
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain | ClinVar | gnomAD | ESM1b | AlphaMissense | REVEL | FoldX | Rosetta | Foldetta | PremPS | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | DOI | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Score | Prediction | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.3125A>G | Q1042R 2D  AIThe SynGAP1 missense variant Q1042R is listed in ClinVar (ID 2662705.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33443677‑A‑G). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta results are unavailable. Overall, the majority of evidence supports a benign impact for Q1042R, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 2 | 6-33443677-A-G | 2 | 1.24e-6 | -2.928 | Likely Benign | 0.413 | Ambiguous | Likely Benign | 0.300 | Likely Benign | -1.39 | Neutral | 0.586 | Possibly Damaging | 0.120 | Benign | 5.48 | Benign | 0.12 | Tolerated | 3.77 | 5 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||
| c.3136C>G | P1046A 2D  AIThe SynGAP1 missense variant P1046A is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443688‑C‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33443688-C-G | 1 | 6.20e-7 | -3.246 | Likely Benign | 0.048 | Likely Benign | Likely Benign | 0.041 | Likely Benign | -1.67 | Neutral | 0.001 | Benign | 0.008 | Benign | 2.39 | Pathogenic | 0.29 | Tolerated | 3.77 | 5 | -1 | 1 | 3.4 | -26.04 | |||||||||||||||||||||||||||
| c.313T>C | S105P 2D  AIThe SynGAP1 missense variant S105P is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only two tools—polyPhen‑2 HumDiv and SIFT—predict a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also indicates likely benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -3.631 | Likely Benign | 0.166 | Likely Benign | Likely Benign | 0.204 | Likely Benign | 0.03 | Neutral | 0.808 | Possibly Damaging | 0.212 | Benign | 4.00 | Benign | 0.00 | Affected | 4.32 | 1 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||
| c.3152G>A | G1051D 2D  AISynGAP1 missense variant G1051D is listed in ClinVar as Benign and is present in gnomAD (variant ID 6‑33443704‑G‑A). Prediction tools that classify the variant as benign include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are polyPhen‑2 HumDiv, ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign versus two pathogenic votes), and Foldetta stability analysis is unavailable. Overall, the balance of evidence favors a benign effect, consistent with the ClinVar annotation and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Benign | 1 | 6-33443704-G-A | 2 | 1.24e-6 | -9.379 | Likely Pathogenic | 0.311 | Likely Benign | Likely Benign | 0.445 | Likely Benign | -0.31 | Neutral | 0.761 | Possibly Damaging | 0.239 | Benign | -0.74 | Pathogenic | 0.39 | Tolerated | 3.77 | 5 | -1 | 1 | -3.1 | 58.04 | ||||||||||||||||||||||||||||
| c.3170G>A | S1057N 2D  AIThe SynGAP1 missense variant S1057N is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All available in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No tool predicts pathogenicity. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the computational evidence overwhelmingly supports a benign effect, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -6.386 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.218 | Likely Benign | -0.41 | Neutral | 0.451 | Benign | 0.129 | Benign | 5.25 | Benign | 0.28 | Tolerated | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||||
| c.3223C>A | Q1075K 2D  AIThe SynGAP1 missense variant Q1075K (ClinVar ID 2762879.0) is listed as “Uncertain” in ClinVar and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign” because three of the four contributing tools predict benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote) as benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -5.135 | Likely Benign | 0.728 | Likely Pathogenic | Likely Benign | 0.134 | Likely Benign | -0.67 | Neutral | 0.963 | Probably Damaging | 0.959 | Probably Damaging | 2.75 | Benign | 1.00 | Tolerated | 3.77 | 5 | 1 | 1 | -0.4 | 0.04 | ||||||||||||||||||||||||||||||
| c.3238G>T | A1080S 2D  AIThe SynGAP1 missense variant A1080S is listed in ClinVar (ID 2703014.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33443790‑G‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign effect, and this conclusion does not contradict the ClinVar designation, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33443790-G-T | 1 | 6.26e-7 | -3.277 | Likely Benign | 0.108 | Likely Benign | Likely Benign | 0.103 | Likely Benign | 0.01 | Neutral | 0.702 | Possibly Damaging | 0.346 | Benign | 4.16 | Benign | 0.08 | Tolerated | 3.77 | 5 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||
| c.3250C>G | P1084A 2D AIThe SynGAP1 missense variant P1084A is listed in ClinVar (ID 2827308.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which itself is “Likely Benign”). In contrast, PROVEAN and polyPhen‑2 HumDiv predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -3.928 | Likely Benign | 0.066 | Likely Benign | Likely Benign | 0.114 | Likely Benign | -2.54 | Deleterious | 0.649 | Possibly Damaging | 0.157 | Benign | 4.05 | Benign | 0.35 | Tolerated | 3.77 | 5 | -1 | 1 | 3.4 | -26.04 | ||||||||||||||||||||||||||||||
| c.3254G>A | R1085Q 2D  AIThe SynGAP1 missense variant R1085Q (ClinVar ID 1729448.0) is listed as ClinVar status Uncertain and is present in gnomAD (6‑33443806‑G‑A). Functional prediction tools show a split opinion: benign predictions come from REVEL, PROVEAN, ESM1b, and FATHMM, whereas pathogenic predictions are reported by polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) also favors benign. No Foldetta stability analysis is available for this variant. Overall, the majority of computational evidence points to a benign effect, which is consistent with the ClinVar uncertain designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33443806-G-A | 5 | 3.16e-6 | -3.843 | Likely Benign | 0.589 | Likely Pathogenic | Likely Benign | 0.224 | Likely Benign | -1.43 | Neutral | 0.998 | Probably Damaging | 0.988 | Probably Damaging | 2.73 | Benign | 0.02 | Affected | 3.77 | 5 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||
| c.3262A>G | S1088G 2D  AIThe SynGAP1 missense variant S1088G is listed in ClinVar (ID 2742833.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as Likely Benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this is consistent with the ClinVar “Uncertain” status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -5.034 | Likely Benign | 0.285 | Likely Benign | Likely Benign | 0.163 | Likely Benign | -1.83 | Neutral | 0.979 | Probably Damaging | 0.973 | Probably Damaging | 2.63 | Benign | 0.03 | Affected | 3.77 | 5 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||
| c.3293G>A | S1098N 2D AIThe SynGAP1 missense variant S1098N is listed in ClinVar (ID 864704.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33443845‑G‑A). All evaluated in‑silico predictors agree on a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the computational evidence strongly supports a benign classification, which is consistent with the ClinVar “Uncertain” status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Conflicting | 2 | 6-33443845-G-A | 6 | 3.89e-6 | -5.120 | Likely Benign | 0.156 | Likely Benign | Likely Benign | 0.063 | Likely Benign | -0.58 | Neutral | 0.369 | Benign | 0.120 | Benign | 2.76 | Benign | 0.36 | Tolerated | 3.77 | 5 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||
| c.3304G>C | A1102P 2D  AIThe SynGAP1 missense variant A1102P is listed in ClinVar (ID 2789225.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only FATHMM predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -5.120 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.118 | Likely Benign | -0.97 | Neutral | 0.000 | Benign | 0.002 | Benign | 2.26 | Pathogenic | 0.13 | Tolerated | 3.77 | 5 | -1 | 1 | -3.4 | 26.04 | ||||||||||||||||||||||||||||||
| c.3310C>T | P1104S 2D  AIThe SynGAP1 missense variant P1104S is listed in ClinVar (ID 2912797.0) as Benign and is present in gnomAD (variant ID 6‑33443862‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign, and AlphaMissense‑Optimized also reports Benign. Foldetta results are not available. Overall, the majority of computational evidence supports a benign classification, which is consistent with the ClinVar status. Thus, the variant is most likely benign and does not contradict the ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 1 | 6-33443862-C-T | 1 | 6.54e-7 | -2.330 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.088 | Likely Benign | -0.30 | Neutral | 0.770 | Possibly Damaging | 0.404 | Benign | 2.77 | Benign | 0.10 | Tolerated | 3.77 | 5 | -1 | 1 | 0.8 | -10.04 | |||||||||||||||||||||||||||
| c.3314G>A | R1105Q 2D AIThe SynGAP1 missense variant R1105Q is listed in ClinVar (ID 1803693.0) with an uncertain significance status and is present in gnomAD (variant ID 6‑33443866‑G‑A). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report benign or tolerated outcomes. Only polyPhen‑2 HumDiv predicts a pathogenic effect. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments further support this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the preponderance of evidence indicates the variant is most likely benign, which is consistent with its ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 2 | 6-33443866-G-A | 3 | 1.96e-6 | -3.666 | Likely Benign | 0.216 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -1.21 | Neutral | 0.958 | Probably Damaging | 0.194 | Benign | 2.50 | Benign | 0.16 | Tolerated | 3.77 | 5 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||
| c.3338G>A | G1113D 2D AIThe SynGAP1 missense variant G1113D is listed in ClinVar with an uncertain significance (ClinVar ID 2766136.0) and is present in gnomAD (variant ID 6‑33443890‑G‑A). Functional prediction tools that agree on a benign outcome include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—all of which classify the substitution as benign. AlphaMissense‑Optimized also predicts a benign effect, whereas AlphaMissense‑Default remains uncertain. No tool predicts a pathogenic effect. The high‑accuracy consensus methods reinforce this benign assessment: the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” and AlphaMissense‑Optimized also indicates benign. Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the collective evidence strongly supports a benign impact, and this conclusion does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33443890-G-A | -4.638 | Likely Benign | 0.354 | Ambiguous | Likely Benign | 0.061 | Likely Benign | -0.72 | Neutral | 0.029 | Benign | 0.017 | Benign | 2.58 | Benign | 0.34 | Tolerated | 4.32 | 2 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.3361A>G | S1121G 2D AIThe SynGAP1 missense variant S1121G is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443913‑A‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also benign. Foldetta results are not available. Overall, the preponderance of evidence indicates that the variant is most likely benign, which does not contradict the current ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33443913-A-G | 1 | 7.00e-7 | -1.220 | Likely Benign | 0.054 | Likely Benign | Likely Benign | 0.067 | Likely Benign | -0.53 | Neutral | 0.003 | Benign | 0.004 | Benign | 6.63 | Benign | 0.00 | Affected | 3.77 | 5 | 0 | 1 | 0.4 | -30.03 | |||||||||||||||||||||||||||
| c.3424T>C | S1142P 2D AIThe SynGAP1 missense variant S1142P is listed in ClinVar (ID 2747352.0) as Benign and is present in gnomAD (variant ID 6‑33444459‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, aligning with the ClinVar classification and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Likely Benign | 1 | 6-33444459-T-C | 1 | 6.20e-7 | -2.713 | Likely Benign | 0.222 | Likely Benign | Likely Benign | 0.107 | Likely Benign | -2.19 | Neutral | 0.918 | Possibly Damaging | 0.761 | Possibly Damaging | 2.64 | Benign | 0.00 | Affected | 4.32 | 4 | -1 | 1 | -0.8 | 10.04 | |||||||||||||||||||||||||||
| c.3434A>G | N1145S 2D  AIThe SynGAP1 missense variant N1145S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33444469‑A‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). In contrast, PolyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33444469-A-G | 2 | 1.24e-6 | -0.989 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.308 | Likely Benign | -1.15 | Neutral | 0.997 | Probably Damaging | 0.989 | Probably Damaging | 5.55 | Benign | 0.89 | Tolerated | 4.32 | 4 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||
| c.3520G>A | E1174K 2D  AIThe SynGAP1 missense variant E1174K is listed in ClinVar with an uncertain significance (ClinVar ID 1905754.0) and is present in gnomAD (variant ID 6‑33444555‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” classification, matching the reported SGM‑Consensus result. AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. Taken together, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Uncertain | 1 | 6-33444555-G-A | 2 | 1.24e-6 | -4.345 | Likely Benign | 0.898 | Likely Pathogenic | Ambiguous | 0.442 | Likely Benign | -1.59 | Neutral | 0.962 | Probably Damaging | 0.367 | Benign | 5.52 | Benign | 0.03 | Affected | 4.32 | 2 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||
| c.3529G>A | E1177K 2D AISynGAP1 missense variant E1177K is listed in ClinVar with an Uncertain significance status and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from PROVEAN, SIFT, ESM1b, and FATHMM, while pathogenic calls come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. High‑accuracy assessments give AlphaMissense‑Optimized as Uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta stability analysis is unavailable. Overall, the balance of evidence leans toward a benign effect, which does not contradict the ClinVar designation of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Uncertain | 1 | -3.413 | Likely Benign | 0.944 | Likely Pathogenic | Ambiguous | 0.560 | Likely Pathogenic | -1.75 | Neutral | 0.905 | Possibly Damaging | 0.637 | Possibly Damaging | 5.44 | Benign | 0.11 | Tolerated | 4.32 | 2 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.3572G>A | R1191Q 2D AIThe SynGAP1 missense variant R1191Q is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33444607‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM, while polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default predict a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain”; the SGM‑Consensus (majority vote) remains “Likely Benign”; and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this is not contradictory to the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Uncertain | 2 | 6-33444607-G-A | 9 | 5.58e-6 | -1.069 | Likely Benign | 0.943 | Likely Pathogenic | Ambiguous | 0.343 | Likely Benign | -1.41 | Neutral | 0.998 | Probably Damaging | 0.992 | Probably Damaging | 2.68 | Benign | 0.08 | Tolerated | 3.82 | 4 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||
| c.3574C>G | L1192V 2D  AIThe SynGAP1 missense variant L1192V is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). In contrast, polyPhen‑2 (both HumDiv and HumVar models) predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM‑Consensus also indicates a likely benign outcome; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Thus, based on current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Uncertain | 1 | -4.132 | Likely Benign | 0.471 | Ambiguous | Likely Benign | 0.041 | Likely Benign | -0.89 | Neutral | 0.779 | Possibly Damaging | 0.527 | Possibly Damaging | 2.69 | Benign | 0.16 | Tolerated | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||
| c.3595G>A | E1199K 2D AIThe SynGAP1 missense variant E1199K (ClinVar ID 1026146.0) is listed as Uncertain in ClinVar and is present in gnomAD (ID 6‑33446587‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, whereas tools that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split; Foldetta results are not available. Overall, the majority of evidence points toward a pathogenic impact, which does not contradict the ClinVar Uncertain classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | Uncertain | 1 | 6-33446587-G-A | 1 | 6.20e-7 | -10.853 | Likely Pathogenic | 0.954 | Likely Pathogenic | Ambiguous | 0.171 | Likely Benign | -2.26 | Neutral | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.52 | Benign | 0.00 | Affected | 3.77 | 5 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||
| c.3638A>G | N1213S 2D AIThe SynGAP1 missense variant N1213S is listed in ClinVar as Benign (ClinVar ID 708250.0) and is present in the gnomAD database (gnomAD ID 6‑33446630‑A‑G). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). In contrast, PolyPhen‑2 (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus also as benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, aligning with the ClinVar classification and indicating no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Benign | 1 | 6-33446630-A-G | 13 | 8.05e-6 | -4.086 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.094 | Likely Benign | -0.56 | Neutral | 0.906 | Possibly Damaging | 0.551 | Possibly Damaging | 2.82 | Benign | 0.68 | Tolerated | 3.77 | 5 | 1 | 1 | 2.7 | -27.03 | 10.1016/j.ajhg.2020.11.011 | |||||||||||||||||||||||||
| c.3662G>A | R1221Q 2D AIThe SynGAP1 missense variant R1221Q is listed in ClinVar with an “Uncertain” status and is present in the gnomAD database (ID 6‑33446654‑G‑A). Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only polyPhen‑2 HumDiv indicates a pathogenic effect. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, SGM Consensus is likely benign, while Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the preponderance of evidence points to a benign impact, which is consistent with the ClinVar “Uncertain” classification and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Conflicting | 2 | 6-33446654-G-A | 4 | 2.48e-6 | -5.491 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.078 | Likely Benign | -1.46 | Neutral | 0.836 | Possibly Damaging | 0.153 | Benign | 2.56 | Benign | 0.12 | Tolerated | 3.77 | 5 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||
