64 | 6 | 0.1063 | 0.6581 | -1 | 0 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||||
| c.2342T>C | M781T 2D AIThe SynGAP1 missense variant M781T is catalogued in gnomAD (ID 6‑33442894‑T‑C) and has no ClinVar entry. Across the evaluated in‑silico tools, every predictor classifies the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool reports a pathogenic or likely pathogenic outcome. The high‑accuracy consensus methods reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability predictor, has no available result for this variant. Based on the unanimous benign predictions and the lack of any ClinVar pathogenic annotation, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.580690 | Disordered | 0.792850 | Binding | 0.342 | 0.889 | 0.625 | 6-33442894-T-C | 1 | 6.21e-7 | -3.774 | Likely Benign | 0.310 | Likely Benign | Likely Benign | 0.175 | Likely Benign | -0.86 | Neutral | 0.015 | Benign | 0.037 | Benign | 2.75 | Benign | 0.59 | Tolerated | 3.64 | 6 | 0.2267 | 0.1599 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.2348T>C | M783T 2D AIThe SynGAP1 missense variant M783T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as tolerated or benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a damaging or pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates likely benign. Foldetta results are not available for this variant. Overall, the preponderance of evidence from multiple prediction algorithms and the high‑accuracy consensus points to a benign effect, with no conflict with ClinVar reporting. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.736850 | Disordered | 0.738119 | Binding | 0.331 | 0.889 | 0.625 | -4.064 | Likely Benign | 0.299 | Likely Benign | Likely Benign | 0.111 | Likely Benign | -2.08 | Neutral | 0.625 | Possibly Damaging | 0.265 | Benign | 2.69 | Benign | 0.03 | Affected | 0.2217 | 0.2308 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2350G>C | A784P 2D AIThe SynGAP1 missense variant A784P is reported in gnomAD (variant ID 6-33442902‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact for A784P. This conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.801317 | Disordered | 0.708872 | Binding | 0.314 | 0.896 | 0.625 | 6-33442902-G-C | 4 | 2.48e-6 | -3.777 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 0.189 | Likely Benign | -0.73 | Neutral | 0.586 | Possibly Damaging | 0.396 | Benign | 2.66 | Benign | 0.19 | Tolerated | 3.64 | 6 | 0.1846 | 0.4771 | -1 | 1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||||||
| c.2359C>T | P787S 2D AIThe SynGAP1 P787S variant is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33442911‑C‑T). Prediction tools that agree on a benign effect include REVEL, ESM1b, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; AlphaMissense‑Default remains uncertain. The high‑accuracy consensus (SGM Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a pathogenic majority. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.901269 | Disordered | 0.613211 | Binding | 0.377 | 0.899 | 0.750 | Uncertain | 1 | 6-33442911-C-T | 3 | 1.86e-6 | -4.203 | Likely Benign | 0.564 | Ambiguous | Likely Benign | 0.221 | Likely Benign | -3.81 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.48 | Pathogenic | 0.02 | Affected | 3.64 | 6 | 0.3420 | 0.4675 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||
| c.2362T>C | S788P 2D AIThe SynGAP1 missense variant S788P is catalogued in gnomAD (ID 6‑33442914‑T‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The AlphaMissense‑Default score is uncertain. A consensus derived from the SGM framework (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is unavailable for this residue. Taken together, the preponderance of evidence from standard predictors and the SGM consensus points to a likely pathogenic effect, which does not contradict any existing ClinVar annotation because none is present. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.956248 | Disordered | 0.573557 | Binding | 0.349 | 0.895 | 0.750 | 6-33442914-T-C | -1.857 | Likely Benign | 0.435 | Ambiguous | Likely Benign | 0.236 | Likely Benign | -3.91 | Deleterious | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 1.51 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.2338 | 0.5537 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||||
| c.2368A>C | T790P 2D  AIThe SynGAP1 missense variant T790P has no ClinVar entry (ClinVar status: not reported) but is present in gnomAD (ID 6‑33442920‑A‑C). Prediction tools that agree on a benign effect include REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of conventional tools (5 pathogenic vs 4 benign) lean toward a pathogenic interpretation, while the single high‑accuracy tool suggests benign. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.964893 | Disordered | 0.509280 | Binding | 0.385 | 0.896 | 0.875 | 6-33442920-A-C | -3.564 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 0.250 | Likely Benign | -3.55 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 2.25 | Pathogenic | 0.01 | Affected | 3.64 | 6 | 0.2147 | 0.4748 | -1 | 0 | -0.9 | -3.99 | ||||||||||||||||||||||||||||||||||||
| c.2372A>C | K791T 2D  AIThe SynGAP1 missense variant K791T is listed in gnomAD (ID 6‑33442924‑A‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). No tool predicts pathogenicity. The high‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the evidence overwhelmingly supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.966441 | Disordered | 0.478670 | Uncertain | 0.356 | 0.896 | 0.875 | 6-33442924-A-C | 2 | 1.24e-6 | -1.578 | Likely Benign | 0.415 | Ambiguous | Likely Benign | 0.033 | Likely Benign | -0.92 | Neutral | 0.032 | Benign | 0.017 | Benign | 4.17 | Benign | 0.16 | Tolerated | 3.64 | 6 | 0.2734 | 0.3340 | -1 | 0 | 3.2 | -27.07 | |||||||||||||||||||||||||||||||||
| c.2375A>C | E792A 2D  AIThe SynGAP1 missense variant E792A is catalogued in gnomAD (ID 6‑33442927‑A‑C) but has no ClinVar submission. Functional prediction tools cluster into two groups: benign (REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized) and pathogenic (PROVEAN, SIFT). The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also favors benign. Foldetta, a protein‑folding stability predictor, has no available result for this residue. Overall, the preponderance of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar pathogenic classification; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.974374 | Disordered | 0.452261 | Uncertain | 0.352 | 0.896 | 0.875 | 6-33442927-A-C | 1 | 6.20e-7 | -3.248 | Likely Benign | 0.515 | Ambiguous | Likely Benign | 0.060 | Likely Benign | -3.35 | Deleterious | 0.000 | Benign | 0.001 | Benign | 3.92 | Benign | 0.01 | Affected | 3.64 | 6 | 0.4586 | 0.7544 | -1 | 0 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||||||
| c.2380C>A | P794T 2D  AIThe SynGAP1 missense variant P794T is catalogued in gnomAD (ID 6‑33442932‑C‑A) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool in the dataset predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction. The Foldetta stability analysis is not available for this variant. Overall, the consensus of all available predictions is benign, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.979741 | Disordered | 0.408951 | Uncertain | 0.550 | 0.898 | 0.875 | 6-33442932-C-A | 1 | 6.20e-7 | -4.838 | Likely Benign | 0.060 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.34 | Neutral | 0.245 | Benign | 0.138 | Benign | 4.25 | Benign | 0.66 | Tolerated | 4.07 | 3 | 0.1739 | 0.6207 | -1 | 0 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||
| c.2383C>T | P795S 2D AIThe SynGAP1 missense variant P795S is catalogued in gnomAD (6‑33442935‑C‑T) but has no entry in ClinVar. Across the spectrum of in‑silico predictors, every tool listed—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently scores the variant as benign. No pathogenic predictions are reported. Grouping by agreement, all available tools fall into the benign category, with no tools indicating pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. Foldetta results are not provided, so they are treated as unavailable. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.972450 | Disordered | 0.410339 | Uncertain | 0.457 | 0.903 | 0.875 | 6-33442935-C-T | 1 | 6.20e-7 | -5.846 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.23 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.30 | Benign | 0.24 | Tolerated | 4.32 | 2 | 0.3517 | 0.5766 | -1 | 1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||||||
| c.2386C>G | P796A 2D AIThe SynGAP1 missense variant P796A is not reported in ClinVar (ClinVar ID: None) but is present in gnomAD (gnomAD ID: 6‑33442938‑C‑G). All evaluated in silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized return benign or benign‑like scores. No tool predicts pathogenicity. Grouping by agreement, the benign‑predicting tools comprise the entire set, while no pathogenic predictions are available. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method, did not provide a result for this variant. Overall, the computational evidence strongly suggests that P796A is most likely benign, and this conclusion does not contradict the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.936162 | Disordered | 0.426363 | Uncertain | 0.427 | 0.900 | 0.875 | 6-33442938-C-G | -6.077 | Likely Benign | 0.050 | Likely Benign | Likely Benign | 0.023 | Likely Benign | -0.31 | Neutral | 0.174 | Benign | 0.063 | Benign | 4.28 | Benign | 0.08 | Tolerated | 4.32 | 2 | 0.3598 | 0.3776 | -1 | 1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||
| c.2399G>A | G800D 2D AIThe SynGAP1 missense variant G800D is catalogued in gnomAD (6-33442951‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the change as benign or likely benign. No tool predicts pathogenicity. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized reports a benign outcome, and the SGM‑Consensus (derived from the majority of the four high‑accuracy predictors) also yields a benign verdict. Foldetta results are unavailable, so they do not influence the assessment. Overall, the evidence strongly supports a benign classification, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.852992 | Disordered | 0.588350 | Binding | 0.303 | 0.884 | 0.625 | 6-33442951-G-A | -5.929 | Likely Benign | 0.400 | Ambiguous | Likely Benign | 0.088 | Likely Benign | -0.84 | Neutral | 0.451 | Benign | 0.265 | Benign | 2.76 | Benign | 0.34 | Tolerated | 4.32 | 2 | 0.1664 | 0.1877 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||
