
Table of SynGAP1 Isoform α2 (UniProt Q96PV0-1) Missense Variants.
| c.dna | Variant | SGM Consensus | Domain and Structure information: based on WT protein | Annotated databases | Deep learning-based pathogenicity predictions | Folding stability-based pathogenicity predictions | Sequence/structure-based pathogenicity predictions | Phase Separation | Evolutionary/physical properties | Molecular Dynamics-based analysis | DOI | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | IUPred2 | ANCHOR2 | AlphaFold | MobiDB | PhosphoSitePlus | ClinVar | gnomAD | ESM1b | AlphaMissense | FoldX | Rosetta | Foldetta | PremPS | REVEL | PROVEAN | PolyPhen-2 HumDiv | PolyPhen-2 HumVar | FATHMM | SIFT | PSMutPred | PAM | Physical | SASA | Normalized B-factor backbone | Normalized B-factor sidechain | SynGAP Structural Annotation | |||||||||||||||||||||||||||||||||||||||||||||
| Score | Prediction | Score | Prediction | pLDDT | disorder | disorder | LTP | HTP | KL | PTM | Clinical Status | Review | Subm. | ID | Allele count | Allele freq. | LLR score | Prediction | Pathogenicity | Class | Optimized | Average ΔΔG | Prediction | StdDev | ΔΔG | Prediction | ΔΔG | Prediction | ΔΔG | Prediction | Score | Prediction | Score | Prediction | pph2_prob | Prediction | pph2_prob | Prediction | Nervous System Score | Prediction | Prediction | Status | Conservation | Sequences | IP RF | SP RF | Prediction | PAM250 | PAM120 | Hydropathy Δ | MW Δ | Average | Δ | Δ | StdDev | Δ | StdDev | Secondary | Tertiary bonds | Inside out | GAP-Ras interface | At membrane | No effect | MD Alert | Verdict | Description | |||||
| c.1707T>A | F569L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F569L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are none; all available predictors that provide a verdict classify the variant as pathogenic, with the exception of FoldX and Foldetta, whose results are uncertain and therefore treated as unavailable. The high‑accuracy predictors give the following: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, is uncertain and thus not considered evidence. Based on the overwhelming consensus of pathogenic predictions and the lack of contrary evidence, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.024393 | Structured | 0.054289 | Uncertain | 0.941 | 0.242 | 0.000 | -9.784 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.86 | Ambiguous | 0.1 | 2.04 | Destabilizing | 1.45 | Ambiguous | 1.28 | Destabilizing | 0.675 | Likely Pathogenic | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -1.13 | Pathogenic | 0.05 | Affected | 0.1977 | 0.2476 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1707T>G | F569L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F569L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are none; all available predictors that provide a verdict classify the variant as pathogenic, with the exception of FoldX and Foldetta, whose outputs are uncertain and therefore treated as unavailable. The high‑accuracy predictors give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, is uncertain. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.024393 | Structured | 0.054289 | Uncertain | 0.941 | 0.242 | 0.000 | -9.784 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.86 | Ambiguous | 0.1 | 2.04 | Destabilizing | 1.45 | Ambiguous | 1.28 | Destabilizing | 0.677 | Likely Pathogenic | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -1.13 | Pathogenic | 0.05 | Affected | 0.1977 | 0.2476 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1708G>A | A570T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A570T is not reported in ClinVar and is absent from gnomAD. Prediction tools that provide a definitive call all indicate a deleterious effect: SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenicity. No tool reports a benign outcome; the remaining predictions (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Optimized) are uncertain and therefore do not influence the overall assessment. High‑accuracy methods specifically show SGM‑Consensus as Likely Pathogenic, AlphaMissense‑Optimized as uncertain, and Foldetta as uncertain. Taken together, the majority of conclusive predictions support a pathogenic effect. Consequently, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.054494 | Uncertain | 0.932 | 0.263 | 0.000 | -11.390 | Likely Pathogenic | 0.801 | Likely Pathogenic | Ambiguous | 1.45 | Ambiguous | 0.3 | 1.67 | Ambiguous | 1.56 | Ambiguous | 0.86 | Ambiguous | 0.568 | Likely Pathogenic | -3.28 | Deleterious | 0.998 | Probably Damaging | 0.993 | Probably Damaging | -1.26 | Pathogenic | 0.05 | Affected | 0.1345 | 0.3874 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.1708G>C | A570P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A570P is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. No tool predicts a benign outcome. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the unanimous pathogenic predictions and the absence of any benign calls, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which simply lacks an entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.054494 | Uncertain | 0.932 | 0.263 | 0.000 | -15.178 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.21 | Destabilizing | 0.5 | 8.45 | Destabilizing | 6.83 | Destabilizing | 1.19 | Destabilizing | 0.832 | Likely Pathogenic | -4.55 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.31 | Pathogenic | 0.02 | Affected | 0.1965 | 0.2578 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1708G>T | A570S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A570S is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The remaining tools (FoldX, Rosetta, Foldetta, premPS, and ESM1b) yield uncertain or inconclusive results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign, while Foldetta remains uncertain. Overall, the majority of reliable predictions indicate a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.046336 | Structured | 0.054494 | Uncertain | 0.932 | 0.263 | 0.000 | -7.893 | In-Between | 0.194 | Likely Benign | Likely Benign | 0.77 | Ambiguous | 0.1 | 1.68 | Ambiguous | 1.23 | Ambiguous | 0.51 | Ambiguous | 0.399 | Likely Benign | -2.26 | Neutral | 0.983 | Probably Damaging | 0.993 | Probably Damaging | -1.19 | Pathogenic | 0.17 | Tolerated | 0.2091 | 0.3256 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||
| c.1709C>A | A570D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A570D is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect, so the benign group is empty. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.054494 | Uncertain | 0.932 | 0.263 | 0.000 | -14.117 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.47 | Destabilizing | 1.2 | 2.33 | Destabilizing | 2.40 | Destabilizing | 1.36 | Destabilizing | 0.805 | Likely Pathogenic | -5.31 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.28 | Pathogenic | 0.03 | Affected | 0.2006 | 0.2206 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.1709C>G | A570G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 A570G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SIFT and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and FATHMM. The remaining tools (FoldX, premPS, ESM1b, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta are unavailable due to mixed or uncertain outputs. Overall, the majority of evaluated tools (seven pathogenic vs. two benign) indicate that the variant is most likely pathogenic, and this assessment does not contradict ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.046336 | Structured | 0.054494 | Uncertain | 0.932 | 0.263 | 0.000 | -7.509 | In-Between | 0.562 | Ambiguous | Likely Benign | 1.34 | Ambiguous | 0.1 | 2.12 | Destabilizing | 1.73 | Ambiguous | 0.99 | Ambiguous | 0.607 | Likely Pathogenic | -3.62 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | -1.30 | Pathogenic | 0.09 | Tolerated | 0.1700 | 0.2499 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||
| c.1709C>T | A570V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 A570V missense variant is catalogued in gnomAD (ID 6‑33440761‑C‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from premPS and SIFT, while pathogenic predictions are made by REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Four tools report uncertainty: FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.054494 | Uncertain | 0.932 | 0.263 | 0.000 | 6-33440761-C-T | 1 | 6.22e-7 | -13.083 | Likely Pathogenic | 0.882 | Likely Pathogenic | Ambiguous | 0.88 | Ambiguous | 0.3 | 1.63 | Ambiguous | 1.26 | Ambiguous | 0.46 | Likely Benign | 0.669 | Likely Pathogenic | -3.75 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | -1.30 | Pathogenic | 0.06 | Tolerated | 3.37 | 35 | 0.1050 | 0.3173 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||
| c.170T>A | L57H 2D  AIThe SynGAP1 missense variant L57H is not reported in ClinVar and has no entry in gnomAD. Prediction tools show a split: benign calls come from REVEL, PROVEAN, ESM1b, and FATHMM, whereas pathogenic calls come from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments further indicate that AlphaMissense‑Optimized is Uncertain, whereas the SGM‑Consensus remains Likely Benign; Foldetta stability analysis is unavailable. Overall, the majority of high‑confidence tools and the consensus score favor a benign effect, and this conclusion does not conflict with the absence of a ClinVar assertion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.254060 | Structured | 0.481044 | Uncertain | 0.554 | 0.642 | 0.000 | -6.251 | Likely Benign | 0.796 | Likely Pathogenic | Ambiguous | 0.173 | Likely Benign | -1.58 | Neutral | 0.984 | Probably Damaging | 0.971 | Probably Damaging | 3.90 | Benign | 0.00 | Affected | 0.1045 | 0.0611 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||||||||||||
| c.170T>C | L57P 2D  AIThe SynGAP1 missense variant L57P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM. Tools that agree on a pathogenic effect include polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (2 pathogenic vs. 2 benign). Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions (six pathogenic vs. three benign) indicate that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.254060 | Structured | 0.481044 | Uncertain | 0.554 | 0.642 | 0.000 | -10.724 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.242 | Likely Benign | -1.77 | Neutral | 0.943 | Possibly Damaging | 0.944 | Probably Damaging | 3.90 | Benign | 0.00 | Affected | 0.3651 | 0.1703 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.170T>G | L57R 2D  AIThe SynGAP1 missense variant L57R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which collectively classify the variant as likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default predict a pathogenic impact. The high‑accuracy AlphaMissense‑Optimized result is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) is not available for this variant. Overall, the balance of evidence leans toward a benign interpretation, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.254060 | Structured | 0.481044 | Uncertain | 0.554 | 0.642 | 0.000 | -6.034 | Likely Benign | 0.810 | Likely Pathogenic | Ambiguous | 0.213 | Likely Benign | -1.55 | Neutral | 0.943 | Possibly Damaging | 0.944 | Probably Damaging | 3.91 | Benign | 0.00 | Affected | 0.1245 | 0.0685 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.1711T>A | S571T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S571T is not reported in ClinVar (ClinVar ID: None) and has no entry in gnomAD (gnomAD ID: None). Prediction tools that indicate a benign effect include FoldX, Rosetta, Foldetta, premPS, SIFT, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools (seven) predict pathogenicity, while six predict benignity, and the high‑accuracy subset is split. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.069024 | Structured | 0.045569 | Uncertain | 0.928 | 0.270 | 0.000 | -8.243 | Likely Pathogenic | 0.431 | Ambiguous | Likely Benign | 0.37 | Likely Benign | 0.1 | -0.21 | Likely Benign | 0.08 | Likely Benign | 0.25 | Likely Benign | 0.564 | Likely Pathogenic | -2.76 | Deleterious | 0.933 | Possibly Damaging | 0.933 | Probably Damaging | -1.25 | Pathogenic | 0.10 | Tolerated | 0.1360 | 0.4014 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1711T>C | S571P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S571P is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while premPS remains inconclusive. High‑accuracy assessments corroborate this trend: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. Consequently, the aggregate evidence strongly supports a pathogenic effect for S571P, and this conclusion does not conflict with the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.069024 | Structured | 0.045569 | Uncertain | 0.928 | 0.270 | 0.000 | Uncertain | 1 | -14.701 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.18 | Destabilizing | 0.2 | 4.89 | Destabilizing | 4.04 | Destabilizing | 0.87 | Ambiguous | 0.814 | Likely Pathogenic | -4.68 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | -1.30 | Pathogenic | 0.02 | Affected | 0.2195 | 0.3760 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||
| c.1711T>G | S571A 2D 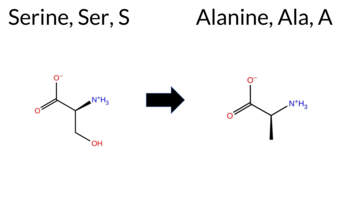 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S571A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, Rosetta, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and FATHMM. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because it yields a 2‑2 split. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta (combining FoldX‑MD and Rosetta outputs) as benign, and the SGM Consensus remains unavailable. Overall, the preponderance of evidence points to a benign impact for S571A, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.069024 | Structured | 0.045569 | Uncertain | 0.928 | 0.270 | 0.000 | -6.344 | Likely Benign | 0.233 | Likely Benign | Likely Benign | -0.44 | Likely Benign | 0.1 | -0.19 | Likely Benign | -0.32 | Likely Benign | 0.51 | Ambiguous | 0.563 | Likely Pathogenic | -2.69 | Deleterious | 0.980 | Probably Damaging | 0.994 | Probably Damaging | -1.22 | Pathogenic | 0.09 | Tolerated | 0.4739 | 0.2671 | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||
| c.1712C>G | S571W 2D 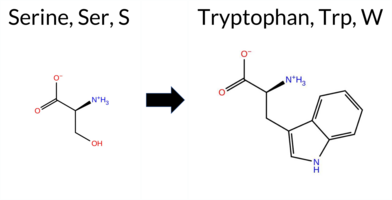 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S571W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that provide a clear verdict overwhelmingly classify the substitution as pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict deleterious effects. No tool in the dataset returned a benign classification; the only non‑conclusive results come from FoldX, Rosetta, Foldetta, and premPS, which are listed as uncertain. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta’s stability analysis is inconclusive. Based on the aggregate predictions, the variant is most likely pathogenic, and this assessment is consistent with the absence of a ClinVar entry (no contradictory status). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.069024 | Structured | 0.045569 | Uncertain | 0.928 | 0.270 | 0.000 | -14.025 | Likely Pathogenic | 0.961 | Likely Pathogenic | Likely Pathogenic | -1.13 | Ambiguous | 0.1 | -1.44 | Ambiguous | -1.29 | Ambiguous | 0.67 | Ambiguous | 0.867 | Likely Pathogenic | -6.64 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.32 | Pathogenic | 0.00 | Affected | 0.0648 | 0.3809 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1712C>T | S571L 2D 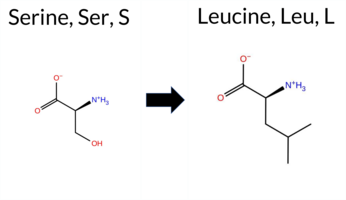 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S571L is listed in ClinVar with an uncertain significance (ClinVar ID 3897075) and is present in gnomAD (ID 6‑33440764‑C‑T). Prediction tools that indicate a benign effect include premPS and AlphaMissense‑Optimized, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) all predict a pathogenic outcome. The high‑accuracy AlphaMissense‑Optimized score is benign, whereas the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—supports pathogenicity. Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, is inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for S571L, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.069024 | Structured | 0.045569 | Uncertain | 0.928 | 0.270 | 0.000 | Uncertain | 2 | 6-33440764-C-T | 1 | 6.23e-7 | -11.651 | Likely Pathogenic | 0.660 | Likely Pathogenic | Likely Benign | -1.53 | Ambiguous | 0.1 | -1.05 | Ambiguous | -1.29 | Ambiguous | 0.27 | Likely Benign | 0.841 | Likely Pathogenic | -5.61 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | -1.25 | Pathogenic | 0.04 | Affected | 3.37 | 35 | 0.0959 | 0.3918 | -2 | -3 | 4.6 | 26.08 | ||||||||||||||||||||||
| c.1715G>T | W572L 2D 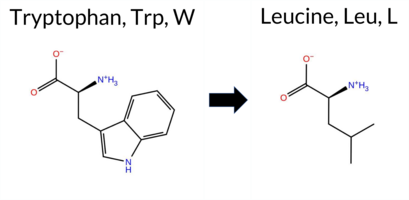 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W572L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only SIFT, which scores the variant as benign. All other evaluated predictors—REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a pathogenic effect. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.150080 | Structured | 0.039626 | Uncertain | 0.935 | 0.256 | 0.000 | -15.765 | Likely Pathogenic | 0.973 | Likely Pathogenic | Likely Pathogenic | 2.62 | Destabilizing | 0.1 | 3.97 | Destabilizing | 3.30 | Destabilizing | 0.90 | Ambiguous | 0.591 | Likely Pathogenic | -11.52 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -0.91 | Pathogenic | 0.31 | Tolerated | 0.2246 | 0.2837 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||||||
| c.1716G>C | W572C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W572C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.150080 | Structured | 0.039626 | Uncertain | 0.935 | 0.256 | 0.000 | -15.589 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.49 | Destabilizing | 0.2 | 5.59 | Destabilizing | 5.54 | Destabilizing | 1.72 | Destabilizing | 0.861 | Likely Pathogenic | -11.82 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.25 | Pathogenic | 0.00 | Affected | 0.3648 | 0.1304 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||||
| c.1716G>T | W572C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W572C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. No inconclusive or missing predictions are present. Based on the consensus of all available computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.150080 | Structured | 0.039626 | Uncertain | 0.935 | 0.256 | 0.000 | -15.589 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.49 | Destabilizing | 0.2 | 5.59 | Destabilizing | 5.54 | Destabilizing | 1.72 | Destabilizing | 0.861 | Likely Pathogenic | -11.82 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.25 | Pathogenic | 0.00 | Affected | 0.3648 | 0.1304 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||||
| c.1717C>G | R573G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R573G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while no tool predicts a benign outcome. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Based on the uniform predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar annotation exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.134866 | Structured | 0.032433 | Uncertain | 0.934 | 0.235 | 0.000 | -15.387 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.71 | Destabilizing | 0.7 | 3.16 | Destabilizing | 3.44 | Destabilizing | 1.37 | Destabilizing | 0.744 | Likely Pathogenic | -6.01 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.45 | Pathogenic | 0.01 | Affected | 0.2931 | 0.2166 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1718G>C | R573P 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R573P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a pathogenic or likely pathogenic outcome. No tool in the dataset predicts a benign effect. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic impact. Based on the uniform predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.134866 | Structured | 0.032433 | Uncertain | 0.934 | 0.235 | 0.000 | -16.684 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 5.76 | Destabilizing | 1.1 | 8.58 | Destabilizing | 7.17 | Destabilizing | 1.21 | Destabilizing | 0.831 | Likely Pathogenic | -5.78 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.47 | Pathogenic | 0.01 | Affected | 0.1961 | 0.3239 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.1720C>A | L574M 2D  AIThe SynGAP1 missense variant L574M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and FATHMM; ESM1b is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as benign. No predictions or stability results are missing or inconclusive. Based on the overall consensus, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.083462 | Structured | 0.026427 | Uncertain | 0.927 | 0.246 | 0.000 | -7.195 | In-Between | 0.098 | Likely Benign | Likely Benign | 0.14 | Likely Benign | 0.2 | 0.34 | Likely Benign | 0.24 | Likely Benign | -0.09 | Likely Benign | 0.113 | Likely Benign | 0.85 | Neutral | 0.691 | Possibly Damaging | 0.278 | Benign | -1.29 | Pathogenic | 0.11 | Tolerated | 0.0894 | 0.3087 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.1720C>G | L574V 2D 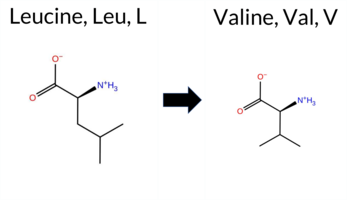 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L574V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result. No other high‑confidence pathogenic predictions are present. Based on the preponderance of benign predictions and the lack of any ClinVar pathogenic annotation, the variant is most likely benign. This conclusion does not contradict any existing ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.083462 | Structured | 0.026427 | Uncertain | 0.927 | 0.246 | 0.000 | -5.559 | Likely Benign | 0.105 | Likely Benign | Likely Benign | 0.78 | Ambiguous | 0.1 | 0.37 | Likely Benign | 0.58 | Ambiguous | 0.25 | Likely Benign | 0.149 | Likely Benign | -0.40 | Neutral | 0.004 | Benign | 0.003 | Benign | -1.27 | Pathogenic | 0.29 | Tolerated | 0.1481 | 0.2978 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1721T>A | L574Q 2D 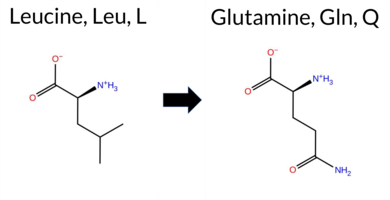 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L574Q resides in the GAP domain. ClinVar contains no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; Rosetta’s assessment is uncertain. High‑accuracy methods give a consistent benign signal: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.083462 | Structured | 0.026427 | Uncertain | 0.927 | 0.246 | 0.000 | -3.015 | Likely Benign | 0.196 | Likely Benign | Likely Benign | 0.26 | Likely Benign | 0.2 | 0.52 | Ambiguous | 0.39 | Likely Benign | -0.18 | Likely Benign | 0.327 | Likely Benign | 2.53 | Neutral | 0.998 | Probably Damaging | 0.937 | Probably Damaging | -1.17 | Pathogenic | 0.65 | Tolerated | 0.1107 | 0.0888 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1721T>C | L574P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L574P is not reported in ClinVar and is present in gnomAD (6-33440773‑T‑C). Prediction tools that indicate a benign effect include REVEL, PROVEAN, and SIFT, whereas the majority of tools predict a pathogenic impact: FoldX, Rosetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, Foldetta, and the SGM Consensus score (likely pathogenic). High‑accuracy methods specifically give a pathogenic verdict: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.083462 | Structured | 0.026427 | Uncertain | 0.927 | 0.246 | 0.000 | 6-33440773-T-C | -13.394 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 2.95 | Destabilizing | 0.5 | 9.19 | Destabilizing | 6.07 | Destabilizing | 0.59 | Ambiguous | 0.442 | Likely Benign | -1.05 | Neutral | 1.000 | Probably Damaging | 0.971 | Probably Damaging | -1.29 | Pathogenic | 0.26 | Tolerated | 3.38 | 32 | 0.3811 | 0.0953 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||
| c.1721T>G | L574R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L574R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, polyPhen‑2 HumVar, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, ESM1b, and FATHMM. Two tools remain inconclusive: Rosetta (Uncertain) and AlphaMissense‑Default (Uncertain). High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of individual predictors (seven benign vs. three pathogenic) support a benign classification, and this conclusion does not contradict the ClinVar status, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.083462 | Structured | 0.026427 | Uncertain | 0.927 | 0.246 | 0.000 | -8.702 | Likely Pathogenic | 0.563 | Ambiguous | Likely Benign | 0.04 | Likely Benign | 0.2 | 0.88 | Ambiguous | 0.46 | Likely Benign | -0.12 | Likely Benign | 0.322 | Likely Benign | 0.30 | Neutral | 0.907 | Possibly Damaging | 0.292 | Benign | -1.22 | Pathogenic | 0.52 | Tolerated | 0.1419 | 0.0530 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||
| c.1723C>A | R575S 2D 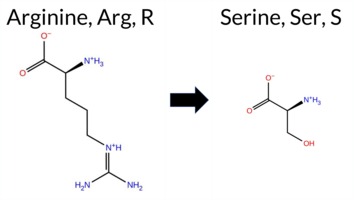 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R575S is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include only SIFT, whereas the majority—REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict pathogenicity. FoldX, Rosetta, Foldetta, and premPS give uncertain results, which are treated as unavailable evidence. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for R575S, and this assessment does not contradict the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.021362 | Uncertain | 0.916 | 0.259 | 0.000 | -11.124 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 1.66 | Ambiguous | 0.1 | 0.55 | Ambiguous | 1.11 | Ambiguous | 0.76 | Ambiguous | 0.582 | Likely Pathogenic | -2.71 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.21 | Pathogenic | 0.33 | Tolerated | 0.2669 | 0.2394 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||
| c.1723C>G | R575G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R575G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on benign effect include only SIFT, whereas the remaining tools—REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus—consistently predict pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Because the majority of evidence points to deleterious impact, the variant is most likely pathogenic; this conclusion does not contradict ClinVar status, which currently has no entry for R575G. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.021362 | Uncertain | 0.916 | 0.259 | 0.000 | -13.104 | Likely Pathogenic | 0.914 | Likely Pathogenic | Ambiguous | 2.18 | Destabilizing | 0.0 | 1.15 | Ambiguous | 1.67 | Ambiguous | 1.23 | Destabilizing | 0.772 | Likely Pathogenic | -4.22 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.31 | Pathogenic | 0.13 | Tolerated | 0.2889 | 0.1755 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1724G>C | R575P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R575P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only SIFT, whereas all other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Based on the overwhelming consensus of pathogenic predictions and the absence of any benign consensus, the variant is most likely pathogenic. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for R575P. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.021362 | Uncertain | 0.916 | 0.259 | 0.000 | -16.008 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 3.50 | Destabilizing | 0.1 | 4.97 | Destabilizing | 4.24 | Destabilizing | 1.13 | Destabilizing | 0.774 | Likely Pathogenic | -3.69 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.33 | Pathogenic | 0.10 | Tolerated | 0.2080 | 0.2670 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.1724G>T | R575L 2D 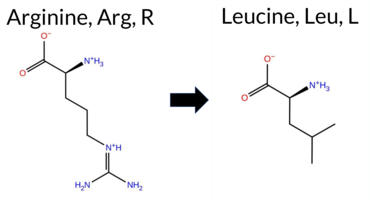 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R575L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include SIFT, FoldX, and Foldetta. Those that predict pathogenicity comprise SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain predictions come from AlphaMissense‑Optimized, Rosetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as Benign. Overall, the majority of tools (10/13) predict pathogenicity, and the high‑accuracy consensus leans toward pathogenic, though Foldetta suggests stability‑preserving benign effects. Thus, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.021362 | Uncertain | 0.916 | 0.259 | 0.000 | -12.442 | Likely Pathogenic | 0.788 | Likely Pathogenic | Ambiguous | -0.04 | Likely Benign | 0.1 | -0.89 | Ambiguous | -0.47 | Likely Benign | 0.59 | Ambiguous | 0.602 | Likely Pathogenic | -4.42 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.24 | Pathogenic | 0.11 | Tolerated | 0.1574 | 0.2991 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.1726T>A | C576S 2D 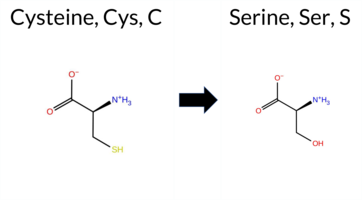 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C576S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions from REVEL and FATHMM, while the majority of other in silico methods (premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict pathogenicity. FoldX and Rosetta give uncertain stability changes, and Foldetta likewise reports no definitive effect. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic. Foldetta remains inconclusive. Overall, the preponderance of evidence from multiple pathogenic‑oriented tools and the high‑accuracy predictions indicates that C576S is most likely pathogenic, which is consistent with the absence of a ClinVar entry and gnomAD data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.017684 | Uncertain | 0.913 | 0.245 | 0.000 | -10.474 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.77 | Ambiguous | 0.1 | 1.57 | Ambiguous | 1.17 | Ambiguous | 1.61 | Destabilizing | 0.414 | Likely Benign | -8.91 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.40 | Benign | 0.02 | Affected | 0.4968 | 0.1464 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.1726T>C | C576R 2D 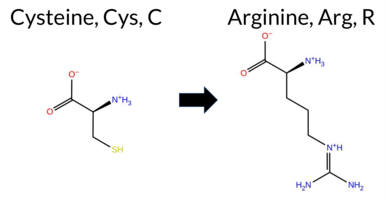 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C576R (ClinVar ID 2780076) is reported as Pathogenic and is not present in gnomAD. Prediction tools that classify the variant as benign include only FATHMM. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy assessments further support this: AlphaMissense‑Optimized returns a pathogenic score; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenicity. No predictions or stability results are missing or inconclusive. Based on the overwhelming agreement among predictive tools and the high‑accuracy methods, the variant is most likely pathogenic, consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.017684 | Uncertain | 0.913 | 0.245 | 0.000 | Pathogenic/Likely path. | 2 | -14.886 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 7.20 | Destabilizing | 1.0 | 4.09 | Destabilizing | 5.65 | Destabilizing | 1.64 | Destabilizing | 0.579 | Likely Pathogenic | -10.88 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.38 | Benign | 0.00 | Affected | 3.37 | 35 | 0.1887 | 0.1279 | -3 | -4 | -7.0 | 53.05 | |||||||||||||||||||||||||
| c.1726T>G | C576G 2D 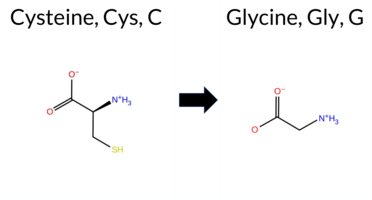 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C576G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from REVEL, FoldX (uncertain), Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a benign outcome. When high‑accuracy methods are considered, AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labels it likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) also predicts pathogenicity. No prediction is inconclusive or missing. Consequently, the variant is most likely pathogenic based on the consensus of available computational evidence, and this assessment does not contradict any ClinVar annotation (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.017684 | Uncertain | 0.913 | 0.245 | 0.000 | -14.809 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 1.66 | Ambiguous | 0.0 | 2.53 | Destabilizing | 2.10 | Destabilizing | 1.67 | Destabilizing | 0.586 | Likely Pathogenic | -10.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.38 | Benign | 0.00 | Affected | 0.3269 | 0.2574 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1727G>A | C576Y 2D 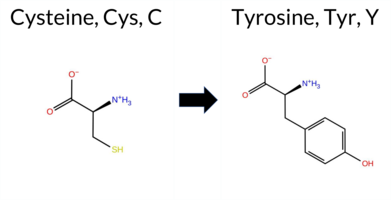 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C576Y is not reported in ClinVar and has no gnomAD allele. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, whereas SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default and AlphaMissense‑Optimized all classify the change as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also indicates pathogenicity. No tool yields an inconclusive result. Based on the consensus of available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.017684 | Uncertain | 0.913 | 0.245 | 0.000 | -13.891 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 8.77 | Destabilizing | 0.5 | 3.90 | Destabilizing | 6.34 | Destabilizing | 0.63 | Ambiguous | 0.612 | Likely Pathogenic | -9.98 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.38 | Benign | 0.00 | Affected | 0.1398 | 0.3009 | 0 | -2 | -3.8 | 60.04 | |||||||||||||||||||||||||||||
| c.1727G>C | C576S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C576S is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect are limited to FATHMM, whereas the remaining 11 tools (SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict a pathogenic or likely pathogenic outcome. High‑accuracy methods reinforce this trend: AlphaMissense‑Optimized reports pathogenic; the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also returns likely pathogenic; Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta, is inconclusive. Folding‑stability tools FoldX and Rosetta individually yield uncertain results and are treated as unavailable. Taken together, the majority of evidence points to a pathogenic effect. This conclusion is consistent with the absence of ClinVar annotation and gnomAD data, so there is no contradiction with existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.017684 | Uncertain | 0.913 | 0.245 | 0.000 | -10.474 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.77 | Ambiguous | 0.1 | 1.57 | Ambiguous | 1.17 | Ambiguous | 1.61 | Destabilizing | 0.523 | Likely Pathogenic | -8.91 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.40 | Benign | 0.02 | Affected | 0.4968 | 0.1464 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.1727G>T | C576F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C576F is not reported in ClinVar (ClinVar ID = None) and has no entries in gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect are limited to FATHMM, which scores the variant as benign. All other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta—consistently predict a pathogenic or likely pathogenic impact. Uncertain predictions from Rosetta and premPS are treated as unavailable. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the overwhelming agreement among high‑confidence tools, the variant is most likely pathogenic, and this assessment does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.017684 | Uncertain | 0.913 | 0.245 | 0.000 | -13.467 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 5.04 | Destabilizing | 0.5 | 1.81 | Ambiguous | 3.43 | Destabilizing | 0.58 | Ambiguous | 0.516 | Likely Pathogenic | -9.93 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.40 | Benign | 0.02 | Affected | 0.1626 | 0.3527 | -4 | -2 | 0.3 | 44.04 | |||||||||||||||||||||||||||||
| c.1728C>G | C576W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C576W is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, while the majority of other in silico predictors (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a pathogenic impact; premPS is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the preponderance of pathogenic predictions and the absence of benign consensus, the variant is most likely pathogenic, with no contradiction to ClinVar status (which has no entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.017684 | Uncertain | 0.913 | 0.245 | 0.000 | -14.796 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 13.87 | Destabilizing | 1.5 | 5.46 | Destabilizing | 9.67 | Destabilizing | 0.60 | Ambiguous | 0.473 | Likely Benign | -10.01 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.38 | Benign | 0.00 | Affected | 0.1894 | 0.3017 | -8 | -2 | -3.4 | 83.07 | |||||||||||||||||||||||||||||
| c.1729G>C | A577P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A577P is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include only SIFT, whereas the remaining evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict a pathogenic outcome; premPS is inconclusive and is therefore not counted. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the consensus of these predictions, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.019074 | Uncertain | 0.913 | 0.239 | 0.000 | -9.009 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.93 | Destabilizing | 0.2 | 9.88 | Destabilizing | 6.91 | Destabilizing | 0.87 | Ambiguous | 0.585 | Likely Pathogenic | -2.72 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.34 | Pathogenic | 0.20 | Tolerated | 0.2256 | 0.4600 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1729G>T | A577S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A577S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote) all indicate benign or likely benign. In contrast, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM predict pathogenicity, while Rosetta remains uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, the SGM‑Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.113710 | Structured | 0.019074 | Uncertain | 0.913 | 0.239 | 0.000 | -4.417 | Likely Benign | 0.151 | Likely Benign | Likely Benign | 0.10 | Likely Benign | 0.0 | 0.60 | Ambiguous | 0.35 | Likely Benign | 0.05 | Likely Benign | 0.342 | Likely Benign | -0.30 | Neutral | 0.981 | Probably Damaging | 0.992 | Probably Damaging | -1.24 | Pathogenic | 0.80 | Tolerated | 0.2763 | 0.5056 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.172A>C | M58L 2D  AIThe SynGAP1 missense variant M58L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign. Only SIFT predicts a deleterious impact. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign outcome. High‑accuracy assessments confirm this: AlphaMissense‑Optimized scores benign, and the SGM‑Consensus result is benign; Foldetta data are not available. Consequently, the aggregate evidence points to a benign effect for M58L, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | -0.661 | Likely Benign | 0.240 | Likely Benign | Likely Benign | 0.208 | Likely Benign | -0.11 | Neutral | 0.006 | Benign | 0.039 | Benign | 4.77 | Benign | 0.00 | Affected | 0.1514 | 0.4621 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.172A>G | M58V 2D  AIThe SynGAP1 missense variant M58V is listed in ClinVar (ID 2962156.0) with an uncertain significance status and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The high‑accuracy consensus from AlphaMissense‑Optimized, SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta (protein‑folding stability) is available only for the first two; Foldetta data are missing. The SGM Consensus, based on a majority of benign predictions, indicates a likely benign outcome. Overall, the majority of evidence points to a benign impact, which does not contradict the ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | Uncertain | 1 | -2.211 | Likely Benign | 0.688 | Likely Pathogenic | Likely Benign | 0.160 | Likely Benign | -0.71 | Neutral | 0.006 | Benign | 0.091 | Benign | 4.19 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2951 | 0.3917 | 1 | 2 | 2.3 | -32.06 | |||||||||||||||||||||||||||||||||||
| c.172A>T | M58L 2D AIThe SynGAP1 missense variant M58L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar annotation—there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | -0.661 | Likely Benign | 0.240 | Likely Benign | Likely Benign | 0.208 | Likely Benign | -0.11 | Neutral | 0.006 | Benign | 0.039 | Benign | 4.77 | Benign | 0.00 | Affected | 0.1514 | 0.4621 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||||||||||||
| c.1730C>A | A577E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 A577E missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, and SIFT, whereas those that agree on a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while Foldetta (combining FoldX‑MD and Rosetta outputs) is also uncertain; Rosetta and premPS are inconclusive. Overall, the majority of evaluated tools (five pathogenic vs. four benign) and the SGM‑Consensus support a pathogenic classification. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.113710 | Structured | 0.019074 | Uncertain | 0.913 | 0.239 | 0.000 | -9.607 | Likely Pathogenic | 0.794 | Likely Pathogenic | Ambiguous | 0.29 | Likely Benign | 0.0 | 0.72 | Ambiguous | 0.51 | Ambiguous | 0.62 | Ambiguous | 0.419 | Likely Benign | -1.91 | Neutral | 0.999 | Probably Damaging | 0.996 | Probably Damaging | -1.12 | Pathogenic | 0.76 | Tolerated | 0.1487 | 0.1799 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||
| c.1730C>T | A577V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A577V is catalogued in gnomAD (ID 6‑33440782‑C‑T) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign; the SGM consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also reports benign. No prediction or stability result is missing or inconclusive in these key analyses. Consequently, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.113710 | Structured | 0.019074 | Uncertain | 0.913 | 0.239 | 0.000 | 6-33440782-C-T | 2 | 1.24e-6 | -4.265 | Likely Benign | 0.432 | Ambiguous | Likely Benign | 0.69 | Ambiguous | 0.1 | 0.00 | Likely Benign | 0.35 | Likely Benign | 0.16 | Likely Benign | 0.417 | Likely Benign | -2.11 | Neutral | 0.997 | Probably Damaging | 0.976 | Probably Damaging | -1.32 | Pathogenic | 0.26 | Tolerated | 3.37 | 34 | 0.1278 | 0.4790 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||
| c.1732G>A | E578K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E578K is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, and SIFT, whereas polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default predict a pathogenic outcome. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show that the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Pathogenic, AlphaMissense‑Optimized remains Uncertain, and Foldetta predicts a benign effect. Overall, the majority of tools (seven benign vs. five pathogenic) suggest a benign impact, and this assessment does not contradict the absence of ClinVar evidence. Thus, the variant is most likely benign based on the current predictions, with no conflicting ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.059222 | Structured | 0.020971 | Uncertain | 0.902 | 0.240 | 0.000 | -13.391 | Likely Pathogenic | 0.870 | Likely Pathogenic | Ambiguous | 0.07 | Likely Benign | 0.1 | -0.19 | Likely Benign | -0.06 | Likely Benign | -0.27 | Likely Benign | 0.450 | Likely Benign | -1.65 | Neutral | 0.996 | Probably Damaging | 0.987 | Probably Damaging | -1.30 | Pathogenic | 0.49 | Tolerated | 0.1954 | 0.5480 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1732G>C | E578Q 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 E578Q missense variant is not reported in ClinVar and is absent from gnomAD. Consensus from most in silico predictors indicates a benign effect: REVEL, FoldX, Rosetta, premPS, PROVEAN, SIFT, AlphaMissense‑Optimized, and Foldetta all predict benign. Pathogenic predictions come from polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. AlphaMissense‑Default remains uncertain. High‑accuracy assessment shows AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—leans pathogenic. Foldetta, a protein‑folding stability method, also predicts benign. Overall, the majority of tools, including the high‑accuracy AlphaMissense‑Optimized and Foldetta, support a benign classification, and this is consistent with the lack of ClinVar evidence. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.059222 | Structured | 0.020971 | Uncertain | 0.902 | 0.240 | 0.000 | -9.771 | Likely Pathogenic | 0.491 | Ambiguous | Likely Benign | 0.01 | Likely Benign | 0.1 | -0.12 | Likely Benign | -0.06 | Likely Benign | -0.16 | Likely Benign | 0.353 | Likely Benign | -1.21 | Neutral | 0.994 | Probably Damaging | 0.986 | Probably Damaging | -1.40 | Pathogenic | 0.36 | Tolerated | 0.1000 | 0.5319 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.1733A>C | E578A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 E578A missense change is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized; pathogenic predictions come from REVEL, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. ESM1b is uncertain. High‑accuracy assessment shows AlphaMissense‑Optimized predicts benign, while the SGM consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) favors pathogenic, and Foldetta also predicts benign. Overall, the majority of conventional tools lean benign, but the high‑accuracy consensus and AlphaMissense‑Optimized suggest a pathogenic effect. Thus, the variant is most likely pathogenic according to the most reliable predictors, and this assessment does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.059222 | Structured | 0.020971 | Uncertain | 0.902 | 0.240 | 0.000 | -7.369 | In-Between | 0.604 | Likely Pathogenic | Likely Benign | 0.34 | Likely Benign | 0.1 | -0.48 | Likely Benign | -0.07 | Likely Benign | -0.02 | Likely Benign | 0.525 | Likely Pathogenic | -2.30 | Neutral | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.40 | Pathogenic | 0.44 | Tolerated | 0.3326 | 0.5118 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||
| c.1733A>G | E578G 2D 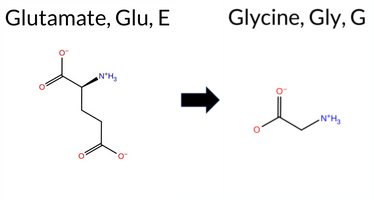 3DClick to see structure in 3D Viewer AIThe SynGAP1 E578G missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are REVEL, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. Four tools give uncertain or inconclusive results (FoldX, Rosetta, Foldetta, ESM1b). High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—predicts pathogenic. Foldetta, a protein‑folding stability method, yields an uncertain result. Overall, the majority of high‑confidence predictions lean toward pathogenicity, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.059222 | Structured | 0.020971 | Uncertain | 0.902 | 0.240 | 0.000 | -7.680 | In-Between | 0.601 | Likely Pathogenic | Likely Benign | 0.98 | Ambiguous | 0.1 | 0.63 | Ambiguous | 0.81 | Ambiguous | 0.08 | Likely Benign | 0.589 | Likely Pathogenic | -2.40 | Neutral | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.49 | Pathogenic | 0.19 | Tolerated | 0.2608 | 0.4843 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||
| c.1733A>T | E578V 2D 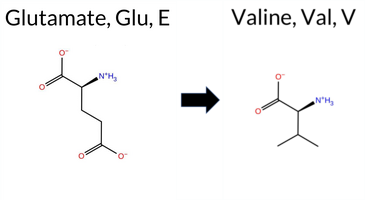 3DClick to see structure in 3D Viewer AISynGAP1 E578V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. In silico predictors cluster into two groups: benign predictions come from Rosetta, Foldetta, premPS, and SIFT, while pathogenic predictions arise from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: FoldX and AlphaMissense‑Optimized. High‑accuracy assessments further show that the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic, AlphaMissense‑Optimized is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the balance of evidence favors a pathogenic effect, and this conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.059222 | Structured | 0.020971 | Uncertain | 0.902 | 0.240 | 0.000 | -11.393 | Likely Pathogenic | 0.881 | Likely Pathogenic | Ambiguous | 0.72 | Ambiguous | 0.1 | 0.02 | Likely Benign | 0.37 | Likely Benign | 0.12 | Likely Benign | 0.607 | Likely Pathogenic | -3.74 | Deleterious | 0.996 | Probably Damaging | 0.991 | Probably Damaging | -1.43 | Pathogenic | 0.13 | Tolerated | 0.0575 | 0.5703 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1734G>C | E578D 2D 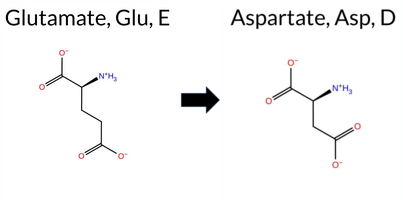 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E578D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) leans toward benign (2 benign vs 1 pathogenic votes), and Foldetta also predicts benign stability. No prediction or folding result is missing or inconclusive. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.059222 | Structured | 0.020971 | Uncertain | 0.902 | 0.240 | 0.000 | -4.366 | Likely Benign | 0.447 | Ambiguous | Likely Benign | 0.46 | Likely Benign | 0.1 | 0.15 | Likely Benign | 0.31 | Likely Benign | -0.05 | Likely Benign | 0.318 | Likely Benign | -0.53 | Neutral | 0.989 | Probably Damaging | 0.979 | Probably Damaging | -1.43 | Pathogenic | 0.33 | Tolerated | 0.1561 | 0.3554 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1734G>T | E578D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E578D missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) resolves to benign (2 benign vs 1 pathogenic votes), and Foldetta predicts a benign stability change. No prediction or stability result is missing or inconclusive. Overall, the majority of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.059222 | Structured | 0.020971 | Uncertain | 0.902 | 0.240 | 0.000 | -4.366 | Likely Benign | 0.447 | Ambiguous | Likely Benign | 0.46 | Likely Benign | 0.1 | 0.15 | Likely Benign | 0.31 | Likely Benign | -0.05 | Likely Benign | 0.318 | Likely Benign | -0.53 | Neutral | 0.989 | Probably Damaging | 0.979 | Probably Damaging | -1.43 | Pathogenic | 0.33 | Tolerated | 0.1561 | 0.3554 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1735C>G | R579G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R579G is reported in gnomAD (ID 6‑33440787‑C‑G) and has no ClinVar entry. Prediction tools that assess pathogenicity uniformly favor a deleterious effect: SGM‑Consensus (Likely Pathogenic), REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenic. No tool in the dataset reports a benign outcome; the only uncertain calls are from FoldX, AlphaMissense‑Optimized, and Foldetta. High‑accuracy assessments further support pathogenicity: the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic, while AlphaMissense‑Optimized and Foldetta remain uncertain. Consequently, the collective evidence indicates that R579G is most likely pathogenic, and this conclusion is not contradicted by ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.053060 | Structured | 0.022872 | Uncertain | 0.877 | 0.244 | 0.000 | 6-33440787-C-G | 1 | 6.20e-7 | -14.298 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 1.43 | Ambiguous | 0.0 | 2.36 | Destabilizing | 1.90 | Ambiguous | 1.32 | Destabilizing | 0.680 | Likely Pathogenic | -5.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.40 | Pathogenic | 0.01 | Affected | 3.37 | 34 | 0.3078 | 0.2554 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1736G>A | R579Q 2D 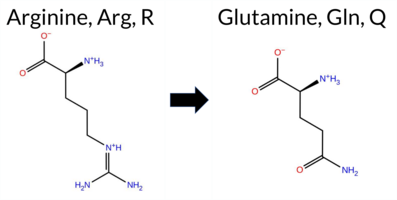 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R579Q is listed in ClinVar with an uncertain significance (ClinVar ID 3964539) and is present in gnomAD (6‑33440788‑G‑A). Prediction tools that indicate a benign effect include SIFT and AlphaMissense‑Optimized, whereas the remaining tools (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. FoldX and Rosetta individually also return uncertain results. Overall, the majority of evidence points to a pathogenic effect, which does not contradict the ClinVar uncertain status. Therefore, the variant is most likely pathogenic based on current computational predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.053060 | Structured | 0.022872 | Uncertain | 0.877 | 0.244 | 0.000 | Uncertain | 2 | 6-33440788-G-A | 18 | 1.12e-5 | -9.193 | Likely Pathogenic | 0.690 | Likely Pathogenic | Likely Benign | 0.65 | Ambiguous | 0.1 | 0.70 | Ambiguous | 0.68 | Ambiguous | 1.13 | Destabilizing | 0.673 | Likely Pathogenic | -3.31 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | -1.34 | Pathogenic | 0.06 | Tolerated | 3.37 | 34 | 0.2677 | 0.1334 | 1 | 1 | 1.0 | -28.06 | ||||||||||||||||||||||
| c.1736G>C | R579P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R579P is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a pathogenic effect include SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a benign outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions or stability results are missing or inconclusive. Based on the unanimous pathogenic predictions and the absence of any ClinVar or gnomAD evidence to the contrary, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.053060 | Structured | 0.022872 | Uncertain | 0.877 | 0.244 | 0.000 | -14.826 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.00 | Destabilizing | 0.2 | 6.36 | Destabilizing | 4.68 | Destabilizing | 0.93 | Ambiguous | 0.821 | Likely Pathogenic | -6.26 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.41 | Pathogenic | 0.01 | Affected | 0.2207 | 0.3081 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.1736G>T | R579L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R579L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, and SIFT, whereas those that predict a pathogenic impact are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. Overall, the majority of evidence (seven pathogenic vs. five benign predictions) points to a pathogenic effect for R579L. This conclusion is not contradicted by ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.053060 | Structured | 0.022872 | Uncertain | 0.877 | 0.244 | 0.000 | -9.290 | Likely Pathogenic | 0.904 | Likely Pathogenic | Ambiguous | -0.24 | Likely Benign | 0.1 | 0.07 | Likely Benign | -0.09 | Likely Benign | 0.48 | Likely Benign | 0.802 | Likely Pathogenic | -6.39 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.36 | Pathogenic | 0.06 | Tolerated | 0.1747 | 0.3259 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.1738G>C | G580R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G580R is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all predict a pathogenic outcome. No tool predicts a benign effect. Several methods return uncertain results—Rosetta, Foldetta (combining FoldX‑MD and Rosetta outputs), and premPS—so these do not influence the overall assessment. High‑accuracy evaluations reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is likely pathogenic, while Foldetta remains uncertain. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.025952 | Uncertain | 0.853 | 0.236 | 0.000 | -11.459 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 2.20 | Destabilizing | 0.1 | 1.26 | Ambiguous | 1.73 | Ambiguous | 0.81 | Ambiguous | 0.623 | Likely Pathogenic | -7.33 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.26 | Pathogenic | 0.03 | Affected | 0.1009 | 0.3197 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1738G>T | G580C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G580C is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a pathogenic outcome include REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools with uncertain or inconclusive results are Rosetta and premPS. High‑accuracy methods all support a deleterious effect: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No tool predicts benign. **Based on the consensus of available predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status (which is currently unreported).** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.025952 | Uncertain | 0.853 | 0.236 | 0.000 | -11.608 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 2.94 | Destabilizing | 0.1 | 1.18 | Ambiguous | 2.06 | Destabilizing | 0.60 | Ambiguous | 0.755 | Likely Pathogenic | -8.66 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.18 | Pathogenic | 0.01 | Affected | 0.1422 | 0.2146 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.1739G>A | G580D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G580D is not reported in ClinVar and has no entries in gnomAD. Consensus from multiple in silico predictors indicates a pathogenic effect: SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic, while premPS and Rosetta are uncertain. High‑accuracy tools reinforce this assessment: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No benign predictions are present. Consequently, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.025952 | Uncertain | 0.853 | 0.236 | 0.000 | -10.086 | Likely Pathogenic | 0.965 | Likely Pathogenic | Likely Pathogenic | 2.85 | Destabilizing | 0.1 | 1.39 | Ambiguous | 2.12 | Destabilizing | 0.83 | Ambiguous | 0.712 | Likely Pathogenic | -6.73 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.25 | Pathogenic | 0.04 | Affected | 0.1793 | 0.1971 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.1739G>C | G580A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G580A is not reported in ClinVar (ClinVar status: None) and has no entries in gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity are unanimous: REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus all predict a deleterious effect. Tools with uncertain or inconclusive results (Rosetta and premPS) are noted as unavailable for pathogenicity inference. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic; and Foldetta, a protein‑folding stability predictor that integrates FoldX‑MD and Rosetta outputs, also classifies the variant as Pathogenic. Consequently, the variant is most likely pathogenic based on the consensus of predictive tools, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.025952 | Uncertain | 0.853 | 0.236 | 0.000 | -10.841 | Likely Pathogenic | 0.956 | Likely Pathogenic | Likely Pathogenic | 2.84 | Destabilizing | 0.1 | 1.55 | Ambiguous | 2.20 | Destabilizing | 0.64 | Ambiguous | 0.646 | Likely Pathogenic | -5.73 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | -1.22 | Pathogenic | 0.05 | Affected | 0.3657 | 0.2862 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1739G>T | G580V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G580V is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess functional impact uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. The only inconclusive result is premPS, which is listed as uncertain. No tool predicts a benign effect. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenic. Based on the unanimous pathogenic predictions and the absence of any benign calls, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.025952 | Uncertain | 0.853 | 0.236 | 0.000 | -13.705 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 4.10 | Destabilizing | 0.1 | 3.89 | Destabilizing | 4.00 | Destabilizing | 0.79 | Ambiguous | 0.750 | Likely Pathogenic | -8.66 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.18 | Pathogenic | 0.04 | Affected | 0.1270 | 0.2909 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.173T>A | M58K 2D  AIThe SynGAP1 missense variant M58K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. There is no ClinVar entry to contradict this assessment, so the variant is most likely benign based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | -6.059 | Likely Benign | 0.939 | Likely Pathogenic | Ambiguous | 0.199 | Likely Benign | -1.52 | Neutral | 0.018 | Benign | 0.184 | Benign | 4.08 | Benign | 0.00 | Affected | 0.1615 | 0.1163 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||||||||||||
| c.173T>C | M58T 2D  AIThe SynGAP1 missense variant M58T is listed in gnomAD (ID 6‑33423582‑T‑C) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | 6-33423582-T-C | 1 | 6.20e-7 | -4.308 | Likely Benign | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.159 | Likely Benign | -1.58 | Neutral | 0.018 | Benign | 0.184 | Benign | 4.09 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1999 | 0.2357 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||||||||||||
| c.173T>G | M58R 2D  AIThe SynGAP1 missense variant M58R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and no Foldetta stability assessment is available. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments therefore indicate a benign likelihood: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus is benign, and Foldetta data are missing. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | -5.035 | Likely Benign | 0.940 | Likely Pathogenic | Ambiguous | 0.237 | Likely Benign | -1.78 | Neutral | 0.042 | Benign | 0.184 | Benign | 4.07 | Benign | 0.00 | Affected | 0.1745 | 0.1113 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||||||||||||
| c.1741C>G | R581G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R581G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates “Likely Pathogenic.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM‑Consensus is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenicity. Uncertain results from Rosetta and premPS are treated as unavailable and do not alter the overall conclusion. **Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status (none reported).** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.029544 | Uncertain | 0.829 | 0.236 | 0.000 | -12.097 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 2.48 | Destabilizing | 0.2 | 1.70 | Ambiguous | 2.09 | Destabilizing | 0.90 | Ambiguous | 0.581 | Likely Pathogenic | -5.93 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.30 | Pathogenic | 0.04 | Affected | 0.3102 | 0.2566 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1742G>C | R581P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R581P is not reported in ClinVar and is present in gnomAD (variant ID 6‑33440794‑G‑C). Functional prediction tools that agree on a benign effect include premPS and SIFT, whereas the majority of tools predict a pathogenic impact: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is pathogenic. No predictions are inconclusive or missing. Overall, the evidence strongly favors a pathogenic classification for R581P, and this is consistent with the absence of a ClinVar entry (no contradiction). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.029544 | Uncertain | 0.829 | 0.236 | 0.000 | 6-33440794-G-C | 1 | 6.20e-7 | -13.309 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.13 | Destabilizing | 0.1 | 3.80 | Destabilizing | 3.97 | Destabilizing | 0.44 | Likely Benign | 0.562 | Likely Pathogenic | -5.68 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.01 | Pathogenic | 0.07 | Tolerated | 3.37 | 34 | 0.2192 | 0.3639 | -2 | 0 | 2.9 | -59.07 | ||||||||||||||||||||||||
| c.1742G>T | R581L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R581L is not reported in ClinVar and is present in gnomAD (ID 6‑33440794‑G‑T). Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, premPS, and SIFT, whereas tools that predict pathogenicity are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy AlphaMissense‑Optimized score is pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenicity. In contrast, the Foldetta stability assessment, which integrates FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the majority of computational evidence supports a pathogenic classification for R581L, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.104810 | Structured | 0.029544 | Uncertain | 0.829 | 0.236 | 0.000 | 6-33440794-G-T | 3 | 1.86e-6 | -10.134 | Likely Pathogenic | 0.958 | Likely Pathogenic | Likely Pathogenic | 0.29 | Likely Benign | 0.1 | -0.20 | Likely Benign | 0.05 | Likely Benign | 0.45 | Likely Benign | 0.654 | Likely Pathogenic | -5.93 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.33 | Pathogenic | 0.08 | Tolerated | 3.37 | 34 | 0.1550 | 0.3483 | -2 | -3 | 8.3 | -43.03 | ||||||||||||||||||||||||
| c.1744G>A | E582K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E582K missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, premPS, PROVEAN, SIFT, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta predicts a benign effect on protein stability. Taken together, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is available). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.102787 | Structured | 0.033838 | Uncertain | 0.845 | 0.235 | 0.000 | -11.826 | Likely Pathogenic | 0.814 | Likely Pathogenic | Ambiguous | 0.20 | Likely Benign | 0.1 | 0.07 | Likely Benign | 0.14 | Likely Benign | 0.02 | Likely Benign | 0.168 | Likely Benign | -1.83 | Neutral | 0.994 | Probably Damaging | 0.994 | Probably Damaging | 3.31 | Benign | 0.33 | Tolerated | 0.2038 | 0.3762 | 0 | 1 | -0.4 | -0.94 | ||||||||||||||||||||||||||||||
| c.1744G>C | E582Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E582Q missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. The SGM‑Consensus, which is a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments—AlphaMissense‑Optimized, SGM‑Consensus, and Foldetta—all predict a benign impact, with Foldetta combining FoldX‑MD and Rosetta stability outputs. In contrast, the two polyPhen‑2 scores and AlphaMissense‑Default suggest pathogenicity, but these are outnumbered by benign predictions. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates that the variant is most likely benign, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.102787 | Structured | 0.033838 | Uncertain | 0.845 | 0.235 | 0.000 | -3.700 | Likely Benign | 0.605 | Likely Pathogenic | Likely Benign | 0.17 | Likely Benign | 0.1 | 0.21 | Likely Benign | 0.19 | Likely Benign | -0.24 | Likely Benign | 0.138 | Likely Benign | -0.61 | Neutral | 0.992 | Probably Damaging | 0.993 | Probably Damaging | 3.22 | Benign | 0.57 | Tolerated | 0.0979 | 0.3402 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1745A>C | E582A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E582A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Two tools (FoldX and ESM1b) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.102787 | Structured | 0.033838 | Uncertain | 0.845 | 0.235 | 0.000 | -7.432 | In-Between | 0.661 | Likely Pathogenic | Likely Benign | 0.78 | Ambiguous | 0.2 | 0.15 | Likely Benign | 0.47 | Likely Benign | 0.27 | Likely Benign | 0.263 | Likely Benign | -2.99 | Deleterious | 0.998 | Probably Damaging | 0.999 | Probably Damaging | 3.19 | Benign | 0.26 | Tolerated | 0.3236 | 0.4000 | 0 | -1 | 5.3 | -58.04 | ||||||||||||||||||||||||||||||
| c.1745A>G | E582G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E582G is not reported in ClinVar (ClinVar ID: None) and has no entry in gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. FoldX, Rosetta, and Foldetta give uncertain or inconclusive results. High‑accuracy methods give mixed signals: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic, and Foldetta’s stability assessment is uncertain. Overall, six tools predict pathogenicity while five predict benign, and the high‑accuracy consensus is split. Thus, the variant is most likely pathogenic based on the preponderance of evidence, and this assessment does not contradict ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.033838 | Uncertain | 0.845 | 0.235 | 0.000 | -9.621 | Likely Pathogenic | 0.630 | Likely Pathogenic | Likely Benign | 1.35 | Ambiguous | 0.2 | 1.24 | Ambiguous | 1.30 | Ambiguous | 0.37 | Likely Benign | 0.224 | Likely Benign | -3.95 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.13 | Benign | 0.13 | Tolerated | 0.2835 | 0.3325 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1745A>T | E582V 2D 3DClick to see structure in 3D Viewer AISynGAP1 E582V is not reported in ClinVar and is absent from gnomAD. Computational predictors show a split: benign calls from REVEL, Rosetta, premPS, SIFT, and FATHMM; pathogenic calls from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default; and two uncertain calls from FoldX and AlphaMissense‑Optimized. High‑accuracy methods give a pathogenic consensus: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is uncertain due to conflicting inputs. Overall, the computational evidence leans toward pathogenicity, and this assessment does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.102787 | Structured | 0.033838 | Uncertain | 0.845 | 0.235 | 0.000 | -10.737 | Likely Pathogenic | 0.842 | Likely Pathogenic | Ambiguous | 0.87 | Ambiguous | 0.1 | -0.13 | Likely Benign | 0.37 | Likely Benign | 0.24 | Likely Benign | 0.251 | Likely Benign | -3.92 | Deleterious | 0.995 | Probably Damaging | 0.996 | Probably Damaging | 3.15 | Benign | 0.10 | Tolerated | 0.0564 | 0.4186 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1746G>C | E582D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E582D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two consensus groups: a benign group comprising REVEL, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, and a pathogenic group containing polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Remaining tools (FoldX, Rosetta, ESM1b, AlphaMissense‑Default) yield uncertain results and are treated as unavailable. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leans toward benign, and Foldetta indicates no significant destabilization. Consequently, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.102787 | Structured | 0.033838 | Uncertain | 0.845 | 0.235 | 0.000 | -7.974 | In-Between | 0.520 | Ambiguous | Likely Benign | 0.57 | Ambiguous | 0.2 | 0.95 | Ambiguous | 0.76 | Ambiguous | 0.40 | Likely Benign | 0.093 | Likely Benign | -1.63 | Neutral | 0.986 | Probably Damaging | 0.989 | Probably Damaging | 3.22 | Benign | 0.27 | Tolerated | 0.1481 | 0.2236 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1746G>T | E582D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E582D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two consensus groups: a benign group comprising REVEL, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized, and a pathogenic group consisting of polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Remaining tools (FoldX, Rosetta, ESM1b, AlphaMissense‑Default) yield uncertain results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive because two of the four inputs are uncertain; Foldetta, which integrates FoldX‑MD and Rosetta outputs, also reports an uncertain stability change. Consequently, the preponderance of evidence points to a benign effect. This conclusion aligns with the absence of a ClinVar assertion, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.102787 | Structured | 0.033838 | Uncertain | 0.845 | 0.235 | 0.000 | -7.974 | In-Between | 0.520 | Ambiguous | Likely Benign | 0.57 | Ambiguous | 0.2 | 0.95 | Ambiguous | 0.76 | Ambiguous | 0.40 | Likely Benign | 0.093 | Likely Benign | -1.63 | Neutral | 0.986 | Probably Damaging | 0.989 | Probably Damaging | 3.22 | Benign | 0.27 | Tolerated | 0.1481 | 0.2236 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1747G>A | D583N 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant D583N is reported in gnomAD (ID 6‑33440799‑G‑A) but has no ClinVar entry. Functional prediction tools show mixed results: benign calls come from FoldX, Rosetta, Foldetta, premPS, and SIFT, whereas pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. The high‑accuracy assessment indicates AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the predictions are split, with a slight tilt toward pathogenicity from the consensus and high‑accuracy tools, while stability‑based methods suggest benign. Therefore, the variant is most likely pathogenic based on the aggregate predictions, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | 6-33440799-G-A | 3 | 1.86e-6 | -8.048 | Likely Pathogenic | 0.856 | Likely Pathogenic | Ambiguous | 0.13 | Likely Benign | 0.1 | 0.00 | Likely Benign | 0.07 | Likely Benign | 0.21 | Likely Benign | 0.632 | Likely Pathogenic | -4.78 | Deleterious | 0.996 | Probably Damaging | 0.995 | Probably Damaging | -1.40 | Pathogenic | 0.09 | Tolerated | 3.37 | 35 | 0.1024 | 0.3884 | 1 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||
| c.1747G>C | D583H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D583H is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include Rosetta and premPS, whereas the majority of tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict a pathogenic outcome. FoldX and Foldetta provide uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Based on the overall consensus of the available predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | -9.191 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 1.22 | Ambiguous | 0.2 | -0.07 | Likely Benign | 0.58 | Ambiguous | -0.22 | Likely Benign | 0.713 | Likely Pathogenic | -6.64 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.43 | Pathogenic | 0.03 | Affected | 0.1217 | 0.4182 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.1747G>T | D583Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D583Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect are Foldetta and premPS, whereas the remaining tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict a pathogenic outcome; FoldX and Rosetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | -12.187 | Likely Pathogenic | 0.970 | Likely Pathogenic | Likely Pathogenic | 0.75 | Ambiguous | 0.2 | -1.10 | Ambiguous | -0.18 | Likely Benign | 0.10 | Likely Benign | 0.760 | Likely Pathogenic | -8.50 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.45 | Pathogenic | 0.01 | Affected | 0.0537 | 0.3868 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1748A>C | D583A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 D583A missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Rosetta, Foldetta, premPS, SIFT, and ESM1b, while those that agree on a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX is uncertain and thus not counted. High‑accuracy methods give AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions (10 pathogenic vs. 5 benign) and the two high‑accuracy pathogenic calls outweigh the single high‑accuracy benign call, indicating that the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | -4.545 | Likely Benign | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.98 | Ambiguous | 0.2 | -0.16 | Likely Benign | 0.41 | Likely Benign | 0.13 | Likely Benign | 0.787 | Likely Pathogenic | -7.64 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.39 | Pathogenic | 0.14 | Tolerated | 0.3025 | 0.3705 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.1748A>G | D583G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 D583G missense change is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include premPS and ESM1b, whereas the majority of algorithms—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Default—classify it as pathogenic. Uncertain results come from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessment shows the SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenicity, and AlphaMissense‑Optimized remains inconclusive; Foldetta also reports no definitive stability change. Overall, the preponderance of evidence points to a pathogenic effect for D583G, and this conclusion does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | -6.765 | Likely Benign | 0.953 | Likely Pathogenic | Ambiguous | 1.10 | Ambiguous | 0.2 | 0.56 | Ambiguous | 0.83 | Ambiguous | 0.10 | Likely Benign | 0.850 | Likely Pathogenic | -6.77 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.43 | Pathogenic | 0.03 | Affected | 0.3072 | 0.4284 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.1748A>T | D583V 2D  3DClick to see structure in 3D Viewer AISynGAP1 D583V is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Rosetta, Foldetta, premPS, and SIFT, whereas pathogenic predictions are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX and ESM1b give uncertain results. High‑accuracy methods give a split view: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) predicts benign. Overall, the majority of tools support a pathogenic effect, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | -7.796 | In-Between | 0.973 | Likely Pathogenic | Likely Pathogenic | 1.20 | Ambiguous | 0.2 | -0.31 | Likely Benign | 0.45 | Likely Benign | 0.12 | Likely Benign | 0.839 | Likely Pathogenic | -8.63 | Deleterious | 0.999 | Probably Damaging | 0.999 | Probably Damaging | -1.40 | Pathogenic | 0.08 | Tolerated | 0.0778 | 0.4090 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1749C>A | D583E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 D583E missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, and SIFT. Those that predict a pathogenic effect are SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and ESM1b. High‑accuracy assessments show SGM‑Consensus as Likely Pathogenic, Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign, and AlphaMissense‑Optimized as Uncertain. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | -7.861 | In-Between | 0.851 | Likely Pathogenic | Ambiguous | 0.25 | Likely Benign | 0.2 | -0.36 | Likely Benign | -0.06 | Likely Benign | -0.20 | Likely Benign | 0.467 | Likely Benign | -3.52 | Deleterious | 0.960 | Probably Damaging | 0.969 | Probably Damaging | -1.18 | Pathogenic | 0.12 | Tolerated | 0.1200 | 0.4037 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.1749C>G | D583E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D583E missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, and SIFT. Those that predict a pathogenic effect are SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: AlphaMissense‑Optimized and ESM1b. High‑accuracy assessments show SGM‑Consensus as Likely Pathogenic, Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign, and AlphaMissense‑Optimized as Uncertain. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.038478 | Uncertain | 0.805 | 0.247 | 0.000 | -7.861 | In-Between | 0.851 | Likely Pathogenic | Ambiguous | 0.25 | Likely Benign | 0.2 | -0.36 | Likely Benign | -0.06 | Likely Benign | -0.20 | Likely Benign | 0.466 | Likely Benign | -3.52 | Deleterious | 0.960 | Probably Damaging | 0.969 | Probably Damaging | -1.18 | Pathogenic | 0.12 | Tolerated | 0.1200 | 0.4037 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.174G>A | M58I 2D  AIThe SynGAP1 missense variant M58I is not reported in ClinVar (ClinVar ID = None) but is present in gnomAD (ID = 6‑33423583‑G‑A). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains likely benign; Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, and this is not contradicted by ClinVar status. Thus, based on the available evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | 6-33423583-G-A | 1 | 6.20e-7 | -2.153 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.078 | Likely Benign | -0.55 | Neutral | 0.006 | Benign | 0.091 | Benign | 4.21 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1397 | 0.3848 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||||||||||||