| c.3731G>A | S1244N 2D AIThe SynGAP1 missense variant S1244N is listed in ClinVar with an “Uncertain” status (ClinVar ID 931075.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, and AlphaMissense‑Optimized, whereas PolyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default all predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—indicates pathogenicity. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the balance of evidence leans toward a pathogenic effect, which does not contradict the ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | Uncertain | 1 | -9.008 | Likely Pathogenic | 0.751 | Likely Pathogenic | Likely Benign | 0.154 | Likely Benign | -1.87 | Neutral | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 2.10 | Pathogenic | 0.15 | Tolerated | 3.77 | 5 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||
| c.3773A>G | Q1258R 2D AIThe SynGAP1 missense variant Q1258R is listed in ClinVar with an uncertain significance (ClinVar ID 3359527.0) and is not observed in gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default, while only REVEL predicts a benign outcome. The high‑accuracy predictors give the following results: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic classification; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no available output for this variant. Based on the preponderance of pathogenic predictions and the SGM Consensus, the variant is most likely pathogenic, which is consistent with its ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | Uncertain | 1 | -10.971 | Likely Pathogenic | 0.931 | Likely Pathogenic | Ambiguous | 0.316 | Likely Benign | -3.19 | Deleterious | 0.994 | Probably Damaging | 0.988 | Probably Damaging | 2.00 | Pathogenic | 0.00 | Affected | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||||
| c.380G>A | R127Q 2D AIThe SynGAP1 missense variant R127Q (ClinVar ID 2898917.0) is listed as ClinVar status Uncertain and is present in gnomAD (6‑33432245‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of computational evidence supports a benign effect, which is consistent with the ClinVar Uncertain status and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33432245-G-A | 6 | 3.72e-6 | -1.711 | Likely Benign | 0.320 | Likely Benign | Likely Benign | 0.037 | Likely Benign | -1.04 | Neutral | 0.006 | Benign | 0.001 | Benign | 4.04 | Benign | 0.02 | Affected | 3.74 | 4 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||
| c.3824G>A | R1275Q 2D AIThe SynGAP1 missense variant R1275Q is listed in ClinVar with an uncertain significance (ClinVar ID 1720188.0) and is present in gnomAD (6‑33447872‑G‑A). Consensus from multiple in‑silico predictors shows a split: benign calls from REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, whereas pathogenic calls come from polyPhen‑2 HumDiv and SIFT. High‑accuracy tools reinforce the benign trend: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports likely benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this assessment does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33447872-G-A | 2 | 1.29e-6 | -4.928 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.103 | Likely Benign | -1.72 | Neutral | 0.898 | Possibly Damaging | 0.147 | Benign | 2.59 | Benign | 0.03 | Affected | 3.77 | 5 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||
| c.3983G>A | R1328Q 2D AIThe SynGAP1 missense variant R1328Q is listed in ClinVar (ID 1805359.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33451857‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this is not in conflict with the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 3 | 6-33451857-G-A | 35 | 1.49e-4 | -2.921 | Likely Benign | 0.273 | Likely Benign | Likely Benign | 0.043 | Likely Benign | -1.02 | Neutral | 0.799 | Possibly Damaging | 0.098 | Benign | 4.12 | Benign | 0.03 | Affected | 3.77 | 5 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||
| c.3G>A | M1I 2D  AIThe SynGAP1 missense variant M1I is listed in ClinVar (ID 833646.0) with an uncertain significance annotation and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM all classify the substitution as benign, while SIFT uniquely predicts it to be pathogenic. The consensus score from the SGM framework, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome. High‑accuracy predictors are incomplete: AlphaMissense‑Optimized and the Foldetta stability assessment are unavailable for this variant. Taking the collective evidence into account, the variant is most likely benign, and this assessment does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Conflicting | 3 | -5.397 | Likely Benign | 0.227 | Likely Benign | -0.17 | Neutral | 0.001 | Benign | 0.000 | Benign | 4.25 | Benign | 0.00 | Affected | 4.32 | 1 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||
| c.4006G>A | E1336K 2D AISynGAP1 missense variant E1336K is listed in ClinVar as benign (ClinVar ID 984837.0) and is present in gnomAD (6‑33451880‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains benign; Foldetta stability analysis is unavailable. Overall, the majority of predictions lean toward a benign impact, aligning with the ClinVar designation, though a single high‑accuracy tool suggests pathogenicity. Thus, the variant is most likely benign, and this conclusion does not contradict its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 2 | 6-33451880-G-A | 6 | 4.20e-6 | -4.697 | Likely Benign | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.272 | Likely Benign | -2.44 | Neutral | 0.748 | Possibly Damaging | 0.079 | Benign | 3.23 | Benign | 0.00 | Affected | 3.77 | 5 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||
| c.4013G>A | R1338Q 2D AIThe SynGAP1 missense variant R1338Q is listed in ClinVar (ID 450879.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33451887‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) which reports it as “Likely Benign.” In contrast, polyPhen‑2 HumDiv and SIFT predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; a Foldetta stability analysis is not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Conflicting | 3 | 6-33451887-G-A | 12 | 8.40e-6 | -3.494 | Likely Benign | 0.317 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -1.87 | Neutral | 0.896 | Possibly Damaging | 0.194 | Benign | 3.81 | Benign | 0.02 | Affected | 3.77 | 5 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||
| c.401G>A | S134N 2D AIThe SynGAP1 missense variant S134N is listed in ClinVar with an “Uncertain” status (ClinVar ID 2819575.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Those that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The remaining tools—AlphaMissense‑Optimized and the SGM‑Consensus—are inconclusive; the consensus score is “Likely Benign” based on a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta) has no available result. Overall, the balance of evidence points to a benign impact, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -5.534 | Likely Benign | 0.813 | Likely Pathogenic | Ambiguous | 0.075 | Likely Benign | -1.62 | Neutral | 0.001 | Benign | 0.002 | Benign | 3.90 | Benign | 0.00 | Affected | 3.61 | 5 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||||||||
| c.4021G>T | A1341S 2D AIThe SynGAP1 missense variant A1341S is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6-33451895-G-T). All available in silico predictors agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is benign; Foldetta results are not available. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33451895-G-T | -2.867 | Likely Benign | 0.078 | Likely Benign | Likely Benign | 0.099 | Likely Benign | 0.80 | Neutral | 0.000 | Benign | 0.001 | Benign | 4.40 | Benign | 1.00 | Tolerated | 3.77 | 5 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.404G>A | R135Q 2D AIThe SynGAP1 missense variant R135Q is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33432701‑G‑A). Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; pathogenic predictions come from SIFT, ESM1b, and AlphaMissense‑Default. The remaining high‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑to‑2 tie, and Foldetta stability analysis is not available. Overall, the balance of evidence favors a benign effect, and this conclusion does not conflict with the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | 6-33432701-G-A | 5 | 3.84e-6 | -8.011 | Likely Pathogenic | 0.853 | Likely Pathogenic | Ambiguous | 0.087 | Likely Benign | -1.94 | Neutral | 0.327 | Benign | 0.100 | Benign | 3.76 | Benign | 0.02 | Affected | 3.61 | 5 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||
| c.407G>A | R136Q 2D AIThe SynGAP1 R136Q variant is listed in ClinVar as benign and is present in gnomAD (6‑33432704‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized returns an uncertain result. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive due to a 2‑vs‑2 split, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no reported result for this variant. Based on the available predictions, the variant is most likely benign, which aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Benign | 1 | 6-33432704-G-A | 13 | 9.17e-6 | -11.146 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 0.190 | Likely Benign | -2.26 | Neutral | 0.957 | Probably Damaging | 0.342 | Benign | 3.52 | Benign | 0.01 | Affected | 3.61 | 5 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||
| c.416G>A | S139N 2D AIThe SynGAP1 missense variant S139N is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33432713‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only AlphaMissense‑Default predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33432713-G-A | 3 | 2.22e-6 | -4.584 | Likely Benign | 0.688 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | -0.75 | Neutral | 0.149 | Benign | 0.047 | Benign | 4.14 | Benign | 0.24 | Tolerated | 3.61 | 5 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||
| c.451G>C | D151H 2D  AIThe SynGAP1 missense variant D151H is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6‑33432748‑G‑C). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as “Likely Pathogenic.” No Foldetta stability result is available for this variant. Overall, the majority of computational evidence indicates that D151H is most likely pathogenic, which does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Uncertain | 1 | 6-33432748-G-C | 2 | 1.26e-6 | -11.747 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.335 | Likely Benign | -3.90 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | 3.86 | Benign | 0.00 | Affected | 3.61 | 5 | -1 | 1 | 0.3 | 22.05 | |||||||||||||||||||||||||||
| c.455G>A | R152Q 2D AIThe SynGAP1 missense variant R152Q is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33432752‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, whereas those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two pathogenic, two benign). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus remains unavailable, and Foldetta (combining FoldX‑MD and Rosetta outputs) has no reported result for this variant. Overall, the preponderance of evidence (seven pathogenic versus three benign predictions) indicates that the variant is most likely pathogenic, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | 6-33432752-G-A | 5 | 3.14e-6 | -10.336 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 0.181 | Likely Benign | -2.34 | Neutral | 0.997 | Probably Damaging | 0.968 | Probably Damaging | 3.89 | Benign | 0.00 | Affected | 3.61 | 5 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||
| c.48G>A | M16I 2D AIThe SynGAP1 missense variant M16I is listed in ClinVar with an “Uncertain” status (ClinVar ID 1424213.0) and is present in gnomAD (6‑33420312‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33420312-G-A | 1 | 6.49e-7 | -2.198 | Likely Benign | 0.722 | Likely Pathogenic | Likely Benign | 0.057 | Likely Benign | -0.15 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.28 | Benign | 0.00 | Affected | 4.32 | 1 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||
| c.491G>A | R164Q 2D AISynGAP1 missense variant R164Q is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33432788‑G‑A). Functional prediction tools show mixed results: benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions come from polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split; Foldetta results are not available. Overall, the balance of evidence slightly favors a benign interpretation, and this does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | 6-33432788-G-A | 2 | 1.24e-6 | -11.208 | Likely Pathogenic | 0.600 | Likely Pathogenic | Likely Benign | 0.184 | Likely Benign | -1.86 | Neutral | 0.957 | Probably Damaging | 0.342 | Benign | 3.82 | Benign | 0.00 | Affected | 3.74 | 4 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||
| c.505G>A | D169N 2D  AIThe SynGAP1 missense variant D169N is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show a split: six methods (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Optimized) predict a benign effect, while three (SIFT, ESM1b, AlphaMissense‑Default) predict pathogenicity. High‑accuracy assessments are mixed: AlphaMissense‑Optimized indicates benign, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive. No Foldetta stability data are available. Overall, the balance of evidence leans toward a benign impact, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | -10.713 | Likely Pathogenic | 0.761 | Likely Pathogenic | Likely Benign | 0.110 | Likely Benign | -2.04 | Neutral | 0.079 | Benign | 0.052 | Benign | 4.07 | Benign | 0.01 | Affected | 3.74 | 4 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||
| c.509G>A | R170Q 2D AISynGAP1 missense variant R170Q is listed in ClinVar as Pathogenic and is not reported in gnomAD. Computational predictors show a split: benign calls come from REVEL, PROVEAN, polyPhen‑2 HumVar, and FATHMM, while pathogenic calls come from polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is Uncertain, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive; Foldetta stability analysis is unavailable. Thus, no single method or high‑accuracy consensus strongly supports pathogenicity. The variant is most likely benign according to the current computational evidence, which contradicts the ClinVar pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Pathogenic/Likely path. | 6 | -9.021 | Likely Pathogenic | 0.798 | Likely Pathogenic | Ambiguous | 0.221 | Likely Benign | -2.31 | Neutral | 0.947 | Possibly Damaging | 0.342 | Benign | 3.91 | Benign | 0.00 | Affected | 3.74 | 4 | 1 | 1 | 1.0 | -28.06 | 10.1016/j.ajhg.2020.11.011 | ||||||||||||||||||||||||||||||
| c.515G>A | R172Q 2D AISynGAP1 missense variant R172Q is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33435157‑G‑A). Functional prediction tools that agree on benign impact include REVEL, PROVEAN, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are polyPhen‑2 HumDiv and SIFT, while ESM1b and AlphaMissense‑Default are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also returns benign. Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | 6-33435157-G-A | 3 | 1.86e-6 | -7.245 | In-Between | 0.465 | Ambiguous | Likely Benign | 0.135 | Likely Benign | -1.72 | Neutral | 0.804 | Possibly Damaging | 0.091 | Benign | 4.04 | Benign | 0.04 | Affected | 3.61 | 5 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||||||||
| c.526A>G | S176G 2D AIThe SynGAP1 missense variant S176G is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33435168‑A‑G). Consensus among most in silico predictors is benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized all report a benign effect. No tool predicts pathogenicity. Two predictors are inconclusive: ESM1b and AlphaMissense‑Default, which are grouped under uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) remains uncertain, and Foldetta stability analysis is unavailable. Overall, the computational evidence overwhelmingly favors a benign impact, which does not contradict the ClinVar uncertain classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | 6-33435168-A-G | 1 | 6.20e-7 | -7.541 | In-Between | 0.360 | Ambiguous | Likely Benign | 0.066 | Likely Benign | -1.08 | Neutral | 0.131 | Benign | 0.039 | Benign | 4.08 | Benign | 0.22 | Tolerated | 3.54 | 6 | 0 | 1 | 0.4 | -30.03 | ||||||||||||||||||||||||||||
| c.5G>A | S2N 2D AIThe SynGAP1 missense variant S2N is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33420269‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the preponderance of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 2 | 6-33420269-G-A | 3 | 1.96e-6 | -4.104 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.092 | Likely Benign | -0.36 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.06 | Benign | 0.00 | Affected | 4.32 | 1 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||
| c.667A>T | T223S 2D  3DClick to see structure in 3D Viewer AISynGAP1 T223S is listed in ClinVar as a variant of uncertain significance and is present in the gnomAD database (ID 6‑33435518‑A‑T). Functional prediction tools that reach consensus classify the variant as benign: FoldX, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict pathogenicity include REVEL, PROVEAN, and SIFT. Predictions that are inconclusive or uncertain are Rosetta, premPS, AlphaMissense‑Default, and ESM1b. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, Foldetta is benign, while the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 1‑to‑1 split between benign and pathogenic calls. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | PH | Conflicting | 2 | 6-33435518-A-T | 3 | 1.86e-6 | -7.714 | In-Between | 0.410 | Ambiguous | Likely Benign | 0.535 | Likely Pathogenic | 0.26 | Likely Benign | 0.1 | 0.50 | Ambiguous | 0.38 | Likely Benign | 0.62 | Ambiguous | -2.86 | Deleterious | 0.421 | Benign | 0.058 | Benign | 5.80 | Benign | 0.02 | Affected | 3.41 | 13 | 1 | 1 | -0.1 | -14.03 | 200.7 | 17.3 | -0.2 | 0.2 | 0.0 | 0.0 | X | Uncertain | The introduced residue Ser223 is located on the outer surface of an anti-parallel β sheet strand (res. Cys219-Thr224). Its hydroxyl group forms hydrogen bonds with nearby residues Thr228 and Lys207 in the variant simulations, similar to the hydroxyl group of Thr223 in the WT simulations. These hydrogen-bonding interactions at the β sheet surface contribute to the stability of the secondary structure element and may prevent it from unfolding. However, since the model ends abruptly at the N-terminus, no definite conclusions can be drawn from the simulations. | |||||||||