| c.2399G>T | G800V 2D AIThe SynGAP1 missense variant G800V is predicted to be benign by all evaluated in‑silico tools. ClinVar contains no entry for this variant, and it is not reported in gnomAD. Consensus predictions from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports a likely benign outcome. Foldetta results are unavailable, so they do not influence the overall assessment. Based on the collective evidence, the variant is most likely benign, and this conclusion is consistent with the absence of a pathogenic ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.852992 | Disordered | 0.588350 | Binding | 0.303 | 0.884 | 0.625 | -4.890 | Likely Benign | 0.113 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.24 | Neutral | 0.451 | Benign | 0.157 | Benign | 2.73 | Benign | 0.25 | Tolerated | 0.1130 | 0.4198 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||
| c.23T>C | I8T 2D AIThe SynGAP1 missense variant I8T is reported in gnomAD (variant ID 6‑33420287‑T‑C) but has no ClinVar entry. In silico prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.543080 | Binding | 0.341 | 0.916 | 0.625 | 6-33420287-T-C | -3.149 | Likely Benign | 0.239 | Likely Benign | Likely Benign | 0.108 | Likely Benign | -0.10 | Neutral | 0.047 | Benign | 0.006 | Benign | 4.03 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0949 | 0.1019 | -1 | 0 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||||||
| c.23T>G | I8S 2D AIThe SynGAP1 missense variant I8S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign prediction: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are not available. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.543080 | Binding | 0.341 | 0.916 | 0.625 | -1.577 | Likely Benign | 0.122 | Likely Benign | Likely Benign | 0.266 | Likely Benign | 0.18 | Neutral | 0.107 | Benign | 0.006 | Benign | 4.02 | Benign | 0.00 | Affected | 0.2726 | 0.0910 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2402G>T | G801V 2D AIThe SynGAP1 missense variant G801V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are not available. Overall, the preponderance of evidence indicates that G801V is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.874069 | Disordered | 0.636323 | Binding | 0.320 | 0.892 | 0.625 | -4.781 | Likely Benign | 0.119 | Likely Benign | Likely Benign | 0.098 | Likely Benign | -1.64 | Neutral | 0.611 | Possibly Damaging | 0.272 | Benign | 2.70 | Benign | 0.11 | Tolerated | 0.1041 | 0.4230 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||
| c.2405G>T | G802V 2D AIThe SynGAP1 missense variant G802V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign. No Foldetta stability analysis is available for this variant. Overall, the consensus of the available predictions indicates that G802V is most likely benign, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | 0.894241 | Disordered | 0.681966 | Binding | 0.294 | 0.898 | 0.625 | -3.871 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.49 | Neutral | 0.411 | Benign | 0.321 | Benign | 2.67 | Benign | 0.00 | Affected | 0.1345 | 0.3533 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||
| c.2411A>G | D804G 2D AIThe SynGAP1 missense variant D804G is catalogued in gnomAD (ID 6‑33442963‑A‑G) but has no ClinVar entry. Prediction tools cluster into two groups: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic, and Foldetta results are unavailable. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.801317 | Disordered | 0.786762 | Binding | 0.294 | 0.900 | 0.625 | 6-33442963-A-G | 1 | 6.20e-7 | -5.051 | Likely Benign | 0.680 | Likely Pathogenic | Likely Benign | 0.287 | Likely Benign | -3.82 | Deleterious | 0.980 | Probably Damaging | 0.858 | Possibly Damaging | 1.21 | Pathogenic | 0.04 | Affected | 3.77 | 5 | 0.3854 | 0.6488 | -1 | 1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||
| c.2416T>G | F806V 2D AIThe SynGAP1 missense variant F806V is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, ESM1b, and AlphaMissense‑Optimized, whereas the majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default) predict it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus remains pathogenic; a Foldetta stability analysis is unavailable. Overall, the balance of evidence from the majority of prediction algorithms points to a pathogenic impact, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | SH3-binding motif | 0.736850 | Disordered | 0.847454 | Binding | 0.276 | 0.904 | 0.500 | -4.548 | Likely Benign | 0.676 | Likely Pathogenic | Likely Benign | 0.192 | Likely Benign | -3.79 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.16 | Pathogenic | 0.02 | Affected | 0.2464 | 0.1325 | -1 | -1 | 1.4 | -48.04 | ||||||||||||||||||||||||||||||||||||||
| c.2423T>G | V808G 2D AIThe SynGAP1 V808G missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are PROVEAN, SIFT, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta results are unavailable. Overall, the majority of individual predictors (five benign vs. three pathogenic) support a benign classification, and this is not contradicted by any ClinVar annotation. Thus, based on the available predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.699094 | Disordered | 0.856438 | Binding | 0.289 | 0.903 | 0.500 | -6.635 | Likely Benign | 0.460 | Ambiguous | Likely Benign | 0.268 | Likely Benign | -2.94 | Deleterious | 0.069 | Benign | 0.135 | Benign | 2.27 | Pathogenic | 0.00 | Affected | 0.1925 | 0.3336 | -1 | -3 | -4.6 | -42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2426G>T | S809I 2D AIThe SynGAP1 missense variant S809I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Consensus from standard in silico predictors shows a split: six tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Optimized) predict a benign effect, while two (SIFT, AlphaMissense‑Default) predict pathogenicity; ESM1b is uncertain. High‑accuracy assessment further supports a benign outcome: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign classification, and Foldetta data are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | SH3-binding motif | 0.626927 | Disordered | 0.853218 | Binding | 0.330 | 0.907 | 0.500 | -7.708 | In-Between | 0.632 | Likely Pathogenic | Likely Benign | 0.087 | Likely Benign | -1.93 | Neutral | 0.065 | Benign | 0.022 | Benign | 2.50 | Benign | 0.01 | Affected | 0.1039 | 0.5984 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2452C>T | P818S 2D AIThe SynGAP1 missense variant P818S is catalogued in gnomAD (ID 6‑33443004‑C‑T) but has no ClinVar entry. Functional prediction tools split in two groups: benign calls come from REVEL and ESM1b, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The consensus predictor SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Pathogenic.” High‑accuracy assessments are limited: AlphaMissense‑Optimized yields an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this residue. Taken together, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.599170 | Disordered | 0.715889 | Binding | 0.371 | 0.893 | 0.625 | 6-33443004-C-T | 1 | 6.20e-7 | -5.740 | Likely Benign | 0.932 | Likely Pathogenic | Ambiguous | 0.203 | Likely Benign | -4.38 | Deleterious | 0.989 | Probably Damaging | 0.824 | Possibly Damaging | 2.04 | Pathogenic | 0.04 | Affected | 3.77 | 5 | 0.3581 | 0.6199 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.2465C>G | T822R 2D AIThe SynGAP1 missense variant T822R is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default) predict a pathogenic impact. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is a 2‑vs‑2 tie and therefore unavailable; Foldetta predictions are not provided. Overall, the balance of evidence (five pathogenic versus three benign predictions) suggests the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.724957 | Disordered | 0.651624 | Binding | 0.295 | 0.881 | 0.750 | 0.414 | Likely Benign | 0.952 | Likely Pathogenic | Ambiguous | 0.237 | Likely Benign | -2.74 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 2.50 | Benign | 0.05 | Affected | 0.1182 | 0.3439 | -1 | -1 | -3.8 | 55.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2465C>T | T822M 2D AIThe SynGAP1 missense variant T822M is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443017‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and ESM1b, whereas those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive because it yields a 2‑to‑2 split. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of available predictions (five pathogenic vs. three benign) lean toward pathogenicity. Thus, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.724957 | Disordered | 0.651624 | Binding | 0.295 | 0.881 | 0.750 | 6-33443017-C-T | 5 | 3.10e-6 | -4.525 | Likely Benign | 0.833 | Likely Pathogenic | Ambiguous | 0.239 | Likely Benign | -2.11 | Neutral | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 2.48 | Pathogenic | 0.03 | Affected | 3.77 | 5 | 0.1580 | 0.6379 | -1 | -1 | 2.6 | 30.09 | |||||||||||||||||||||||||||||||||||
| c.2468G>T | S823I 2D AIThe SynGAP1 missense variant S823I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only REVEL predicts a benign outcome, while ESM1b remains uncertain. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, and the SGM‑Consensus is labeled Likely Pathogenic. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.685117 | Disordered | 0.627336 | Binding | 0.358 | 0.884 | 0.750 | -7.332 | In-Between | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.287 | Likely Benign | -4.26 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 1.92 | Pathogenic | 0.00 | Affected | 0.0964 | 0.5848 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2471G>T | S824I 2D AIThe SynGAP1 missense variant S824I has no ClinVar record and is not listed in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign, reflecting the majority of benign calls. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and Foldetta results are unavailable. Overall, the balance of evidence—five benign versus three pathogenic calls, a benign SGM‑Consensus, and no conflicting ClinVar annotation—suggests that the variant is most likely benign. This conclusion does not contradict any existing ClinVar status, as none is present. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.611272 | Binding | 0.314 | 0.884 | 0.750 | -6.799 | Likely Benign | 0.950 | Likely Pathogenic | Ambiguous | 0.124 | Likely Benign | -1.18 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.60 | Benign | 0.11 | Tolerated | 0.1177 | 0.6045 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2480T>G | I827S 2D AIThe SynGAP1 missense variant I827S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign, and AlphaMissense‑Optimized is classified as Uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.636272 | Binding | 0.383 | 0.884 | 0.625 | -3.693 | Likely Benign | 0.849 | Likely Pathogenic | Ambiguous | 0.140 | Likely Benign | -0.42 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.70 | Benign | 0.29 | Tolerated | 0.2891 | 0.0512 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2483C>G | T828R 2D AIThe SynGAP1 missense variant T828R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact for T828R, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.604312 | Disordered | 0.631236 | Binding | 0.321 | 0.879 | 0.500 | -4.887 | Likely Benign | 0.773 | Likely Pathogenic | Likely Benign | 0.184 | Likely Benign | -1.38 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.64 | Benign | 0.47 | Tolerated | 0.0815 | 0.2530 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||||||||||||