| c.174G>C | M58I 2D AIThe SynGAP1 missense variant M58I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | -2.153 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.078 | Likely Benign | -0.55 | Neutral | 0.006 | Benign | 0.091 | Benign | 4.21 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1397 | 0.3848 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||
| c.174G>T | M58I 2D AIThe SynGAP1 missense variant M58I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome; Foldetta results are unavailable. Overall, the majority of predictions (seven benign vs. three pathogenic) suggest the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.188120 | Structured | 0.484415 | Uncertain | 0.515 | 0.665 | 0.000 | -2.153 | Likely Benign | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.078 | Likely Benign | -0.55 | Neutral | 0.006 | Benign | 0.091 | Benign | 4.21 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1397 | 0.3848 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||||||||||||
| c.1750A>C | I584L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I584L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and FATHMM, while premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the preponderance of evidence points to a benign impact. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.059222 | Structured | 0.046673 | Uncertain | 0.846 | 0.244 | 0.000 | -8.266 | Likely Pathogenic | 0.285 | Likely Benign | Likely Benign | -0.18 | Likely Benign | 0.1 | -0.30 | Likely Benign | -0.24 | Likely Benign | 0.84 | Ambiguous | 0.420 | Likely Benign | -1.74 | Neutral | 0.008 | Benign | 0.046 | Benign | -1.23 | Pathogenic | 0.18 | Tolerated | 0.0927 | 0.2817 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1750A>G | I584V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I584V is catalogued in gnomAD (ID 6‑33440802‑A‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are premPS, polyPhen‑2 HumDiv, and FATHMM. Two tools (FoldX and ESM1b) returned uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign (2 benign vs. 1 pathogenic vote), and Foldetta predicts benign stability. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar classification is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.059222 | Structured | 0.046673 | Uncertain | 0.846 | 0.244 | 0.000 | 6-33440802-A-G | 1 | 6.20e-7 | -7.562 | In-Between | 0.234 | Likely Benign | Likely Benign | 0.67 | Ambiguous | 0.1 | 0.29 | Likely Benign | 0.48 | Likely Benign | 1.16 | Destabilizing | 0.405 | Likely Benign | -0.95 | Neutral | 0.642 | Possibly Damaging | 0.349 | Benign | -1.18 | Pathogenic | 0.18 | Tolerated | 3.37 | 34 | 0.1007 | 0.2659 | 3 | 4 | -0.3 | -14.03 | |||||||||||||||||||||||||
| c.1750A>T | I584F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 I584F missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only Rosetta, whereas the remaining pathogenic‑predicating tools—REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently classify the variant as deleterious. High‑accuracy assessments further support a pathogenic interpretation: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic,” while AlphaMissense‑Optimized and Foldetta yield uncertain results and are therefore not used as evidence. No other folding‑stability methods provide definitive support. Overall, the preponderance of evidence points to a pathogenic effect, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.059222 | Structured | 0.046673 | Uncertain | 0.846 | 0.244 | 0.000 | -13.582 | Likely Pathogenic | 0.833 | Likely Pathogenic | Ambiguous | 3.20 | Destabilizing | 0.2 | 0.28 | Likely Benign | 1.74 | Ambiguous | 0.66 | Ambiguous | 0.618 | Likely Pathogenic | -3.47 | Deleterious | 0.980 | Probably Damaging | 0.808 | Possibly Damaging | -1.26 | Pathogenic | 0.04 | Affected | 0.0641 | 0.2150 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.1751T>A | I584N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I584N is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic. No tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a pathogenic impact. All available predictions are concordant and supportive. Based on these computational assessments, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.059222 | Structured | 0.046673 | Uncertain | 0.846 | 0.244 | 0.000 | -13.153 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 2.70 | Destabilizing | 0.1 | 2.13 | Destabilizing | 2.42 | Destabilizing | 2.08 | Destabilizing | 0.706 | Likely Pathogenic | -6.57 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.18 | Pathogenic | 0.01 | Affected | 0.0693 | 0.0470 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1751T>C | I584T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I584T is not reported in ClinVar and is absent from gnomAD. Consensus from most in silico predictors indicates a pathogenic effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify it as likely pathogenic. Only AlphaMissense‑Optimized predicts a benign outcome, while Rosetta and Foldetta are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. With the overwhelming majority of tools supporting pathogenicity and no ClinVar entry to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.059222 | Structured | 0.046673 | Uncertain | 0.846 | 0.244 | 0.000 | -10.413 | Likely Pathogenic | 0.765 | Likely Pathogenic | Likely Benign | 2.05 | Destabilizing | 0.1 | 1.70 | Ambiguous | 1.88 | Ambiguous | 1.66 | Destabilizing | 0.748 | Likely Pathogenic | -4.63 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | -1.11 | Pathogenic | 0.02 | Affected | 0.0911 | 0.0608 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.1751T>G | I584S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 I584S missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on pathogenicity include SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign effect. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as pathogenic. No contradictory evidence is present. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not conflict with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.059222 | Structured | 0.046673 | Uncertain | 0.846 | 0.244 | 0.000 | -13.379 | Likely Pathogenic | 0.945 | Likely Pathogenic | Ambiguous | 3.15 | Destabilizing | 0.1 | 2.53 | Destabilizing | 2.84 | Destabilizing | 1.70 | Destabilizing | 0.710 | Likely Pathogenic | -5.54 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | -1.18 | Pathogenic | 0.03 | Affected | 0.2391 | 0.0858 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1753G>A | A585T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A585T is reported in gnomAD (ID 6‑33440805‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, PROVEAN, and SIFT, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic effect for A585T. This conclusion is not contradicted by ClinVar, which contains no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.055884 | Uncertain | 0.880 | 0.244 | 0.000 | 6-33440805-G-A | 13 | 8.05e-6 | -10.063 | Likely Pathogenic | 0.876 | Likely Pathogenic | Ambiguous | 1.66 | Ambiguous | 0.2 | 1.97 | Ambiguous | 1.82 | Ambiguous | 0.23 | Likely Benign | 0.465 | Likely Benign | -1.73 | Neutral | 1.000 | Probably Damaging | 0.994 | Probably Damaging | -1.30 | Pathogenic | 0.26 | Tolerated | 3.37 | 35 | 0.1132 | 0.4212 | 0 | 1 | -2.5 | 30.03 | ||||||||||||||||||||||||
| c.1753G>C | A585P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A585P is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools cluster into two groups: the single benign prediction comes from SIFT, while all other evaluated algorithms—REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict pathogenicity. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels it “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic outcome. Based on the convergence of these predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.055884 | Uncertain | 0.880 | 0.244 | 0.000 | -10.999 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 5.44 | Destabilizing | 0.1 | 5.92 | Destabilizing | 5.68 | Destabilizing | 0.77 | Ambiguous | 0.549 | Likely Pathogenic | -3.09 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.33 | Pathogenic | 0.16 | Tolerated | 0.1884 | 0.2943 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1753G>T | A585S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A585S is not reported in ClinVar (no entry) and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. FoldX, Rosetta, and Foldetta are inconclusive. High‑accuracy methods give a benign consensus: AlphaMissense‑Optimized predicts benign, SGM‑Consensus predicts Likely Benign, while Foldetta remains uncertain. Overall, the majority of evidence supports a benign classification, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.055884 | Uncertain | 0.880 | 0.244 | 0.000 | -6.332 | Likely Benign | 0.246 | Likely Benign | Likely Benign | 0.91 | Ambiguous | 0.2 | 1.44 | Ambiguous | 1.18 | Ambiguous | 0.02 | Likely Benign | 0.326 | Likely Benign | 0.39 | Neutral | 0.993 | Probably Damaging | 0.996 | Probably Damaging | -1.27 | Pathogenic | 0.98 | Tolerated | 0.2121 | 0.3388 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1754C>A | A585E 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A585E is not reported in ClinVar (status: None) and is absent from gnomAD. Prediction tools cluster into two groups: the single benign call comes from SIFT, while all other evaluated algorithms—including REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—label the change as pathogenic or likely pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports pathogenic. Thus, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.055884 | Uncertain | 0.880 | 0.244 | 0.000 | -14.715 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 5.38 | Destabilizing | 0.7 | 3.70 | Destabilizing | 4.54 | Destabilizing | 1.42 | Destabilizing | 0.539 | Likely Pathogenic | -2.59 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.30 | Pathogenic | 0.28 | Tolerated | 0.1160 | 0.1212 | 0 | -1 | -5.3 | 58.04 | |||||||||||||||||||||||||||||
| c.1754C>G | A585G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A585G is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, whereas the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta reports an uncertain stability change. No evidence from these high‑confidence tools supports pathogenicity. Overall, the balance of evidence favors a benign effect for A585G, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.060549 | Structured | 0.055884 | Uncertain | 0.880 | 0.244 | 0.000 | -3.879 | Likely Benign | 0.629 | Likely Pathogenic | Likely Benign | 1.62 | Ambiguous | 0.0 | 1.94 | Ambiguous | 1.78 | Ambiguous | 0.59 | Ambiguous | 0.384 | Likely Benign | -1.16 | Neutral | 0.999 | Probably Damaging | 0.995 | Probably Damaging | -1.33 | Pathogenic | 0.24 | Tolerated | 0.1724 | 0.2299 | 1 | 0 | -2.2 | -14.03 | ||||||||||||||||||||||||||||||
| c.1754C>T | A585V 2D AIThe SynGAP1 missense variant A585V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and SIFT, whereas a majority of tools predict a pathogenic impact: SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools with uncertain or inconclusive results (FoldX, Rosetta, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.055884 | Uncertain | 0.880 | 0.244 | 0.000 | -10.843 | Likely Pathogenic | 0.970 | Likely Pathogenic | Likely Pathogenic | 0.95 | Ambiguous | 1.4 | 1.12 | Ambiguous | 1.04 | Ambiguous | 0.51 | Ambiguous | 0.420 | Likely Benign | -3.35 | Deleterious | 0.999 | Probably Damaging | 0.988 | Probably Damaging | -1.27 | Pathogenic | 0.10 | Tolerated | 0.1004 | 0.3496 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.1756G>A | D586N 2D AIThe SynGAP1 D586N missense variant is not reported in ClinVar and has no gnomAD entry. Consensus prediction tools that agree on benign impact include FoldX, Rosetta, Foldetta, premPS, SIFT, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, more tools predict pathogenicity than benign, and the high‑accuracy consensus leans toward pathogenic. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -9.497 | Likely Pathogenic | 0.767 | Likely Pathogenic | Likely Benign | 0.09 | Likely Benign | 0.8 | 0.24 | Likely Benign | 0.17 | Likely Benign | 0.19 | Likely Benign | 0.523 | Likely Pathogenic | -2.52 | Deleterious | 0.992 | Probably Damaging | 0.995 | Probably Damaging | -1.25 | Pathogenic | 0.23 | Tolerated | 0.1124 | 0.5253 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1756G>C | D586H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D586H missense variant is not reported in ClinVar and has no gnomAD entry. Prediction tools cluster into two groups: benign predictions come from premPS and SIFT, while the remaining nine tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all indicate pathogenicity. The high‑accuracy methods reinforce this trend: AlphaMissense‑Optimized scores the variant as pathogenic, and the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—also predicts pathogenicity. Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result. Overall, the preponderance of evidence points to a pathogenic effect. This conclusion is consistent with the absence of ClinVar annotation, as there is no conflicting status to contradict the prediction. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -10.104 | Likely Pathogenic | 0.974 | Likely Pathogenic | Likely Pathogenic | 1.00 | Ambiguous | 0.3 | 0.89 | Ambiguous | 0.95 | Ambiguous | 0.26 | Likely Benign | 0.672 | Likely Pathogenic | -3.44 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.23 | Pathogenic | 0.17 | Tolerated | 0.1307 | 0.5558 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.1756G>T | D586Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D586Y missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that indicate a benign effect include FoldX, SIFT, and premPS, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic outcome. Rosetta and Foldetta, which assess protein‑folding stability, return uncertain results and are therefore not considered evidence for either direction. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also as pathogenic, and Foldetta as uncertain. Overall, the preponderance of pathogenic predictions (13 vs. 3 benign) suggests that D586Y is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -12.916 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 0.37 | Likely Benign | 0.4 | 1.09 | Ambiguous | 0.73 | Ambiguous | -0.49 | Likely Benign | 0.712 | Likely Pathogenic | -5.38 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.16 | Pathogenic | 0.41 | Tolerated | 0.0623 | 0.5634 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1757A>C | D586A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant D586A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include FoldX, SIFT, and premPS, whereas a majority of predictors (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labels it Likely Pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains inconclusive. No evidence from the uncertain tools (Rosetta, Foldetta) alters this consensus. Consequently, the variant is most likely pathogenic, and this prediction does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -9.955 | Likely Pathogenic | 0.960 | Likely Pathogenic | Likely Pathogenic | 0.41 | Likely Benign | 0.2 | 0.88 | Ambiguous | 0.65 | Ambiguous | 0.25 | Likely Benign | 0.693 | Likely Pathogenic | -4.58 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.22 | Pathogenic | 0.31 | Tolerated | 0.3397 | 0.4673 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.1757A>G | D586G 2D 3DClick to see structure in 3D Viewer AISynGAP1 D586G is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include premPS and SIFT, whereas the majority of tools predict it to be pathogenic: SGM‑Consensus, REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a unanimous vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -8.497 | Likely Pathogenic | 0.942 | Likely Pathogenic | Ambiguous | 1.37 | Ambiguous | 0.3 | 2.49 | Destabilizing | 1.93 | Ambiguous | 0.34 | Likely Benign | 0.833 | Likely Pathogenic | -4.67 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.26 | Pathogenic | 0.17 | Tolerated | 0.3334 | 0.5052 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.1757A>T | D586V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D586V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, premPS, and SIFT. Tools that predict pathogenicity include SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of predictions (nine pathogenic vs. five benign) indicate a pathogenic effect. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -12.409 | Likely Pathogenic | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.40 | Likely Benign | 0.2 | 0.03 | Likely Benign | 0.22 | Likely Benign | 0.18 | Likely Benign | 0.801 | Likely Pathogenic | -5.58 | Deleterious | 0.998 | Probably Damaging | 0.999 | Probably Damaging | -1.23 | Pathogenic | 0.24 | Tolerated | 0.0826 | 0.5458 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1758C>A | D586E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D586E missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Optimized, and Foldetta. Those that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default; Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of evidence points to a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -3.233 | Likely Benign | 0.683 | Likely Pathogenic | Likely Benign | -0.42 | Likely Benign | 0.1 | 0.88 | Ambiguous | 0.23 | Likely Benign | 0.38 | Likely Benign | 0.367 | Likely Benign | -0.12 | Neutral | 0.929 | Possibly Damaging | 0.969 | Probably Damaging | -1.20 | Pathogenic | 1.00 | Tolerated | 0.1304 | 0.5126 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||
| c.1758C>G | D586E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D586E missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Default; Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.060549 | Structured | 0.066018 | Uncertain | 0.866 | 0.241 | 0.000 | -3.233 | Likely Benign | 0.683 | Likely Pathogenic | Likely Benign | -0.42 | Likely Benign | 0.1 | 0.88 | Ambiguous | 0.23 | Likely Benign | 0.38 | Likely Benign | 0.367 | Likely Benign | -0.12 | Neutral | 0.929 | Possibly Damaging | 0.969 | Probably Damaging | -1.20 | Pathogenic | 1.00 | Tolerated | 0.1304 | 0.5126 | 3 | 2 | 0.0 | 14.03 | ||||||||||||||||||||||||||||||
| c.1759A>G | R587G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R587G is not reported in ClinVar and is present in gnomAD (ID 6‑33440811‑A‑G). Prediction tools that agree on a benign effect include SIFT and AlphaMissense‑Optimized, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) classify the change as pathogenic. High‑accuracy assessments further support a pathogenic interpretation: AlphaMissense‑Optimized predicts benign, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—returns pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the preponderance of evidence from multiple independent predictors indicates that R587G is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | 6-33440811-A-G | 2 | 1.24e-6 | -13.780 | Likely Pathogenic | 0.780 | Likely Pathogenic | Likely Benign | 1.55 | Ambiguous | 0.2 | 2.43 | Destabilizing | 1.99 | Ambiguous | 1.55 | Destabilizing | 0.578 | Likely Pathogenic | -6.07 | Deleterious | 1.000 | Probably Damaging | 0.972 | Probably Damaging | -1.28 | Pathogenic | 0.07 | Tolerated | 3.37 | 35 | 0.3183 | 0.3401 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1759A>T | R587W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R587W is not reported in ClinVar and is present in gnomAD (ID 6‑33440811‑A‑T). Functional prediction tools show a split assessment: benign predictions come from FoldX, Rosetta, and Foldetta, whereas pathogenic predictions are reported by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: premPS and AlphaMissense‑Optimized. High‑accuracy consensus methods further clarify the picture: the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect, whereas Foldetta, a protein‑folding stability predictor combining FoldX‑MD and Rosetta outputs, classifies the variant as benign. AlphaMissense‑Optimized remains inconclusive. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not conflict with the ClinVar status, which currently lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | 6-33440811-A-T | 1 | 6.20e-7 | -15.383 | Likely Pathogenic | 0.879 | Likely Pathogenic | Ambiguous | -0.01 | Likely Benign | 0.1 | -0.44 | Likely Benign | -0.23 | Likely Benign | 0.76 | Ambiguous | 0.692 | Likely Pathogenic | -7.17 | Deleterious | 1.000 | Probably Damaging | 0.985 | Probably Damaging | -1.33 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.1326 | 0.3992 | -3 | 2 | 3.6 | 30.03 | ||||||||||||||||||||||||
| c.175C>A | L59M 2D AIThe SynGAP1 missense variant L59M is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evaluated predictors lean toward a benign interpretation. Consequently, the variant is most likely benign based on current computational evidence, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.212910 | Structured | 0.484882 | Uncertain | 0.510 | 0.668 | 0.000 | -3.394 | Likely Benign | 0.618 | Likely Pathogenic | Likely Benign | 0.088 | Likely Benign | -0.65 | Neutral | 0.824 | Possibly Damaging | 0.910 | Probably Damaging | 3.30 | Benign | 0.00 | Affected | 0.0772 | 0.3311 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.175C>G | L59V 2D AIThe SynGAP1 missense variant L59V is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for this variant. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.212910 | Structured | 0.484882 | Uncertain | 0.510 | 0.668 | 0.000 | -4.465 | Likely Benign | 0.588 | Likely Pathogenic | Likely Benign | 0.049 | Likely Benign | -1.15 | Neutral | 0.458 | Possibly Damaging | 0.745 | Possibly Damaging | 3.36 | Benign | 0.00 | Affected | 0.1508 | 0.3217 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1760G>A | R587K 2D  3DClick to see structure in 3D Viewer AISynGAP1 R587K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions from SIFT and AlphaMissense‑Optimized, and pathogenic predictions from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—indicates likely pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. Overall, the majority of evidence points toward a pathogenic effect, which is consistent with the SGM‑Consensus prediction but contradicts the benign calls from SIFT and AlphaMissense‑Optimized. Thus, the variant is most likely pathogenic, and this conclusion aligns with the lack of ClinVar annotation rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | -10.220 | Likely Pathogenic | 0.433 | Ambiguous | Likely Benign | 0.63 | Ambiguous | 0.1 | 1.14 | Ambiguous | 0.89 | Ambiguous | 0.88 | Ambiguous | 0.539 | Likely Pathogenic | -2.55 | Deleterious | 0.967 | Probably Damaging | 0.955 | Probably Damaging | -1.16 | Pathogenic | 0.07 | Tolerated | 0.5313 | 0.3897 | Weaken | 3 | 2 | 0.6 | -28.01 | ||||||||||||||||||||||||||||
| c.1760G>T | R587M 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant R587M is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign calls from FoldX, Rosetta, and Foldetta; pathogenic calls from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default; and two uncertain calls from premPS and AlphaMissense‑Optimized. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts pathogenicity; and Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the majority of evidence points to a pathogenic impact for R587M, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | -15.106 | Likely Pathogenic | 0.931 | Likely Pathogenic | Ambiguous | 0.23 | Likely Benign | 0.0 | -0.10 | Likely Benign | 0.07 | Likely Benign | 0.84 | Ambiguous | 0.787 | Likely Pathogenic | -5.24 | Deleterious | 1.000 | Probably Damaging | 0.979 | Probably Damaging | -1.30 | Pathogenic | 0.02 | Affected | 0.1734 | 0.3910 | 0 | -1 | 6.4 | -24.99 | |||||||||||||||||||||||||||||
| c.1761G>C | R587S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R587S is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: the single benign prediction comes from SIFT, while the remaining tools—REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus—indicate pathogenicity. High‑accuracy assessments further support a deleterious effect: the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Pathogenic”; AlphaMissense‑Optimized is “Uncertain”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is also “Uncertain.” Taken together, the preponderance of evidence points to a pathogenic impact for R587S. This conclusion is not contradicted by ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | -12.264 | Likely Pathogenic | 0.830 | Likely Pathogenic | Ambiguous | 0.84 | Ambiguous | 0.1 | 1.79 | Ambiguous | 1.32 | Ambiguous | 1.17 | Destabilizing | 0.508 | Likely Pathogenic | -4.84 | Deleterious | 0.990 | Probably Damaging | 0.779 | Possibly Damaging | -1.20 | Pathogenic | 0.09 | Tolerated | 3.37 | 35 | 0.2852 | 0.4165 | -1 | 0 | 3.7 | -69.11 | |||||||||||||||||||||||||||
| c.1761G>T | R587S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 R587S missense variant is catalogued in gnomAD (ID 6‑33440813‑G‑T) but has no ClinVar entry. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default, while only SIFT predicts a benign outcome. Uncertain results are reported by FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized remains uncertain, but the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as pathogenic, and Foldetta likewise yields an uncertain stability change. Overall, the preponderance of evidence indicates that R587S is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.054297 | Structured | 0.077330 | Uncertain | 0.862 | 0.216 | 0.000 | 6-33440813-G-T | 4 | 2.48e-6 | -12.264 | Likely Pathogenic | 0.830 | Likely Pathogenic | Ambiguous | 0.84 | Ambiguous | 0.1 | 1.79 | Ambiguous | 1.32 | Ambiguous | 1.17 | Destabilizing | 0.508 | Likely Pathogenic | -4.84 | Deleterious | 0.990 | Probably Damaging | 0.779 | Possibly Damaging | -1.20 | Pathogenic | 0.09 | Tolerated | 3.37 | 35 | 0.2852 | 0.4165 | -1 | 0 | 3.7 | -69.11 | ||||||||||||||||||||||||
| c.1762C>A | L588I 2D  AISynGAP1 missense variant L588I has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify the variant as benign include PROVEAN. Those that predict pathogenicity are SGM‑Consensus, REVEL, FoldX, Foldetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results come from Rosetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of evidence supports a pathogenic effect, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.082229 | Uncertain | 0.887 | 0.214 | 0.000 | -12.454 | Likely Pathogenic | 0.874 | Likely Pathogenic | Ambiguous | 2.54 | Destabilizing | 1.2 | 1.80 | Ambiguous | 2.17 | Destabilizing | 0.88 | Ambiguous | 0.607 | Likely Pathogenic | -1.99 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | -1.27 | Pathogenic | 0.04 | Affected | 0.0949 | 0.2433 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.1762C>G | L588V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L588V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: SIFT classifies it as benign, whereas the remaining 13 tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) predict pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Taken together, the overwhelming majority of evidence points to a pathogenic effect for L588V. This conclusion is consistent with the absence of a ClinVar entry, so there is no contradiction with existing clinical annotations. Thus, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.082229 | Uncertain | 0.887 | 0.214 | 0.000 | -10.374 | Likely Pathogenic | 0.897 | Likely Pathogenic | Ambiguous | 3.61 | Destabilizing | 0.4 | 2.81 | Destabilizing | 3.21 | Destabilizing | 1.24 | Destabilizing | 0.533 | Likely Pathogenic | -2.99 | Deleterious | 0.998 | Probably Damaging | 0.992 | Probably Damaging | -1.28 | Pathogenic | 0.08 | Tolerated | 0.1422 | 0.2228 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1762C>T | L588F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L588F is reported in gnomAD (variant ID 6‑33440814‑C‑T) but has no ClinVar entry. Across the available in‑silico predictors, every tool examined classifies the substitution as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return pathogenic or likely pathogenic scores. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Because every available prediction converges on a deleterious effect and there is no ClinVar annotation to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.082229 | Uncertain | 0.887 | 0.214 | 0.000 | 6-33440814-C-T | -14.050 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 2.90 | Destabilizing | 0.2 | 2.55 | Destabilizing | 2.73 | Destabilizing | 1.12 | Destabilizing | 0.736 | Likely Pathogenic | -3.98 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | -1.38 | Pathogenic | 0.04 | Affected | 3.38 | 34 | 0.0816 | 0.2007 | 0 | 2 | -1.0 | 34.02 | ||||||||||||||||||||||||||
| c.1763T>C | L588P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L588P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. All available in silico predictors classify the change as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict the ClinVar status, which remains uncertain. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.082229 | Uncertain | 0.887 | 0.214 | 0.000 | Uncertain | 2 | -14.771 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 5.61 | Destabilizing | 0.5 | 12.91 | Destabilizing | 9.26 | Destabilizing | 2.33 | Destabilizing | 0.932 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.42 | Pathogenic | 0.00 | Affected | 3.38 | 34 | 0.3533 | 0.0992 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||
| c.1763T>G | L588R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L588R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity uniformly classify the variant as pathogenic: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy methods corroborate this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. All available evidence points to a deleterious impact. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.082229 | Uncertain | 0.887 | 0.214 | 0.000 | -15.602 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 6.96 | Destabilizing | 0.4 | 6.14 | Destabilizing | 6.55 | Destabilizing | 2.07 | Destabilizing | 0.942 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.42 | Pathogenic | 0.00 | Affected | 0.1209 | 0.0530 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1765A>C | I589L 2D AIThe SynGAP1 missense variant I589L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only PROVEAN, whereas the remaining tools (REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus) all predict a pathogenic impact. High‑accuracy assessments further support a deleterious outcome: the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic”; AlphaMissense‑Optimized and Foldetta are inconclusive and therefore not considered evidence. Taken together, the preponderance of evidence points to a pathogenic effect for I589L. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.018415 | Structured | 0.084536 | Uncertain | 0.927 | 0.214 | 0.000 | -11.337 | Likely Pathogenic | 0.850 | Likely Pathogenic | Ambiguous | 0.95 | Ambiguous | 1.1 | 1.44 | Ambiguous | 1.20 | Ambiguous | 0.95 | Ambiguous | 0.728 | Likely Pathogenic | -1.99 | Neutral | 0.955 | Possibly Damaging | 0.985 | Probably Damaging | -1.76 | Pathogenic | 0.02 | Affected | 0.1243 | 0.3430 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.1765A>G | I589V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I589V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, polyPhen‑2 (HumDiv and HumVar), and FATHMM. The remaining tools—FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Default—return uncertain or inconclusive results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta is uncertain. Taken together, the majority of evidence, including the high‑accuracy predictions, points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.018415 | Structured | 0.084536 | Uncertain | 0.927 | 0.214 | 0.000 | -6.966 | Likely Benign | 0.464 | Ambiguous | Likely Benign | 0.98 | Ambiguous | 0.0 | 0.78 | Ambiguous | 0.88 | Ambiguous | 0.96 | Ambiguous | 0.535 | Likely Pathogenic | -1.00 | Neutral | 0.969 | Probably Damaging | 0.960 | Probably Damaging | -1.50 | Pathogenic | 0.31 | Tolerated | 0.1356 | 0.3568 | 4 | 3 | -0.3 | -14.03 | ||||||||||||||||||||||||||||||
| c.1765A>T | I589F 2D AIThe SynGAP1 missense variant I589F is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic. No tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a pathogenic impact. All available predictions are concordant and supportive. Therefore, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.018415 | Structured | 0.084536 | Uncertain | 0.927 | 0.214 | 0.000 | -15.300 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 7.38 | Destabilizing | 3.4 | 2.05 | Destabilizing | 4.72 | Destabilizing | 1.04 | Destabilizing | 0.905 | Likely Pathogenic | -3.98 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | -1.98 | Pathogenic | 0.00 | Affected | 0.0888 | 0.2574 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.1766T>A | I589N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I589N is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. All available predictions are consistent and conclusive. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.018415 | Structured | 0.084536 | Uncertain | 0.927 | 0.214 | 0.000 | -15.539 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.72 | Destabilizing | 0.2 | 3.13 | Destabilizing | 3.43 | Destabilizing | 2.69 | Destabilizing | 0.930 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.99 | Pathogenic | 0.00 | Affected | 0.0968 | 0.0742 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1766T>C | I589T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I589T is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. All available predictions are consistent and conclusive. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.018415 | Structured | 0.084536 | Uncertain | 0.927 | 0.214 | 0.000 | -12.128 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 2.60 | Destabilizing | 0.0 | 2.54 | Destabilizing | 2.57 | Destabilizing | 2.07 | Destabilizing | 0.935 | Likely Pathogenic | -4.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.97 | Pathogenic | 0.02 | Affected | 0.1153 | 0.1231 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.1766T>G | I589S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I589S is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate a pathogenic or likely pathogenic outcome. No tool in the dataset predicts a benign effect. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also labels it pathogenic. Based on the uniform predictions, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.018415 | Structured | 0.084536 | Uncertain | 0.927 | 0.214 | 0.000 | -15.223 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.62 | Destabilizing | 0.1 | 4.12 | Destabilizing | 3.87 | Destabilizing | 2.21 | Destabilizing | 0.954 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.99 | Pathogenic | 0.00 | Affected | 0.2786 | 0.1130 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1768A>C | S590R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S590R is not reported in ClinVar (status: None) and has no entries in gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus (likely pathogenic), REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No predictions are inconclusive or missing. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.022667 | Structured | 0.088943 | Uncertain | 0.918 | 0.199 | 0.000 | -18.228 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.69 | Ambiguous | 0.6 | 3.21 | Destabilizing | 2.45 | Destabilizing | 1.28 | Destabilizing | 0.640 | Likely Pathogenic | -4.88 | Deleterious | 1.000 | Probably Damaging | 0.979 | Probably Damaging | 3.11 | Benign | 0.01 | Affected | 0.0983 | 0.3388 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1768A>T | S590C 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant S590C has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show a split assessment: benign predictions come from FoldX, Rosetta, Foldetta, SIFT, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and the SGM‑Consensus. The high‑accuracy subset indicates that AlphaMissense‑Optimized predicts a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta both support a pathogenic or neutral outcome, respectively. Overall, the majority of tools lean toward a pathogenic interpretation, and this aligns with the SGM‑Consensus designation. Because there is no ClinVar classification, the predictions do not contradict existing clinical data. Thus, based on the available computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.022667 | Structured | 0.088943 | Uncertain | 0.918 | 0.199 | 0.000 | -10.823 | Likely Pathogenic | 0.698 | Likely Pathogenic | Likely Benign | -0.09 | Likely Benign | 0.1 | 0.35 | Likely Benign | 0.13 | Likely Benign | 0.56 | Ambiguous | 0.631 | Likely Pathogenic | -4.48 | Deleterious | 1.000 | Probably Damaging | 0.968 | Probably Damaging | 3.08 | Benign | 0.07 | Tolerated | 0.1094 | 0.5332 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.1769G>A | S590N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S590N is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, while the majority of other in silico predictors (PolyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, PROVEAN, premPS, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus) indicate a pathogenic or likely pathogenic impact. FoldX and Foldetta, which assess protein‑folding stability, return uncertain results and are therefore not considered evidence for either outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of pathogenic predictions strongly suggests that S590N is most likely pathogenic, a conclusion that is consistent with the absence of ClinVar annotation and gnomAD data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.022667 | Structured | 0.088943 | Uncertain | 0.918 | 0.199 | 0.000 | -15.149 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 1.27 | Ambiguous | 0.1 | 2.10 | Destabilizing | 1.69 | Ambiguous | 1.48 | Destabilizing | 0.411 | Likely Benign | -2.96 | Deleterious | 0.921 | Possibly Damaging | 0.598 | Possibly Damaging | 3.14 | Benign | 0.01 | Affected | 0.1254 | 0.4202 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||
| c.1769G>C | S590T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S590T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and ESM1b. Two tools (premPS and AlphaMissense‑Default) return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.022667 | Structured | 0.088943 | Uncertain | 0.918 | 0.199 | 0.000 | -12.472 | Likely Pathogenic | 0.458 | Ambiguous | Likely Benign | 0.23 | Likely Benign | 0.1 | 0.42 | Likely Benign | 0.33 | Likely Benign | 0.64 | Ambiguous | 0.459 | Likely Benign | -2.89 | Deleterious | 0.183 | Benign | 0.050 | Benign | 3.15 | Benign | 0.08 | Tolerated | 0.1420 | 0.5723 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||
| c.1769G>T | S590I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S590I is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include FoldX, premPS, and FATHMM, whereas the majority of tools predict it to be pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools give uncertain results: Rosetta and Foldetta. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence from multiple independent predictors indicates that S590I is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.022667 | Structured | 0.088943 | Uncertain | 0.918 | 0.199 | 0.000 | -17.956 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | -0.04 | Likely Benign | 0.5 | 1.15 | Ambiguous | 0.56 | Ambiguous | 0.31 | Likely Benign | 0.686 | Likely Pathogenic | -5.78 | Deleterious | 0.998 | Probably Damaging | 0.948 | Probably Damaging | 3.05 | Benign | 0.02 | Affected | 0.0975 | 0.5025 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.176T>A | L59Q 2D AIThe SynGAP1 missense variant L59Q is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools are mixed: benign calls come from SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, and FATHMM, whereas pathogenic calls are made by polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further highlight this discordance: AlphaMissense‑Optimized predicts a pathogenic effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. No Foldetta stability analysis is available for this residue. Overall, the majority of high‑confidence tools lean toward a pathogenic interpretation, but the presence of several strong benign predictions and the lack of ClinVar evidence suggest uncertainty. The variant is most likely pathogenic based on the prevailing predictions, and this does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.212910 | Structured | 0.484882 | Uncertain | 0.510 | 0.668 | 0.000 | -3.841 | Likely Benign | 0.977 | Likely Pathogenic | Likely Pathogenic | 0.287 | Likely Benign | -2.21 | Neutral | 0.943 | Possibly Damaging | 0.944 | Probably Damaging | 3.26 | Benign | 0.00 | Affected | 0.1103 | 0.0758 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.176T>C | L59P 2D AIThe SynGAP1 missense variant L59P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.212910 | Structured | 0.484882 | Uncertain | 0.510 | 0.668 | 0.000 | -7.076 | In-Between | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.362 | Likely Benign | -2.74 | Deleterious | 0.943 | Possibly Damaging | 0.944 | Probably Damaging | 3.25 | Benign | 0.00 | Affected | 0.3787 | 0.1382 | -3 | -3 | -5.4 | -16.04 | ||||||||||||||||||||||||||||||||||||||||
| c.176T>G | L59R 2D AISynGAP1 missense variant L59R is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome; Foldetta results are unavailable. The predictions are therefore split, with an equal number of benign and pathogenic calls, and the high‑accuracy tools provide contradictory signals. Consequently, the variant is most likely pathogenic based on the presence of multiple pathogenic predictions and the high‑accuracy AlphaMissense‑Optimized result, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.212910 | Structured | 0.484882 | Uncertain | 0.510 | 0.668 | 0.000 | -4.037 | Likely Benign | 0.960 | Likely Pathogenic | Likely Pathogenic | 0.266 | Likely Benign | -2.09 | Neutral | 0.943 | Possibly Damaging | 0.944 | Probably Damaging | 3.26 | Benign | 0.00 | Affected | 0.1298 | 0.0600 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.1770C>A | S590R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S590R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools (AlphaMissense‑Default, AlphaMissense‑Optimized, SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, Rosetta, and Foldetta) all predict a pathogenic impact; FoldX is uncertain and therefore not counted. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.022667 | Structured | 0.088943 | Uncertain | 0.918 | 0.199 | 0.000 | -18.228 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.69 | Ambiguous | 0.6 | 3.21 | Destabilizing | 2.45 | Destabilizing | 1.28 | Destabilizing | 0.391 | Likely Benign | -4.88 | Deleterious | 1.000 | Probably Damaging | 0.979 | Probably Damaging | 3.11 | Benign | 0.01 | Affected | 0.0983 | 0.3388 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1770C>G | S590R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S590R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining tools (AlphaMissense‑Default, AlphaMissense‑Optimized, SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, Rosetta, and Foldetta) all predict a pathogenic impact; FoldX is uncertain and therefore not counted. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.022667 | Structured | 0.088943 | Uncertain | 0.918 | 0.199 | 0.000 | -18.228 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.69 | Ambiguous | 0.6 | 3.21 | Destabilizing | 2.45 | Destabilizing | 1.28 | Destabilizing | 0.389 | Likely Benign | -4.88 | Deleterious | 1.000 | Probably Damaging | 0.979 | Probably Damaging | 3.11 | Benign | 0.01 | Affected | 0.0983 | 0.3388 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1771G>T | A591S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A591S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign effect, and the Foldetta stability analysis is inconclusive (unavailable). Consequently, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.018787 | Structured | 0.093848 | Uncertain | 0.882 | 0.185 | 0.000 | -7.535 | In-Between | 0.126 | Likely Benign | Likely Benign | 0.58 | Ambiguous | 0.1 | 1.21 | Ambiguous | 0.90 | Ambiguous | 0.49 | Likely Benign | 0.083 | Likely Benign | -2.11 | Neutral | 0.034 | Benign | 0.082 | Benign | 3.52 | Benign | 0.19 | Tolerated | 0.2405 | 0.3304 | 1 | 1 | -2.6 | 16.00 | |||||||||||||||||||||||||||||
| c.1772C>A | A591D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A591D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the remaining evaluated algorithms (AlphaMissense‑Default, ESM1b, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, premPS, FoldX‑MD, Rosetta, Foldetta, and the SGM Consensus) uniformly predict a pathogenic or likely pathogenic outcome; FoldX‑MD is uncertain but does not counter the overall trend. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the preponderance of pathogenic predictions and the absence of benign evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.018787 | Structured | 0.093848 | Uncertain | 0.882 | 0.185 | 0.000 | -14.747 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.55 | Ambiguous | 0.2 | 2.77 | Destabilizing | 2.16 | Destabilizing | 1.40 | Destabilizing | 0.414 | Likely Benign | -5.02 | Deleterious | 0.919 | Possibly Damaging | 0.495 | Possibly Damaging | 3.42 | Benign | 0.00 | Affected | 0.1550 | 0.1741 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.1772C>G | A591G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A591G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only PROVEAN predicts a pathogenic outcome. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact for A591G, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.018787 | Structured | 0.093848 | Uncertain | 0.882 | 0.185 | 0.000 | -6.596 | Likely Benign | 0.233 | Likely Benign | Likely Benign | 1.22 | Ambiguous | 0.2 | 1.74 | Ambiguous | 1.48 | Ambiguous | 0.83 | Ambiguous | 0.142 | Likely Benign | -2.77 | Deleterious | 0.007 | Benign | 0.009 | Benign | 3.60 | Benign | 0.16 | Tolerated | 0.2117 | 0.2228 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1772C>T | A591V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A591V missense variant is not reported in ClinVar and is present in gnomAD (ID 6‑33440824‑C‑T). Functional prediction tools show discordant results: benign calls come from REVEL, polyPhen‑2 HumVar, and FATHMM, while pathogenic calls are made by PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain, and the SGM Consensus remains Likely Pathogenic. Overall, the majority of evidence points toward a pathogenic effect, and this assessment is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.018787 | Structured | 0.093848 | Uncertain | 0.882 | 0.185 | 0.000 | 6-33440824-C-T | 2 | 1.24e-6 | -12.282 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 1.35 | Ambiguous | 0.4 | 0.98 | Ambiguous | 1.17 | Ambiguous | 0.86 | Ambiguous | 0.321 | Likely Benign | -3.79 | Deleterious | 0.970 | Probably Damaging | 0.373 | Benign | 3.35 | Benign | 0.02 | Affected | 3.37 | 35 | 0.1128 | 0.4228 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||
| c.1774T>A | S592T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S592T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include Foldetta, SIFT, and AlphaMissense‑Optimized, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) classify the change as pathogenic. High‑accuracy assessments further support this dichotomy: AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus—derived from a unanimous pathogenic vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—indicates pathogenicity; Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, also reports a benign effect. Predictions from FoldX, Rosetta, and premPS are inconclusive and are treated as unavailable. Overall, the preponderance of evidence from multiple independent predictors points to a pathogenic effect for S592T, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.012270 | Structured | 0.100070 | Uncertain | 0.913 | 0.182 | 0.000 | -11.670 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | 0.86 | Ambiguous | 0.1 | -0.88 | Ambiguous | -0.01 | Likely Benign | 0.61 | Ambiguous | 0.819 | Likely Pathogenic | -2.79 | Deleterious | 0.933 | Possibly Damaging | 0.933 | Probably Damaging | -1.26 | Pathogenic | 0.17 | Tolerated | 0.1531 | 0.4684 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1774T>C | S592P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S592P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include only SIFT, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No predictions are inconclusive or missing. Based on the overwhelming agreement among pathogenic predictors and the absence of benign evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.012270 | Structured | 0.100070 | Uncertain | 0.913 | 0.182 | 0.000 | -14.621 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 4.26 | Destabilizing | 0.1 | 2.36 | Destabilizing | 3.31 | Destabilizing | 1.24 | Destabilizing | 0.909 | Likely Pathogenic | -4.77 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | -1.29 | Pathogenic | 0.11 | Tolerated | 0.2412 | 0.4259 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.1774T>G | S592A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S592A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX, SIFT, and AlphaMissense‑Optimized, whereas a majority (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic outcome. Tools with uncertain or inconclusive results are Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic classification. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.012270 | Structured | 0.100070 | Uncertain | 0.913 | 0.182 | 0.000 | -10.902 | Likely Pathogenic | 0.572 | Likely Pathogenic | Likely Benign | -0.48 | Likely Benign | 0.1 | -0.93 | Ambiguous | -0.71 | Ambiguous | 0.79 | Ambiguous | 0.661 | Likely Pathogenic | -2.72 | Deleterious | 0.980 | Probably Damaging | 0.994 | Probably Damaging | -1.25 | Pathogenic | 0.44 | Tolerated | 0.4878 | 0.3318 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.1775C>T | S592L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S592L is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools show a predominance of pathogenic calls: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict deleterious effects, whereas benign predictions are limited to Foldetta, premPS, and SIFT. Uncertain results are reported only by FoldX and Rosetta. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Pathogenic. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenicity, SGM Consensus confirms a pathogenic status, and Foldetta, a protein‑folding stability method, predicts a benign effect. Overall, the preponderance of evidence indicates that S592L is most likely pathogenic, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.012270 | Structured | 0.100070 | Uncertain | 0.913 | 0.182 | 0.000 | -12.948 | Likely Pathogenic | 0.960 | Likely Pathogenic | Likely Pathogenic | 0.94 | Ambiguous | 0.3 | -1.89 | Ambiguous | -0.48 | Likely Benign | 0.50 | Likely Benign | 0.711 | Likely Pathogenic | -5.57 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | -1.12 | Pathogenic | 0.25 | Tolerated | 0.1089 | 0.4367 | -3 | -2 | 4.6 | 26.08 | |||||||||||||||||||||||||||||
| c.1777C>A | L593I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L593I missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are FoldX, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Predictions that are uncertain are Rosetta, Foldetta, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign effect. The variant’s predicted benign status does not contradict its ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.009728 | Structured | 0.110534 | Uncertain | 0.941 | 0.151 | 0.000 | -8.617 | Likely Pathogenic | 0.448 | Ambiguous | Likely Benign | 2.16 | Destabilizing | 0.3 | 0.76 | Ambiguous | 1.46 | Ambiguous | 0.39 | Likely Benign | 0.169 | Likely Benign | -1.59 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 3.14 | Benign | 0.17 | Tolerated | 0.1071 | 0.3582 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1777C>G | L593V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 L593V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Uncertain or inconclusive results come from Rosetta, Foldetta, and AlphaMissense‑Default. High‑accuracy assessments: AlphaMissense‑Optimized classifies the variant as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a pathogenic prediction; Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain. Overall, the majority of evidence (six pathogenic vs. four benign) points to a pathogenic effect. This conclusion is consistent with the lack of ClinVar annotation and gnomAD presence, so there is no contradiction with existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.009728 | Structured | 0.110534 | Uncertain | 0.941 | 0.151 | 0.000 | -10.327 | Likely Pathogenic | 0.551 | Ambiguous | Likely Benign | 2.81 | Destabilizing | 0.2 | 1.10 | Ambiguous | 1.96 | Ambiguous | 1.39 | Destabilizing | 0.268 | Likely Benign | -2.59 | Deleterious | 0.998 | Probably Damaging | 0.992 | Probably Damaging | 3.04 | Benign | 0.12 | Tolerated | 0.1599 | 0.3577 | 2 | 1 | 0.4 | -14.03 | ||||||||||||||||||||||||||||||
| c.1777C>T | L593F 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L593F is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and FATHMM, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Predictions that are inconclusive—FoldX, Foldetta, premPS, and AlphaMissense‑Optimized—are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.110534 | Uncertain | 0.941 | 0.151 | 0.000 | -12.723 | Likely Pathogenic | 0.954 | Likely Pathogenic | Ambiguous | 1.46 | Ambiguous | 0.4 | 0.24 | Likely Benign | 0.85 | Ambiguous | 0.62 | Ambiguous | 0.304 | Likely Benign | -3.78 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.95 | Benign | 0.01 | Affected | 0.0884 | 0.2631 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1778T>C | L593P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L593P is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic outcome. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, a conclusion that contradicts its current ClinVar “Uncertain” status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.110534 | Uncertain | 0.941 | 0.151 | 0.000 | Uncertain | 1 | -13.961 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.75 | Destabilizing | 0.9 | 10.77 | Destabilizing | 8.26 | Destabilizing | 2.43 | Destabilizing | 0.777 | Likely Pathogenic | -6.77 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.77 | Benign | 0.00 | Affected | 0.3783 | 0.1274 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||
| c.1778T>G | L593R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L593R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a destabilizing, pathogenic effect. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.110534 | Uncertain | 0.941 | 0.151 | 0.000 | -16.139 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.27 | Destabilizing | 0.3 | 3.39 | Destabilizing | 3.83 | Destabilizing | 2.11 | Destabilizing | 0.740 | Likely Pathogenic | -5.87 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.77 | Benign | 0.00 | Affected | 0.1440 | 0.0615 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1780T>A | F594I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F594I is not reported in ClinVar and has no entries in gnomAD. In silico assessment shows that all evaluated pathogenicity predictors—REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—classify the substitution as pathogenic, while FoldX, Rosetta, and Foldetta provide inconclusive results. High‑accuracy tools further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta remains uncertain. Consequently, the overwhelming majority of predictions point to a pathogenic impact. The variant is therefore most likely pathogenic, and this assessment does not contradict any ClinVar annotation, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009187 | Structured | 0.120166 | Uncertain | 0.946 | 0.147 | 0.000 | -11.271 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.93 | Ambiguous | 0.1 | 1.67 | Ambiguous | 1.80 | Ambiguous | 1.61 | Destabilizing | 0.935 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | -1.91 | Pathogenic | 0.02 | Affected | 0.1694 | 0.1925 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1780T>C | F594L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F594L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: SGM‑Consensus, REVEL, FoldX (uncertain), Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while only FoldX and Foldetta are inconclusive. In the high‑accuracy subset, AlphaMissense‑Optimized remains pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta is uncertain. No tools predict a benign outcome. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009187 | Structured | 0.120166 | Uncertain | 0.946 | 0.147 | 0.000 | -11.270 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.50 | Ambiguous | 0.1 | 2.41 | Destabilizing | 1.96 | Ambiguous | 1.56 | Destabilizing | 0.940 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -1.98 | Pathogenic | 0.03 | Affected | 0.1790 | 0.2439 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1780T>G | F594V 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F594V lies within the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that assess pathogenicity unanimously favor a deleterious effect: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity; only Rosetta is uncertain. No tool predicts a benign outcome. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which simply lacks an entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009187 | Structured | 0.120166 | Uncertain | 0.946 | 0.147 | 0.000 | -12.648 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 2.91 | Destabilizing | 0.2 | 1.90 | Ambiguous | 2.41 | Destabilizing | 1.80 | Destabilizing | 0.931 | Likely Pathogenic | -6.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.91 | Pathogenic | 0.01 | Affected | 0.1813 | 0.1575 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1781T>A | F594Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F594Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to Rosetta, which scores the substitution as benign. In contrast, the majority of tools predict a pathogenic impact: REVEL, SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, SGM‑Consensus, and premPS all classify the variant as pathogenic. FoldX and Foldetta are uncertain, and AlphaMissense‑Optimized is also uncertain, so these results are treated as unavailable. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic, while AlphaMissense‑Optimized and Foldetta remain inconclusive. Overall, the preponderance of evidence indicates that F594Y is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009187 | Structured | 0.120166 | Uncertain | 0.946 | 0.147 | 0.000 | -13.692 | Likely Pathogenic | 0.935 | Likely Pathogenic | Ambiguous | 1.30 | Ambiguous | 0.2 | 0.41 | Likely Benign | 0.86 | Ambiguous | 1.21 | Destabilizing | 0.929 | Likely Pathogenic | -2.99 | Deleterious | 0.993 | Probably Damaging | 0.976 | Probably Damaging | -1.98 | Pathogenic | 0.01 | Affected | 0.1264 | 0.0731 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1781T>C | F594S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F594S is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify it as pathogenic: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. With unanimous pathogenic predictions and no ClinVar evidence to contradict, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009187 | Structured | 0.120166 | Uncertain | 0.946 | 0.147 | 0.000 | -15.930 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.70 | Destabilizing | 0.3 | 5.20 | Destabilizing | 4.95 | Destabilizing | 2.60 | Destabilizing | 0.952 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -2.04 | Pathogenic | 0.00 | Affected | 0.3804 | 0.0200 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1781T>G | F594C 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F594C is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. All available evidence points to a damaging effect. Therefore, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009187 | Structured | 0.120166 | Uncertain | 0.946 | 0.147 | 0.000 | -14.591 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.97 | Destabilizing | 0.0 | 4.35 | Destabilizing | 4.16 | Destabilizing | 1.36 | Destabilizing | 0.943 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -2.05 | Pathogenic | 0.03 | Affected | 0.2595 | 0.0615 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.1782C>A | F594L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F594L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: SGM‑Consensus, REVEL, FoldX (uncertain), Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while only FoldX and Foldetta are inconclusive. In the high‑accuracy subset, AlphaMissense‑Optimized remains pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta is uncertain. No tools predict a benign outcome. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009187 | Structured | 0.120166 | Uncertain | 0.946 | 0.147 | 0.000 | -11.270 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.50 | Ambiguous | 0.1 | 2.41 | Destabilizing | 1.96 | Ambiguous | 1.56 | Destabilizing | 0.893 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -1.98 | Pathogenic | 0.03 | Affected | 0.1790 | 0.2439 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1782C>G | F594L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F594L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity; SGM‑Consensus also indicates a likely pathogenic outcome. No tool in the dataset predicts a benign effect. Stability‑focused methods give mixed results: FoldX is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also uncertain, so these do not provide decisive evidence. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is likely pathogenic, while Foldetta remains uncertain. Overall, the consensus of available predictions strongly suggests that F594L is most likely pathogenic, and this conclusion is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009187 | Structured | 0.120166 | Uncertain | 0.946 | 0.147 | 0.000 | -11.270 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.50 | Ambiguous | 0.1 | 2.41 | Destabilizing | 1.96 | Ambiguous | 1.56 | Destabilizing | 0.893 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -1.98 | Pathogenic | 0.03 | Affected | 0.1790 | 0.2439 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1783C>A | L595M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L595M is not reported in ClinVar (ClinVar ID: None) and has no entries in gnomAD (gnomAD ID: None). Functional prediction tools show a split consensus: benign predictions come from REVEL, FoldX, Rosetta, PROVEAN, and FATHMM, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta predicts a benign effect on protein stability. No prediction or folding result is missing; all available outputs are reported. Based on the balanced distribution of benign and pathogenic calls, the variant is most likely benign, but the evidence is conflicting and does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.015344 | Structured | 0.128444 | Uncertain | 0.920 | 0.150 | 0.000 | -11.325 | Likely Pathogenic | 0.840 | Likely Pathogenic | Ambiguous | 0.32 | Likely Benign | 0.0 | 0.41 | Likely Benign | 0.37 | Likely Benign | 0.90 | Ambiguous | 0.387 | Likely Benign | -1.99 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.74 | Benign | 0.02 | Affected | 0.0930 | 0.3140 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.1783C>G | L595V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L595V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, Rosetta, and FATHMM, while pathogenic calls are made by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Predictions marked uncertain include FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy consensus methods give a clearer picture: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also inconclusive. Overall, the majority of evidence points toward a pathogenic effect, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.015344 | Structured | 0.128444 | Uncertain | 0.920 | 0.150 | 0.000 | -13.490 | Likely Pathogenic | 0.905 | Likely Pathogenic | Ambiguous | 1.29 | Ambiguous | 0.1 | 0.24 | Likely Benign | 0.77 | Ambiguous | 1.01 | Destabilizing | 0.398 | Likely Benign | -2.99 | Deleterious | 0.998 | Probably Damaging | 0.992 | Probably Damaging | 2.78 | Benign | 0.01 | Affected | 0.1441 | 0.3406 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1784T>A | L595Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L595Q is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that classify the variant as benign include only FATHMM. All other evaluated algorithms—REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized—predict a pathogenic effect, and the SGM‑Consensus score indicates a likely pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized returns a pathogenic prediction, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a likely pathogenic result, while Foldetta’s stability analysis is inconclusive. Overall, the majority of computational evidence points to a pathogenic effect, which does not contradict the ClinVar designation of uncertain significance but suggests a higher likelihood of pathogenicity. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.015344 | Structured | 0.128444 | Uncertain | 0.920 | 0.150 | 0.000 | Uncertain | 1 | -15.101 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 0.79 | Ambiguous | 0.1 | 1.40 | Ambiguous | 1.10 | Ambiguous | 1.99 | Destabilizing | 0.733 | Likely Pathogenic | -5.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.75 | Benign | 0.00 | Affected | 3.37 | 35 | 0.1074 | 0.1563 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||
| c.1784T>C | L595P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L595P is listed in ClinVar with an “Uncertain” status (ClinVar ID 3172762.0) and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict a pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.015344 | Structured | 0.128444 | Uncertain | 0.920 | 0.150 | 0.000 | Uncertain | 1 | -11.856 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.09 | Destabilizing | 0.8 | 5.88 | Destabilizing | 3.99 | Destabilizing | 1.78 | Destabilizing | 0.747 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.72 | Benign | 0.00 | Affected | 3.37 | 35 | 0.3336 | 0.1713 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||
| c.1784T>G | L595R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L595R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, while the majority of tools (SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; FoldX and Foldetta are uncertain. High‑accuracy methods give a pathogenic signal: AlphaMissense‑Optimized is pathogenic, SGM‑Consensus is likely pathogenic, and Foldetta remains uncertain. Overall, the evidence strongly favors a pathogenic effect, and this conclusion does not contradict any ClinVar annotation, as no ClinVar record exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.015344 | Structured | 0.128444 | Uncertain | 0.920 | 0.150 | 0.000 | -14.601 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 1.22 | Ambiguous | 0.0 | 2.20 | Destabilizing | 1.71 | Ambiguous | 1.52 | Destabilizing | 0.707 | Likely Pathogenic | -5.97 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.76 | Benign | 0.00 | Affected | 0.1344 | 0.1005 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1786C>A | R596S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R596S is not reported in ClinVar and is absent from gnomAD. In silico assessment shows a consensus of pathogenicity: all evaluated tools (REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a deleterious effect, while Rosetta remains uncertain. Grouping by agreement, no tool predicts benign; the pathogenic group includes 13 predictions, with Rosetta excluded as inconclusive. High‑accuracy methods further support a damaging outcome: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Consequently, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.017797 | Structured | 0.135423 | Uncertain | 0.918 | 0.134 | 0.000 | -13.921 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.51 | Destabilizing | 0.2 | 1.53 | Ambiguous | 2.52 | Destabilizing | 1.17 | Destabilizing | 0.626 | Likely Pathogenic | -5.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.42 | Pathogenic | 0.00 | Affected | 0.3129 | 0.2680 | 0 | -1 | 3.7 | -69.11 | |||||||||||||||||||||||||||||
| c.1786C>G | R596G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R596G is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity largely agree: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a deleterious effect, while premPS remains uncertain. No tools predict a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Based on the consensus of these predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.017797 | Structured | 0.135423 | Uncertain | 0.918 | 0.134 | 0.000 | -13.525 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 4.02 | Destabilizing | 0.1 | 2.36 | Destabilizing | 3.19 | Destabilizing | 0.81 | Ambiguous | 0.629 | Likely Pathogenic | -6.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.41 | Pathogenic | 0.00 | Affected | 0.3214 | 0.2316 | -3 | -2 | 4.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1787G>C | R596P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R596P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while no tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized indicates pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No contradictory evidence is present. Based on the uniform predictions, the variant is most likely pathogenic, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.017797 | Structured | 0.135423 | Uncertain | 0.918 | 0.134 | 0.000 | -13.786 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 6.80 | Destabilizing | 0.1 | 4.78 | Destabilizing | 5.79 | Destabilizing | 1.15 | Destabilizing | 0.780 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.42 | Pathogenic | 0.00 | Affected | 0.2335 | 0.3356 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.1789T>A | F597I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F597I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All available evidence points to a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion is consistent with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | -13.674 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 3.62 | Destabilizing | 0.9 | 2.15 | Destabilizing | 2.89 | Destabilizing | 1.45 | Destabilizing | 0.951 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | -2.18 | Pathogenic | 0.01 | Affected | 0.2008 | 0.1815 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1789T>C | F597L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F597L is listed in ClinVar with an uncertain significance (ClinVar ID 3658115.0) and is not reported in gnomAD. Prediction tools that classify the variant as benign include only SIFT, whereas the remaining tools—SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. The high‑accuracy AlphaMissense‑Optimized score is pathogenic, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for F597L, which is consistent with its ClinVar uncertain status rather than contradicting it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | Uncertain | 1 | -10.173 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.74 | Ambiguous | 0.1 | 2.12 | Destabilizing | 1.43 | Ambiguous | 1.20 | Destabilizing | 0.929 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -2.06 | Pathogenic | 0.13 | Tolerated | 0.2232 | 0.2596 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||