| c.718G>A | D240N 2D AIThe SynGAP1 missense variant D240N is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Benign predictions are provided by FoldX, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions come from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus. High‑accuracy methods give a split: AlphaMissense‑Optimized predicts benign, SGM‑Consensus predicts pathogenic, and Foldetta predicts benign. Overall, the majority of tools favor a benign effect, and this consensus does not contradict the ClinVar uncertain status. Thus, the variant is most likely benign based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | PH | Uncertain | 1 | -12.942 | Likely Pathogenic | 0.755 | Likely Pathogenic | Likely Benign | 0.701 | Likely Pathogenic | 0.22 | Likely Benign | 0.9 | 0.47 | Likely Benign | 0.35 | Likely Benign | 0.37 | Likely Benign | -4.37 | Deleterious | 0.993 | Probably Damaging | 0.984 | Probably Damaging | 5.88 | Benign | 0.01 | Affected | 2 | 1 | 0.0 | -0.98 | ||||||||||||||||||||||
| c.74G>A | R25Q 2D AIThe SynGAP1 missense variant R25Q is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33423483‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33423483-G-A | 15 | 9.29e-6 | -4.126 | Likely Benign | 0.212 | Likely Benign | Likely Benign | 0.038 | Likely Benign | -0.70 | Neutral | 0.829 | Possibly Damaging | 0.614 | Possibly Damaging | 4.01 | Benign | 0.00 | Affected | 4.32 | 1 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||
| c.767A>G | N256S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N256S is listed in ClinVar as Pathogenic (ClinVar ID 2584352.0) and is not reported in gnomAD. Functional prediction tools show a split: benign calls come from FoldX, Rosetta, Foldetta, premPS, and FATHMM, while pathogenic calls come from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy subset gives AlphaMissense‑Optimized as Uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of predictions support a pathogenic effect, aligning with the ClinVar classification. Therefore, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | Likely Pathogenic | 1 | -10.640 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 0.707 | Likely Pathogenic | 0.31 | Likely Benign | 0.2 | 0.36 | Likely Benign | 0.34 | Likely Benign | 0.48 | Likely Benign | -4.33 | Deleterious | 0.997 | Probably Damaging | 0.970 | Probably Damaging | 5.87 | Benign | 0.02 | Affected | 3.39 | 15 | 1 | 1 | 2.7 | -27.03 | ||||||||||||||||||||
| c.815G>A | R272Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R272Q is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33437720‑G‑A). Prediction tools that classify the variant as benign include REVEL, Rosetta, Foldetta, AlphaMissense‑Default, AlphaMissense‑Optimized, and PROVEAN. Those that predict pathogenicity are premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and FATHMM. The high‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive; and Foldetta predicts benign. With the majority of high‑accuracy tools supporting a benign effect, the variant is most likely benign, which does not contradict its current ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | Uncertain | 2 | 6-33437720-G-A | 14 | 8.67e-6 | -9.559 | Likely Pathogenic | 0.286 | Likely Benign | Likely Benign | 0.321 | Likely Benign | 0.73 | Ambiguous | 0.1 | 0.15 | Likely Benign | 0.44 | Likely Benign | 1.00 | Destabilizing | -1.81 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.88 | Pathogenic | 0.03 | Affected | 3.38 | 19 | 1 | 1 | 1.0 | -28.06 | 255.7 | 52.9 | 0.0 | 0.0 | -0.2 | 0.1 | X | Uncertain | The guanidinium group of Arg272, located at the end of an anti-parallel β sheet strand (res. Arg259-Arg272), is stably maintained in an upright and outward position via stacking with the indole ring of the Trp362 side chain in another β strand (res. Thr359-Pro364). In the WT simulations, Arg272 forms hydrogen bonds with the glycine-rich Ω loop residues (res. Val365-Pro398, e.g., Gly380) and creates a salt bridge with the carboxylate group of the Asp304 side chain.In the variant simulations, the carboxamide group of the Gln272 side chain does not stack with the indole ring of Trp362 as stably as the guanidinium group of Arg272 in the WT. Consequently, the Gln272 side chain is freer to interact with the loop residues than Arg272, potentially negatively affecting the dynamic SynGAP-membrane association. Additionally, Arg272 faces the RasGTPase interface, so the residue swap could impact the SynGAP-Ras complex formation and GTPase activation. | |||||||||
| c.865A>G | M289V 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant M289V is reported in ClinVar as Benign (ClinVar ID 2122760.0) and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus all predict benign, while only FATHMM predicts pathogenic. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also indicates benign. No prediction or stability result is inconclusive. Overall, the computational evidence strongly supports a benign classification, consistent with the ClinVar status and showing no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | Benign | 1 | -4.239 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.150 | Likely Benign | 1.09 | Ambiguous | 0.1 | -0.27 | Likely Benign | 0.41 | Likely Benign | 0.24 | Likely Benign | -0.36 | Neutral | 0.136 | Benign | 0.054 | Benign | 1.80 | Pathogenic | 1.00 | Tolerated | 3.38 | 23 | 2 | 1 | 2.3 | -32.06 | 204.2 | 51.0 | 0.0 | 0.0 | 0.2 | 0.0 | X | Potentially Benign | The hydrophobic residue Met289, located in a β hairpin linking two anti-parallel β sheet strands (res. Met289-Arg299, res. Arg272-Leu286), is swapped for another hydrophobic residue, valine. In the variant simulations, the branched hydrocarbon side chain of Val289 packs against the phenol group of the Tyr291 side chain but is unable to form methionine-aromatic interactions. β hairpins are potential nucleation sites during the initial stages of protein folding, so even minor changes in them could be significant. However, based on the simulations, the residue swap does not cause adverse effects on the structure. | |||||||||||
| c.886T>G | S296A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S296A (ClinVar ID 469166.0) is listed as ClinVar status Uncertain and is not present in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign or neutral. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict pathogenic. Grouping by consensus, the benign‑predicting tools outnumber the pathogenic ones. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a benign effect. No evidence from the available tools suggests a deleterious impact. Therefore, the variant is most likely benign, and this conclusion is consistent with its ClinVar status of Uncertain rather than pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | Uncertain | 1 | -6.847 | Likely Benign | 0.247 | Likely Benign | Likely Benign | 0.209 | Likely Benign | 0.50 | Ambiguous | 0.3 | -0.26 | Likely Benign | 0.12 | Likely Benign | 0.35 | Likely Benign | -1.79 | Neutral | 0.992 | Probably Damaging | 0.987 | Probably Damaging | 1.97 | Pathogenic | 0.65 | Tolerated | 3.40 | 16 | 1 | 1 | 2.6 | -16.00 | 182.5 | 26.6 | -0.2 | 0.1 | -0.5 | 0.0 | X | Potentially Pathogenic | The hydroxyl group of the Ser296 side chain, located in an anti-parallel β sheet strand (res. Met289-Pro298), stably hydrogen bonds with the carboxylate group of Asp330 in a neighboring β strand (res. Ala322-Asp332). The backbone carbonyl group of Ser296 also hydrogen bonds with the guanidinium group of Arg279 in another nearby β strand (res. Arg279-Cys285). In the variant simulations, the methyl group of the Ala296 side chain cannot hydrogen bond with Asp330, causing the carboxylate group positioning to fluctuate more than in the WT simulations.Although the residue swap does not seem to affect the anti-parallel β sheet assembly during the simulations, it is possible that the Ser296-Asp330 hydrogen bond plays a crucial role in maintaining the C2 domain fold. Notably, because Ser296 is located near the membrane interface, the potential effect of the residue swap on the SynGAP-membrane association cannot be addressed by solvent-only simulations. | |||||||||||
| c.892C>T | P298S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P298S is listed in ClinVar as Benign (ClinVar ID 2965590.0) and is present in gnomAD (ID 6‑33437797‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. No evidence from FoldX, Rosetta, or premPS is available to support either outcome. Overall, the majority of predictions support a benign impact, aligning with the ClinVar designation. Thus, the variant is most likely benign and does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | Benign | 1 | 6-33437797-C-T | 5 | 3.10e-6 | -6.342 | Likely Benign | 0.144 | Likely Benign | Likely Benign | 0.189 | Likely Benign | 1.38 | Ambiguous | 0.2 | 1.41 | Ambiguous | 1.40 | Ambiguous | 0.58 | Ambiguous | -1.20 | Neutral | 0.991 | Probably Damaging | 0.898 | Possibly Damaging | 2.03 | Pathogenic | 0.85 | Tolerated | 3.39 | 20 | -1 | 1 | 0.8 | -10.04 | |||||||||||||||||
| c.928G>A | E310K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E310K is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that assess pathogenicity all return a deleterious signal: REVEL, FoldX (uncertain), Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenic or likely pathogenic. No tool in the dataset predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. Consequently, the variant is most likely pathogenic based on the available predictions, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | Conflicting | 4 | -14.601 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.764 | Likely Pathogenic | 1.97 | Ambiguous | 1.2 | 3.66 | Destabilizing | 2.82 | Destabilizing | 1.02 | Destabilizing | -3.68 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 1.19 | Pathogenic | 0.01 | Affected | 3.38 | 19 | 0 | 1 | -0.4 | -0.94 | 213.4 | 58.0 | 0.1 | 0.0 | 0.2 | 0.1 | X | Potentially Pathogenic | The carboxylate group of Glu310, located in an anti-parallel β sheet strand (res. Thr305-Asn315), is ideally positioned to interact with the side chain hydroxyl and backbone amide groups of Thr295 on a twisted anti-parallel β strand (res. Met289-Arg299). Because the carboxylate group can also interact with the GAP domain residues (e.g., Gln612, Tyr614), Glu310 plays a key role in maintaining the tertiary assembly between the C2 and GAP domains. In the variant simulations, the amino group of the Lys310 side chain hydrogen bonds with the GAP domain residues and forms a salt bridge with Glu613. Although no apparent negative effects are seen due to the residue swap, it is possible that the loss of hydrogen bonding with the hydroxyl group of the Thr295 side chain causes problems during folding, potentially compromising the twisting of the β sheet. | |||||||||||
| c.92G>A | R31Q 2D AIThe SynGAP1 missense variant R31Q is listed in ClinVar (ID 1977609.0) with an “Uncertain” status and is present in gnomAD (6‑33423501‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, which does not contradict the ClinVar “Uncertain” classification and suggests the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33423501-G-A | 7 | 4.34e-6 | -4.434 | Likely Benign | 0.136 | Likely Benign | Likely Benign | 0.051 | Likely Benign | -0.92 | Neutral | 0.829 | Possibly Damaging | 0.614 | Possibly Damaging | 4.01 | Benign | 0.00 | Affected | 4.32 | 1 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||||||||||||
| c.937G>A | E313K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E313K is listed in ClinVar as Benign (ClinVar ID 3695040.0) and is not reported in gnomAD. Prediction tools that report a benign effect are absent; all available predictors that provide a definitive call classify the variant as pathogenic. These include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts Pathogenic, the SGM‑Consensus indicates Likely Pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) is Uncertain. Based on the overwhelming pathogenic predictions, the variant is most likely pathogenic, which contradicts its ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | Likely Benign | 1 | -12.902 | Likely Pathogenic | 0.959 | Likely Pathogenic | Likely Pathogenic | 0.575 | Likely Pathogenic | 0.64 | Ambiguous | 0.6 | 1.40 | Ambiguous | 1.02 | Ambiguous | 0.75 | Ambiguous | -3.31 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 1.90 | Pathogenic | 0.02 | Affected | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||
| c.958G>C | V320L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V320L is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33437863‑G‑C). Functional prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM. Predictions that are inconclusive are Rosetta, Foldetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | Uncertain | 1 | 6-33437863-G-C | 6 | 3.72e-6 | -6.207 | Likely Benign | 0.362 | Ambiguous | Likely Benign | 0.096 | Likely Benign | -0.26 | Likely Benign | 0.2 | 1.33 | Ambiguous | 0.54 | Ambiguous | 0.51 | Ambiguous | -1.02 | Neutral | 0.900 | Possibly Damaging | 0.373 | Benign | 1.78 | Pathogenic | 0.92 | Tolerated | 3.38 | 23 | 2 | 1 | -0.4 | 14.03 | 245.8 | -10.2 | 0.3 | 0.9 | 0.1 | 0.3 | X | Potentially Benign | The isopropyl side chain of Val310, located in a β hairpin loop linking two anti-parallel β sheet strands (res. Thr305-Asn315, res. Ala322-Asp330), hydrophobically packs with the side chains of nearby residues (e.g., Leu286, Val350, Pro318). The hydrophobic Leu320 side chain mostly forms the same interactions; hence, the residue swap does not seem to negatively affect the protein structure based on the variant simulations. | |||||||||
| c.971G>A | R324Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R324Q is listed in ClinVar with an uncertain significance (ClinVar ID 2572558.0) and is present in gnomAD (ID 6‑33437876‑G‑A). Prediction tools that classify the variant as benign include REVEL, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict pathogenicity are premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The high‑accuracy AlphaMissense‑Optimized score is benign, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also indicates a likely benign outcome. Protein‑stability predictions from FoldX, Rosetta, and the combined Foldetta method are all uncertain. Overall, the consensus of available computational evidence points to a benign effect for R324Q, which is consistent with its ClinVar status of uncertain significance rather than a pathogenic designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | Uncertain | 3 | 6-33437876-G-A | 3 | 1.86e-6 | -5.001 | Likely Benign | 0.173 | Likely Benign | Likely Benign | 0.307 | Likely Benign | 0.56 | Ambiguous | 0.1 | 0.63 | Ambiguous | 0.60 | Ambiguous | 1.02 | Destabilizing | -1.17 | Neutral | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 1.92 | Pathogenic | 0.41 | Tolerated | 3.39 | 22 | 1 | 1 | 1.0 | -28.06 | |||||||||||||||||
| c.1067G>A | R356H 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant R356H is recorded in ClinVar as benign (ClinVar ID 2984966.0) and is present in the gnomAD database (6‑33437972‑G‑A). Prediction tools that indicate a benign effect include REVEL, Rosetta, Foldetta, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Default, with the SGM‑Consensus also labeling it likely pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions support a pathogenic impact, which contradicts the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | Likely Benign | 1 | 6-33437972-G-A | 9 | 5.66e-6 | -11.453 | Likely Pathogenic | 0.614 | Likely Pathogenic | Likely Benign | 0.314 | Likely Benign | 0.59 | Ambiguous | 0.1 | -0.27 | Likely Benign | 0.16 | Likely Benign | 1.17 | Destabilizing | -4.43 | Deleterious | 0.999 | Probably Damaging | 0.987 | Probably Damaging | 1.70 | Pathogenic | 0.01 | Affected | 3.39 | 22 | 0 | 2 | 1.3 | -19.05 | |||||||||||||||||
| c.106C>T | H36Y 2D  AIThe SynGAP1 missense variant H36Y is listed in ClinVar with an uncertain significance (ClinVar ID 2089635.0) and is present in the gnomAD database (gnomAD ID 6‑33423515‑C‑T). Functional prediction tools largely agree that the substitution is benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all report a benign effect. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is labeled Likely Benign. No Foldetta stability prediction is available. Overall, the computational evidence overwhelmingly supports a benign classification, which is consistent with the ClinVar designation of uncertain significance rather than a pathogenic claim. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33423515-C-T | 2 | 1.24e-6 | -3.461 | Likely Benign | 0.139 | Likely Benign | Likely Benign | 0.023 | Likely Benign | -1.03 | Neutral | 0.219 | Benign | 0.066 | Benign | 4.16 | Benign | 0.00 | Affected | 4.32 | 1 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||