| c.248G>C | R83T 2D AIThe SynGAP1 missense variant R83T has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign calls (REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus “Likely Benign”) and pathogenic calls (polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized). High‑accuracy assessments further split the verdict: AlphaMissense‑Optimized predicts pathogenic, whereas the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign; Foldetta results are unavailable. With five tools supporting benign and five supporting pathogenic, the evidence is evenly divided. Consequently, the variant’s clinical significance remains uncertain and is not contradicted by any ClinVar annotation, which has no classification. Overall, the variant is most likely pathogenic, and this does not contradict ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.522784 | Binding | 0.275 | 0.895 | 0.250 | -3.585 | Likely Benign | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.124 | Likely Benign | -2.15 | Neutral | 0.909 | Possibly Damaging | 0.587 | Possibly Damaging | 3.18 | Benign | 0.00 | Affected | 0.1671 | 0.3360 | -1 | -1 | 3.8 | -55.08 | |||||||||||||||||||||||||||||||||||||||
| c.2501T>C | M834T 2D AIThe SynGAP1 missense variant M834T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for M834T, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.585406 | Disordered | 0.640801 | Binding | 0.258 | 0.863 | 0.375 | -1.525 | Likely Benign | 0.332 | Likely Benign | Likely Benign | 0.137 | Likely Benign | -2.03 | Neutral | 0.224 | Benign | 0.085 | Benign | 2.45 | Pathogenic | 0.00 | Affected | 0.1757 | 0.1799 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2507G>T | S836I 2D AIThe SynGAP1 missense variant S836I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Consensus from standard in silico predictors shows five tools (REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Optimized) predict a benign effect, while two tools (PROVEAN, SIFT) predict pathogenicity. Two additional predictors (ESM1b, AlphaMissense‑Default) return uncertain results. High‑accuracy assessment further indicates a benign prediction from AlphaMissense‑Optimized; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (one benign, one pathogenic, two uncertain). Foldetta stability analysis is unavailable. Overall, the balance of evidence favors a benign classification, and this conclusion does not conflict with the absence of a ClinVar annotation. Therefore, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.525368 | Disordered | 0.634582 | Binding | 0.269 | 0.859 | 0.250 | -7.751 | In-Between | 0.517 | Ambiguous | Likely Benign | 0.149 | Likely Benign | -3.33 | Deleterious | 0.057 | Benign | 0.053 | Benign | 2.51 | Benign | 0.03 | Affected | 0.0756 | 0.5023 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2509G>T | V837F 2D AIThe SynGAP1 missense variant V837F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.626284 | Binding | 0.318 | 0.871 | 0.125 | -5.223 | Likely Benign | 0.322 | Likely Benign | Likely Benign | 0.164 | Likely Benign | -1.41 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.60 | Benign | 0.08 | Tolerated | 0.0749 | 0.3744 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2510T>G | V837G 2D AIThe SynGAP1 missense variant V837G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta results are not available, so they do not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.525368 | Disordered | 0.626284 | Binding | 0.318 | 0.871 | 0.125 | -2.599 | Likely Benign | 0.315 | Likely Benign | Likely Benign | 0.187 | Likely Benign | 0.93 | Neutral | 0.997 | Probably Damaging | 0.999 | Probably Damaging | 3.18 | Benign | 1.00 | Tolerated | 0.2260 | 0.2422 | -1 | -3 | -4.6 | -42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2519G>T | S840I 2D AIThe SynGAP1 missense variant S840I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated predictors—PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—classify the variant as pathogenic or likely pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus indicates likely pathogenic. Foldetta results are not available, so they do not influence the overall assessment. Based on the consensus of the majority of prediction tools and the high‑accuracy methods, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.622677 | Disordered | 0.611356 | Binding | 0.259 | 0.865 | 0.250 | -12.509 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.357 | Likely Benign | -4.31 | Deleterious | 0.998 | Probably Damaging | 0.967 | Probably Damaging | 1.51 | Pathogenic | 0.00 | Affected | 0.0788 | 0.5251 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2522T>G | V841G 2D AIThe SynGAP1 missense variant V841G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are REVEL and FATHMM, while the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, whereas the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) favors pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect for V841G, and this conclusion does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.622677 | Disordered | 0.616495 | Binding | 0.261 | 0.873 | 0.125 | -10.054 | Likely Pathogenic | 0.913 | Likely Pathogenic | Ambiguous | 0.288 | Likely Benign | -4.11 | Deleterious | 0.997 | Probably Damaging | 0.999 | Probably Damaging | 2.51 | Benign | 0.00 | Affected | 0.2351 | 0.2422 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2528T>C | M843T 2D AIThe SynGAP1 missense variant M843T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from REVEL and FATHMM, and pathogenic predictions from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b remains uncertain. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic majority, and Foldetta data are unavailable. Overall, the preponderance of evidence indicates that M843T is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.585406 | Disordered | 0.617934 | Binding | 0.327 | 0.854 | 0.375 | -7.297 | In-Between | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.295 | Likely Benign | -3.22 | Deleterious | 0.968 | Probably Damaging | 0.954 | Probably Damaging | 2.62 | Benign | 0.00 | Affected | 0.2235 | 0.2037 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||||||||
| c.2549G>T | G850V 2D AIThe SynGAP1 missense variant G850V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools converge on a benign effect: REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate a likely benign outcome. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; no Foldetta stability data are available, so it does not influence the assessment. Overall, the preponderance of evidence points to a benign effect for G850V, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.648219 | Disordered | 0.540897 | Binding | 0.312 | 0.820 | 0.500 | -5.379 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.213 | Likely Benign | -1.74 | Neutral | 0.960 | Probably Damaging | 0.679 | Possibly Damaging | 4.21 | Benign | 0.02 | Affected | 0.1255 | 0.4077 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.254C>G | T85R 2D AIThe SynGAP1 missense variant T85R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools show a split: benign calls come from REVEL, PROVEAN, polyPhen‑2 HumVar, and FATHMM, while pathogenic calls come from polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; ESM1b is uncertain. High‑accuracy assessment gives AlphaMissense‑Optimized a pathogenic score, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default (pathogenic), ESM1b (uncertain), FATHMM (benign), and PROVEAN (benign)—leans toward benign. Foldetta, a protein‑folding stability predictor, has no available result for this variant. Overall, the balance of evidence tilts toward a benign interpretation, with no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign, though the presence of pathogenic predictions from several tools indicates that further functional validation would be prudent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.680603 | Disordered | 0.542004 | Binding | 0.288 | 0.888 | 0.500 | -7.936 | In-Between | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.116 | Likely Benign | -2.32 | Neutral | 0.813 | Possibly Damaging | 0.072 | Benign | 3.91 | Benign | 0.00 | Affected | 0.0754 | 0.2482 | -1 | -1 | -3.8 | 55.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2555G>A | G852D 2D AIThe SynGAP1 missense variant G852D is catalogued in gnomAD (ID 6‑33443107‑G‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or tolerated outcomes, while the single pathogenic signal comes from SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates a benign likelihood; Foldetta results are not available. Overall, the preponderance of evidence points to a benign impact for G852D, and this conclusion is not contradicted by any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.506063 | Binding | 0.276 | 0.816 | 0.625 | 6-33443107-G-A | 1 | 6.20e-7 | -5.588 | Likely Benign | 0.203 | Likely Benign | Likely Benign | 0.154 | Likely Benign | 0.67 | Neutral | 0.001 | Benign | 0.005 | Benign | 4.18 | Benign | 0.01 | Affected | 3.77 | 5 | 0.1855 | 0.2175 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||
| c.2555G>T | G852V 2D AIThe SynGAP1 missense variant G852V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for G852V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.733139 | Disordered | 0.506063 | Binding | 0.276 | 0.816 | 0.625 | -5.629 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.30 | Neutral | 0.918 | Possibly Damaging | 0.697 | Possibly Damaging | 4.19 | Benign | 0.02 | Affected | 0.1205 | 0.4173 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2558G>T | G853V 2D AIThe SynGAP1 missense variant G853V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for G853V, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.657645 | Disordered | 0.496246 | Uncertain | 0.284 | 0.815 | 0.625 | -5.688 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.129 | Likely Benign | -1.53 | Neutral | 0.611 | Possibly Damaging | 0.502 | Possibly Damaging | 4.14 | Benign | 0.01 | Affected | 0.1188 | 0.4609 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.256G>T | V86F 2D AIThe SynGAP1 missense variant V86F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as pathogenic, while the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome; Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, and this conclusion does not contradict ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.685117 | Disordered | 0.552911 | Binding | 0.295 | 0.887 | 0.500 | -5.011 | Likely Benign | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.126 | Likely Benign | -1.54 | Neutral | 0.824 | Possibly Damaging | 0.403 | Benign | 3.75 | Benign | 0.00 | Affected | 0.0755 | 0.4078 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2570G>T | S857I 2D AIThe SynGAP1 missense variant S857I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S857I, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.728858 | Disordered | 0.475747 | Uncertain | 0.288 | 0.826 | 0.375 | -7.092 | In-Between | 0.192 | Likely Benign | Likely Benign | 0.198 | Likely Benign | -0.44 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 4.04 | Benign | 0.05 | Affected | 0.1070 | 0.6208 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2573G>T | S858I 2D AIThe SynGAP1 missense variant S858I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which classifies the variant as Likely Benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized returns Benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability analysis is available for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.482724 | Uncertain | 0.305 | 0.833 | 0.375 | -6.973 | Likely Benign | 0.234 | Likely Benign | Likely Benign | 0.125 | Likely Benign | -1.53 | Neutral | 0.818 | Possibly Damaging | 0.932 | Probably Damaging | 4.10 | Benign | 0.01 | Affected | 0.1016 | 0.5680 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2576G>T | S859I 2D AIThe SynGAP1 missense variant S859I is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of computational evidence indicates a benign impact, and this conclusion does not contradict any ClinVar annotation because no ClinVar record exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.648219 | Disordered | 0.497075 | Uncertain | 0.288 | 0.819 | 0.375 | -8.342 | Likely Pathogenic | 0.351 | Ambiguous | Likely Benign | 0.256 | Likely Benign | -1.94 | Neutral | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 3.99 | Benign | 0.02 | Affected | 0.1094 | 0.5867 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2578G>T | V860F 2D AIThe SynGAP1 missense variant V860F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy methods give a benign consensus: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign outcome. Foldetta results are not available for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.545602 | Disordered | 0.518121 | Binding | 0.269 | 0.803 | 0.250 | -4.576 | Likely Benign | 0.143 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -2.25 | Neutral | 0.846 | Possibly Damaging | 0.522 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 0.0717 | 0.4138 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2579T>G | V860G 2D AIThe SynGAP1 missense variant V860G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools cluster into two groups: the benign group includes REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; the pathogenic group contains only SIFT. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, the SGM Consensus (derived from the majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign, while Foldetta results are unavailable. Overall, the collective evidence indicates that V860G is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.545602 | Disordered | 0.518121 | Binding | 0.269 | 0.803 | 0.250 | -3.471 | Likely Benign | 0.173 | Likely Benign | Likely Benign | 0.198 | Likely Benign | -1.54 | Neutral | 0.012 | Benign | 0.009 | Benign | 4.11 | Benign | 0.00 | Affected | 0.2312 | 0.2547 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.257T>G | V86G 2D AIThe SynGAP1 missense variant V86G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, ESM1b, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split. Foldetta, which evaluates protein‑folding stability via FoldX‑MD and Rosetta, has no available result for this variant. Overall, the majority of evidence (five pathogenic versus four benign predictions) indicates that V86G is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.685117 | Disordered | 0.552911 | Binding | 0.295 | 0.887 | 0.500 | -4.212 | Likely Benign | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.130 | Likely Benign | -2.50 | Deleterious | 0.307 | Benign | 0.824 | Possibly Damaging | 3.74 | Benign | 0.00 | Affected | 0.2321 | 0.2547 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2597T>G | V866G 2D AIThe SynGAP1 missense variant V866G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact for V866G, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.599170 | Disordered | 0.638070 | Binding | 0.266 | 0.788 | 0.250 | -1.519 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -2.30 | Neutral | 0.912 | Possibly Damaging | 0.993 | Probably Damaging | 2.69 | Benign | 0.07 | Tolerated | 0.2548 | 0.2157 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2600G>T | G867V 2D AIThe SynGAP1 missense variant G867V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Two tools—ESM1b and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split between benign and uncertain calls. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the balance of evidence (five benign predictions versus two pathogenic predictions, with no decisive high‑accuracy support for pathogenicity) indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.517562 | Disordered | 0.657954 | Binding | 0.285 | 0.801 | 0.250 | -7.049 | In-Between | 0.441 | Ambiguous | Likely Benign | 0.111 | Likely Benign | -2.23 | Neutral | 0.999 | Probably Damaging | 0.958 | Probably Damaging | 2.70 | Benign | 0.10 | Tolerated | 0.1125 | 0.4613 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2602G>C | D868H 2D AIThe SynGAP1 missense variant D868H is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (variant ID 6‑33443154‑G‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs. 2 pathogenic), and Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, and this is not contradicted by ClinVar status. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.525368 | Disordered | 0.676362 | Binding | 0.262 | 0.815 | 0.250 | 6-33443154-G-C | 1 | 6.20e-7 | -3.358 | Likely Benign | 0.763 | Likely Pathogenic | Likely Benign | 0.212 | Likely Benign | -2.34 | Neutral | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.49 | Pathogenic | 0.08 | Tolerated | 3.82 | 4 | 0.2258 | 0.7837 | -1 | 1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||
| c.2618G>T | S873I 2D AIThe SynGAP1 missense variant S873I has no ClinVar record and is not reported in gnomAD. Functional prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the majority of other in‑silico predictors (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) indicate a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. AlphaMissense‑Optimized returns an uncertain result, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available prediction for this residue. High‑accuracy assessment therefore points to a Likely Pathogenic status from SGM‑Consensus, with AlphaMissense‑Optimized inconclusive and Foldetta missing. Overall, the preponderance of evidence suggests the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.414856 | Structured | 0.649816 | Binding | 0.283 | 0.866 | 0.125 | -9.412 | Likely Pathogenic | 0.945 | Likely Pathogenic | Ambiguous | 0.305 | Likely Benign | -3.44 | Deleterious | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 2.66 | Benign | 0.02 | Affected | 0.1012 | 0.5560 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2621A>C | Q874P 2D AIThe SynGAP1 missense variant Q874P is reported in gnomAD (ID 6‑33443173‑A‑C) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar classification (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.490133 | Structured | 0.635258 | Binding | 0.289 | 0.873 | 0.250 | 6-33443173-A-C | 9 | 5.58e-6 | -4.622 | Likely Benign | 0.125 | Likely Benign | Likely Benign | 0.219 | Likely Benign | -2.89 | Deleterious | 0.996 | Probably Damaging | 0.992 | Probably Damaging | 2.77 | Benign | 0.00 | Affected | 3.77 | 5 | 0.2373 | 0.5911 | -1 | 0 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||
| c.2633C>G | T878R 2D AIThe SynGAP1 missense variant T878R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.622677 | Disordered | 0.628767 | Binding | 0.288 | 0.878 | 0.250 | -4.289 | Likely Benign | 0.496 | Ambiguous | Likely Benign | 0.119 | Likely Benign | -1.39 | Neutral | 0.611 | Possibly Damaging | 0.398 | Benign | 2.64 | Benign | 0.00 | Affected | 0.1006 | 0.2920 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||||||||||||
| c.2635_2636delinsAA | A879K 2D  AIThe SynGAP1 missense variant A879K is listed in ClinVar (ID 575856.0) as benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this consensus aligns with the ClinVar designation, with no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.622695 | Binding | 0.277 | 0.874 | 0.250 | Likely Benign | 1 | -5.877 | Likely Benign | 0.757 | Likely Pathogenic | Likely Benign | -0.71 | Neutral | 0.969 | Probably Damaging | 0.593 | Possibly Damaging | 2.69 | Benign | 0.21 | Tolerated | 3.77 | 5 | 0.0930 | 0.2569 | -1 | -1 | -5.7 | 57.10 | |||||||||||||||||||||||||||||||||||||
| c.2636C>A | A879E 2D  AIThe SynGAP1 missense variant A879E is reported in gnomAD (variant ID 6‑33443188‑C‑A) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. High‑accuracy predictions specifically show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta data is missing. Overall, the majority of evidence points to a benign effect, and this assessment does not contradict any ClinVar status because none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.608892 | Disordered | 0.622695 | Binding | 0.277 | 0.874 | 0.250 | 6-33443188-C-A | 1 | 6.20e-7 | -3.786 | Likely Benign | 0.503 | Ambiguous | Likely Benign | 0.070 | Likely Benign | -0.27 | Neutral | 0.872 | Possibly Damaging | 0.659 | Possibly Damaging | 2.70 | Benign | 0.27 | Tolerated | 3.77 | 5 | 0.1120 | 0.1903 | -1 | 0 | -5.3 | 58.04 | ||||||||||||||||||||||||||||||||||
| c.263T>G | V88G 2D AIThe SynGAP1 missense variant V88G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2) and Foldetta results are unavailable. Overall, the majority of predictions (5 benign vs. 4 pathogenic) and the lack of a ClinVar pathogenic classification suggest that V88G is most likely benign, with no contradiction to existing ClinVar data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.703578 | Disordered | 0.552910 | Binding | 0.323 | 0.870 | 0.500 | -8.588 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.084 | Likely Benign | -2.40 | Neutral | 0.024 | Benign | 0.208 | Benign | 3.68 | Benign | 0.00 | Affected | 0.2608 | 0.2609 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2645G>T | G882V 2D AIThe SynGAP1 missense variant G882V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation—there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.632888 | Binding | 0.306 | 0.878 | 0.250 | -5.893 | Likely Benign | 0.257 | Likely Benign | Likely Benign | 0.115 | Likely Benign | -2.41 | Neutral | 0.224 | Benign | 0.066 | Benign | 2.06 | Pathogenic | 0.02 | Affected | 0.1178 | 0.4717 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2662G>C | A888P 2D AIThe SynGAP1 missense variant A888P is reported in gnomAD (variant ID 6‑33443214‑G‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the preponderance of evidence supports a benign classification for A888P, and this conclusion is not contradicted by any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.784345 | Disordered | 0.575860 | Binding | 0.352 | 0.928 | 0.625 | 6-33443214-G-C | 5 | 3.10e-6 | -2.368 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.050 | Likely Benign | -0.02 | Neutral | 0.000 | Benign | 0.000 | Benign | 2.56 | Benign | 0.00 | Affected | 4.32 | 4 | 0.1859 | 0.5764 | -1 | 1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||||||