| c.1789T>G | F597V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F597V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All available evidence points to a pathogenic effect. Therefore, the variant is most likely pathogenic, and this conclusion is consistent with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | -13.883 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 3.75 | Destabilizing | 0.7 | 2.02 | Destabilizing | 2.89 | Destabilizing | 1.60 | Destabilizing | 0.939 | Likely Pathogenic | -6.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -2.16 | Pathogenic | 0.01 | Affected | 0.2237 | 0.1583 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.178G>A | D60N 2D AIThe SynGAP1 D60N missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also as benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -3.610 | Likely Benign | 0.577 | Likely Pathogenic | Likely Benign | 0.128 | Likely Benign | -0.22 | Neutral | 0.805 | Possibly Damaging | 0.857 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.1219 | 0.8168 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.178G>C | D60H 2D AIThe SynGAP1 missense variant D60H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are not available. Overall, the balance of evidence leans toward a benign interpretation, with no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -5.257 | Likely Benign | 0.934 | Likely Pathogenic | Ambiguous | 0.165 | Likely Benign | -1.59 | Neutral | 0.972 | Probably Damaging | 0.969 | Probably Damaging | 3.91 | Benign | 0.00 | Affected | 0.1433 | 0.8401 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.178G>T | D60Y 2D AIThe SynGAP1 D60Y missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. ESM1b is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict (2 pathogenic vs. 1 benign). Foldetta, a protein‑folding stability method, has no available result for this variant. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the D60Y variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -7.748 | In-Between | 0.971 | Likely Pathogenic | Likely Pathogenic | 0.221 | Likely Benign | -2.60 | Deleterious | 0.972 | Probably Damaging | 0.969 | Probably Damaging | 3.90 | Benign | 0.00 | Affected | 0.0517 | 0.7790 | -4 | -3 | 2.2 | 48.09 | ||||||||||||||||||||||||||||||||||||||||
| c.1790T>A | F597Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F597Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include ESM1b and Rosetta, whereas a majority of tools (REVEL, SIFT, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, PROVEAN, premPS, and AlphaMissense‑Default) predict a pathogenic impact. Predictions that are inconclusive or unavailable are FoldX, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, AlphaMissense‑Optimized as Uncertain, and Foldetta as Uncertain. Overall, the preponderance of evidence points to a pathogenic effect for F597Y. This conclusion is not contradicted by ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | -5.869 | Likely Benign | 0.796 | Likely Pathogenic | Ambiguous | 1.41 | Ambiguous | 0.1 | 0.37 | Likely Benign | 0.89 | Ambiguous | 1.11 | Destabilizing | 0.877 | Likely Pathogenic | -2.99 | Deleterious | 0.993 | Probably Damaging | 0.976 | Probably Damaging | -1.99 | Pathogenic | 0.02 | Affected | 0.1471 | 0.1494 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1790T>C | F597S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F597S is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. All available predictions are consistent and conclusive. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | -14.943 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.53 | Destabilizing | 0.3 | 4.90 | Destabilizing | 4.22 | Destabilizing | 2.18 | Destabilizing | 0.953 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -2.19 | Pathogenic | 0.00 | Affected | 0.4035 | 0.0200 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1790T>G | F597C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F597C is reported in gnomAD (variant ID 6-33440842‑T‑G) but has no ClinVar entry. Functional prediction tools uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool in the dataset predicts a benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. Based on the unanimous pathogenic predictions and the absence of any benign calls, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | 6-33440842-T-G | 1 | 6.19e-7 | -12.099 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 3.77 | Destabilizing | 0.2 | 4.17 | Destabilizing | 3.97 | Destabilizing | 1.97 | Destabilizing | 0.953 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -2.19 | Pathogenic | 0.00 | Affected | 3.37 | 35 | 0.2618 | 0.0783 | -2 | -4 | -0.3 | -44.04 | ||||||||||||||||||||||||
| c.1791C>A | F597L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F597L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: the benign group contains only SIFT, while the pathogenic group includes SGM‑Consensus (Likely Pathogenic), REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX and Foldetta are inconclusive, providing no definitive evidence. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts a damaging effect, SGM‑Consensus concurs with a likely pathogenic classification, and Foldetta remains uncertain. Taken together, the overwhelming majority of predictions indicate a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | -10.173 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.74 | Ambiguous | 0.1 | 2.12 | Destabilizing | 1.43 | Ambiguous | 1.20 | Destabilizing | 0.879 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -2.06 | Pathogenic | 0.13 | Tolerated | 0.2232 | 0.2596 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1791C>G | F597L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F597L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: the benign group contains only SIFT, while the pathogenic group includes SGM‑Consensus (Likely Pathogenic), REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX and Foldetta are inconclusive, providing no definitive evidence. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts a damaging effect, SGM‑Consensus concurs with a likely pathogenic classification, and Foldetta remains uncertain. Taken together, the overwhelming majority of predictions indicate a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.142961 | Uncertain | 0.944 | 0.151 | 0.000 | -10.173 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.74 | Ambiguous | 0.1 | 2.12 | Destabilizing | 1.43 | Ambiguous | 1.20 | Destabilizing | 0.879 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -2.06 | Pathogenic | 0.13 | Tolerated | 0.2232 | 0.2596 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1792C>A | L598I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L598I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar) and ESM1b. The remaining tools (FoldX, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of reliable predictions indicate a benign impact. This conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.007259 | Structured | 0.147872 | Uncertain | 0.953 | 0.154 | 0.000 | -9.396 | Likely Pathogenic | 0.512 | Ambiguous | Likely Benign | 0.80 | Ambiguous | 0.0 | 0.38 | Likely Benign | 0.59 | Ambiguous | 0.78 | Ambiguous | 0.246 | Likely Benign | -1.96 | Neutral | 0.988 | Probably Damaging | 0.910 | Probably Damaging | 3.47 | Benign | 0.10 | Tolerated | 0.0792 | 0.2000 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1792C>T | L598F 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L598F is not reported in ClinVar and is absent from gnomAD. Computational predictors show mixed results: benign calls come from REVEL, Rosetta, premPS, SIFT, and FATHMM; pathogenic calls come from SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. FoldX and AlphaMissense‑Optimized returned uncertain results, which are treated as unavailable evidence. High‑accuracy tools give a split view: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus majority vote (AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts Likely Pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the balance of evidence leans toward pathogenicity, with six pathogenic versus five benign predictions and no ClinVar entry to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.007259 | Structured | 0.147872 | Uncertain | 0.953 | 0.154 | 0.000 | -10.649 | Likely Pathogenic | 0.820 | Likely Pathogenic | Ambiguous | 1.12 | Ambiguous | 0.7 | -0.24 | Likely Benign | 0.44 | Likely Benign | 0.47 | Likely Benign | 0.283 | Likely Benign | -3.91 | Deleterious | 0.999 | Probably Damaging | 0.946 | Probably Damaging | 3.62 | Benign | 0.22 | Tolerated | 0.0630 | 0.1414 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1793T>A | L598H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L598H is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized returns a pathogenic score; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.007259 | Structured | 0.147872 | Uncertain | 0.953 | 0.154 | 0.000 | -17.210 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 2.52 | Destabilizing | 0.2 | 2.37 | Destabilizing | 2.45 | Destabilizing | 2.24 | Destabilizing | 0.648 | Likely Pathogenic | -6.87 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.10 | Benign | 0.00 | Affected | 0.1060 | 0.0879 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1793T>C | L598P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L598P is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: pathogenic predictions come from SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a benign outcome. High‑accuracy methods reinforce the pathogenic view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates it is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also classifies it as pathogenic. No prediction or stability result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.007259 | Structured | 0.147872 | Uncertain | 0.953 | 0.154 | 0.000 | -12.621 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 8.31 | Destabilizing | 0.5 | 12.04 | Destabilizing | 10.18 | Destabilizing | 2.02 | Destabilizing | 0.705 | Likely Pathogenic | -6.87 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.10 | Benign | 0.00 | Affected | 0.2742 | 0.1192 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1793T>G | L598R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L598R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.007259 | Structured | 0.147872 | Uncertain | 0.953 | 0.154 | 0.000 | -17.392 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.70 | Destabilizing | 0.5 | 3.06 | Destabilizing | 2.88 | Destabilizing | 2.23 | Destabilizing | 0.682 | Likely Pathogenic | -5.87 | Deleterious | 0.999 | Probably Damaging | 0.973 | Probably Damaging | 3.10 | Benign | 0.00 | Affected | 0.1297 | 0.0679 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1795T>A | C599S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C599S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only SIFT, which scores the variant as benign. All other evaluated algorithms—REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus—predict a pathogenic or likely pathogenic outcome. High‑accuracy assessments show the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, AlphaMissense‑Optimized as Uncertain, and Foldetta (combining FoldX‑MD and Rosetta) as Uncertain. No prediction or folding stability result is missing; all available data are reported. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009865 | Structured | 0.151725 | Uncertain | 0.960 | 0.151 | 0.000 | -13.336 | Likely Pathogenic | 0.954 | Likely Pathogenic | Ambiguous | 0.85 | Ambiguous | 0.0 | 1.57 | Ambiguous | 1.21 | Ambiguous | 1.27 | Destabilizing | 0.919 | Likely Pathogenic | -9.95 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.45 | Pathogenic | 0.06 | Tolerated | 0.3414 | 0.1415 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.1795T>C | C599R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C599R is not reported in ClinVar and is absent from gnomAD. In silico prediction tools uniformly indicate a deleterious effect: REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, PROVEAN, ESM1b, FATHMM, premPS, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic, while FoldX reports an uncertain outcome. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely pathogenic status; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009865 | Structured | 0.151725 | Uncertain | 0.960 | 0.151 | 0.000 | -15.968 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.00 | Ambiguous | 0.3 | 3.29 | Destabilizing | 2.15 | Destabilizing | 1.54 | Destabilizing | 0.909 | Likely Pathogenic | -11.95 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.50 | Pathogenic | 0.00 | Affected | 0.1519 | 0.1566 | -4 | -3 | -7.0 | 53.05 | |||||||||||||||||||||||||||||
| c.1795T>G | C599G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a deleterious effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts a benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the evidence strongly favors a pathogenic effect, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009865 | Structured | 0.151725 | Uncertain | 0.960 | 0.151 | 0.000 | -13.342 | Likely Pathogenic | 0.949 | Likely Pathogenic | Ambiguous | 1.40 | Ambiguous | 0.0 | 0.90 | Ambiguous | 1.15 | Ambiguous | 1.43 | Destabilizing | 0.923 | Likely Pathogenic | -11.95 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.50 | Pathogenic | 0.03 | Affected | 0.2444 | 0.2130 | -3 | -3 | -2.9 | -46.09 | |||||||||||||||||||||||||||||
| c.1796G>A | C599Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C599Y is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that indicate a benign effect include only premPS, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is labeled Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among the majority of tools, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009865 | Structured | 0.151725 | Uncertain | 0.960 | 0.151 | 0.000 | -13.563 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 4.00 | Destabilizing | 1.4 | 2.80 | Destabilizing | 3.40 | Destabilizing | -0.29 | Likely Benign | 0.905 | Likely Pathogenic | -10.95 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.49 | Pathogenic | 0.00 | Affected | 0.1092 | 0.2555 | 0 | -2 | -3.8 | 60.04 | |||||||||||||||||||||||||||||
| c.1796G>C | C599S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C599S is not reported in ClinVar and has no entries in gnomAD. Prediction tools cluster into two groups: the sole benign prediction comes from SIFT, whereas the remaining nine tools—REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—indicate pathogenicity. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized is uncertain, SGM‑Consensus predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. FoldX and Rosetta individually report uncertain stability changes. Overall, the majority of evidence points to a deleterious effect, so the variant is most likely pathogenic, with no ClinVar annotation to contradict this assessment. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009865 | Structured | 0.151725 | Uncertain | 0.960 | 0.151 | 0.000 | -13.336 | Likely Pathogenic | 0.954 | Likely Pathogenic | Ambiguous | 0.85 | Ambiguous | 0.0 | 1.57 | Ambiguous | 1.21 | Ambiguous | 1.27 | Destabilizing | 0.866 | Likely Pathogenic | -9.95 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.45 | Pathogenic | 0.06 | Tolerated | 0.3414 | 0.1415 | 0 | -1 | -3.3 | -16.06 | |||||||||||||||||||||||||||||
| c.1796G>T | C599F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C599F is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that assess the variant’s effect largely agree on a deleterious outcome: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic or likely pathogenic. Only premPS predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized reports pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or folding‑stability result is missing or inconclusive. Based on the overwhelming majority of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009865 | Structured | 0.151725 | Uncertain | 0.960 | 0.151 | 0.000 | -13.969 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 3.72 | Destabilizing | 1.3 | 2.70 | Destabilizing | 3.21 | Destabilizing | -0.42 | Likely Benign | 0.902 | Likely Pathogenic | -10.95 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.47 | Pathogenic | 0.00 | Affected | 0.1319 | 0.3073 | -4 | -2 | 0.3 | 44.04 | |||||||||||||||||||||||||||||
| c.1797C>G | C599W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant C599W is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only premPS; all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009865 | Structured | 0.151725 | Uncertain | 0.960 | 0.151 | 0.000 | -17.500 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.85 | Destabilizing | 0.6 | 3.32 | Destabilizing | 3.59 | Destabilizing | 0.16 | Likely Benign | 0.715 | Likely Pathogenic | -10.95 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.52 | Pathogenic | 0.00 | Affected | 0.1500 | 0.2363 | -8 | -2 | -3.4 | 83.07 | |||||||||||||||||||||||||||||
| c.1798C>A | P600T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P600T is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include Foldetta, Rosetta, and premPS. Those that predict pathogenicity are REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, while Foldetta indicates a benign effect on protein folding stability. Overall, the majority of computational evidence supports a pathogenic effect for P600T, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.162960 | Uncertain | 0.947 | 0.147 | 0.000 | -11.945 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 2.61 | Destabilizing | 0.1 | -2.90 | Stabilizing | -0.15 | Likely Benign | 0.24 | Likely Benign | 0.737 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.34 | Pathogenic | 0.01 | Affected | 0.1871 | 0.4954 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||
| c.1798C>G | P600A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P600A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from Rosetta and premPS, while the remaining 11 tools (REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and SGM Consensus) predict pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it “Likely Pathogenic,” and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports an uncertain result. Taken together, the overwhelming majority of evidence indicates that P600A is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.162960 | Uncertain | 0.947 | 0.147 | 0.000 | -11.385 | Likely Pathogenic | 0.974 | Likely Pathogenic | Likely Pathogenic | 2.16 | Destabilizing | 0.0 | -3.30 | Stabilizing | -0.57 | Ambiguous | 0.39 | Likely Benign | 0.581 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.38 | Pathogenic | 0.03 | Affected | 0.3692 | 0.4132 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.1798C>T | P600S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P600S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that classify the variant as benign include Rosetta and Foldetta, whereas the majority of other in‑silico predictors (REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict it to be pathogenic. High‑accuracy assessments further show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) as benign. No contradictory evidence exists in ClinVar. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.162960 | Uncertain | 0.947 | 0.147 | 0.000 | -12.002 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 2.54 | Destabilizing | 0.1 | -2.60 | Stabilizing | -0.03 | Likely Benign | 0.52 | Ambiguous | 0.717 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.40 | Pathogenic | 0.01 | Affected | 0.3744 | 0.4200 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||
| c.1799C>A | P600Q 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P600Q is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions are limited to Rosetta, whereas the remaining tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—all classify the variant as pathogenic. Uncertain results come from FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence indicates that P600Q is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.162960 | Uncertain | 0.947 | 0.147 | 0.000 | -14.187 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.76 | Ambiguous | 0.0 | -3.09 | Stabilizing | -0.67 | Ambiguous | 0.53 | Ambiguous | 0.739 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 1.34 | Pathogenic | 0.00 | Affected | 0.1740 | 0.3970 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||
| c.1799C>G | P600R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P600R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include Rosetta and premPS, whereas the majority of tools—REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic outcome; FoldX and Foldetta are inconclusive. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta remains uncertain. Overall, the computational evidence strongly favors a pathogenic classification, and this does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.162960 | Uncertain | 0.947 | 0.147 | 0.000 | -15.304 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.48 | Ambiguous | 0.1 | -2.48 | Stabilizing | -0.50 | Ambiguous | 0.37 | Likely Benign | 0.756 | Likely Pathogenic | -8.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.34 | Pathogenic | 0.00 | Affected | 0.1604 | 0.2734 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.1799C>T | P600L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P600L is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include Rosetta and premPS, whereas the majority of tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact. FoldX and Foldetta give uncertain results. High‑accuracy methods specifically show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta remains inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for P600L, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.009728 | Structured | 0.162960 | Uncertain | 0.947 | 0.147 | 0.000 | -13.209 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.36 | Ambiguous | 0.1 | -3.58 | Stabilizing | -1.11 | Ambiguous | -0.49 | Likely Benign | 0.734 | Likely Pathogenic | -9.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.35 | Pathogenic | 0.00 | Affected | 0.2391 | 0.5555 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.179A>C | D60A 2D AIThe SynGAP1 D60A missense variant has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are unavailable. Overall, the majority of evaluated tools lean toward a benign interpretation, with no evidence of pathogenicity from the high‑confidence methods. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status, as none exists for this allele. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -4.500 | Likely Benign | 0.918 | Likely Pathogenic | Ambiguous | 0.167 | Likely Benign | -2.17 | Neutral | 0.909 | Possibly Damaging | 0.857 | Possibly Damaging | 3.96 | Benign | 0.00 | Affected | 0.3851 | 0.7405 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.179A>G | D60G 2D AIThe SynGAP1 D60G missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -4.423 | Likely Benign | 0.775 | Likely Pathogenic | Likely Benign | 0.128 | Likely Benign | -1.67 | Neutral | 0.805 | Possibly Damaging | 0.857 | Possibly Damaging | 3.94 | Benign | 0.00 | Affected | 0.4143 | 0.7069 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.179A>T | D60V 2D AIThe SynGAP1 D60V missense variant is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL, ESM1b, and FATHMM. Tools that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 vs 2), and Foldetta results are unavailable. Overall, the majority of evaluated predictors (7 of 10) indicate pathogenicity, while only three suggest benignity. Therefore, the variant is most likely pathogenic based on current computational evidence, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -6.576 | Likely Benign | 0.980 | Likely Pathogenic | Likely Pathogenic | 0.254 | Likely Benign | -2.72 | Deleterious | 0.972 | Probably Damaging | 0.954 | Probably Damaging | 3.91 | Benign | 0.00 | Affected | 0.0765 | 0.7962 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||||||||||||
| c.17C>A | A6D 2D AIThe SynGAP1 missense variant A6D is reported in gnomAD (ID 6‑33420281‑C‑A) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also likely benign; Foldetta results are not available. Overall, the consensus of the majority of tools, including the high‑accuracy methods, indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.566480 | Disordered | 0.549054 | Binding | 0.377 | 0.920 | 0.875 | 6-33420281-C-A | -3.340 | Likely Benign | 0.210 | Likely Benign | Likely Benign | 0.211 | Likely Benign | 0.34 | Neutral | 0.117 | Benign | 0.010 | Benign | 4.07 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1945 | 0.2530 | -2 | 0 | -5.3 | 44.01 | ||||||||||||||||||||||||||||||||||||
| c.17C>G | A6G 2D AIThe SynGAP1 missense variant A6G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.566480 | Disordered | 0.549054 | Binding | 0.377 | 0.920 | 0.875 | -2.786 | Likely Benign | 0.097 | Likely Benign | Likely Benign | 0.107 | Likely Benign | 0.31 | Neutral | 0.052 | Benign | 0.004 | Benign | 4.08 | Benign | 0.00 | Affected | 0.2139 | 0.4369 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.17C>T | A6V 2D AIThe SynGAP1 A6V missense variant is reported in gnomAD (ID 6‑33420281‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts it to be pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labels it “Likely Benign.” High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign, and the SGM‑Consensus is also benign; Foldetta results are not available. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.566480 | Disordered | 0.549054 | Binding | 0.377 | 0.920 | 0.875 | 6-33420281-C-T | -3.781 | Likely Benign | 0.191 | Likely Benign | Likely Benign | 0.123 | Likely Benign | 0.32 | Neutral | 0.117 | Benign | 0.007 | Benign | 4.09 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0987 | 0.5799 | 0 | 0 | 2.4 | 28.05 | ||||||||||||||||||||||||||||||||||||
| c.1801G>A | A601T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A601T missense variant is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect are FATHMM and AlphaMissense‑Optimized, whereas the remaining pathogenic‑predicting tools—SGM‑Consensus, REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently flag the variant as deleterious. Two tools, FoldX and premPS, return uncertain results and are not considered evidence for either side. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as pathogenic. Taken together, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.008895 | Structured | 0.174517 | Uncertain | 0.955 | 0.156 | 0.000 | -10.635 | Likely Pathogenic | 0.662 | Likely Pathogenic | Likely Benign | 1.55 | Ambiguous | 0.1 | 2.66 | Destabilizing | 2.11 | Destabilizing | 0.76 | Ambiguous | 0.642 | Likely Pathogenic | -3.98 | Deleterious | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 2.57 | Benign | 0.00 | Affected | 0.1564 | 0.5554 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.1801G>C | A601P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A601P is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while the remaining 12 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify it as pathogenic. The high‑accuracy methods—AlphaMissense‑Optimized, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta (combining FoldX‑MD and Rosetta outputs)—all report pathogenicity. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.008895 | Structured | 0.174517 | Uncertain | 0.955 | 0.156 | 0.000 | -14.584 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 4.90 | Destabilizing | 0.6 | 8.84 | Destabilizing | 6.87 | Destabilizing | 0.87 | Ambiguous | 0.713 | Likely Pathogenic | -4.98 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.56 | Benign | 0.01 | Affected | 0.2176 | 0.4328 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1801G>T | A601S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A601S missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The remaining tools—FoldX, Rosetta, Foldetta, and premPS—return uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is a 2‑vs‑2 tie and thus inconclusive. Foldetta, which integrates FoldX‑MD and Rosetta outputs, also yields an uncertain result. Overall, more tools (six) predict pathogenicity than benign (three), and no ClinVar evidence contradicts this assessment. Therefore, the variant is most likely pathogenic based on the available predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.008895 | Structured | 0.174517 | Uncertain | 0.955 | 0.156 | 0.000 | -11.248 | Likely Pathogenic | 0.136 | Likely Benign | Likely Benign | 0.79 | Ambiguous | 0.1 | 1.63 | Ambiguous | 1.21 | Ambiguous | 0.79 | Ambiguous | 0.541 | Likely Pathogenic | -2.99 | Deleterious | 0.983 | Probably Damaging | 0.993 | Probably Damaging | 2.56 | Benign | 0.01 | Affected | 0.2732 | 0.4730 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||
| c.1802C>G | A601G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A601G missense variant is listed in gnomAD (ID 6‑33440854‑C‑G) but has no ClinVar entry. Prediction tools that agree on a benign effect are FATHMM and AlphaMissense‑Optimized; those that agree on a pathogenic effect are REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The high‑accuracy consensus methods give a pathogenic verdict: the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also pathogenic. AlphaMissense‑Default and premPS are uncertain, and no evidence is available from other folding‑stability tools. Overall, the preponderance of evidence points to a pathogenic impact for A601G, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.008895 | Structured | 0.174517 | Uncertain | 0.955 | 0.156 | 0.000 | 6-33440854-C-G | 1 | 6.19e-7 | -11.772 | Likely Pathogenic | 0.543 | Ambiguous | Likely Benign | 2.03 | Destabilizing | 0.0 | 2.31 | Destabilizing | 2.17 | Destabilizing | 0.94 | Ambiguous | 0.511 | Likely Pathogenic | -3.98 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.55 | Benign | 0.01 | Affected | 3.37 | 35 | 0.2337 | 0.4370 | 0 | 1 | -2.2 | -14.03 | |||||||||||||||||||||||||
| c.1804A>C | I602L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I602L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Foldetta, PROVEAN, and AlphaMissense‑Optimized, whereas a majority of tools predict pathogenicity: REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Three tools (Rosetta, premPS, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the balance of evidence leans toward pathogenicity, with no conflict with the ClinVar status because the variant is not yet classified in that database. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.010221 | Structured | 0.186541 | Uncertain | 0.963 | 0.171 | 0.000 | -9.660 | Likely Pathogenic | 0.558 | Ambiguous | Likely Benign | -0.15 | Likely Benign | 0.1 | 1.12 | Ambiguous | 0.49 | Likely Benign | 0.91 | Ambiguous | 0.631 | Likely Pathogenic | -1.99 | Neutral | 0.645 | Possibly Damaging | 0.718 | Possibly Damaging | -1.54 | Pathogenic | 0.04 | Affected | 0.1044 | 0.3199 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1804A>G | I602V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I602V missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the change as benign include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are premPS, polyPhen‑2 HumDiv, SIFT, and FATHMM. Uncertain or inconclusive results come from FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts benign, while Foldetta’s stability analysis remains uncertain. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by any ClinVar annotation, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.010221 | Structured | 0.186541 | Uncertain | 0.963 | 0.171 | 0.000 | -6.317 | Likely Benign | 0.075 | Likely Benign | Likely Benign | 1.24 | Ambiguous | 0.0 | 0.78 | Ambiguous | 1.01 | Ambiguous | 1.01 | Destabilizing | 0.309 | Likely Benign | -0.84 | Neutral | 0.528 | Possibly Damaging | 0.179 | Benign | -1.89 | Pathogenic | 0.03 | Affected | 0.1173 | 0.3326 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||
| c.1804A>T | I602F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I602F missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign outcome. Predictions that are uncertain or inconclusive are Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Uncertain. Taken together, the overwhelming majority of evidence points to a pathogenic effect. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010221 | Structured | 0.186541 | Uncertain | 0.963 | 0.171 | 0.000 | -13.974 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 2.47 | Destabilizing | 1.1 | -0.61 | Ambiguous | 0.93 | Ambiguous | 0.87 | Ambiguous | 0.822 | Likely Pathogenic | -3.98 | Deleterious | 0.999 | Probably Damaging | 0.937 | Probably Damaging | -1.82 | Pathogenic | 0.00 | Affected | 0.0708 | 0.2343 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.1805T>A | I602N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I602N is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All available evidence points to a damaging impact. Therefore, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010221 | Structured | 0.186541 | Uncertain | 0.963 | 0.171 | 0.000 | -16.033 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 2.94 | Destabilizing | 0.2 | 3.34 | Destabilizing | 3.14 | Destabilizing | 2.31 | Destabilizing | 0.880 | Likely Pathogenic | -6.89 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -2.01 | Pathogenic | 0.00 | Affected | 0.0718 | 0.0700 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1805T>C | I602T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I602T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify it as pathogenic or likely pathogenic. No tool reports a benign outcome. High‑accuracy assessments corroborate this trend: AlphaMissense‑Optimized is uncertain, SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenicity. Consequently, the variant is most likely pathogenic according to the available computational evidence, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010221 | Structured | 0.186541 | Uncertain | 0.963 | 0.171 | 0.000 | -12.238 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 2.39 | Destabilizing | 0.1 | 2.14 | Destabilizing | 2.27 | Destabilizing | 1.94 | Destabilizing | 0.931 | Likely Pathogenic | -4.82 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | -2.00 | Pathogenic | 0.00 | Affected | 0.0890 | 0.0989 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.1805T>G | I602S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I602S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. No conflicting benign evidence is available. Therefore, based on the consensus of all available predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010221 | Structured | 0.186541 | Uncertain | 0.963 | 0.171 | 0.000 | -15.558 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 3.50 | Destabilizing | 0.1 | 3.79 | Destabilizing | 3.65 | Destabilizing | 2.04 | Destabilizing | 0.935 | Likely Pathogenic | -5.89 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -2.01 | Pathogenic | 0.00 | Affected | 0.2213 | 0.1087 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1806T>G | I602M 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 I602M variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. In contrast, the majority of tools predict it to be pathogenic: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show a split: AlphaMissense‑Optimized and Foldetta both predict benign, whereas the SGM‑Consensus predicts pathogenic. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010221 | Structured | 0.186541 | Uncertain | 0.963 | 0.171 | 0.000 | -10.839 | Likely Pathogenic | 0.638 | Likely Pathogenic | Likely Benign | 0.03 | Likely Benign | 0.1 | 0.48 | Likely Benign | 0.26 | Likely Benign | 1.10 | Destabilizing | 0.675 | Likely Pathogenic | -2.91 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.97 | Pathogenic | 0.00 | Affected | 0.0801 | 0.2321 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||
| c.1807A>C | M603L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 M603L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include FoldX, premPS, PROVEAN, SIFT, AlphaMissense‑Optimized, and the protein‑folding stability method Foldetta. Those that predict pathogenicity are REVEL, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. Two tools give uncertain results: Rosetta and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, more evidence supports a benign effect (six benign versus five pathogenic predictions, with two uncertain). Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -7.134 | In-Between | 0.590 | Likely Pathogenic | Likely Benign | 0.04 | Likely Benign | 0.1 | 0.86 | Ambiguous | 0.45 | Likely Benign | -0.18 | Likely Benign | 0.548 | Likely Pathogenic | -2.16 | Neutral | 0.484 | Possibly Damaging | 0.654 | Possibly Damaging | -0.98 | Pathogenic | 0.52 | Tolerated | 0.1345 | 0.3227 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||
| c.1807A>G | M603V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M603V is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from premPS and SIFT, while pathogenic predictions are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. The remaining tools—FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized—return uncertain or inconclusive results. High‑accuracy assessments reinforce the pathogenic signal: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic, and AlphaMissense‑Optimized is uncertain; Foldetta also remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for M603V. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -10.442 | Likely Pathogenic | 0.800 | Likely Pathogenic | Ambiguous | 1.34 | Ambiguous | 0.2 | 0.62 | Ambiguous | 0.98 | Ambiguous | -0.15 | Likely Benign | 0.668 | Likely Pathogenic | -3.48 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | -0.92 | Pathogenic | 0.09 | Tolerated | 0.2698 | 0.2601 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||
| c.1807A>T | M603L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 M603L missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Foldetta, premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. Rosetta and ESM1b give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools lean toward a benign interpretation, but the SGM Consensus indicates pathogenicity, leaving the variant’s clinical significance uncertain. This assessment does not contradict any ClinVar status, as no ClinVar entry exists for M603L. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -7.134 | In-Between | 0.590 | Likely Pathogenic | Likely Benign | 0.04 | Likely Benign | 0.1 | 0.86 | Ambiguous | 0.45 | Likely Benign | -0.18 | Likely Benign | 0.548 | Likely Pathogenic | -2.16 | Neutral | 0.484 | Possibly Damaging | 0.654 | Possibly Damaging | -0.98 | Pathogenic | 0.52 | Tolerated | 0.1345 | 0.3227 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||
| c.1808T>A | M603K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M603K is not reported in ClinVar and has no entries in gnomAD. Prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, SGM Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized, while only FoldX reports a benign outcome. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. No other tools provide a clear benign signal. Consequently, the variant is most likely pathogenic, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -15.561 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 0.17 | Likely Benign | 0.0 | 1.19 | Ambiguous | 0.68 | Ambiguous | 0.67 | Ambiguous | 0.933 | Likely Pathogenic | -5.64 | Deleterious | 0.923 | Possibly Damaging | 0.922 | Probably Damaging | -1.35 | Pathogenic | 0.00 | Affected | 0.1268 | 0.0488 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||
| c.1808T>C | M603T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M603T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that provide a clear verdict all classify the substitution as pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool in the dataset reports a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus indicates likely pathogenic, while the Foldetta stability analysis is inconclusive and therefore not considered evidence. No contradictory evidence is present in ClinVar. Consequently, the variant is most likely pathogenic based on the available predictions, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -13.152 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | 1.71 | Ambiguous | 0.2 | 1.71 | Ambiguous | 1.71 | Ambiguous | 0.66 | Ambiguous | 0.903 | Likely Pathogenic | -5.48 | Deleterious | 0.996 | Probably Damaging | 0.985 | Probably Damaging | -1.29 | Pathogenic | 0.00 | Affected | 0.1883 | 0.1495 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.1808T>G | M603R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M603R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FoldX, which scores the variant as benign. All other evaluated predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus also indicates likely pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result. No other tools provide conflicting evidence. Based on the preponderance of pathogenic predictions and the absence of benign consensus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -14.968 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 0.08 | Likely Benign | 0.0 | 1.13 | Ambiguous | 0.61 | Ambiguous | 0.65 | Ambiguous | 0.935 | Likely Pathogenic | -5.64 | Deleterious | 0.963 | Probably Damaging | 0.943 | Probably Damaging | -1.35 | Pathogenic | 0.00 | Affected | 0.1516 | 0.0837 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||
| c.1809G>A | M603I 2D AIThe SynGAP1 missense variant M603I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that classify the variant as benign include Rosetta, Foldetta, and premPS, whereas the majority of tools predict it to be pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX reports an uncertain effect. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the balance of evidence points to a pathogenic effect for M603I, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -10.086 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.81 | Ambiguous | 0.6 | 0.07 | Likely Benign | 0.44 | Likely Benign | -0.07 | Likely Benign | 0.699 | Likely Pathogenic | -3.32 | Deleterious | 0.833 | Possibly Damaging | 0.886 | Possibly Damaging | -1.21 | Pathogenic | 0.03 | Affected | 0.1208 | 0.2350 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1809G>C | M603I 2D AIThe SynGAP1 missense variant M603I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include Rosetta, Foldetta, and premPS, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX reports an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the preponderance of evidence points to a pathogenic effect for M603I. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -10.086 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.81 | Ambiguous | 0.6 | 0.07 | Likely Benign | 0.44 | Likely Benign | -0.07 | Likely Benign | 0.699 | Likely Pathogenic | -3.32 | Deleterious | 0.833 | Possibly Damaging | 0.886 | Possibly Damaging | -1.21 | Pathogenic | 0.03 | Affected | 0.1208 | 0.2350 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1809G>T | M603I 2D AIThe SynGAP1 missense variant M603I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include Rosetta, Foldetta, and premPS, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX reports an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the preponderance of evidence points to a pathogenic effect for M603I. This conclusion is not contradicted by ClinVar status, as the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.011342 | Structured | 0.197847 | Uncertain | 0.942 | 0.176 | 0.000 | -10.086 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.81 | Ambiguous | 0.6 | 0.07 | Likely Benign | 0.44 | Likely Benign | -0.07 | Likely Benign | 0.698 | Likely Pathogenic | -3.32 | Deleterious | 0.833 | Possibly Damaging | 0.886 | Possibly Damaging | -1.21 | Pathogenic | 0.03 | Affected | 0.1208 | 0.2350 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.180T>A | D60E 2D AIThe SynGAP1 D60E missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -3.818 | Likely Benign | 0.780 | Likely Pathogenic | Likely Benign | 0.089 | Likely Benign | -0.90 | Neutral | 0.643 | Possibly Damaging | 0.785 | Possibly Damaging | 4.05 | Benign | 0.00 | Affected | 0.1342 | 0.7869 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.180T>G | D60E 2D AIThe SynGAP1 D60E missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also resolves to benign. Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.284882 | Structured | 0.480942 | Uncertain | 0.521 | 0.676 | 0.000 | -3.818 | Likely Benign | 0.780 | Likely Pathogenic | Likely Benign | 0.089 | Likely Benign | -0.90 | Neutral | 0.643 | Possibly Damaging | 0.785 | Possibly Damaging | 4.05 | Benign | 0.00 | Affected | 0.1342 | 0.7869 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1810T>A | S604T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S604T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Foldetta, premPS, SIFT, and FATHMM, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Predictions that are uncertain or inconclusive (FoldX, Rosetta, AlphaMissense‑Optimized) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall pattern of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.192527 | Uncertain | 0.911 | 0.195 | 0.000 | -9.674 | Likely Pathogenic | 0.897 | Likely Pathogenic | Ambiguous | 0.64 | Ambiguous | 0.1 | -0.58 | Ambiguous | 0.03 | Likely Benign | -0.16 | Likely Benign | 0.337 | Likely Benign | -2.99 | Deleterious | 0.826 | Possibly Damaging | 0.872 | Possibly Damaging | 3.19 | Benign | 0.08 | Tolerated | 0.1687 | 0.4919 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1810T>C | S604P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S604P is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include premPS and FATHMM, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict it to be pathogenic. High‑accuracy methods give consistent results: AlphaMissense‑Optimized indicates pathogenicity; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also labels the variant as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a destabilizing, pathogenic effect. Taken together, the overwhelming majority of computational evidence supports a pathogenic classification for S604P, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.192527 | Uncertain | 0.911 | 0.195 | 0.000 | -10.953 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.76 | Destabilizing | 0.3 | 8.40 | Destabilizing | 6.08 | Destabilizing | 0.04 | Likely Benign | 0.512 | Likely Pathogenic | -4.98 | Deleterious | 0.997 | Probably Damaging | 0.986 | Probably Damaging | 3.10 | Benign | 0.01 | Affected | 0.2524 | 0.4829 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.1810T>G | S604A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S604A has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and Foldetta. Those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Rosetta is uncertain and does not contribute to a consensus. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. With two of the three high‑accuracy tools indicating benign and no ClinVar evidence to contradict, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.192527 | Uncertain | 0.911 | 0.195 | 0.000 | -10.017 | Likely Pathogenic | 0.608 | Likely Pathogenic | Likely Benign | 0.02 | Likely Benign | 0.1 | -0.60 | Ambiguous | -0.29 | Likely Benign | 0.11 | Likely Benign | 0.378 | Likely Benign | -2.99 | Deleterious | 0.944 | Possibly Damaging | 0.987 | Probably Damaging | 3.25 | Benign | 0.08 | Tolerated | 0.5198 | 0.3530 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||
| c.1811C>G | S604W 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S604W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Benign predictions are provided by Rosetta, premPS, and FATHMM. Stability‑based methods give mixed results: FoldX is uncertain, while Foldetta is also uncertain. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors pathogenic. Foldetta remains inconclusive. Overall, the consensus of the majority of tools indicates a pathogenic effect. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing clinical classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.010926 | Structured | 0.192527 | Uncertain | 0.911 | 0.195 | 0.000 | -19.117 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | -1.23 | Ambiguous | 0.1 | -2.05 | Stabilizing | -1.64 | Ambiguous | -0.19 | Likely Benign | 0.645 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.07 | Benign | 0.00 | Affected | 0.0818 | 0.4602 | -2 | -3 | -0.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1813C>A | P605T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P605T is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a pathogenic effect include SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a benign outcome. High‑accuracy methods specifically show pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No predictions or stability results are missing or inconclusive. Based on the unanimous pathogenic predictions and the absence of any ClinVar or gnomAD evidence to the contrary, the variant is most likely pathogenic and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.023087 | Structured | 0.192737 | Uncertain | 0.929 | 0.231 | 0.000 | -11.533 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 2.87 | Destabilizing | 0.4 | 2.14 | Destabilizing | 2.51 | Destabilizing | 0.72 | Ambiguous | 0.801 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 0.69 | Pathogenic | 0.00 | Affected | 0.1626 | 0.5228 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||
| c.1813C>G | P605A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P605A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as pathogenic, while premPS remains uncertain. High‑accuracy assessments corroborate this trend: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates pathogenic. No tool predicts a benign outcome. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.023087 | Structured | 0.192737 | Uncertain | 0.929 | 0.231 | 0.000 | -10.085 | Likely Pathogenic | 0.962 | Likely Pathogenic | Likely Pathogenic | 2.58 | Destabilizing | 0.3 | 2.42 | Destabilizing | 2.50 | Destabilizing | 0.91 | Ambiguous | 0.744 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 0.75 | Pathogenic | 0.00 | Affected | 0.3432 | 0.4607 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.1814C>A | P605H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P605H is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a benign outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. All available predictions are consistent and indicate a deleterious effect. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.023087 | Structured | 0.192737 | Uncertain | 0.929 | 0.231 | 0.000 | -13.846 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 8.80 | Destabilizing | 2.7 | 6.69 | Destabilizing | 7.75 | Destabilizing | 0.87 | Ambiguous | 0.828 | Likely Pathogenic | -8.95 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 0.68 | Pathogenic | 0.00 | Affected | 0.1778 | 0.4142 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||
| c.1814C>T | P605L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P605L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only premPS. All other evaluated tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, AlphaMissense‑Default, AlphaMissense‑Optimized, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, indicates a pathogenic effect. Based on the overwhelming agreement among these predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.023087 | Structured | 0.192737 | Uncertain | 0.929 | 0.231 | 0.000 | -12.114 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.65 | Destabilizing | 1.1 | 2.74 | Destabilizing | 2.70 | Destabilizing | -0.10 | Likely Benign | 0.814 | Likely Pathogenic | -9.95 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 0.69 | Pathogenic | 0.00 | Affected | 0.2232 | 0.6158 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1816A>C | S606R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S606R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, SIFT, and FATHMM, whereas a separate group predicts pathogenicity: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Tools with uncertain outcomes are Foldetta, premPS, and Rosetta. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta remains inconclusive. Overall, the majority of high‑confidence predictions and the consensus vote indicate a pathogenic effect. Because ClinVar contains no entry for this variant, there is no contradiction between the computational evidence and clinical annotation. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.191720 | Uncertain | 0.875 | 0.247 | 0.000 | -12.900 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.15 | Likely Benign | 0.4 | 1.80 | Ambiguous | 0.98 | Ambiguous | 0.82 | Ambiguous | 0.252 | Likely Benign | -4.98 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 3.38 | Benign | 0.08 | Tolerated | 0.0819 | 0.2750 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1816A>G | S606G 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant S606G is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result and is treated as unavailable. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.191720 | Uncertain | 0.875 | 0.247 | 0.000 | -12.281 | Likely Pathogenic | 0.603 | Likely Pathogenic | Likely Benign | 0.43 | Likely Benign | 0.1 | 1.42 | Ambiguous | 0.93 | Ambiguous | 0.84 | Ambiguous | 0.229 | Likely Benign | -3.98 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | 3.35 | Benign | 0.04 | Affected | 0.2286 | 0.3279 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||
| c.1816A>T | S606C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S606C is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign, and the SGM‑Consensus as Likely Pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overall distribution of predictions, the variant is most likely benign, although the SGM‑Consensus suggests pathogenicity; this does not contradict any ClinVar status because the variant is not yet classified in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.191720 | Uncertain | 0.875 | 0.247 | 0.000 | -11.122 | Likely Pathogenic | 0.774 | Likely Pathogenic | Likely Benign | -0.34 | Likely Benign | 0.0 | -0.31 | Likely Benign | -0.33 | Likely Benign | 0.49 | Likely Benign | 0.348 | Likely Benign | -4.98 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 3.31 | Benign | 0.00 | Affected | 0.0986 | 0.4580 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.1817G>A | S606N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S606N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions (REVEL, FoldX, Rosetta, SIFT, FATHMM) and pathogenic predictions (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default). Two tools give uncertain results: premPS and AlphaMissense‑Optimized. High‑accuracy assessments show SGM‑Consensus as Likely Pathogenic, AlphaMissense‑Optimized as Uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Because the majority of individual predictors lean toward pathogenic and the SGM‑Consensus, a high‑confidence consensus, also indicates pathogenicity, the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.191720 | Uncertain | 0.875 | 0.247 | 0.000 | -11.352 | Likely Pathogenic | 0.919 | Likely Pathogenic | Ambiguous | 0.11 | Likely Benign | 0.1 | 0.20 | Likely Benign | 0.16 | Likely Benign | 0.76 | Ambiguous | 0.136 | Likely Benign | -2.99 | Deleterious | 0.920 | Possibly Damaging | 0.955 | Probably Damaging | 3.37 | Benign | 0.10 | Tolerated | 0.1137 | 0.3218 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||
| c.1817G>C | S606T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S606T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, FATHMM, and AlphaMissense‑Optimized, whereas a pathogenic consensus is reached by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Uncertain results come from Rosetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools predict pathogenicity, and the SGM Consensus supports this view, while the high‑accuracy methods give mixed results. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.041405 | Structured | 0.191720 | Uncertain | 0.875 | 0.247 | 0.000 | -11.052 | Likely Pathogenic | 0.554 | Ambiguous | Likely Benign | 0.06 | Likely Benign | 0.1 | -0.91 | Ambiguous | -0.43 | Likely Benign | 0.57 | Ambiguous | 0.203 | Likely Benign | -2.99 | Deleterious | 0.826 | Possibly Damaging | 0.933 | Probably Damaging | 3.34 | Benign | 0.03 | Affected | 0.1260 | 0.4513 | 1 | 1 | 0.1 | 14.03 | ||||||||||||||||||||||||||||||
| c.1817G>T | S606I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S606I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, Rosetta, Foldetta, premPS, and FATHMM, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points toward a pathogenic impact for S606I. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.191720 | Uncertain | 0.875 | 0.247 | 0.000 | -14.552 | Likely Pathogenic | 0.976 | Likely Pathogenic | Likely Pathogenic | -0.60 | Ambiguous | 0.1 | -0.08 | Likely Benign | -0.34 | Likely Benign | 0.41 | Likely Benign | 0.288 | Likely Benign | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 3.31 | Benign | 0.01 | Affected | 0.0891 | 0.4162 | -1 | -2 | 5.3 | 26.08 | |||||||||||||||||||||||||||||
| c.1818T>A | S606R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S606R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, SIFT, and FATHMM, whereas a separate group predicts pathogenicity: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Tools with uncertain outcomes are Foldetta, premPS, and Rosetta. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta remains inconclusive. Overall, the majority of high‑confidence predictions and the consensus vote indicate a pathogenic effect. Because ClinVar contains no entry for this variant, there is no contradiction between the computational evidence and clinical annotation. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.191720 | Uncertain | 0.875 | 0.247 | 0.000 | -12.900 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.15 | Likely Benign | 0.4 | 1.80 | Ambiguous | 0.98 | Ambiguous | 0.82 | Ambiguous | 0.246 | Likely Benign | -4.98 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 3.38 | Benign | 0.08 | Tolerated | 0.0819 | 0.2750 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1818T>G | S606R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S606R is not reported in ClinVar or gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, SIFT, and FATHMM, whereas a majority predict pathogenicity: PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain. High‑accuracy methods confirm the pathogenic trend: AlphaMissense‑Optimized is pathogenic, SGM Consensus is Likely Pathogenic, and Foldetta remains uncertain. Overall, the evidence points to the variant being most likely pathogenic, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.191720 | Uncertain | 0.875 | 0.247 | 0.000 | -12.900 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.15 | Likely Benign | 0.4 | 1.80 | Ambiguous | 0.98 | Ambiguous | 0.82 | Ambiguous | 0.246 | Likely Benign | -4.98 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 3.38 | Benign | 0.08 | Tolerated | 0.0819 | 0.2750 | 0 | -1 | -3.7 | 69.11 | |||||||||||||||||||||||||||||
| c.1819C>A | L607I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L607I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions come from SGM‑Consensus, REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default, while benign calls are made by PROVEAN and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Stability predictions from FoldX, Rosetta, and premPS are inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for L607I. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.194229 | Uncertain | 0.869 | 0.250 | 0.000 | -12.061 | Likely Pathogenic | 0.644 | Likely Pathogenic | Likely Benign | 0.63 | Ambiguous | 0.1 | 1.25 | Ambiguous | 0.94 | Ambiguous | 0.82 | Ambiguous | 0.727 | Likely Pathogenic | -1.99 | Neutral | 0.992 | Probably Damaging | 0.997 | Probably Damaging | -1.54 | Pathogenic | 0.01 | Affected | 0.1079 | 0.3767 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.1819C>T | L607F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L607F is catalogued in gnomAD (6‑33440871‑C‑T) but has no ClinVar entry. Functional prediction tools largely agree on a deleterious effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all report pathogenic or likely pathogenic. Only FoldX predicts a benign outcome, while Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments show the SGM‑Consensus as likely pathogenic, AlphaMissense‑Optimized as uncertain, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for L607F, and this conclusion is not contradicted by ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.194229 | Uncertain | 0.869 | 0.250 | 0.000 | 6-33440871-C-T | 1 | 6.19e-7 | -13.654 | Likely Pathogenic | 0.948 | Likely Pathogenic | Ambiguous | 0.23 | Likely Benign | 0.1 | 1.20 | Ambiguous | 0.72 | Ambiguous | 0.61 | Ambiguous | 0.758 | Likely Pathogenic | -3.98 | Deleterious | 0.998 | Probably Damaging | 0.997 | Probably Damaging | -1.54 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.0872 | 0.2816 | 0 | 2 | -1.0 | 34.02 | ||||||||||||||||||||||||
| c.181G>A | E61K 2D AIThe SynGAP1 E61K missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools cluster into two consensus groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus, SGM‑Consensus, classifies the variant as Likely Benign, and AlphaMissense‑Optimized also predicts Benign. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the aggregate evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.352862 | Structured | 0.477329 | Uncertain | 0.518 | 0.699 | 0.125 | -4.953 | Likely Benign | 0.425 | Ambiguous | Likely Benign | 0.120 | Likely Benign | -0.22 | Neutral | 0.458 | Possibly Damaging | 0.678 | Possibly Damaging | 4.34 | Benign | 0.00 | Affected | 0.2736 | 0.5691 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||||||
| c.181G>C | E61Q 2D AIThe SynGAP1 missense variant E61Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 (HumDiv and HumVar) and SIFT all predict a pathogenic impact. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Benign. No Foldetta stability result is available. Overall, the majority of evidence points to a benign effect for E61Q, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.352862 | Structured | 0.477329 | Uncertain | 0.518 | 0.699 | 0.125 | -5.443 | Likely Benign | 0.267 | Likely Benign | Likely Benign | 0.058 | Likely Benign | -0.41 | Neutral | 0.659 | Possibly Damaging | 0.775 | Possibly Damaging | 4.18 | Benign | 0.00 | Affected | 0.1344 | 0.5617 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||||||
| c.1820T>A | L607H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L607H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on pathogenicity include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; no tool predicts a benign effect. Uncertain predictions come from FoldX, Rosetta, and Foldetta. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, while Foldetta’s stability analysis is inconclusive. Taken together, the overwhelming majority of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.194229 | Uncertain | 0.869 | 0.250 | 0.000 | -14.775 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.76 | Ambiguous | 0.1 | 1.88 | Ambiguous | 1.32 | Ambiguous | 1.38 | Destabilizing | 0.906 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.48 | Pathogenic | 0.01 | Affected | 0.1199 | 0.0541 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1820T>C | L607P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L607P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while the SGM‑Consensus score is “Likely Pathogenic.” No tool in the dataset predicts a benign outcome; the only inconclusive result is FoldX, which is listed as uncertain and therefore does not influence the overall assessment. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. Consequently, the variant is most likely pathogenic based on the available predictions, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.194229 | Uncertain | 0.869 | 0.250 | 0.000 | -14.059 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 1.11 | Ambiguous | 0.7 | 6.93 | Destabilizing | 4.02 | Destabilizing | 1.29 | Destabilizing | 0.922 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.54 | Pathogenic | 0.00 | Affected | 0.3710 | 0.1274 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1820T>G | L607R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L607R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FoldX, which scores the variant as benign. All other evaluated algorithms—REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. Rosetta and Foldetta provide uncertain results and are therefore treated as unavailable evidence. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Pathogenic,” while Foldetta remains uncertain. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.194229 | Uncertain | 0.869 | 0.250 | 0.000 | -14.234 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | -0.15 | Likely Benign | 0.1 | 1.48 | Ambiguous | 0.67 | Ambiguous | 1.24 | Destabilizing | 0.920 | Likely Pathogenic | -5.98 | Deleterious | 0.998 | Probably Damaging | 0.998 | Probably Damaging | -1.51 | Pathogenic | 0.01 | Affected | 0.1490 | 0.0615 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1822T>A | F608I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F608I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenic or likely pathogenic. The only tool with an inconclusive result is FoldX, which is listed as uncertain. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is consistent with the absence of any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.106997 | Structured | 0.197190 | Uncertain | 0.891 | 0.247 | 0.000 | -14.939 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 1.92 | Ambiguous | 0.1 | 2.14 | Destabilizing | 2.03 | Destabilizing | 1.24 | Destabilizing | 0.904 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.997 | Probably Damaging | -1.62 | Pathogenic | 0.00 | Affected | 0.2115 | 0.2361 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1822T>C | F608L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F608L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. FoldX and Foldetta report uncertain stability changes, but these are not considered evidence against pathogenicity. When predictions are grouped, no tool predicts a benign outcome; all available evidence supports a harmful effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (derived from the majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic, and Foldetta remains inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.106997 | Structured | 0.197190 | Uncertain | 0.891 | 0.247 | 0.000 | -12.274 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.86 | Ambiguous | 0.2 | 2.59 | Destabilizing | 1.73 | Ambiguous | 1.24 | Destabilizing | 0.883 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -1.50 | Pathogenic | 0.01 | Affected | 0.2231 | 0.2874 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1822T>G | F608V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F608V is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; all of these return a pathogenic or likely pathogenic label. No tool in the dataset predicts a benign outcome. Uncertain results are reported only for Rosetta and Foldetta, which are treated as unavailable evidence. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta remains uncertain. Taken together, the overwhelming majority of predictions indicate that F608V is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.106997 | Structured | 0.197190 | Uncertain | 0.891 | 0.247 | 0.000 | -16.084 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 2.21 | Destabilizing | 0.1 | 1.39 | Ambiguous | 1.80 | Ambiguous | 1.47 | Destabilizing | 0.917 | Likely Pathogenic | -6.97 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.59 | Pathogenic | 0.01 | Affected | 0.2193 | 0.2199 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1823T>A | F608Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F608Y is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a pathogenic effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus all predict pathogenicity, while only Rosetta predicts a benign effect. Tools with uncertain outcomes—FoldX, Foldetta, and AlphaMissense‑Optimized—do not provide decisive evidence. High‑accuracy assessments further support a deleterious impact: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic, whereas AlphaMissense‑Optimized and Foldetta remain inconclusive. Overall, the preponderance of evidence indicates that F608Y is most likely pathogenic, and this conclusion is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.106997 | Structured | 0.197190 | Uncertain | 0.891 | 0.247 | 0.000 | -13.249 | Likely Pathogenic | 0.812 | Likely Pathogenic | Ambiguous | 0.62 | Ambiguous | 0.1 | 0.41 | Likely Benign | 0.52 | Ambiguous | 1.05 | Destabilizing | 0.747 | Likely Pathogenic | -2.99 | Deleterious | 0.993 | Probably Damaging | 0.976 | Probably Damaging | -1.44 | Pathogenic | 0.00 | Affected | 0.1504 | 0.1346 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1823T>C | F608S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F608S is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a pathogenic effect. No tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. All available predictions are consistent and conclusive. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.106997 | Structured | 0.197190 | Uncertain | 0.891 | 0.247 | 0.000 | -15.181 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 3.16 | Destabilizing | 0.2 | 4.00 | Destabilizing | 3.58 | Destabilizing | 1.66 | Destabilizing | 0.917 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.59 | Pathogenic | 0.02 | Affected | 0.4366 | 0.0400 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1823T>G | F608C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F608C is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, every tool examined (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) classifies the change as pathogenic; no tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a pathogenic effect. Because all evidence points to a deleterious impact and there is no ClinVar entry to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.106997 | Structured | 0.197190 | Uncertain | 0.891 | 0.247 | 0.000 | -14.409 | Likely Pathogenic | 0.980 | Likely Pathogenic | Likely Pathogenic | 2.46 | Destabilizing | 0.2 | 3.04 | Destabilizing | 2.75 | Destabilizing | 1.77 | Destabilizing | 0.920 | Likely Pathogenic | -7.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.67 | Pathogenic | 0.00 | Affected | 0.2738 | 0.1050 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.1824T>A | F608L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F608L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: REVEL, polyPhen‑2 (HumDiv and HumVar), SIFT, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. FoldX and Foldetta report uncertain stability changes, but these are not considered evidence against pathogenicity. When predictions are grouped, no tool predicts a benign outcome; all available evidence supports a harmful effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (derived from the majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic, and Foldetta remains inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.106997 | Structured | 0.197190 | Uncertain | 0.891 | 0.247 | 0.000 | -12.274 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.86 | Ambiguous | 0.2 | 2.59 | Destabilizing | 1.73 | Ambiguous | 1.24 | Destabilizing | 0.747 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -1.50 | Pathogenic | 0.01 | Affected | 0.2231 | 0.2874 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1824T>G | F608L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F608L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect: SGM‑Consensus, REVEL, FoldX‑MD (uncertain), Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while only FoldX and Foldetta are inconclusive. High‑accuracy assessments corroborate this trend: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labels it likely pathogenic, and Foldetta reports an uncertain stability change. No tool predicts a benign outcome. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.106997 | Structured | 0.197190 | Uncertain | 0.891 | 0.247 | 0.000 | -12.274 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 0.86 | Ambiguous | 0.2 | 2.59 | Destabilizing | 1.73 | Ambiguous | 1.24 | Destabilizing | 0.747 | Likely Pathogenic | -5.97 | Deleterious | 0.999 | Probably Damaging | 0.994 | Probably Damaging | -1.50 | Pathogenic | 0.01 | Affected | 0.2231 | 0.2874 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1825G>A | G609R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G609R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta, SIFT, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Predictions that remain uncertain are Foldetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evaluated tools (eight pathogenic vs. three benign) predict a pathogenic impact for G609R. This conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.203786 | Uncertain | 0.851 | 0.252 | 0.000 | -10.172 | Likely Pathogenic | 0.543 | Ambiguous | Likely Benign | 2.09 | Destabilizing | 0.1 | 0.37 | Likely Benign | 1.23 | Ambiguous | 0.60 | Ambiguous | 0.520 | Likely Pathogenic | -2.68 | Deleterious | 0.974 | Probably Damaging | 0.818 | Possibly Damaging | -1.41 | Pathogenic | 0.07 | Tolerated | 0.1158 | 0.4348 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1825G>C | G609R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G609R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include Rosetta, SIFT, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. Predictions that remain uncertain are Foldetta, premPS, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evaluated tools (eight pathogenic vs. three benign) predict a pathogenic impact for G609R. This conclusion is not contradicted by ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.203786 | Uncertain | 0.851 | 0.252 | 0.000 | -10.172 | Likely Pathogenic | 0.543 | Ambiguous | Likely Benign | 2.09 | Destabilizing | 0.1 | 0.37 | Likely Benign | 1.23 | Ambiguous | 0.60 | Ambiguous | 0.520 | Likely Pathogenic | -2.68 | Deleterious | 0.974 | Probably Damaging | 0.818 | Possibly Damaging | -1.41 | Pathogenic | 0.07 | Tolerated | 0.1158 | 0.4348 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1825G>T | G609W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G609W (ClinVar ID 4327030) is not found in gnomAD. Prediction tools that classify the variant as benign include premPS and AlphaMissense‑Optimized, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM) predict it to be pathogenic; Rosetta, Foldetta, and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence from pathogenic‑oriented tools and the SGM Consensus supports a pathogenic effect, which is consistent with the ClinVar classification and does not contradict it. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.203786 | Uncertain | 0.851 | 0.252 | 0.000 | 1 | -13.074 | Likely Pathogenic | 0.525 | Ambiguous | Likely Benign | 3.14 | Destabilizing | 0.7 | -0.87 | Ambiguous | 1.14 | Ambiguous | 0.28 | Likely Benign | 0.566 | Likely Pathogenic | -4.70 | Deleterious | 0.999 | Probably Damaging | 0.976 | Probably Damaging | -1.47 | Pathogenic | 0.01 | Affected | 0.0935 | 0.3181 | -7 | -2 | -0.5 | 129.16 | ||||||||||||||||||||||||||||