| c.1131G>A | M377I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M377I (ClinVar ID 3803473.0, status = Uncertain) is present in gnomAD (ID = 6‑33438036‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” while Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. Overall, the computational evidence strongly favors a benign classification, which does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | Uncertain | 1 | 6-33438036-G-A | 1 | 6.23e-7 | -2.895 | Likely Benign | 0.212 | Likely Benign | Likely Benign | 0.227 | Likely Benign | 0.76 | Ambiguous | 0.3 | 0.54 | Ambiguous | 0.65 | Ambiguous | 0.24 | Likely Benign | -0.41 | Neutral | 0.000 | Benign | 0.001 | Benign | 5.46 | Benign | 0.26 | Tolerated | 4.32 | 12 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||
| c.1339G>C | V447L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V447L is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict pathogenicity are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Uncertain results are reported by FoldX, Rosetta, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta as Benign. Overall, the majority of evidence points to a benign effect, and this consensus does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | Uncertain | 1 | -5.136 | Likely Benign | 0.491 | Ambiguous | Likely Benign | 0.180 | Likely Benign | -1.13 | Ambiguous | 0.1 | 0.54 | Ambiguous | -0.30 | Likely Benign | 0.03 | Likely Benign | -0.29 | Neutral | 0.947 | Possibly Damaging | 0.851 | Possibly Damaging | 3.61 | Benign | 0.90 | Tolerated | 3.37 | 32 | 1 | 2 | -0.4 | 14.03 | ||||||||||||||||||||
| c.1402A>G | M468V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M468V is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from PROVEAN, SIFT, and AlphaMissense‑Optimized, while pathogenic predictions are made by REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. The remaining tools, premPS and AlphaMissense‑Default, return uncertain results. High‑accuracy assessments further clarify the variant’s impact: AlphaMissense‑Optimized predicts a benign effect; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates pathogenicity; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also classifies the variant as pathogenic. Overall, the preponderance of evidence points to a pathogenic effect, which does not contradict the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | Uncertain | 1 | -9.461 | Likely Pathogenic | 0.361 | Ambiguous | Likely Benign | 0.570 | Likely Pathogenic | 2.69 | Destabilizing | 0.1 | 2.20 | Destabilizing | 2.45 | Destabilizing | 0.89 | Ambiguous | -1.66 | Neutral | 0.998 | Probably Damaging | 0.993 | Probably Damaging | -1.21 | Pathogenic | 0.08 | Tolerated | 3.37 | 31 | 1 | 2 | 2.3 | -32.06 | |||||||||||||||||||||
| c.1404G>A | M468I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M468I is listed in ClinVar with an uncertain significance (ClinVar ID 3657719.0) and is present in gnomAD (6‑33438436‑G‑A). Functional prediction tools cluster into two groups: benign predictions come from premPS, PROVEAN, and SIFT, while pathogenic predictions arise from REVEL, FoldX, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools report uncertainty: AlphaMissense‑Optimized and Rosetta. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is inconclusive, SGM Consensus is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Overall, the preponderance of evidence indicates a pathogenic impact for M468I, which does not contradict the ClinVar uncertain status but suggests a likely pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | 6-33438436-G-A | 1 | 6.20e-7 | -8.583 | Likely Pathogenic | 0.907 | Likely Pathogenic | Ambiguous | 0.508 | Likely Pathogenic | 2.53 | Destabilizing | 0.2 | 1.89 | Ambiguous | 2.21 | Destabilizing | 0.37 | Likely Benign | -1.06 | Neutral | 0.748 | Possibly Damaging | 0.886 | Possibly Damaging | -1.10 | Pathogenic | 0.07 | Tolerated | 3.37 | 31 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||
| c.1408A>C | M470L 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant M470L is listed in ClinVar as benign (ClinVar ID 536996.0) and is present in gnomAD (variant ID 6‑33438440‑A‑C). Functional prediction tools cluster into two groups: benign predictions come from SIFT and AlphaMissense‑Optimized, while pathogenic predictions are made by REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as benign, SGM Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No definitive folding‑stability change is reported by FoldX or Rosetta individually. Overall, the majority of predictive algorithms favor a pathogenic effect, directly contradicting the benign classification in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Likely Benign | 1 | 6-33438440-A-C | 1 | 6.20e-7 | -8.993 | Likely Pathogenic | 0.406 | Ambiguous | Likely Benign | 0.678 | Likely Pathogenic | 0.73 | Ambiguous | 0.1 | 0.84 | Ambiguous | 0.79 | Ambiguous | 1.04 | Destabilizing | -2.72 | Deleterious | 0.484 | Possibly Damaging | 0.654 | Possibly Damaging | -1.22 | Pathogenic | 0.16 | Tolerated | 3.37 | 34 | 4 | 2 | 1.9 | -18.03 | 225.3 | 17.9 | 0.0 | 0.0 | -0.8 | 0.5 | X | Potentially Benign | The thioether group of Met470, located in the middle of an α helix (res. Ala461–Phe476), interacts with hydrophobic residues in the inter-helix space (e.g., Val473, Leu558) formed by two other α helices (res. Ser604–Arg581, res. Pro562–Arg579). In the WT simulations, Met470 also packs against the positively charged guanidinium groups of Arg575, Arg429, and Arg579, which form salt bridges with the negatively charged carboxylate groups of the Asp474 and Asp467 side chains at the protein surface. In the variant simulations, the iso-butyl side chain of Leu470 packs similarly with the hydrophobic residues as methionine, resulting in no negative effects on the protein structure during the simulation. | ||||||||
| c.1408A>G | M470V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M470V is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Consensus from most in silico predictors indicates a pathogenic effect: SGM‑Consensus, REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM all score it as deleterious. Only two tools—SIFT and AlphaMissense‑Optimized—classify it as benign, while Rosetta and AlphaMissense‑Default remain inconclusive. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized reports a benign outcome, but the SGM‑Consensus (derived from a majority of pathogenic calls among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) and Foldetta (combining pathogenic FoldX with uncertain Rosetta) both predict pathogenicity. Overall, the preponderance of evidence supports a likely pathogenic classification, which does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | -8.856 | Likely Pathogenic | 0.478 | Ambiguous | Likely Benign | 0.770 | Likely Pathogenic | 2.73 | Destabilizing | 0.1 | 1.88 | Ambiguous | 2.31 | Destabilizing | 1.31 | Destabilizing | -3.58 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | -1.20 | Pathogenic | 0.15 | Tolerated | 3.37 | 34 | 1 | 2 | 2.3 | -32.06 | ||||||||||||||||||||
| c.1441C>T | H481Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant H481Y is listed in ClinVar as benign (ClinVar ID 1543764.0) and is present in the gnomAD database (gnomAD ID 6‑33438473‑C‑T). Prediction tools that classify the variant as benign include REVEL, Rosetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. FoldX and Foldetta report uncertain stability effects. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Taking all available evidence together, the variant is most likely benign, which is consistent with its ClinVar benign annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Likely Benign | 1 | 6-33438473-C-T | 16 | 9.91e-6 | -10.910 | Likely Pathogenic | 0.565 | Likely Pathogenic | Likely Benign | 0.256 | Likely Benign | -0.53 | Ambiguous | 0.1 | -0.46 | Likely Benign | -0.50 | Ambiguous | 0.20 | Likely Benign | -3.32 | Deleterious | 0.988 | Probably Damaging | 0.979 | Probably Damaging | 3.40 | Benign | 0.59 | Tolerated | 3.37 | 33 | 0 | 2 | 1.9 | 26.03 | 256.5 | -44.4 | 0.0 | 0.0 | 0.2 | 0.2 | X | X | Uncertain | The imidazole ring of the His481 side chain is located in a short helical structure (res. Glu480-Leu482) within an α-α loop connecting the two α-helices (res. Ala461-Phe476 and Leu489-Glu519) at the GAP-Ras interface. In the WT simulations, His481 alternately stacks against Arg485, Arg587, and Glu480 without a definite role. In the variant simulations, Tyr481 also alternately stacks with nearby arginine residues, including Arg485, Arg587, and Arg479. The interaction between Tyr481 and Arg479 affects the α-α loop, causing it to fold into a distorted helical structure, an effect that might be more pronounced during protein folding. Finally, the potential effect of the residue swap on SynGAP-Ras complex formation or GTPase activation cannot be fully addressed using the SynGAP solvent-only simulations. | |||||||
| c.1454G>A | R485H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R485H missense variant is listed in ClinVar as Benign (ClinVar ID 3707943.0) and is present in the gnomAD database (gnomAD ID 6‑33438486‑G‑A). Functional prediction tools that agree on a benign effect are Rosetta and Foldetta, while the majority of tools (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the preponderance of evidence points to a pathogenic effect, which contradicts the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Likely Benign | 1 | 6-33438486-G-A | 13 | 8.05e-6 | -13.628 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 0.618 | Likely Pathogenic | 0.77 | Ambiguous | 0.1 | 0.12 | Likely Benign | 0.45 | Likely Benign | 1.13 | Destabilizing | -4.97 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.93 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0 | 2 | 1.3 | -19.05 | |||||||||||||||||
| c.1485A>C | E495D 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant E495D is listed in ClinVar with an uncertain significance (ClinVar ID 2000233.0) and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions from SIFT and ESM1b, and pathogenic predictions from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus (SGM Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN classifies the variant as likely pathogenic. AlphaMissense‑Optimized also predicts pathogenicity, whereas Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. Overall, the majority of evidence points to a pathogenic effect, which does not contradict the ClinVar uncertain status but suggests a higher likelihood of deleterious impact. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Conflicting | 2 | -3.574 | Likely Benign | 0.958 | Likely Pathogenic | Likely Pathogenic | 0.566 | Likely Pathogenic | 1.39 | Ambiguous | 0.1 | 1.03 | Ambiguous | 1.21 | Ambiguous | 0.98 | Ambiguous | -2.52 | Deleterious | 0.998 | Probably Damaging | 0.989 | Probably Damaging | -1.41 | Pathogenic | 0.17 | Tolerated | 3.37 | 35 | 3 | 2 | 0.0 | -14.03 | 220.6 | 38.8 | 0.0 | 0.0 | 0.1 | 0.1 | X | X | Uncertain | Glu495 is located in the α-helix (res. Leu489-Glu519), and its carboxylate group forms salt bridges with the neighboring Lys492 and with Arg596 on an opposing α-helix (res. Glu582-Met603) in the WT simulations. In the variant simulations, the acidic carboxylate side chain of Asp495 can also form salt bridges with both Lys492 and Arg596. However, the shorter side chain of aspartate tends to favor forming a salt bridge with the nearby Arg499 on the same α-helix instead. Asp495 might not maintain the salt bridge with Arg596 on the opposing α-helix as efficiently as Glu495 in the WT, potentially weakening the tertiary structure. Regardless, the potential negative effect is likely to be minor, with no deleterious effects observed on the protein structure during the simulations. However, due to its location at the GAP-Ras interface, the effect of the residue swap on SynGAP-Ras complex formation or GTPase activation cannot be fully addressed using the SynGAP solvent-only simulations. | ||||||||||
| c.1516C>T | L506F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L506F is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact; premPS and AlphaMissense‑Optimized are inconclusive. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain, the SGM Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, which is consistent with its ClinVar “Uncertain” classification and does not contradict the available data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | -11.262 | Likely Pathogenic | 0.883 | Likely Pathogenic | Ambiguous | 0.464 | Likely Benign | 4.92 | Destabilizing | 0.8 | 5.76 | Destabilizing | 5.34 | Destabilizing | 0.91 | Ambiguous | -3.98 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.62 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0 | 2 | -1.0 | 34.02 | ||||||||||||||||||||
| c.1552T>C | Y518H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y518H is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic impact: FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, and ESM1b. Predictions that are inconclusive are Foldetta, AlphaMissense‑Optimized, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for Y518H, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | -9.797 | Likely Pathogenic | 0.943 | Likely Pathogenic | Ambiguous | 0.496 | Likely Benign | 2.39 | Destabilizing | 0.4 | 0.82 | Ambiguous | 1.61 | Ambiguous | 1.31 | Destabilizing | -4.74 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.40 | Benign | 0.08 | Tolerated | 0 | 2 | -1.9 | -26.03 | ||||||||||||||||||||||
| c.1635G>A | M545I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M545I is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions arise from FoldX, Rosetta, and SIFT, whereas pathogenic predictions come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default; premPS remains inconclusive. High‑accuracy methods provide mixed evidence: AlphaMissense‑Optimized indicates pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also suggests likely pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a benign effect. Overall, the preponderance of conventional tools and the SGM Consensus lean toward pathogenicity, whereas the Foldetta result is an outlier. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict its ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | -8.348 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.592 | Likely Pathogenic | 0.47 | Likely Benign | 0.1 | 0.14 | Likely Benign | 0.31 | Likely Benign | 0.63 | Ambiguous | -3.61 | Deleterious | 0.935 | Possibly Damaging | 0.941 | Probably Damaging | -1.27 | Pathogenic | 0.28 | Tolerated | 3.37 | 35 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||
| c.1651C>A | L551M 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L551M is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33438894‑C‑A). Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, PROVEAN, SIFT, and AlphaMissense‑Optimized, while those that predict pathogenicity are REVEL, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Two tools report an uncertain outcome: premPS and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of predictions lean toward a benign effect, and this does not contradict the ClinVar “Uncertain” classification. Thus, the variant is most likely benign based on the current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | Uncertain | 1 | 6-33438894-C-A | 7 | 4.34e-6 | -9.937 | Likely Pathogenic | 0.480 | Ambiguous | Likely Benign | 0.544 | Likely Pathogenic | -0.07 | Likely Benign | 0.1 | 0.13 | Likely Benign | 0.03 | Likely Benign | 0.71 | Ambiguous | -0.56 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.48 | Pathogenic | 0.06 | Tolerated | 3.37 | 35 | 4 | 2 | -1.9 | 18.03 | 246.5 | -18.6 | 0.0 | 0.0 | 0.3 | 0.0 | X | Potentially Benign | L551 is located on an α-helix (res. Ala533-Val560). The iso-butyl side chain of Leu551 hydrophobically packs with nearby hydrophobic residues such as Cys547, Phe652, Leu633, and Ile630 in the inter-helix space. In the variant simulations, the thioether side chain of Met551 can maintain similar hydrophobic interactions as Leu551 in the WT, thus causing no negative effect on the protein structure during the simulations. | |||||||||
| c.1673A>G | H558R 2D 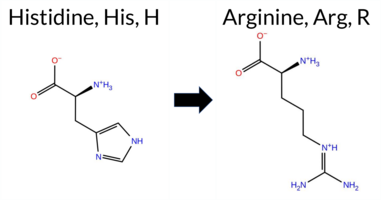 3DClick to see structure in 3D Viewer AISynGAP1 missense variant H558R is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from AlphaMissense‑Optimized, Rosetta, SIFT, and polyPhen‑2 HumVar, while pathogenic predictions arise from REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and FATHMM. Four tools give inconclusive results: AlphaMissense‑Default, SGM‑Consensus, FoldX, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign effect, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains uncertain. Overall, the majority of evidence points toward a pathogenic impact, which does not conflict with the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | -14.445 | Likely Pathogenic | 0.554 | Ambiguous | Likely Benign | 0.587 | Likely Pathogenic | -1.14 | Ambiguous | 0.1 | -0.23 | Likely Benign | -0.69 | Ambiguous | 1.03 | Destabilizing | -4.94 | Deleterious | 0.677 | Possibly Damaging | 0.239 | Benign | -1.24 | Pathogenic | 0.14 | Tolerated | 3.37 | 35 | 0 | 2 | -1.3 | 19.05 | ||||||||||||||||||||
| c.1702G>T | V568L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V568L is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Among the available in‑silico predictors, eight tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict a pathogenic effect, whereas three tools (FoldX, Foldetta, and polyPhen‑2 HumVar) predict a benign outcome; the remaining three (Rosetta, premPS, AlphaMissense‑Optimized) are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as benign. Overall, the preponderance of evidence points to a pathogenic impact, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | -9.503 | Likely Pathogenic | 0.921 | Likely Pathogenic | Ambiguous | 0.651 | Likely Pathogenic | -0.30 | Likely Benign | 0.3 | 0.57 | Ambiguous | 0.14 | Likely Benign | 0.56 | Ambiguous | -2.69 | Deleterious | 0.511 | Possibly Damaging | 0.147 | Benign | -1.23 | Pathogenic | 0.04 | Affected | 3.37 | 35 | 1 | 2 | -0.4 | 14.03 | ||||||||||||||||||||