| c.2666G>T | G889V 2D AIThe SynGAP1 missense variant G889V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and FATHMM. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for G889V, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.690604 | Disordered | 0.552581 | Binding | 0.331 | 0.928 | 0.625 | -5.781 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -2.11 | Neutral | 0.611 | Possibly Damaging | 0.243 | Benign | 2.39 | Pathogenic | 0.03 | Affected | 0.1198 | 0.4523 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2674T>C | S892P 2D AIThe SynGAP1 missense variant S892P is reported in gnomAD (ID 6‑33443226‑T‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign impact. This conclusion is not contradicted by ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.473390 | Uncertain | 0.319 | 0.926 | 0.875 | 6-33443226-T-C | 1 | 6.20e-7 | -3.247 | Likely Benign | 0.222 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -0.87 | Neutral | 0.057 | Benign | 0.047 | Benign | 2.60 | Benign | 0.01 | Affected | 4.32 | 4 | 0.2230 | 0.5251 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||
| c.2678A>C | Q893P 2D AIThe SynGAP1 missense variant Q893P is reported in gnomAD (ID 6‑33443230‑A‑C) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a “Likely Benign” verdict. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign. No Foldetta stability data are available, so it does not influence the assessment. Overall, the majority of predictions, including the high‑confidence tools, support a benign classification, and this is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.754692 | Disordered | 0.447267 | Uncertain | 0.310 | 0.925 | 0.750 | 6-33443230-A-C | 1 | 6.20e-7 | -2.463 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.084 | Likely Benign | -0.70 | Neutral | 0.802 | Possibly Damaging | 0.432 | Benign | 2.69 | Benign | 0.05 | Affected | 4.32 | 4 | 0.2271 | 0.5151 | -1 | 0 | 1.9 | -31.01 | ||||||||||||||||||||||||||||||||||
| c.2681G>T | G894V 2D AIThe SynGAP1 missense variant G894V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also leans benign, and Foldetta results are unavailable. Overall, the majority of high‑confidence tools predict a benign impact, and there is no conflict with the ClinVar status. Thus, the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.788093 | Disordered | 0.425700 | Uncertain | 0.310 | 0.925 | 0.750 | -5.047 | Likely Benign | 0.395 | Ambiguous | Likely Benign | 0.188 | Likely Benign | -2.61 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.64 | Benign | 0.02 | Affected | 0.1187 | 0.4091 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2683A>C | S895R 2D AIThe SynGAP1 missense variant S895R is reported in ClinVar as not yet classified and is present in gnomAD (ID 6‑33443235‑A‑C). Consensus from multiple in silico predictors shows a split: benign calls come from REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while pathogenic calls arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. When grouping by agreement, the benign group contains five tools, whereas the pathogenic group contains four. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts pathogenicity, whereas the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points toward a benign effect, and this conclusion does not conflict with the current ClinVar status, which remains unclassified. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.414977 | Uncertain | 0.294 | 0.925 | 0.750 | 6-33443235-A-C | 1 | 6.20e-7 | -4.746 | Likely Benign | 0.973 | Likely Pathogenic | Likely Pathogenic | 0.167 | Likely Benign | -1.72 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.64 | Benign | 0.10 | Tolerated | 4.32 | 4 | 0.0949 | 0.4046 | -1 | 0 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||||||
| c.2684G>T | S895I 2D AIThe SynGAP1 missense variant S895I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for S895I, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.414977 | Uncertain | 0.294 | 0.925 | 0.750 | -6.315 | Likely Benign | 0.697 | Likely Pathogenic | Likely Benign | 0.144 | Likely Benign | -2.34 | Neutral | 0.997 | Probably Damaging | 0.996 | Probably Damaging | 2.65 | Benign | 0.04 | Affected | 0.1010 | 0.6248 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2685T>A | S895R 2D AIThe SynGAP1 missense variant S895R is not reported in ClinVar and has no gnomAD entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign effect, and this is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.414977 | Uncertain | 0.294 | 0.925 | 0.750 | -4.746 | Likely Benign | 0.973 | Likely Pathogenic | Likely Pathogenic | 0.123 | Likely Benign | -1.72 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.64 | Benign | 0.10 | Tolerated | 4.32 | 4 | 0.0949 | 0.4046 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2685T>G | S895R 2D AIThe SynGAP1 missense variant S895R is not reported in ClinVar and has no gnomAD entry. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign. No Foldetta stability prediction is available. Overall, the majority of evidence points to a benign impact, and this is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.788093 | Disordered | 0.414977 | Uncertain | 0.294 | 0.925 | 0.750 | -4.746 | Likely Benign | 0.973 | Likely Pathogenic | Likely Pathogenic | 0.123 | Likely Benign | -1.72 | Neutral | 0.997 | Probably Damaging | 0.995 | Probably Damaging | 2.64 | Benign | 0.10 | Tolerated | 4.32 | 4 | 0.0949 | 0.4046 | -1 | 0 | -3.7 | 69.11 | |||||||||||||||||||||||||||||||||||||
| c.2693C>G | S898C 2D AIThe SynGAP1 missense variant S898C is catalogued in gnomAD (ID 6‑33443245‑C‑G) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM, while ESM1b remains uncertain. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores the variant as benign, and the SGM Consensus also indicates benign. Foldetta, a protein‑folding stability predictor that integrates FoldX‑MD and Rosetta outputs, did not return a result for this variant, so its stability impact is unavailable. Overall, the preponderance of evidence points to a benign effect, and this assessment does not conflict with ClinVar, which currently has no classification for S898C. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.690604 | Disordered | 0.426070 | Uncertain | 0.305 | 0.922 | 0.500 | 6-33443245-C-G | 1 | 6.20e-7 | -7.007 | In-Between | 0.257 | Likely Benign | Likely Benign | 0.146 | Likely Benign | -2.43 | Neutral | 0.999 | Probably Damaging | 0.986 | Probably Damaging | 2.43 | Pathogenic | 0.01 | Affected | 4.32 | 4 | 0.1487 | 0.6166 | -1 | 0 | 3.3 | 16.06 | |||||||||||||||||||||||||||||||||||
| c.2696T>C | I899T 2D AIThe SynGAP1 missense variant I899T is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33443248‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.443727 | Uncertain | 0.292 | 0.928 | 0.375 | 6-33443248-T-C | 2 | 1.24e-6 | -2.472 | Likely Benign | 0.521 | Ambiguous | Likely Benign | 0.103 | Likely Benign | -0.25 | Neutral | 0.245 | Benign | 0.096 | Benign | 2.75 | Benign | 0.01 | Affected | 4.32 | 4 | 0.1006 | 0.1014 | -1 | 0 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||||
| c.2696T>G | I899S 2D AIThe SynGAP1 missense variant I899S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not conflict with the ClinVar record, which contains no assertion for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.443727 | Uncertain | 0.292 | 0.928 | 0.375 | -0.857 | Likely Benign | 0.340 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -0.38 | Neutral | 0.003 | Benign | 0.004 | Benign | 2.88 | Benign | 0.00 | Affected | 0.2838 | 0.0840 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2699C>G | T900R 2D AIThe SynGAP1 missense variant T900R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized, while polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. No Foldetta stability prediction is available, so it does not influence the assessment. Overall, the majority of computational evidence points to a benign effect, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.465347 | Uncertain | 0.289 | 0.924 | 0.375 | -3.340 | Likely Benign | 0.617 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | 0.34 | Neutral | 0.782 | Possibly Damaging | 0.530 | Possibly Damaging | 2.68 | Benign | 0.83 | Tolerated | 0.1027 | 0.2851 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||||||||||||
| c.2699C>T | T900M 2D AIThe SynGAP1 missense variant T900M is listed in ClinVar (ID 1063691.0) with an “Uncertain” clinical significance and is present in the gnomAD database (gnomAD ID 6‑33443251‑C‑T). Functional prediction tools uniformly classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign effects. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. Foldetta results are unavailable. Overall, the consensus of all available predictions points to a benign impact, which does not contradict the ClinVar “Uncertain” status. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.675549 | Disordered | 0.465347 | Uncertain | 0.289 | 0.924 | 0.375 | Conflicting | 2 | 6-33443251-C-T | 14 | 8.68e-6 | -3.852 | Likely Benign | 0.176 | Likely Benign | Likely Benign | 0.015 | Likely Benign | -0.81 | Neutral | 0.060 | Benign | 0.016 | Benign | 2.79 | Benign | 0.08 | Tolerated | 4.32 | 4 | 0.1356 | 0.6533 | -1 | -1 | 2.6 | 30.09 | ||||||||||||||||||||||||||||||||
| c.269T>G | V90G 2D AIThe SynGAP1 missense variant V90G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign verdict. Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.570702 | Disordered | 0.542047 | Binding | 0.343 | 0.873 | 0.500 | -2.617 | Likely Benign | 0.358 | Ambiguous | Likely Benign | 0.094 | Likely Benign | -0.55 | Neutral | 0.024 | Benign | 0.208 | Benign | 4.00 | Benign | 0.00 | Affected | 0.2047 | 0.2936 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2708G>T | G903V 2D AIThe SynGAP1 missense variant G903V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts benign, while Foldetta results are unavailable. Overall, the balance of evidence (five benign predictions versus three pathogenic predictions, with no conflicting ClinVar annotation) indicates that the variant is most likely benign. This conclusion does not contradict any ClinVar status, as the variant is not currently catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.680603 | Disordered | 0.549818 | Binding | 0.291 | 0.917 | 0.375 | -4.750 | Likely Benign | 0.491 | Ambiguous | Likely Benign | 0.168 | Likely Benign | -1.93 | Neutral | 0.997 | Probably Damaging | 0.959 | Probably Damaging | 2.44 | Pathogenic | 0.95 | Tolerated | 0.1269 | 0.4266 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2711T>C | M904T 2D AIThe SynGAP1 missense variant M904T is listed in ClinVar (ID 1311496.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.589073 | Binding | 0.350 | 0.920 | 0.250 | Uncertain | 1 | -2.721 | Likely Benign | 0.668 | Likely Pathogenic | Likely Benign | 0.042 | Likely Benign | -1.15 | Neutral | 0.277 | Benign | 0.103 | Benign | 2.78 | Benign | 0.18 | Tolerated | 3.77 | 5 | 0.2394 | 0.2568 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||