| c.1826G>A | G609E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G609E has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. The remaining tools (Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.179055 | Structured | 0.203786 | Uncertain | 0.851 | 0.252 | 0.000 | -10.470 | Likely Pathogenic | 0.421 | Ambiguous | Likely Benign | 2.95 | Destabilizing | 0.4 | -1.65 | Ambiguous | 0.65 | Ambiguous | 0.61 | Ambiguous | 0.531 | Likely Pathogenic | -2.28 | Neutral | 0.916 | Possibly Damaging | 0.588 | Possibly Damaging | -1.41 | Pathogenic | 0.17 | Tolerated | 0.1709 | 0.4491 | 0 | -2 | -3.1 | 72.06 | ||||||||||||||||||||||||||||||
| c.1826G>C | G609A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G609A is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, PROVEAN, and FATHMM. One tool, Foldetta, yields an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.179055 | Structured | 0.203786 | Uncertain | 0.851 | 0.252 | 0.000 | -6.790 | Likely Benign | 0.153 | Likely Benign | Likely Benign | 2.38 | Destabilizing | 0.3 | 0.01 | Likely Benign | 1.20 | Ambiguous | 0.43 | Likely Benign | 0.494 | Likely Benign | -2.65 | Deleterious | 0.282 | Benign | 0.164 | Benign | -1.43 | Pathogenic | 0.10 | Tolerated | 0.3812 | 0.3896 | 1 | 0 | 2.2 | 14.03 | ||||||||||||||||||||||||||||||
| c.1826G>T | G609V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G609V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from premPS and AlphaMissense‑Optimized, whereas the remaining 10 tools (SGM Consensus, REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM) all predict pathogenicity. High‑accuracy assessments further highlight the discordance: AlphaMissense‑Optimized reports a benign effect, whereas the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta outputs) both indicate pathogenicity. With the majority of evidence pointing to deleterious impact, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.179055 | Structured | 0.203786 | Uncertain | 0.851 | 0.252 | 0.000 | -11.049 | Likely Pathogenic | 0.374 | Ambiguous | Likely Benign | 4.17 | Destabilizing | 0.3 | 3.77 | Destabilizing | 3.97 | Destabilizing | 0.34 | Likely Benign | 0.734 | Likely Pathogenic | -4.47 | Deleterious | 0.974 | Probably Damaging | 0.818 | Possibly Damaging | -1.48 | Pathogenic | 0.02 | Affected | 0.1269 | 0.3317 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1828C>A | L610I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L610I is listed in gnomAD (ID 6‑33440880‑C‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from PROVEAN, SIFT, ESM1b, and AlphaMissense‑Optimized, while pathogenic predictions arise from REVEL, polyPhen‑2 (HumDiv and HumVar) and FATHMM. Four tools are uncertain (FoldX, Foldetta, premPS, AlphaMissense‑Default). High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores benign, the SGM consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, whereas Foldetta’s stability estimate is unavailable. Overall, the balance of evidence points to a benign effect for L610I, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.271506 | Structured | 0.209504 | Uncertain | 0.888 | 0.253 | 0.000 | 6-33440880-C-A | 1 | 6.19e-7 | -6.362 | Likely Benign | 0.389 | Ambiguous | Likely Benign | 1.50 | Ambiguous | 0.2 | 0.18 | Likely Benign | 0.84 | Ambiguous | 0.76 | Ambiguous | 0.544 | Likely Pathogenic | -1.86 | Neutral | 0.992 | Probably Damaging | 0.997 | Probably Damaging | -1.34 | Pathogenic | 0.15 | Tolerated | 3.37 | 35 | 0.0999 | 0.3219 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||
| c.1828C>G | L610V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L610V is reported in gnomAD (ID 6‑33440880‑C‑G) but has no ClinVar entry. Functional prediction tools largely agree on a deleterious effect: pathogenic calls are made by REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM, while only AlphaMissense‑Optimized predicts a benign outcome. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments further support this view: AlphaMissense‑Optimized indicates benign, whereas the SGM Consensus remains pathogenic and Foldetta (combining FoldX‑MD and Rosetta) is inconclusive. With the overwhelming majority of tools predicting pathogenicity and no ClinVar evidence to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.209504 | Uncertain | 0.888 | 0.253 | 0.000 | 6-33440880-C-G | 3 | 1.86e-6 | -11.304 | Likely Pathogenic | 0.474 | Ambiguous | Likely Benign | 2.24 | Destabilizing | 0.3 | 0.76 | Ambiguous | 1.50 | Ambiguous | 1.21 | Destabilizing | 0.740 | Likely Pathogenic | -2.86 | Deleterious | 0.985 | Probably Damaging | 0.992 | Probably Damaging | -1.46 | Pathogenic | 0.01 | Affected | 3.37 | 35 | 0.1515 | 0.3039 | 1 | 2 | 0.4 | -14.03 | ||||||||||||||||||||||||
| c.1828C>T | L610F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L610F is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a pathogenic effect include SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—all of which classify the variant as pathogenic. No tools predict a benign outcome. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic effect. Based on the consensus of these predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.209504 | Uncertain | 0.888 | 0.253 | 0.000 | -13.244 | Likely Pathogenic | 0.808 | Likely Pathogenic | Ambiguous | 6.24 | Destabilizing | 1.1 | 3.40 | Destabilizing | 4.82 | Destabilizing | 0.68 | Ambiguous | 0.782 | Likely Pathogenic | -3.92 | Deleterious | 0.998 | Probably Damaging | 0.997 | Probably Damaging | -1.52 | Pathogenic | 0.01 | Affected | 0.0834 | 0.2816 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1829T>A | L610H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L610H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess pathogenicity all agree that the variant is deleterious: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as pathogenic. No tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a destabilizing, pathogenic effect. All available predictions are concordant and supportive. Based on these computational assessments, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.209504 | Uncertain | 0.888 | 0.253 | 0.000 | -14.573 | Likely Pathogenic | 0.960 | Likely Pathogenic | Likely Pathogenic | 4.83 | Destabilizing | 0.5 | 4.98 | Destabilizing | 4.91 | Destabilizing | 2.03 | Destabilizing | 0.927 | Likely Pathogenic | -6.91 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.59 | Pathogenic | 0.00 | Affected | 0.1067 | 0.0741 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1829T>C | L610P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L610P is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, every tool examined (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) classifies the substitution as pathogenic; no tool predicts a benign effect. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a pathogenic effect. Because all evidence points to a deleterious impact and there is no ClinVar entry to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.209504 | Uncertain | 0.888 | 0.253 | 0.000 | -14.863 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 6.02 | Destabilizing | 0.2 | 8.15 | Destabilizing | 7.09 | Destabilizing | 1.99 | Destabilizing | 0.934 | Likely Pathogenic | -6.91 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.59 | Pathogenic | 0.00 | Affected | 0.3599 | 0.1113 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1829T>G | L610R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L610R is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity are unanimous: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. No tool in the dataset predicts a benign effect. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports a pathogenic outcome; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, predicts a pathogenic impact. Consequently, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.271506 | Structured | 0.209504 | Uncertain | 0.888 | 0.253 | 0.000 | -13.855 | Likely Pathogenic | 0.956 | Likely Pathogenic | Likely Pathogenic | 5.14 | Destabilizing | 0.5 | 4.60 | Destabilizing | 4.87 | Destabilizing | 1.89 | Destabilizing | 0.949 | Likely Pathogenic | -5.94 | Deleterious | 0.998 | Probably Damaging | 0.998 | Probably Damaging | -1.58 | Pathogenic | 0.00 | Affected | 0.1334 | 0.0615 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.182A>C | E61A 2D AIThe SynGAP1 missense variant E61A is listed in ClinVar (ID 3767543.0) with an *Uncertain* clinical significance and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign, while Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the current ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.352862 | Structured | 0.477329 | Uncertain | 0.518 | 0.699 | 0.125 | Uncertain | 1 | -5.235 | Likely Benign | 0.453 | Ambiguous | Likely Benign | 0.074 | Likely Benign | -1.52 | Neutral | 0.458 | Possibly Damaging | 0.678 | Possibly Damaging | 4.12 | Benign | 0.00 | Affected | 0.4499 | 0.5878 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||
| c.182A>G | E61G 2D AIThe SynGAP1 missense variant E61G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar) and SIFT. AlphaMissense‑Default remains uncertain. The high‑accuracy consensus methods reinforce the benign assessment: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports “Likely Benign,” and the protein‑folding stability tool Foldetta is not available for this variant. Overall, the majority of evidence points to a benign effect. There is no ClinVar entry to contradict this conclusion. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.352862 | Structured | 0.477329 | Uncertain | 0.518 | 0.699 | 0.125 | -5.574 | Likely Benign | 0.469 | Ambiguous | Likely Benign | 0.072 | Likely Benign | -1.68 | Neutral | 0.659 | Possibly Damaging | 0.775 | Possibly Damaging | 4.08 | Benign | 0.00 | Affected | 0.3593 | 0.4965 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.182A>T | E61V 2D AIThe SynGAP1 E61V missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence favors a benign classification, and this conclusion does not contradict the ClinVar status, which currently has no record for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.352862 | Structured | 0.477329 | Uncertain | 0.518 | 0.699 | 0.125 | -5.723 | Likely Benign | 0.769 | Likely Pathogenic | Likely Benign | 0.115 | Likely Benign | -1.80 | Neutral | 0.824 | Possibly Damaging | 0.775 | Possibly Damaging | 4.07 | Benign | 0.00 | Affected | 0.0803 | 0.6413 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.1831A>C | M611L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M611L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote). Only FATHMM predicts a pathogenic outcome, while Rosetta, Foldetta, and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none exists). Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | -6.721 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.39 | Likely Benign | 0.1 | 0.65 | Ambiguous | 0.52 | Ambiguous | 0.56 | Ambiguous | 0.229 | Likely Benign | -1.29 | Neutral | 0.059 | Benign | 0.017 | Benign | -1.07 | Pathogenic | 0.76 | Tolerated | 0.1130 | 0.3547 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1831A>G | M611V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M611V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), ESM1b, and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—yields an inconclusive result (two benign, two pathogenic). Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports an uncertain effect, so its result is treated as unavailable. Overall, the balance of evidence leans toward a benign impact, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | -8.057 | Likely Pathogenic | 0.176 | Likely Benign | Likely Benign | 1.42 | Ambiguous | 0.4 | 1.56 | Ambiguous | 1.49 | Ambiguous | 0.66 | Ambiguous | 0.315 | Likely Benign | -2.06 | Neutral | 0.960 | Probably Damaging | 0.474 | Possibly Damaging | -1.04 | Pathogenic | 0.49 | Tolerated | 0.2324 | 0.2721 | 2 | 1 | 2.3 | -32.06 | ||||||||||||||||||||||||||||||
| c.1831A>T | M611L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M611L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus (Likely Benign), REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Only FATHMM predicts pathogenic, while Rosetta, Foldetta, and premPS are uncertain. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates Likely Benign; Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. Overall, the preponderance of evidence points to a benign impact for M611L, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | -6.721 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.39 | Likely Benign | 0.1 | 0.65 | Ambiguous | 0.52 | Ambiguous | 0.56 | Ambiguous | 0.229 | Likely Benign | -1.29 | Neutral | 0.059 | Benign | 0.017 | Benign | -1.07 | Pathogenic | 0.76 | Tolerated | 0.1130 | 0.3547 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1832T>A | M611K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 M611K variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Optimized. The majority of other in silico predictors (SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) indicate a pathogenic effect; FoldX is uncertain and therefore not considered evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (derived from the unanimous pathogenic vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) and Foldetta (combining FoldX‑MD and Rosetta outputs) both predict pathogenicity. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | -13.021 | Likely Pathogenic | 0.630 | Likely Pathogenic | Likely Benign | 1.86 | Ambiguous | 0.6 | 2.65 | Destabilizing | 2.26 | Destabilizing | 1.65 | Destabilizing | 0.665 | Likely Pathogenic | -4.10 | Deleterious | 0.250 | Benign | 0.120 | Benign | -1.26 | Pathogenic | 0.03 | Affected | 0.1193 | 0.0688 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||
| c.1832T>C | M611T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M611T is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33440884‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome. Four tools (FoldX, Rosetta, Foldetta, premPS) return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | Uncertain | 1 | 6-33440884-T-C | 1 | 6.19e-7 | -5.696 | Likely Benign | 0.101 | Likely Benign | Likely Benign | 1.98 | Ambiguous | 0.2 | 0.94 | Ambiguous | 1.46 | Ambiguous | 0.87 | Ambiguous | 0.240 | Likely Benign | -2.40 | Neutral | 0.034 | Benign | 0.038 | Benign | -1.19 | Pathogenic | 0.29 | Tolerated | 3.37 | 35 | 0.1635 | 0.1415 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||
| c.1832T>G | M611R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M611R is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are AlphaMissense‑Optimized, polyPhen2_HumVar, and SIFT. Tools that agree on a pathogenic effect include SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen2_HumDiv, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, whereas the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is reported as uncertain. No prediction or folding result is missing; all available data are considered. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | -11.050 | Likely Pathogenic | 0.642 | Likely Pathogenic | Likely Benign | 1.80 | Ambiguous | 0.8 | 2.00 | Destabilizing | 1.90 | Ambiguous | 1.58 | Destabilizing | 0.644 | Likely Pathogenic | -4.10 | Deleterious | 0.779 | Possibly Damaging | 0.159 | Benign | -1.21 | Pathogenic | 0.21 | Tolerated | 0.1399 | 0.0837 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||
| c.1833G>A | M611I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M611I is reported in gnomAD (ID 6‑33440885‑G‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized; pathogenic predictions arise from SGM‑Consensus, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further clarify the variant’s likely effect: AlphaMissense‑Optimized classifies it as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates pathogenicity, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain stability change. No folding‑stability method provides definitive evidence. Overall, the majority of predictions lean toward pathogenicity, and this conclusion does not conflict with ClinVar status, which lacks an entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | 6-33440885-G-A | 1 | 6.19e-7 | -8.552 | Likely Pathogenic | 0.736 | Likely Pathogenic | Likely Benign | 1.45 | Ambiguous | 0.4 | 1.36 | Ambiguous | 1.41 | Ambiguous | 0.72 | Ambiguous | 0.292 | Likely Benign | -2.10 | Neutral | 0.250 | Benign | 0.091 | Benign | -1.14 | Pathogenic | 0.38 | Tolerated | 3.37 | 35 | 0.1009 | 0.2302 | 1 | 2 | 2.6 | -18.03 | ||||||||||||||||||||||||
| c.1833G>C | M611I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M611I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect are SGM‑Consensus, ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are inconclusive are FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain. Overall, the majority of evidence points toward a pathogenic classification, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | -8.552 | Likely Pathogenic | 0.736 | Likely Pathogenic | Likely Benign | 1.45 | Ambiguous | 0.4 | 1.36 | Ambiguous | 1.41 | Ambiguous | 0.72 | Ambiguous | 0.292 | Likely Benign | -2.10 | Neutral | 0.250 | Benign | 0.091 | Benign | -1.14 | Pathogenic | 0.38 | Tolerated | 3.37 | 35 | 0.1009 | 0.2302 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||
| c.1833G>T | M611I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M611I is not reported in ClinVar and is absent from gnomAD. In silico predictors that classify the variant as benign include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Optimized. Predictors that classify it as pathogenic are SGM‑Consensus, ESM1b, FATHMM, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, premPS, and Foldetta) provide uncertain or unavailable results. High‑accuracy assessment shows AlphaMissense‑Optimized predicts benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenic; Foldetta’s stability output is unavailable. Overall, the majority of predictions lean toward a benign effect, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.210791 | Uncertain | 0.870 | 0.253 | 0.000 | -8.552 | Likely Pathogenic | 0.736 | Likely Pathogenic | Likely Benign | 1.45 | Ambiguous | 0.4 | 1.36 | Ambiguous | 1.41 | Ambiguous | 0.72 | Ambiguous | 0.292 | Likely Benign | -2.10 | Neutral | 0.250 | Benign | 0.091 | Benign | -1.14 | Pathogenic | 0.38 | Tolerated | 3.37 | 35 | 0.1009 | 0.2302 | 1 | 2 | 2.6 | -18.03 | |||||||||||||||||||||||||||
| c.1834C>A | Q612K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q612K is not reported in ClinVar (ClinVar status: not listed) and has no entry in gnomAD (gnomAD ID: none). Prediction tools that indicate a benign effect include FoldX, Rosetta, Foldetta, and SIFT, whereas pathogenic predictions come from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default; premPS and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools predict pathogenicity, and the high‑accuracy consensus also leans pathogenic, while the folding‑stability method suggests benign. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.203988 | Uncertain | 0.822 | 0.263 | 0.000 | -12.393 | Likely Pathogenic | 0.849 | Likely Pathogenic | Ambiguous | 0.15 | Likely Benign | 0.1 | 0.48 | Likely Benign | 0.32 | Likely Benign | 0.81 | Ambiguous | 0.619 | Likely Pathogenic | -3.88 | Deleterious | 0.931 | Possibly Damaging | 0.931 | Probably Damaging | -1.22 | Pathogenic | 0.19 | Tolerated | 0.1810 | 0.3641 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||
| c.1834C>G | Q612E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q612E is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). In silico predictors that agree on a benign effect include REVEL, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Predictors that agree on a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. FoldX, Rosetta, and Foldetta are inconclusive, with Foldetta specifically yielding an uncertain stability change. High‑accuracy tools give a mixed picture: AlphaMissense‑Optimized predicts benign, SGM‑Consensus predicts pathogenic, and Foldetta remains uncertain. Overall, the majority of consensus‑based and high‑accuracy predictions lean toward pathogenicity. Thus, the variant is most likely pathogenic, and this assessment does not contradict the current ClinVar status, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.203988 | Uncertain | 0.822 | 0.263 | 0.000 | -12.179 | Likely Pathogenic | 0.338 | Likely Benign | Likely Benign | 0.52 | Ambiguous | 0.4 | 1.01 | Ambiguous | 0.77 | Ambiguous | 1.03 | Destabilizing | 0.423 | Likely Benign | -2.89 | Deleterious | 0.995 | Probably Damaging | 0.981 | Probably Damaging | -1.17 | Pathogenic | 0.26 | Tolerated | 0.1393 | 0.2099 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.1835A>C | Q612P 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q612P is listed in ClinVar (ID 3660462.0) with an uncertain significance annotation and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from FoldX, SIFT, and AlphaMissense‑Optimized; pathogenic predictions arise from REVEL, PolyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, PROVEAN, and the SGM Consensus score (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). The high‑accuracy AlphaMissense‑Optimized predicts benign, whereas the SGM Consensus predicts likely pathogenic; Foldetta, a folding‑stability method combining FoldX‑MD and Rosetta outputs, returns an uncertain result and is therefore not factored into the consensus. Overall, the majority of evidence supports a pathogenic effect, which contrasts with the ClinVar uncertain classification. Thus, based on current predictions, the variant is most likely pathogenic, contradicting the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.203988 | Uncertain | 0.822 | 0.263 | 0.000 | Uncertain | 1 | -9.684 | Likely Pathogenic | 0.673 | Likely Pathogenic | Likely Benign | -0.19 | Likely Benign | 0.3 | 3.06 | Destabilizing | 1.44 | Ambiguous | 0.56 | Ambiguous | 0.671 | Likely Pathogenic | -5.84 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.31 | Pathogenic | 0.19 | Tolerated | 0.2252 | 0.4050 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||
| c.1835A>G | Q612R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q612R is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are SIFT and FoldX. Tools that agree on a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Predictions that are uncertain or inconclusive are Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts pathogenicity, while AlphaMissense‑Optimized and Foldetta are uncertain. No high‑accuracy tool provides a benign prediction. Overall, the majority of available evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.203988 | Uncertain | 0.822 | 0.263 | 0.000 | -10.571 | Likely Pathogenic | 0.837 | Likely Pathogenic | Ambiguous | -0.35 | Likely Benign | 0.2 | 1.56 | Ambiguous | 0.61 | Ambiguous | 0.78 | Ambiguous | 0.683 | Likely Pathogenic | -3.85 | Deleterious | 0.956 | Probably Damaging | 0.969 | Probably Damaging | -1.29 | Pathogenic | 0.10 | Tolerated | 0.1480 | 0.2196 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||
| c.1835A>T | Q612L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q612L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, Foldetta, premPS, and SIFT, whereas those that predict a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain (unavailable), SGM Consensus as likely pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta) as benign. Overall, the majority of conventional tools lean toward pathogenicity, and the SGM Consensus supports this, while the high‑accuracy Foldetta result is contradictory. Thus, the variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.203988 | Uncertain | 0.822 | 0.263 | 0.000 | -12.076 | Likely Pathogenic | 0.799 | Likely Pathogenic | Ambiguous | -0.12 | Likely Benign | 0.1 | 0.12 | Likely Benign | 0.00 | Likely Benign | 0.44 | Likely Benign | 0.730 | Likely Pathogenic | -6.84 | Deleterious | 0.971 | Probably Damaging | 0.954 | Probably Damaging | -1.33 | Pathogenic | 0.08 | Tolerated | 0.0763 | 0.4756 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||
| c.1836G>C | Q612H 2D  AIThe SynGAP1 missense variant Q612H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to Rosetta, which scores the variant as benign. All other evaluated predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, SGM Consensus confirms a likely pathogenic status, while Foldetta (combining FoldX‑MD and Rosetta outputs) remains uncertain. FoldX and premPS are inconclusive, and Foldetta is unavailable for definitive interpretation. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.203988 | Uncertain | 0.822 | 0.263 | 0.000 | -9.733 | Likely Pathogenic | 0.956 | Likely Pathogenic | Likely Pathogenic | 0.70 | Ambiguous | 0.9 | 0.39 | Likely Benign | 0.55 | Ambiguous | 0.79 | Ambiguous | 0.550 | Likely Pathogenic | -4.55 | Deleterious | 0.991 | Probably Damaging | 0.986 | Probably Damaging | -1.26 | Pathogenic | 0.02 | Affected | 0.1379 | 0.3084 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.1836G>T | Q612H 2D AIThe SynGAP1 missense variant Q612H is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to Rosetta, which scores the variant as benign. All other evaluated predictors—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, SGM Consensus confirms a likely pathogenic status, while Foldetta (combining FoldX‑MD and Rosetta outputs) remains uncertain. FoldX and premPS are inconclusive, and Foldetta is unavailable for definitive interpretation. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.203988 | Uncertain | 0.822 | 0.263 | 0.000 | -9.733 | Likely Pathogenic | 0.956 | Likely Pathogenic | Likely Pathogenic | 0.70 | Ambiguous | 0.9 | 0.39 | Likely Benign | 0.55 | Ambiguous | 0.79 | Ambiguous | 0.550 | Likely Pathogenic | -4.55 | Deleterious | 0.991 | Probably Damaging | 0.986 | Probably Damaging | -1.26 | Pathogenic | 0.02 | Affected | 0.1379 | 0.3084 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.1837G>A | E613K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E613K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from FoldX, Foldetta, and premPS, whereas pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is also pathogenic. High‑accuracy assessments give AlphaMissense‑Optimized as pathogenic, SGM Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction is inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.193489 | Uncertain | 0.816 | 0.254 | 0.000 | -11.892 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.34 | Likely Benign | 0.6 | -0.54 | Ambiguous | -0.10 | Likely Benign | -0.08 | Likely Benign | 0.567 | Likely Pathogenic | -3.72 | Deleterious | 0.996 | Probably Damaging | 0.987 | Probably Damaging | -1.15 | Pathogenic | 0.04 | Affected | 0.2979 | 0.5942 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1837G>C | E613Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E613Q missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, and SIFT, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic impact. Two tools give uncertain results: AlphaMissense‑Optimized and Rosetta. High‑accuracy assessments show that the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts a likely pathogenic outcome, AlphaMissense‑Optimized is uncertain, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a benign effect. Overall, the balance of evidence leans toward pathogenicity, and this assessment does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.193489 | Uncertain | 0.816 | 0.254 | 0.000 | -9.245 | Likely Pathogenic | 0.887 | Likely Pathogenic | Ambiguous | 0.41 | Likely Benign | 0.4 | -0.84 | Ambiguous | -0.22 | Likely Benign | 0.11 | Likely Benign | 0.495 | Likely Benign | -2.79 | Deleterious | 0.994 | Probably Damaging | 0.986 | Probably Damaging | -1.28 | Pathogenic | 0.09 | Tolerated | 0.1650 | 0.6181 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1838A>C | E613A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E613A missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include Rosetta, premPS, and Foldetta, whereas the majority of tools predict a pathogenic impact: REVEL, SIFT, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus score. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as benign. Overall, the preponderance of evidence (10 pathogenic vs. 3 benign predictions) indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.193489 | Uncertain | 0.816 | 0.254 | 0.000 | -10.841 | Likely Pathogenic | 0.906 | Likely Pathogenic | Ambiguous | 0.90 | Ambiguous | 0.5 | -0.17 | Likely Benign | 0.37 | Likely Benign | 0.32 | Likely Benign | 0.688 | Likely Pathogenic | -5.57 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.26 | Pathogenic | 0.02 | Affected | 0.4696 | 0.5929 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1838A>G | E613G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E613G is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include only premPS, whereas the remaining tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) uniformly predict a pathogenic impact. The high‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain. No prediction or stability assessment is missing or inconclusive beyond the uncertain labels. Overall, the preponderance of evidence points to a pathogenic effect for E613G, and this assessment does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.193489 | Uncertain | 0.816 | 0.254 | 0.000 | -12.417 | Likely Pathogenic | 0.911 | Likely Pathogenic | Ambiguous | 1.49 | Ambiguous | 0.3 | 1.34 | Ambiguous | 1.42 | Ambiguous | 0.08 | Likely Benign | 0.641 | Likely Pathogenic | -6.56 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.26 | Pathogenic | 0.01 | Affected | 0.3422 | 0.5266 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1838A>T | E613V 2D 3DClick to see structure in 3D Viewer AISynGAP1 E613V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are Foldetta and premPS, whereas the remaining tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—consistently predict a pathogenic impact. FoldX and Rosetta give uncertain results and are not included in the agreement groups. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as benign. Because the majority of evidence points to a deleterious effect, the variant is most likely pathogenic, which is consistent with the lack of ClinVar annotation and gnomAD observation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.193489 | Uncertain | 0.816 | 0.254 | 0.000 | -12.799 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.72 | Ambiguous | 0.5 | -0.76 | Ambiguous | -0.02 | Likely Benign | 0.31 | Likely Benign | 0.767 | Likely Pathogenic | -6.57 | Deleterious | 0.996 | Probably Damaging | 0.991 | Probably Damaging | -1.25 | Pathogenic | 0.03 | Affected | 0.0935 | 0.6565 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1839G>C | E613D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E613D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, premPS, and SIFT, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Three tools—FoldX, Foldetta, and AlphaMissense‑Optimized—return uncertain or inconclusive results. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Overall, the majority of evidence points to a pathogenic impact for E613D. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.193489 | Uncertain | 0.816 | 0.254 | 0.000 | -8.795 | Likely Pathogenic | 0.842 | Likely Pathogenic | Ambiguous | 0.67 | Ambiguous | 0.3 | 0.48 | Likely Benign | 0.58 | Ambiguous | 0.13 | Likely Benign | 0.474 | Likely Benign | -2.79 | Deleterious | 0.989 | Probably Damaging | 0.979 | Probably Damaging | -1.27 | Pathogenic | 0.15 | Tolerated | 0.1998 | 0.4000 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1839G>T | E613D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E613D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, premPS, and SIFT, while pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Default. Three tools—FoldX, Foldetta, and AlphaMissense‑Optimized—return uncertain or inconclusive results. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Overall, the majority of evidence points to a pathogenic impact for E613D. This conclusion is consistent with the lack of ClinVar annotation and does not contradict any existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.275179 | Structured | 0.193489 | Uncertain | 0.816 | 0.254 | 0.000 | -8.795 | Likely Pathogenic | 0.842 | Likely Pathogenic | Ambiguous | 0.67 | Ambiguous | 0.3 | 0.48 | Likely Benign | 0.58 | Ambiguous | 0.13 | Likely Benign | 0.474 | Likely Benign | -2.79 | Deleterious | 0.989 | Probably Damaging | 0.979 | Probably Damaging | -1.27 | Pathogenic | 0.15 | Tolerated | 0.1998 | 0.4000 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.183G>C | E61D 2D AIThe SynGAP1 missense variant E61D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumVar and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant’s predicted benign status does not contradict ClinVar, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.352862 | Structured | 0.477329 | Uncertain | 0.518 | 0.699 | 0.125 | -4.394 | Likely Benign | 0.231 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -0.29 | Neutral | 0.267 | Benign | 0.585 | Possibly Damaging | 4.11 | Benign | 0.00 | Affected | 0.1895 | 0.3851 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.183G>T | E61D 2D AIThe SynGAP1 missense variant E61D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumVar and SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. The variant’s predicted benign status does not contradict ClinVar, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.352862 | Structured | 0.477329 | Uncertain | 0.518 | 0.699 | 0.125 | -4.394 | Likely Benign | 0.231 | Likely Benign | Likely Benign | 0.035 | Likely Benign | -0.29 | Neutral | 0.267 | Benign | 0.585 | Possibly Damaging | 4.11 | Benign | 0.00 | Affected | 0.1895 | 0.3851 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1840T>A | Y614N 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant Y614N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas the majority of other in silico predictors (Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a pathogenic impact; FoldX is inconclusive and is treated as unavailable. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Taken together, the preponderance of evidence points to a pathogenic effect for Y614N, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.182134 | Uncertain | 0.852 | 0.254 | 0.000 | -13.004 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 1.32 | Ambiguous | 0.5 | 3.16 | Destabilizing | 2.24 | Destabilizing | 1.58 | Destabilizing | 0.471 | Likely Benign | -8.86 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.42 | Benign | 0.06 | Tolerated | 0.1944 | 0.0573 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.1840T>C | Y614H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y614H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic impact: AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and Rosetta. Tools with uncertain or inconclusive results (FoldX, Foldetta, premPS) are not considered evidence for either side. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.182134 | Uncertain | 0.852 | 0.254 | 0.000 | -9.678 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.17 | Ambiguous | 0.5 | 2.38 | Destabilizing | 1.78 | Ambiguous | 0.91 | Ambiguous | 0.397 | Likely Benign | -4.95 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.58 | Benign | 0.27 | Tolerated | 0.1948 | 0.0373 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||
| c.1840T>G | Y614D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y614D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM predicts it to be benign, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify it as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.182134 | Uncertain | 0.852 | 0.254 | 0.000 | -15.073 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 2.14 | Destabilizing | 0.6 | 4.16 | Destabilizing | 3.15 | Destabilizing | 1.69 | Destabilizing | 0.597 | Likely Pathogenic | -9.83 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.40 | Benign | 0.02 | Affected | 0.4013 | 0.0573 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1841A>C | Y614S 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant Y614S is not reported in ClinVar and is present in gnomAD (ID 6‑33440893‑A‑C). Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas the majority of algorithms—AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, PROVEAN, polyPhen‑2 (HumDiv and HumVar), premPS, Rosetta, Foldetta, and the SGM Consensus—indicate pathogenicity; FoldX remains uncertain. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized scores the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labels it likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic effect. Overall, the preponderance of evidence points to a pathogenic classification, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.182134 | Uncertain | 0.852 | 0.254 | 0.000 | 6-33440893-A-C | 1 | 6.20e-7 | -12.709 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.74 | Ambiguous | 0.4 | 3.25 | Destabilizing | 2.50 | Destabilizing | 2.05 | Destabilizing | 0.482 | Likely Benign | -8.83 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.48 | Benign | 0.09 | Tolerated | 3.37 | 35 | 0.4155 | 0.1220 | -2 | -3 | 0.5 | -76.10 | ||||||||||||||||||||||||
| c.1841A>G | Y614C 2D  AIThe SynGAP1 missense variant Y614C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels it likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.314870 | Structured | 0.182134 | Uncertain | 0.852 | 0.254 | 0.000 | -11.716 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 1.56 | Ambiguous | 0.7 | 3.21 | Destabilizing | 2.39 | Destabilizing | 1.76 | Destabilizing | 0.539 | Likely Pathogenic | -8.83 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.41 | Benign | 0.02 | Affected | 0.3081 | 0.1679 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||
| c.1841A>T | Y614F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y614F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Uncertain or unavailable results come from Foldetta, premPS, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta is unavailable. Overall, the majority of reliable predictors indicate a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.314870 | Structured | 0.182134 | Uncertain | 0.852 | 0.254 | 0.000 | -5.584 | Likely Benign | 0.630 | Likely Pathogenic | Likely Benign | 0.09 | Likely Benign | 0.2 | 0.98 | Ambiguous | 0.54 | Ambiguous | 0.78 | Ambiguous | 0.364 | Likely Benign | -3.75 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.42 | Benign | 0.07 | Tolerated | 0.1909 | 0.2762 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||
| c.1843C>A | P615T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P615T is not reported in ClinVar and is absent from gnomAD. Among the evaluated in‑silico predictors, SIFT is the sole tool that predicts a benign effect, whereas the remaining majority—including REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—predict a pathogenic impact. Predictions from Rosetta, Foldetta, and premPS are uncertain and do not provide definitive evidence. High‑accuracy assessments further support a deleterious interpretation: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates it is likely pathogenic, while Foldetta’s stability analysis remains inconclusive. Based on the preponderance of pathogenic predictions and the lack of benign consensus, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.179032 | Uncertain | 0.879 | 0.255 | 0.000 | -13.764 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 3.02 | Destabilizing | 0.3 | 0.95 | Ambiguous | 1.99 | Ambiguous | 0.75 | Ambiguous | 0.823 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.26 | Pathogenic | 0.06 | Tolerated | 0.1602 | 0.3806 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||
| c.1843C>G | P615A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 P615A variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include SIFT and Rosetta, whereas the majority of predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score (Likely Pathogenic) all suggest a pathogenic impact. Uncertain results are reported by FoldX, Foldetta, and premPS. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta’s stability analysis is inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for P615A, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.179032 | Uncertain | 0.879 | 0.255 | 0.000 | -12.156 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | 1.73 | Ambiguous | 0.3 | 0.35 | Likely Benign | 1.04 | Ambiguous | 0.85 | Ambiguous | 0.693 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.25 | Pathogenic | 0.10 | Tolerated | 0.3169 | 0.3214 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||
| c.1843C>T | P615S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P615S is not reported in ClinVar (ClinVar status: not reported) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a pathogenic effect include REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; the SGM Consensus also indicates a likely pathogenic outcome. No tools predict a benign effect. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, while Foldetta’s stability prediction is uncertain and therefore not taken as evidence. Overall, the preponderance of evidence points to the variant being most likely pathogenic, with no contradiction to the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.179032 | Uncertain | 0.879 | 0.255 | 0.000 | -12.566 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.46 | Destabilizing | 0.3 | 1.28 | Ambiguous | 1.87 | Ambiguous | 0.98 | Ambiguous | 0.780 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.19 | Pathogenic | 0.04 | Affected | 0.3191 | 0.3310 | 1 | -1 | 0.8 | -10.04 | |||||||||||||||||||||||||||||
| c.1844C>A | P615Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change P615Q is not listed in ClinVar and has no allele record in gnomAD. Functional prediction tools that assess sequence conservation and structural impact uniformly classify the variant as pathogenic: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only Rosetta and the combined Foldetta stability assessment are inconclusive. Grouping the predictions, the benign category contains no tools, while the pathogenic category includes all the above. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports Likely Pathogenic, and Foldetta remains uncertain. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.179032 | Uncertain | 0.879 | 0.255 | 0.000 | -13.247 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.16 | Destabilizing | 0.3 | 1.28 | Ambiguous | 1.72 | Ambiguous | 1.33 | Destabilizing | 0.742 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | -1.28 | Pathogenic | 0.01 | Affected | 0.1395 | 0.3445 | 0 | -1 | -1.9 | 31.01 | |||||||||||||||||||||||||||||
| c.1844C>G | P615R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P615R is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. No tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, indicates a destabilizing, pathogenic effect. No predictions or stability results are missing or inconclusive. Based on the unanimous pathogenic predictions and the absence of any ClinVar or gnomAD evidence to the contrary, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.179032 | Uncertain | 0.879 | 0.255 | 0.000 | -15.628 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.62 | Destabilizing | 0.5 | 2.69 | Destabilizing | 2.66 | Destabilizing | 1.14 | Destabilizing | 0.871 | Likely Pathogenic | -8.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.29 | Pathogenic | 0.01 | Affected | 0.1463 | 0.2603 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||
| c.1844C>T | P615L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant P615L is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include premPS and Rosetta, whereas the remaining tools—REVEL, FoldX, PolyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, PROVEAN, and the SGM Consensus—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenicity, and Foldetta yields an uncertain result. Taken together, the overwhelming majority of computational evidence indicates that P615L is likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.236433 | Structured | 0.179032 | Uncertain | 0.879 | 0.255 | 0.000 | -11.884 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.03 | Destabilizing | 0.4 | -0.01 | Likely Benign | 1.01 | Ambiguous | 0.50 | Likely Benign | 0.699 | Likely Pathogenic | -9.96 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.13 | Pathogenic | 0.04 | Affected | 0.1949 | 0.4692 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||
| c.1846G>A | D616N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D616N missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into benign (REVEL, premPS, SIFT, FATHMM, AlphaMissense‑Optimized) and pathogenic (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b). Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) favors pathogenic, and Foldetta remains uncertain. Overall, the majority of conventional predictors lean toward a benign effect, whereas the SGM Consensus suggests pathogenicity, leaving the variant’s clinical significance ambiguous. Based on the prevailing evidence, the variant is most likely benign, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | -8.292 | Likely Pathogenic | 0.349 | Ambiguous | Likely Benign | 0.54 | Ambiguous | 0.2 | 1.05 | Ambiguous | 0.80 | Ambiguous | 0.03 | Likely Benign | 0.149 | Likely Benign | -3.74 | Deleterious | 0.875 | Possibly Damaging | 0.581 | Possibly Damaging | 3.41 | Benign | 0.11 | Tolerated | 0.1053 | 0.3976 | 2 | 1 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.1846G>C | D616H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D616H missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM. Those that agree on a pathogenic effect comprise SGM‑Consensus, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Two tools give uncertain results—Rosetta and AlphaMissense‑Optimized—so their outputs are treated as unavailable for inference. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is Pathogenic. Overall, the majority of evidence points to a pathogenic effect. The variant’s predicted pathogenicity does not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | -9.815 | Likely Pathogenic | 0.904 | Likely Pathogenic | Ambiguous | 2.13 | Destabilizing | 0.2 | 1.89 | Ambiguous | 2.01 | Destabilizing | 0.45 | Likely Benign | 0.316 | Likely Benign | -5.57 | Deleterious | 0.999 | Probably Damaging | 0.952 | Probably Damaging | 3.30 | Benign | 0.03 | Affected | 0.1330 | 0.4273 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.1846G>T | D616Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D616Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM, whereas a majority of tools predict a pathogenic outcome: SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as likely pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No evidence from FoldX or Rosetta alone is conclusive. Overall, the balance of evidence favors a pathogenic interpretation, and this conclusion does not contradict any existing ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | -12.638 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 1.70 | Ambiguous | 0.3 | 1.32 | Ambiguous | 1.51 | Ambiguous | 0.35 | Likely Benign | 0.374 | Likely Benign | -7.43 | Deleterious | 0.999 | Probably Damaging | 0.970 | Probably Damaging | 3.28 | Benign | 0.01 | Affected | 0.0465 | 0.4069 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1847A>C | D616A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D616A missense variant is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, FATHMM, and polyPhen‑2 HumVar, while pathogenic predictions arise from SGM‑Consensus (Likely Pathogenic), Rosetta, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized model classifies the variant as benign, whereas the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates pathogenicity. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the majority of evidence points toward a pathogenic effect, and this assessment does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | -11.386 | Likely Pathogenic | 0.664 | Likely Pathogenic | Likely Benign | 1.76 | Ambiguous | 0.2 | 2.07 | Destabilizing | 1.92 | Ambiguous | 0.41 | Likely Benign | 0.126 | Likely Benign | -6.13 | Deleterious | 0.539 | Possibly Damaging | 0.122 | Benign | 3.32 | Benign | 0.10 | Tolerated | 0.3543 | 0.4207 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.1847A>G | D616G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D616G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Tools with uncertain or inconclusive results—FoldX, AlphaMissense‑Default, and Foldetta—are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the predictions are split evenly between benign and pathogenic, with no clear consensus. Thus, the variant is most likely of uncertain significance; it does not contradict any ClinVar status because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | -10.310 | Likely Pathogenic | 0.547 | Ambiguous | Likely Benign | 1.48 | Ambiguous | 0.1 | 2.13 | Destabilizing | 1.81 | Ambiguous | 0.49 | Likely Benign | 0.144 | Likely Benign | -5.60 | Deleterious | 0.985 | Probably Damaging | 0.800 | Possibly Damaging | 3.37 | Benign | 0.06 | Tolerated | 0.3496 | 0.4386 | 1 | -1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||
| c.1847A>T | D616V 2D 3DClick to see structure in 3D Viewer AISynGAP1 D616V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, premPS, and FATHMM, while pathogenic calls are made by FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus. Uncertain results are reported by Rosetta and AlphaMissense‑Optimized. High‑accuracy assessments give a pathogenic signal: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the majority of evidence, including the high‑accuracy tools, supports a pathogenic effect for D616V. This conclusion is not contradicted by ClinVar, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | -13.992 | Likely Pathogenic | 0.919 | Likely Pathogenic | Ambiguous | 2.41 | Destabilizing | 0.2 | 1.95 | Ambiguous | 2.18 | Destabilizing | 0.36 | Likely Benign | 0.268 | Likely Benign | -7.36 | Deleterious | 0.972 | Probably Damaging | 0.682 | Possibly Damaging | 3.26 | Benign | 0.00 | Affected | 0.0699 | 0.4393 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.1848T>A | D616E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D616E missense variant is catalogued in gnomAD (ID 6‑33440900‑T‑A) but has no ClinVar submission. Functional prediction tools show a split assessment: benign calls come from REVEL, both polyPhen‑2 HumDiv and HumVar, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls arise from PROVEAN, SIFT, and AlphaMissense‑Default. The remaining predictors (FoldX, Rosetta, Foldetta, premPS, ESM1b) are inconclusive. A high‑accuracy consensus (SGM) that aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN yields a pathogenic verdict. AlphaMissense‑Optimized remains benign, and Foldetta, which evaluates protein‑folding stability, is uncertain. Overall, the majority of evidence leans toward pathogenicity, and this conclusion does not conflict with ClinVar because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | 6-33440900-T-A | 1 | 6.20e-7 | -7.250 | In-Between | 0.695 | Likely Pathogenic | Likely Benign | 0.96 | Ambiguous | 0.1 | 1.52 | Ambiguous | 1.24 | Ambiguous | 0.58 | Ambiguous | 0.092 | Likely Benign | -2.85 | Deleterious | 0.421 | Benign | 0.232 | Benign | 3.32 | Benign | 0.03 | Affected | 3.37 | 35 | 0.1225 | 0.4128 | 2 | 3 | 0.0 | 14.03 | |||||||||||||||||||||||||
| c.1848T>G | D616E 2D 3DClick to see structure in 3D Viewer AISynGAP1 D616E is not reported in ClinVar but is present in gnomAD (ID 6‑33440900‑T‑G). Functional prediction tools that agree on benign impact include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, SIFT, and AlphaMissense‑Default. The remaining tools (FoldX, Rosetta, Foldetta, premPS, ESM1b) are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of conventional predictors lean toward a benign effect, but the high‑accuracy consensus is split, leaving the variant’s clinical significance unresolved. Thus, the variant is most likely benign based on the bulk of predictions, and this does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.166689 | Uncertain | 0.867 | 0.252 | 0.000 | 6-33440900-T-G | 3 | 1.86e-6 | -7.250 | In-Between | 0.695 | Likely Pathogenic | Likely Benign | 0.96 | Ambiguous | 0.1 | 1.52 | Ambiguous | 1.24 | Ambiguous | 0.58 | Ambiguous | 0.092 | Likely Benign | -2.85 | Deleterious | 0.421 | Benign | 0.232 | Benign | 3.32 | Benign | 0.03 | Affected | 3.37 | 35 | 0.1225 | 0.4128 | 2 | 3 | 0.0 | 14.03 | |||||||||||||||||||||||||
| c.1849G>A | E617K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E617K is not reported in ClinVar but is present in gnomAD (6‑33440901‑G‑A). Functional prediction tools cluster into two groups: benign predictions come from FoldX, premPS, and SIFT, while pathogenic predictions arise from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. A third set of methods (Foldetta, AlphaMissense‑Optimized, Rosetta) yield uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic effect for E617K, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.155123 | Uncertain | 0.877 | 0.240 | 0.000 | 6-33440901-G-A | 1 | 6.20e-7 | -10.702 | Likely Pathogenic | 0.910 | Likely Pathogenic | Ambiguous | 0.37 | Likely Benign | 0.1 | 1.19 | Ambiguous | 0.78 | Ambiguous | 0.17 | Likely Benign | 0.534 | Likely Pathogenic | -3.32 | Deleterious | 0.997 | Probably Damaging | 0.987 | Probably Damaging | -1.34 | Pathogenic | 0.48 | Tolerated | 3.37 | 35 | 0.1981 | 0.6282 | 1 | 0 | -0.4 | -0.94 | ||||||||||||||||||||||||
| c.1849G>C | E617Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E617Q missense variant is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Foldetta and Rosetta give uncertain results, which are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM‑Consensus (derived from a majority of high‑confidence predictors) indicates a likely pathogenic outcome. Foldetta’s stability prediction is inconclusive. Overall, the balance of evidence leans toward a pathogenic impact for E617Q, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.155123 | Uncertain | 0.877 | 0.240 | 0.000 | -8.650 | Likely Pathogenic | 0.747 | Likely Pathogenic | Likely Benign | 0.20 | Likely Benign | 0.2 | 1.01 | Ambiguous | 0.61 | Ambiguous | 0.21 | Likely Benign | 0.481 | Likely Benign | -2.39 | Neutral | 0.996 | Probably Damaging | 0.986 | Probably Damaging | -1.39 | Pathogenic | 0.29 | Tolerated | 0.1050 | 0.5951 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.184G>A | D62N 2D AIThe SynGAP1 missense variant D62N is reported in gnomAD (variant ID 6‑33423593‑G‑A) but has no ClinVar entry. In silico prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign status. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are not available. Overall, the consensus of the majority of tools indicates that D62N is most likely benign, and this assessment does not contradict any ClinVar classification because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | 6-33423593-G-A | 1 | 6.20e-7 | -4.607 | Likely Benign | 0.207 | Likely Benign | Likely Benign | 0.075 | Likely Benign | -1.08 | Neutral | 0.028 | Benign | 0.032 | Benign | 4.11 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1670 | 0.6154 | 1 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||||||
| c.184G>C | D62H 2D AIThe SynGAP1 missense variant D62H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all classify the change as benign, while the only pathogenic call comes from SIFT. AlphaMissense‑Default remains uncertain, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as likely benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts benign, SGM‑Consensus indicates likely benign, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Based on the collective predictions, D62H is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -5.253 | Likely Benign | 0.511 | Ambiguous | Likely Benign | 0.070 | Likely Benign | -1.53 | Neutral | 0.172 | Benign | 0.248 | Benign | 4.05 | Benign | 0.00 | Affected | 0.2059 | 0.6579 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||||||||||||
| c.184G>T | D62Y 2D AIThe SynGAP1 missense variant D62Y is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates Likely Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -6.313 | Likely Benign | 0.569 | Likely Pathogenic | Likely Benign | 0.109 | Likely Benign | -2.17 | Neutral | 0.388 | Benign | 0.328 | Benign | 4.03 | Benign | 0.00 | Affected | 0.0657 | 0.5588 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||||||||||||
| c.1850A>C | E617A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E617A is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include FoldX, premPS, SIFT, and AlphaMissense‑Optimized, whereas a majority of tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default) predict a pathogenic outcome; Rosetta and Foldetta are uncertain. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized indicates benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates pathogenic, and Foldetta remains uncertain. Overall, the balance of evidence favors a pathogenic interpretation. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.155123 | Uncertain | 0.877 | 0.240 | 0.000 | -8.704 | Likely Pathogenic | 0.769 | Likely Pathogenic | Likely Benign | 0.37 | Likely Benign | 0.1 | 0.89 | Ambiguous | 0.63 | Ambiguous | 0.18 | Likely Benign | 0.627 | Likely Pathogenic | -4.75 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.37 | Pathogenic | 0.46 | Tolerated | 0.3033 | 0.5317 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1850A>G | E617G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E617G is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from premPS, SIFT, and AlphaMissense‑Optimized, whereas pathogenic predictions are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. Uncertain results are reported by FoldX, Rosetta, Foldetta, and ESM1b. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta is uncertain. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.155123 | Uncertain | 0.877 | 0.240 | 0.000 | -7.984 | In-Between | 0.777 | Likely Pathogenic | Likely Benign | 0.59 | Ambiguous | 0.2 | 1.60 | Ambiguous | 1.10 | Ambiguous | 0.22 | Likely Benign | 0.701 | Likely Pathogenic | -4.99 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.41 | Pathogenic | 0.18 | Tolerated | 0.2344 | 0.5442 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1850A>T | E617V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E617V has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into benign (premPS, SIFT) and pathogenic (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default). Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments reinforce the pathogenic signal: the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic, while AlphaMissense‑Optimized remains uncertain and Foldetta is also uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E617V. This conclusion is not contradicted by ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.111485 | Structured | 0.155123 | Uncertain | 0.877 | 0.240 | 0.000 | -10.826 | Likely Pathogenic | 0.907 | Likely Pathogenic | Ambiguous | 0.60 | Ambiguous | 0.1 | 0.92 | Ambiguous | 0.76 | Ambiguous | 0.28 | Likely Benign | 0.816 | Likely Pathogenic | -5.71 | Deleterious | 0.998 | Probably Damaging | 0.991 | Probably Damaging | -1.47 | Pathogenic | 0.13 | Tolerated | 0.0587 | 0.6503 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1851G>C | E617D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change E617D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that classify the variant as benign include REVEL, FoldX, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized scores the variant as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a “Likely Benign” verdict; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also reports a benign effect. Based on the preponderance of evidence from both general and high‑accuracy predictors, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.111485 | Structured | 0.155123 | Uncertain | 0.877 | 0.240 | 0.000 | -1.349 | Likely Benign | 0.241 | Likely Benign | Likely Benign | 0.12 | Likely Benign | 0.1 | 0.80 | Ambiguous | 0.46 | Likely Benign | 0.07 | Likely Benign | 0.322 | Likely Benign | -0.01 | Neutral | 0.994 | Probably Damaging | 0.979 | Probably Damaging | -1.35 | Pathogenic | 0.88 | Tolerated | 3.37 | 35 | 0.1854 | 0.3386 | 2 | 3 | 0.0 | -14.03 | |||||||||||||||||||||||||||
| c.1851G>T | E617D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E617D is listed in ClinVar with an uncertain significance (ID 2584916.0) and is not reported in gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Foldetta, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score all indicate benign or likely benign. In contrast, polyPhen‑2 (HumDiv and HumVar) and FATHMM predict a pathogenic impact, while Rosetta remains inconclusive. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is likely benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Overall, the preponderance of evidence supports a benign classification, which does not contradict the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.111485 | Structured | 0.155123 | Uncertain | 0.877 | 0.240 | 0.000 | Uncertain | 1 | -1.349 | Likely Benign | 0.241 | Likely Benign | Likely Benign | 0.12 | Likely Benign | 0.1 | 0.80 | Ambiguous | 0.46 | Likely Benign | 0.07 | Likely Benign | 0.322 | Likely Benign | -0.01 | Neutral | 0.994 | Probably Damaging | 0.979 | Probably Damaging | -1.35 | Pathogenic | 0.88 | Tolerated | 3.37 | 35 | 0.1854 | 0.3386 | 2 | 3 | 0.0 | -14.03 | |||||||||||||||||||||||||
| c.1852C>A | Q618K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q618K is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33440904‑C‑A). Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while ESM1b is uncertain. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign result; and Foldetta also predicts benign stability. No predictions or folding stability results are missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.138725 | Uncertain | 0.904 | 0.240 | 0.000 | 6-33440904-C-A | 24 | 1.49e-5 | -7.708 | In-Between | 0.229 | Likely Benign | Likely Benign | 0.02 | Likely Benign | 0.0 | 0.16 | Likely Benign | 0.09 | Likely Benign | -0.46 | Likely Benign | 0.281 | Likely Benign | -0.05 | Neutral | 0.338 | Benign | 0.111 | Benign | -1.21 | Pathogenic | 0.29 | Tolerated | 3.37 | 35 | 0.1413 | 0.2750 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||
| c.1852C>G | Q618E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q618E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and FATHMM. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the preponderance of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and gnomAD presence, and there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.138725 | Uncertain | 0.904 | 0.240 | 0.000 | -12.535 | Likely Pathogenic | 0.162 | Likely Benign | Likely Benign | 0.46 | Likely Benign | 0.1 | 0.50 | Ambiguous | 0.48 | Likely Benign | 0.33 | Likely Benign | 0.233 | Likely Benign | -1.16 | Neutral | 0.046 | Benign | 0.021 | Benign | -1.17 | Pathogenic | 0.26 | Tolerated | 0.1003 | 0.1469 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.1853A>C | Q618P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q618P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are SGM Consensus, ESM1b, FATHMM, PROVEAN, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) as uncertain. Overall, the majority of predictions lean toward a benign effect, though a subset of high‑confidence tools suggest pathogenicity. The variant’s status is not contradicted by ClinVar, which has no entry for this change. Based on the collective evidence, Q618P is most likely benign, albeit with some conflicting pathogenic signals from high‑accuracy predictors. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.138725 | Uncertain | 0.904 | 0.240 | 0.000 | -8.273 | Likely Pathogenic | 0.248 | Likely Benign | Likely Benign | -0.11 | Likely Benign | 0.1 | 3.41 | Destabilizing | 1.65 | Ambiguous | 0.23 | Likely Benign | 0.449 | Likely Benign | -3.32 | Deleterious | 0.261 | Benign | 0.246 | Benign | -1.35 | Pathogenic | 0.08 | Tolerated | 0.1990 | 0.4011 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||
| c.1853A>G | Q618R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q618R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a pathogenic outcome, while Rosetta and premPS are inconclusive. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.175930 | Structured | 0.138725 | Uncertain | 0.904 | 0.240 | 0.000 | -2.513 | Likely Benign | 0.207 | Likely Benign | Likely Benign | -0.43 | Likely Benign | 0.1 | 0.60 | Ambiguous | 0.09 | Likely Benign | -0.66 | Ambiguous | 0.285 | Likely Benign | 1.22 | Neutral | 0.005 | Benign | 0.009 | Benign | -1.24 | Pathogenic | 1.00 | Tolerated | 0.1218 | 0.1163 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||
| c.1853A>T | Q618L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q618L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, and polyPhen‑2 HumVar, while those that predict a pathogenic effect are SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, ESM1b, and FATHMM. AlphaMissense‑Default is uncertain, whereas AlphaMissense‑Optimized predicts benign. High‑accuracy methods give the following results: AlphaMissense‑Optimized – benign; SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) – Likely Pathogenic; Foldetta – benign. Overall, the majority of tools (nine benign vs. five pathogenic) predict a benign impact. Thus, the variant is most likely benign based on current predictions, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.138725 | Uncertain | 0.904 | 0.240 | 0.000 | -8.561 | Likely Pathogenic | 0.423 | Ambiguous | Likely Benign | -0.07 | Likely Benign | 0.1 | 0.20 | Likely Benign | 0.07 | Likely Benign | 0.31 | Likely Benign | 0.479 | Likely Benign | -3.94 | Deleterious | 0.712 | Possibly Damaging | 0.268 | Benign | -1.28 | Pathogenic | 0.09 | Tolerated | 0.0612 | 0.4146 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||
| c.1854G>C | Q618H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q618H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, premPS, PROVEAN, and AlphaMissense‑Optimized; pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Four tools (FoldX, Foldetta, ESM1b, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive and Foldetta is uncertain. Overall, the majority of evidence points toward a benign effect, and this conclusion does not contradict the lack of ClinVar annotation or gnomAD presence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.138725 | Uncertain | 0.904 | 0.240 | 0.000 | -7.092 | In-Between | 0.489 | Ambiguous | Likely Benign | 0.59 | Ambiguous | 0.1 | 0.44 | Likely Benign | 0.52 | Ambiguous | 0.34 | Likely Benign | 0.475 | Likely Benign | -1.99 | Neutral | 0.946 | Possibly Damaging | 0.638 | Possibly Damaging | -1.35 | Pathogenic | 0.04 | Affected | 0.1015 | 0.2655 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||
| c.1854G>T | Q618H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q618H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, premPS, PROVEAN, and AlphaMissense‑Optimized, while pathogenic predictions arise from polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Four tools (FoldX, Foldetta, ESM1b, AlphaMissense‑Default) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta is also uncertain. Overall, the balance of evidence leans toward a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.138725 | Uncertain | 0.904 | 0.240 | 0.000 | -7.092 | In-Between | 0.489 | Ambiguous | Likely Benign | 0.59 | Ambiguous | 0.1 | 0.44 | Likely Benign | 0.52 | Ambiguous | 0.34 | Likely Benign | 0.475 | Likely Benign | -1.99 | Neutral | 0.946 | Possibly Damaging | 0.638 | Possibly Damaging | -1.35 | Pathogenic | 0.04 | Affected | 0.1015 | 0.2655 | 3 | 0 | 0.3 | 9.01 | ||||||||||||||||||||||||||||||
| c.1855A>C | T619P 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant T619P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from FoldX and SIFT, while pathogenic predictions are made by REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. Two tools report uncertainty: premPS and AlphaMissense‑Optimized. High‑accuracy assessments further clarify the variant’s impact: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic effect; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. Overall, the majority of evidence points to a pathogenic effect for T619P, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.219301 | Structured | 0.119723 | Uncertain | 0.929 | 0.237 | 0.000 | -12.879 | Likely Pathogenic | 0.933 | Likely Pathogenic | Ambiguous | 0.43 | Likely Benign | 0.2 | 4.81 | Destabilizing | 2.62 | Destabilizing | 0.82 | Ambiguous | 0.860 | Likely Pathogenic | -5.51 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.39 | Pathogenic | 0.09 | Tolerated | 0.1940 | 0.3806 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1855A>G | T619A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 T619A missense variant is not reported in ClinVar and has no gnomAD entry. Consensus prediction tools cluster into two groups: benign predictions come from SIFT, ESM1b, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default. Uncertain results are reported by FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized classifying the change as benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels it as likely pathogenic; Foldetta’s stability analysis is inconclusive. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not contradict the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.219301 | Structured | 0.119723 | Uncertain | 0.929 | 0.237 | 0.000 | -6.341 | Likely Benign | 0.672 | Likely Pathogenic | Likely Benign | 0.59 | Ambiguous | 0.1 | 0.62 | Ambiguous | 0.61 | Ambiguous | 0.53 | Ambiguous | 0.613 | Likely Pathogenic | -4.21 | Deleterious | 0.996 | Probably Damaging | 0.989 | Probably Damaging | -1.27 | Pathogenic | 0.17 | Tolerated | 0.3915 | 0.3109 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1855A>T | T619S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T619S is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Prediction tools that agree on a benign effect include only AlphaMissense‑Optimized. All other evaluated algorithms—SGM‑Consensus (Likely Pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a pathogenic impact. High‑accuracy assessments further support this view: AlphaMissense‑Optimized reports a benign outcome, whereas the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates pathogenicity. Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, yields an uncertain result. Overall, the majority of evidence points to a pathogenic effect for T619S, and this conclusion does not contradict the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.219301 | Structured | 0.119723 | Uncertain | 0.929 | 0.237 | 0.000 | Uncertain | 1 | -8.608 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | 1.09 | Ambiguous | 0.2 | 1.35 | Ambiguous | 1.22 | Ambiguous | 0.85 | Ambiguous | 0.602 | Likely Pathogenic | -3.42 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.30 | Pathogenic | 0.05 | Affected | 3.37 | 35 | 0.3255 | 0.2860 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||
| c.1856C>A | T619N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T619N has no ClinVar entry and is not reported in gnomAD. Prediction tools that assess pathogenicity all lean toward a deleterious effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict pathogenic. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. No tool predicts a benign effect. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Based on the overall consensus of the available predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.219301 | Structured | 0.119723 | Uncertain | 0.929 | 0.237 | 0.000 | -11.796 | Likely Pathogenic | 0.900 | Likely Pathogenic | Ambiguous | 0.61 | Ambiguous | 0.1 | 0.96 | Ambiguous | 0.79 | Ambiguous | 1.23 | Destabilizing | 0.715 | Likely Pathogenic | -4.61 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.38 | Pathogenic | 0.05 | Affected | 0.0998 | 0.2690 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.1856C>G | T619S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T619S missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include only AlphaMissense‑Optimized. All other evaluated predictors (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default) uniformly predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized remains benign, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as Likely Pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, yields an uncertain result. No other high‑confidence stability predictions are available. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status, as none is assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.219301 | Structured | 0.119723 | Uncertain | 0.929 | 0.237 | 0.000 | -8.608 | Likely Pathogenic | 0.677 | Likely Pathogenic | Likely Benign | 1.09 | Ambiguous | 0.2 | 1.35 | Ambiguous | 1.22 | Ambiguous | 0.85 | Ambiguous | 0.523 | Likely Pathogenic | -3.42 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.30 | Pathogenic | 0.05 | Affected | 3.37 | 35 | 0.3255 | 0.2860 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||