| c.172A>G | M58V 2D AIThe SynGAP1 missense variant M58V is listed in ClinVar (ID 2962156.0) with an uncertain significance status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy consensus from AlphaMissense‑Optimized, SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta (protein‑folding stability) is available only for the first two; Foldetta data are missing. The SGM Consensus, based on a majority of benign predictions, indicates a likely benign outcome. Overall, the majority of evidence points to a benign impact, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -2.211 | Likely Benign | 0.688 | Likely Pathogenic | Likely Benign | 0.160 | Likely Benign | -0.71 | Neutral | 0.006 | Benign | 0.091 | Benign | 4.19 | Benign | 0.00 | Affected | 4.32 | 1 | 1 | 2 | 2.3 | -32.06 | ||||||||||||||||||||||||||||||
| c.187G>C | E63Q 2D 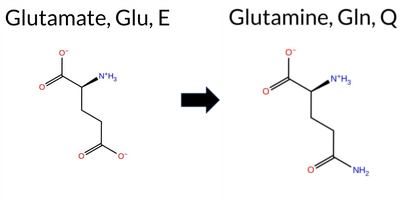 AIThe SynGAP1 missense variant E63Q is listed in ClinVar (ID 2132335.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (majority of the four high‑accuracy tools) also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of predictions points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -4.038 | Likely Benign | 0.687 | Likely Pathogenic | Likely Benign | 0.078 | Likely Benign | -0.85 | Neutral | 0.659 | Possibly Damaging | 0.775 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 4.32 | 1 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.1966G>C | E656Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E656Q is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33441225‑G‑C). Functional prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default; Rosetta reports an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑2 split. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | Uncertain | 1 | 6-33441225-G-C | 1 | 6.20e-7 | -9.145 | Likely Pathogenic | 0.766 | Likely Pathogenic | Likely Benign | 0.249 | Likely Benign | -0.14 | Likely Benign | 0.0 | -0.81 | Ambiguous | -0.48 | Likely Benign | 0.25 | Likely Benign | -2.29 | Neutral | 0.980 | Probably Damaging | 0.528 | Possibly Damaging | 3.46 | Benign | 0.02 | Affected | 3.39 | 24 | 2 | 2 | 0.0 | -0.98 | 224.3 | 1.7 | 0.0 | 0.1 | 0.1 | 0.0 | X | Potentially Benign | The carboxylate side chain of Glu656, located on an α helix (res. Ser641-Glu666), frequently forms a hydrogen bond with the nearby residue Ser659 on the same α helix. In the variant simulations, the carboxamide side chain of Gln656 alternatively forms a hydrogen bond with either Ser659 or Glu548 on an opposing helix (res. Ala533-Val560).Although the frequent interaction between Gln656 and Glu548 may strengthen or stabilize the tertiary structure assembly, the effect is likely to be marginal. | |||||||||
| c.1998G>C | E666D 2D 3DClick to see structure in 3D Viewer AISynGAP1 E666D is listed in ClinVar with an uncertain significance (ID 587483.0) and is not reported in gnomAD. Functional prediction tools show a mixed signal: benign calls come from REVEL, SIFT, FATHMM, AlphaMissense‑Optimized, and Rosetta; pathogenic calls come from premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as likely pathogenic. High‑accuracy assessments give AlphaMissense‑Optimized a benign prediction, while the SGM Consensus remains pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the balance of evidence slightly favors a pathogenic interpretation, but the predictions are not unequivocal. Thus, the variant is most likely pathogenic according to the current computational data, and this does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | -8.820 | Likely Pathogenic | 0.704 | Likely Pathogenic | Likely Benign | 0.197 | Likely Benign | 0.88 | Ambiguous | 0.0 | 0.37 | Likely Benign | 0.63 | Ambiguous | 1.05 | Destabilizing | -2.69 | Deleterious | 0.992 | Probably Damaging | 0.603 | Possibly Damaging | 3.43 | Benign | 0.06 | Tolerated | 3.38 | 28 | 3 | 2 | 0.0 | -14.03 | 237.2 | 16.5 | 0.0 | 0.0 | -0.3 | 0.1 | X | Potentially Pathogenic | The carboxylate group of Glu666, located on the α-helix (res. Ser641-Glu666), is involved in a highly coordinated hydrogen-bonding network between residues from two α-helices (res. Ser641-Glu666 and res. Arg563-Glu578) and from the α-α loop connecting the two α-helices (res. Ser641-Glu666 and res. Leu685-Val699), such as Lys566, Thr672, and Asn669, in the WT simulations. In the variant simulations, the shorter side chain of Asp666 cannot maintain these interactions as efficiently as Glu666 in the WT, resulting in a less coordinated hydrogen-bond network. | |||||||||||
| c.2086C>G | L696V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L696V variant is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized, whereas the majority of other in silico predictors (FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) report a pathogenic outcome; Rosetta remains inconclusive. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts benign, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Overall, the preponderance of evidence points to a pathogenic effect for the variant, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | -11.909 | Likely Pathogenic | 0.745 | Likely Pathogenic | Likely Benign | 0.351 | Likely Benign | 2.35 | Destabilizing | 0.1 | 1.85 | Ambiguous | 2.10 | Destabilizing | 1.46 | Destabilizing | -2.79 | Deleterious | 0.992 | Probably Damaging | 0.970 | Probably Damaging | 3.16 | Benign | 0.00 | Affected | 3.46 | 13 | 1 | 2 | 0.4 | -14.03 | ||||||||||||||||||||
| c.2131C>G | L711V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L711V is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33441596‑C‑G). Prediction tools that indicate a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. The majority of other in silico predictors—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—classify the change as pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as likely pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the preponderance of evidence points to a pathogenic effect, which does not conflict with the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Uncertain | 1 | 6-33441596-C-G | 1 | 6.20e-7 | -10.045 | Likely Pathogenic | 0.709 | Likely Pathogenic | Likely Benign | 0.170 | Likely Benign | 3.48 | Destabilizing | 0.1 | 2.22 | Destabilizing | 2.85 | Destabilizing | 1.40 | Destabilizing | -2.59 | Deleterious | 0.992 | Probably Damaging | 0.970 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 3.50 | 9 | 1 | 2 | 0.4 | -14.03 | |||||||||||||||||
| c.2158G>A | D720N 2D 3DClick to see structure in 3D Viewer AISynGAP1 D720N is listed in ClinVar as benign (ClinVar ID 2837618.0) and is present in gnomAD (ID 6‑33441623‑G‑A). Prediction tools that indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus as pathogenic. With seven pathogenic versus six benign predictions overall, the variant is most likely pathogenic according to in‑silico evidence, which contradicts the benign classification in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | Likely Benign | 1 | 6-33441623-G-A | 5 | 3.10e-6 | -9.135 | Likely Pathogenic | 0.654 | Likely Pathogenic | Likely Benign | 0.289 | Likely Benign | 0.01 | Likely Benign | 0.0 | -0.20 | Likely Benign | -0.10 | Likely Benign | 0.46 | Likely Benign | -3.74 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.18 | Pathogenic | 0.01 | Affected | 3.50 | 9 | 1 | 2 | 0.0 | -0.98 | |||||||||||||||||
| c.2195G>A | R732K 2D 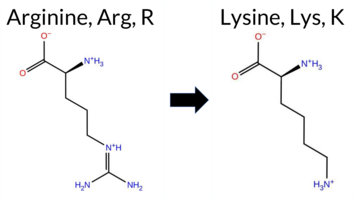 AIThe SynGAP1 missense variant R732K is listed in ClinVar (ID 537019.0) with an “Uncertain” clinical significance and is present in gnomAD (6‑33441660‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; no Foldetta stability result is available. Overall, the majority of evidence points to a benign impact, and this consensus does not conflict with the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Conflicting | 2 | 6-33441660-G-A | 4 | 2.48e-6 | -5.278 | Likely Benign | 0.240 | Likely Benign | Likely Benign | 0.045 | Likely Benign | -0.82 | Neutral | 0.973 | Probably Damaging | 0.943 | Probably Damaging | 2.69 | Benign | 0.21 | Tolerated | 3.59 | 7 | 3 | 2 | 0.6 | -28.01 | |||||||||||||||||||||||||||
| c.2215G>C | E739Q 2D AIThe SynGAP1 missense variant E739Q is listed in ClinVar (ID 2429558.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -2.846 | Likely Benign | 0.161 | Likely Benign | Likely Benign | 0.071 | Likely Benign | -1.06 | Neutral | 0.801 | Possibly Damaging | 0.339 | Benign | 2.57 | Benign | 0.00 | Affected | 4.32 | 2 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.2217G>C | E739D 2D AIThe SynGAP1 missense variant E739D is listed in ClinVar (ID 3661302.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this is not in conflict with the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -3.369 | Likely Benign | 0.062 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -0.49 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.59 | Benign | 0.00 | Affected | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||||
| c.2224C>T | R742W 2D 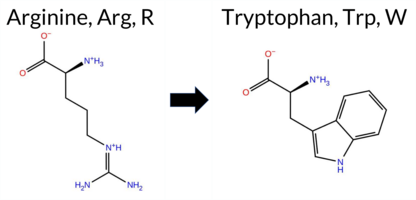 AIThe SynGAP1 missense variant R742W is listed in ClinVar (ID 2581888.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33441689‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign impact, which is consistent with the ClinVar “Uncertain” classification and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33441689-C-T | 6 | 3.72e-6 | -7.725 | In-Between | 0.133 | Likely Benign | Likely Benign | 0.079 | Likely Benign | -1.71 | Neutral | 0.992 | Probably Damaging | 0.684 | Possibly Damaging | 2.66 | Benign | 0.01 | Affected | 4.32 | 2 | -3 | 2 | 3.6 | 30.03 | |||||||||||||||||||||||||||
| c.2275A>C | M759L 2D AIThe SynGAP1 missense variant M759L is listed in ClinVar with an uncertain significance (ClinVar ID 942432.0) and is present in gnomAD (gnomAD ID 6‑33441740‑A‑C). All evaluated in‑silico predictors agree on a benign effect: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign classifications. No tool predicts pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the computational evidence strongly supports a benign impact, which is consistent with the ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33441740-A-C | 2 | 1.24e-6 | -2.431 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.048 | Likely Benign | -0.53 | Neutral | 0.002 | Benign | 0.005 | Benign | 2.84 | Benign | 1.00 | Tolerated | 3.99 | 5 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||
| c.2277G>A | M759I 2D AIThe SynGAP1 missense variant M759I is listed in ClinVar (ID 3686687.0) with an “Uncertain” status and is present in gnomAD (variant ID 6‑33441742‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33441742-G-A | 1 | 6.20e-7 | -4.058 | Likely Benign | 0.393 | Ambiguous | Likely Benign | 0.075 | Likely Benign | -0.88 | Neutral | 0.454 | Possibly Damaging | 0.192 | Benign | 2.83 | Benign | 0.34 | Tolerated | 3.99 | 5 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||
| c.2302G>A | D768N 2D AIThe SynGAP1 missense variant D768N is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33442460‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). No tool predicts a pathogenic outcome; AlphaMissense‑Default is uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus is “Likely Benign,” and Foldetta data are unavailable. Overall, the consensus of available predictions indicates that the variant is most likely benign, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33442460-G-A | 2 | 2.57e-6 | -6.892 | Likely Benign | 0.453 | Ambiguous | Likely Benign | 0.048 | Likely Benign | -0.77 | Neutral | 0.106 | Benign | 0.009 | Benign | 4.07 | Benign | 0.96 | Tolerated | 3.64 | 6 | 1 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||
| c.2343G>A | M781I 2D AIThe SynGAP1 missense variant M781I is listed in ClinVar (ID 2802065.0) as Benign and is not reported in gnomAD. All available in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the predictions strongly support a benign effect, consistent with the ClinVar designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 1 | -2.484 | Likely Benign | 0.323 | Likely Benign | Likely Benign | 0.101 | Likely Benign | 0.05 | Neutral | 0.000 | Benign | 0.001 | Benign | 2.89 | Benign | 1.00 | Tolerated | 3.64 | 6 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||
| c.2349G>A | M783I 2D AIThe SynGAP1 missense variant M783I is listed in ClinVar as a benign alteration (ClinVar ID 3618151.0) and is present in the gnomAD database (gnomAD ID 6‑33442901‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). No tool in the dataset predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a likely benign effect. The Foldetta protein‑folding stability analysis is not available for this variant. Overall, the computational evidence strongly suggests that the variant is most likely benign, in agreement with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 1 | 6-33442901-G-A | 6 | 3.72e-6 | -3.560 | Likely Benign | 0.418 | Ambiguous | Likely Benign | 0.042 | Likely Benign | -0.54 | Neutral | 0.004 | Benign | 0.006 | Benign | 2.87 | Benign | 0.22 | Tolerated | 3.64 | 6 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||
| c.2408A>G | K803R 2D  AIThe SynGAP1 missense variant K803R is listed in ClinVar (ID 834618.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only two tools—SIFT and FATHMM—predict pathogenicity. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence indicates the variant is most likely benign, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | Uncertain | 1 | -2.281 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.018 | Likely Benign | -1.52 | Neutral | 0.103 | Benign | 0.038 | Benign | 2.38 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||||
| c.2493G>C | E831D 2D AIThe SynGAP1 missense variant E831D is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33443045‑G‑C). All available in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic or likely‑pathogenic outcome. Grouping by agreement, the benign‑predicting tools comprise the entire set, while no pathogenic predictions are present. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign classification. Foldetta results are unavailable. Overall, the computational evidence strongly supports a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33443045-G-C | 1 | 6.19e-7 | -3.055 | Likely Benign | 0.063 | Likely Benign | Likely Benign | 0.073 | Likely Benign | 1.23 | Neutral | 0.002 | Benign | 0.002 | Benign | 2.64 | Benign | 0.77 | Tolerated | 3.77 | 5 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||
| c.2502G>C | M834I 2D AIThe SynGAP1 missense variant M834I is listed in ClinVar (ID 3007819.0) with an uncertain significance designation and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only SIFT classifies the change as pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the collective predictions indicate that M834I is most likely benign, which is consistent with its ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -3.377 | Likely Benign | 0.291 | Likely Benign | Likely Benign | 0.055 | Likely Benign | -1.21 | Neutral | 0.026 | Benign | 0.009 | Benign | 2.56 | Benign | 0.00 | Affected | 4.32 | 4 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||
| c.2521G>A | V841M 2D  AISynGAP1 variant V841M is listed in ClinVar with an uncertain significance and is present in gnomAD (6-33443073-G-A). Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions from polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. The ESM1b score is inconclusive. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, while Foldetta stability analysis is unavailable. Taken together, the majority of evidence, including the high‑accuracy tools, points to a benign effect for V841M. This conclusion does not conflict with the ClinVar uncertain status, which reflects the current lack of definitive clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | 6-33443073-G-A | 3 | 1.86e-6 | -7.000 | In-Between | 0.651 | Likely Pathogenic | Likely Benign | 0.119 | Likely Benign | -0.74 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.54 | Benign | 0.02 | Affected | 3.77 | 5 | 1 | 2 | -2.3 | 32.06 | ||||||||||||||||||||||||||||
| c.2650C>T | R884W 2D AIThe SynGAP1 missense variant R884W is listed in ClinVar with an “Uncertain” status and is present in the gnomAD database (gnomAD ID 6‑33443202‑C‑T). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. No Foldetta (protein‑folding stability) result is available for this variant. Overall, the majority of computational predictions, including the high‑accuracy tools, indicate that R884W is most likely benign, which is consistent with the ClinVar “Uncertain” status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33443202-C-T | 5 | 3.10e-6 | -3.785 | Likely Benign | 0.332 | Likely Benign | Likely Benign | 0.151 | Likely Benign | 0.26 | Neutral | 0.995 | Probably Damaging | 0.812 | Possibly Damaging | 2.56 | Benign | 0.05 | Affected | 4.32 | 4 | -3 | 2 | 3.6 | 30.03 | |||||||||||||||||||||||||||
| c.2881C>T | H961Y 2D AIThe SynGAP1 missense variant H961Y is listed in ClinVar with an uncertain significance (ClinVar ID 862637.0) and is present in gnomAD (ID 6‑33443433‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is not in conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Conflicting | 2 | 6-33443433-C-T | 3 | 1.86e-6 | -8.051 | Likely Pathogenic | 0.157 | Likely Benign | Likely Benign | 0.102 | Likely Benign | -1.07 | Neutral | 0.716 | Possibly Damaging | 0.147 | Benign | 4.10 | Benign | 0.55 | Tolerated | 3.77 | 5 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||