| c.2713C>A | R905S 2D AIThe SynGAP1 missense variant R905S is catalogued in gnomAD (ID 6‑33443265‑C‑A) but has no ClinVar entry. Consensus from multiple in‑silico predictors shows a split: benign‑oriented tools—REVEL, PROVEAN, SIFT, ESM1b, and FATHMM—all classify the change as benign, while pathogenic‑oriented tools—polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default—label it pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign status. High‑accuracy assessments are mixed: AlphaMissense‑Optimized returns an uncertain result, SGM‑Consensus remains likely benign, and Foldetta data are unavailable. Overall, the majority of evidence points toward a benign effect, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.618085 | Binding | 0.291 | 0.920 | 0.250 | 6-33443265-C-A | 1 | 6.20e-7 | -2.382 | Likely Benign | 0.903 | Likely Pathogenic | Ambiguous | 0.133 | Likely Benign | -1.39 | Neutral | 0.999 | Probably Damaging | 0.962 | Probably Damaging | 2.71 | Benign | 0.15 | Tolerated | 3.77 | 5 | 0.2806 | 0.4124 | -1 | 0 | 3.7 | -69.11 | ||||||||||||||||||||||||||||||||||
| c.2720G>T | S907I 2D AIThe SynGAP1 missense variant S907I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus (majority vote) as Likely Benign, and Foldetta results are unavailable. Based on the overall balance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.637480 | Disordered | 0.661854 | Binding | 0.336 | 0.920 | 0.250 | -6.082 | Likely Benign | 0.795 | Likely Pathogenic | Ambiguous | 0.229 | Likely Benign | -2.20 | Neutral | 0.998 | Probably Damaging | 0.967 | Probably Damaging | 2.62 | Benign | 0.03 | Affected | 0.1006 | 0.5853 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2726T>C | M909T 2D AIThe SynGAP1 missense variant M909T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the majority of evidence points to a benign impact for M909T, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.696196 | Binding | 0.314 | 0.914 | 0.250 | -3.053 | Likely Benign | 0.647 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | -0.74 | Neutral | 0.901 | Possibly Damaging | 0.430 | Benign | 2.86 | Benign | 1.00 | Tolerated | 0.2157 | 0.2085 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2731G>T | V911F 2D AIThe SynGAP1 missense variant V911F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence points to a benign effect for V911F, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.685117 | Disordered | 0.724137 | Binding | 0.327 | 0.914 | 0.375 | -4.454 | Likely Benign | 0.169 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -1.08 | Neutral | 0.977 | Probably Damaging | 0.721 | Possibly Damaging | 2.68 | Benign | 0.04 | Affected | 0.0749 | 0.4380 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2732T>G | V911G 2D AIThe SynGAP1 missense variant V911G is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). All available in silico predictors classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity, so the benign group includes every listed predictor, while the pathogenic group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign verdict. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is not available for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.685117 | Disordered | 0.724137 | Binding | 0.327 | 0.914 | 0.375 | -2.754 | Likely Benign | 0.222 | Likely Benign | Likely Benign | 0.124 | Likely Benign | -0.04 | Neutral | 0.004 | Benign | 0.003 | Benign | 2.74 | Benign | 0.16 | Tolerated | 0.2307 | 0.3360 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2735C>T | T912I 2D AIThe SynGAP1 missense variant T912I is listed in gnomAD (ID 6‑33443287‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default is uncertain, and Foldetta (FoldX‑MD/Rosetta stability assessment) has no available result for this variant. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta data is missing. Overall, the majority of reliable predictors indicate a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.724957 | Disordered | 0.740671 | Binding | 0.285 | 0.909 | 0.250 | 6-33443287-C-T | 2 | 1.24e-6 | -4.461 | Likely Benign | 0.522 | Ambiguous | Likely Benign | 0.117 | Likely Benign | -1.27 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 3.95 | Benign | 0.00 | Affected | 3.77 | 5 | 0.0945 | 0.5615 | -1 | 0 | 5.2 | 12.05 | ||||||||||||||||||||||||||||||||||
| c.2738C>G | T913R 2D AIThe SynGAP1 missense variant T913R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.763517 | Binding | 0.339 | 0.899 | 0.375 | -3.218 | Likely Benign | 0.494 | Ambiguous | Likely Benign | 0.152 | Likely Benign | -0.97 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.68 | Benign | 0.03 | Affected | 0.1025 | 0.3373 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||||||||||||
| c.2746G>T | V916F 2D AIThe SynGAP1 missense variant V916F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—polyPhen‑2 HumDiv and SIFT—suggest a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.642678 | Disordered | 0.835395 | Binding | 0.308 | 0.879 | 0.250 | -4.011 | Likely Benign | 0.123 | Likely Benign | Likely Benign | 0.121 | Likely Benign | -1.18 | Neutral | 0.642 | Possibly Damaging | 0.265 | Benign | 2.67 | Benign | 0.02 | Affected | 0.0729 | 0.4593 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2747T>G | V916G 2D AIThe SynGAP1 missense variant V916G is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.642678 | Disordered | 0.835395 | Binding | 0.308 | 0.879 | 0.250 | -1.950 | Likely Benign | 0.091 | Likely Benign | Likely Benign | 0.093 | Likely Benign | -0.63 | Neutral | 0.666 | Possibly Damaging | 0.917 | Probably Damaging | 2.69 | Benign | 0.05 | Affected | 0.2420 | 0.2681 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2749C>G | P917A 2D AIThe SynGAP1 missense variant P917A is not reported in ClinVar and is present in gnomAD (ID 6‑33443301‑C‑G). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all classify the change as benign or likely benign. Only SIFT predicts a pathogenic effect. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized reports benign, and the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective predictions point to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no entry for P917A. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.863949 | Binding | 0.314 | 0.862 | 0.375 | 6-33443301-C-G | 1 | 6.20e-7 | -3.681 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.053 | Likely Benign | -1.33 | Neutral | 0.065 | Benign | 0.037 | Benign | 2.72 | Benign | 0.00 | Affected | 3.77 | 5 | 0.3458 | 0.4772 | -1 | 1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||
| c.2749C>T | P917S 2D AIThe SynGAP1 missense variant P917S is catalogued in gnomAD (ID 6‑33443301‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign. Only SIFT predicts pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote) is benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant, so it provides no additional evidence. Overall, the preponderance of predictions indicates that P917S is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.599170 | Disordered | 0.863949 | Binding | 0.314 | 0.862 | 0.375 | 6-33443301-C-T | 1 | 6.20e-7 | -3.562 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.056 | Likely Benign | -0.54 | Neutral | 0.003 | Benign | 0.002 | Benign | 2.70 | Benign | 0.00 | Affected | 3.77 | 5 | 0.3478 | 0.5121 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.275G>T | G92V 2D AIThe SynGAP1 missense variant G92V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts Benign, and the SGM‑Consensus (majority vote) also indicates Likely Benign. Foldetta results are unavailable. Overall, the majority of computational evidence points to a benign impact for G92V, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.575842 | Disordered | 0.537848 | Binding | 0.337 | 0.874 | 0.625 | -4.627 | Likely Benign | 0.338 | Likely Benign | Likely Benign | 0.164 | Likely Benign | -2.62 | Deleterious | 0.999 | Probably Damaging | 0.972 | Probably Damaging | 4.00 | Benign | 0.00 | Affected | 0.1171 | 0.4363 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2768T>G | I923S 2D AIThe SynGAP1 missense variant I923S is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for I923S. This conclusion is not contradicted by ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.562014 | Disordered | 0.964857 | Binding | 0.292 | 0.852 | 0.250 | -0.002 | Likely Benign | 0.760 | Likely Pathogenic | Likely Benign | 0.094 | Likely Benign | -0.61 | Neutral | 0.912 | Possibly Damaging | 0.529 | Possibly Damaging | 2.73 | Benign | 0.33 | Tolerated | 0.3300 | 0.1455 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||||||||||||
| c.2779T>G | F927V 2D AIThe SynGAP1 missense variant F927V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL and ESM1b, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, and the SGM‑Consensus also indicates Likely Pathogenic. Foldetta results are unavailable and therefore not considered. Overall, the preponderance of evidence from multiple in silico predictors and high‑accuracy tools indicates that the variant is most likely pathogenic, with no ClinVar annotation to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.529623 | Disordered | 0.985043 | Binding | 0.324 | 0.854 | 0.250 | -4.919 | Likely Benign | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.470 | Likely Benign | -5.21 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.34 | Pathogenic | 0.00 | Affected | 0.2207 | 0.1206 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2794T>G | F932V 2D AIThe SynGAP1 missense variant F932V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL and ESM1b, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic outcome. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely pathogenic status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.675549 | Disordered | 0.989197 | Binding | 0.293 | 0.858 | 0.500 | -5.728 | Likely Benign | 0.976 | Likely Pathogenic | Likely Pathogenic | 0.392 | Likely Benign | -3.68 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.32 | Pathogenic | 0.01 | Affected | 0.2137 | 0.3082 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||||||||||||