| c.1856C>T | T619I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T619I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess pathogenicity unanimously classify the variant as pathogenic: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect; the remaining tools (FoldX, Rosetta, Foldetta, premPS) return uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. Based on the consensus of pathogenic predictions and the lack of benign evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.219301 | Structured | 0.119723 | Uncertain | 0.929 | 0.237 | 0.000 | -11.760 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | -1.06 | Ambiguous | 0.1 | -1.35 | Ambiguous | -1.21 | Ambiguous | 0.58 | Ambiguous | 0.735 | Likely Pathogenic | -5.54 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | -1.40 | Pathogenic | 0.03 | Affected | 0.0869 | 0.4377 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.1858T>A | S620T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S620T has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify it as benign include Foldetta, premPS, PROVEAN, SIFT, AlphaMissense‑Optimized, and Rosetta. Those that predict pathogenicity are SGM‑Consensus, REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicating likely pathogenic, and Foldetta predicting benign stability. No prediction or folding result is missing or inconclusive. Overall, the majority of tools lean toward a pathogenic effect, and this assessment does not contradict the ClinVar status, which is currently unclassified. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.139895 | Structured | 0.100377 | Uncertain | 0.936 | 0.219 | 0.000 | -9.171 | Likely Pathogenic | 0.660 | Likely Pathogenic | Likely Benign | 0.52 | Ambiguous | 0.1 | -0.40 | Likely Benign | 0.06 | Likely Benign | 0.30 | Likely Benign | 0.551 | Likely Pathogenic | -1.99 | Neutral | 0.896 | Possibly Damaging | 0.933 | Probably Damaging | -1.25 | Pathogenic | 0.14 | Tolerated | 0.1288 | 0.5199 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1858T>C | S620P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S620P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly favor a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while premPS remains uncertain. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Consequently, the variant is most likely pathogenic, and this prediction is consistent with the absence of a ClinVar entry (no contradiction). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.139895 | Structured | 0.100377 | Uncertain | 0.936 | 0.219 | 0.000 | -12.208 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | 4.89 | Destabilizing | 0.5 | 12.23 | Destabilizing | 8.56 | Destabilizing | 0.73 | Ambiguous | 0.834 | Likely Pathogenic | -3.62 | Deleterious | 0.998 | Probably Damaging | 0.993 | Probably Damaging | -1.35 | Pathogenic | 0.02 | Affected | 0.2147 | 0.4776 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.1858T>G | S620A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S620A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No evidence from these analyses suggests a deleterious effect. Consequently, the variant is most likely benign based on the aggregate predictions, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.139895 | Structured | 0.100377 | Uncertain | 0.936 | 0.219 | 0.000 | -4.637 | Likely Benign | 0.088 | Likely Benign | Likely Benign | -0.42 | Likely Benign | 0.0 | -0.90 | Ambiguous | -0.66 | Ambiguous | -0.19 | Likely Benign | 0.375 | Likely Benign | -0.52 | Neutral | 0.968 | Probably Damaging | 0.994 | Probably Damaging | -1.27 | Pathogenic | 0.66 | Tolerated | 0.4950 | 0.3636 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.1859C>T | S620L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S620L is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include only premPS, whereas the remaining tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict pathogenicity. FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are inconclusive. High‑accuracy assessments further support a deleterious outcome: the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic; AlphaMissense‑Optimized remains uncertain; and Foldetta is also uncertain. Overall, the preponderance of evidence indicates that S620L is most likely pathogenic, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.139895 | Structured | 0.100377 | Uncertain | 0.936 | 0.219 | 0.000 | -13.939 | Likely Pathogenic | 0.856 | Likely Pathogenic | Ambiguous | -1.51 | Ambiguous | 0.1 | -1.08 | Ambiguous | -1.30 | Ambiguous | 0.26 | Likely Benign | 0.736 | Likely Pathogenic | -3.71 | Deleterious | 0.999 | Probably Damaging | 0.995 | Probably Damaging | -1.33 | Pathogenic | 0.02 | Affected | 0.0976 | 0.4696 | -3 | -2 | 4.6 | 26.08 | |||||||||||||||||||||||||||||
| c.185A>C | D62A 2D AIThe SynGAP1 D62A missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Benign and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Based on the preponderance of evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -3.983 | Likely Benign | 0.318 | Likely Benign | Likely Benign | 0.070 | Likely Benign | -1.67 | Neutral | 0.006 | Benign | 0.023 | Benign | 4.09 | Benign | 0.00 | Affected | 0.4152 | 0.5972 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||||||||||||
| c.185A>G | D62G 2D AIThe SynGAP1 D62G missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -4.047 | Likely Benign | 0.316 | Likely Benign | Likely Benign | 0.097 | Likely Benign | -1.76 | Neutral | 0.012 | Benign | 0.032 | Benign | 4.07 | Benign | 0.00 | Affected | 0.4016 | 0.6081 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.185A>T | D62V 2D AIThe SynGAP1 missense variant D62V is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools cluster around a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized all indicate a benign outcome, while the sole pathogenic signal comes from SIFT. AlphaMissense‑Default remains uncertain, and no Foldetta stability assessment is available. High‑accuracy methods reinforce the benign consensus: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports Likely Benign, and Foldetta data are missing. Taken together, the preponderance of evidence supports a benign classification for D62V, and this assessment does not conflict with the absence of a ClinVar entry. Therefore, the variant is most likely benign, and this conclusion does not contradict the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -4.417 | Likely Benign | 0.489 | Ambiguous | Likely Benign | 0.118 | Likely Benign | -2.04 | Neutral | 0.028 | Benign | 0.088 | Benign | 4.04 | Benign | 0.00 | Affected | 0.1039 | 0.6129 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||||||||||||
| c.1861C>G | R621G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R621G is reported in gnomAD (ID 6‑33440913‑C‑G) but has no ClinVar entry. In silico predictors largely agree on a deleterious effect: pathogenic calls come from REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The only benign prediction is from FATHMM; FoldX and Foldetta give uncertain results. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence indicates the variant is most likely pathogenic, and this conclusion is not contradicted by ClinVar data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.084420 | Uncertain | 0.945 | 0.216 | 0.000 | 6-33440913-C-G | 1 | 6.20e-7 | -16.611 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.18 | Ambiguous | 0.3 | 2.07 | Destabilizing | 1.63 | Ambiguous | 1.17 | Destabilizing | 0.558 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.84 | Benign | 0.01 | Affected | 3.37 | 35 | 0.3109 | 0.2651 | -2 | -3 | 4.1 | -99.14 | ||||||||||||||||||||||||
| c.1862G>C | R621P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant R621P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, whereas the remaining eleven tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the change as pathogenic. The high‑accuracy subset further supports this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also indicates pathogenic. No prediction is inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, which currently contains no entry for R621P. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.084420 | Uncertain | 0.945 | 0.216 | 0.000 | -17.022 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 4.94 | Destabilizing | 0.4 | 9.39 | Destabilizing | 7.17 | Destabilizing | 0.96 | Ambiguous | 0.745 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.81 | Benign | 0.01 | Affected | 0.2143 | 0.3692 | 0 | -2 | 2.9 | -59.07 | |||||||||||||||||||||||||||||
| c.1862G>T | R621L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant R621L has no ClinVar entry and is absent from gnomAD. Prediction tools that classify it as benign include Rosetta, premPS, and FATHMM, whereas the majority—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus—label it pathogenic or likely pathogenic. FoldX and Foldetta return uncertain results and are not considered evidence. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, and Foldetta remains inconclusive. Overall, the consensus of the available predictions points to a pathogenic effect for R621L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.084420 | Uncertain | 0.945 | 0.216 | 0.000 | -16.055 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 1.70 | Ambiguous | 0.3 | -0.22 | Likely Benign | 0.74 | Ambiguous | 0.34 | Likely Benign | 0.718 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.83 | Benign | 0.00 | Affected | 0.1424 | 0.3568 | -3 | -2 | 8.3 | -43.03 | |||||||||||||||||||||||||||||
| c.1864A>C | T622P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T622P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess functional impact uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as pathogenic. No tool predicts a benign outcome; the only inconclusive result is premPS, which is listed as uncertain. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) also indicates pathogenic. Consequently, the variant is most likely pathogenic, and this conclusion is consistent with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.071403 | Uncertain | 0.957 | 0.198 | 0.000 | -16.410 | Likely Pathogenic | 0.964 | Likely Pathogenic | Likely Pathogenic | 2.81 | Destabilizing | 0.4 | 8.90 | Destabilizing | 5.86 | Destabilizing | 0.78 | Ambiguous | 0.924 | Likely Pathogenic | -5.57 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.57 | Pathogenic | 0.01 | Affected | 0.1700 | 0.3876 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1864A>G | T622A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T622A is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FoldX, which scores the variant as benign. In contrast, the majority of tools predict a pathogenic impact: REVEL, SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools with uncertain or inconclusive results—AlphaMissense‑Optimized, Rosetta, Foldetta, and premPS—are treated as unavailable for pathogenicity assessment. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for T622A, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.071403 | Uncertain | 0.957 | 0.198 | 0.000 | -10.953 | Likely Pathogenic | 0.827 | Likely Pathogenic | Ambiguous | 0.03 | Likely Benign | 0.0 | 1.04 | Ambiguous | 0.54 | Ambiguous | 0.75 | Ambiguous | 0.893 | Likely Pathogenic | -4.58 | Deleterious | 0.998 | Probably Damaging | 0.989 | Probably Damaging | -1.52 | Pathogenic | 0.01 | Affected | 0.3191 | 0.3091 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1864A>T | T622S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T622S is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from SIFT and AlphaMissense‑Optimized, while pathogenic predictions arise from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant as benign, whereas the SGM‑Consensus predicts it to be likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result. No other folding‑stability tools provide conclusive evidence. Overall, the preponderance of predictions, including the SGM‑Consensus, indicates that T622S is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.071403 | Uncertain | 0.957 | 0.198 | 0.000 | -9.840 | Likely Pathogenic | 0.669 | Likely Pathogenic | Likely Benign | 0.78 | Ambiguous | 0.1 | 1.36 | Ambiguous | 1.07 | Ambiguous | 0.80 | Ambiguous | 0.705 | Likely Pathogenic | -3.52 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.50 | Pathogenic | 0.09 | Tolerated | 0.2523 | 0.3183 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1865C>A | T622N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant T622N has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect are SIFT and AlphaMissense‑Optimized, whereas a majority of tools predict a pathogenic impact: REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and the SGM‑Consensus. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No prediction or stability result is missing; all available data are considered. Overall, the preponderance of evidence points to a pathogenic effect for T622N, and this conclusion is not contradicted by ClinVar status, which currently lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.071403 | Uncertain | 0.957 | 0.198 | 0.000 | -7.962 | In-Between | 0.693 | Likely Pathogenic | Likely Benign | 1.00 | Ambiguous | 0.1 | 2.37 | Destabilizing | 1.69 | Ambiguous | 0.94 | Ambiguous | 0.564 | Likely Pathogenic | -4.14 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | -1.24 | Pathogenic | 0.13 | Tolerated | 0.0943 | 0.2848 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.1865C>G | T622S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T622S is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from SIFT and AlphaMissense‑Optimized, while pathogenic predictions arise from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized labeling the variant as benign, whereas the SGM‑Consensus predicts it to be likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result. No other folding‑stability tools provide conclusive evidence. Overall, the preponderance of predictions, including the SGM‑Consensus, indicates that T622S is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.071403 | Uncertain | 0.957 | 0.198 | 0.000 | -9.840 | Likely Pathogenic | 0.669 | Likely Pathogenic | Likely Benign | 0.78 | Ambiguous | 0.1 | 1.36 | Ambiguous | 1.07 | Ambiguous | 0.80 | Ambiguous | 0.595 | Likely Pathogenic | -3.52 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | -1.50 | Pathogenic | 0.09 | Tolerated | 0.2523 | 0.3183 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1865C>T | T622I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T622I is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign are Foldetta and premPS, whereas the remaining tools (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict it to be pathogenic; FoldX and Rosetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall distribution of predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.268042 | Structured | 0.071403 | Uncertain | 0.957 | 0.198 | 0.000 | -13.276 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | -1.47 | Ambiguous | 0.1 | 0.67 | Ambiguous | -0.40 | Likely Benign | 0.45 | Likely Benign | 0.905 | Likely Pathogenic | -5.57 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | -1.57 | Pathogenic | 0.00 | Affected | 0.0855 | 0.4926 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.1867C>A | L623I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L623I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign calls come only from REVEL and PROVEAN, while pathogenic calls are made by FoldX, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus (which is derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Uncertain results are reported by Rosetta and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is inconclusive, the SGM‑Consensus is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenicity. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.060667 | Uncertain | 0.962 | 0.211 | 0.000 | -11.952 | Likely Pathogenic | 0.911 | Likely Pathogenic | Ambiguous | 3.15 | Destabilizing | 0.5 | 1.90 | Ambiguous | 2.53 | Destabilizing | 1.13 | Destabilizing | 0.398 | Likely Benign | -1.99 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 1.63 | Pathogenic | 0.02 | Affected | 0.1109 | 0.3767 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.1867C>G | L623V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L623V is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only REVEL, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Rosetta and AlphaMissense‑Optimized give uncertain results. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic, and AlphaMissense‑Optimized remains uncertain. Overall, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.060667 | Uncertain | 0.962 | 0.211 | 0.000 | -12.802 | Likely Pathogenic | 0.896 | Likely Pathogenic | Ambiguous | 3.96 | Destabilizing | 0.3 | 1.84 | Ambiguous | 2.90 | Destabilizing | 1.45 | Destabilizing | 0.416 | Likely Benign | -2.99 | Deleterious | 0.998 | Probably Damaging | 0.992 | Probably Damaging | 1.60 | Pathogenic | 0.01 | Affected | 0.1653 | 0.3588 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1867C>T | L623F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L623F is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated tools—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN)—predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result. No predictions are missing or inconclusive. Based on the overwhelming agreement among pathogenic predictions and the lack of a benign signal, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.060667 | Uncertain | 0.962 | 0.211 | 0.000 | -11.655 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 1.22 | Ambiguous | 0.2 | 0.92 | Ambiguous | 1.07 | Ambiguous | 0.57 | Ambiguous | 0.440 | Likely Benign | -3.98 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 1.65 | Pathogenic | 0.02 | Affected | 0.0838 | 0.3016 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1868T>A | L623H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L623H is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity all converge on a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, while no tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized indicates pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No contradictory evidence is present. Based on the unanimous computational predictions, the variant is most likely pathogenic, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.060667 | Uncertain | 0.962 | 0.211 | 0.000 | -14.631 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.76 | Destabilizing | 0.3 | 2.22 | Destabilizing | 2.49 | Destabilizing | 2.40 | Destabilizing | 0.793 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.55 | Pathogenic | 0.00 | Affected | 0.1099 | 0.0541 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1868T>C | L623P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L623P is not reported in ClinVar and is absent from gnomAD. Prediction tools uniformly indicate a deleterious effect: pathogenic predictions are returned by REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the change as pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels it likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a pathogenic effect. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.060667 | Uncertain | 0.962 | 0.211 | 0.000 | -12.385 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 6.70 | Destabilizing | 0.2 | 11.18 | Destabilizing | 8.94 | Destabilizing | 2.26 | Destabilizing | 0.788 | Likely Pathogenic | -6.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 1.56 | Pathogenic | 0.00 | Affected | 0.3961 | 0.1474 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1868T>G | L623R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L623R is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All available evidence points to a pathogenic impact. Thus, the variant is most likely pathogenic, with no contradiction to ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.060667 | Uncertain | 0.962 | 0.211 | 0.000 | -13.718 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 4.08 | Destabilizing | 1.4 | 3.28 | Destabilizing | 3.68 | Destabilizing | 2.31 | Destabilizing | 0.795 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 1.55 | Pathogenic | 0.00 | Affected | 0.1327 | 0.0615 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.186T>A | D62E 2D AIThe SynGAP1 missense variant D62E is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic prediction comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions point to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -2.653 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -0.19 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.49 | Benign | 0.00 | Affected | 0.1884 | 0.5800 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.186T>G | D62E 2D AIThe SynGAP1 missense variant D62E is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from SIFT. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments confirm this: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta, has no available result for this variant. Overall, the consensus of the majority of tools and the high‑accuracy predictions point to a benign effect, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.425610 | Structured | 0.476010 | Uncertain | 0.575 | 0.720 | 0.125 | -2.653 | Likely Benign | 0.095 | Likely Benign | Likely Benign | 0.065 | Likely Benign | -0.19 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.49 | Benign | 0.00 | Affected | 0.1884 | 0.5800 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1870A>C | T624P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T624P is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a deleterious effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic, while premPS remains uncertain. No tool predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. Consequently, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.137348 | Structured | 0.052894 | Uncertain | 0.962 | 0.217 | 0.000 | -15.681 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | 3.64 | Destabilizing | 0.2 | 9.04 | Destabilizing | 6.34 | Destabilizing | 0.83 | Ambiguous | 0.914 | Likely Pathogenic | -5.94 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | -1.55 | Pathogenic | 0.01 | Affected | 0.1500 | 0.3569 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1870A>G | T624A 2D AIThe SynGAP1 T624A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are FoldX and Foldetta, while the majority of tools—REVEL, SIFT, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and the SGM‑Consensus—predict a pathogenic outcome. Uncertain predictions come from AlphaMissense‑Optimized, Rosetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the preponderance of evidence points to a pathogenic effect for T624A, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.137348 | Structured | 0.052894 | Uncertain | 0.962 | 0.217 | 0.000 | -12.967 | Likely Pathogenic | 0.902 | Likely Pathogenic | Ambiguous | -0.38 | Likely Benign | 0.4 | 0.51 | Ambiguous | 0.07 | Likely Benign | 0.80 | Ambiguous | 0.895 | Likely Pathogenic | -4.94 | Deleterious | 0.962 | Probably Damaging | 0.694 | Possibly Damaging | -1.45 | Pathogenic | 0.03 | Affected | 0.2903 | 0.2872 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1870A>T | T624S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T624S missense variant is not reported in ClinVar (status: None) and has no entry in gnomAD. Prediction tools that agree on a benign effect include FoldX, Foldetta, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Rosetta and premPS are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.137348 | Structured | 0.052894 | Uncertain | 0.962 | 0.217 | 0.000 | -13.314 | Likely Pathogenic | 0.766 | Likely Pathogenic | Likely Benign | -0.10 | Likely Benign | 0.1 | 0.95 | Ambiguous | 0.43 | Likely Benign | 0.69 | Ambiguous | 0.761 | Likely Pathogenic | -3.93 | Deleterious | 0.826 | Possibly Damaging | 0.789 | Possibly Damaging | -1.43 | Pathogenic | 0.01 | Affected | 0.2326 | 0.2813 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1871C>A | T624N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T624N is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include FoldX and Foldetta, whereas the majority of tools predict a pathogenic impact: REVEL, SIFT, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and the SGM Consensus (which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). Uncertain results come from AlphaMissense‑Optimized, Rosetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus as likely pathogenic, and Foldetta as benign. Overall, the balance of evidence favors a pathogenic classification for T624N, and this conclusion does not contradict any ClinVar annotation because no ClinVar status is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.137348 | Structured | 0.052894 | Uncertain | 0.962 | 0.217 | 0.000 | -14.658 | Likely Pathogenic | 0.915 | Likely Pathogenic | Ambiguous | -0.24 | Likely Benign | 0.0 | 0.87 | Ambiguous | 0.32 | Likely Benign | 0.97 | Ambiguous | 0.848 | Likely Pathogenic | -4.94 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | -1.53 | Pathogenic | 0.00 | Affected | 0.0773 | 0.2694 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.1871C>G | T624S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T624S missense variant is not reported in ClinVar (status: None) and has no entry in gnomAD. Prediction tools that agree on a benign effect include FoldX, Foldetta, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Rosetta and premPS are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict any ClinVar annotation (none is present). Thus, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.137348 | Structured | 0.052894 | Uncertain | 0.962 | 0.217 | 0.000 | -13.314 | Likely Pathogenic | 0.766 | Likely Pathogenic | Likely Benign | -0.10 | Likely Benign | 0.1 | 0.95 | Ambiguous | 0.43 | Likely Benign | 0.69 | Ambiguous | 0.734 | Likely Pathogenic | -3.93 | Deleterious | 0.826 | Possibly Damaging | 0.789 | Possibly Damaging | -1.43 | Pathogenic | 0.01 | Affected | 0.2326 | 0.2813 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.1871C>T | T624I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T624I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include Rosetta, premPS, and SIFT, whereas the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). FoldX and Foldetta are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for T624I. This prediction is consistent with the lack of ClinVar annotation and does not contradict any existing ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.137348 | Structured | 0.052894 | Uncertain | 0.962 | 0.217 | 0.000 | -10.999 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | -0.74 | Ambiguous | 0.1 | -0.39 | Likely Benign | -0.57 | Ambiguous | 0.14 | Likely Benign | 0.865 | Likely Pathogenic | -5.95 | Deleterious | 1.000 | Probably Damaging | 0.972 | Probably Damaging | -1.43 | Pathogenic | 0.08 | Tolerated | 0.0728 | 0.4734 | 0 | -1 | 5.2 | 12.05 | |||||||||||||||||||||||||||||
| c.1873C>A | L625I 2D AIThe SynGAP1 missense variant L625I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, and FATHMM, whereas a majority of tools (premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. Tools with uncertain or inconclusive results—FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized—are treated as unavailable. High‑accuracy assessments are likewise inconclusive: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑vs‑2 tie, and Foldetta is uncertain. Consequently, the overall evidence leans toward pathogenicity, with no conflict with ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.229226 | Structured | 0.045896 | Uncertain | 0.966 | 0.215 | 0.000 | -11.713 | Likely Pathogenic | 0.866 | Likely Pathogenic | Ambiguous | 0.75 | Ambiguous | 0.6 | 0.72 | Ambiguous | 0.74 | Ambiguous | 1.09 | Destabilizing | 0.412 | Likely Benign | -1.96 | Neutral | 0.999 | Probably Damaging | 0.997 | Probably Damaging | 3.06 | Benign | 0.01 | Affected | 0.0864 | 0.3588 | 2 | 2 | 0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1873C>G | L625V 2D AISynGAP1 missense variant L625V is listed in ClinVar with an uncertain significance (ClinVar ID 3392716.0) and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL and FATHMM, while pathogenic predictions are made by premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points toward a pathogenic effect, which does not contradict the ClinVar uncertain status but suggests a higher likelihood of pathogenicity. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.045896 | Uncertain | 0.966 | 0.215 | 0.000 | Uncertain | 1 | -11.319 | Likely Pathogenic | 0.833 | Likely Pathogenic | Ambiguous | 1.80 | Ambiguous | 0.7 | 1.69 | Ambiguous | 1.75 | Ambiguous | 1.42 | Destabilizing | 0.480 | Likely Benign | -2.96 | Deleterious | 0.998 | Probably Damaging | 0.992 | Probably Damaging | 3.07 | Benign | 0.01 | Affected | 0.1306 | 0.3427 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||
| c.1873C>T | L625F 2D AIThe SynGAP1 missense variant L625F is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM. Tools that predict a pathogenic effect include SGM‑Consensus (Likely Pathogenic), Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Predictions that are uncertain or inconclusive are FoldX, premPS, and Foldetta. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts Pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts Likely Pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is Uncertain. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.045896 | Uncertain | 0.966 | 0.215 | 0.000 | -12.989 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 1.70 | Ambiguous | 1.3 | 2.26 | Destabilizing | 1.98 | Ambiguous | 0.74 | Ambiguous | 0.479 | Likely Benign | -3.82 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.01 | Benign | 0.01 | Affected | 0.0586 | 0.2998 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1874T>A | L625H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L625H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.045896 | Uncertain | 0.966 | 0.215 | 0.000 | -14.264 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 2.85 | Destabilizing | 0.3 | 2.96 | Destabilizing | 2.91 | Destabilizing | 2.02 | Destabilizing | 0.738 | Likely Pathogenic | -6.87 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.98 | Benign | 0.00 | Affected | 0.0954 | 0.0541 | -2 | -3 | -7.0 | 23.98 | |||||||||||||||||||||||||||||
| c.1874T>C | L625P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L625P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy methods give consistent results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.045896 | Uncertain | 0.966 | 0.215 | 0.000 | -14.819 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 5.47 | Destabilizing | 0.6 | 12.49 | Destabilizing | 8.98 | Destabilizing | 1.98 | Destabilizing | 0.746 | Likely Pathogenic | -6.92 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.98 | Benign | 0.00 | Affected | 0.3294 | 0.1313 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1874T>G | L625R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L625R is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.229226 | Structured | 0.045896 | Uncertain | 0.966 | 0.215 | 0.000 | -14.507 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 2.31 | Destabilizing | 0.7 | 4.21 | Destabilizing | 3.26 | Destabilizing | 1.88 | Destabilizing | 0.733 | Likely Pathogenic | -5.93 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.99 | Benign | 0.00 | Affected | 0.1091 | 0.0615 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1876A>C | I626L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I626L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, SIFT, and FATHMM, whereas polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default predict a pathogenic outcome. Three tools—Foldetta, premPS, and Rosetta—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta also yields an uncertain stability prediction. Overall, the balance of evidence leans toward a benign interpretation, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.109221 | Structured | 0.040732 | Uncertain | 0.970 | 0.223 | 0.000 | -10.696 | Likely Pathogenic | 0.622 | Likely Pathogenic | Likely Benign | 0.32 | Likely Benign | 0.1 | 1.00 | Ambiguous | 0.66 | Ambiguous | 0.83 | Ambiguous | 0.311 | Likely Benign | -1.86 | Neutral | 0.955 | Possibly Damaging | 0.985 | Probably Damaging | 3.52 | Benign | 0.12 | Tolerated | 0.0741 | 0.2461 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1876A>G | I626V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I626V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and polyPhen‑2 HumVar. Predictions that are uncertain or unavailable are AlphaMissense‑Default, FoldX, Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus also indicates a likely benign outcome, while Foldetta’s stability analysis is inconclusive. Based on the overall consensus of the available predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.109221 | Structured | 0.040732 | Uncertain | 0.970 | 0.223 | 0.000 | -6.636 | Likely Benign | 0.398 | Ambiguous | Likely Benign | 1.24 | Ambiguous | 0.0 | 1.06 | Ambiguous | 1.15 | Ambiguous | 0.80 | Ambiguous | 0.182 | Likely Benign | -0.80 | Neutral | 0.969 | Probably Damaging | 0.960 | Probably Damaging | 3.33 | Benign | 0.13 | Tolerated | 0.1003 | 0.2891 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||
| c.1876A>T | I626F 2D 3DClick to see structure in 3D Viewer AISynGAP1 I626F is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS (uncertain), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—predict it to be pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, but the SGM‑Consensus (derived from a majority of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. Taken together, the overwhelming majority of evidence indicates that I626F is likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.040732 | Uncertain | 0.970 | 0.223 | 0.000 | -14.483 | Likely Pathogenic | 0.952 | Likely Pathogenic | Ambiguous | 4.37 | Destabilizing | 0.3 | 2.12 | Destabilizing | 3.25 | Destabilizing | 0.66 | Ambiguous | 0.631 | Likely Pathogenic | -3.78 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.07 | Benign | 0.00 | Affected | 0.0481 | 0.1964 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.1877T>A | I626N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I626N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods further support this: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the unanimous agreement of the majority of tools and the corroborating high‑accuracy predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.040732 | Uncertain | 0.970 | 0.223 | 0.000 | -15.240 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.49 | Destabilizing | 0.1 | 3.56 | Destabilizing | 3.53 | Destabilizing | 2.36 | Destabilizing | 0.621 | Likely Pathogenic | -6.41 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.03 | Benign | 0.00 | Affected | 0.0880 | 0.0270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1877T>C | I626T 2D AISynGAP1 missense variant I626T is listed in ClinVar with an uncertain significance (ClinVar ID 3359331.0) and is not reported in gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic predictions are returned by REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Only FATHMM predicts a benign outcome, while AlphaMissense‑Optimized is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is inconclusive, SGM‑Consensus indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Overall, the consensus of the majority of tools points to a pathogenic effect, contradicting the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.040732 | Uncertain | 0.970 | 0.223 | 0.000 | Uncertain | 1 | -10.420 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 2.94 | Destabilizing | 0.1 | 2.70 | Destabilizing | 2.82 | Destabilizing | 2.23 | Destabilizing | 0.640 | Likely Pathogenic | -4.18 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.04 | Benign | 0.00 | Affected | 0.1000 | 0.0840 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||
| c.1877T>G | I626S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I626S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.040732 | Uncertain | 0.970 | 0.223 | 0.000 | -14.449 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 3.80 | Destabilizing | 0.0 | 4.54 | Destabilizing | 4.17 | Destabilizing | 2.16 | Destabilizing | 0.706 | Likely Pathogenic | -5.41 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.04 | Benign | 0.00 | Affected | 0.2308 | 0.0858 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.1878T>G | I626M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I626M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX, Rosetta, Foldetta, and premPS give uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts likely pathogenic; Foldetta’s stability prediction is uncertain. Overall, the majority of evidence points to a pathogenic impact. This conclusion does not contradict ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.109221 | Structured | 0.040732 | Uncertain | 0.970 | 0.223 | 0.000 | -11.407 | Likely Pathogenic | 0.618 | Likely Pathogenic | Likely Benign | 0.72 | Ambiguous | 0.1 | 1.78 | Ambiguous | 1.25 | Ambiguous | 0.92 | Ambiguous | 0.405 | Likely Benign | -2.76 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.06 | Benign | 0.02 | Affected | 0.0661 | 0.1959 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||
| c.1879G>A | A627T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A627T is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently predict a pathogenic impact. Two tools, premPS and AlphaMissense‑Optimized, return uncertain results. High‑accuracy assessments further support pathogenicity: the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict, and Foldetta also predicts a destabilizing, pathogenic effect. AlphaMissense‑Optimized remains uncertain. Overall, the preponderance of evidence indicates that A627T is most likely pathogenic, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.037862 | Uncertain | 0.970 | 0.210 | 0.000 | -8.613 | Likely Pathogenic | 0.937 | Likely Pathogenic | Ambiguous | 2.27 | Destabilizing | 0.3 | 2.33 | Destabilizing | 2.30 | Destabilizing | 0.80 | Ambiguous | 0.505 | Likely Pathogenic | -3.95 | Deleterious | 0.994 | Probably Damaging | 0.807 | Possibly Damaging | 2.56 | Benign | 0.01 | Affected | 0.1069 | 0.5403 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.1879G>C | A627P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A627P is not reported in ClinVar and is absent from gnomAD. Prediction tools were grouped by consensus: Benign – none; Pathogenic – SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized. High‑accuracy methods specifically: AlphaMissense‑Optimized predicts pathogenicity; SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic; Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. All available evidence points to a deleterious effect. Therefore, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.037862 | Uncertain | 0.970 | 0.210 | 0.000 | -15.404 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 5.78 | Destabilizing | 0.3 | 7.84 | Destabilizing | 6.81 | Destabilizing | 1.13 | Destabilizing | 0.740 | Likely Pathogenic | -4.96 | Deleterious | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 2.43 | Pathogenic | 0.01 | Affected | 0.1752 | 0.3422 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1879G>T | A627S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A627S missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are Rosetta, PROVEAN, and ESM1b. The remaining tools—FoldX, Foldetta, and premPS—return uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split, and Foldetta is uncertain. Overall, the majority of available predictions lean toward a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.100716 | Structured | 0.037862 | Uncertain | 0.970 | 0.210 | 0.000 | -10.782 | Likely Pathogenic | 0.329 | Likely Benign | Likely Benign | 1.11 | Ambiguous | 0.2 | 2.05 | Destabilizing | 1.58 | Ambiguous | 0.71 | Ambiguous | 0.316 | Likely Benign | -2.94 | Deleterious | 0.411 | Benign | 0.387 | Benign | 2.78 | Benign | 0.11 | Tolerated | 0.2266 | 0.4224 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||
| c.187G>A | E63K 2D AIThe SynGAP1 E63K missense variant (ClinVar ID 2830630.0) is listed as “Uncertain” and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default all predict a pathogenic outcome. AlphaMissense‑Optimized is inconclusive, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. The SGM‑Consensus, which aggregates the majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. Overall, the high‑accuracy consensus leans toward a benign effect, and this assessment does not contradict the ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.474807 | Uncertain | 0.494 | 0.739 | 0.125 | Uncertain | 1 | -4.976 | Likely Benign | 0.894 | Likely Pathogenic | Ambiguous | 0.103 | Likely Benign | -0.70 | Neutral | 0.458 | Possibly Damaging | 0.678 | Possibly Damaging | 3.98 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1995 | 0.7261 | 1 | 0 | -0.4 | -0.94 | |||||||||||||||||||||||||||||||||||
| c.187G>C | E63Q 2D AIThe SynGAP1 missense variant E63Q is listed in ClinVar (ID 2132335.0) with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as benign, and the SGM‑Consensus (majority of the four high‑accuracy tools) also indicates a benign outcome. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the preponderance of predictions points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.474807 | Uncertain | 0.494 | 0.739 | 0.125 | Uncertain | 1 | -4.038 | Likely Benign | 0.687 | Likely Pathogenic | Likely Benign | 0.078 | Likely Benign | -0.85 | Neutral | 0.659 | Possibly Damaging | 0.775 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0970 | 0.6787 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||||||||
| c.1880C>A | A627D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A627D is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (gnomAD ID: none). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool in the dataset predicts a benign effect. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts pathogenic. All available evidence points to a damaging impact. Consequently, the variant is most likely pathogenic based on current predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.037862 | Uncertain | 0.970 | 0.210 | 0.000 | -16.603 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 6.09 | Destabilizing | 1.3 | 5.83 | Destabilizing | 5.96 | Destabilizing | 1.58 | Destabilizing | 0.726 | Likely Pathogenic | -5.93 | Deleterious | 0.999 | Probably Damaging | 0.961 | Probably Damaging | 2.43 | Pathogenic | 0.00 | Affected | 0.1502 | 0.1816 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.1880C>G | A627G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A627G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect comprise SGM‑Consensus, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX and Foldetta are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta remains uncertain. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.037862 | Uncertain | 0.970 | 0.210 | 0.000 | -11.716 | Likely Pathogenic | 0.640 | Likely Pathogenic | Likely Benign | 1.38 | Ambiguous | 0.1 | 2.07 | Destabilizing | 1.73 | Ambiguous | 1.25 | Destabilizing | 0.458 | Likely Benign | -3.96 | Deleterious | 0.997 | Probably Damaging | 0.876 | Possibly Damaging | 2.51 | Benign | 0.01 | Affected | 0.1921 | 0.2990 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1880C>T | A627V 2D AIThe SynGAP1 missense variant A627V is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity unanimously classify the variant as deleterious: REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. No tool in the dataset predicts a benign effect. Two tools return uncertain results: Rosetta and premPS. High‑accuracy assessments reinforce the pathogenic prediction: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta is pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.037862 | Uncertain | 0.970 | 0.210 | 0.000 | -12.150 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 2.64 | Destabilizing | 1.5 | 1.59 | Ambiguous | 2.12 | Destabilizing | 0.74 | Ambiguous | 0.549 | Likely Pathogenic | -3.98 | Deleterious | 0.999 | Probably Damaging | 0.900 | Possibly Damaging | 2.47 | Pathogenic | 0.00 | Affected | 0.0856 | 0.4551 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||||
| c.1882A>C | K628Q 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K628Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include FoldX, Rosetta, and Foldetta, all of which score the variant as benign. In contrast, the majority of tools predict a pathogenic impact: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). The premPS tool yields an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, and Foldetta as benign. Overall, the balance of evidence favors a pathogenic classification, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | -12.263 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 0.46 | Likely Benign | 0.0 | -0.16 | Likely Benign | 0.15 | Likely Benign | 0.95 | Ambiguous | 0.587 | Likely Pathogenic | -3.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.46 | Pathogenic | 0.00 | Affected | 0.3444 | 0.1358 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1882A>G | K628E 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 K628E missense variant is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that are inconclusive or uncertain are FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Uncertain. Overall, the majority of evidence points to a deleterious effect. The variant is most likely pathogenic based on these predictions, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | -14.658 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 1.04 | Ambiguous | 0.2 | 0.52 | Ambiguous | 0.78 | Ambiguous | 1.06 | Destabilizing | 0.652 | Likely Pathogenic | -3.98 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.37 | Pathogenic | 0.00 | Affected | 0.2909 | 0.0810 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1883A>C | K628T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K628T is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools largely converge on a deleterious effect: the majority—including REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus—label the change as pathogenic or likely pathogenic. Only Rosetta predicts a benign outcome, while FoldX, Foldetta, and premPS are inconclusive. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized scores the variant as pathogenic, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic, and Foldetta remains uncertain. Taken together, the preponderance of evidence indicates that K628T is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | -14.675 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 1.08 | Ambiguous | 0.1 | 0.07 | Likely Benign | 0.58 | Ambiguous | 0.61 | Ambiguous | 0.661 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.36 | Pathogenic | 0.00 | Affected | 0.1622 | 0.3541 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1883A>G | K628R 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant K628R is reported in gnomAD (variant ID 6‑33440935‑A‑G) but has no ClinVar entry. Functional prediction tools show a split assessment: benign calls come from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized, whereas pathogenic calls are made by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. Two tools remain inconclusive (premPS and AlphaMissense‑Default). The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, yields a likely pathogenic verdict, while the high‑accuracy AlphaMissense‑Optimized predicts benign. Foldetta, a protein‑folding stability method that combines FoldX‑MD and Rosetta outputs, also predicts benign. Overall, the majority of evidence leans toward pathogenicity, and this conclusion does not conflict with ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | 6-33440935-A-G | 1 | 6.20e-7 | -11.324 | Likely Pathogenic | 0.476 | Ambiguous | Likely Benign | 0.22 | Likely Benign | 0.1 | -0.11 | Likely Benign | 0.06 | Likely Benign | 0.94 | Ambiguous | 0.592 | Likely Pathogenic | -2.99 | Deleterious | 0.996 | Probably Damaging | 0.990 | Probably Damaging | 2.49 | Pathogenic | 0.00 | Affected | 3.37 | 34 | 0.3873 | 0.0873 | 2 | 3 | -0.6 | 28.01 | ||||||||||||||||||||||||
| c.1883A>T | K628M 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 K628M missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FoldX and premPS, whereas the majority of tools—REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic impact. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) yields an inconclusive result, which is treated as unavailable evidence. Overall, the preponderance of predictions points to a pathogenic effect for K628M, and this conclusion does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | -14.949 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | -0.41 | Likely Benign | 0.2 | -0.76 | Ambiguous | -0.59 | Ambiguous | 0.37 | Likely Benign | 0.669 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.34 | Pathogenic | 0.00 | Affected | 0.0802 | 0.3875 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1884G>C | K628N 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K628N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only REVEL, which scores the variant as benign. All other evaluated algorithms—premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN)—classify the variant as pathogenic or likely pathogenic. FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability predictions and are therefore treated as unavailable evidence. High‑accuracy assessments specifically show AlphaMissense‑Optimized predicting pathogenicity, the SGM‑Consensus indicating a likely pathogenic status, and Foldetta yielding an uncertain result. Based on the overwhelming agreement among the majority of prediction tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | -12.284 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.78 | Ambiguous | 0.1 | 0.59 | Ambiguous | 0.69 | Ambiguous | 1.11 | Destabilizing | 0.403 | Likely Benign | -4.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.37 | Pathogenic | 0.00 | Affected | 3.37 | 34 | 0.2740 | 0.1254 | 0 | 1 | 0.4 | -14.07 | |||||||||||||||||||||||||||
| c.1884G>T | K628N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K628N is catalogued in gnomAD (6‑33440936‑G‑T) but has no ClinVar entry. Functional prediction tools cluster into two groups: a single benign prediction from REVEL, and a consensus of nine pathogenic predictions from premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also labels the variant as Likely Pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus confirms a likely pathogenic status, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result and therefore does not alter the overall interpretation. Consequently, the variant is most likely pathogenic according to the available computational evidence, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.035486 | Uncertain | 0.957 | 0.229 | 0.000 | 6-33440936-G-T | 1 | 6.20e-7 | -12.284 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 0.78 | Ambiguous | 0.1 | 0.59 | Ambiguous | 0.69 | Ambiguous | 1.11 | Destabilizing | 0.403 | Likely Benign | -4.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.37 | Pathogenic | 0.00 | Affected | 3.37 | 34 | 0.2740 | 0.1254 | 0 | 1 | 0.4 | -14.07 | ||||||||||||||||||||||||
| c.1885G>A | V629I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V629I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, AlphaMissense‑Optimized, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar) and SIFT, with ESM1b also indicating pathogenicity. AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, and Foldetta also predicts a benign outcome. Taken together, the preponderance of evidence supports a benign classification for V629I, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -9.876 | Likely Pathogenic | 0.462 | Ambiguous | Likely Benign | -0.22 | Likely Benign | 0.1 | 0.37 | Likely Benign | 0.08 | Likely Benign | 0.21 | Likely Benign | 0.305 | Likely Benign | -1.00 | Neutral | 0.975 | Probably Damaging | 0.958 | Probably Damaging | 3.26 | Benign | 0.04 | Affected | 0.0650 | 0.2919 | 4 | 3 | 0.3 | 14.03 | ||||||||||||||||||||||||||||||
| c.1885G>C | V629L 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V629L has no ClinVar entry and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, and FATHMM, whereas the majority of algorithms predict a pathogenic outcome: Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools report uncertainty: Foldetta and premPS. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus also indicates likely pathogenic, while Foldetta remains uncertain. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -12.785 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | 0.06 | Likely Benign | 0.2 | 2.26 | Destabilizing | 1.16 | Ambiguous | 0.54 | Ambiguous | 0.391 | Likely Benign | -2.79 | Deleterious | 0.975 | Probably Damaging | 0.958 | Probably Damaging | 3.22 | Benign | 0.02 | Affected | 0.0805 | 0.3336 | 2 | 1 | -0.4 | 14.03 | |||||||||||||||||||||||||||||
| c.1885G>T | V629F 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V629F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact; premPS is uncertain and is not counted as evidence. High‑accuracy methods further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta is pathogenic. No prediction is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions and the absence of any ClinVar annotation, the variant is most likely pathogenic, with no contradiction from ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -13.484 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 3.41 | Destabilizing | 0.5 | 4.40 | Destabilizing | 3.91 | Destabilizing | 0.64 | Ambiguous | 0.642 | Likely Pathogenic | -4.68 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.07 | Benign | 0.00 | Affected | 0.0560 | 0.2915 | -1 | -1 | -1.4 | 48.04 | |||||||||||||||||||||||||||||
| c.1886T>A | V629D 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant V629D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all return pathogenic or likely pathogenic calls, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized indicates pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) predicts pathogenic. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -17.143 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 3.85 | Destabilizing | 0.1 | 3.82 | Destabilizing | 3.84 | Destabilizing | 2.17 | Destabilizing | 0.713 | Likely Pathogenic | -6.37 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.06 | Benign | 0.00 | Affected | 0.1284 | 0.0541 | -2 | -3 | -7.7 | 15.96 | |||||||||||||||||||||||||||||
| c.1886T>C | V629A 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 V629A missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. Those that predict a pathogenic effect comprise SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. Two tools give uncertain results: Rosetta and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as Pathogenic (combining FoldX‑MD and Rosetta outputs). Overall, the majority of predictions (8 pathogenic vs. 3 benign) indicate that V629A is most likely pathogenic, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -8.652 | Likely Pathogenic | 0.926 | Likely Pathogenic | Ambiguous | 2.24 | Destabilizing | 0.1 | 1.96 | Ambiguous | 2.10 | Destabilizing | 1.58 | Destabilizing | 0.492 | Likely Benign | -3.58 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.18 | Benign | 0.11 | Tolerated | 0.2518 | 0.2124 | 0 | 0 | -2.4 | -28.05 | |||||||||||||||||||||||||||||
| c.1886T>G | V629G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 V629G missense change is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining eleven tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) uniformly predict a pathogenic outcome; AlphaMissense‑Optimized is uncertain. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. Taken together, the overwhelming majority of evidence indicates that V629G is likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.040537 | Structured | 0.034796 | Uncertain | 0.970 | 0.236 | 0.000 | -13.150 | Likely Pathogenic | 0.871 | Likely Pathogenic | Ambiguous | 3.81 | Destabilizing | 0.0 | 4.52 | Destabilizing | 4.17 | Destabilizing | 1.94 | Destabilizing | 0.678 | Likely Pathogenic | -6.47 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.07 | Benign | 0.00 | Affected | 0.1903 | 0.2240 | -1 | -3 | -4.6 | -42.08 | ||||||||||||||||||||||||||||||
| c.1888A>C | I630L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I630L is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33440940‑A‑C). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are ESM1b and FATHMM. The SGM Consensus, which takes a majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as benign and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign; the SGM Consensus remains unavailable. Based on the overall predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.040537 | Structured | 0.036106 | Uncertain | 0.966 | 0.236 | 0.000 | 6-33440940-A-C | -8.949 | Likely Pathogenic | 0.277 | Likely Benign | Likely Benign | -0.39 | Likely Benign | 0.0 | 0.23 | Likely Benign | -0.08 | Likely Benign | 0.33 | Likely Benign | 0.165 | Likely Benign | -1.30 | Neutral | 0.102 | Benign | 0.108 | Benign | -0.81 | Pathogenic | 0.27 | Tolerated | 3.37 | 34 | 0.0688 | 0.2461 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||
| c.1888A>T | I630F 2D 3DClick to see structure in 3D Viewer AISynGAP1 I630F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only AlphaMissense‑Optimized. Those that agree on a pathogenic effect comprise SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results are reported by Rosetta, Foldetta, and premPS and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicting likely pathogenic, and Foldetta yielding an uncertain stability change. Overall, the preponderance of evidence points to a pathogenic impact for I630F. This conclusion is not contradicted by ClinVar, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.036106 | Uncertain | 0.966 | 0.236 | 0.000 | -13.669 | Likely Pathogenic | 0.705 | Likely Pathogenic | Likely Benign | 2.52 | Destabilizing | 0.3 | 0.76 | Ambiguous | 1.64 | Ambiguous | 0.75 | Ambiguous | 0.782 | Likely Pathogenic | -3.12 | Deleterious | 0.935 | Possibly Damaging | 0.473 | Possibly Damaging | -1.46 | Pathogenic | 0.01 | Affected | 0.0443 | 0.1964 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.1889T>A | I630N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I630N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a deleterious effect. Benign predictions: none. Pathogenic predictions: SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts pathogenic. Based on the consensus of all available predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.036106 | Uncertain | 0.966 | 0.236 | 0.000 | -14.259 | Likely Pathogenic | 0.979 | Likely Pathogenic | Likely Pathogenic | 2.96 | Destabilizing | 0.0 | 2.55 | Destabilizing | 2.76 | Destabilizing | 2.39 | Destabilizing | 0.825 | Likely Pathogenic | -4.86 | Deleterious | 1.000 | Probably Damaging | 0.994 | Probably Damaging | -1.49 | Pathogenic | 0.00 | Affected | 0.0853 | 0.0270 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.1889T>C | I630T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I630T has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include PROVEAN, SIFT, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Default; ESM1b remains uncertain. High‑accuracy assessments further support a deleterious outcome: AlphaMissense‑Optimized indicates benign, whereas the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic majority, and Foldetta also predicts pathogenic. No prediction is missing or inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for I630T, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.040537 | Structured | 0.036106 | Uncertain | 0.966 | 0.236 | 0.000 | -7.780 | In-Between | 0.754 | Likely Pathogenic | Likely Benign | 2.77 | Destabilizing | 0.1 | 2.27 | Destabilizing | 2.52 | Destabilizing | 1.94 | Destabilizing | 0.734 | Likely Pathogenic | -2.15 | Neutral | 0.997 | Probably Damaging | 0.961 | Probably Damaging | -1.46 | Pathogenic | 0.35 | Tolerated | 0.0985 | 0.0640 | 0 | -1 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||
| c.1889T>G | I630S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I630S is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a pathogenic effect include SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. No tool predicts a benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as pathogenic. No contradictory evidence is present. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not conflict with the ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.040537 | Structured | 0.036106 | Uncertain | 0.966 | 0.236 | 0.000 | -12.527 | Likely Pathogenic | 0.952 | Likely Pathogenic | Ambiguous | 3.61 | Destabilizing | 0.1 | 3.74 | Destabilizing | 3.68 | Destabilizing | 2.25 | Destabilizing | 0.851 | Likely Pathogenic | -3.86 | Deleterious | 1.000 | Probably Damaging | 0.981 | Probably Damaging | -1.44 | Pathogenic | 0.00 | Affected | 0.2363 | 0.0858 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.188A>C | E63A 2D AIThe SynGAP1 missense variant E63A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on benign impact include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain and the SGM‑Consensus as Likely Benign; Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign effect, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.474807 | Uncertain | 0.494 | 0.739 | 0.125 | -3.426 | Likely Benign | 0.850 | Likely Pathogenic | Ambiguous | 0.120 | Likely Benign | -1.84 | Neutral | 0.458 | Possibly Damaging | 0.678 | Possibly Damaging | 3.90 | Benign | 0.00 | Affected | 0.3281 | 0.6649 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||||||||||||
| c.188A>G | E63G 2D AIThe SynGAP1 missense variant E63G is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. AlphaMissense‑Optimized yields an uncertain result, and no Foldetta stability assessment is available. Overall, the majority of individual predictors and the SGM‑Consensus lean toward a benign interpretation, with no conflicting evidence from ClinVar. Thus, the variant is most likely benign based on current computational predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.474807 | Uncertain | 0.494 | 0.739 | 0.125 | -3.450 | Likely Benign | 0.898 | Likely Pathogenic | Ambiguous | 0.150 | Likely Benign | -2.24 | Neutral | 0.659 | Possibly Damaging | 0.775 | Possibly Damaging | 3.87 | Benign | 0.00 | Affected | 0.2705 | 0.5786 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||||||||||||
| c.188A>T | E63V 2D AIThe SynGAP1 E63V missense variant has no ClinVar record and is not present in gnomAD. Prediction tools that agree on benign impact include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Benign. AlphaMissense‑Optimized returns an Uncertain result, and no Foldetta stability data are available. Overall, the balance of evidence leans toward a benign effect, with several high‑confidence predictors supporting pathogenicity, leaving the assessment inconclusive. The predictions do not contradict any ClinVar status, as none is assigned. Based on the aggregate predictions, the variant is most likely benign, and this is consistent with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.474807 | Uncertain | 0.494 | 0.739 | 0.125 | -3.588 | Likely Benign | 0.921 | Likely Pathogenic | Ambiguous | 0.143 | Likely Benign | -2.15 | Neutral | 0.824 | Possibly Damaging | 0.775 | Possibly Damaging | 3.85 | Benign | 0.00 | Affected | 0.0559 | 0.7584 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||||||||||||
| c.1890C>G | I630M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I630M is listed in gnomAD (ID 6‑33440942‑C‑G) but has no ClinVar entry. Functional prediction tools show a split: benign calls come from Rosetta, Foldetta, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic calls come from REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM. FoldX is uncertain, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. High‑accuracy assessments give AlphaMissense‑Optimized benign, Foldetta benign, and an inconclusive SGM Consensus. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.040537 | Structured | 0.036106 | Uncertain | 0.966 | 0.236 | 0.000 | 6-33440942-C-G | 1 | 6.20e-7 | -10.586 | Likely Pathogenic | 0.259 | Likely Benign | Likely Benign | -0.55 | Ambiguous | 0.1 | 0.32 | Likely Benign | -0.12 | Likely Benign | 1.06 | Destabilizing | 0.508 | Likely Pathogenic | -1.90 | Neutral | 0.833 | Possibly Damaging | 0.700 | Possibly Damaging | -1.38 | Pathogenic | 0.02 | Affected | 3.37 | 34 | 0.0618 | 0.1759 | 1 | 2 | -2.6 | 18.03 | |||||||||||||||||||||||||
| c.1891C>A | Q631K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q631K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from FoldX, Foldetta, and FATHMM, while pathogenic predictions arise from REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score. Uncertain or inconclusive results are reported by Rosetta, premPS, and AlphaMissense‑Optimized. High‑accuracy methods give a mixed picture: Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a benign effect; the SGM‑Consensus, a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates a likely pathogenic outcome; AlphaMissense‑Optimized remains uncertain. Overall, the majority of evidence points toward a pathogenic impact, and this assessment does not conflict with the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.038963 | Uncertain | 0.948 | 0.230 | 0.000 | -15.194 | Likely Pathogenic | 0.953 | Likely Pathogenic | Ambiguous | -0.37 | Likely Benign | 0.1 | 1.13 | Ambiguous | 0.38 | Likely Benign | 0.88 | Ambiguous | 0.596 | Likely Pathogenic | -3.98 | Deleterious | 0.958 | Probably Damaging | 0.931 | Probably Damaging | 2.79 | Benign | 0.01 | Affected | 0.1288 | 0.2157 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||
| c.1891C>G | Q631E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q631E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that indicate a benign effect include FoldX, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifying it as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) yielding an uncertain result. Overall, the majority of evidence (eight pathogenic predictions versus three benign) points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, which is currently unavailable. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.038963 | Uncertain | 0.948 | 0.230 | 0.000 | -15.628 | Likely Pathogenic | 0.782 | Likely Pathogenic | Likely Benign | 0.04 | Likely Benign | 0.1 | 1.55 | Ambiguous | 0.80 | Ambiguous | 0.95 | Ambiguous | 0.532 | Likely Pathogenic | -2.99 | Deleterious | 0.997 | Probably Damaging | 0.981 | Probably Damaging | 2.78 | Benign | 0.01 | Affected | 0.1068 | 0.1264 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.1892A>C | Q631P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q631P is not reported in ClinVar and has no entry in gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while the remaining eleven tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) all classify the change as pathogenic. The high‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also indicates pathogenic. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.038963 | Uncertain | 0.948 | 0.230 | 0.000 | -16.914 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 4.98 | Destabilizing | 0.3 | 11.18 | Destabilizing | 8.08 | Destabilizing | 0.56 | Ambiguous | 0.641 | Likely Pathogenic | -5.97 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.75 | Benign | 0.00 | Affected | 0.1999 | 0.2982 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||
| c.1892A>G | Q631R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q631R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions come from FoldX, Foldetta, and FATHMM, while pathogenic predictions are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; Rosetta and premPS are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic impact for Q631R, and this conclusion is not contradicted by any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.038963 | Uncertain | 0.948 | 0.230 | 0.000 | -14.881 | Likely Pathogenic | 0.958 | Likely Pathogenic | Likely Pathogenic | -0.34 | Likely Benign | 0.1 | 0.83 | Ambiguous | 0.25 | Likely Benign | 0.77 | Ambiguous | 0.627 | Likely Pathogenic | -3.98 | Deleterious | 0.973 | Probably Damaging | 0.969 | Probably Damaging | 2.77 | Benign | 0.01 | Affected | 0.1193 | 0.0653 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||
| c.1892A>T | Q631L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q631L is not reported in ClinVar (ClinVar status: None) and has no entry in gnomAD (gnomAD status: None). Prediction tools that agree on a benign effect include FATHMM and Foldetta, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus (likely pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic, and Foldetta as benign; these results are reported but not used as definitive evidence when inconclusive. Overall, the preponderance of evidence from consensus and individual predictors indicates a pathogenic effect for Q631L. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.038963 | Uncertain | 0.948 | 0.230 | 0.000 | -14.727 | Likely Pathogenic | 0.901 | Likely Pathogenic | Ambiguous | -1.23 | Ambiguous | 0.0 | 0.95 | Ambiguous | -0.14 | Likely Benign | 0.51 | Ambiguous | 0.619 | Likely Pathogenic | -6.97 | Deleterious | 0.982 | Probably Damaging | 0.954 | Probably Damaging | 2.85 | Benign | 0.05 | Affected | 0.0627 | 0.3150 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||
| c.1893G>C | Q631H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q631H is reported in gnomAD (6‑33440945‑G‑C) but has no ClinVar entry. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors (Rosetta, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score (Likely Pathogenic) all indicate a pathogenic impact. Uncertain results come from FoldX, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as Likely Pathogenic, and Foldetta as uncertain. Based on the preponderance of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.038963 | Uncertain | 0.948 | 0.230 | 0.000 | 6-33440945-G-C | 2 | 1.24e-6 | -13.282 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.84 | Ambiguous | 0.2 | 2.21 | Destabilizing | 1.53 | Ambiguous | 0.84 | Ambiguous | 0.475 | Likely Benign | -4.98 | Deleterious | 0.995 | Probably Damaging | 0.986 | Probably Damaging | 2.75 | Benign | 0.00 | Affected | 3.37 | 34 | 0.0834 | 0.1757 | 0 | 3 | 0.3 | 9.01 | ||||||||||||||||||||||||
| c.1893G>T | Q631H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q631H is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign predictions come from REVEL and FATHMM, whereas the majority of algorithms—including AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, Rosetta, and the SGM Consensus—classify the change as pathogenic. Predictions from FoldX, Foldetta, and premPS are inconclusive. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, while Foldetta’s stability analysis is uncertain. Overall, the preponderance of evidence points to a pathogenic impact for Q631H, which is consistent with the absence of a benign ClinVar annotation and the lack of population data in gnomAD. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.038963 | Uncertain | 0.948 | 0.230 | 0.000 | -13.282 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.84 | Ambiguous | 0.2 | 2.21 | Destabilizing | 1.53 | Ambiguous | 0.84 | Ambiguous | 0.475 | Likely Benign | -4.98 | Deleterious | 0.995 | Probably Damaging | 0.986 | Probably Damaging | 2.75 | Benign | 0.00 | Affected | 3.37 | 34 | 0.0834 | 0.1757 | 0 | 3 | 0.3 | 9.01 | |||||||||||||||||||||||||||
| c.1894A>C | N632H 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N632H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are AlphaMissense‑Optimized and Foldetta, whereas the remaining pathogenic‑oriented tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default—consistently predict a deleterious impact. FoldX, Rosetta, and premPS give uncertain results and are therefore not considered evidence for either side. High‑accuracy assessments further support this dichotomy: AlphaMissense‑Optimized reports a benign outcome, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a pathogenic verdict, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the preponderance of evidence points to a pathogenic classification for N632H, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.041437 | Uncertain | 0.938 | 0.254 | 0.000 | -14.273 | Likely Pathogenic | 0.587 | Likely Pathogenic | Likely Benign | 0.55 | Ambiguous | 0.3 | -0.58 | Ambiguous | -0.02 | Likely Benign | 0.59 | Ambiguous | 0.827 | Likely Pathogenic | -4.48 | Deleterious | 0.998 | Probably Damaging | 0.937 | Probably Damaging | -1.53 | Pathogenic | 0.00 | Affected | 0.1467 | 0.6406 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.1894A>G | N632D 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant N632D is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity largely agree: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default all predict a pathogenic effect, while SGM‑Consensus also indicates a likely pathogenic outcome. No tool in the dataset predicts a benign effect. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain, SGM‑Consensus remains likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain. Because the majority of evidence points to deleterious impact and there is no ClinVar annotation to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.041437 | Uncertain | 0.938 | 0.254 | 0.000 | -14.117 | Likely Pathogenic | 0.921 | Likely Pathogenic | Ambiguous | 1.84 | Ambiguous | 0.4 | 1.50 | Ambiguous | 1.67 | Ambiguous | 1.09 | Destabilizing | 0.827 | Likely Pathogenic | -4.31 | Deleterious | 0.985 | Probably Damaging | 0.776 | Possibly Damaging | -1.53 | Pathogenic | 0.01 | Affected | 0.1791 | 0.3854 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.1894A>T | N632Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N632Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from FoldX and premPS, while pathogenic calls are made by SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. Uncertain results are reported by Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments further indicate that AlphaMissense‑Optimized is inconclusive, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is also inconclusive. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.041437 | Uncertain | 0.938 | 0.254 | 0.000 | -15.245 | Likely Pathogenic | 0.840 | Likely Pathogenic | Ambiguous | -0.05 | Likely Benign | 0.7 | -1.02 | Ambiguous | -0.54 | Ambiguous | 0.28 | Likely Benign | 0.844 | Likely Pathogenic | -7.04 | Deleterious | 0.999 | Probably Damaging | 0.960 | Probably Damaging | -1.55 | Pathogenic | 0.00 | Affected | 0.0698 | 0.5959 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.1895A>C | N632T 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N632T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are REVEL, PROVEAN, and FATHMM. The remaining tools—FoldX, Rosetta, and AlphaMissense‑Default—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of predictions lean toward a benign impact, and this does not contradict any ClinVar annotation (none is available). Thus, based on the current computational evidence, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.042364 | Structured | 0.041437 | Uncertain | 0.938 | 0.254 | 0.000 | -6.797 | Likely Benign | 0.371 | Ambiguous | Likely Benign | 1.14 | Ambiguous | 0.2 | -0.58 | Ambiguous | 0.28 | Likely Benign | 0.39 | Likely Benign | 0.541 | Likely Pathogenic | -4.48 | Deleterious | 0.214 | Benign | 0.062 | Benign | -1.49 | Pathogenic | 0.13 | Tolerated | 0.1295 | 0.6809 | 0 | 0 | 2.8 | -13.00 | ||||||||||||||||||||||||||||||
| c.1895A>G | N632S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N632S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen2_HumVar. Those that predict a pathogenic effect are PROVEAN, polyPhen2_HumDiv, and FATHMM. The remaining tools—FoldX, Rosetta, Foldetta, and ESM1b—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of available predictions (six benign vs. three pathogenic) support a benign classification, and this does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.042364 | Structured | 0.041437 | Uncertain | 0.938 | 0.254 | 0.000 | -7.677 | In-Between | 0.291 | Likely Benign | Likely Benign | 0.81 | Ambiguous | 0.1 | 0.78 | Ambiguous | 0.80 | Ambiguous | 0.41 | Likely Benign | 0.469 | Likely Benign | -3.85 | Deleterious | 0.718 | Possibly Damaging | 0.086 | Benign | -1.37 | Pathogenic | 0.12 | Tolerated | 0.3302 | 0.6486 | 1 | 1 | 2.7 | -27.03 | ||||||||||||||||||||||||||||||