| c.291G>T | E97D 2D AIThe SynGAP1 missense variant E97D is listed in ClinVar (ID 1313570.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33425899‑G‑T). Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Pathogenic predictions come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect. This consensus does not contradict the ClinVar “Uncertain” status, which remains unresolved. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 3 | 6-33425899-G-T | -3.239 | Likely Benign | 0.077 | Likely Benign | Likely Benign | 0.081 | Likely Benign | -0.49 | Neutral | 0.880 | Possibly Damaging | 0.636 | Possibly Damaging | 4.12 | Benign | 0.00 | Affected | 4.32 | 1 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.3049T>C | F1017L 2D  AIThe SynGAP1 missense variant F1017L is listed in ClinVar (ID 3719654.0) as benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and the SGM‑Consensus score (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus (majority vote) is benign. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence points to a benign impact, aligning with the ClinVar classification and showing no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 1 | -2.048 | Likely Benign | 0.934 | Likely Pathogenic | Ambiguous | 0.157 | Likely Benign | -2.38 | Neutral | 0.798 | Possibly Damaging | 0.373 | Benign | 2.65 | Benign | 0.72 | Tolerated | 3.77 | 5 | 0 | 2 | 1.0 | -34.02 | ||||||||||||||||||||||||||||||
| c.3209G>A | R1070K 2D AIThe SynGAP1 missense variant R1070K is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All evaluated in‑silico predictors agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” High‑accuracy tools reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign. Foldetta results are unavailable. Based on the unanimous benign predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Conflicting | 2 | -5.093 | Likely Benign | 0.326 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -1.42 | Neutral | 0.049 | Benign | 0.048 | Benign | 3.86 | Benign | 0.09 | Tolerated | 3.77 | 5 | 3 | 2 | 0.6 | -28.01 | ||||||||||||||||||||||||||||||
| c.323A>G | K108R 2D AIThe SynGAP1 missense variant K108R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33432188‑A‑G). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments further support benignity: AlphaMissense‑Optimized is benign, and the SGM‑Consensus itself is benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33432188-A-G | 6 | 3.72e-6 | -2.892 | Likely Benign | 0.148 | Likely Benign | Likely Benign | 0.184 | Likely Benign | 0.37 | Neutral | 0.993 | Probably Damaging | 0.956 | Probably Damaging | 4.22 | Benign | 1.00 | Tolerated | 3.61 | 5 | 3 | 2 | -0.6 | 28.01 | |||||||||||||||||||||||||||
| c.3253C>T | R1085W 2D AISynGAP1 missense variant R1085W is listed in ClinVar as Uncertain and is present in gnomAD (ID 6‑33443805‑C‑T). Prediction tools that classify the variant as benign include REVEL, ESM1b, and FATHMM, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is Uncertain, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta stability assessment are unavailable. Overall, the majority of available predictions (five pathogenic vs. three benign) indicate a pathogenic effect. Thus, the variant is most likely pathogenic, which contradicts the ClinVar designation of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | 6-33443805-C-T | 2 | 1.26e-6 | -6.339 | Likely Benign | 0.821 | Likely Pathogenic | Ambiguous | 0.202 | Likely Benign | -3.15 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.70 | Benign | 0.00 | Affected | 3.77 | 5 | -3 | 2 | 3.6 | 30.03 | ||||||||||||||||||||||||||||
| c.3313C>T | R1105W 2D AIThe SynGAP1 missense variant R1105W is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33443865‑C‑T). Prediction tools show mixed results: benign calls come from REVEL, ESM1b, and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. The AlphaMissense‑Default tool remains uncertain. A consensus analysis (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a pathogenic majority. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts benign, whereas the SGM Consensus predicts pathogenic; Foldetta results are unavailable. Overall, the preponderance of evidence points to a pathogenic effect for R1105W, which does not conflict with the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | 6-33443865-C-T | 6 | 3.93e-6 | -6.911 | Likely Benign | 0.488 | Ambiguous | Likely Benign | 0.133 | Likely Benign | -4.34 | Deleterious | 0.999 | Probably Damaging | 0.696 | Possibly Damaging | 2.42 | Pathogenic | 0.02 | Affected | 3.77 | 5 | -3 | 2 | 3.6 | 30.03 | ||||||||||||||||||||||||||||
| c.3442A>T | M1148L 2D AIThe SynGAP1 missense variant M1148L is listed in ClinVar (ID 1010061.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign effect, and this is consistent with the ClinVar “Uncertain” designation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -1.777 | Likely Benign | 0.093 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -1.13 | Neutral | 0.016 | Benign | 0.016 | Benign | 2.62 | Benign | 0.00 | Affected | 4.32 | 2 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||
| c.3567G>C | E1189D 2D AIThe SynGAP1 missense variant E1189D (gnomAD ID 6-33444602‑G‑C) is listed in ClinVar as Benign (ClinVar ID 833989.0). In silico predictors that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Predictors that indicate a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. The high‑accuracy AlphaMissense‑Optimized tool classifies the variant as benign, and the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also favors a benign outcome. No Foldetta stability assessment is available for this residue. Overall, the majority of evidence points to a benign impact, and this conclusion aligns with the ClinVar designation, showing no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Likely Benign | 1 | 6-33444602-G-C | 3 | 1.86e-6 | -3.582 | Likely Benign | 0.461 | Ambiguous | Likely Benign | 0.359 | Likely Benign | -1.42 | Neutral | 0.992 | Probably Damaging | 0.989 | Probably Damaging | 5.30 | Benign | 0.25 | Tolerated | 3.82 | 4 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||
| c.3631A>G | M1211V 2D AIThe SynGAP1 missense variant M1211V is listed in ClinVar as Benign (ClinVar ID 3674914.0) and is present in gnomAD (ID 6‑33446623‑A‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, aligning with the ClinVar classification and not contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Benign | 1 | 6-33446623-A-G | 3 | 1.86e-6 | -2.101 | Likely Benign | 0.258 | Likely Benign | Likely Benign | 0.412 | Likely Benign | -0.29 | Neutral | 0.932 | Possibly Damaging | 0.949 | Probably Damaging | 5.43 | Benign | 0.72 | Tolerated | 3.77 | 5 | 1 | 2 | 2.3 | -32.06 | ||||||||||||||||||||||||||
| c.3633G>A | M1211I 2D AIThe SynGAP1 missense variant M1211I is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6-33446625-G-A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are AlphaMissense‑Default, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. High‑accuracy predictions therefore point to a benign outcome: AlphaMissense‑Optimized is benign, and the SGM‑Consensus also indicates benign. Based on the aggregate predictions, the variant is most likely benign, which does not contradict the ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Uncertain | 1 | 6-33446625-G-A | 3 | 1.86e-6 | -1.537 | Likely Benign | 0.764 | Likely Pathogenic | Likely Benign | 0.298 | Likely Benign | -0.42 | Neutral | 0.969 | Probably Damaging | 0.968 | Probably Damaging | 5.40 | Benign | 1.00 | Tolerated | 3.77 | 5 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||
| c.3655T>C | Y1219H 2D AIThe SynGAP1 missense variant Y1219H is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that indicate a benign effect include only REVEL, whereas the remaining tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic)—all predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as Likely Pathogenic. No Foldetta stability prediction is available for this variant. Overall, the preponderance of evidence from both general and high‑accuracy tools indicates that the variant is most likely pathogenic, which does not contradict its current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | Coiled-coil | Uncertain | 1 | -9.511 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.363 | Likely Benign | -3.62 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 2.15 | Pathogenic | 0.00 | Affected | 3.77 | 5 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||
| c.3705G>A | M1235I 2D AIThe SynGAP1 missense variant M1235I is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as “Likely Benign”; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Coiled-coil | Uncertain | 1 | -4.312 | Likely Benign | 0.310 | Likely Benign | Likely Benign | 0.027 | Likely Benign | -1.44 | Neutral | 0.139 | Benign | 0.056 | Benign | 2.69 | Benign | 0.04 | Affected | 3.77 | 5 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.3721C>A | L1241M 2D AISynGAP1 missense variant L1241M is listed in ClinVar with an Uncertain significance and is not reported in gnomAD. Functional prediction tools show a split verdict: benign calls come from REVEL, PROVEAN, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic calls are made by polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is unresolved (2 benign vs. 2 pathogenic). Foldetta, a protein‑folding stability predictor that combines FoldX‑MD and Rosetta outputs, has no available result for this variant. Consequently, the high‑accuracy tools do not converge on a single interpretation. Overall, the predictions are balanced between benign and pathogenic, leaving the variant’s effect uncertain, which aligns with its ClinVar designation of Uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Coiled-coil | Uncertain | 1 | -5.881 | Likely Benign | 0.782 | Likely Pathogenic | Likely Benign | 0.167 | Likely Benign | -1.43 | Neutral | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.65 | Pathogenic | 0.00 | Affected | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||||
| c.3821G>A | R1274H 2D AIThe SynGAP1 missense variant R1274H (ClinVar ID 2803246.0) is classified as Benign in ClinVar and is present in gnomAD (ID 6‑33447869‑G‑A). Functional prediction tools show mixed results: benign calls come from REVEL, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, whereas the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields a tie and is therefore inconclusive. No Foldetta stability prediction is available for this residue. Overall, the majority of conventional tools predict pathogenicity, and the high‑accuracy AlphaMissense‑Optimized result is benign, leaving the evidence mixed. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, contradicting its ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 1 | 6-33447869-G-A | 4 | 2.58e-6 | -5.259 | Likely Benign | 0.256 | Likely Benign | Likely Benign | 0.149 | Likely Benign | -3.20 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.49 | Pathogenic | 0.01 | Affected | 3.77 | 5 | 0 | 2 | 1.3 | -19.05 | ||||||||||||||||||||||||||||
| c.3846G>C | E1282D 2D AIThe SynGAP1 missense variant E1282D is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6-33447894-G-C). All available in silico predictors classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool reports a pathogenic prediction. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote) is likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no reported result for this variant, so its status is unavailable. Overall, the computational evidence overwhelmingly supports a benign effect, which is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33447894-G-C | 1 | 6.44e-7 | -3.879 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.104 | Likely Benign | -1.26 | Neutral | 0.112 | Benign | 0.036 | Benign | 2.70 | Benign | 0.39 | Tolerated | 3.77 | 5 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||
| c.3858A>T | E1286D 2D AIThe SynGAP1 missense variant E1286D is listed in ClinVar (ID 469159.0) with an “Uncertain” clinical significance and is present in gnomAD (variant ID 6‑33447906‑A‑T). All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. No tool in the set predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the computational evidence strongly supports a benign effect, and this conclusion does not contradict the current ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Conflicting | 4 | 6-33447906-A-T | 143 | 9.22e-5 | -4.010 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.036 | Likely Benign | 1.02 | Neutral | 0.001 | Benign | 0.004 | Benign | 2.96 | Benign | 1.00 | Tolerated | 3.77 | 5 | 3 | 2 | 0.0 | -14.03 | 10.1016/j.ajhg.2020.11.011 | ||||||||||||||||||||||||||
| c.4000A>G | N1334D 2D  AIThe SynGAP1 missense variant N1334D (ClinVar ID 3653769.0) is listed as Uncertain in ClinVar and is present in gnomAD (ID 6‑33451874‑A‑G). Functional prediction tools show a split: benign calls come from REVEL, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default. High‑accuracy assessments give a benign result from AlphaMissense‑Optimized, an inconclusive SGM Consensus (a 2‑vs‑2 majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and no available Foldetta stability data. Overall, the majority of predictions (5/10) indicate pathogenicity, and the high‑accuracy tools do not overturn this trend. Therefore, the variant is most likely pathogenic, which does not contradict its ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Uncertain | 1 | 6-33451874-A-G | -4.584 | Likely Benign | 0.674 | Likely Pathogenic | Likely Benign | 0.126 | Likely Benign | -3.06 | Deleterious | 0.886 | Possibly Damaging | 0.522 | Possibly Damaging | 3.55 | Benign | 0.00 | Affected | 3.77 | 5 | 1 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.453C>A | D151E 2D  AIThe SynGAP1 D151E variant is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, and FATHMM. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain,” SGM‑Consensus as “Likely Benign,” and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) has no available result for this variant. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -5.662 | Likely Benign | 0.886 | Likely Pathogenic | Ambiguous | 0.142 | Likely Benign | -2.02 | Neutral | 0.984 | Probably Damaging | 0.967 | Probably Damaging | 3.99 | Benign | 0.11 | Tolerated | 3.61 | 5 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||
| c.502C>T | H168Y 2D AIThe SynGAP1 missense variant H168Y is listed in ClinVar (ID 956914.0) as benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Tools that predict a pathogenic effect are SIFT and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; the Foldetta protein‑folding stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is consistent with the ClinVar benign classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 1 | -8.914 | Likely Pathogenic | 0.264 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -1.53 | Neutral | 0.192 | Benign | 0.062 | Benign | 4.18 | Benign | 0.01 | Affected | 4.32 | 3 | 0 | 2 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||
| c.603T>A | D201E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D201E missense variant (ClinVar ID 3004688.0) is classified as **Benign** in ClinVar and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only polyPhen‑2 HumDiv predicts a pathogenic outcome, while Rosetta, Foldetta, and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as **Benign**, the SGM‑Consensus as **Likely Benign**, and Foldetta as **Uncertain**. Taken together, the overwhelming majority of evidence points to a benign impact, and this conclusion aligns with the ClinVar designation. Thus, the variant is most likely benign, with no contradiction to its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | Benign | 1 | -2.640 | Likely Benign | 0.406 | Ambiguous | Likely Benign | 0.165 | Likely Benign | 0.42 | Likely Benign | 0.2 | 1.99 | Ambiguous | 1.21 | Ambiguous | 0.23 | Likely Benign | -0.69 | Neutral | 0.633 | Possibly Damaging | 0.108 | Benign | 4.30 | Benign | 1.00 | Tolerated | 3.46 | 9 | 3 | 2 | 0.0 | 14.03 | 258.7 | -24.8 | 0.9 | 0.1 | -0.3 | 0.2 | X | Uncertain | Asp201, an acidic residue located in the N-terminal loop before the first anti-parallel β sheet strand (res. Ile205-Pro208), is replaced by another acidic residue, glutamate. The carboxylate groups of both Asp201 and Glu201 side chains form hydrogen bonds with the hydroxyl group of Ser221 in the simulations. Due to its shorter side chain, Asp201 can also hydrogen bond with the backbone amide groups of neighboring loop residues Ser204 and Asp203. However, since the model ends abruptly at the N-terminus, no definite conclusions can be drawn from the simulations. | |||||||||||
| c.603T>G | D201E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D201E missense variant (gnomAD ID 6‑33435245‑T‑G) is listed in ClinVar with an uncertain significance. Across a broad panel of in silico predictors, the majority indicate a benign effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized all score benign. Only polyPhen‑2 HumDiv predicts pathogenicity, while Rosetta, Foldetta, and AlphaMissense‑Default remain inconclusive. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to “likely benign.” High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta is inconclusive. Taken together, the preponderance of evidence points to a benign impact, which does not conflict with the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | PH | Conflicting | 2 | 6-33435245-T-G | 20 | 1.24e-5 | -2.640 | Likely Benign | 0.406 | Ambiguous | Likely Benign | 0.165 | Likely Benign | 0.42 | Likely Benign | 0.2 | 1.99 | Ambiguous | 1.21 | Ambiguous | 0.23 | Likely Benign | -0.69 | Neutral | 0.633 | Possibly Damaging | 0.108 | Benign | 4.30 | Benign | 1.00 | Tolerated | 3.46 | 9 | 3 | 2 | 0.0 | 14.03 | 258.7 | -24.8 | 0.9 | 0.1 | -0.3 | 0.2 | X | Uncertain | Asp201, an acidic residue located in the N-terminal loop before the first anti-parallel β sheet strand (res. Ile205-Pro208), is replaced by another acidic residue, glutamate. The carboxylate groups of both Asp201 and Glu201 side chains form hydrogen bonds with the hydroxyl group of Ser221 in the simulations. Due to its shorter side chain, Asp201 can also hydrogen bond with the backbone amide groups of neighboring loop residues Ser204 and Asp203. However, since the model ends abruptly at the N-terminus, no definite conclusions can be drawn from the simulations. | ||||||||