| c.2801T>C | M934T 2D AIThe SynGAP1 missense variant M934T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, and ESM1b, while those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Pathogenic, and the Foldetta stability analysis is unavailable. Based on the majority of predictions and the consensus call, the variant is most likely pathogenic; this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | 0.762850 | Disordered | 0.984677 | Binding | 0.290 | 0.867 | 0.625 | -2.579 | Likely Benign | 0.823 | Likely Pathogenic | Ambiguous | 0.270 | Likely Benign | -3.33 | Deleterious | 0.811 | Possibly Damaging | 0.424 | Benign | 2.39 | Pathogenic | 0.19 | Tolerated | 0.2331 | 0.2267 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||||||||||||
| c.2809G>C | D937H 2D AIThe SynGAP1 D937H missense variant (ClinVar ID 2825773.0) is listed as “Uncertain” and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote) is benign, and Foldetta (protein‑folding stability analysis combining FoldX‑MD and Rosetta) data are unavailable. Based on the preponderance of evidence from both general and high‑accuracy predictors, the variant is most likely benign, which is consistent with its ClinVar “Uncertain” status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.819762 | Disordered | 0.963385 | Binding | 0.348 | 0.883 | 0.625 | Uncertain | 1 | -0.733 | Likely Benign | 0.677 | Likely Pathogenic | Likely Benign | 0.150 | Likely Benign | -1.74 | Neutral | 1.000 | Probably Damaging | 0.975 | Probably Damaging | 2.68 | Benign | 0.13 | Tolerated | 3.77 | 5 | 0.2792 | 0.8228 | -1 | 1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||
| c.280C>A | P94T 2D AIThe SynGAP1 missense variant P94T is reported in gnomAD (variant ID 6‑33425888‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign status. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Consequently, the collective evidence indicates that P94T is most likely benign, and this assessment does not contradict any ClinVar classification because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.671169 | Disordered | 0.570978 | Binding | 0.350 | 0.869 | 0.625 | 6-33425888-C-A | 1 | 6.20e-7 | -4.254 | Likely Benign | 0.085 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -2.35 | Neutral | 0.198 | Benign | 0.015 | Benign | 4.12 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1326 | 0.4789 | -1 | 0 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||
| c.2813G>T | G938V 2D AIThe SynGAP1 missense variant G938V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which is “Likely Benign”). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. No Foldetta stability result is available, so it does not influence the assessment. Overall, the majority of evidence indicates that G938V is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.905695 | Disordered | 0.949795 | Binding | 0.318 | 0.883 | 0.625 | -6.112 | Likely Benign | 0.153 | Likely Benign | Likely Benign | 0.123 | Likely Benign | -0.81 | Neutral | 0.999 | Probably Damaging | 0.985 | Probably Damaging | 2.83 | Benign | 0.31 | Tolerated | 0.1230 | 0.3811 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2819G>A | G940D 2D AIThe SynGAP1 missense variant G940D is not reported in ClinVar (ClinVar status: none) but is present in gnomAD (ID 6‑33443371‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Two tools, AlphaMissense‑Default and ESM1b, return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it receives two benign and two uncertain votes, and Foldetta’s protein‑folding stability analysis is unavailable. Overall, the balance of evidence favors a benign classification, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.889439 | Disordered | 0.920635 | Binding | 0.383 | 0.902 | 0.625 | 6-33443371-G-A | 6 | 3.72e-6 | -7.311 | In-Between | 0.397 | Ambiguous | Likely Benign | 0.076 | Likely Benign | -0.68 | Neutral | 0.770 | Possibly Damaging | 0.583 | Possibly Damaging | 2.72 | Benign | 0.17 | Tolerated | 3.77 | 5 | 0.1837 | 0.2252 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||||||||
| c.2819G>T | G940V 2D AIThe SynGAP1 missense variant G940V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is Likely Benign; Foldetta results are unavailable. Overall, the consensus of available predictions points to a benign impact, and this is consistent with the lack of ClinVar evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.889439 | Disordered | 0.920635 | Binding | 0.383 | 0.902 | 0.625 | -6.252 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.110 | Likely Benign | 0.36 | Neutral | 0.626 | Possibly Damaging | 0.419 | Benign | 2.92 | Benign | 0.31 | Tolerated | 0.1222 | 0.3822 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2821C>A | P941T 2D AIThe SynGAP1 missense variant P941T is listed in gnomAD (ID 6‑33443373‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it to be pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” consensus. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.900790 | Binding | 0.403 | 0.906 | 0.625 | 6-33443373-C-A | 2 | 1.24e-6 | -6.193 | Likely Benign | 0.065 | Likely Benign | Likely Benign | 0.033 | Likely Benign | 0.19 | Neutral | 0.144 | Benign | 0.085 | Benign | 2.77 | Benign | 0.05 | Affected | 4.32 | 4 | 0.1612 | 0.5607 | -1 | 0 | 0.9 | 3.99 | ||||||||||||||||||||||||||||||||||
| c.2821C>G | P941A 2D AIThe SynGAP1 missense variant P941A is reported in gnomAD (variant ID 6-33443373-C-G) but has no ClinVar entry. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a benign effect. No tool predicts pathogenicity. The high‑accuracy consensus, SGM‑Consensus, also reports the variant as Likely Benign, and AlphaMissense‑Optimized concurs with a benign prediction. Foldetta, a protein‑folding stability method, did not provide a result for this variant, so its status remains unavailable. Overall, the collective evidence strongly supports a benign classification, and this assessment is consistent with the absence of a ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.900790 | Binding | 0.403 | 0.906 | 0.625 | 6-33443373-C-G | 2 | 1.24e-6 | -4.313 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.046 | Likely Benign | 0.10 | Neutral | 0.000 | Benign | 0.002 | Benign | 2.80 | Benign | 0.10 | Tolerated | 4.32 | 4 | 0.2844 | 0.5190 | -1 | 1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||||
| c.2821C>T | P941S 2D AIThe SynGAP1 missense variant P941S is reported in gnomAD (ID 6‑33443373‑C‑T) but has no ClinVar entry. Functional prediction tools uniformly classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, which evaluates protein‑folding stability via FoldX‑MD and Rosetta, has no available result for this variant. Overall, the consensus of all available predictions indicates that P941S is most likely benign, and this assessment does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.874069 | Disordered | 0.900790 | Binding | 0.403 | 0.906 | 0.625 | 6-33443373-C-T | 1 | 6.20e-7 | -5.099 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.039 | Likely Benign | 0.58 | Neutral | 0.036 | Benign | 0.039 | Benign | 3.05 | Benign | 0.95 | Tolerated | 4.32 | 4 | 0.2759 | 0.5576 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.2824C>T | P942S 2D AIThe SynGAP1 missense variant P942S is catalogued in gnomAD (ID 6‑33443376‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.919029 | Disordered | 0.878102 | Binding | 0.365 | 0.915 | 0.625 | 6-33443376-C-T | 1 | 6.20e-7 | -3.919 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.036 | Likely Benign | -0.80 | Neutral | 0.011 | Benign | 0.015 | Benign | 2.43 | Pathogenic | 0.00 | Affected | 4.32 | 4 | 0.3226 | 0.4806 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.2828G>T | G943V 2D AIThe SynGAP1 missense variant G943V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. The only tool with an uncertain outcome is ESM1b, which does not alter the overall consensus. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign classification. High‑accuracy predictors corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise indicates likely benign; Foldetta results are not available. Taken together, the evidence overwhelmingly supports a benign interpretation, and there is no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.973328 | Disordered | 0.860437 | Binding | 0.372 | 0.910 | 0.750 | -7.658 | In-Between | 0.092 | Likely Benign | Likely Benign | 0.265 | Likely Benign | -0.43 | Neutral | 0.224 | Benign | 0.062 | Benign | 2.85 | Benign | 0.16 | Tolerated | 0.1399 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2837G>T | G946V 2D AIThe SynGAP1 missense variant G946V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. ESM1b is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for G946V, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985417 | Disordered | 0.845792 | Binding | 0.357 | 0.920 | 0.750 | -7.528 | In-Between | 0.109 | Likely Benign | Likely Benign | 0.368 | Likely Benign | -0.30 | Neutral | 0.901 | Possibly Damaging | 0.516 | Possibly Damaging | 4.57 | Benign | 0.00 | Affected | 0.1524 | 0.3707 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.283C>G | H95D 2D  AIThe SynGAP1 missense variant H95D is not listed in ClinVar and is present in gnomAD (variant ID 6‑33425891‑C‑G). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign or likely benign. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus also reports likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the computational evidence overwhelmingly supports a benign classification, and this is consistent with the absence of a ClinVar pathogenic report. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.661982 | Disordered | 0.590542 | Binding | 0.335 | 0.875 | 0.625 | 6-33425891-C-G | 3 | 1.86e-6 | -2.387 | Likely Benign | 0.188 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -0.81 | Neutral | 0.084 | Benign | 0.009 | Benign | 4.22 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2759 | 0.1801 | -1 | 1 | -0.3 | -22.05 | ||||||||||||||||||||||||||||||||||