| c.1895A>T | N632I 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N632I is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include premPS and Foldetta, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus (likely pathogenic), REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Default. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, remains likely pathogenic; and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, indicates a benign effect. Overall, the preponderance of evidence points to a pathogenic classification for N632I, and this conclusion does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.041437 | Uncertain | 0.938 | 0.254 | 0.000 | -12.620 | Likely Pathogenic | 0.881 | Likely Pathogenic | Ambiguous | 1.33 | Ambiguous | 0.3 | -1.24 | Ambiguous | 0.05 | Likely Benign | 0.20 | Likely Benign | 0.839 | Likely Pathogenic | -7.76 | Deleterious | 0.987 | Probably Damaging | 0.887 | Possibly Damaging | -1.56 | Pathogenic | 0.02 | Affected | 0.0712 | 0.5973 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.1896C>A | N632K 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N632K is not reported in ClinVar and is present in gnomAD. Prediction tools that indicate a benign effect include FoldX, Rosetta, and Foldetta, whereas the majority of other in silico predictors (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) classify it as pathogenic; premPS is uncertain. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) predicts benign. Overall, the preponderance of evidence points to a pathogenic effect for N632K, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.041437 | Uncertain | 0.938 | 0.254 | 0.000 | 6-33440948-C-A | -13.266 | Likely Pathogenic | 0.974 | Likely Pathogenic | Likely Pathogenic | -0.06 | Likely Benign | 0.2 | 0.12 | Likely Benign | 0.03 | Likely Benign | 0.95 | Ambiguous | 0.766 | Likely Pathogenic | -5.14 | Deleterious | 0.983 | Probably Damaging | 0.714 | Possibly Damaging | -1.43 | Pathogenic | 0.01 | Affected | 3.37 | 34 | 0.2375 | 0.4897 | 0 | 1 | -0.4 | 14.07 | ||||||||||||||||||||||||||
| c.1896C>G | N632K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N632K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include FoldX, Rosetta, and Foldetta, whereas the majority of other in‑silico predictors (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict a pathogenic outcome; premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Based on the overall consensus of the majority of tools, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.042364 | Structured | 0.041437 | Uncertain | 0.938 | 0.254 | 0.000 | -13.266 | Likely Pathogenic | 0.974 | Likely Pathogenic | Likely Pathogenic | -0.06 | Likely Benign | 0.2 | 0.12 | Likely Benign | 0.03 | Likely Benign | 0.95 | Ambiguous | 0.766 | Likely Pathogenic | -5.14 | Deleterious | 0.983 | Probably Damaging | 0.714 | Possibly Damaging | -1.43 | Pathogenic | 0.01 | Affected | 3.37 | 34 | 0.2375 | 0.4897 | 0 | 1 | -0.4 | 14.07 | |||||||||||||||||||||||||||
| c.1897C>A | L633M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L633M has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, Foldetta, premPS) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta is also inconclusive. Overall, the majority of definitive predictions (5 pathogenic vs. 4 benign) lean toward a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.045352 | Structured | 0.045407 | Uncertain | 0.952 | 0.252 | 0.000 | -10.300 | Likely Pathogenic | 0.718 | Likely Pathogenic | Likely Benign | 0.61 | Ambiguous | 0.1 | 1.47 | Ambiguous | 1.04 | Ambiguous | 0.90 | Ambiguous | 0.355 | Likely Benign | -1.99 | Neutral | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.77 | Benign | 0.01 | Affected | 0.0984 | 0.2683 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.1897C>G | L633V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L633V is not reported in ClinVar and is present in the gnomAD database (ID 6‑33440949‑C‑G). Prediction tools that indicate a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect comprise FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, SGM‑Consensus, and Foldetta; the Rosetta score is uncertain and therefore not considered. High‑accuracy methods give a pathogenic consensus: AlphaMissense‑Optimized predicts benign, but the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) and Foldetta both predict pathogenic. Overall, the majority of evidence supports a pathogenic impact for L633V, and this assessment does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.045407 | Uncertain | 0.952 | 0.252 | 0.000 | 6-33440949-C-G | 1 | 6.20e-7 | -9.992 | Likely Pathogenic | 0.760 | Likely Pathogenic | Likely Benign | 2.32 | Destabilizing | 0.2 | 1.71 | Ambiguous | 2.02 | Destabilizing | 1.32 | Destabilizing | 0.327 | Likely Benign | -2.99 | Deleterious | 0.996 | Probably Damaging | 0.992 | Probably Damaging | 2.86 | Benign | 0.03 | Affected | 3.37 | 34 | 0.1517 | 0.2766 | 1 | 2 | 0.4 | -14.03 | ||||||||||||||||||||||||
| c.1898T>A | L633Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L633Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.045407 | Uncertain | 0.952 | 0.252 | 0.000 | -14.303 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 2.98 | Destabilizing | 0.1 | 2.97 | Destabilizing | 2.98 | Destabilizing | 2.23 | Destabilizing | 0.712 | Likely Pathogenic | -5.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.69 | Benign | 0.00 | Affected | 0.1333 | 0.0876 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1898T>G | L633R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L633R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. Based on the overwhelming consensus of pathogenic predictions and the absence of any benign signal, the variant is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.045407 | Uncertain | 0.952 | 0.252 | 0.000 | -14.360 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 4.41 | Destabilizing | 0.2 | 4.85 | Destabilizing | 4.63 | Destabilizing | 2.15 | Destabilizing | 0.667 | Likely Pathogenic | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 2.70 | Benign | 0.00 | Affected | 0.1674 | 0.0518 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.189G>C | E63D 2D AIThe SynGAP1 missense variant E63D is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely benign outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores the variant as benign, and the SGM‑Consensus also reports it as likely benign. The Foldetta protein‑folding stability analysis is unavailable for this variant. Overall, the majority of computational evidence points to a benign effect, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.474807 | Uncertain | 0.494 | 0.739 | 0.125 | -3.821 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 0.066 | Likely Benign | -0.83 | Neutral | 0.267 | Benign | 0.585 | Possibly Damaging | 3.98 | Benign | 0.00 | Affected | 0.1524 | 0.4130 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.189G>T | E63D 2D AIThe SynGAP1 missense variant E63D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions include REVEL, PROVEAN, polyPhen‑2 HumDiv, ESM1b, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Benign. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, and the SGM‑Consensus (majority of the high‑accuracy tools) also indicates benign. Foldetta results are unavailable, so they do not influence the assessment. Overall, the collective evidence points to a benign effect for E63D, and this conclusion does not conflict with ClinVar, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.429200 | Structured | 0.474807 | Uncertain | 0.494 | 0.739 | 0.125 | -3.821 | Likely Benign | 0.594 | Likely Pathogenic | Likely Benign | 0.066 | Likely Benign | -0.83 | Neutral | 0.267 | Benign | 0.585 | Possibly Damaging | 3.98 | Benign | 0.00 | Affected | 0.1524 | 0.4130 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.1900G>A | A634T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A634T is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors indicates a pathogenic effect: SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all predict pathogenicity, while only FATHMM predicts benign. Two tools give uncertain results: AlphaMissense‑Optimized and Foldetta. High‑accuracy assessment shows that the SGM‑Consensus (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) supports a pathogenic classification, whereas AlphaMissense‑Optimized and Foldetta remain inconclusive. Overall, the preponderance of evidence points to a pathogenic impact for A634T, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.085092 | Structured | 0.052058 | Uncertain | 0.932 | 0.242 | 0.000 | -12.451 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 2.56 | Destabilizing | 0.1 | 1.11 | Ambiguous | 1.84 | Ambiguous | 1.34 | Destabilizing | 0.603 | Likely Pathogenic | -3.98 | Deleterious | 0.994 | Probably Damaging | 0.986 | Probably Damaging | 2.51 | Benign | 0.01 | Affected | 0.1397 | 0.5240 | 1 | 0 | -2.5 | 30.03 | |||||||||||||||||||||||||||||
| c.1900G>C | A634P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A634P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.085092 | Structured | 0.052058 | Uncertain | 0.932 | 0.242 | 0.000 | -15.372 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 4.17 | Destabilizing | 0.2 | 7.72 | Destabilizing | 5.95 | Destabilizing | 1.39 | Destabilizing | 0.745 | Likely Pathogenic | -4.98 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.50 | Benign | 0.01 | Affected | 0.2090 | 0.3429 | 1 | -1 | -3.4 | 26.04 | |||||||||||||||||||||||||||||
| c.1900G>T | A634S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 A634S variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are FATHMM and AlphaMissense‑Optimized; those that agree on a pathogenic effect are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points to a pathogenic impact, and this conclusion does not contradict the ClinVar status, which simply lacks an entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.085092 | Structured | 0.052058 | Uncertain | 0.932 | 0.242 | 0.000 | -9.706 | Likely Pathogenic | 0.434 | Ambiguous | Likely Benign | 0.91 | Ambiguous | 0.1 | 1.28 | Ambiguous | 1.10 | Ambiguous | 0.77 | Ambiguous | 0.506 | Likely Pathogenic | -2.99 | Deleterious | 0.953 | Possibly Damaging | 0.985 | Probably Damaging | 2.67 | Benign | 0.05 | Affected | 0.2607 | 0.4231 | 1 | 1 | -2.6 | 16.00 | ||||||||||||||||||||||||||||||
| c.1901C>A | A634D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A634D is not reported in ClinVar (ClinVar status: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess pathogenicity all agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. No tool predicts a benign outcome. High‑accuracy methods reinforce this consensus: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates pathogenicity; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic effect. No predictions or stability results are missing or inconclusive. Based on the unanimous computational evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.085092 | Structured | 0.052058 | Uncertain | 0.932 | 0.242 | 0.000 | -16.727 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 5.26 | Destabilizing | 0.5 | 4.24 | Destabilizing | 4.75 | Destabilizing | 1.79 | Destabilizing | 0.731 | Likely Pathogenic | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 2.49 | Pathogenic | 0.00 | Affected | 0.1790 | 0.1816 | 0 | -2 | -5.3 | 44.01 | |||||||||||||||||||||||||||||
| c.1901C>G | A634G 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant A634G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls are made by Rosetta, premPS, PROVEAN, both polyPhen‑2 versions, ESM1b, and AlphaMissense‑Default. FoldX and Foldetta give uncertain results. High‑accuracy assessments indicate AlphaMissense‑Optimized predicts benign, whereas the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta remains uncertain. Overall, the majority of tools lean toward pathogenicity, and the high‑accuracy consensus also supports a pathogenic interpretation. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.085092 | Structured | 0.052058 | Uncertain | 0.932 | 0.242 | 0.000 | -10.685 | Likely Pathogenic | 0.613 | Likely Pathogenic | Likely Benign | 1.63 | Ambiguous | 0.1 | 2.27 | Destabilizing | 1.95 | Ambiguous | 1.09 | Destabilizing | 0.418 | Likely Benign | -3.98 | Deleterious | 0.997 | Probably Damaging | 0.990 | Probably Damaging | 2.69 | Benign | 0.15 | Tolerated | 0.2182 | 0.3187 | 1 | 0 | -2.2 | -14.03 | |||||||||||||||||||||||||||||
| c.1901C>T | A634V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant A634V is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools largely converge on a deleterious effect: pathogenic calls are made by REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score, while only FATHMM predicts a benign outcome; Rosetta remains inconclusive. High‑accuracy assessments reinforce the pathogenic trend: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. Taken together, the overwhelming majority of evidence supports a pathogenic effect for A634V, which is in contrast to its current ClinVar classification of uncertain significance. Therefore, the variant is most likely pathogenic, contradicting its ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.085092 | Structured | 0.052058 | Uncertain | 0.932 | 0.242 | 0.000 | Uncertain | 1 | -12.612 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 2.67 | Destabilizing | 0.2 | 1.44 | Ambiguous | 2.06 | Destabilizing | 1.14 | Destabilizing | 0.631 | Likely Pathogenic | -3.98 | Deleterious | 0.997 | Probably Damaging | 0.976 | Probably Damaging | 2.55 | Benign | 0.01 | Affected | 0.1215 | 0.4371 | 0 | 0 | 2.4 | 28.05 | |||||||||||||||||||||||||||
| c.1903A>C | N635H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N635H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Three tools (FoldX, premPS, AlphaMissense‑Default) give uncertain results. High‑accuracy methods give the following: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic; and Foldetta predicts benign. No prediction or folding‑stability result is missing or inconclusive. Based on the available evidence, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.039760 | Structured | 0.060246 | Uncertain | 0.900 | 0.252 | 0.000 | -12.507 | Likely Pathogenic | 0.419 | Ambiguous | Likely Benign | 1.07 | Ambiguous | 0.2 | -0.10 | Likely Benign | 0.49 | Likely Benign | 0.91 | Ambiguous | 0.429 | Likely Benign | -4.78 | Deleterious | 0.993 | Probably Damaging | 0.879 | Possibly Damaging | 2.90 | Benign | 0.00 | Affected | 0.1184 | 0.3720 | 2 | 1 | 0.3 | 23.04 | ||||||||||||||||||||||||||||||
| c.1903A>G | N635D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N635D is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, and FATHMM. Those that predict a pathogenic effect are premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). Predictions that are inconclusive are Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy methods give an uncertain result for AlphaMissense‑Optimized, a Likely Pathogenic verdict from the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and an uncertain outcome from Foldetta. Overall, the majority of computational evidence points to a pathogenic effect, and this assessment is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.039760 | Structured | 0.060246 | Uncertain | 0.900 | 0.252 | 0.000 | -14.453 | Likely Pathogenic | 0.799 | Likely Pathogenic | Ambiguous | 0.47 | Likely Benign | 0.1 | 0.73 | Ambiguous | 0.60 | Ambiguous | 1.26 | Destabilizing | 0.432 | Likely Benign | -4.71 | Deleterious | 0.955 | Possibly Damaging | 0.628 | Possibly Damaging | 2.92 | Benign | 0.01 | Affected | 0.1725 | 0.1937 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.1903A>T | N635Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N635Y has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include Foldetta, premPS, FATHMM, AlphaMissense‑Optimized, and Rosetta. Those that predict a pathogenic impact are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX is uncertain and therefore treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicating likely pathogenic, and Foldetta predicting a benign outcome. Overall, the majority of tools lean toward a benign interpretation, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.039760 | Structured | 0.060246 | Uncertain | 0.900 | 0.252 | 0.000 | -14.931 | Likely Pathogenic | 0.662 | Likely Pathogenic | Likely Benign | 0.73 | Ambiguous | 0.3 | -0.11 | Likely Benign | 0.31 | Likely Benign | -0.16 | Likely Benign | 0.554 | Likely Pathogenic | -7.64 | Deleterious | 0.998 | Probably Damaging | 0.922 | Probably Damaging | 2.88 | Benign | 0.00 | Affected | 0.0750 | 0.3632 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.1904A>C | N635T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N635T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, and ESM1b; polyPhen‑2 HumVar, however, classifies it as benign. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain, and FoldX and premPS also yield uncertain results. Overall, the majority of evidence (five benign vs. four pathogenic) supports a benign classification. This conclusion is consistent with the lack of ClinVar annotation and gnomAD presence, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.039760 | Structured | 0.060246 | Uncertain | 0.900 | 0.252 | 0.000 | -11.705 | Likely Pathogenic | 0.256 | Likely Benign | Likely Benign | 1.50 | Ambiguous | 0.1 | 0.44 | Likely Benign | 0.97 | Ambiguous | 0.81 | Ambiguous | 0.240 | Likely Benign | -5.58 | Deleterious | 0.536 | Possibly Damaging | 0.184 | Benign | 2.98 | Benign | 0.04 | Affected | 0.1206 | 0.4032 | 0 | 0 | 2.8 | -13.00 | ||||||||||||||||||||||||||||||
| c.1904A>T | N635I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N635I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX is uncertain. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, while the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—classifies the variant as pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts a benign impact. Overall, the majority of tools lean toward a pathogenic interpretation, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.039760 | Structured | 0.060246 | Uncertain | 0.900 | 0.252 | 0.000 | -15.012 | Likely Pathogenic | 0.608 | Likely Pathogenic | Likely Benign | 0.94 | Ambiguous | 0.1 | -0.05 | Likely Benign | 0.45 | Likely Benign | -0.35 | Likely Benign | 0.363 | Likely Benign | -8.56 | Deleterious | 0.980 | Probably Damaging | 0.889 | Possibly Damaging | 2.88 | Benign | 0.00 | Affected | 0.0736 | 0.3776 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.1905C>A | N635K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N635K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict pathogenicity: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic outcome. Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for N635K, and this conclusion does not contradict any existing ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.039760 | Structured | 0.060246 | Uncertain | 0.900 | 0.252 | 0.000 | -13.144 | Likely Pathogenic | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.60 | Ambiguous | 0.1 | 0.75 | Ambiguous | 0.68 | Ambiguous | 0.85 | Ambiguous | 0.332 | Likely Benign | -5.64 | Deleterious | 0.949 | Possibly Damaging | 0.550 | Possibly Damaging | 2.92 | Benign | 0.00 | Affected | 0.2125 | 0.2510 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.1905C>G | N635K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N635K is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also classifies the variant as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic and the SGM Consensus as Likely Pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) yields an uncertain result. Overall, the preponderance of evidence from multiple in silico predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.039760 | Structured | 0.060246 | Uncertain | 0.900 | 0.252 | 0.000 | -13.144 | Likely Pathogenic | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.60 | Ambiguous | 0.1 | 0.75 | Ambiguous | 0.68 | Ambiguous | 0.85 | Ambiguous | 0.332 | Likely Benign | -5.64 | Deleterious | 0.949 | Possibly Damaging | 0.550 | Possibly Damaging | 2.92 | Benign | 0.00 | Affected | 0.2125 | 0.2510 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.1906T>A | F636I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F636I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy tools, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | -14.031 | Likely Pathogenic | 0.983 | Likely Pathogenic | Likely Pathogenic | 2.96 | Destabilizing | 0.2 | 4.57 | Destabilizing | 3.77 | Destabilizing | 1.10 | Destabilizing | 0.501 | Likely Pathogenic | -5.78 | Deleterious | 0.994 | Probably Damaging | 0.977 | Probably Damaging | 3.44 | Benign | 0.03 | Affected | 0.1660 | 0.2153 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1906T>C | F636L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F636L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic, the SGM Consensus confirms Likely Pathogenic, and the Foldetta stability analysis is inconclusive. Other stability tools (FoldX, Rosetta, premPS) are uncertain. Based on the preponderance of pathogenic predictions and the consensus from high‑accuracy methods, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | -11.871 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.88 | Ambiguous | 0.1 | 0.69 | Ambiguous | 0.79 | Ambiguous | 0.71 | Ambiguous | 0.478 | Likely Benign | -5.78 | Deleterious | 0.994 | Probably Damaging | 0.952 | Probably Damaging | 3.60 | Benign | 0.22 | Tolerated | 0.1853 | 0.3024 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1906T>G | F636V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F636V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | -14.603 | Likely Pathogenic | 0.975 | Likely Pathogenic | Likely Pathogenic | 3.06 | Destabilizing | 0.1 | 5.13 | Destabilizing | 4.10 | Destabilizing | 1.07 | Destabilizing | 0.533 | Likely Pathogenic | -6.74 | Deleterious | 0.991 | Probably Damaging | 0.985 | Probably Damaging | 3.44 | Benign | 0.04 | Affected | 0.1841 | 0.2030 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1907T>A | F636Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F636Y is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. The SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, favors a pathogenic outcome (3/4 votes). High‑accuracy assessments further reveal AlphaMissense‑Optimized as benign, SGM Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as inconclusive. Stability‑based tools FoldX, Rosetta, and premPS are uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | -9.638 | Likely Pathogenic | 0.695 | Likely Pathogenic | Likely Benign | 0.84 | Ambiguous | 0.1 | 0.51 | Ambiguous | 0.68 | Ambiguous | 0.90 | Ambiguous | 0.394 | Likely Benign | -2.89 | Deleterious | 0.927 | Possibly Damaging | 0.836 | Possibly Damaging | 3.40 | Benign | 0.08 | Tolerated | 0.1297 | 0.1866 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1907T>C | F636S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F636S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, while the remaining 13 tools (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus) all predict a pathogenic impact. High‑accuracy assessments reinforce this: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic. No prediction or folding‑stability result is missing or inconclusive. Based on the overwhelming agreement among the majority of tools and the high‑accuracy methods, the variant is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | -14.290 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 2.08 | Destabilizing | 0.1 | 2.66 | Destabilizing | 2.37 | Destabilizing | 1.51 | Destabilizing | 0.559 | Likely Pathogenic | -7.50 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.41 | Benign | 0.03 | Affected | 0.3877 | 0.0200 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1907T>G | F636C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F636C is reported in gnomAD (ID 6‑33440959‑T‑G) but has no ClinVar entry. Prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). Only FATHMM predicts a benign outcome; FoldX is uncertain and therefore not counted as evidence. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming majority of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | 6-33440959-T-G | 3 | 1.86e-6 | -13.287 | Likely Pathogenic | 0.972 | Likely Pathogenic | Likely Pathogenic | 1.74 | Ambiguous | 0.1 | 2.65 | Destabilizing | 2.20 | Destabilizing | 1.22 | Destabilizing | 0.612 | Likely Pathogenic | -7.67 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.40 | Benign | 0.04 | Affected | 3.37 | 34 | 0.2301 | 0.0830 | -2 | -4 | -0.3 | -44.04 | ||||||||||||||||||||||||
| c.1908T>A | F636L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F636L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas pathogenic predictions are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic, the SGM Consensus confirms Likely Pathogenic, and the Foldetta stability analysis is inconclusive. Other stability tools (FoldX, Rosetta, premPS) are uncertain. Based on the preponderance of pathogenic predictions and the consensus from high‑accuracy methods, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | -11.871 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.88 | Ambiguous | 0.1 | 0.69 | Ambiguous | 0.79 | Ambiguous | 0.71 | Ambiguous | 0.268 | Likely Benign | -5.78 | Deleterious | 0.994 | Probably Damaging | 0.952 | Probably Damaging | 3.60 | Benign | 0.22 | Tolerated | 0.1853 | 0.3024 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1908T>G | F636L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F636L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, SIFT, and FATHMM, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus confirms a Likely Pathogenic status, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result. No evidence from FoldX‑MD, Rosetta, or premPS is available to alter this view. Overall, the majority of reliable predictors indicate a pathogenic impact, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.041405 | Structured | 0.071525 | Uncertain | 0.913 | 0.264 | 0.000 | -11.871 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.88 | Ambiguous | 0.1 | 0.69 | Ambiguous | 0.79 | Ambiguous | 0.71 | Ambiguous | 0.268 | Likely Benign | -5.78 | Deleterious | 0.994 | Probably Damaging | 0.952 | Probably Damaging | 3.60 | Benign | 0.22 | Tolerated | 0.1853 | 0.3024 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1909T>A | S637T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S637T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive result is from Rosetta, which is treated as unavailable. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.076542 | Structured | 0.083482 | Uncertain | 0.920 | 0.253 | 0.000 | -4.116 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.36 | Likely Benign | 0.1 | -0.91 | Ambiguous | -0.28 | Likely Benign | -0.39 | Likely Benign | 0.067 | Likely Benign | -0.19 | Neutral | 0.086 | Benign | 0.019 | Benign | 3.45 | Benign | 0.19 | Tolerated | 0.1675 | 0.4572 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1909T>C | S637P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S637P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, premPS, polyPhen‑2 HumVar, and FATHMM, while pathogenic calls arise from FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments give a pathogenic signal: the SGM Consensus predicts likely pathogenic, Foldetta predicts destabilizing pathogenic effects, whereas AlphaMissense‑Optimized remains uncertain. Overall, the balance of evidence favors a pathogenic interpretation, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.076542 | Structured | 0.083482 | Uncertain | 0.920 | 0.253 | 0.000 | -11.455 | Likely Pathogenic | 0.793 | Likely Pathogenic | Ambiguous | 6.73 | Destabilizing | 0.1 | 6.36 | Destabilizing | 6.55 | Destabilizing | 0.44 | Likely Benign | 0.192 | Likely Benign | -3.12 | Deleterious | 0.946 | Possibly Damaging | 0.360 | Benign | 3.36 | Benign | 0.03 | Affected | 0.2374 | 0.4094 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.1909T>G | S637A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S637A is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, SIFT, polyPhen‑2 (HumDiv and HumVar), REVEL, FoldX, and premPS. No tool predicts a pathogenic outcome. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is inconclusive (Uncertain). Taken together, the overwhelming majority of evidence indicates a benign effect. The variant’s status is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.076542 | Structured | 0.083482 | Uncertain | 0.920 | 0.253 | 0.000 | -4.186 | Likely Benign | 0.137 | Likely Benign | Likely Benign | 0.31 | Likely Benign | 0.1 | 0.99 | Ambiguous | 0.65 | Ambiguous | 0.22 | Likely Benign | 0.078 | Likely Benign | -0.64 | Neutral | 0.120 | Benign | 0.182 | Benign | 3.41 | Benign | 1.00 | Tolerated | 0.4853 | 0.2996 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.190A>C | I64L 2D AIThe SynGAP1 missense variant I64L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus indicates Likely Benign, and Foldetta results are unavailable. Overall, the preponderance of evidence points to a benign impact for I64L, and this conclusion is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | -2.498 | Likely Benign | 0.437 | Ambiguous | Likely Benign | 0.087 | Likely Benign | -0.27 | Neutral | 0.010 | Benign | 0.001 | Benign | 4.13 | Benign | 0.00 | Affected | 0.0628 | 0.3030 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.190A>G | I64V 2D AIThe SynGAP1 missense variant I64V is reported as “Likely Benign” in ClinVar and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of computational evidence supports a benign classification, and this is consistent with the ClinVar status (no conflicting pathogenic annotation). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | -2.616 | Likely Benign | 0.154 | Likely Benign | Likely Benign | 0.086 | Likely Benign | 0.12 | Neutral | 0.000 | Benign | 0.000 | Benign | 4.24 | Benign | 0.00 | Affected | 0.0994 | 0.3302 | 4 | 3 | -0.3 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.190A>T | I64L 2D AIThe SynGAP1 missense variant I64L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized predicts Benign, SGM‑Consensus indicates Likely Benign, and Foldetta results are unavailable. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | -2.498 | Likely Benign | 0.437 | Ambiguous | Likely Benign | 0.087 | Likely Benign | -0.27 | Neutral | 0.010 | Benign | 0.001 | Benign | 4.13 | Benign | 0.00 | Affected | 0.0628 | 0.3030 | 2 | 2 | -0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.1910C>A | S637Y 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S637Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that indicate a benign effect include REVEL, FoldX, premPS, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as uncertain. Overall, the majority of individual predictors (seven versus five) lean toward pathogenicity, and the consensus‑based SGM‑Consensus also supports a likely pathogenic classification. Therefore, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.076542 | Structured | 0.083482 | Uncertain | 0.920 | 0.253 | 0.000 | -12.633 | Likely Pathogenic | 0.770 | Likely Pathogenic | Likely Benign | 0.13 | Likely Benign | 0.1 | 1.52 | Ambiguous | 0.83 | Ambiguous | 0.50 | Likely Benign | 0.209 | Likely Benign | -3.78 | Deleterious | 0.985 | Probably Damaging | 0.681 | Possibly Damaging | 3.35 | Benign | 0.00 | Affected | 0.0968 | 0.4071 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||
| c.1910C>G | S637C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S637C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that classify the variant as benign include REVEL, FoldX, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts benign; Foldetta predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence supports a benign impact, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.076542 | Structured | 0.083482 | Uncertain | 0.920 | 0.253 | 0.000 | -10.040 | Likely Pathogenic | 0.138 | Likely Benign | Likely Benign | 0.25 | Likely Benign | 0.0 | 0.54 | Ambiguous | 0.40 | Likely Benign | 0.22 | Likely Benign | 0.169 | Likely Benign | -2.83 | Deleterious | 0.985 | Probably Damaging | 0.533 | Possibly Damaging | 3.34 | Benign | 0.01 | Affected | 0.1327 | 0.4439 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||
| c.1910C>T | S637F 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant S637F is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, FoldX, Foldetta, premPS, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Uncertain results are reported by Rosetta and AlphaMissense‑Optimized. The high‑accuracy consensus (SGM‑Consensus) aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN and yields a pathogenic verdict. Foldetta, a protein‑folding stability method that integrates FoldX‑MD and Rosetta outputs, predicts a benign effect. AlphaMissense‑Optimized remains inconclusive. Overall, the majority of evidence points toward a pathogenic impact, and this assessment does not contradict the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.076542 | Structured | 0.083482 | Uncertain | 0.920 | 0.253 | 0.000 | -13.266 | Likely Pathogenic | 0.831 | Likely Pathogenic | Ambiguous | 0.01 | Likely Benign | 0.1 | 0.96 | Ambiguous | 0.49 | Likely Benign | 0.49 | Likely Benign | 0.222 | Likely Benign | -3.92 | Deleterious | 0.985 | Probably Damaging | 0.681 | Possibly Damaging | 3.35 | Benign | 0.01 | Affected | 0.0780 | 0.4126 | -3 | -2 | 3.6 | 60.10 | |||||||||||||||||||||||||||||
| c.1912A>C | K638Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K638Q missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.045352 | Structured | 0.098064 | Uncertain | 0.937 | 0.260 | 0.000 | -9.561 | Likely Pathogenic | 0.556 | Ambiguous | Likely Benign | 0.45 | Likely Benign | 0.0 | 0.37 | Likely Benign | 0.41 | Likely Benign | 0.22 | Likely Benign | 0.421 | Likely Benign | -3.60 | Deleterious | 0.997 | Probably Damaging | 0.991 | Probably Damaging | 3.42 | Benign | 0.12 | Tolerated | 0.3623 | 0.0920 | 1 | 1 | 0.4 | -0.04 | ||||||||||||||||||||||||||||||
| c.1912A>G | K638E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K638E is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic impact: SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Predictions from FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized are uncertain or unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic classification; this conclusion does not contradict ClinVar status, which currently contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.098064 | Uncertain | 0.937 | 0.260 | 0.000 | -13.390 | Likely Pathogenic | 0.927 | Likely Pathogenic | Ambiguous | 0.57 | Ambiguous | 0.0 | 1.00 | Ambiguous | 0.79 | Ambiguous | 0.32 | Likely Benign | 0.363 | Likely Benign | -3.70 | Deleterious | 0.995 | Probably Damaging | 0.947 | Probably Damaging | 3.50 | Benign | 0.12 | Tolerated | 0.3036 | 0.0790 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1913A>C | K638T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K638T is not reported in ClinVar and is absent from gnomAD. Consensus from standard in silico predictors shows a split: benign calls come from REVEL, Rosetta, premPS, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic calls arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default; FoldX and Foldetta are inconclusive. High‑accuracy assessments give AlphaMissense‑Optimized a benign score, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta remains uncertain. Overall, the balance of evidence favors a pathogenic effect for K638T. This prediction is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.098064 | Uncertain | 0.937 | 0.260 | 0.000 | -8.856 | Likely Pathogenic | 0.775 | Likely Pathogenic | Likely Benign | 0.87 | Ambiguous | 0.0 | 0.23 | Likely Benign | 0.55 | Ambiguous | 0.07 | Likely Benign | 0.404 | Likely Benign | -5.39 | Deleterious | 0.999 | Probably Damaging | 0.998 | Probably Damaging | 3.52 | Benign | 0.03 | Affected | 0.1632 | 0.2619 | 0 | -1 | 3.2 | -27.07 | |||||||||||||||||||||||||||||
| c.1913A>G | K638R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K638R is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that uniformly indicate a benign effect include REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Benign). In contrast, PROVEAN and polyPhen‑2 HumDiv predict a pathogenic impact, while premPS remains inconclusive. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, is benign. Overall, the majority of evidence points to a benign effect, which does not contradict the ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.045352 | Structured | 0.098064 | Uncertain | 0.937 | 0.260 | 0.000 | Uncertain | 1 | -2.700 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.09 | Likely Benign | 0.1 | -0.04 | Likely Benign | 0.03 | Likely Benign | 0.53 | Ambiguous | 0.216 | Likely Benign | -2.55 | Deleterious | 0.649 | Possibly Damaging | 0.240 | Benign | 3.41 | Benign | 0.13 | Tolerated | 3.37 | 31 | 0.4026 | 0.0975 | 2 | 3 | -0.6 | 28.01 | |||||||||||||||||||||||||
| c.1913A>T | K638M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K638M missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include FoldX, FATHMM, premPS, and Foldetta. Those that predict a pathogenic effect comprise SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized and Rosetta give uncertain results, which are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of evidence points to a pathogenic impact, and this conclusion is not contradicted by the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.098064 | Uncertain | 0.937 | 0.260 | 0.000 | -9.702 | Likely Pathogenic | 0.882 | Likely Pathogenic | Ambiguous | -0.21 | Likely Benign | 0.0 | 0.61 | Ambiguous | 0.20 | Likely Benign | 0.09 | Likely Benign | 0.526 | Likely Pathogenic | -5.19 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.41 | Benign | 0.01 | Affected | 0.0929 | 0.2896 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1914G>C | K638N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K638N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic classification; this conclusion does not contradict ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.098064 | Uncertain | 0.937 | 0.260 | 0.000 | -9.420 | Likely Pathogenic | 0.947 | Likely Pathogenic | Ambiguous | 0.74 | Ambiguous | 0.1 | 0.90 | Ambiguous | 0.82 | Ambiguous | 0.40 | Likely Benign | 0.256 | Likely Benign | -4.59 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 3.40 | Benign | 0.02 | Affected | 0.2875 | 0.1126 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1914G>T | K638N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K638N is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as uncertain. Overall, the balance of evidence favors a pathogenic classification; this conclusion does not contradict ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.045352 | Structured | 0.098064 | Uncertain | 0.937 | 0.260 | 0.000 | -9.420 | Likely Pathogenic | 0.947 | Likely Pathogenic | Ambiguous | 0.74 | Ambiguous | 0.1 | 0.90 | Ambiguous | 0.82 | Ambiguous | 0.40 | Likely Benign | 0.256 | Likely Benign | -4.59 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 3.40 | Benign | 0.02 | Affected | 0.2875 | 0.1126 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1915T>A | F639I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F639I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include FATHMM and Rosetta, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. No prediction or stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.117665 | Uncertain | 0.942 | 0.284 | 0.000 | -12.548 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 4.32 | Destabilizing | 0.3 | 0.47 | Likely Benign | 2.40 | Destabilizing | 1.44 | Destabilizing | 0.545 | Likely Pathogenic | -5.98 | Deleterious | 0.994 | Probably Damaging | 0.659 | Possibly Damaging | 3.08 | Benign | 0.01 | Affected | 0.2070 | 0.1755 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1915T>C | F639L 2D AIThe SynGAP1 missense variant F639L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, polyPhen2_HumVar, and FATHMM, whereas pathogenic predictions are returned by SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen2_HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Rosetta and Foldetta provide uncertain results. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, while Foldetta’s stability analysis is inconclusive. Based on the majority of predictions, the variant is most likely pathogenic, which is consistent with the lack of ClinVar annotation and gnomAD absence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.117665 | Uncertain | 0.942 | 0.284 | 0.000 | -11.660 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.12 | Destabilizing | 0.7 | 1.15 | Ambiguous | 1.64 | Ambiguous | 1.43 | Destabilizing | 0.499 | Likely Benign | -5.98 | Deleterious | 0.994 | Probably Damaging | 0.380 | Benign | 3.12 | Benign | 0.03 | Affected | 0.2347 | 0.2726 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1915T>G | F639V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F639V is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, while the remaining 12 tools (SGM‑Consensus, REVEL, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all predict pathogenicity; Rosetta is uncertain and is therefore not counted. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized reports pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.117665 | Uncertain | 0.942 | 0.284 | 0.000 | -12.910 | Likely Pathogenic | 0.965 | Likely Pathogenic | Likely Pathogenic | 4.17 | Destabilizing | 0.1 | 1.18 | Ambiguous | 2.68 | Destabilizing | 1.47 | Destabilizing | 0.560 | Likely Pathogenic | -6.97 | Deleterious | 0.992 | Probably Damaging | 0.756 | Possibly Damaging | 3.08 | Benign | 0.02 | Affected | 0.2074 | 0.1583 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1916T>A | F639Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F639Y is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions are returned by SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments give a mixed signal: AlphaMissense‑Optimized classifies the change as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the balance of evidence leans toward pathogenicity, and this assessment does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.117665 | Uncertain | 0.942 | 0.284 | 0.000 | -11.762 | Likely Pathogenic | 0.670 | Likely Pathogenic | Likely Benign | 1.92 | Ambiguous | 0.1 | 0.99 | Ambiguous | 1.46 | Ambiguous | 1.20 | Destabilizing | 0.330 | Likely Benign | -2.99 | Deleterious | 0.930 | Possibly Damaging | 0.263 | Benign | 3.06 | Benign | 0.03 | Affected | 0.1530 | 0.1635 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1916T>C | F639S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F639S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.117665 | Uncertain | 0.942 | 0.284 | 0.000 | -11.511 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 4.92 | Destabilizing | 0.3 | 3.08 | Destabilizing | 4.00 | Destabilizing | 1.87 | Destabilizing | 0.561 | Likely Pathogenic | -7.97 | Deleterious | 0.965 | Probably Damaging | 0.457 | Possibly Damaging | 3.12 | Benign | 0.04 | Affected | 0.3813 | 0.0200 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1916T>G | F639C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F639C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (both HumDiv and HumVar), SIFT, and ESM1b all indicate pathogenicity, while only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the change as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic effect. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.117665 | Uncertain | 0.942 | 0.284 | 0.000 | -12.532 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 4.02 | Destabilizing | 0.2 | 2.80 | Destabilizing | 3.41 | Destabilizing | 1.38 | Destabilizing | 0.615 | Likely Pathogenic | -7.97 | Deleterious | 1.000 | Probably Damaging | 0.935 | Probably Damaging | 3.05 | Benign | 0.01 | Affected | 0.2451 | 0.0783 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.1917T>A | F639L 2D AIThe SynGAP1 missense variant F639L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, polyPhen2_HumVar, and FATHMM, whereas pathogenic predictions are returned by SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen2_HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Rosetta and Foldetta provide uncertain results. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, while Foldetta’s stability analysis is inconclusive. Based on the majority of predictions, the variant is most likely pathogenic, which is consistent with the lack of ClinVar annotation and gnomAD absence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.117665 | Uncertain | 0.942 | 0.284 | 0.000 | -11.660 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.12 | Destabilizing | 0.7 | 1.15 | Ambiguous | 1.64 | Ambiguous | 1.43 | Destabilizing | 0.336 | Likely Benign | -5.98 | Deleterious | 0.994 | Probably Damaging | 0.380 | Benign | 3.12 | Benign | 0.03 | Affected | 0.2347 | 0.2726 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1917T>G | F639L 2D AIThe SynGAP1 missense variant F639L is not reported in ClinVar or gnomAD. Prediction tools that indicate a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas a majority of tools predict pathogenicity: SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results come from Rosetta and Foldetta. High‑accuracy methods specifically show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta as inconclusive. Overall, the balance of evidence points to a pathogenic effect for F639L, and this assessment does not conflict with any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.038042 | Structured | 0.117665 | Uncertain | 0.942 | 0.284 | 0.000 | -11.660 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 2.12 | Destabilizing | 0.7 | 1.15 | Ambiguous | 1.64 | Ambiguous | 1.43 | Destabilizing | 0.336 | Likely Benign | -5.98 | Deleterious | 0.994 | Probably Damaging | 0.380 | Benign | 3.12 | Benign | 0.03 | Affected | 0.2347 | 0.2726 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1918A>C | T640P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T640P is not reported in ClinVar and is absent from gnomAD. In silico predictors uniformly favor a benign effect: REVEL, Rosetta, premPS, PROVEAN, polyPhen2_HumDiv, polyPhen2_HumVar, SIFT, FATHMM, AlphaMissense-Default, and AlphaMissense-Optimized all indicate benign. No tool predicts pathogenicity. FoldX and ESM1b were inconclusive. High‑accuracy methods corroborate this: AlphaMissense-Optimized is benign, the SGM Consensus (majority vote of AlphaMissense-Default, ESM1b, FATHMM, PROVEAN) is benign, and Foldetta (combining FoldX‑MD and Rosetta) also predicts benign. Consequently, the variant is most likely benign, and this assessment does not contradict the ClinVar status, which currently has no entry for T640P. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.066181 | Structured | 0.137043 | Uncertain | 0.893 | 0.284 | 0.000 | -7.594 | In-Between | 0.077 | Likely Benign | Likely Benign | 0.98 | Ambiguous | 0.3 | -0.38 | Likely Benign | 0.30 | Likely Benign | 0.31 | Likely Benign | 0.313 | Likely Benign | -0.91 | Neutral | 0.004 | Benign | 0.002 | Benign | 3.47 | Benign | 0.14 | Tolerated | 0.2179 | 0.4722 | 0 | -1 | -0.9 | -3.99 | |||||||||||||||||||||||||||||
| c.1918A>G | T640A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T640A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN, REVEL, FoldX, premPS, polyPhen‑2 (HumDiv and HumVar), and SIFT all score the substitution as benign. No tool predicts pathogenicity; only Rosetta yields an uncertain result. High‑accuracy assessments corroborate this benign trend: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled likely benign, while Foldetta (combining FoldX‑MD and Rosetta outputs) remains uncertain. Taken together, the evidence overwhelmingly supports a benign classification for T640A, and this conclusion does not contradict any ClinVar annotation (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.066181 | Structured | 0.137043 | Uncertain | 0.893 | 0.284 | 0.000 | -3.068 | Likely Benign | 0.074 | Likely Benign | Likely Benign | 0.14 | Likely Benign | 0.1 | 0.93 | Ambiguous | 0.54 | Ambiguous | -0.17 | Likely Benign | 0.078 | Likely Benign | 0.51 | Neutral | 0.009 | Benign | 0.001 | Benign | 3.51 | Benign | 0.42 | Tolerated | 0.3834 | 0.3202 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.1918A>T | T640S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T640S is listed in ClinVar as Benign (ClinVar ID 2980241.0) and is present in the gnomAD database (gnomAD ID 6‑33441177‑A‑T). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive result is from FoldX, which is treated as unavailable. High‑accuracy assessments confirm benignity: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is benign. Overall, the variant is most likely benign, and this conclusion is consistent with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.066181 | Structured | 0.137043 | Uncertain | 0.893 | 0.284 | 0.000 | Benign | 1 | 6-33441177-A-T | 1 | 6.20e-7 | -2.371 | Likely Benign | 0.062 | Likely Benign | Likely Benign | -0.78 | Ambiguous | 0.1 | 0.43 | Likely Benign | -0.18 | Likely Benign | -0.30 | Likely Benign | 0.088 | Likely Benign | 0.92 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.60 | Benign | 0.33 | Tolerated | 3.37 | 30 | 0.3406 | 0.3484 | 1 | 1 | -0.1 | -14.03 | ||||||||||||||||||||||
| c.1919C>A | T640N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T640N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as tolerated or benign. Only the protein‑stability predictors FoldX and Rosetta returned uncertain results, which are treated as unavailable evidence. High‑accuracy assessments corroborate the benign prediction: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also reports a benign effect. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.066181 | Structured | 0.137043 | Uncertain | 0.893 | 0.284 | 0.000 | -4.258 | Likely Benign | 0.107 | Likely Benign | Likely Benign | -0.50 | Ambiguous | 0.2 | 0.53 | Ambiguous | 0.02 | Likely Benign | 0.03 | Likely Benign | 0.097 | Likely Benign | 0.51 | Neutral | 0.229 | Benign | 0.084 | Benign | 3.45 | Benign | 0.25 | Tolerated | 0.1518 | 0.4079 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.1919C>G | T640S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T640S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, premPS, Foldetta, Rosetta, REVEL, and the SGM‑Consensus score (Likely Benign) all classify the change as tolerated. No tool predicts pathogenicity; the only inconclusive result is from FoldX, which is listed as Uncertain. High‑accuracy methods corroborate the benign assessment: AlphaMissense‑Optimized predicts Benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports Benign. Consequently, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.066181 | Structured | 0.137043 | Uncertain | 0.893 | 0.284 | 0.000 | -2.371 | Likely Benign | 0.062 | Likely Benign | Likely Benign | -0.78 | Ambiguous | 0.1 | 0.43 | Likely Benign | -0.18 | Likely Benign | -0.30 | Likely Benign | 0.109 | Likely Benign | 0.92 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.60 | Benign | 0.33 | Tolerated | 3.37 | 30 | 0.3406 | 0.3484 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||
| c.1919C>T | T640I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T640I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, and AlphaMissense‑Optimized. Only PROVEAN predicts a pathogenic outcome. The remaining tools (FoldX, Rosetta, ESM1b, AlphaMissense‑Default, and Foldetta) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is unavailable due to a tie, and Foldetta also yields an uncertain stability change. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.066181 | Structured | 0.137043 | Uncertain | 0.893 | 0.284 | 0.000 | -7.256 | In-Between | 0.358 | Ambiguous | Likely Benign | 0.70 | Ambiguous | 0.1 | 0.66 | Ambiguous | 0.68 | Ambiguous | 0.28 | Likely Benign | 0.193 | Likely Benign | -2.75 | Deleterious | 0.322 | Benign | 0.022 | Benign | 3.46 | Benign | 0.12 | Tolerated | 0.1030 | 0.4793 | 0 | -1 | 5.2 | 12.05 | ||||||||||||||||||||||||||||||
| c.191T>A | I64K 2D  AIThe SynGAP1 missense variant I64K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus remains benign; Foldetta results are unavailable. Overall, the majority of predictions lean toward a benign impact, and this conclusion does not contradict the ClinVar status, which contains no report for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | -3.206 | Likely Benign | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.159 | Likely Benign | -0.47 | Neutral | 0.334 | Benign | 0.029 | Benign | 4.07 | Benign | 0.00 | Affected | 0.0878 | 0.0740 | -2 | -3 | -8.4 | 15.01 | |||||||||||||||||||||||||||||||||||||||
| c.191T>C | I64T 2D AIThe SynGAP1 missense variant I64T is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33425799‑T‑C). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Those that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a Likely Benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Benign, and the Foldetta protein‑folding stability analysis is unavailable. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | 6-33425799-T-C | 1 | 6.20e-7 | -3.183 | Likely Benign | 0.943 | Likely Pathogenic | Ambiguous | 0.075 | Likely Benign | -0.51 | Neutral | 0.092 | Benign | 0.007 | Benign | 4.08 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0978 | 0.0851 | -1 | 0 | -5.2 | -12.05 | ||||||||||||||||||||||||||||||||||
| c.191T>G | I64R 2D  AIThe SynGAP1 missense variant I64R is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which classifies the variant as “Likely Benign.” Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized score is uncertain, and the Foldetta protein‑folding stability assessment is unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar assertion, so there is no contradiction with existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | -2.108 | Likely Benign | 0.936 | Likely Pathogenic | Ambiguous | 0.165 | Likely Benign | -0.54 | Neutral | 0.842 | Possibly Damaging | 0.068 | Benign | 4.05 | Benign | 0.00 | Affected | 0.1103 | 0.0940 | -2 | -3 | -9.0 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.1921T>A | S641T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S641T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all score the variant as benign. Rosetta also predicts a benign outcome, while FoldX and Foldetta are inconclusive (marked “Uncertain”). No tool predicts pathogenicity. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; Foldetta remains uncertain. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.125101 | Structured | 0.157322 | Uncertain | 0.786 | 0.270 | 0.000 | -6.637 | Likely Benign | 0.087 | Likely Benign | Likely Benign | 0.83 | Ambiguous | 0.3 | 0.41 | Likely Benign | 0.62 | Ambiguous | 0.13 | Likely Benign | 0.043 | Likely Benign | -1.50 | Neutral | 0.008 | Benign | 0.003 | Benign | 3.39 | Benign | 0.09 | Tolerated | 0.1505 | 0.5888 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1921T>C | S641P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S641P is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN and SIFT. The remaining tools (FoldX, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leaning toward benign, and Foldetta as uncertain. Overall, the majority of reliable predictions indicate a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.125101 | Structured | 0.157322 | Uncertain | 0.786 | 0.270 | 0.000 | -6.907 | Likely Benign | 0.378 | Ambiguous | Likely Benign | 1.73 | Ambiguous | 1.3 | 0.07 | Likely Benign | 0.90 | Ambiguous | 0.59 | Ambiguous | 0.205 | Likely Benign | -3.05 | Deleterious | 0.000 | Benign | 0.000 | Benign | 3.34 | Benign | 0.04 | Affected | 0.2289 | 0.5791 | 1 | -1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||
| c.1921T>G | S641A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S641A is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, every tool examined (REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly classifies the substitution as benign. No pathogenic predictions are present. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. Consequently, the variant is most likely benign based on current predictions, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.125101 | Structured | 0.157322 | Uncertain | 0.786 | 0.270 | 0.000 | -6.103 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.37 | Likely Benign | 0.2 | -0.28 | Likely Benign | 0.05 | Likely Benign | 0.27 | Likely Benign | 0.067 | Likely Benign | -2.07 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.43 | Benign | 0.17 | Tolerated | 0.4873 | 0.4680 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.1922C>T | S641L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S641L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields an equal split of benign and pathogenic calls. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict any ClinVar annotation, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.125101 | Structured | 0.157322 | Uncertain | 0.786 | 0.270 | 0.000 | -9.501 | Likely Pathogenic | 0.237 | Likely Benign | Likely Benign | 0.33 | Likely Benign | 0.3 | -0.14 | Likely Benign | 0.10 | Likely Benign | 0.32 | Likely Benign | 0.103 | Likely Benign | -4.33 | Deleterious | 0.115 | Benign | 0.014 | Benign | 3.39 | Benign | 0.07 | Tolerated | 0.1248 | 0.5376 | -3 | -2 | 4.6 | 26.08 | ||||||||||||||||||||||||||||||
| c.1924A>C | K642Q 2D 3DClick to see structure in 3D Viewer AISynGAP1 K642Q is not reported in ClinVar and has no allele in gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, and polyPhen‑2 HumVar; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy assessment shows AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as Benign. No other high‑confidence predictions are available. Overall, the balance of evidence leans toward a benign effect, with the single high‑accuracy pathogenic signal from SGM‑Consensus not contradicting the lack of ClinVar annotation. Thus, the variant is most likely benign, and this assessment does not conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.181468 | Uncertain | 0.806 | 0.289 | 0.000 | -12.186 | Likely Pathogenic | 0.862 | Likely Pathogenic | Ambiguous | 0.08 | Likely Benign | 0.0 | 0.17 | Likely Benign | 0.13 | Likely Benign | 0.42 | Likely Benign | 0.380 | Likely Benign | -3.88 | Deleterious | 0.576 | Possibly Damaging | 0.383 | Benign | 2.87 | Benign | 0.02 | Affected | 0.4477 | 0.1253 | 1 | 1 | 0.4 | -0.04 | |||||||||||||||||||||||||||||
| c.1924A>G | K642E 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant K642E is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions (REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM) and pathogenic predictions (SGM‑Consensus, PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized). High‑accuracy methods give a mixed signal: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, whereas Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. No prediction is inconclusive. Overall, the majority of tools lean toward pathogenicity, and this is consistent with the lack of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.181468 | Uncertain | 0.806 | 0.289 | 0.000 | -11.287 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 0.26 | Likely Benign | 0.1 | -0.13 | Likely Benign | 0.07 | Likely Benign | 0.40 | Likely Benign | 0.404 | Likely Benign | -3.92 | Deleterious | 0.116 | Benign | 0.011 | Benign | 2.96 | Benign | 0.03 | Affected | 0.3865 | 0.1013 | 0 | 1 | 0.4 | 0.94 | |||||||||||||||||||||||||||||
| c.1925A>G | K642R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K642R is not reported in ClinVar and has no entries in gnomAD. Prediction tools that assess pathogenicity largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all return benign. In contrast, PROVEAN, SIFT, and ESM1b predict a pathogenic impact. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign, Foldetta is benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the balance of evidence favors a benign classification, and this conclusion does not conflict with the absence of a ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.061840 | Structured | 0.181468 | Uncertain | 0.806 | 0.289 | 0.000 | -10.562 | Likely Pathogenic | 0.192 | Likely Benign | Likely Benign | -0.13 | Likely Benign | 0.1 | 0.39 | Likely Benign | 0.13 | Likely Benign | 0.04 | Likely Benign | 0.319 | Likely Benign | -2.79 | Deleterious | 0.001 | Benign | 0.001 | Benign | 2.91 | Benign | 0.02 | Affected | 0.4962 | 0.1262 | 3 | 2 | -0.6 | 28.01 | ||||||||||||||||||||||||||||||
| c.1925A>T | K642M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K642M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that assess the variant’s effect fall into two broad groups: benign predictions come from FoldX, premPS, and FATHMM; pathogenic predictions come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools give uncertain results: Rosetta and Foldetta. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. High‑accuracy assessments are as follows: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus also predicts pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. Overall, the preponderance of evidence from multiple independent predictors indicates that K642M is most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.181468 | Uncertain | 0.806 | 0.289 | 0.000 | -13.557 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 0.43 | Likely Benign | 0.1 | 0.62 | Ambiguous | 0.53 | Ambiguous | 0.21 | Likely Benign | 0.510 | Likely Pathogenic | -5.88 | Deleterious | 1.000 | Probably Damaging | 0.941 | Probably Damaging | 2.81 | Benign | 0.00 | Affected | 0.1256 | 0.3589 | 0 | -1 | 5.8 | 3.02 | |||||||||||||||||||||||||||||
| c.1926G>C | K642N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 K642N missense variant is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, and polyPhen‑2 HumVar. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy methods give a pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. With two of the three high‑accuracy tools supporting pathogenicity and an overall balance of predictions, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.181468 | Uncertain | 0.806 | 0.289 | 0.000 | -11.423 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.25 | Likely Benign | 0.0 | 0.05 | Likely Benign | 0.15 | Likely Benign | 0.48 | Likely Benign | 0.273 | Likely Benign | -4.88 | Deleterious | 0.958 | Probably Damaging | 0.392 | Benign | 2.88 | Benign | 0.00 | Affected | 0.3553 | 0.1590 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1926G>T | K642N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant K642N is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM, while those that predict a pathogenic impact are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and polyPhen‑2 HumVar (benign). High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as benign. Overall, seven tools favor pathogenicity versus six favoring benign, and the high‑accuracy predictions are mixed. Thus, the variant is most likely pathogenic based on the preponderance of evidence, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.061840 | Structured | 0.181468 | Uncertain | 0.806 | 0.289 | 0.000 | -11.423 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 0.25 | Likely Benign | 0.0 | 0.05 | Likely Benign | 0.15 | Likely Benign | 0.48 | Likely Benign | 0.274 | Likely Benign | -4.88 | Deleterious | 0.958 | Probably Damaging | 0.392 | Benign | 2.88 | Benign | 0.00 | Affected | 0.3553 | 0.1590 | 1 | 0 | 0.4 | -14.07 | |||||||||||||||||||||||||||||
| c.1927G>A | E643K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E643K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split opinion: benign calls come from REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), and FATHMM, while pathogenic calls arise from SGM‑Consensus (Likely Pathogenic), PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. Four tools (Foldetta, premPS, AlphaMissense‑Optimized, and Rosetta) give uncertain results. High‑accuracy assessments focus on AlphaMissense‑Optimized (Uncertain), SGM‑Consensus (Likely Pathogenic), and Foldetta (Uncertain). Because the consensus of the most reliable predictors leans toward pathogenicity, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.033407 | Structured | 0.215915 | Uncertain | 0.871 | 0.315 | 0.000 | -14.318 | Likely Pathogenic | 0.868 | Likely Pathogenic | Ambiguous | 0.39 | Likely Benign | 0.2 | 1.44 | Ambiguous | 0.92 | Ambiguous | 0.82 | Ambiguous | 0.449 | Likely Benign | -3.79 | Deleterious | 0.042 | Benign | 0.004 | Benign | 2.95 | Benign | 0.04 | Affected | 0.2961 | 0.6269 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1927G>C | E643Q 2D AIThe SynGAP1 missense variant E643Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SGM‑Consensus (Likely Pathogenic), PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized predicts a benign outcome, while premPS is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as benign. Overall, the majority of predictions (8 benign vs. 5 pathogenic) and the two of three high‑accuracy tools favor a benign classification. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.033407 | Structured | 0.215915 | Uncertain | 0.871 | 0.315 | 0.000 | -12.404 | Likely Pathogenic | 0.688 | Likely Pathogenic | Likely Benign | 0.49 | Likely Benign | 0.6 | 0.15 | Likely Benign | 0.32 | Likely Benign | 0.83 | Ambiguous | 0.341 | Likely Benign | -2.86 | Deleterious | 0.446 | Benign | 0.038 | Benign | 2.94 | Benign | 0.01 | Affected | 0.1603 | 0.6308 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1928A>C | E643A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E643A is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. FoldX and Foldetta give uncertain results. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta remains uncertain. Overall, the majority of tools lean toward a benign interpretation, and this assessment does not contradict ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.033407 | Structured | 0.215915 | Uncertain | 0.871 | 0.315 | 0.000 | -12.562 | Likely Pathogenic | 0.733 | Likely Pathogenic | Likely Benign | 0.80 | Ambiguous | 0.2 | 0.39 | Likely Benign | 0.60 | Ambiguous | 0.21 | Likely Benign | 0.469 | Likely Benign | -5.81 | Deleterious | 0.771 | Possibly Damaging | 0.233 | Benign | 2.92 | Benign | 0.01 | Affected | 0.4074 | 0.6096 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1928A>G | E643G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E643G is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic impact comprise SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default; FoldX and Foldetta are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic. Foldetta’s stability prediction is uncertain. Overall, the majority of reliable tools predict pathogenicity, and this conclusion does not contradict the lack of ClinVar annotation. Thus, the variant is most likely pathogenic based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.033407 | Structured | 0.215915 | Uncertain | 0.871 | 0.315 | 0.000 | -12.503 | Likely Pathogenic | 0.707 | Likely Pathogenic | Likely Benign | 1.45 | Ambiguous | 0.3 | 2.06 | Destabilizing | 1.76 | Ambiguous | 1.01 | Destabilizing | 0.520 | Likely Pathogenic | -6.81 | Deleterious | 0.983 | Probably Damaging | 0.390 | Benign | 2.94 | Benign | 0.00 | Affected | 0.2821 | 0.5319 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1928A>T | E643V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E643V missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions come from Rosetta, premPS, polyPhen‑2 HumVar, and FATHMM, while pathogenic predictions arise from SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. Three tools (FoldX, Foldetta, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments show that the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic (3 pathogenic vs. 1 benign). AlphaMissense‑Optimized remains uncertain, and Foldetta also yields an uncertain stability change. Overall, the preponderance of evidence points to a pathogenic effect for E643V, and this conclusion does not contradict any existing ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.033407 | Structured | 0.215915 | Uncertain | 0.871 | 0.315 | 0.000 | -12.975 | Likely Pathogenic | 0.893 | Likely Pathogenic | Ambiguous | 1.13 | Ambiguous | 0.1 | -0.06 | Likely Benign | 0.54 | Ambiguous | -0.28 | Likely Benign | 0.554 | Likely Pathogenic | -6.85 | Deleterious | 0.727 | Possibly Damaging | 0.145 | Benign | 2.89 | Benign | 0.00 | Affected | 0.0948 | 0.6637 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1929G>C | E643D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E643D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, AlphaMissense‑Default, AlphaMissense‑Optimized, FATHMM, and polyPhen2_HumVar. Tools that predict a pathogenic effect are premPS, PROVEAN, polyPhen2_HumDiv, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of predictions (8 benign vs. 5 pathogenic) support a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.033407 | Structured | 0.215915 | Uncertain | 0.871 | 0.315 | 0.000 | -8.083 | Likely Pathogenic | 0.223 | Likely Benign | Likely Benign | 0.46 | Likely Benign | 0.2 | -0.34 | Likely Benign | 0.06 | Likely Benign | 1.09 | Destabilizing | 0.292 | Likely Benign | -2.96 | Deleterious | 0.694 | Possibly Damaging | 0.064 | Benign | 2.98 | Benign | 0.01 | Affected | 0.2040 | 0.4276 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.1929G>T | E643D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E643D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, AlphaMissense‑Default, AlphaMissense‑Optimized, FATHMM, and polyPhen2_HumVar. Tools that predict a pathogenic effect are premPS, PROVEAN, polyPhen2_HumDiv, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of predictions (8 benign vs. 5 pathogenic) support a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.033407 | Structured | 0.215915 | Uncertain | 0.871 | 0.315 | 0.000 | -8.083 | Likely Pathogenic | 0.223 | Likely Benign | Likely Benign | 0.46 | Likely Benign | 0.2 | -0.34 | Likely Benign | 0.06 | Likely Benign | 1.09 | Destabilizing | 0.292 | Likely Benign | -2.96 | Deleterious | 0.694 | Possibly Damaging | 0.064 | Benign | 2.98 | Benign | 0.01 | Affected | 0.2040 | 0.4276 | 3 | 2 | 0.0 | -14.03 | ||||||||||||||||||||||||||||||
| c.192A>G | I64M 2D AIThe SynGAP1 missense variant I64M is listed in gnomAD (ID 6‑33425800‑A‑G) but has no ClinVar entry. Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic outcome are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.422041 | Structured | 0.475481 | Uncertain | 0.478 | 0.747 | 0.125 | 6-33425800-A-G | 2 | 1.24e-6 | -4.327 | Likely Benign | 0.523 | Ambiguous | Likely Benign | 0.047 | Likely Benign | -0.05 | Neutral | 0.637 | Possibly Damaging | 0.047 | Benign | 4.04 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0568 | 0.2310 | 1 | 2 | -2.6 | 18.03 | ||||||||||||||||||||||||||||||||||
| c.1930G>A | D644N 2D AIThe SynGAP1 missense variant D644N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—all classifying the substitution as benign. No tool predicts pathogenicity. The only inconclusive result is AlphaMissense‑Default, which is listed as uncertain and therefore does not influence the overall assessment. High‑accuracy methods further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports benign. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -4.389 | Likely Benign | 0.360 | Ambiguous | Likely Benign | 0.06 | Likely Benign | 0.3 | -0.28 | Likely Benign | -0.11 | Likely Benign | 0.02 | Likely Benign | 0.124 | Likely Benign | -2.28 | Neutral | 0.007 | Benign | 0.001 | Benign | 3.45 | Benign | 0.25 | Tolerated | 0.1368 | 0.6261 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1930G>C | D644H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D644H missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that predict a pathogenic effect are PROVEAN, polyPhen2_HumDiv, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and Foldetta as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. No other high‑accuracy predictions are available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -6.786 | Likely Benign | 0.771 | Likely Pathogenic | Likely Benign | 0.34 | Likely Benign | 0.1 | -0.83 | Ambiguous | -0.25 | Likely Benign | 0.09 | Likely Benign | 0.284 | Likely Benign | -2.93 | Deleterious | 0.789 | Possibly Damaging | 0.158 | Benign | 3.43 | Benign | 0.07 | Tolerated | 0.1656 | 0.6306 | 1 | -1 | 0.3 | 22.05 | ||||||||||||||||||||||||||||||
| c.1930G>T | D644Y 2D 3DClick to see structure in 3D Viewer AISynGAP1 D644Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, premPS, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default; the SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports it as Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, SGM‑Consensus as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Uncertain. The remaining tools, FoldX, REVEL, premPS, polyPhen‑2 HumVar, and FATHMM, support a benign effect, while PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default support a pathogenic effect. Overall, the majority of evidence leans toward pathogenicity, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -10.143 | Likely Pathogenic | 0.721 | Likely Pathogenic | Likely Benign | 0.03 | Likely Benign | 0.1 | -1.18 | Ambiguous | -0.58 | Ambiguous | -0.04 | Likely Benign | 0.318 | Likely Benign | -4.93 | Deleterious | 0.968 | Probably Damaging | 0.311 | Benign | 3.44 | Benign | 0.02 | Affected | 0.0679 | 0.6094 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.1931A>C | D644A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D644A missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, FoldX, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, Foldetta, and premPS. Only PROVEAN predicts it as pathogenic, while Rosetta and AlphaMissense‑Default are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign; and Foldetta also predicts benign stability. Based on the aggregate evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -3.358 | Likely Benign | 0.502 | Ambiguous | Likely Benign | -0.14 | Likely Benign | 0.1 | -0.83 | Ambiguous | -0.49 | Likely Benign | -0.18 | Likely Benign | 0.274 | Likely Benign | -3.90 | Deleterious | 0.311 | Benign | 0.032 | Benign | 3.51 | Benign | 0.39 | Tolerated | 0.3883 | 0.5481 | 0 | -2 | 5.3 | -44.01 | ||||||||||||||||||||||||||||||
| c.1931A>G | D644G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D644G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and AlphaMissense‑Default (polyPhen‑2 HumVar is benign). High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -4.496 | Likely Benign | 0.602 | Likely Pathogenic | Likely Benign | 0.13 | Likely Benign | 0.0 | -0.18 | Likely Benign | -0.03 | Likely Benign | 0.04 | Likely Benign | 0.271 | Likely Benign | -4.38 | Deleterious | 0.456 | Possibly Damaging | 0.069 | Benign | 3.46 | Benign | 0.14 | Tolerated | 0.4035 | 0.5610 | 1 | -1 | 3.1 | -58.04 | ||||||||||||||||||||||||||||||
| c.1931A>T | D644V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D644V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split. Overall, the majority of evidence points to a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -6.230 | Likely Benign | 0.636 | Likely Pathogenic | Likely Benign | 0.50 | Ambiguous | 0.0 | -0.35 | Likely Benign | 0.08 | Likely Benign | -0.03 | Likely Benign | 0.347 | Likely Benign | -4.96 | Deleterious | 0.198 | Benign | 0.052 | Benign | 3.49 | Benign | 0.11 | Tolerated | 0.0951 | 0.6267 | -2 | -3 | 7.7 | -15.96 | ||||||||||||||||||||||||||||||