| c.73C>T | R25W 2D AIThe SynGAP1 missense variant R25W is listed in ClinVar with an “Uncertain” status (ClinVar ID 2993054.0) and is present in gnomAD (ID 6‑33423482‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this is not in conflict with the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 2 | 6-33423482-C-T | 6 | 3.72e-6 | -5.133 | Likely Benign | 0.549 | Ambiguous | Likely Benign | 0.158 | Likely Benign | -1.60 | Neutral | 0.994 | Probably Damaging | 0.919 | Probably Damaging | 3.92 | Benign | 0.00 | Affected | 4.32 | 1 | -3 | 2 | 3.6 | 30.03 | |||||||||||||||||||||||||||
| c.819G>T | E273D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E273D is listed in ClinVar as Benign (ClinVar ID 1471608.0) and is present in gnomAD (variant ID 6‑33437724‑G‑T). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while premPS is uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is Likely Benign. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote) is benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. No prediction contradicts the ClinVar benign status; overall, the evidence strongly supports that E273D is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | Benign | 1 | 6-33437724-G-T | 2 | 1.24e-6 | -1.811 | Likely Benign | 0.058 | Likely Benign | Likely Benign | 0.092 | Likely Benign | 0.26 | Likely Benign | 0.1 | -0.48 | Likely Benign | -0.11 | Likely Benign | -0.63 | Ambiguous | 1.99 | Neutral | 0.004 | Benign | 0.010 | Benign | 2.00 | Pathogenic | 1.00 | Tolerated | 3.38 | 18 | 3 | 2 | 0.0 | -14.03 | 223.1 | 22.1 | 0.2 | 0.0 | 0.0 | 0.1 | X | Potentially Benign | The negatively charged residue Glu273, located in a β hairpin loop (res. Glu273-Lys278) that connects two anti-parallel β sheet strands, is replaced with another negatively charged residue, aspartate. Because the C2 domain loop faces the membrane surface, the potentially crucial role of the carboxylate group of Glu273 or Asp273 on SynGAP-membrane association cannot be fully explored via solvent-only simulations.As a minor note, the neighboring residue Arg272, which stacks with the indole ring of the Trp362 side chain and directly faces RasGTPase, forms a salt bridge more often with Asp273 than with the non-mutated Glu273 in the simulations. Regardless, due to the similar physicochemical properties of the WT and variant residues at the membrane interface, the residue swap is likely to be well tolerated. | ||||||||
| c.862G>A | D288N 2D 3DClick to see structure in 3D Viewer AISynGAP1 D288N is listed in ClinVar with an uncertain significance (ClinVar ID 2572204.0) and is present in gnomAD (6‑33437767‑G‑A). Computational predictors are divided: benign calls come from REVEL, FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized, while pathogenic calls come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM; AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic. Because the majority of high‑accuracy tools predict benign and the overall split of predictions is even, the variant is most likely benign, which does not contradict the ClinVar status of uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | Uncertain | 1 | 6-33437767-G-A | 2 | 1.24e-6 | -10.535 | Likely Pathogenic | 0.521 | Ambiguous | Likely Benign | 0.321 | Likely Benign | -0.39 | Likely Benign | 0.1 | 0.01 | Likely Benign | -0.19 | Likely Benign | -0.03 | Likely Benign | -3.73 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.78 | Pathogenic | 0.05 | Affected | 3.38 | 23 | 1 | 2 | 0.0 | -0.98 | |||||||||||||||||
| c.88C>T | H30Y 2D AIThe SynGAP1 H30Y missense variant (ClinVar ID 972248.0) is listed as “Uncertain” and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumVar and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as “Likely Benign”; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -3.047 | Likely Benign | 0.115 | Likely Benign | Likely Benign | 0.082 | Likely Benign | -1.84 | Neutral | 0.273 | Benign | 0.478 | Possibly Damaging | 3.99 | Benign | 0.00 | Affected | 4.32 | 1 | 0 | 2 | 1.9 | 26.03 | ||||||||||||||||||||||||||||||
| c.910G>A | D304N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant D304N is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a benign impact, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | C2 | Uncertain | 1 | -6.194 | Likely Benign | 0.391 | Ambiguous | Likely Benign | 0.345 | Likely Benign | 0.30 | Likely Benign | 0.1 | -0.08 | Likely Benign | 0.11 | Likely Benign | 0.21 | Likely Benign | -4.18 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.81 | Pathogenic | 0.03 | Affected | 3.38 | 23 | 1 | 2 | 0.0 | -0.98 | |||||||||||||||||||||
| c.930G>C | E310D 2D 3DClick to see structure in 3D Viewer AISynGAP1 E310D is reported in ClinVar (ID 975473.0) as Pathogenic and is not found in gnomAD. Prediction tools that assess the variant’s effect all converge on a deleterious outcome: REVEL, FoldX (uncertain), Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate pathogenicity, leaving no tool in the benign category. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also reports Pathogenic. The single uncertain result from FoldX is treated as unavailable. Overall, the variant is most likely pathogenic, and this assessment is consistent with its ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | Likely Pathogenic | 1 | -11.218 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.666 | Likely Pathogenic | 1.87 | Ambiguous | 0.5 | 2.39 | Destabilizing | 2.13 | Destabilizing | 1.04 | Destabilizing | -2.76 | Deleterious | 0.997 | Probably Damaging | 0.992 | Probably Damaging | 1.19 | Pathogenic | 0.02 | Affected | 3.38 | 19 | 3 | 2 | 0.0 | -14.03 | 232.6 | 27.2 | 0.1 | 0.0 | 0.1 | 0.1 | X | Potentially Benign | The carboxylate group of Glu310, located in an anti-parallel β sheet strand (res. Thr305-Asn315), is ideally positioned to interact with the hydroxyl and backbone amide groups of Thr295 on a twisted anti-parallel β strand. Because the carboxylate group can also interact with the GAP domain residues (e.g., Gln612, Tyr614), Glu310 potentially plays a key role in maintaining the tertiary assembly between the C2 and GAP domains. In the variant simulations, the carboxylate group of Asp310 can form the same interactions as glutamate; however, due to its one hydrocarbon shorter length, the connections are less stable or less optimal. | |||||||||||
| c.1027G>A | V343I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V343I is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33437932‑G‑A). Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to “Likely Benign” (3 benign vs. 1 pathogenic). High‑accuracy assessments are consistent: AlphaMissense‑Optimized is benign; the SGM‑Consensus is likely benign; and Foldetta, combining FoldX‑MD and Rosetta outputs, is benign. Overall, the collective evidence strongly supports a benign classification, and this does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | Uncertain | 2 | 6-33437932-G-A | 1 | 6.20e-7 | -6.020 | Likely Benign | 0.117 | Likely Benign | Likely Benign | 0.020 | Likely Benign | -0.27 | Likely Benign | 0.0 | -0.04 | Likely Benign | -0.16 | Likely Benign | -0.39 | Likely Benign | -0.14 | Neutral | 0.159 | Benign | 0.084 | Benign | 1.98 | Pathogenic | 0.27 | Tolerated | 3.37 | 25 | 4 | 3 | 0.3 | 14.03 | 240.2 | -26.9 | -0.2 | 0.2 | -0.2 | 0.2 | X | Potentially Benign | The iso-propyl side chain of Val343, located in an anti-parallel β sheet strand (res. Gly341-Pro349), is packing against multiple hydrophobic residues of the C2 domain (e.g., Leu327, Leu274, Val365). In the variant simulations, the sec-butyl side chain of Ile343 is basically able to form the same interactions as valine due to its similar hydrophobic profile. The residue swap also does not seem to cause negative effects on the protein structure based on the simulations. | ||||||||
| c.1221G>T | Q407H 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q407H is listed in ClinVar with an uncertain significance (ClinVar ID 2772184.0) and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and FATHMM, while pathogenic predictions are made by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Predictions marked as uncertain include FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for Q407H. This conclusion does not conflict with the ClinVar designation of uncertain significance, which remains unresolved pending further evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | C2 | Uncertain | 1 | -10.526 | Likely Pathogenic | 0.830 | Likely Pathogenic | Ambiguous | 0.206 | Likely Benign | 0.59 | Ambiguous | 0.0 | 0.61 | Ambiguous | 0.60 | Ambiguous | 1.10 | Destabilizing | -4.51 | Deleterious | 0.982 | Probably Damaging | 0.947 | Probably Damaging | 3.88 | Benign | 0.01 | Affected | 3.38 | 28 | 0 | 3 | 0.3 | 9.01 | ||||||||||||||||||||
| c.1231A>G | I411V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I411V is reported in ClinVar as benign (ClinVar ID 1654508.0) and is not found in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Two tools predict a pathogenic outcome: PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. Predictions that are inconclusive or unavailable are AlphaMissense‑Default, FoldX, Rosetta, premPS, and Foldetta. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta) is uncertain. Overall, the preponderance of evidence points to a benign effect for I411V, which is consistent with its ClinVar classification and does not contradict the reported status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | Likely Benign | 1 | -6.290 | Likely Benign | 0.385 | Ambiguous | Likely Benign | 0.212 | Likely Benign | 0.74 | Ambiguous | 0.0 | 0.82 | Ambiguous | 0.78 | Ambiguous | 0.99 | Ambiguous | -0.86 | Neutral | 0.935 | Possibly Damaging | 0.858 | Possibly Damaging | 3.90 | Benign | 0.27 | Tolerated | 3.38 | 28 | 4 | 3 | -0.3 | -14.03 | 233.3 | 28.2 | -0.2 | 0.0 | -0.2 | 0.0 | X | Potentially Benign | The sec-butyl side chain of Ile411, located in the hydrophobic space between an anti-parallel β sheet strand (res. Pro398-Ile411) and an α helix (res. Asp684-Gln702), packs against multiple residues (e.g., Met409, Arg259). In the variant simulations, the side chain of Val411 is able to favorably fill the same hydrophobic niche despite its slightly smaller size. In short, the residue swap has no apparent negative effect on the structure based on the simulations. | |||||||||||
| c.1300G>A | V434I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V434I (ClinVar ID 212346.0, status Uncertain) is present in gnomAD (ID 6‑33438205‑G‑A). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No predictions or stability results are missing or inconclusive. Based on the collective evidence, the variant is most likely benign, which does not contradict the ClinVar status of Uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | Uncertain | 1 | 6-33438205-G-A | 1 | 6.19e-7 | -6.999 | Likely Benign | 0.129 | Likely Benign | Likely Benign | 0.192 | Likely Benign | -0.04 | Likely Benign | 0.0 | 0.22 | Likely Benign | 0.09 | Likely Benign | 0.31 | Likely Benign | -0.82 | Neutral | 0.947 | Possibly Damaging | 0.851 | Possibly Damaging | 3.53 | Benign | 0.18 | Tolerated | 3.37 | 29 | 4 | 3 | 0.3 | 14.03 | 246.7 | -27.7 | 0.0 | 0.0 | 0.1 | 0.0 | X | Potentially Benign | The iso-propyl side chain of Val434, located at the end of an α helix (res. Met414-Glu436), packs against hydrophobic residues in an interhelix space (e.g., Met430, Ala707, Leu711). In the variant simulations, the sec-butyl group of Ile434 is able to form the same hydrophobic interactions. Accordingly, the residue swap does not negatively affect the protein structure based on the simulations. | ||||||||
| c.1354G>A | V452I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V452I is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar) and SIFT, while ESM1b also predicts pathogenicity. Uncertain predictions come from Rosetta and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Taken together, the majority of evidence points to a benign impact. This conclusion does not contradict the ClinVar “Uncertain” classification, which remains inconclusive. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | Uncertain | 1 | -8.985 | Likely Pathogenic | 0.361 | Ambiguous | Likely Benign | 0.218 | Likely Benign | -0.08 | Likely Benign | 0.1 | 0.51 | Ambiguous | 0.22 | Likely Benign | 0.25 | Likely Benign | -0.99 | Neutral | 0.947 | Possibly Damaging | 0.851 | Possibly Damaging | 3.26 | Benign | 0.05 | Affected | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||
| c.1480A>G | I494V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant I494V is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33438512‑A‑G). Functional prediction tools that agree on benign impact include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Pathogenic predictions come from premPS and FATHMM. Predictions that are inconclusive are FoldX, Rosetta, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign; Foldetta remains uncertain. Overall, the majority of evidence supports a benign effect, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | Conflicting | 2 | 6-33438512-A-G | 36 | 2.23e-5 | -7.102 | In-Between | 0.112 | Likely Benign | Likely Benign | 0.439 | Likely Benign | 1.16 | Ambiguous | 0.0 | 0.71 | Ambiguous | 0.94 | Ambiguous | 1.02 | Destabilizing | -0.83 | Neutral | 0.278 | Benign | 0.179 | Benign | -1.30 | Pathogenic | 0.07 | Tolerated | 3.37 | 35 | 4 | 3 | -0.3 | -14.03 | 248.6 | 29.3 | 0.0 | 0.0 | -1.1 | 0.5 | X | Potentially Benign | The sec-butyl side chain of Ile494, located in an α-helix (res. Leu489-Glu519), packs against hydrophobic residues (e.g., Phe484, Leu465, Trp572, Ala493, Met468) in an inter-helix space (res. Leu489-Glu519 and res. Ala461-Phe476). In the variant simulations, the hydrophobic iso-propyl side chain of Val494, which is of a similar size and has similar physicochemical properties to Ile494 in the WT, resides similarly in the inter-helix hydrophobic space. Thus, no negative effects on the protein structure are observed. | |||||||||
| c.1511A>G | K504R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K504R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33438543‑A‑G). Consensus from most in‑silico predictors is benign: REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all report a benign effect, while only FATHMM predicts pathogenicity. Uncertain calls come from Rosetta and premPS. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | Uncertain | 1 | 6-33438543-A-G | 2 | 1.24e-6 | -4.365 | Likely Benign | 0.088 | Likely Benign | Likely Benign | 0.238 | Likely Benign | 0.13 | Likely Benign | 0.1 | 0.51 | Ambiguous | 0.32 | Likely Benign | 0.94 | Ambiguous | -2.16 | Neutral | 0.002 | Benign | 0.015 | Benign | -1.41 | Pathogenic | 0.11 | Tolerated | 3.37 | 35 | 2 | 3 | -0.6 | 28.01 | |||||||||||||||||
| c.1663G>A | V555I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V555I is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Only FATHMM predicts a pathogenic outcome, while FoldX, Foldetta, and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | Uncertain | 1 | -4.544 | Likely Benign | 0.084 | Likely Benign | Likely Benign | 0.253 | Likely Benign | -0.82 | Ambiguous | 0.0 | -0.41 | Likely Benign | -0.62 | Ambiguous | -0.55 | Ambiguous | 0.45 | Neutral | 0.002 | Benign | 0.002 | Benign | -1.26 | Pathogenic | 1.00 | Tolerated | 4 | 3 | 0.3 | 14.03 | ||||||||||||||||||||||
| c.1851G>T | E617D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E617D is listed in ClinVar with an uncertain significance (ID 2584916.0) and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score all indicate benign or likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact, while Rosetta remains inconclusive. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is likely benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Overall, the preponderance of evidence supports a benign classification, which does not contradict the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | Uncertain | 1 | -1.349 | Likely Benign | 0.241 | Likely Benign | Likely Benign | 0.322 | Likely Benign | 0.12 | Likely Benign | 0.1 | 0.80 | Ambiguous | 0.46 | Likely Benign | 0.07 | Likely Benign | -0.01 | Neutral | 0.994 | Probably Damaging | 0.979 | Probably Damaging | -1.35 | Pathogenic | 0.88 | Tolerated | 3.37 | 35 | 2 | 3 | 0.0 | -14.03 | ||||||||||||||||||||
| c.1888A>G | I630V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I630V is listed in ClinVar as Benign and is present in gnomAD (variant ID 6‑33440940‑A‑G). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome; all other tools (FoldX, Rosetta, Foldetta, premPS, ESM1b) return uncertain or inconclusive results. The high‑accuracy consensus (SGM Consensus) derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a benign majority (2 benign vs. 1 pathogenic, 1 uncertain). AlphaMissense‑Optimized also predicts benign. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is uncertain. Taken together, the overwhelming majority of predictions support a benign effect, and this conclusion aligns with the ClinVar designation. Thus, the variant is most likely benign, with no contradiction to the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | Benign/Likely benign | 4 | 6-33440940-A-G | 59 | 3.66e-5 | -7.264 | In-Between | 0.145 | Likely Benign | Likely Benign | 0.143 | Likely Benign | 1.33 | Ambiguous | 0.0 | 0.94 | Ambiguous | 1.14 | Ambiguous | 0.64 | Ambiguous | -0.38 | Neutral | 0.018 | Benign | 0.011 | Benign | -1.37 | Pathogenic | 0.35 | Tolerated | 3.37 | 34 | 4 | 3 | -0.3 | -14.03 | 235.0 | 26.2 | -0.1 | 0.0 | -0.3 | 0.1 | X | Potentially Benign | The sec-butyl side chain of Ile630, located in an α helix (res. Glu617-Asn635), packs with hydrophobic residues (e.g., Phe594, Leu633, Ile626, Ile602) in the hydrophobic inter-helix space between two α helices (res. Glu617-Asn635 and res. Glu582-Met603).In the variant simulations, the iso-propyl side chain of Val630, which shares a similar size and physicochemical properties with Ile630 in the WT, maintains similar interactions in the inter-helix space. Although no negative structural effects are observed during the simulations, the implications of the residue swap on the complex formation with the GTPase, due to its location, cannot be investigated using solvent-only simulations. | |||||||||