| c.2840G>T | G947V 2D AIThe SynGAP1 missense variant G947V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate benign, whereas polyPhen‑2 HumDiv and SIFT predict pathogenicity; ESM1b remains uncertain. High‑accuracy methods reinforce the benign assessment: AlphaMissense‑Optimized scores benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” and Foldetta data are not available. Overall, the preponderance of evidence points to a benign impact for G947V, and this conclusion is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988695 | Disordered | 0.850554 | Binding | 0.358 | 0.919 | 0.750 | -7.171 | In-Between | 0.105 | Likely Benign | Likely Benign | 0.296 | Likely Benign | -1.11 | Neutral | 0.586 | Possibly Damaging | 0.303 | Benign | 4.93 | Benign | 0.01 | Affected | 0.1443 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2843G>T | G948V 2D AIThe SynGAP1 missense variant G948V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also reports Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the consensus of the majority of prediction tools and the high‑accuracy methods indicates that G948V is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.862121 | Binding | 0.365 | 0.919 | 0.750 | -6.541 | Likely Benign | 0.102 | Likely Benign | Likely Benign | 0.256 | Likely Benign | -0.48 | Neutral | 0.901 | Possibly Damaging | 0.435 | Benign | 4.52 | Benign | 0.06 | Tolerated | 0.1464 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2846G>T | G949V 2D AIThe SynGAP1 missense variant G949V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors a benign outcome. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.988861 | Disordered | 0.874971 | Binding | 0.365 | 0.923 | 0.750 | -7.364 | In-Between | 0.102 | Likely Benign | Likely Benign | 0.328 | Likely Benign | -0.55 | Neutral | 0.992 | Probably Damaging | 0.834 | Possibly Damaging | 2.21 | Pathogenic | 0.01 | Affected | 0.1493 | 0.3165 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2849G>T | G950V 2D AIThe SynGAP1 missense variant G950V is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and FATHMM, while ESM1b remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that G950V is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.987317 | Disordered | 0.888649 | Binding | 0.368 | 0.923 | 0.750 | -7.796 | In-Between | 0.093 | Likely Benign | Likely Benign | 0.428 | Likely Benign | -0.91 | Neutral | 0.411 | Benign | 0.239 | Benign | 2.26 | Pathogenic | 0.01 | Affected | 0.1392 | 0.3507 | -1 | -3 | 4.6 | 42.08 | ||||||||||||||||||||||||||||||||||||||||
| c.2855G>T | G952V 2D AIThe SynGAP1 missense variant G952V is listed in ClinVar (ID 2055482.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while ESM1b remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as “Likely Benign.” High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates that G952V is most likely benign, and this conclusion does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.910621 | Binding | 0.341 | 0.926 | 0.750 | Uncertain | 1 | -7.074 | In-Between | 0.078 | Likely Benign | Likely Benign | 0.231 | Likely Benign | -0.33 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.20 | Benign | 0.02 | Affected | 3.77 | 5 | 0.1538 | 0.3507 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||
| c.2875C>G | H959D 2D AIThe SynGAP1 missense variant H959D is listed in gnomAD (ID 6‑33443427‑C‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only ESM1b predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to “Likely Benign” (three benign votes versus one pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar status (none reported). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.985964 | Disordered | 0.980566 | Binding | 0.333 | 0.905 | 0.750 | 6-33443427-C-G | 1 | 6.20e-7 | -12.060 | Likely Pathogenic | 0.235 | Likely Benign | Likely Benign | 0.176 | Likely Benign | -0.73 | Neutral | 0.144 | Benign | 0.058 | Benign | 4.14 | Benign | 0.29 | Tolerated | 3.77 | 5 | 0.2586 | 0.3066 | -1 | 1 | -0.3 | -22.05 | ||||||||||||||||||||||||||||||||||
| c.287G>T | G96V 2D AIThe SynGAP1 missense variant G96V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect. The variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.657645 | Disordered | 0.599491 | Binding | 0.335 | 0.871 | 0.625 | -4.266 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.109 | Likely Benign | -1.43 | Neutral | 0.334 | Benign | 0.029 | Benign | 4.18 | Benign | 0.00 | Affected | 0.1575 | 0.3833 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2893C>G | H965D 2D AIThe SynGAP1 missense variant H965D is reported in gnomAD (6‑33443445‑C‑G) and has no ClinVar entry. Consensus from most in silico predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classifies the change as benign, while only the ESM1b model flags it as pathogenic. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized returns a benign score, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, did not provide a result for this residue, so its status is unavailable. Overall, the preponderance of evidence indicates that H965D is most likely benign, and this assessment does not contradict any ClinVar classification because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.988505 | Disordered | 0.978700 | Binding | 0.342 | 0.882 | 0.750 | 6-33443445-C-G | 1 | 6.20e-7 | -9.827 | Likely Pathogenic | 0.192 | Likely Benign | Likely Benign | 0.147 | Likely Benign | -0.94 | Neutral | 0.007 | Benign | 0.018 | Benign | 4.09 | Benign | 0.62 | Tolerated | 3.77 | 5 | 0.2697 | 0.3066 | -1 | 1 | -0.3 | -22.05 | ||||||||||||||||||||||||||||||||||
| c.2896C>G | H966D 2D AIThe SynGAP1 missense variant H966D is listed in gnomAD (ID 6‑33443448‑C‑G) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the change as benign or likely benign. Only two tools predict a damaging outcome—polyPhen‑2 HumDiv and ESM1b—which are outliers relative to the consensus. High‑accuracy assessments confirm the benign trend: AlphaMissense‑Optimized reports a benign effect, and the SGM‑Consensus also indicates a likely benign status. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions support a benign impact, and this is consistent with the absence of a pathogenic ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.976962 | Disordered | 0.974672 | Binding | 0.378 | 0.879 | 0.750 | 6-33443448-C-G | 1 | 6.20e-7 | -8.426 | Likely Pathogenic | 0.201 | Likely Benign | Likely Benign | 0.182 | Likely Benign | -1.09 | Neutral | 0.494 | Possibly Damaging | 0.170 | Benign | 4.05 | Benign | 0.93 | Tolerated | 4.32 | 2 | 0.2504 | 0.3066 | -1 | 1 | -0.3 | -22.05 | ||||||||||||||||||||||||||||||||||
| c.2903G>A | G968D 2D AIThe SynGAP1 missense variant G968D is catalogued in gnomAD (ID 6‑33443455‑G‑A) but has no ClinVar entry. Across a broad panel of in‑silico predictors, every tool reports a benign outcome: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity, so the pathogenic‑prediction group is empty. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta results are not available for this variant. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | 6-33443455-G-A | 1 | 6.20e-7 | -5.134 | Likely Benign | 0.179 | Likely Benign | Likely Benign | 0.157 | Likely Benign | -0.48 | Neutral | 0.440 | Benign | 0.198 | Benign | 4.19 | Benign | 0.21 | Tolerated | 4.32 | 2 | 0.1965 | 0.3243 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||||||||
| c.2903G>T | G968V 2D AIThe SynGAP1 missense variant G968V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The predictions do not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.978316 | Disordered | 0.961360 | Binding | 0.327 | 0.896 | 0.750 | -6.152 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.239 | Likely Benign | -1.08 | Neutral | 0.918 | Possibly Damaging | 0.525 | Possibly Damaging | 4.19 | Benign | 0.11 | Tolerated | 0.1252 | 0.4036 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2906G>T | G969V 2D AIThe SynGAP1 missense variant G969V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.956572 | Binding | 0.405 | 0.898 | 0.750 | -5.946 | Likely Benign | 0.089 | Likely Benign | Likely Benign | 0.116 | Likely Benign | -0.92 | Neutral | 0.761 | Possibly Damaging | 0.239 | Benign | 4.18 | Benign | 0.01 | Affected | 0.1497 | 0.3847 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
| c.2911C>T | P971S 2D AIThe SynGAP1 missense variant P971S is catalogued in gnomAD (ID 6‑33443463‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) all indicate a benign or likely benign outcome. Only SIFT classifies the change as pathogenic, representing a minority opinion. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus likewise reports likely benign. No Foldetta stability analysis is available, so it does not influence the overall assessment. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.950334 | Disordered | 0.951523 | Binding | 0.545 | 0.905 | 0.625 | 6-33443463-C-T | 1 | 6.20e-7 | -4.188 | Likely Benign | 0.061 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.51 | Neutral | 0.002 | Benign | 0.003 | Benign | 3.99 | Benign | 0.00 | Affected | 4.32 | 2 | 0.3009 | 0.5667 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||||
| c.2914C>G | P972A 2D AIThe SynGAP1 missense variant P972A is listed in ClinVar with an uncertain significance (ClinVar ID 3172763.0) and is present in the gnomAD database (gnomAD ID 6‑33443466‑C‑G). All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the computational evidence strongly suggests the variant is most likely benign, and this conclusion does not contradict the current ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.954150 | Binding | 0.472 | 0.904 | 0.625 | Uncertain | 1 | 6-33443466-C-G | 1 | 6.20e-7 | -0.167 | Likely Benign | 0.045 | Likely Benign | Likely Benign | 0.046 | Likely Benign | -0.89 | Neutral | 0.016 | Benign | 0.011 | Benign | 4.29 | Benign | 0.07 | Tolerated | 4.32 | 2 | 0.3212 | 0.4753 | -1 | 1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||||
| c.2914C>T | P972S 2D AIThe SynGAP1 missense variant P972S (ClinVar ID 3361353.0) is listed as Uncertain in ClinVar and is present in gnomAD (ID 6‑33443466‑C‑T). Consensus among most in silico predictors indicates a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the substitution as benign. Only SIFT classifies it as pathogenic, representing the sole discordant prediction. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign.” No Foldetta stability analysis is available for this residue. Overall, the preponderance of computational evidence points to a benign effect, which is consistent with the ClinVar designation of uncertainty rather than pathogenicity. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.891961 | Disordered | 0.954150 | Binding | 0.472 | 0.904 | 0.625 | Uncertain | 1 | 6-33443466-C-T | 4 | 2.48e-6 | -4.008 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.074 | Likely Benign | -0.38 | Neutral | 0.001 | Benign | 0.002 | Benign | 4.28 | Benign | 0.05 | Affected | 4.32 | 2 | 0.3168 | 0.4822 | -1 | 1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||
| c.2918G>T | G973V 2D AIThe SynGAP1 missense variant G973V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while only SIFT predicts it as pathogenic. Grouping by consensus, the benign‑predicting tools outnumber the single pathogenic prediction. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized reports a benign outcome, and the SGM Consensus—derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also yields a benign classification. Foldetta results are unavailable, so they do not influence the assessment. Overall, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.887230 | Disordered | 0.959498 | Binding | 0.390 | 0.897 | 0.625 | -4.688 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -0.76 | Neutral | 0.224 | Benign | 0.091 | Benign | 4.18 | Benign | 0.01 | Affected | 0.1392 | 0.3684 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||||||||||||
Found 8751 rows. Show 800 rows per page. Page 3/11 « Previous | Next »