| c.1932C>A | D644E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D644E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts a pathogenic outcome; the only inconclusive result is from Rosetta, which is treated as unavailable. High‑accuracy assessments confirm a benign impact: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -1.778 | Likely Benign | 0.178 | Likely Benign | Likely Benign | -0.17 | Likely Benign | 0.1 | -0.51 | Ambiguous | -0.34 | Likely Benign | -0.47 | Likely Benign | 0.132 | Likely Benign | 1.31 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.55 | Benign | 1.00 | Tolerated | 0.1528 | 0.6186 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.1932C>G | D644E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D644E is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts a pathogenic outcome; the only inconclusive result is from Rosetta, which is treated as unavailable. High‑accuracy assessments confirm a benign impact: AlphaMissense‑Optimized is benign, SGM‑Consensus is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Based on the collective predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.066181 | Structured | 0.248888 | Uncertain | 0.883 | 0.320 | 0.000 | -1.778 | Likely Benign | 0.178 | Likely Benign | Likely Benign | -0.17 | Likely Benign | 0.1 | -0.51 | Ambiguous | -0.34 | Likely Benign | -0.47 | Likely Benign | 0.132 | Likely Benign | 1.31 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.55 | Benign | 1.00 | Tolerated | 0.1528 | 0.6186 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.1933T>A | F645I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F645I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM, while pathogenic predictions are given by premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) report uncertain or inconclusive outcomes. High‑accuracy assessments indicate that AlphaMissense‑Optimized is uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, and Foldetta is uncertain. Overall, the majority of evaluated predictors (five pathogenic vs four benign) lean toward a pathogenic effect. Therefore, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, which currently has no entry for F645I. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -13.055 | Likely Pathogenic | 0.878 | Likely Pathogenic | Ambiguous | 1.12 | Ambiguous | 0.2 | 1.68 | Ambiguous | 1.40 | Ambiguous | 1.01 | Destabilizing | 0.299 | Likely Benign | -4.24 | Deleterious | 0.190 | Benign | 0.019 | Benign | 3.44 | Benign | 0.04 | Affected | 0.1920 | 0.2891 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1933T>C | F645L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F645L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; pathogenic predictions arise from SGM‑Consensus (Likely Pathogenic), PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. No evidence from FoldX, Rosetta, or premPS is available to alter this view. Overall, the majority of computational evidence points to a pathogenic impact for F645L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -10.624 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.98 | Ambiguous | 0.1 | 1.25 | Ambiguous | 1.12 | Ambiguous | 0.99 | Ambiguous | 0.264 | Likely Benign | -4.24 | Deleterious | 0.116 | Benign | 0.008 | Benign | 3.44 | Benign | 0.03 | Affected | 0.2214 | 0.3873 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1933T>G | F645V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F645V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM. Tools that agree on a pathogenic effect are premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy assessments give an uncertain result from AlphaMissense‑Optimized, a pathogenic outcome from the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and an uncertain outcome from Foldetta (combining FoldX‑MD and Rosetta). No definitive folding‑stability change is reported. Overall, the majority of evidence points to a pathogenic impact for F645V, and this conclusion does not contradict any ClinVar annotation because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -12.175 | Likely Pathogenic | 0.810 | Likely Pathogenic | Ambiguous | 1.98 | Ambiguous | 0.1 | 1.70 | Ambiguous | 1.84 | Ambiguous | 1.02 | Destabilizing | 0.316 | Likely Benign | -4.41 | Deleterious | 0.036 | Benign | 0.018 | Benign | 3.40 | Benign | 0.02 | Affected | 0.2163 | 0.2919 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1934T>A | F645Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F645Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools with inconclusive results are FoldX (Uncertain) and premPS (Uncertain). High‑accuracy assessments show AlphaMissense‑Optimized predicting Benign, the SGM‑Consensus indicating Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicting Benign. No tool predicts pathogenicity. Consequently, the variant is most likely benign based on the collective predictions, and this assessment does not contradict any ClinVar status, as the variant is not currently classified in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -3.544 | Likely Benign | 0.131 | Likely Benign | Likely Benign | 0.52 | Ambiguous | 0.2 | 0.21 | Likely Benign | 0.37 | Likely Benign | -0.61 | Ambiguous | 0.141 | Likely Benign | 2.40 | Neutral | 0.000 | Benign | 0.000 | Benign | 3.61 | Benign | 1.00 | Tolerated | 0.1506 | 0.2189 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1934T>C | F645S 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F645S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign calls come from REVEL, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic calls are made by FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while both the SGM Consensus and Foldetta predict pathogenicity. No evidence from ClinVar contradicts these findings. Overall, the preponderance of predictions supports a pathogenic classification for F645S. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -9.748 | Likely Pathogenic | 0.947 | Likely Pathogenic | Ambiguous | 2.49 | Destabilizing | 0.2 | 2.30 | Destabilizing | 2.40 | Destabilizing | 1.57 | Destabilizing | 0.326 | Likely Benign | -5.34 | Deleterious | 0.755 | Possibly Damaging | 0.112 | Benign | 3.37 | Benign | 0.03 | Affected | 0.3899 | 0.0547 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1934T>G | F645C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F645C is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas the majority of tools predict a pathogenic outcome: FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is inconclusive. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized remains uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also pathogenic. Based on the preponderance of pathogenic predictions and the high‑accuracy consensus, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -9.182 | Likely Pathogenic | 0.828 | Likely Pathogenic | Ambiguous | 2.25 | Destabilizing | 0.1 | 2.44 | Destabilizing | 2.35 | Destabilizing | 1.38 | Destabilizing | 0.286 | Likely Benign | -5.41 | Deleterious | 0.967 | Probably Damaging | 0.389 | Benign | 3.35 | Benign | 0.01 | Affected | 0.2421 | 0.1372 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.1935T>A | F645L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F645L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM; pathogenic predictions arise from SGM‑Consensus (Likely Pathogenic), PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is inconclusive. No evidence from FoldX, Rosetta, or premPS is available to alter this view. Overall, the majority of computational evidence points to a pathogenic impact for F645L, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -10.624 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.98 | Ambiguous | 0.1 | 1.25 | Ambiguous | 1.12 | Ambiguous | 0.99 | Ambiguous | 0.159 | Likely Benign | -4.24 | Deleterious | 0.116 | Benign | 0.008 | Benign | 3.44 | Benign | 0.03 | Affected | 0.2214 | 0.3873 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1935T>G | F645L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F645L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that assess sequence conservation and structural impact fall into two groups: benign predictions are made by REVEL, polyPhen‑2 (HumDiv and HumVar), and FATHMM; pathogenic predictions are made by SGM‑Consensus (Likely Pathogenic), PROVEAN, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates Likely Pathogenic, while Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. No evidence from FoldX‑MD, Rosetta, or premPS is available to alter this conclusion. Overall, the majority of computational evidence points to a pathogenic impact for F645L, and this assessment is consistent with the absence of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.276445 | Uncertain | 0.921 | 0.325 | 0.000 | -10.624 | Likely Pathogenic | 0.987 | Likely Pathogenic | Likely Pathogenic | 0.98 | Ambiguous | 0.1 | 1.25 | Ambiguous | 1.12 | Ambiguous | 0.99 | Ambiguous | 0.159 | Likely Benign | -4.24 | Deleterious | 0.116 | Benign | 0.008 | Benign | 3.44 | Benign | 0.03 | Affected | 0.2214 | 0.3873 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1936C>A | L646M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L646M is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome. Predictions that are inconclusive are FoldX, Rosetta, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign, while Foldetta’s stability analysis remains uncertain. Overall, the evidence overwhelmingly supports a benign classification for this variant, and this conclusion does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.303751 | Uncertain | 0.941 | 0.344 | 0.000 | -1.911 | Likely Benign | 0.152 | Likely Benign | Likely Benign | 0.70 | Ambiguous | 0.1 | 1.66 | Ambiguous | 1.18 | Ambiguous | -1.10 | Stabilizing | 0.106 | Likely Benign | 1.86 | Neutral | 0.211 | Benign | 0.055 | Benign | 3.57 | Benign | 1.00 | Tolerated | 0.2041 | 0.3427 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.1936C>G | L646V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L646V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and FATHMM, whereas pathogenic predictions arise from FoldX, Rosetta, premPS, SIFT, and ESM1b. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, resolves to “Likely Benign” (3 benign vs 1 pathogenic votes). High‑accuracy assessments further indicate AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of tools, including the high‑accuracy methods, lean toward a benign effect, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.303751 | Uncertain | 0.941 | 0.344 | 0.000 | -8.995 | Likely Pathogenic | 0.332 | Likely Benign | Likely Benign | 3.82 | Destabilizing | 0.2 | 2.01 | Destabilizing | 2.92 | Destabilizing | 1.41 | Destabilizing | 0.125 | Likely Benign | -1.40 | Neutral | 0.040 | Benign | 0.021 | Benign | 3.21 | Benign | 0.03 | Affected | 0.2200 | 0.3767 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1937T>A | L646Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L646Q lies in the GAP domain. ClinVar has no entry for this variant, and it is not present in gnomAD. Prediction tools that agree on a benign effect are REVEL, FATHMM, and AlphaMissense‑Optimized. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Taken together, the overwhelming majority of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.303751 | Uncertain | 0.941 | 0.344 | 0.000 | -12.735 | Likely Pathogenic | 0.752 | Likely Pathogenic | Likely Benign | 3.23 | Destabilizing | 0.1 | 3.04 | Destabilizing | 3.14 | Destabilizing | 2.12 | Destabilizing | 0.462 | Likely Benign | -2.78 | Deleterious | 0.994 | Probably Damaging | 0.790 | Possibly Damaging | 3.17 | Benign | 0.00 | Affected | 0.2090 | 0.1963 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1937T>C | L646P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L646P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.303751 | Uncertain | 0.941 | 0.344 | 0.000 | -12.956 | Likely Pathogenic | 0.970 | Likely Pathogenic | Likely Pathogenic | 7.34 | Destabilizing | 0.5 | 12.08 | Destabilizing | 9.71 | Destabilizing | 2.72 | Destabilizing | 0.604 | Likely Pathogenic | -4.55 | Deleterious | 0.999 | Probably Damaging | 0.926 | Probably Damaging | 3.17 | Benign | 0.00 | Affected | 0.3481 | 0.1874 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1937T>G | L646R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L646R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic calls are made by FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is uncertain, SGM Consensus is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a pathogenic impact. Overall, the preponderance of evidence points to a pathogenic effect for L646R, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.303751 | Uncertain | 0.941 | 0.344 | 0.000 | -12.393 | Likely Pathogenic | 0.920 | Likely Pathogenic | Ambiguous | 4.44 | Destabilizing | 0.2 | 3.94 | Destabilizing | 4.19 | Destabilizing | 2.75 | Destabilizing | 0.455 | Likely Benign | -3.56 | Deleterious | 0.014 | Benign | 0.002 | Benign | 3.17 | Benign | 0.03 | Affected | 0.2304 | 0.1405 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1939G>A | G647S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G647S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome; the only inconclusive results come from Rosetta and premPS, which are treated as unavailable. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized scores benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Overall, the evidence strongly favors a benign classification, and this is consistent with its absence from ClinVar. The variant is most likely benign, and this is consistent with its absence from ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -2.175 | Likely Benign | 0.082 | Likely Benign | Likely Benign | 0.05 | Likely Benign | 0.1 | -0.75 | Ambiguous | -0.35 | Likely Benign | -0.56 | Ambiguous | 0.037 | Likely Benign | 0.65 | Neutral | 0.002 | Benign | 0.004 | Benign | 3.48 | Benign | 0.89 | Tolerated | 0.2409 | 0.3959 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.1939G>C | G647R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G647R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign or likely benign. In contrast, AlphaMissense‑Default and polyPhen‑2 HumDiv predict pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus as likely benign, and Foldetta (combining FoldX‑MD and Rosetta) is inconclusive. No evidence from FoldX or Rosetta alone is available. Overall, the preponderance of evidence supports a benign classification, and this is consistent with the lack of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -6.590 | Likely Benign | 0.636 | Likely Pathogenic | Likely Benign | -0.51 | Ambiguous | 0.1 | -0.97 | Ambiguous | -0.74 | Ambiguous | 0.29 | Likely Benign | 0.097 | Likely Benign | -1.81 | Neutral | 0.787 | Possibly Damaging | 0.349 | Benign | 3.49 | Benign | 0.17 | Tolerated | 0.1020 | 0.3712 | -3 | -2 | -4.1 | 99.14 | |||||||||||||||||||||||||||||
| c.1939G>T | G647C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G647C is not reported in ClinVar and is absent from gnomAD. Across the available in‑silico predictors, the majority (REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus “Likely Benign”) uniformly predict a benign effect, whereas only SIFT classifies the change as pathogenic. High‑accuracy tools give consistent benign results: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a benign stability change. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -2.955 | Likely Benign | 0.112 | Likely Benign | Likely Benign | 0.41 | Likely Benign | 0.0 | -0.20 | Likely Benign | 0.11 | Likely Benign | 0.05 | Likely Benign | 0.112 | Likely Benign | -1.63 | Neutral | 0.003 | Benign | 0.004 | Benign | 3.44 | Benign | 0.02 | Affected | 0.1194 | 0.3250 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.193C>A | H65N 2D  AIThe SynGAP1 missense variant H65N is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are AlphaMissense‑Default and SIFT. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely benign; a Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this is consistent with the lack of ClinVar annotation. Thus, the variant is most likely benign and does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.476188 | Uncertain | 0.458 | 0.758 | 0.125 | -2.788 | Likely Benign | 0.771 | Likely Pathogenic | Likely Benign | 0.038 | Likely Benign | -1.42 | Neutral | 0.273 | Benign | 0.107 | Benign | 4.18 | Benign | 0.00 | Affected | 0.1280 | 0.2200 | 2 | 1 | -0.3 | -23.04 | |||||||||||||||||||||||||||||||||||||||
| c.193C>G | H65D 2D  AIThe SynGAP1 H65D missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Benign, and AlphaMissense‑Optimized is classified as Uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.476188 | Uncertain | 0.458 | 0.758 | 0.125 | -2.240 | Likely Benign | 0.937 | Likely Pathogenic | Ambiguous | 0.126 | Likely Benign | -1.78 | Neutral | 0.462 | Possibly Damaging | 0.227 | Benign | 4.17 | Benign | 0.00 | Affected | 0.2142 | 0.1828 | 1 | -1 | -0.3 | -22.05 | |||||||||||||||||||||||||||||||||||||||
| c.193C>T | H65Y 2D  AIThe SynGAP1 missense variant H65Y has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and FATHMM. Tools that predict a pathogenic effect are SIFT and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely benign, and Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar pathogenic designation and the lack of population frequency data. The variant is most likely benign based on predictions, and this does not contradict ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.476188 | Uncertain | 0.458 | 0.758 | 0.125 | -3.644 | Likely Benign | 0.852 | Likely Pathogenic | Ambiguous | 0.037 | Likely Benign | -1.11 | Neutral | 0.273 | Benign | 0.152 | Benign | 4.15 | Benign | 0.00 | Affected | 0.0788 | 0.3628 | 0 | 2 | 1.9 | 26.03 | |||||||||||||||||||||||||||||||||||||||
| c.1940G>A | G647D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G647D is reported in ClinVar as having no entry and is present in gnomAD (6‑33441199‑G‑A). Functional prediction tools largely agree on a benign effect: REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized all predict benign. Only AlphaMissense‑Default predicts pathogenic, while FoldX, Foldetta, premPS, and ESM1b are uncertain. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields benign; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. Overall, the consensus of most tools and the high‑accuracy predictions indicate that G647D is most likely benign, and this is consistent with the absence of a pathogenic ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | 6-33441199-G-A | 1 | 6.20e-7 | -7.643 | In-Between | 0.582 | Likely Pathogenic | Likely Benign | 0.68 | Ambiguous | 0.1 | 0.33 | Likely Benign | 0.51 | Ambiguous | 0.56 | Ambiguous | 0.098 | Likely Benign | -1.63 | Neutral | 0.282 | Benign | 0.128 | Benign | 3.45 | Benign | 0.24 | Tolerated | 3.37 | 30 | 0.1630 | 0.1372 | -1 | 1 | -3.1 | 58.04 | |||||||||||||||||||||||||
| c.1940G>C | G647A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G647A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome. Predictions that are inconclusive are Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta as Uncertain. Taken together, the overwhelming majority of evidence indicates a benign effect for G647A, and this conclusion is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -0.266 | Likely Benign | 0.078 | Likely Benign | Likely Benign | -0.18 | Likely Benign | 0.0 | -0.99 | Ambiguous | -0.59 | Ambiguous | -0.69 | Ambiguous | 0.037 | Likely Benign | 0.48 | Neutral | 0.000 | Benign | 0.002 | Benign | 3.55 | Benign | 0.81 | Tolerated | 0.3558 | 0.4136 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1940G>T | G647V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G647V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that assess sequence conservation and structural impact uniformly classify the substitution as benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, premPS, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a tolerated change. No tool predicts pathogenicity; only FoldX and Rosetta report uncertain stability effects, which are treated as unavailable evidence. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a benign effect. Therefore, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.048328 | Structured | 0.325524 | Uncertain | 0.936 | 0.356 | 0.000 | -4.713 | Likely Benign | 0.126 | Likely Benign | Likely Benign | 0.85 | Ambiguous | 0.1 | -0.59 | Ambiguous | 0.13 | Likely Benign | -0.13 | Likely Benign | 0.115 | Likely Benign | -1.77 | Neutral | 0.178 | Benign | 0.073 | Benign | 3.52 | Benign | 0.12 | Tolerated | 0.1318 | 0.3946 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1942T>A | F648I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F648I resides in the GAP domain. ClinVar contains no entry for this change, and it is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM. All other evaluated algorithms—FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—report a pathogenic or likely pathogenic outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) also yields a pathogenic prediction. Taken together, the evidence overwhelmingly supports a pathogenic classification for F648I, and this conclusion does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.346782 | Uncertain | 0.943 | 0.339 | 0.000 | -11.912 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.49 | Destabilizing | 0.1 | 2.49 | Destabilizing | 2.49 | Destabilizing | 1.05 | Destabilizing | 0.472 | Likely Benign | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 3.41 | Benign | 0.04 | Affected | 0.1673 | 0.1955 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1942T>C | F648L 2D AISynGAP1 missense variant F648L is listed in ClinVar with an uncertain significance (ClinVar ID 3383902.0) and is not reported in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, whereas the remaining tools—FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b—consistently predict pathogenicity. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized scores pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a destabilizing, pathogenic change. Taken together, the preponderance of evidence points to a pathogenic impact for F648L, which contradicts the current ClinVar uncertain status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.346782 | Uncertain | 0.943 | 0.339 | 0.000 | Uncertain | 1 | -9.296 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.71 | Destabilizing | 0.8 | 2.08 | Destabilizing | 2.40 | Destabilizing | 1.04 | Destabilizing | 0.468 | Likely Benign | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.976 | Probably Damaging | 3.45 | Benign | 0.08 | Tolerated | 0.1961 | 0.3126 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||
| c.1942T>G | F648V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F648V is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include SIFT and FATHMM, whereas the remaining tools—REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and the SGM‑Consensus—predict it to be pathogenic. High‑accuracy methods give consistent results: AlphaMissense‑Optimized indicates pathogenicity; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a destabilizing, pathogenic effect. No prediction is missing or inconclusive. Overall, the evidence overwhelmingly supports a pathogenic classification, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.346782 | Uncertain | 0.943 | 0.339 | 0.000 | -11.574 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 3.11 | Destabilizing | 0.1 | 2.80 | Destabilizing | 2.96 | Destabilizing | 1.14 | Destabilizing | 0.514 | Likely Pathogenic | -6.98 | Deleterious | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 3.49 | Benign | 0.06 | Tolerated | 0.1848 | 0.1983 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1943T>A | F648Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F648Y is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (gnomAD ID: 6‑33441202‑T‑A). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic impact: premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, AlphaMissense‑Default, and ESM1b. Predictions that are inconclusive or unavailable are FoldX, Rosetta, Foldetta, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the balance of evidence favors a pathogenic classification for F648Y. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.346782 | Uncertain | 0.943 | 0.339 | 0.000 | 6-33441202-T-A | 4 | 2.48e-6 | -8.632 | Likely Pathogenic | 0.889 | Likely Pathogenic | Ambiguous | 0.74 | Ambiguous | 0.1 | 0.94 | Ambiguous | 0.84 | Ambiguous | 1.11 | Destabilizing | 0.407 | Likely Benign | -2.99 | Deleterious | 0.984 | Probably Damaging | 0.913 | Probably Damaging | 3.41 | Benign | 0.11 | Tolerated | 3.37 | 30 | 0.1307 | 0.1396 | 3 | 7 | -4.1 | 16.00 | ||||||||||||||||||||||||
| c.1943T>C | F648S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F648S is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: benign calls are limited to SIFT and FATHMM, whereas the remaining 12 predictors—including REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, both polyPhen‑2 versions, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the substitution as pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports a likely pathogenic outcome, and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also indicates a pathogenic effect. No prediction is inconclusive or missing. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.346782 | Uncertain | 0.943 | 0.339 | 0.000 | -10.356 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.36 | Destabilizing | 0.0 | 3.23 | Destabilizing | 3.30 | Destabilizing | 1.35 | Destabilizing | 0.604 | Likely Pathogenic | -7.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 3.48 | Benign | 0.06 | Tolerated | 0.3680 | 0.0200 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1943T>G | F648C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F648C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools largely agree on a deleterious effect: all evaluated algorithms except FATHMM predict pathogenicity, while FATHMM alone predicts benign. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also reports a pathogenic effect. In summary, the overwhelming consensus from both general and high‑accuracy predictors is that F648C is pathogenic. This conclusion is consistent with the absence of any ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.346782 | Uncertain | 0.943 | 0.339 | 0.000 | -10.547 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.28 | Destabilizing | 0.1 | 3.35 | Destabilizing | 3.32 | Destabilizing | 1.22 | Destabilizing | 0.621 | Likely Pathogenic | -7.98 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 3.40 | Benign | 0.03 | Affected | 0.2283 | 0.0783 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.1944C>A | F648L 2D AIThe SynGAP1 missense variant F648L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, while the remaining 12 tools—including FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—predict a pathogenic effect. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized is pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, is pathogenic; and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic. Because the majority of evidence points to a deleterious impact and there is no ClinVar annotation to contradict this, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.346782 | Uncertain | 0.943 | 0.339 | 0.000 | -9.296 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.71 | Destabilizing | 0.8 | 2.08 | Destabilizing | 2.40 | Destabilizing | 1.04 | Destabilizing | 0.319 | Likely Benign | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.976 | Probably Damaging | 3.45 | Benign | 0.08 | Tolerated | 0.1961 | 0.3126 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1944C>G | F648L 2D AIThe SynGAP1 missense variant F648L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. In contrast, the majority of tools predict a pathogenic impact: FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), AlphaMissense‑Default, AlphaMissense‑Optimized, and ESM1b all indicate pathogenicity, and the SGM Consensus score is “Likely Pathogenic.” High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion is consistent with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.346782 | Uncertain | 0.943 | 0.339 | 0.000 | -9.296 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 2.71 | Destabilizing | 0.8 | 2.08 | Destabilizing | 2.40 | Destabilizing | 1.04 | Destabilizing | 0.319 | Likely Benign | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.976 | Probably Damaging | 3.45 | Benign | 0.08 | Tolerated | 0.1961 | 0.3126 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1945A>C | M649L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M649L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions arise from PROVEAN and AlphaMissense‑Default, while premPS remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta predicts benign stability. Overall, the preponderance of evidence points to a benign effect. This conclusion is consistent with the absence of a ClinVar assertion, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.051831 | Structured | 0.360413 | Uncertain | 0.962 | 0.345 | 0.000 | -5.210 | Likely Benign | 0.745 | Likely Pathogenic | Likely Benign | 0.19 | Likely Benign | 0.4 | 0.26 | Likely Benign | 0.23 | Likely Benign | 0.64 | Ambiguous | 0.294 | Likely Benign | -2.99 | Deleterious | 0.009 | Benign | 0.007 | Benign | 3.73 | Benign | 0.15 | Tolerated | 0.1361 | 0.4025 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||
| c.1945A>G | M649V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M649V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools predict a pathogenic impact: FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. The high‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. With 8 pathogenic versus 3 benign predictions, the overall evidence favors a deleterious effect. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.360413 | Uncertain | 0.962 | 0.345 | 0.000 | -9.907 | Likely Pathogenic | 0.887 | Likely Pathogenic | Ambiguous | 2.84 | Destabilizing | 0.3 | 2.55 | Destabilizing | 2.70 | Destabilizing | 1.05 | Destabilizing | 0.370 | Likely Benign | -3.99 | Deleterious | 0.997 | Probably Damaging | 0.735 | Possibly Damaging | 3.40 | Benign | 0.06 | Tolerated | 0.2389 | 0.3121 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||
| c.1945A>T | M649L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M649L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN and AlphaMissense‑Default, while premPS remains uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta (combining FoldX‑MD and Rosetta outputs) as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.051831 | Structured | 0.360413 | Uncertain | 0.962 | 0.345 | 0.000 | -5.210 | Likely Benign | 0.745 | Likely Pathogenic | Likely Benign | 0.19 | Likely Benign | 0.4 | 0.26 | Likely Benign | 0.23 | Likely Benign | 0.64 | Ambiguous | 0.294 | Likely Benign | -2.99 | Deleterious | 0.009 | Benign | 0.007 | Benign | 3.73 | Benign | 0.15 | Tolerated | 0.1361 | 0.4025 | 4 | 2 | 1.9 | -18.03 | ||||||||||||||||||||||||||||||
| c.1946T>A | M649K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M649K is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: pathogenic predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Benign calls are limited to polyPhen‑2 (HumVar) and FATHMM. High‑accuracy methods reinforce the pathogenic consensus: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenic. No prediction or stability result is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.360413 | Uncertain | 0.962 | 0.345 | 0.000 | -14.533 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.42 | Destabilizing | 0.0 | 6.19 | Destabilizing | 4.81 | Destabilizing | 1.79 | Destabilizing | 0.614 | Likely Pathogenic | -5.98 | Deleterious | 0.968 | Probably Damaging | 0.382 | Benign | 3.34 | Benign | 0.01 | Affected | 0.1438 | 0.1079 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||
| c.1946T>C | M649T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M649T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also classifies the variant as pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.360413 | Uncertain | 0.962 | 0.345 | 0.000 | -11.916 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 3.19 | Destabilizing | 0.2 | 3.45 | Destabilizing | 3.32 | Destabilizing | 1.92 | Destabilizing | 0.531 | Likely Pathogenic | -5.98 | Deleterious | 0.999 | Probably Damaging | 0.779 | Possibly Damaging | 3.35 | Benign | 0.00 | Affected | 0.1732 | 0.1615 | -1 | -1 | -2.6 | -30.09 | |||||||||||||||||||||||||||||
| c.1946T>G | M649R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M649R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; and Foldetta, which integrates FoldX‑MD and Rosetta stability predictions, also reports a pathogenic effect. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.051831 | Structured | 0.360413 | Uncertain | 0.962 | 0.345 | 0.000 | -14.827 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 5.02 | Destabilizing | 0.1 | 5.97 | Destabilizing | 5.50 | Destabilizing | 2.09 | Destabilizing | 0.595 | Likely Pathogenic | -5.98 | Deleterious | 0.985 | Probably Damaging | 0.464 | Possibly Damaging | 3.33 | Benign | 0.00 | Affected | 0.1635 | 0.1228 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||
| c.1948A>C | N650H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N650H lies in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, FoldX, and the SGM‑Consensus (majority vote). Uncertain or inconclusive results come from Rosetta, Foldetta, and premPS. High‑accuracy assessments: AlphaMissense‑Optimized predicts pathogenicity; the SGM‑Consensus also indicates likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive. Based on the preponderance of pathogenic predictions and the high‑accuracy tools’ results, the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.086953 | Structured | 0.361944 | Uncertain | 0.961 | 0.357 | 0.000 | -14.187 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 2.14 | Destabilizing | 0.3 | 1.79 | Ambiguous | 1.97 | Ambiguous | 0.55 | Ambiguous | 0.465 | Likely Benign | -4.98 | Deleterious | 0.999 | Probably Damaging | 0.929 | Probably Damaging | 3.01 | Benign | 0.05 | Affected | 0.1990 | 0.4784 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.1948A>G | N650D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N650D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect comprise SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Stability‑based methods (FoldX, Rosetta, premPS, Foldetta) yield uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as Uncertain. Overall, the majority of available predictions support a pathogenic impact. The variant is most likely pathogenic based on the consensus of predictive tools, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.086953 | Structured | 0.361944 | Uncertain | 0.961 | 0.357 | 0.000 | -14.267 | Likely Pathogenic | 0.971 | Likely Pathogenic | Likely Pathogenic | 1.14 | Ambiguous | 0.3 | 0.61 | Ambiguous | 0.88 | Ambiguous | 0.91 | Ambiguous | 0.389 | Likely Benign | -4.98 | Deleterious | 0.591 | Possibly Damaging | 0.185 | Benign | 2.99 | Benign | 0.03 | Affected | 0.2320 | 0.2973 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.1948A>T | N650Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N650Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include premPS and FATHMM, whereas the majority of tools (SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; FoldX, Rosetta, and Foldetta are inconclusive. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, and Foldetta remains uncertain. Overall, the evidence strongly favors a pathogenic classification for the variant, and this conclusion does not contradict the ClinVar status, which currently contains no entry for this mutation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.086953 | Structured | 0.361944 | Uncertain | 0.961 | 0.357 | 0.000 | -16.923 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 1.56 | Ambiguous | 0.3 | 1.32 | Ambiguous | 1.44 | Ambiguous | 0.16 | Likely Benign | 0.587 | Likely Pathogenic | -7.98 | Deleterious | 1.000 | Probably Damaging | 0.984 | Probably Damaging | 3.03 | Benign | 0.03 | Affected | 0.0852 | 0.3865 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.1949A>C | N650T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N650T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, and FATHMM, whereas a larger group predicts pathogenicity: SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results come from Foldetta, premPS, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta as inconclusive. Overall, the majority of evidence points to a pathogenic effect. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.086953 | Structured | 0.361944 | Uncertain | 0.961 | 0.357 | 0.000 | -13.626 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.31 | Likely Benign | 0.1 | 0.73 | Ambiguous | 0.52 | Ambiguous | 0.69 | Ambiguous | 0.471 | Likely Benign | -5.98 | Deleterious | 0.986 | Probably Damaging | 0.710 | Possibly Damaging | 3.04 | Benign | 0.01 | Affected | 0.1700 | 0.4843 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.1949A>G | N650S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N650S lies in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). Uncertain or inconclusive results come from AlphaMissense‑Optimized, FoldX, Rosetta, Foldetta, and premPS. For high‑accuracy methods, AlphaMissense‑Optimized is uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta is uncertain. Overall, the majority of available predictions support a pathogenic effect. The variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which is currently absent. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.086953 | Structured | 0.361944 | Uncertain | 0.961 | 0.357 | 0.000 | -11.291 | Likely Pathogenic | 0.916 | Likely Pathogenic | Ambiguous | 0.79 | Ambiguous | 0.2 | 1.43 | Ambiguous | 1.11 | Ambiguous | 0.77 | Ambiguous | 0.395 | Likely Benign | -4.98 | Deleterious | 0.996 | Probably Damaging | 0.606 | Possibly Damaging | 3.06 | Benign | 0.02 | Affected | 0.3791 | 0.5090 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||||||
| c.1949A>T | N650I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N650I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split assessment: benign predictions come from REVEL, Rosetta, Foldetta, premPS, and FATHMM; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. FoldX reports an uncertain outcome. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the balance of evidence favors a pathogenic effect for the variant, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.086953 | Structured | 0.361944 | Uncertain | 0.961 | 0.357 | 0.000 | -15.940 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 0.50 | Ambiguous | 0.2 | 0.21 | Likely Benign | 0.36 | Likely Benign | 0.21 | Likely Benign | 0.485 | Likely Benign | -8.97 | Deleterious | 0.999 | Probably Damaging | 0.955 | Probably Damaging | 3.02 | Benign | 0.00 | Affected | 0.0907 | 0.4339 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.194A>C | H65P 2D  AIThe SynGAP1 missense variant H65P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT. AlphaMissense‑Default is uncertain, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for H65P, and this conclusion does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.476188 | Uncertain | 0.458 | 0.758 | 0.125 | -1.732 | Likely Benign | 0.479 | Ambiguous | Likely Benign | 0.103 | Likely Benign | -1.47 | Neutral | 0.676 | Possibly Damaging | 0.227 | Benign | 4.16 | Benign | 0.00 | Affected | 0.1915 | 0.3701 | 0 | -2 | 1.6 | -40.02 | |||||||||||||||||||||||||||||||||||||||
| c.194A>G | H65R 2D  AIThe SynGAP1 missense variant H65R is listed in ClinVar with an “Uncertain” status and is present in gnomAD (ID 6‑33425802‑A‑G). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, whereas the SGM‑Consensus remains benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the ClinVar “Uncertain” designation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.476188 | Uncertain | 0.458 | 0.758 | 0.125 | Uncertain | 1 | 6-33425802-A-G | 1 | 6.20e-7 | -1.980 | Likely Benign | 0.967 | Likely Pathogenic | Likely Pathogenic | 0.073 | Likely Benign | -1.60 | Neutral | 0.462 | Possibly Damaging | 0.227 | Benign | 4.19 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1483 | 0.1671 | 2 | 0 | -1.3 | 19.05 | ||||||||||||||||||||||||||||||||
| c.194A>T | H65L 2D  AIThe SynGAP1 H65L missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that predict a pathogenic outcome are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is “Likely Benign.” AlphaMissense‑Optimized returns an uncertain result, and Foldetta stability analysis is unavailable. Overall, the majority of evidence points to a benign impact, and this conclusion is not contradicted by any ClinVar annotation. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.476188 | Uncertain | 0.458 | 0.758 | 0.125 | -1.889 | Likely Benign | 0.836 | Likely Pathogenic | Ambiguous | 0.159 | Likely Benign | -1.65 | Neutral | 0.462 | Possibly Damaging | 0.227 | Benign | 4.22 | Benign | 0.00 | Affected | 0.0718 | 0.4760 | -2 | -3 | 7.0 | -23.98 | |||||||||||||||||||||||||||||||||||||||
| c.1950T>A | N650K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N650K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, FoldX, Foldetta, and FATHMM, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results are reported by Rosetta and premPS. High‑accuracy assessments give a pathogenic verdict from AlphaMissense‑Optimized and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), while Foldetta predicts a benign effect on protein stability. Overall, the majority of evidence points to a pathogenic impact, and this conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.086953 | Structured | 0.361944 | Uncertain | 0.961 | 0.357 | 0.000 | -13.078 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | -0.33 | Likely Benign | 0.1 | 0.66 | Ambiguous | 0.17 | Likely Benign | 0.92 | Ambiguous | 0.269 | Likely Benign | -5.98 | Deleterious | 0.995 | Probably Damaging | 0.807 | Possibly Damaging | 3.02 | Benign | 0.03 | Affected | 3.37 | 30 | 0.3028 | 0.3360 | 0 | 1 | -0.4 | 14.07 | |||||||||||||||||||||||||||
| c.1950T>G | N650K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N650K is not reported in ClinVar and is present in gnomAD (ID 6‑33441209‑T‑G). Prediction tools that indicate a benign effect include REVEL, FoldX, FATHMM, and Foldetta, whereas a majority of tools predict pathogenicity: SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools (premPS and Rosetta) give uncertain results. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of predictions lean toward pathogenicity, indicating the variant is most likely pathogenic, and this is consistent with the lack of a ClinVar entry; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.086953 | Structured | 0.361944 | Uncertain | 0.961 | 0.357 | 0.000 | 6-33441209-T-G | -13.078 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | -0.33 | Likely Benign | 0.1 | 0.66 | Ambiguous | 0.17 | Likely Benign | 0.92 | Ambiguous | 0.269 | Likely Benign | -5.98 | Deleterious | 0.995 | Probably Damaging | 0.807 | Possibly Damaging | 3.02 | Benign | 0.03 | Affected | 3.37 | 30 | 0.3028 | 0.3360 | 0 | 1 | -0.4 | 14.07 | ||||||||||||||||||||||||||
| c.1951G>A | E651K 2D AIThe SynGAP1 E651K missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign calls (REVEL, FoldX, premPS, polyPhen‑2 HumVar, SIFT, FATHMM) and pathogenic calls (PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default). Three tools (Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized is uncertain; the SGM Consensus—derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN—labels the variant as likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, remains uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.365409 | Uncertain | 0.955 | 0.340 | 0.000 | -8.714 | Likely Pathogenic | 0.818 | Likely Pathogenic | Ambiguous | 0.11 | Likely Benign | 0.4 | 1.15 | Ambiguous | 0.63 | Ambiguous | 0.08 | Likely Benign | 0.211 | Likely Benign | -2.92 | Deleterious | 0.921 | Possibly Damaging | 0.303 | Benign | 3.39 | Benign | 0.17 | Tolerated | 0.2768 | 0.5803 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1951G>C | E651Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E651Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM—all classifying the change as benign. No tool predicts a pathogenic outcome. The high‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign effect; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign result. Consequently, the variant is most likely benign based on the available predictions, and this assessment does not contradict any ClinVar status (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.088832 | Structured | 0.365409 | Uncertain | 0.955 | 0.340 | 0.000 | -6.308 | Likely Benign | 0.476 | Ambiguous | Likely Benign | 0.13 | Likely Benign | 0.1 | 0.15 | Likely Benign | 0.14 | Likely Benign | -0.13 | Likely Benign | 0.158 | Likely Benign | -1.23 | Neutral | 0.244 | Benign | 0.075 | Benign | 3.34 | Benign | 0.58 | Tolerated | 0.1438 | 0.5893 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1952A>C | E651A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E651A missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and Foldetta. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Rosetta is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicting it as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicting a benign impact. Overall, the balance of evidence leans toward a benign classification, and this conclusion does not contradict the absence of a ClinVar record. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.365409 | Uncertain | 0.955 | 0.340 | 0.000 | -8.694 | Likely Pathogenic | 0.610 | Likely Pathogenic | Likely Benign | 0.38 | Likely Benign | 0.1 | 0.52 | Ambiguous | 0.45 | Likely Benign | 0.23 | Likely Benign | 0.396 | Likely Benign | -4.54 | Deleterious | 0.986 | Probably Damaging | 0.875 | Possibly Damaging | 3.33 | Benign | 0.13 | Tolerated | 0.3926 | 0.5551 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1952A>G | E651G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E651G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and AlphaMissense‑Default. Predictions that are uncertain or unavailable are FoldX, Rosetta, ESM1b, and Foldetta. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as unavailable. Overall, the majority of tools (five benign vs. four pathogenic) lean toward a benign interpretation, and this does not contradict the lack of ClinVar annotation. Thus, based on the available predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.088832 | Structured | 0.365409 | Uncertain | 0.955 | 0.340 | 0.000 | -7.596 | In-Between | 0.591 | Likely Pathogenic | Likely Benign | 0.66 | Ambiguous | 0.1 | 1.61 | Ambiguous | 1.14 | Ambiguous | 0.32 | Likely Benign | 0.444 | Likely Benign | -4.78 | Deleterious | 0.999 | Probably Damaging | 0.935 | Probably Damaging | 3.31 | Benign | 0.06 | Tolerated | 0.2908 | 0.4488 | 0 | -2 | 3.1 | -72.06 | ||||||||||||||||||||||||||||||
| c.1952A>T | E651V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E651V missense change is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools split in their assessment: benign‑predicted scores include REVEL, Rosetta, Foldetta, premPS, and FATHMM, while pathogenic‑predicted scores come from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized and FoldX give uncertain results. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized is uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, leans pathogenic (3 pathogenic vs. 1 benign); Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a benign effect. Overall, the majority of tools (including the high‑accuracy SGM Consensus) suggest a pathogenic impact, whereas Foldetta and several other predictors indicate benign. The variant is most likely pathogenic based on the prevailing predictions, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.088832 | Structured | 0.365409 | Uncertain | 0.955 | 0.340 | 0.000 | -10.025 | Likely Pathogenic | 0.865 | Likely Pathogenic | Ambiguous | 0.63 | Ambiguous | 0.1 | 0.25 | Likely Benign | 0.44 | Likely Benign | 0.21 | Likely Benign | 0.467 | Likely Benign | -5.53 | Deleterious | 0.988 | Probably Damaging | 0.734 | Possibly Damaging | 3.27 | Benign | 0.05 | Affected | 0.0824 | 0.6136 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1953G>C | E651D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E651D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen2_HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only polyPhen2_HumDiv predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores the variant as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign effect; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a benign prediction. No prediction tool or stability analysis is inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.088832 | Structured | 0.365409 | Uncertain | 0.955 | 0.340 | 0.000 | -3.220 | Likely Benign | 0.231 | Likely Benign | Likely Benign | 0.16 | Likely Benign | 0.1 | 0.33 | Likely Benign | 0.25 | Likely Benign | -0.29 | Likely Benign | 0.153 | Likely Benign | -0.13 | Neutral | 0.906 | Possibly Damaging | 0.429 | Benign | 3.48 | Benign | 0.53 | Tolerated | 0.1813 | 0.3762 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1953G>T | E651D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E651D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen2_HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. Only polyPhen2_HumDiv predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized scores the variant as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign effect; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also reports a benign prediction. No prediction tool or stability analysis is inconclusive or missing. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.088832 | Structured | 0.365409 | Uncertain | 0.955 | 0.340 | 0.000 | -3.220 | Likely Benign | 0.231 | Likely Benign | Likely Benign | 0.16 | Likely Benign | 0.1 | 0.33 | Likely Benign | 0.25 | Likely Benign | -0.29 | Likely Benign | 0.153 | Likely Benign | -0.13 | Neutral | 0.906 | Possibly Damaging | 0.429 | Benign | 3.48 | Benign | 0.53 | Tolerated | 0.1813 | 0.3762 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1954T>A | F652I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F652I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. No prediction or stability assessment is missing or inconclusive. Based on the consensus of the available tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.356594 | Uncertain | 0.966 | 0.338 | 0.000 | -12.085 | Likely Pathogenic | 0.991 | Likely Pathogenic | Likely Pathogenic | 2.15 | Destabilizing | 0.1 | 3.40 | Destabilizing | 2.78 | Destabilizing | 1.40 | Destabilizing | 0.574 | Likely Pathogenic | -5.71 | Deleterious | 0.996 | Probably Damaging | 0.776 | Possibly Damaging | 3.06 | Benign | 0.00 | Affected | 0.1553 | 0.1955 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1954T>C | F652L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F652L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions are made by REVEL and FATHMM, whereas the remaining evaluated algorithms (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) all indicate pathogenicity. Stability‑based assessments from FoldX and Rosetta are inconclusive, and Foldetta likewise yields an uncertain result. High‑accuracy methods give a clearer picture: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labels the variant as likely pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for F652L. This conclusion is consistent with the absence of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.356594 | Uncertain | 0.966 | 0.338 | 0.000 | -9.026 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.31 | Ambiguous | 0.1 | 1.88 | Ambiguous | 1.60 | Ambiguous | 1.35 | Destabilizing | 0.467 | Likely Benign | -5.65 | Deleterious | 0.997 | Probably Damaging | 0.619 | Possibly Damaging | 3.09 | Benign | 0.00 | Affected | 0.1750 | 0.2926 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1954T>G | F652V 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F652V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, whereas only FATHMM predicts a benign outcome. The high‑accuracy subset confirms this trend: AlphaMissense‑Optimized is pathogenic; SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is pathogenic. No prediction or stability result is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.356594 | Uncertain | 0.966 | 0.338 | 0.000 | -11.576 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 2.66 | Destabilizing | 0.1 | 3.21 | Destabilizing | 2.94 | Destabilizing | 1.49 | Destabilizing | 0.595 | Likely Pathogenic | -6.71 | Deleterious | 0.995 | Probably Damaging | 0.885 | Possibly Damaging | 3.05 | Benign | 0.00 | Affected | 0.1656 | 0.1783 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1955T>A | F652Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F652Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, polyPhen‑2 HumVar, ESM1b, and FATHMM, while those that agree on a pathogenic effect are premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. Three tools (FoldX, Foldetta, AlphaMissense‑Optimized) give uncertain results. High‑accuracy methods provide no definitive verdict: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a tie (2 pathogenic, 2 benign) and thus inconclusive; Foldetta is uncertain. Consequently, the overall prediction landscape is balanced, with an equal number of benign and pathogenic calls and several uncertain results. The variant is therefore most likely **inconclusive** in terms of pathogenicity, and this lack of consensus does not contradict any ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.098513 | Structured | 0.356594 | Uncertain | 0.966 | 0.338 | 0.000 | -5.437 | Likely Benign | 0.890 | Likely Pathogenic | Ambiguous | 1.05 | Ambiguous | 0.2 | 0.27 | Likely Benign | 0.66 | Ambiguous | 1.24 | Destabilizing | 0.363 | Likely Benign | -2.92 | Deleterious | 0.957 | Probably Damaging | 0.390 | Benign | 3.13 | Benign | 0.00 | Affected | 0.1085 | 0.1584 | 7 | 3 | -4.1 | 16.00 | ||||||||||||||||||||||||||||||
| c.1955T>C | F652S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F652S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.356594 | Uncertain | 0.966 | 0.338 | 0.000 | -13.679 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 3.16 | Destabilizing | 0.2 | 4.92 | Destabilizing | 4.04 | Destabilizing | 2.16 | Destabilizing | 0.665 | Likely Pathogenic | -7.78 | Deleterious | 1.000 | Probably Damaging | 0.948 | Probably Damaging | 3.05 | Benign | 0.00 | Affected | 0.3777 | 0.0200 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1955T>G | F652C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F652C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all indicate pathogenicity, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the change as pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic effect. No prediction is inconclusive or missing. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.356594 | Uncertain | 0.966 | 0.338 | 0.000 | -12.023 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.27 | Destabilizing | 0.1 | 4.35 | Destabilizing | 3.81 | Destabilizing | 2.54 | Destabilizing | 0.695 | Likely Pathogenic | -7.74 | Deleterious | 1.000 | Probably Damaging | 0.983 | Probably Damaging | 2.99 | Benign | 0.00 | Affected | 0.2293 | 0.0783 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.1956T>A | F652L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F652L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions are made by REVEL and FATHMM, whereas the remaining evaluated algorithms (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) all indicate pathogenicity. Stability‑based assessments from FoldX and Rosetta are inconclusive, and Foldetta likewise yields an uncertain result. High‑accuracy methods give a clearer picture: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labels the variant as likely pathogenic, and Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for F652L. This conclusion is consistent with the absence of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.356594 | Uncertain | 0.966 | 0.338 | 0.000 | -9.026 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.31 | Ambiguous | 0.1 | 1.88 | Ambiguous | 1.60 | Ambiguous | 1.35 | Destabilizing | 0.306 | Likely Benign | -5.65 | Deleterious | 0.997 | Probably Damaging | 0.619 | Possibly Damaging | 3.09 | Benign | 0.00 | Affected | 0.1750 | 0.2926 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1956T>G | F652L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F652L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) indicate a pathogenic impact. FoldX and Rosetta give uncertain results, and Foldetta likewise reports an uncertain stability change. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) labeling it Likely Pathogenic, and Foldetta remaining uncertain. Overall, the preponderance of evidence from multiple pathogenic‑predicting tools and the high‑accuracy methods points to a likely pathogenic effect for F652L, with no conflict from ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.356594 | Uncertain | 0.966 | 0.338 | 0.000 | -9.026 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 1.31 | Ambiguous | 0.1 | 1.88 | Ambiguous | 1.60 | Ambiguous | 1.35 | Destabilizing | 0.305 | Likely Benign | -5.65 | Deleterious | 0.997 | Probably Damaging | 0.619 | Possibly Damaging | 3.09 | Benign | 0.00 | Affected | 0.1750 | 0.2926 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1957C>A | L653M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L653M has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, FATHMM, and AlphaMissense‑Optimized, while those that predict a pathogenic outcome are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Tools with uncertain or missing results (FoldX, Rosetta, Foldetta, premPS) are not considered evidence for either side. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (two pathogenic vs. two benign votes), and Foldetta is also inconclusive. Overall, the majority of standard predictors lean toward pathogenicity, but the most reliable high‑accuracy tool indicates a benign effect, leaving the variant’s impact uncertain. No ClinVar entry exists, so there is no contradiction with clinical annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.049374 | Structured | 0.335213 | Uncertain | 0.963 | 0.332 | 0.000 | -8.838 | Likely Pathogenic | 0.713 | Likely Pathogenic | Likely Benign | 0.99 | Ambiguous | 0.2 | 1.70 | Ambiguous | 1.35 | Ambiguous | 0.94 | Ambiguous | 0.259 | Likely Benign | -1.76 | Neutral | 1.000 | Probably Damaging | 0.991 | Probably Damaging | 3.13 | Benign | 0.01 | Affected | 0.0935 | 0.3227 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.1957C>G | L653V 2D AIThe SynGAP1 missense variant L653V is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that classify the variant as benign include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are FoldX, Rosetta, and premPS, while ESM1b is inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as pathogenic. Overall, the majority of evidence points to a benign impact, and this does not contradict the ClinVar “Uncertain” classification. Thus, based on current predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.049374 | Structured | 0.335213 | Uncertain | 0.963 | 0.332 | 0.000 | Uncertain | 1 | -7.050 | In-Between | 0.301 | Likely Benign | Likely Benign | 3.28 | Destabilizing | 0.3 | 2.18 | Destabilizing | 2.73 | Destabilizing | 1.32 | Destabilizing | 0.146 | Likely Benign | -2.25 | Neutral | 0.227 | Benign | 0.039 | Benign | 3.28 | Benign | 0.08 | Tolerated | 0.1436 | 0.3557 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||
| c.1958T>A | L653Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L653Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity largely agree: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (both HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a deleterious effect. Only FATHMM predicts a benign outcome. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No predictions are missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.049374 | Structured | 0.335213 | Uncertain | 0.963 | 0.332 | 0.000 | -14.347 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 2.71 | Destabilizing | 0.1 | 3.26 | Destabilizing | 2.99 | Destabilizing | 1.98 | Destabilizing | 0.614 | Likely Pathogenic | -5.65 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 3.09 | Benign | 0.01 | Affected | 0.1195 | 0.1363 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.1958T>C | L653P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L653P is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy methods give the following results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.049374 | Structured | 0.335213 | Uncertain | 0.963 | 0.332 | 0.000 | -17.675 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 6.02 | Destabilizing | 0.8 | 9.20 | Destabilizing | 7.61 | Destabilizing | 2.56 | Destabilizing | 0.700 | Likely Pathogenic | -6.51 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.09 | Benign | 0.00 | Affected | 0.3251 | 0.1874 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.1958T>G | L653R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L653R lies in the GAP domain. ClinVar has no entry for this variant, and it is not reported in gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) uniformly predict a pathogenic impact. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts a pathogenic effect. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.049374 | Structured | 0.335213 | Uncertain | 0.963 | 0.332 | 0.000 | -16.078 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 6.92 | Destabilizing | 1.1 | 5.84 | Destabilizing | 6.38 | Destabilizing | 2.55 | Destabilizing | 0.649 | Likely Pathogenic | -5.75 | Deleterious | 0.997 | Probably Damaging | 0.806 | Possibly Damaging | 3.09 | Benign | 0.00 | Affected | 0.1501 | 0.0805 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.195C>A | H65Q 2D  AIThe SynGAP1 missense variant H65Q is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33425803‑C‑A). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as “Likely Benign.” High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain,” SGM‑Consensus as “Likely Benign,” and Foldetta results are unavailable. Based on the overall distribution of predictions, the variant is most likely benign, and this assessment does not contradict the ClinVar status (which has no entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.476188 | Uncertain | 0.458 | 0.758 | 0.125 | 6-33425803-C-A | 1 | 6.20e-7 | -2.966 | Likely Benign | 0.953 | Likely Pathogenic | Ambiguous | 0.035 | Likely Benign | -1.46 | Neutral | 0.462 | Possibly Damaging | 0.227 | Benign | 4.19 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1059 | 0.2900 | 0 | 3 | -0.3 | -9.01 | ||||||||||||||||||||||||||||||||||
| c.195C>G | H65Q 2D AIThe SynGAP1 H65Q missense variant has no ClinVar record and is not listed in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and AlphaMissense‑Default. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is benign, while AlphaMissense‑Optimized is uncertain. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for H65Q. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.411940 | Structured | 0.476188 | Uncertain | 0.458 | 0.758 | 0.125 | -2.966 | Likely Benign | 0.953 | Likely Pathogenic | Ambiguous | 0.035 | Likely Benign | -1.46 | Neutral | 0.462 | Possibly Damaging | 0.227 | Benign | 4.19 | Benign | 0.00 | Affected | 4.32 | 1 | 0.1059 | 0.2900 | 0 | 3 | -0.3 | -9.01 | |||||||||||||||||||||||||||||||||||||
| c.1960G>A | E654K 2D AIThe SynGAP1 missense variant E654K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, polyPhen‑2 HumVar, SIFT, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Pathogenic). The high‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, Foldetta predicts benign, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenic. Overall, the majority of high‑confidence predictions lean toward pathogenicity, and the variant is not contradicted by any ClinVar annotation. Thus, based on the available computational evidence, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.026892 | Structured | 0.303029 | Uncertain | 0.957 | 0.311 | 0.000 | -12.587 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 0.12 | Likely Benign | 0.3 | 0.53 | Ambiguous | 0.33 | Likely Benign | -0.17 | Likely Benign | 0.435 | Likely Benign | -3.80 | Deleterious | 0.921 | Possibly Damaging | 0.303 | Benign | 3.44 | Benign | 0.11 | Tolerated | 0.2365 | 0.4224 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1960G>C | E654Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E654Q missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), and FATHMM, while those that predict a pathogenic outcome are SGM‑Consensus, PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicating likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicting a benign effect. Overall, the majority of tools (nine benign vs. five pathogenic) suggest the variant is most likely benign. This conclusion does not contradict ClinVar status, as no ClinVar assertion is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.026892 | Structured | 0.303029 | Uncertain | 0.957 | 0.311 | 0.000 | -10.368 | Likely Pathogenic | 0.699 | Likely Pathogenic | Likely Benign | 0.00 | Likely Benign | 0.0 | 0.18 | Likely Benign | 0.09 | Likely Benign | -0.12 | Likely Benign | 0.298 | Likely Benign | -2.79 | Deleterious | 0.244 | Benign | 0.075 | Benign | 3.33 | Benign | 0.02 | Affected | 0.1140 | 0.3913 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.1961A>C | E654A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E654A has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign predictions come from FoldX, Rosetta, Foldetta, premPS, and FATHMM, while pathogenic predictions arise from REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta as Benign. Overall, the balance of evidence favors pathogenicity, and this conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.026892 | Structured | 0.303029 | Uncertain | 0.957 | 0.311 | 0.000 | -10.797 | Likely Pathogenic | 0.873 | Likely Pathogenic | Ambiguous | 0.49 | Likely Benign | 0.1 | 0.20 | Likely Benign | 0.35 | Likely Benign | 0.25 | Likely Benign | 0.512 | Likely Pathogenic | -5.70 | Deleterious | 0.986 | Probably Damaging | 0.875 | Possibly Damaging | 3.34 | Benign | 0.02 | Affected | 0.3367 | 0.3971 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1961A>G | E654G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E654G missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on benign impact are premPS and FATHMM, while the majority (REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus) predict pathogenicity. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E654G. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.026892 | Structured | 0.303029 | Uncertain | 0.957 | 0.311 | 0.000 | -12.487 | Likely Pathogenic | 0.898 | Likely Pathogenic | Ambiguous | 1.29 | Ambiguous | 0.2 | 1.62 | Ambiguous | 1.46 | Ambiguous | 0.34 | Likely Benign | 0.547 | Likely Pathogenic | -6.73 | Deleterious | 0.999 | Probably Damaging | 0.935 | Probably Damaging | 3.26 | Benign | 0.01 | Affected | 0.2818 | 0.3109 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1961A>T | E654V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E654V missense variant is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include Rosetta, Foldetta, premPS, and FATHMM, while those that agree on a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX and AlphaMissense‑Optimized are inconclusive. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta predicts benign. Overall, the majority of tools (8 pathogenic vs. 4 benign) and the SGM‑Consensus result support a pathogenic classification. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.026892 | Structured | 0.303029 | Uncertain | 0.957 | 0.311 | 0.000 | -12.306 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 0.55 | Ambiguous | 0.0 | -0.33 | Likely Benign | 0.11 | Likely Benign | 0.21 | Likely Benign | 0.540 | Likely Pathogenic | -6.66 | Deleterious | 0.988 | Probably Damaging | 0.734 | Possibly Damaging | 3.31 | Benign | 0.01 | Affected | 0.0686 | 0.4757 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1962G>C | E654D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E654D missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. Two tools give uncertain results: Rosetta and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the predictions are mixed, with a slight majority leaning toward benign, but the high‑accuracy tools conflict. The variant is most likely benign based on the aggregate predictions, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.026892 | Structured | 0.303029 | Uncertain | 0.957 | 0.311 | 0.000 | -10.454 | Likely Pathogenic | 0.886 | Likely Pathogenic | Ambiguous | 0.44 | Likely Benign | 0.0 | 0.52 | Ambiguous | 0.48 | Likely Benign | 0.29 | Likely Benign | 0.166 | Likely Benign | -2.87 | Deleterious | 0.906 | Possibly Damaging | 0.429 | Benign | 3.38 | Benign | 0.11 | Tolerated | 0.1678 | 0.2583 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1962G>T | E654D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E654D has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, FATHMM, and polyPhen‑2 HumVar. Those that predict a pathogenic effect are SGM Consensus, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show that AlphaMissense‑Optimized is uncertain (treated as unavailable), SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts a benign effect. No other folding‑stability results are available. Overall, the balance of evidence leans toward a benign interpretation, and this assessment does not contradict ClinVar status, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.026892 | Structured | 0.303029 | Uncertain | 0.957 | 0.311 | 0.000 | -10.454 | Likely Pathogenic | 0.886 | Likely Pathogenic | Ambiguous | 0.44 | Likely Benign | 0.0 | 0.52 | Ambiguous | 0.48 | Likely Benign | 0.29 | Likely Benign | 0.166 | Likely Benign | -2.87 | Deleterious | 0.906 | Possibly Damaging | 0.429 | Benign | 3.38 | Benign | 0.11 | Tolerated | 0.1678 | 0.2583 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1963C>A | L655M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L655M is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify the change as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts pathogenicity. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign; the SGM Consensus indicates “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Based on the unanimous benign predictions and the absence of any pathogenic calls, the variant is most likely benign, and this conclusion does not contradict the ClinVar status (which has no entry). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.015344 | Structured | 0.268808 | Uncertain | 0.961 | 0.274 | 0.000 | -3.077 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.0 | 0.25 | Likely Benign | 0.23 | Likely Benign | -0.22 | Likely Benign | 0.076 | Likely Benign | 0.58 | Neutral | 0.048 | Benign | 0.037 | Benign | 3.43 | Benign | 0.18 | Tolerated | 0.0979 | 0.3252 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||
| c.1963C>G | L655V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L655V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, premPS, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, PROVEAN, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also reports a likely benign outcome. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign, SGM‑Consensus indicates likely benign, while Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain result. No tools predict pathogenicity, and the uncertain stability assessment does not alter the overall benign inference. Therefore, based on the available predictions, the variant is most likely benign, and this conclusion is consistent with the absence of a ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.015344 | Structured | 0.268808 | Uncertain | 0.961 | 0.274 | 0.000 | -6.743 | Likely Benign | 0.127 | Likely Benign | Likely Benign | 0.78 | Ambiguous | 0.0 | 0.58 | Ambiguous | 0.68 | Ambiguous | 0.38 | Likely Benign | 0.058 | Likely Benign | -0.62 | Neutral | 0.008 | Benign | 0.003 | Benign | 3.45 | Benign | 0.44 | Tolerated | 0.1551 | 0.3190 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1964T>C | L655P 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L655P is catalogued in gnomAD (ID 6‑33441223‑T‑C) but has no ClinVar entry. Functional prediction tools show a split: benign calls come from REVEL, PROVEAN, SIFT, and FATHMM, whereas pathogenic calls are made by Rosetta, Foldetta, both polyPhen‑2 versions, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; FoldX and premPS are uncertain. High‑accuracy assessments give AlphaMissense‑Optimized as pathogenic, Foldetta as pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive. Overall, the majority of evidence points to a pathogenic effect. This conclusion is consistent with the absence of a ClinVar classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.015344 | Structured | 0.268808 | Uncertain | 0.961 | 0.274 | 0.000 | 6-33441223-T-C | 1 | 6.20e-7 | -9.771 | Likely Pathogenic | 0.982 | Likely Pathogenic | Likely Pathogenic | 1.33 | Ambiguous | 0.5 | 6.52 | Destabilizing | 3.93 | Destabilizing | 0.57 | Ambiguous | 0.126 | Likely Benign | -1.36 | Neutral | 0.981 | Probably Damaging | 0.772 | Possibly Damaging | 3.47 | Benign | 0.32 | Tolerated | 3.39 | 24 | 0.3598 | 0.1274 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||