| c.1913A>G | K638R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K638R is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). In contrast, PROVEAN and polyPhen‑2 HumDiv predict a pathogenic impact, while premPS remains inconclusive. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is benign. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | Uncertain | 1 | -2.700 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.216 | Likely Benign | 0.09 | Likely Benign | 0.1 | -0.04 | Likely Benign | 0.03 | Likely Benign | 0.53 | Ambiguous | -2.55 | Deleterious | 0.649 | Possibly Damaging | 0.240 | Benign | 3.41 | Benign | 0.13 | Tolerated | 3.37 | 31 | 2 | 3 | -0.6 | 28.01 | ||||||||||||||||||||
| c.2047A>G | I683V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I683V is listed in ClinVar with an uncertain significance and is present in gnomAD (6‑33441306‑A‑G). Across a panel of in silico predictors, the majority indicate a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (derived from a majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only polyPhen‑2 HumDiv classifies the change as pathogenic. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote) is benign, and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is inconclusive and therefore not considered evidence. No other tool provides a pathogenic signal. Consequently, the variant is most likely benign, and this assessment does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | Uncertain | 1 | 6-33441306-A-G | 2 | 1.24e-6 | -7.588 | In-Between | 0.138 | Likely Benign | Likely Benign | 0.112 | Likely Benign | 0.90 | Ambiguous | 0.0 | 0.60 | Ambiguous | 0.75 | Ambiguous | 0.76 | Ambiguous | -0.78 | Neutral | 0.538 | Possibly Damaging | 0.080 | Benign | 3.35 | Benign | 0.14 | Tolerated | 3.42 | 17 | 4 | 3 | -0.3 | -14.03 | 215.6 | 29.1 | 0.0 | 0.0 | -0.7 | 0.1 | X | Potentially Benign | The sec-butyl side chain of Ile683, located in an entangled α-α loop connecting the two α-helices (res. Ser641-Glu666 and res. Leu685-Val699), is sterically packed against His453 and Glu688. In the variant simulations, the iso-propyl side chain of Val683 has similar size and physicochemical properties as Ile630 in the WT, and thus, it is able to maintain similar interactions in the inter-helix space. Consequently, no negative structural effects are observed during the simulations due to the residue swap. | ||||||||
| c.218G>A | R73K 2D AIThe SynGAP1 missense variant R73K is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33425826‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized returns a benign prediction, and the SGM‑Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33425826-G-A | 2 | 1.24e-6 | -4.033 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.077 | Likely Benign | -0.46 | Neutral | 0.053 | Benign | 0.007 | Benign | 4.14 | Benign | 0.00 | Affected | 4.32 | 1 | 2 | 3 | 0.6 | -28.01 | |||||||||||||||||||||||||||
| c.2299A>G | I767V 2D AIThe SynGAP1 missense variant I767V is listed in ClinVar (ID 1402700.0) with an “Uncertain” clinical significance and is not reported in gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores. The SGM‑Consensus, which aggregates the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a “Likely Benign” outcome. No tool predicts pathogenicity. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, while Foldetta (combining FoldX‑MD and Rosetta stability predictions) has no available result for this variant. Overall, the computational evidence strongly supports a benign effect, and this conclusion does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -2.791 | Likely Benign | 0.064 | Likely Benign | Likely Benign | 0.096 | Likely Benign | 0.10 | Neutral | 0.072 | Benign | 0.029 | Benign | 4.21 | Benign | 1.00 | Tolerated | 3.64 | 6 | 4 | 3 | -0.3 | -14.03 | ||||||||||||||||||||||||||||||
| c.2420A>T | Y807F 2D  AIThe SynGAP1 missense variant Y807F is listed in ClinVar (ID 1491782.0) with an “Uncertain” status and is not reported in gnomAD. All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign scores, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a likely benign effect. No tool in the set predicts pathogenicity. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (derived from the four high‑accuracy predictors) is benign. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, has no available result for this variant. Overall, the computational evidence strongly supports a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | SH3-binding motif | Uncertain | 1 | -3.667 | Likely Benign | 0.073 | Likely Benign | Likely Benign | 0.057 | Likely Benign | 0.14 | Neutral | 0.012 | Benign | 0.022 | Benign | 2.92 | Benign | 0.98 | Tolerated | 3.77 | 5 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||
| c.256G>A | V86I 2D AIThe SynGAP1 missense variant V86I is listed in ClinVar (ID 588267.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the variant as benign or likely benign. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is “Likely Benign.” No Foldetta (FoldX‑MD/Rosetta stability) result is available, so it does not influence the assessment. Overall, the majority of predictions indicate that V86I is most likely benign, which is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -4.726 | Likely Benign | 0.338 | Likely Benign | Likely Benign | 0.076 | Likely Benign | -0.31 | Neutral | 0.267 | Benign | 0.097 | Benign | 3.94 | Benign | 0.00 | Affected | 4.32 | 1 | 4 | 3 | 0.3 | 14.03 | ||||||||||||||||||||||||||||||
| c.2578G>A | V860I 2D AIThe SynGAP1 missense variant V860I is catalogued in ClinVar as a benign alteration (ClinVar ID 411591.0) and is present in the gnomAD database (gnomAD ID 6‑33443130‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized returns benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” No Foldetta stability prediction is available for this variant. Overall, the consensus of computational evidence strongly favors a benign impact, aligning with the ClinVar designation and showing no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 1 | 6-33443130-G-A | 21 | 1.30e-5 | -4.516 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.039 | Likely Benign | -0.42 | Neutral | 0.009 | Benign | 0.006 | Benign | 4.24 | Benign | 0.00 | Affected | 3.77 | 5 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||
| c.2596G>A | V866I 2D AIThe SynGAP1 missense variant V866I is listed in ClinVar with an “Uncertain” status (ClinVar ID 536995.0) and is present in gnomAD (6‑33443148‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Conflicting | 3 | 6-33443148-G-A | 5 | 3.10e-6 | -4.652 | Likely Benign | 0.118 | Likely Benign | Likely Benign | 0.059 | Likely Benign | -0.39 | Neutral | 0.957 | Probably Damaging | 0.541 | Possibly Damaging | 2.69 | Benign | 0.27 | Tolerated | 3.82 | 4 | 4 | 3 | 0.3 | 14.03 | |||||||||||||||||||||||||||
| c.2695A>G | I899V 2D AIThe SynGAP1 missense variant I899V is listed in ClinVar as a benign alteration (ClinVar ID 1003653.0) and is present in the gnomAD database (gnomAD ID 6‑33443247‑A‑G). All evaluated in‑silico predictors classify the variant as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity, so the pathogenic‑prediction group is empty. High‑accuracy assessments further support a benign effect: AlphaMissense‑Optimized reports benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant. Overall, the computational evidence strongly suggests the variant is benign, consistent with its ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 1 | 6-33443247-A-G | 6 | 3.72e-6 | -2.569 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.040 | Likely Benign | 0.09 | Neutral | 0.220 | Benign | 0.078 | Benign | 2.75 | Benign | 0.92 | Tolerated | 4.32 | 4 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||
| c.3048C>A | D1016E 2D AIThe SynGAP1 missense variant D1016E is reported in ClinVar (ID 3803472.0) as benign and is present in gnomAD (variant ID 6‑33443600‑C‑A). All evaluated in‑silico predictors classify the substitution as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Benign.” Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, did not provide a result for this variant, so its status is unavailable. Overall, the computational evidence overwhelmingly supports a benign effect, aligning with the ClinVar benign classification and showing no contradiction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Likely Benign | 1 | 6-33443600-C-A | 2 | 1.24e-6 | -3.422 | Likely Benign | 0.216 | Likely Benign | Likely Benign | 0.017 | Likely Benign | -0.37 | Neutral | 0.008 | Benign | 0.028 | Benign | 2.64 | Benign | 0.65 | Tolerated | 3.77 | 5 | 2 | 3 | 0.0 | 14.03 | |||||||||||||||||||||||||||
| c.3397A>G | I1133V 2D AIThe SynGAP1 missense variant I1133V is listed in ClinVar as Benign (ClinVar ID 999690.0) and is present in the gnomAD database (gnomAD ID 6‑33443949‑A‑G). All evaluated in‑silico predictors classify the change as benign: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign. Foldetta results are unavailable. Consequently, the variant is most likely benign, and this prediction aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 1 | 6-33443949-A-G | 22 | 1.48e-5 | -3.362 | Likely Benign | 0.067 | Likely Benign | Likely Benign | 0.180 | Likely Benign | 0.06 | Neutral | 0.007 | Benign | 0.007 | Benign | 5.47 | Benign | 0.58 | Tolerated | 4.32 | 3 | 4 | 3 | -0.3 | -14.03 | 10.1016/j.ajhg.2020.11.011 | ||||||||||||||||||||||||||
| c.3502A>G | I1168V 2D AIThe SynGAP1 missense variant I1168V is listed in ClinVar (ID 936001.0) with an “Uncertain” status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. AlphaMissense‑Default is uncertain, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports a “Likely Benign” outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this consensus does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -3.263 | Likely Benign | 0.524 | Ambiguous | Likely Benign | 0.363 | Likely Benign | -0.14 | Neutral | 0.876 | Possibly Damaging | 0.643 | Possibly Damaging | 5.47 | Benign | 0.84 | Tolerated | 3.88 | 3 | 4 | 3 | -0.3 | -14.03 | ||||||||||||||||||||||||||||||
| c.37A>G | I13V 2D AIThe SynGAP1 I13V missense variant is listed in ClinVar (ID 3364831.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as benign; Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for I13V, and this conclusion does not conflict with the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | -2.497 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.110 | Likely Benign | 0.01 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.25 | Benign | 0.00 | Affected | 4 | 3 | -0.3 | -14.03 | ||||||||||||||||||||||||||||||||
| c.4008G>C | E1336D 2D AIThe SynGAP1 missense variant E1336D is listed in ClinVar (ID 3323942.0) as benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus result is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yielding a benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, consistent with the ClinVar benign designation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Likely Benign | 1 | -3.344 | Likely Benign | 0.596 | Likely Pathogenic | Likely Benign | 0.062 | Likely Benign | -1.92 | Neutral | 0.001 | Benign | 0.003 | Benign | 3.30 | Benign | 0.00 | Affected | 3.77 | 5 | 2 | 3 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.103G>A | V35I 2D AIThe SynGAP1 missense variant V35I is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33423512‑G‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates a benign outcome. No Foldetta stability data are available. Overall, the majority of evidence points to a benign impact, and this is consistent with the ClinVar “Uncertain” classification rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33423512-G-A | 5 | 3.10e-6 | -3.764 | Likely Benign | 0.081 | Likely Benign | Likely Benign | 0.017 | Likely Benign | -0.32 | Neutral | 0.672 | Possibly Damaging | 0.369 | Benign | 4.16 | Benign | 0.00 | Affected | 4.32 | 1 | 3 | 4 | 0.3 | 14.03 | |||||||||||||||||||||||||||
| c.1417G>A | V473I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V473I is listed in ClinVar with an uncertain significance and is present in gnomAD (ID 6‑33438449‑G‑A). Functional prediction tools that agree on benign impact include REVEL, FoldX, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Pathogenic predictions are provided by both polyPhen‑2 HumDiv and HumVar. Predictions that are inconclusive are AlphaMissense‑Default, ESM1b, Foldetta, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is unavailable due to no majority, and Foldetta is uncertain. Overall, the balance of evidence favors a benign effect for V473I, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | Uncertain | 1 | 6-33438449-G-A | 1 | 6.20e-7 | -7.481 | In-Between | 0.418 | Ambiguous | Likely Benign | 0.203 | Likely Benign | -0.12 | Likely Benign | 0.0 | 1.20 | Ambiguous | 0.54 | Ambiguous | -0.06 | Likely Benign | -0.91 | Neutral | 0.929 | Possibly Damaging | 0.917 | Probably Damaging | 3.74 | Benign | 0.18 | Tolerated | 3.37 | 34 | 3 | 4 | 0.3 | 14.03 | ||||||||||||||||||
| c.1447A>G | I483V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I483V is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, PROVEAN, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions are reported by premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Predictions marked as uncertain include FoldX, Foldetta, and AlphaMissense‑Default. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields benign, whereas Foldetta remains uncertain. Overall, the balance of evidence from both general and high‑accuracy tools leans toward a benign effect, which does not contradict the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | Conflicting | 2 | -10.121 | Likely Pathogenic | 0.523 | Ambiguous | Likely Benign | 0.228 | Likely Benign | 1.00 | Ambiguous | 0.0 | 0.27 | Likely Benign | 0.64 | Ambiguous | 1.02 | Destabilizing | -0.86 | Neutral | 0.914 | Possibly Damaging | 0.921 | Probably Damaging | 3.23 | Benign | 0.03 | Affected | 3.37 | 32 | 3 | 4 | -0.3 | -14.03 | |||||||||||||||||||||
| c.2503C>A | L835M 2D AIThe SynGAP1 missense variant L835M is listed in ClinVar (ID 2731331.0) as benign and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are PolyPhen‑2 HumDiv and PolyPhen‑2 HumVar. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, the SGM‑Consensus also indicating a likely benign outcome, while Foldetta (combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the preponderance of evidence—including the high‑accuracy tools—supports a benign classification, which is consistent with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Benign | 1 | -4.153 | Likely Benign | 0.121 | Likely Benign | Likely Benign | 0.068 | Likely Benign | -0.45 | Neutral | 0.999 | Probably Damaging | 0.977 | Probably Damaging | 2.67 | Benign | 0.12 | Tolerated | 3.77 | 5 | 2 | 4 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.2998A>G | I1000V 2D AIThe SynGAP1 missense variant I1000V is listed in ClinVar (ID 2572013.0) with an “Uncertain” clinical significance and is not reported in gnomAD. Functional prediction tools that assess evolutionary conservation and structural impact (REVEL, SIFT, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default) all converge on a benign outcome. No tool in the dataset predicts pathogenicity. High‑accuracy predictors reinforce this consensus: AlphaMissense‑Optimized reports a benign effect, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 2 | -4.102 | Likely Benign | 0.098 | Likely Benign | Likely Benign | 0.086 | Likely Benign | -0.20 | Neutral | 0.437 | Benign | 0.170 | Benign | 2.76 | Benign | 0.81 | Tolerated | 4.32 | 4 | 3 | 4 | -0.3 | -14.03 | ||||||||||||||||||||||||||||||
| c.70G>A | V24I 2D AIThe SynGAP1 missense variant V24I is listed in ClinVar with an “Uncertain” status and is present in gnomAD (variant ID 6-33423479-G-A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Only SIFT predicts a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Taken together, the overwhelming majority of computational evidence supports a benign impact for V24I, and this conclusion does not contradict the ClinVar designation, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | Uncertain | 1 | 6-33423479-G-A | 9 | 5.58e-6 | -3.701 | Likely Benign | 0.137 | Likely Benign | Likely Benign | 0.069 | Likely Benign | -0.25 | Neutral | 0.043 | Benign | 0.031 | Benign | 3.96 | Benign | 0.00 | Affected | 4.32 | 1 | 3 | 4 | 0.3 | 14.03 | |||||||||||||||||||||||||||
| c.958G>A | V320I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V320I is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while Rosetta remains inconclusive. High‑accuracy assessments corroborate the benign trend: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Overall, the preponderance of evidence points to a benign effect for V320I, and this conclusion does not conflict with the current ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | C2 | Uncertain | 1 | -5.220 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.027 | Likely Benign | -0.27 | Likely Benign | 0.2 | 0.66 | Ambiguous | 0.20 | Likely Benign | 0.01 | Likely Benign | -0.21 | Neutral | 0.198 | Benign | 0.114 | Benign | 1.77 | Pathogenic | 0.45 | Tolerated | 3.38 | 23 | 3 | 4 | 0.3 | 14.03 | ||||||||||||||||||||
Found 757 rows. Show 200 rows per page. Page 4/4 « Previous |