| c.1964T>G | L655R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L655R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools uniformly indicate a benign effect: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, and the majority of consensus methods (AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all score the variant as benign. The only inconclusive result comes from Rosetta, which is treated as unavailable. High‑accuracy assessments reinforce this benign prediction: AlphaMissense‑Optimized reports benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Thus, based on the available predictions, the variant is most likely benign, with no conflict with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.015344 | Structured | 0.268808 | Uncertain | 0.961 | 0.274 | 0.000 | -4.848 | Likely Benign | 0.284 | Likely Benign | Likely Benign | -0.40 | Likely Benign | 0.0 | 0.54 | Ambiguous | 0.07 | Likely Benign | -0.09 | Likely Benign | 0.160 | Likely Benign | 0.46 | Neutral | 0.006 | Benign | 0.001 | Benign | 3.50 | Benign | 0.60 | Tolerated | 0.1589 | 0.0615 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.1966G>A | E656K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E656K has no ClinVar entry and is absent from gnomAD. Prediction tools that classify it as benign include Rosetta, premPS, and FATHMM. Those that predict pathogenicity are REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; the SGM‑Consensus score is “Likely Pathogenic.” FoldX and Foldetta return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates likely pathogenic, while Foldetta’s stability prediction is inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this assessment does not conflict with the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.032017 | Structured | 0.242242 | Uncertain | 0.963 | 0.264 | 0.000 | -13.833 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | -1.06 | Ambiguous | 0.0 | 0.02 | Likely Benign | -0.52 | Ambiguous | 0.16 | Likely Benign | 0.502 | Likely Pathogenic | -3.49 | Deleterious | 0.985 | Probably Damaging | 0.553 | Possibly Damaging | 3.44 | Benign | 0.03 | Affected | 0.3001 | 0.6406 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1967A>C | E656A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E656A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as likely pathogenic. High‑accuracy assessments further reveal that AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus also indicates likely pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. No prediction or stability result is missing or inconclusive. Based on the overall evidence, the variant is most likely pathogenic; this assessment does not contradict ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.032017 | Structured | 0.242242 | Uncertain | 0.963 | 0.264 | 0.000 | -14.373 | Likely Pathogenic | 0.974 | Likely Pathogenic | Likely Pathogenic | 0.12 | Likely Benign | 0.0 | 0.10 | Likely Benign | 0.11 | Likely Benign | -0.13 | Likely Benign | 0.499 | Likely Benign | -5.38 | Deleterious | 0.985 | Probably Damaging | 0.755 | Possibly Damaging | 3.46 | Benign | 0.03 | Affected | 0.4244 | 0.6271 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1967A>G | E656G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E656G missense variant has no ClinVar entry and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Only premPS and FATHMM predict a benign outcome, while FoldX and Foldetta are inconclusive. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenicity; Foldetta remains uncertain. Overall, the preponderance of evidence points to a pathogenic effect for E656G, and this conclusion is not contradicted by ClinVar status, which currently lacks an entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.032017 | Structured | 0.242242 | Uncertain | 0.963 | 0.264 | 0.000 | -14.112 | Likely Pathogenic | 0.978 | Likely Pathogenic | Likely Pathogenic | 0.96 | Ambiguous | 0.1 | 2.02 | Destabilizing | 1.49 | Ambiguous | 0.22 | Likely Benign | 0.534 | Likely Pathogenic | -6.28 | Deleterious | 1.000 | Probably Damaging | 0.941 | Probably Damaging | 3.44 | Benign | 0.05 | Affected | 0.3216 | 0.5208 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.1967A>T | E656V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E656V has no ClinVar entry and is not reported in gnomAD. Prediction tools that classify the variant as benign include FoldX, Rosetta, Foldetta, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts benign. Overall, the majority of tools (8 of 13) indicate a pathogenic impact, and this conclusion does not contradict any ClinVar status, as none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.032017 | Structured | 0.242242 | Uncertain | 0.963 | 0.264 | 0.000 | -15.252 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | -0.26 | Likely Benign | 0.0 | -0.10 | Likely Benign | -0.18 | Likely Benign | -0.77 | Ambiguous | 0.509 | Likely Pathogenic | -6.38 | Deleterious | 0.784 | Possibly Damaging | 0.223 | Benign | 3.46 | Benign | 0.02 | Affected | 0.0990 | 0.7057 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1968A>C | E656D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant E656D has no ClinVar entry and is not reported in gnomAD. Prediction tools show mixed results: benign predictions come from REVEL, FoldX, premPS, polyPhen‑2 HumVar, SIFT, and FATHMM, while pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts Pathogenic, SGM‑Consensus confirms Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is Uncertain. No evidence from Foldetta or Rosetta is available to refute pathogenicity. Overall, the majority of high‑confidence tools predict a pathogenic impact, and this is consistent with the absence of ClinVar annotation; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.032017 | Structured | 0.242242 | Uncertain | 0.963 | 0.264 | 0.000 | -11.992 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.22 | Likely Benign | 0.1 | 1.02 | Ambiguous | 0.62 | Ambiguous | 0.39 | Likely Benign | 0.275 | Likely Benign | -2.72 | Deleterious | 0.985 | Probably Damaging | 0.426 | Benign | 3.41 | Benign | 0.06 | Tolerated | 0.2052 | 0.4120 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1968A>T | E656D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E656D missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools cluster into two groups: benign calls (REVEL, FoldX, premPS, polyPhen‑2 HumVar, SIFT, FATHMM) and pathogenic calls (PROVEAN, polyPhen‑2 HumDiv, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized). The SGM‑Consensus score is “Likely Pathogenic,” while Foldetta and Rosetta outputs are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates pathogenicity, and Foldetta remains inconclusive. Overall, the majority of evidence points to a pathogenic effect, and this conclusion does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.032017 | Structured | 0.242242 | Uncertain | 0.963 | 0.264 | 0.000 | -11.992 | Likely Pathogenic | 0.990 | Likely Pathogenic | Likely Pathogenic | 0.22 | Likely Benign | 0.1 | 1.02 | Ambiguous | 0.62 | Ambiguous | 0.39 | Likely Benign | 0.273 | Likely Benign | -2.72 | Deleterious | 0.985 | Probably Damaging | 0.426 | Benign | 3.41 | Benign | 0.06 | Tolerated | 0.2052 | 0.4120 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.1969T>A | W657R 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W657R is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools show a split: benign calls come from REVEL, SIFT, and FATHMM, whereas pathogenic calls are made by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus score, which is labeled Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates Likely Pathogenic, and the Foldetta stability analysis is inconclusive. Overall, the majority of evidence points to a pathogenic impact for W657R, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | -13.391 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 1.56 | Ambiguous | 0.2 | 1.64 | Ambiguous | 1.60 | Ambiguous | 1.29 | Destabilizing | 0.461 | Likely Benign | -11.96 | Deleterious | 0.999 | Probably Damaging | 0.964 | Probably Damaging | 3.48 | Benign | 0.07 | Tolerated | 0.4684 | 0.0000 | 2 | -3 | -3.6 | -30.03 | |||||||||||||||||||||||||||||
| c.1969T>C | W657R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W657R is not reported in ClinVar and has no entry in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, SIFT, and FATHMM, while pathogenic predictions are made by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is classified as Likely Pathogenic. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority of the four high‑accuracy tools) is pathogenic, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive. FoldX and Rosetta individually report uncertain effects on protein stability. Overall, the preponderance of evidence from both general and high‑accuracy predictors indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | -13.391 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 1.56 | Ambiguous | 0.2 | 1.64 | Ambiguous | 1.60 | Ambiguous | 1.29 | Destabilizing | 0.461 | Likely Benign | -11.96 | Deleterious | 0.999 | Probably Damaging | 0.964 | Probably Damaging | 3.48 | Benign | 0.07 | Tolerated | 0.4684 | 0.0000 | 2 | -3 | -3.6 | -30.03 | |||||||||||||||||||||||||||||
| c.1969T>G | W657G 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W657G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM. In contrast, the majority of other in silico predictors (AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FoldX, Rosetta, premPS, Foldetta) all classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. No predictions are missing or inconclusive. Based on the preponderance of evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | -12.559 | Likely Pathogenic | 0.968 | Likely Pathogenic | Likely Pathogenic | 3.04 | Destabilizing | 0.2 | 2.80 | Destabilizing | 2.92 | Destabilizing | 1.28 | Destabilizing | 0.389 | Likely Benign | -11.16 | Deleterious | 0.999 | Probably Damaging | 0.941 | Probably Damaging | 3.46 | Benign | 0.15 | Tolerated | 0.4470 | 0.0885 | -7 | -2 | 0.5 | -129.16 | |||||||||||||||||||||||||||||
| c.196C>A | P66T 2D AIThe SynGAP1 missense variant P66T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which classifies the variant as Likely Benign. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The high‑accuracy AlphaMissense‑Optimized result is uncertain, and the Foldetta protein‑folding stability assessment is unavailable. Overall, the balance of evidence leans toward a benign impact, with one high‑accuracy tool (SGM‑Consensus) supporting this view and no ClinVar entry to contradict it. Thus, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.394753 | Structured | 0.474132 | Uncertain | 0.455 | 0.762 | 0.125 | -3.373 | Likely Benign | 0.954 | Likely Pathogenic | Ambiguous | 0.139 | Likely Benign | -1.81 | Neutral | 0.909 | Possibly Damaging | 0.641 | Possibly Damaging | 4.00 | Benign | 0.00 | Affected | 0.1747 | 0.5866 | 0 | -1 | 0.9 | 3.99 | |||||||||||||||||||||||||||||||||||||||
| c.196C>G | P66A 2D AIThe SynGAP1 P66A missense variant (ClinVar ID 1303518.0) is listed as “Uncertain” and is not reported in gnomAD. Functional prediction tools that agree on benign impact include REVEL, PROVEAN, ESM1b, and FATHMM, while polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default all predict pathogenicity. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” status. Separately, the high‑accuracy AlphaMissense‑Optimized result is “Uncertain,” the SGM‑Consensus remains “Likely Benign,” and Foldetta (a protein‑folding stability method combining FoldX‑MD and Rosetta outputs) has no available result for this variant. Overall, the predictions are mixed, but the majority of high‑confidence tools lean toward a benign effect. Thus, the variant is most likely benign based on current computational evidence, and this assessment does not contradict the ClinVar status of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.394753 | Structured | 0.474132 | Uncertain | 0.455 | 0.762 | 0.125 | Uncertain | 1 | -2.845 | Likely Benign | 0.891 | Likely Pathogenic | Ambiguous | 0.091 | Likely Benign | -1.56 | Neutral | 0.805 | Possibly Damaging | 0.539 | Possibly Damaging | 4.04 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3467 | 0.5138 | 1 | -1 | 3.4 | -26.04 | |||||||||||||||||||||||||||||||||||
| c.196C>T | P66S 2D AIThe SynGAP1 missense variant P66S is listed in ClinVar (ID 1915017.0) as benign and is present in gnomAD (variant ID 6‑33425804‑C‑T). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, while the SGM‑Consensus remains likely benign; Foldetta results are unavailable. Overall, the balance of evidence favors a benign interpretation, which is consistent with the ClinVar designation and does not contradict the reported status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.394753 | Structured | 0.474132 | Uncertain | 0.455 | 0.762 | 0.125 | Benign | 1 | 6-33425804-C-T | 2 | 1.24e-6 | -2.760 | Likely Benign | 0.929 | Likely Pathogenic | Ambiguous | 0.081 | Likely Benign | -1.69 | Neutral | 0.909 | Possibly Damaging | 0.641 | Possibly Damaging | 4.01 | Benign | 0.00 | Affected | 4.32 | 1 | 0.3417 | 0.5463 | 1 | -1 | 0.8 | -10.04 | ||||||||||||||||||||||||||||||||
| c.1970G>C | W657S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant W657S is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, SIFT, and FATHMM, whereas a majority of tools (SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta) predict a pathogenic impact; Rosetta remains uncertain. High‑accuracy methods all support pathogenicity: AlphaMissense‑Optimized scores the variant as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status, as no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | -11.817 | Likely Pathogenic | 0.986 | Likely Pathogenic | Likely Pathogenic | 2.27 | Destabilizing | 0.2 | 1.87 | Ambiguous | 2.07 | Destabilizing | 1.26 | Destabilizing | 0.334 | Likely Benign | -11.82 | Deleterious | 0.999 | Probably Damaging | 0.947 | Probably Damaging | 3.52 | Benign | 0.09 | Tolerated | 0.4299 | 0.0489 | -2 | -3 | 0.1 | -99.14 | |||||||||||||||||||||||||||||
| c.1970G>T | W657L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant W657L is listed in ClinVar with an uncertain significance (ClinVar ID 2767440.0) and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, FoldX, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Tools that agree on a pathogenic effect are PROVEAN, ESM1b, and AlphaMissense‑Default. Uncertain predictions come from Foldetta, premPS, and Rosetta. High‑accuracy assessments show AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, and Foldetta predicts a benign folding‑stability change. Overall, the majority of evidence points toward a pathogenic impact, which is consistent with the ClinVar uncertain status but leans toward pathogenic rather than benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | Uncertain | 1 | -14.411 | Likely Pathogenic | 0.960 | Likely Pathogenic | Likely Pathogenic | 0.14 | Likely Benign | 0.1 | 0.73 | Ambiguous | 0.44 | Likely Benign | 0.87 | Ambiguous | 0.213 | Likely Benign | -10.86 | Deleterious | 0.277 | Benign | 0.078 | Benign | 3.52 | Benign | 0.14 | Tolerated | 3.39 | 24 | 0.2302 | 0.2049 | -2 | -2 | 4.7 | -73.05 | |||||||||||||||||||||||||
| c.1971G>C | W657C 2D AISynGAP1 missense variant W657C is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools that classify the variant as benign include REVEL and FATHMM. Those that predict a deleterious effect are FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized; Rosetta reports an uncertain outcome. High‑accuracy assessments further support a damaging interpretation: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Overall, the preponderance of evidence indicates that W657C is most likely pathogenic, which does not contradict the current ClinVar status of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | Uncertain | 1 | -12.035 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.74 | Destabilizing | 0.3 | 1.69 | Ambiguous | 2.22 | Destabilizing | 1.30 | Destabilizing | 0.463 | Likely Benign | -11.06 | Deleterious | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 3.43 | Benign | 0.03 | Affected | 0.3834 | 0.0766 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||
| c.1971G>T | W657C 2D AIThe SynGAP1 W657C missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are REVEL and FATHMM, whereas the majority of tools (SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; Foldetta also supports pathogenicity, while Rosetta remains uncertain. High‑accuracy assessments further reinforce a deleterious outcome: AlphaMissense‑Optimized classifies the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts a pathogenic effect. Overall, the preponderance of evidence points to the variant being most likely pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.030611 | Structured | 0.208729 | Uncertain | 0.941 | 0.245 | 0.000 | -12.035 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 2.74 | Destabilizing | 0.3 | 1.69 | Ambiguous | 2.22 | Destabilizing | 1.30 | Destabilizing | 0.463 | Likely Benign | -11.06 | Deleterious | 1.000 | Probably Damaging | 0.982 | Probably Damaging | 3.43 | Benign | 0.03 | Affected | 0.3834 | 0.0766 | -8 | -2 | 3.4 | -83.07 | |||||||||||||||||||||||||||||
| c.1972G>A | G658S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G658S is reported in gnomAD (variant ID 6-33441231‑G‑A) but has no entry in ClinVar. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. No tool predicts pathogenicity; the only inconclusive result comes from Rosetta, which is treated as unavailable. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts benign. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.029376 | Structured | 0.180299 | Uncertain | 0.942 | 0.251 | 0.000 | 6-33441231-G-A | 8 | 4.96e-6 | -3.445 | Likely Benign | 0.077 | Likely Benign | Likely Benign | -0.12 | Likely Benign | 0.0 | -0.50 | Ambiguous | -0.31 | Likely Benign | -0.11 | Likely Benign | 0.070 | Likely Benign | -0.97 | Neutral | 0.209 | Benign | 0.087 | Benign | 3.58 | Benign | 0.43 | Tolerated | 3.39 | 24 | 0.2783 | 0.3576 | 0 | 1 | -0.4 | 30.03 | ||||||||||||||||||||||||
| c.1972G>C | G658R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G658R is not reported in ClinVar and has no entries in gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized, while pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar) and AlphaMissense‑Default. Four tools (FoldX, Foldetta, premPS, ESM1b) return uncertain results. High‑accuracy methods give a mixed signal: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic, and Foldetta is uncertain. Because the majority of conventional predictors lean benign and no ClinVar evidence contradicts this, the variant is most likely benign, though the conflicting high‑accuracy predictions leave some uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.029376 | Structured | 0.180299 | Uncertain | 0.942 | 0.251 | 0.000 | -7.725 | In-Between | 0.618 | Likely Pathogenic | Likely Benign | -1.17 | Ambiguous | 0.1 | -0.46 | Likely Benign | -0.82 | Ambiguous | 0.64 | Ambiguous | 0.120 | Likely Benign | -3.11 | Deleterious | 0.955 | Possibly Damaging | 0.591 | Possibly Damaging | 3.40 | Benign | 0.19 | Tolerated | 0.1157 | 0.3888 | -3 | -2 | -4.1 | 99.14 | ||||||||||||||||||||||||||||||
| c.1972G>T | G658C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G658C is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. One tool, ESM1b, yields an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also favors benign (2 benign vs. 1 pathogenic, 1 uncertain), and Foldetta predicts a benign impact on protein stability. Overall, the majority of evidence points to a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.029376 | Structured | 0.180299 | Uncertain | 0.942 | 0.251 | 0.000 | -7.577 | In-Between | 0.170 | Likely Benign | Likely Benign | 0.05 | Likely Benign | 0.0 | 0.04 | Likely Benign | 0.05 | Likely Benign | 0.46 | Likely Benign | 0.146 | Likely Benign | -3.49 | Deleterious | 0.989 | Probably Damaging | 0.544 | Possibly Damaging | 3.37 | Benign | 0.04 | Affected | 0.1509 | 0.3141 | -3 | -3 | 2.9 | 46.09 | ||||||||||||||||||||||||||||||
| c.1973G>C | G658A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G658A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly classify the substitution as benign: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict a neutral effect, and the SGM‑Consensus score indicates a likely benign outcome. No tool predicts pathogenicity. High‑accuracy assessments corroborate this view: AlphaMissense‑Optimized returns a benign prediction, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports a likely benign result, while Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, yields an uncertain outcome. Taken together, the evidence overwhelmingly supports a benign impact for G658A, and this conclusion does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.029376 | Structured | 0.180299 | Uncertain | 0.942 | 0.251 | 0.000 | -4.303 | Likely Benign | 0.083 | Likely Benign | Likely Benign | -0.34 | Likely Benign | 0.0 | -0.75 | Ambiguous | -0.55 | Ambiguous | -0.18 | Likely Benign | 0.072 | Likely Benign | -0.93 | Neutral | 0.002 | Benign | 0.001 | Benign | 3.47 | Benign | 0.36 | Tolerated | 0.3937 | 0.3352 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.1973G>T | G658V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G658V is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are PROVEAN and polyPhen‑2 HumDiv; Rosetta’s output is uncertain and therefore not used as evidence. High‑accuracy assessments show AlphaMissense‑Optimized as Benign, the SGM‑Consensus as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta) as Benign. Taken together, the majority of reliable predictors indicate a benign impact. Thus, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.029376 | Structured | 0.180299 | Uncertain | 0.942 | 0.251 | 0.000 | -6.815 | Likely Benign | 0.166 | Likely Benign | Likely Benign | -0.07 | Likely Benign | 0.0 | -0.71 | Ambiguous | -0.39 | Likely Benign | 0.34 | Likely Benign | 0.117 | Likely Benign | -3.23 | Deleterious | 0.841 | Possibly Damaging | 0.264 | Benign | 3.38 | Benign | 0.13 | Tolerated | 0.1337 | 0.3330 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.1975T>A | S659T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S659T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all classify the change as benign. Only Rosetta reports an uncertain outcome, which is treated as unavailable evidence. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, the SGM Consensus is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No tool predicts pathogenicity. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.067594 | Structured | 0.154597 | Uncertain | 0.954 | 0.283 | 0.000 | -5.693 | Likely Benign | 0.157 | Likely Benign | Likely Benign | -0.15 | Likely Benign | 0.0 | -0.74 | Ambiguous | -0.45 | Likely Benign | 0.42 | Likely Benign | 0.106 | Likely Benign | -2.04 | Neutral | 0.069 | Benign | 0.011 | Benign | 3.41 | Benign | 0.39 | Tolerated | 0.1505 | 0.5976 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.1975T>C | S659P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S659P is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are SGM‑Consensus, Rosetta, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. Tools with uncertain or inconclusive results—FoldX, premPS, and Foldetta—are treated as unavailable. High‑accuracy methods give mixed signals: AlphaMissense‑Optimized reports a benign change, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta’s stability assessment is uncertain. Overall, the balance of evidence leans toward a pathogenic effect, but the presence of a strong benign prediction from AlphaMissense‑Optimized and the lack of ClinVar annotation means the variant’s clinical significance remains uncertain and does not contradict existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.154597 | Uncertain | 0.954 | 0.283 | 0.000 | -10.461 | Likely Pathogenic | 0.609 | Likely Pathogenic | Likely Benign | -0.73 | Ambiguous | 0.3 | 2.98 | Destabilizing | 1.13 | Ambiguous | 0.73 | Ambiguous | 0.224 | Likely Benign | -3.26 | Deleterious | 0.932 | Possibly Damaging | 0.245 | Benign | 3.39 | Benign | 0.18 | Tolerated | 0.2207 | 0.5553 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.1975T>G | S659A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S659A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign, while ESM1b remains uncertain. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.067594 | Structured | 0.154597 | Uncertain | 0.954 | 0.283 | 0.000 | -7.066 | In-Between | 0.117 | Likely Benign | Likely Benign | -0.31 | Likely Benign | 0.0 | 0.02 | Likely Benign | -0.15 | Likely Benign | 0.14 | Likely Benign | 0.088 | Likely Benign | -2.22 | Neutral | 0.097 | Benign | 0.114 | Benign | 3.50 | Benign | 0.64 | Tolerated | 0.4842 | 0.3813 | 1 | 1 | 2.6 | -16.00 | |||||||||||||||||||||||||||||
| c.1976C>A | S659Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S659Y is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, ESM1b, and AlphaMissense‑Default; FoldX is uncertain and therefore not considered evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of individual predictors lean toward a benign impact, and there is no ClinVar entry to contradict this assessment. Thus, based on the available predictions, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.067594 | Structured | 0.154597 | Uncertain | 0.954 | 0.283 | 0.000 | -10.832 | Likely Pathogenic | 0.600 | Likely Pathogenic | Likely Benign | -0.64 | Ambiguous | 0.1 | 0.00 | Likely Benign | -0.32 | Likely Benign | 0.29 | Likely Benign | 0.173 | Likely Benign | -4.24 | Deleterious | 0.084 | Benign | 0.014 | Benign | 3.38 | Benign | 0.04 | Affected | 0.0993 | 0.5962 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||
| c.1976C>G | S659C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S659C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Foldetta, premPS, AlphaMissense‑Default, AlphaMissense‑Optimized, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and SIFT. Two tools (Rosetta and ESM1b) return uncertain results and are not counted as evidence for either side. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a benign majority; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.067594 | Structured | 0.154597 | Uncertain | 0.954 | 0.283 | 0.000 | -7.133 | In-Between | 0.161 | Likely Benign | Likely Benign | 0.09 | Likely Benign | 0.0 | 0.69 | Ambiguous | 0.39 | Likely Benign | 0.36 | Likely Benign | 0.173 | Likely Benign | -4.12 | Deleterious | 0.981 | Probably Damaging | 0.397 | Benign | 3.36 | Benign | 0.04 | Affected | 0.1130 | 0.5384 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||
| c.1978A>C | M660L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M660L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, FATHMM, polyPhen‑2 HumVar, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No predictions are missing or inconclusive. Overall, the balance of evidence leans toward a benign classification, and this conclusion does not contradict any ClinVar status because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | -12.576 | Likely Pathogenic | 0.766 | Likely Pathogenic | Likely Benign | 0.16 | Likely Benign | 0.0 | -0.10 | Likely Benign | 0.03 | Likely Benign | 0.97 | Ambiguous | 0.330 | Likely Benign | -2.99 | Deleterious | 0.596 | Possibly Damaging | 0.101 | Benign | 3.56 | Benign | 0.03 | Affected | 0.1337 | 0.3891 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1978A>G | M660V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M660V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Functional prediction tools cluster into three groups: benign predictions come from REVEL, SIFT, and FATHMM; pathogenic predictions arise from SGM‑Consensus, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default; the remaining tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. Overall, the majority of evaluated predictors lean toward a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because no ClinVar status is available. Thus, based on the current computational evidence, the M660V variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | -11.006 | Likely Pathogenic | 0.836 | Likely Pathogenic | Ambiguous | 1.83 | Ambiguous | 0.1 | 0.77 | Ambiguous | 1.30 | Ambiguous | 1.05 | Destabilizing | 0.419 | Likely Benign | -3.99 | Deleterious | 1.000 | Probably Damaging | 0.927 | Probably Damaging | 3.56 | Benign | 0.13 | Tolerated | 0.2614 | 0.2937 | 2 | 1 | 2.3 | -32.06 | |||||||||||||||||||||||||||||
| c.1978A>T | M660L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M660L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, FATHMM, polyPhen‑2 HumVar, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No predictions are missing or inconclusive. Overall, the balance of evidence leans toward a benign classification, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | -12.576 | Likely Pathogenic | 0.766 | Likely Pathogenic | Likely Benign | 0.16 | Likely Benign | 0.0 | -0.10 | Likely Benign | 0.03 | Likely Benign | 0.97 | Ambiguous | 0.330 | Likely Benign | -2.99 | Deleterious | 0.596 | Possibly Damaging | 0.101 | Benign | 3.56 | Benign | 0.03 | Affected | 0.1337 | 0.3891 | 4 | 2 | 1.9 | -18.03 | |||||||||||||||||||||||||||||
| c.1979T>A | M660K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M660K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are polyPhen‑2 HumVar and FATHMM. All other evaluated predictors—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenicity; the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also indicates pathogenicity; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a pathogenic effect. Based on the overwhelming consensus of pathogenic predictions and the corroborating high‑accuracy tools, the variant is most likely pathogenic, and this conclusion does not contradict the current ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | -14.123 | Likely Pathogenic | 0.995 | Likely Pathogenic | Likely Pathogenic | 3.13 | Destabilizing | 0.1 | 4.46 | Destabilizing | 3.80 | Destabilizing | 2.36 | Destabilizing | 0.604 | Likely Pathogenic | -5.99 | Deleterious | 0.862 | Possibly Damaging | 0.216 | Benign | 3.34 | Benign | 0.00 | Affected | 0.1389 | 0.0856 | 0 | -1 | -5.8 | -3.02 | |||||||||||||||||||||||||||||
| c.1979T>C | M660T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M660T is catalogued in gnomAD (ID 6‑33441238‑T‑C) but has no ClinVar entry, so its clinical status is currently unreported. In silico prediction tools largely agree that the substitution is deleterious: pathogenic predictions come from SGM‑Consensus (Likely Pathogenic), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only FATHMM predicts a benign effect. High‑accuracy assessments reinforce the pathogenic view: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding result is missing or inconclusive. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | 6-33441238-T-C | 1 | 6.20e-7 | -9.791 | Likely Pathogenic | 0.989 | Likely Pathogenic | Likely Pathogenic | 3.62 | Destabilizing | 0.1 | 2.05 | Destabilizing | 2.84 | Destabilizing | 1.91 | Destabilizing | 0.561 | Likely Pathogenic | -5.99 | Deleterious | 0.967 | Probably Damaging | 0.633 | Possibly Damaging | 3.36 | Benign | 0.02 | Affected | 3.38 | 28 | 0.1811 | 0.1630 | -1 | -1 | -2.6 | -30.09 | ||||||||||||||||||||||||
| c.1979T>G | M660R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant M660R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenic. No prediction is inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | -15.985 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.95 | Destabilizing | 0.2 | 3.52 | Destabilizing | 3.24 | Destabilizing | 1.96 | Destabilizing | 0.588 | Likely Pathogenic | -5.99 | Deleterious | 0.976 | Probably Damaging | 0.464 | Possibly Damaging | 3.34 | Benign | 0.00 | Affected | 0.1612 | 0.0957 | 0 | -1 | -6.4 | 24.99 | |||||||||||||||||||||||||||||
| c.197C>A | P66H 2D AIThe SynGAP1 missense variant P66H is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, and the SGM‑Consensus result is benign; Foldetta predictions are unavailable. Overall, the balance of evidence leans toward a benign interpretation, with no conflict with ClinVar status because no ClinVar assertion exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.394753 | Structured | 0.474132 | Uncertain | 0.455 | 0.762 | 0.125 | -4.134 | Likely Benign | 0.941 | Likely Pathogenic | Ambiguous | 0.239 | Likely Benign | -2.26 | Neutral | 0.992 | Probably Damaging | 0.893 | Possibly Damaging | 3.89 | Benign | 0.00 | Affected | 0.1914 | 0.4675 | 0 | -2 | -1.6 | 40.02 | |||||||||||||||||||||||||||||||||||||||
| c.197C>G | P66R 2D AIThe SynGAP1 missense variant P66R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are not available. Overall, the majority of evidence points toward a benign impact, and there is no ClinVar entry to contradict this assessment. Thus, the variant is most likely benign based on current predictive data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.394753 | Structured | 0.474132 | Uncertain | 0.455 | 0.762 | 0.125 | -2.708 | Likely Benign | 0.940 | Likely Pathogenic | Ambiguous | 0.221 | Likely Benign | -2.23 | Neutral | 0.972 | Probably Damaging | 0.804 | Possibly Damaging | 3.92 | Benign | 0.00 | Affected | 0.1636 | 0.3928 | 0 | -2 | -2.9 | 59.07 | |||||||||||||||||||||||||||||||||||||||
| c.197C>T | P66L 2D AIThe SynGAP1 missense variant P66L is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, PROVEAN, ESM1b, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized calling the variant pathogenic, while the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates a likely benign outcome; Foldetta results are not available. Overall, the predictions are split evenly between benign and pathogenic, with high‑accuracy tools providing opposing conclusions. Consequently, the variant’s impact remains uncertain and does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.394753 | Structured | 0.474132 | Uncertain | 0.455 | 0.762 | 0.125 | -2.437 | Likely Benign | 0.972 | Likely Pathogenic | Likely Pathogenic | 0.194 | Likely Benign | -2.48 | Neutral | 0.909 | Possibly Damaging | 0.713 | Possibly Damaging | 3.92 | Benign | 0.00 | Affected | 0.2412 | 0.6687 | -3 | -3 | 5.4 | 16.04 | |||||||||||||||||||||||||||||||||||||||
| c.1980G>A | M660I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M660I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, polyPhen‑2 HumVar, and FATHMM, whereas pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized predicts pathogenic, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, and the protein‑folding stability method Foldetta returns an uncertain result. Stability predictions from FoldX, Rosetta, and premPS are also inconclusive. Overall, the majority of evidence points to a pathogenic effect for M660I. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | -11.150 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.15 | Ambiguous | 0.1 | 0.71 | Ambiguous | 0.93 | Ambiguous | 0.95 | Ambiguous | 0.464 | Likely Benign | -3.99 | Deleterious | 0.887 | Possibly Damaging | 0.289 | Benign | 3.52 | Benign | 0.04 | Affected | 0.1170 | 0.2878 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1980G>C | M660I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M660I (GAP domain) is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL, polyPhen‑2 HumVar, and FATHMM. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. No other stability predictions are available. Overall, the preponderance of evidence points to a pathogenic effect for M660I. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | -11.150 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.15 | Ambiguous | 0.1 | 0.71 | Ambiguous | 0.93 | Ambiguous | 0.95 | Ambiguous | 0.464 | Likely Benign | -3.99 | Deleterious | 0.887 | Possibly Damaging | 0.289 | Benign | 3.52 | Benign | 0.04 | Affected | 0.1170 | 0.2878 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1980G>T | M660I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant M660I (GAP domain) is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL, polyPhen‑2 HumVar, and FATHMM. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is inconclusive. No other stability predictions are available. Overall, the preponderance of evidence points to a pathogenic effect for M660I. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.047319 | Structured | 0.134270 | Uncertain | 0.944 | 0.289 | 0.000 | -11.150 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 1.15 | Ambiguous | 0.1 | 0.71 | Ambiguous | 0.93 | Ambiguous | 0.95 | Ambiguous | 0.464 | Likely Benign | -3.99 | Deleterious | 0.887 | Possibly Damaging | 0.289 | Benign | 3.52 | Benign | 0.04 | Affected | 0.1170 | 0.2878 | 2 | 1 | 2.6 | -18.03 | |||||||||||||||||||||||||||||
| c.1981C>A | Q661K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q661K is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Only ESM1b predicts a pathogenic outcome, while AlphaMissense‑Default remains uncertain. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority (2 benign vs. 1 pathogenic, with one uncertain), and Foldetta also predicts benign stability. Overall, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.048328 | Structured | 0.117089 | Uncertain | 0.924 | 0.309 | 0.000 | -10.581 | Likely Pathogenic | 0.400 | Ambiguous | Likely Benign | -0.01 | Likely Benign | 0.0 | -0.18 | Likely Benign | -0.10 | Likely Benign | 0.04 | Likely Benign | 0.108 | Likely Benign | -1.89 | Neutral | 0.098 | Benign | 0.030 | Benign | 3.59 | Benign | 0.42 | Tolerated | 0.1756 | 0.3592 | 1 | 1 | -0.4 | 0.04 | ||||||||||||||||||||||||||||||
| c.1981C>G | Q661E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q661E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Four tools (FoldX, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of available predictions lean toward a benign impact. This conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.048328 | Structured | 0.117089 | Uncertain | 0.924 | 0.309 | 0.000 | -12.121 | Likely Pathogenic | 0.370 | Ambiguous | Likely Benign | 0.64 | Ambiguous | 0.1 | 0.35 | Likely Benign | 0.50 | Ambiguous | 0.61 | Ambiguous | 0.141 | Likely Benign | -2.15 | Neutral | 0.988 | Probably Damaging | 0.619 | Possibly Damaging | 3.42 | Benign | 0.03 | Affected | 0.1199 | 0.2439 | 2 | 2 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.1982A>C | Q661P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q661P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from premPS and FATHMM, whereas the remaining 12 tools (REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score) all classify the change as pathogenic or likely pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a pathogenic effect. No prediction is inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.117089 | Uncertain | 0.924 | 0.309 | 0.000 | -17.549 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 4.11 | Destabilizing | 0.1 | 5.97 | Destabilizing | 5.04 | Destabilizing | 0.45 | Likely Benign | 0.531 | Likely Pathogenic | -4.58 | Deleterious | 1.000 | Probably Damaging | 0.994 | Probably Damaging | 3.41 | Benign | 0.01 | Affected | 0.1811 | 0.3850 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||
| c.1982A>G | Q661R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q661R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, SIFT, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that predict a pathogenic effect are polyPhen2_HumDiv and ESM1b. FoldX, Rosetta, Foldetta, and AlphaMissense‑Default are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also yields a benign prediction, while Foldetta remains uncertain. Overall, the majority of available evidence points to a benign impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.048328 | Structured | 0.117089 | Uncertain | 0.924 | 0.309 | 0.000 | -10.386 | Likely Pathogenic | 0.479 | Ambiguous | Likely Benign | -1.12 | Ambiguous | 0.0 | -0.59 | Ambiguous | -0.86 | Ambiguous | -0.15 | Likely Benign | 0.281 | Likely Benign | -2.06 | Neutral | 0.812 | Possibly Damaging | 0.251 | Benign | 3.52 | Benign | 0.18 | Tolerated | 0.1502 | 0.2196 | 1 | 1 | -1.0 | 28.06 | ||||||||||||||||||||||||||||||
| c.1982A>T | Q661L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q661L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain, so it is not used as evidence. Overall, the majority of predictions support a pathogenic impact, and this is consistent with the lack of ClinVar annotation. Therefore, the variant is most likely pathogenic, with no contradiction from ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.117089 | Uncertain | 0.924 | 0.309 | 0.000 | -9.003 | Likely Pathogenic | 0.613 | Likely Pathogenic | Likely Benign | -0.19 | Likely Benign | 0.1 | -1.12 | Ambiguous | -0.66 | Ambiguous | 0.36 | Likely Benign | 0.391 | Likely Benign | -5.11 | Deleterious | 0.976 | Probably Damaging | 0.567 | Possibly Damaging | 3.49 | Benign | 0.02 | Affected | 0.0834 | 0.4675 | -2 | -2 | 7.3 | -14.97 | |||||||||||||||||||||||||||||
| c.1983G>C | Q661H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q661H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Uncertain calls are made by FoldX and AlphaMissense‑Optimized. High‑accuracy assessments indicate that the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts a pathogenic effect, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a benign outcome. Overall, the balance of evidence leans toward a pathogenic interpretation, and this assessment does not conflict with ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.117089 | Uncertain | 0.924 | 0.309 | 0.000 | -9.938 | Likely Pathogenic | 0.886 | Likely Pathogenic | Ambiguous | 0.78 | Ambiguous | 0.1 | 0.07 | Likely Benign | 0.43 | Likely Benign | 0.09 | Likely Benign | 0.282 | Likely Benign | -3.78 | Deleterious | 0.993 | Probably Damaging | 0.819 | Possibly Damaging | 3.42 | Benign | 0.02 | Affected | 0.1276 | 0.3424 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.1983G>T | Q661H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Q661H is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions are reported by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Uncertain calls are made by FoldX and AlphaMissense‑Optimized. High‑accuracy assessments indicate that the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts a pathogenic effect, while Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a benign outcome. Overall, the balance of evidence leans toward a pathogenic interpretation, and this assessment does not conflict with ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.048328 | Structured | 0.117089 | Uncertain | 0.924 | 0.309 | 0.000 | -9.938 | Likely Pathogenic | 0.886 | Likely Pathogenic | Ambiguous | 0.78 | Ambiguous | 0.1 | 0.07 | Likely Benign | 0.43 | Likely Benign | 0.09 | Likely Benign | 0.282 | Likely Benign | -3.78 | Deleterious | 0.993 | Probably Damaging | 0.819 | Possibly Damaging | 3.42 | Benign | 0.02 | Affected | 0.1276 | 0.3424 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.1984C>A | Q662K 2D AIThe SynGAP1 missense variant Q662K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign, while only ESM1b predicts pathogenicity. When high‑accuracy methods are considered separately, AlphaMissense‑Optimized remains benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields a Likely Benign verdict, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a benign effect. No conflicting evidence is present. Therefore, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.046336 | Structured | 0.103446 | Uncertain | 0.932 | 0.323 | 0.000 | -8.892 | Likely Pathogenic | 0.309 | Likely Benign | Likely Benign | -0.02 | Likely Benign | 0.2 | 0.03 | Likely Benign | 0.01 | Likely Benign | -0.04 | Likely Benign | 0.108 | Likely Benign | -0.80 | Neutral | 0.321 | Benign | 0.030 | Benign | 3.49 | Benign | 0.37 | Tolerated | 0.2391 | 0.3198 | 1 | 1 | -0.4 | 0.04 | |||||||||||||||||||||||||||||
| c.1984C>G | Q662E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q662E is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus, REVEL, FoldX, Foldetta, premPS, PROVEAN, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. High‑accuracy methods all support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta is benign. No prediction or folding‑stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.046336 | Structured | 0.103446 | Uncertain | 0.932 | 0.323 | 0.000 | -12.750 | Likely Pathogenic | 0.191 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.0 | 0.64 | Ambiguous | 0.48 | Likely Benign | 0.24 | Likely Benign | 0.083 | Likely Benign | -1.25 | Neutral | 0.876 | Possibly Damaging | 0.147 | Benign | 3.52 | Benign | 0.27 | Tolerated | 0.1660 | 0.1823 | 2 | 2 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.1985A>C | Q662P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q662P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, and FATHMM, while pathogenic predictions are made by SGM‑Consensus, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. High‑accuracy methods give a consistent pathogenic signal: the SGM‑Consensus score (derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates “Likely Pathogenic,” and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic effect. AlphaMissense‑Optimized returns an uncertain result, which is treated as unavailable. Overall, the majority of evidence points to a pathogenic impact for Q662P, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.046336 | Structured | 0.103446 | Uncertain | 0.932 | 0.323 | 0.000 | -13.174 | Likely Pathogenic | 0.922 | Likely Pathogenic | Ambiguous | 2.15 | Destabilizing | 0.0 | 9.37 | Destabilizing | 5.76 | Destabilizing | 0.36 | Likely Benign | 0.268 | Likely Benign | -3.42 | Deleterious | 0.999 | Probably Damaging | 0.962 | Probably Damaging | 3.40 | Benign | 0.08 | Tolerated | 0.2621 | 0.4364 | 0 | -1 | 1.9 | -31.01 | |||||||||||||||||||||||||||||
| c.1985A>G | Q662R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q662R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that assess sequence conservation, structural impact, or functional effect all converge on a benign outcome: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, FATHMM, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, FoldX, and Rosetta all predict benign. No tool in the dataset indicates pathogenicity. High‑accuracy assessments reinforce this consensus: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts benign. Thus, based on the available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.046336 | Structured | 0.103446 | Uncertain | 0.932 | 0.323 | 0.000 | -6.678 | Likely Benign | 0.300 | Likely Benign | Likely Benign | -0.41 | Likely Benign | 0.0 | 0.10 | Likely Benign | -0.16 | Likely Benign | -0.42 | Likely Benign | 0.159 | Likely Benign | 0.49 | Neutral | 0.428 | Benign | 0.095 | Benign | 3.48 | Benign | 1.00 | Tolerated | 0.2023 | 0.1391 | 1 | 1 | -1.0 | 28.06 | |||||||||||||||||||||||||||||
| c.1985A>T | Q662L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q662L is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen2_HumVar, and FATHMM. Tools that predict a pathogenic effect are PROVEAN, polyPhen2_HumDiv, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign and Foldetta as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic). Overall, the majority of evidence supports a benign impact. This conclusion is consistent with the lack of ClinVar annotation and gnomAD presence, so there is no contradiction with existing database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.046336 | Structured | 0.103446 | Uncertain | 0.932 | 0.323 | 0.000 | -10.291 | Likely Pathogenic | 0.300 | Likely Benign | Likely Benign | 0.04 | Likely Benign | 0.0 | 0.12 | Likely Benign | 0.08 | Likely Benign | 0.17 | Likely Benign | 0.186 | Likely Benign | -4.09 | Deleterious | 0.699 | Possibly Damaging | 0.057 | Benign | 3.42 | Benign | 0.10 | Tolerated | 0.1070 | 0.5255 | -2 | -2 | 7.3 | -14.97 | ||||||||||||||||||||||||||||||
| c.1986G>C | Q662H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q662H is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools overwhelmingly indicate a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign, while the single pathogenic call comes from polyPhen‑2 HumDiv. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” status. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign, SGM‑Consensus (majority vote) predicts likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. FoldX alone is uncertain, but this does not alter the overall consensus. Based on the collective predictions, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.046336 | Structured | 0.103446 | Uncertain | 0.932 | 0.323 | 0.000 | -6.357 | Likely Benign | 0.331 | Likely Benign | Likely Benign | 0.53 | Ambiguous | 0.0 | 0.18 | Likely Benign | 0.36 | Likely Benign | -0.11 | Likely Benign | 0.159 | Likely Benign | -2.00 | Neutral | 0.891 | Possibly Damaging | 0.243 | Benign | 3.42 | Benign | 0.15 | Tolerated | 0.1674 | 0.3186 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.1986G>T | Q662H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Q662H is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools overwhelmingly indicate a benign effect: REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign, while the single pathogenic call comes from polyPhen‑2 HumDiv. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” status, and FoldX is inconclusive. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Based on the preponderance of evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.046336 | Structured | 0.103446 | Uncertain | 0.932 | 0.323 | 0.000 | -6.357 | Likely Benign | 0.331 | Likely Benign | Likely Benign | 0.53 | Ambiguous | 0.0 | 0.18 | Likely Benign | 0.36 | Likely Benign | -0.11 | Likely Benign | 0.159 | Likely Benign | -2.00 | Neutral | 0.891 | Possibly Damaging | 0.243 | Benign | 3.42 | Benign | 0.15 | Tolerated | 0.1674 | 0.3186 | 3 | 0 | 0.3 | 9.01 | |||||||||||||||||||||||||||||
| c.1987T>A | F663I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F663I is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.056825 | Structured | 0.093963 | Uncertain | 0.944 | 0.355 | 0.000 | -15.006 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 2.63 | Destabilizing | 0.1 | 4.51 | Destabilizing | 3.57 | Destabilizing | 1.60 | Destabilizing | 0.641 | Likely Pathogenic | -5.99 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 3.06 | Benign | 0.00 | Affected | 0.1740 | 0.1955 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.1987T>C | F663L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F663L is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, which scores the variant as benign. All other evaluated algorithms—SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also predicts pathogenicity, while Foldetta’s stability analysis is inconclusive and therefore unavailable. FoldX and Foldetta are treated as unavailable due to uncertain outputs. Based on the overwhelming agreement among pathogenic predictors and the lack of benign evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.056825 | Structured | 0.093963 | Uncertain | 0.944 | 0.355 | 0.000 | -11.996 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.91 | Ambiguous | 0.1 | 2.01 | Destabilizing | 1.46 | Ambiguous | 1.51 | Destabilizing | 0.540 | Likely Pathogenic | -5.99 | Deleterious | 0.999 | Probably Damaging | 0.976 | Probably Damaging | 3.07 | Benign | 0.01 | Affected | 0.1973 | 0.2936 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1987T>G | F663V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F663V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b all indicate pathogenicity, while only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the change as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports it as likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts a pathogenic effect. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.056825 | Structured | 0.093963 | Uncertain | 0.944 | 0.355 | 0.000 | -14.196 | Likely Pathogenic | 0.988 | Likely Pathogenic | Likely Pathogenic | 3.04 | Destabilizing | 0.1 | 3.31 | Destabilizing | 3.18 | Destabilizing | 1.75 | Destabilizing | 0.655 | Likely Pathogenic | -6.98 | Deleterious | 0.998 | Probably Damaging | 0.993 | Probably Damaging | 3.02 | Benign | 0.00 | Affected | 0.1844 | 0.1983 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.1988T>A | F663Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F663Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, and FATHMM. Those that predict a pathogenic effect comprise premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. Overall, the majority of tools and the SGM‑Consensus lean toward pathogenicity, while a minority suggest benign impact. Thus, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status, as none is currently assigned. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.056825 | Structured | 0.093963 | Uncertain | 0.944 | 0.355 | 0.000 | -12.749 | Likely Pathogenic | 0.853 | Likely Pathogenic | Ambiguous | 0.37 | Likely Benign | 0.1 | 0.19 | Likely Benign | 0.28 | Likely Benign | 1.03 | Destabilizing | 0.424 | Likely Benign | -2.99 | Deleterious | 0.984 | Probably Damaging | 0.913 | Probably Damaging | 3.11 | Benign | 0.05 | Affected | 0.1243 | 0.1584 | 7 | 3 | -4.1 | 16.00 | |||||||||||||||||||||||||||||
| c.1988T>C | F663S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F663S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming agreement among both general and high‑accuracy predictors, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.056825 | Structured | 0.093963 | Uncertain | 0.944 | 0.355 | 0.000 | -14.800 | Likely Pathogenic | 1.000 | Likely Pathogenic | Likely Pathogenic | 4.15 | Destabilizing | 0.0 | 4.69 | Destabilizing | 4.42 | Destabilizing | 2.50 | Destabilizing | 0.800 | Likely Pathogenic | -7.98 | Deleterious | 1.000 | Probably Damaging | 1.000 | Probably Damaging | 2.98 | Benign | 0.00 | Affected | 0.3965 | 0.0200 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.1988T>G | F663C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F663C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy assessments further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.056825 | Structured | 0.093963 | Uncertain | 0.944 | 0.355 | 0.000 | -13.544 | Likely Pathogenic | 0.997 | Likely Pathogenic | Likely Pathogenic | 3.83 | Destabilizing | 0.1 | 4.54 | Destabilizing | 4.19 | Destabilizing | 1.91 | Destabilizing | 0.772 | Likely Pathogenic | -7.98 | Deleterious | 1.000 | Probably Damaging | 0.998 | Probably Damaging | 2.96 | Benign | 0.00 | Affected | 0.2343 | 0.0783 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.1989T>A | F663L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F663L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors (PolyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, SGM Consensus, premPS, and Rosetta) indicate a pathogenic impact. FoldX‑MD and Rosetta‑based stability calculations are inconclusive, yielding an uncertain Foldetta result. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM Consensus as likely pathogenic, and Foldetta as uncertain. Taken together, the preponderance of evidence from consensus and high‑accuracy tools points to a pathogenic effect for F663L. This conclusion is not contradicted by ClinVar status, as no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.056825 | Structured | 0.093963 | Uncertain | 0.944 | 0.355 | 0.000 | -11.996 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.91 | Ambiguous | 0.1 | 2.01 | Destabilizing | 1.46 | Ambiguous | 1.51 | Destabilizing | 0.423 | Likely Benign | -5.99 | Deleterious | 0.999 | Probably Damaging | 0.976 | Probably Damaging | 3.07 | Benign | 0.01 | Affected | 0.1973 | 0.2936 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1989T>G | F663L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F663L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas the majority of other in silico predictors (PolyPhen‑2 HumDiv/HumVar, SIFT, ESM1b, PROVEAN, AlphaMissense‑Default, AlphaMissense‑Optimized, premPS, and the SGM Consensus) indicate a pathogenic impact. FoldX‑MD and Rosetta give conflicting stability results, with FoldX uncertain and Rosetta pathogenic; Foldetta, which integrates both, is also uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus as likely pathogenic, and Foldetta as inconclusive. Overall, the preponderance of evidence points to a pathogenic effect for F663L. This conclusion is not contradicted by ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.056825 | Structured | 0.093963 | Uncertain | 0.944 | 0.355 | 0.000 | -11.996 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 0.91 | Ambiguous | 0.1 | 2.01 | Destabilizing | 1.46 | Ambiguous | 1.51 | Destabilizing | 0.423 | Likely Benign | -5.99 | Deleterious | 0.999 | Probably Damaging | 0.976 | Probably Damaging | 3.07 | Benign | 0.01 | Affected | 0.1973 | 0.2936 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.1990T>A | L664M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L664M is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, and FATHMM, while those that agree on a pathogenic effect are polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments are mixed: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is also uncertain due to a 2‑vs‑2 split; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is uncertain. Overall, the majority of conventional tools predict a pathogenic impact, whereas the high‑accuracy methods do not provide a definitive verdict. Consequently, the variant is most likely pathogenic based on the available predictions, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.100716 | Structured | 0.089318 | Uncertain | 0.937 | 0.339 | 0.000 | -11.819 | Likely Pathogenic | 0.807 | Likely Pathogenic | Ambiguous | 0.49 | Likely Benign | 0.1 | 1.35 | Ambiguous | 0.92 | Ambiguous | 0.96 | Ambiguous | 0.429 | Likely Benign | -2.00 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 2.92 | Benign | 0.01 | Affected | 0.0620 | 0.2541 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.1990T>G | L664V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L664V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include SGM‑Consensus, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Rosetta and Foldetta are uncertain, providing no definitive evidence. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic (3 pathogenic vs 1 benign). Foldetta’s stability prediction is uncertain. Overall, the majority of reliable tools predict a pathogenic impact. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.089318 | Uncertain | 0.937 | 0.339 | 0.000 | -11.515 | Likely Pathogenic | 0.755 | Likely Pathogenic | Likely Benign | 2.01 | Destabilizing | 0.1 | 1.13 | Ambiguous | 1.57 | Ambiguous | 1.37 | Destabilizing | 0.378 | Likely Benign | -2.99 | Deleterious | 0.993 | Probably Damaging | 0.776 | Possibly Damaging | 2.91 | Benign | 0.01 | Affected | 0.0984 | 0.2628 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.1991T>G | L664W 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L664W is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: benign calls are made only by REVEL and FATHMM, whereas the remaining 13 predictors—including FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the substitution as pathogenic. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a pathogenic effect. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.089318 | Uncertain | 0.937 | 0.339 | 0.000 | -17.438 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 7.81 | Destabilizing | 0.5 | 3.17 | Destabilizing | 5.49 | Destabilizing | 1.57 | Destabilizing | 0.479 | Likely Benign | -5.99 | Deleterious | 1.000 | Probably Damaging | 0.984 | Probably Damaging | 2.84 | Benign | 0.00 | Affected | 0.0543 | 0.2272 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||
| c.1992G>C | L664F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L664F resides in the GAP domain. ClinVar lists no entry for this variant, and it is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM. Those that predict a pathogenic effect include SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Predictions that are uncertain (FoldX, Rosetta, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as Uncertain. Overall, the majority of available predictions support a pathogenic impact. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.089318 | Uncertain | 0.937 | 0.339 | 0.000 | -12.834 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 1.98 | Ambiguous | 0.3 | 1.03 | Ambiguous | 1.51 | Ambiguous | 0.55 | Ambiguous | 0.355 | Likely Benign | -3.99 | Deleterious | 0.999 | Probably Damaging | 0.895 | Possibly Damaging | 3.05 | Benign | 0.03 | Affected | 0.0537 | 0.2311 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1992G>T | L664F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L664F resides in the GAP domain. ClinVar lists no entry for this variant, and it is absent from gnomAD. Prediction tools that agree on a benign effect are REVEL and FATHMM. Those that predict a pathogenic effect include SGM‑Consensus (Likely Pathogenic), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Predictions that are uncertain (FoldX, Rosetta, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as Pathogenic, SGM‑Consensus as Likely Pathogenic, and Foldetta as Uncertain. Overall, the majority of available predictions support a pathogenic impact. Therefore, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.100716 | Structured | 0.089318 | Uncertain | 0.937 | 0.339 | 0.000 | -12.834 | Likely Pathogenic | 0.984 | Likely Pathogenic | Likely Pathogenic | 1.98 | Ambiguous | 0.3 | 1.03 | Ambiguous | 1.51 | Ambiguous | 0.55 | Ambiguous | 0.355 | Likely Benign | -3.99 | Deleterious | 0.999 | Probably Damaging | 0.895 | Possibly Damaging | 3.05 | Benign | 0.03 | Affected | 0.0537 | 0.2311 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
| c.1993T>A | Y665N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 Y665N variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Four tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Default) give uncertain results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of conventional predictors lean toward a benign impact, and this assessment does not contradict ClinVar status (none). Thus, the variant is most likely benign based on the prevailing predictions. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.098513 | Structured | 0.086641 | Uncertain | 0.922 | 0.361 | 0.000 | -11.189 | Likely Pathogenic | 0.470 | Ambiguous | Likely Benign | 1.11 | Ambiguous | 0.1 | 1.43 | Ambiguous | 1.27 | Ambiguous | 0.50 | Likely Benign | 0.219 | Likely Benign | -3.03 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.50 | Benign | 0.88 | Tolerated | 0.1929 | 0.0573 | -2 | -2 | -2.2 | -49.07 | ||||||||||||||||||||||||||||||
| c.1993T>C | Y665H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y665H is not reported in ClinVar (ClinVar status: not reported) but is present in gnomAD (gnomAD ID: 6‑33441252‑T‑C). Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are premPS, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Tools with uncertain or inconclusive results are FoldX, Foldetta, and AlphaMissense‑Default. High‑accuracy methods give a benign consensus: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign, and Foldetta remains uncertain. Overall, the majority of predictions (7 benign vs. 4 pathogenic) and the high‑accuracy consensus support a benign classification. This conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.098513 | Structured | 0.086641 | Uncertain | 0.922 | 0.361 | 0.000 | 6-33441252-T-C | 2 | 1.24e-6 | -9.303 | Likely Pathogenic | 0.389 | Ambiguous | Likely Benign | 0.85 | Ambiguous | 0.0 | 0.28 | Likely Benign | 0.57 | Ambiguous | 1.12 | Destabilizing | 0.129 | Likely Benign | -2.00 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.45 | Benign | 0.48 | Tolerated | 3.38 | 28 | 0.2122 | 0.0513 | 2 | 0 | -1.9 | -26.03 | |||||||||||||||||||||||||
| c.1993T>G | Y665D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y665D is not reported in ClinVar and is absent from gnomAD. Prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and Foldetta as Uncertain. No evidence from these tools contradicts the ClinVar status, which is currently unreported. Overall, the balance of evidence—seven pathogenic versus four benign predictions, with a pathogenic consensus from SGM‑Consensus—suggests the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.098513 | Structured | 0.086641 | Uncertain | 0.922 | 0.361 | 0.000 | -10.101 | Likely Pathogenic | 0.737 | Likely Pathogenic | Likely Benign | 1.15 | Ambiguous | 0.2 | 1.30 | Ambiguous | 1.23 | Ambiguous | 0.15 | Likely Benign | 0.227 | Likely Benign | -3.67 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.75 | Benign | 1.00 | Tolerated | 0.3838 | 0.0573 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.1994A>C | Y665S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y665S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) leans pathogenic, and Foldetta is uncertain (treated as unavailable). Overall, the balance of evidence, particularly the pathogenic signal from the SGM Consensus and the equal split among standard predictors, indicates that the variant is most likely pathogenic. This conclusion does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.098513 | Structured | 0.086641 | Uncertain | 0.922 | 0.361 | 0.000 | -9.110 | Likely Pathogenic | 0.453 | Ambiguous | Likely Benign | 1.24 | Ambiguous | 0.2 | 1.40 | Ambiguous | 1.32 | Ambiguous | 0.87 | Ambiguous | 0.202 | Likely Benign | -2.50 | Deleterious | 1.000 | Probably Damaging | 0.994 | Probably Damaging | 3.55 | Benign | 0.62 | Tolerated | 0.3995 | 0.1793 | -3 | -2 | 0.5 | -76.10 | ||||||||||||||||||||||||||||||
| c.1994A>G | Y665C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y665C is listed in ClinVar with no assertion (status: None) and is present in gnomAD (ID 6‑33441253‑A‑G). Prediction tools that agree on a benign effect include REVEL, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. FoldX, Rosetta, and Foldetta give uncertain results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta is also unavailable. Overall, the evidence is mixed, but the majority of high‑confidence tools lean toward a benign interpretation. Thus, the variant is most likely benign based on current predictions, and this does not contradict the ClinVar status, which remains unclassified. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.098513 | Structured | 0.086641 | Uncertain | 0.922 | 0.361 | 0.000 | 6-33441253-A-G | 1 | 6.20e-7 | -9.007 | Likely Pathogenic | 0.261 | Likely Benign | Likely Benign | 1.05 | Ambiguous | 0.1 | 1.60 | Ambiguous | 1.33 | Ambiguous | 1.12 | Destabilizing | 0.210 | Likely Benign | -3.22 | Deleterious | 1.000 | Probably Damaging | 0.981 | Probably Damaging | 3.45 | Benign | 0.14 | Tolerated | 3.38 | 28 | 0.2562 | 0.2019 | -2 | 0 | 3.8 | -60.04 | |||||||||||||||||||||||||
| c.1994A>T | Y665F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y665F is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only ESM1b predicts a pathogenic outcome, while Rosetta and Foldetta are uncertain. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” status. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the preponderance of evidence points to a benign impact. This conclusion is consistent with the absence of a ClinVar claim; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.098513 | Structured | 0.086641 | Uncertain | 0.922 | 0.361 | 0.000 | -8.460 | Likely Pathogenic | 0.100 | Likely Benign | Likely Benign | 0.07 | Likely Benign | 0.1 | 1.03 | Ambiguous | 0.55 | Ambiguous | 0.38 | Likely Benign | 0.106 | Likely Benign | -1.23 | Neutral | 0.159 | Benign | 0.036 | Benign | 3.45 | Benign | 0.47 | Tolerated | 0.2277 | 0.3076 | 7 | 3 | 4.1 | -16.00 | |||||||||||||||||||||||||||||
| c.1996G>A | E666K 2D AIThe SynGAP1 missense variant E666K is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM. Tools that agree on a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The high‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the majority of predictions lean toward pathogenicity, and this assessment does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.086870 | Uncertain | 0.925 | 0.387 | 0.000 | -11.367 | Likely Pathogenic | 0.946 | Likely Pathogenic | Ambiguous | 0.20 | Likely Benign | 0.6 | 0.30 | Likely Benign | 0.25 | Likely Benign | 0.43 | Likely Benign | 0.401 | Likely Benign | -3.26 | Deleterious | 0.992 | Probably Damaging | 0.717 | Possibly Damaging | 3.46 | Benign | 0.05 | Affected | 0.2953 | 0.5300 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.1996G>C | E666Q 2D AIThe SynGAP1 E666Q missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default; FoldX is uncertain and therefore not counted. High‑accuracy assessments show AlphaMissense‑Optimized as benign, while the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is inconclusive due to a 2‑to‑2 split and is treated as unavailable. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts a benign effect. Overall, the majority of reliable predictions indicate a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.155435 | Structured | 0.086870 | Uncertain | 0.925 | 0.387 | 0.000 | -10.963 | Likely Pathogenic | 0.737 | Likely Pathogenic | Likely Benign | 0.54 | Ambiguous | 0.5 | 0.20 | Likely Benign | 0.37 | Likely Benign | 0.46 | Likely Benign | 0.268 | Likely Benign | -2.29 | Neutral | 0.997 | Probably Damaging | 0.696 | Possibly Damaging | 3.46 | Benign | 0.13 | Tolerated | 0.1437 | 0.4788 | 2 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||||||
| c.1997A>C | E666A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E666A missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, and FATHMM, while those that agree on a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Two tools give inconclusive results: FoldX (Uncertain) and AlphaMissense‑Optimized (Uncertain). High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, predicts Pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is inconclusive because FoldX is Uncertain while Rosetta is Benign. Overall, the majority of available predictions (six pathogenic vs. five benign) lean toward a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.086870 | Uncertain | 0.925 | 0.387 | 0.000 | -11.122 | Likely Pathogenic | 0.921 | Likely Pathogenic | Ambiguous | 0.57 | Ambiguous | 0.1 | 0.18 | Likely Benign | 0.38 | Likely Benign | 0.50 | Likely Benign | 0.407 | Likely Benign | -5.15 | Deleterious | 0.992 | Probably Damaging | 0.863 | Possibly Damaging | 3.45 | Benign | 0.07 | Tolerated | 0.3950 | 0.4878 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.1997A>T | E666V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E666V missense variant has no ClinVar entry and is not reported in gnomAD. Prediction tools show mixed results: benign calls come from REVEL, Rosetta, Foldetta, premPS, polyPhen‑2 HumVar, and FATHMM, while pathogenic calls come from PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, reports a benign effect. FoldX alone is uncertain. Overall, the majority of tools and the high‑accuracy consensus favor a pathogenic interpretation, with no conflict from ClinVar status because no classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.155435 | Structured | 0.086870 | Uncertain | 0.925 | 0.387 | 0.000 | -10.870 | Likely Pathogenic | 0.981 | Likely Pathogenic | Likely Pathogenic | 0.61 | Ambiguous | 0.1 | 0.08 | Likely Benign | 0.35 | Likely Benign | 0.31 | Likely Benign | 0.476 | Likely Benign | -5.95 | Deleterious | 0.575 | Possibly Damaging | 0.214 | Benign | 3.44 | Benign | 0.03 | Affected | 0.0818 | 0.5613 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.1999A>C | I667L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I667L is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are polyPhen‑2 HumDiv and ESM1b. The remaining tools—FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Default—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation and gnomAD presence, indicating no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.142424 | Structured | 0.083597 | Uncertain | 0.927 | 0.379 | 0.000 | -10.452 | Likely Pathogenic | 0.459 | Ambiguous | Likely Benign | 0.93 | Ambiguous | 0.2 | 0.72 | Ambiguous | 0.83 | Ambiguous | 0.75 | Ambiguous | 0.300 | Likely Benign | -1.97 | Neutral | 0.457 | Possibly Damaging | 0.392 | Benign | 3.36 | Benign | 0.13 | Tolerated | 0.0980 | 0.3145 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.1999A>G | I667V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I667V missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Only ESM1b predicts a pathogenic outcome; all other tools (FoldX, Rosetta, premPS, AlphaMissense‑Default, Foldetta) are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM Consensus (derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign majority vote. Foldetta’s stability prediction is uncertain. Overall, the preponderance of evidence points to a benign effect. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.142424 | Structured | 0.083597 | Uncertain | 0.927 | 0.379 | 0.000 | -8.482 | Likely Pathogenic | 0.406 | Ambiguous | Likely Benign | 1.32 | Ambiguous | 0.0 | 1.06 | Ambiguous | 1.19 | Ambiguous | 0.85 | Ambiguous | 0.234 | Likely Benign | -0.86 | Neutral | 0.341 | Benign | 0.052 | Benign | 3.35 | Benign | 0.15 | Tolerated | 0.1178 | 0.3059 | 4 | 3 | -0.3 | -14.03 | ||||||||||||||||||||||||||||||
| c.1999A>T | I667F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I667F missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign calls are limited to FATHMM, while the remaining 11 predictors—SGM‑Consensus, REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta—classify the change as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) reports a pathogenic effect. No prediction is inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.083597 | Uncertain | 0.927 | 0.379 | 0.000 | -14.389 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 7.55 | Destabilizing | 0.3 | 1.80 | Ambiguous | 4.68 | Destabilizing | 0.86 | Ambiguous | 0.569 | Likely Pathogenic | -3.98 | Deleterious | 0.998 | Probably Damaging | 0.790 | Possibly Damaging | 2.96 | Benign | 0.00 | Affected | 0.0668 | 0.2648 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.199C>A | L67I 2D AIThe SynGAP1 missense variant L67I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 HumDiv and SIFT predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for the variant, and this is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.473668 | Uncertain | 0.428 | 0.761 | 0.125 | -4.387 | Likely Benign | 0.307 | Likely Benign | Likely Benign | 0.084 | Likely Benign | -0.29 | Neutral | 0.458 | Possibly Damaging | 0.364 | Benign | 4.10 | Benign | 0.00 | Affected | 0.0680 | 0.2919 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||||||||||||
| c.199C>G | L67V 2D AIThe SynGAP1 missense variant L67V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. In contrast, polyPhen‑2 HumDiv and SIFT predict pathogenic. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign classification. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign and the SGM‑Consensus is likely benign; Foldetta results are unavailable. Overall, the majority of evidence points to a benign impact for the L67V substitution, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.473668 | Uncertain | 0.428 | 0.761 | 0.125 | -3.617 | Likely Benign | 0.285 | Likely Benign | Likely Benign | 0.122 | Likely Benign | -0.31 | Neutral | 0.458 | Possibly Damaging | 0.364 | Benign | 4.15 | Benign | 0.00 | Affected | 0.1138 | 0.2817 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.19T>A | S7T 2D AIThe SynGAP1 missense variant S7T is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign effect. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as likely benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is present). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | -4.182 | Likely Benign | 0.110 | Likely Benign | Likely Benign | 0.101 | Likely Benign | -0.26 | Neutral | 0.024 | Benign | 0.007 | Benign | 4.13 | Benign | 0.00 | Affected | 0.1688 | 0.5919 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||||||||||||
| c.19T>C | S7P 2D AIThe SynGAP1 missense variant S7P is reported in gnomAD (ID 6‑33420283‑T‑C) but has no ClinVar entry. In silico prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign. Only SIFT predicts a pathogenic outcome. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely benign status. High‑accuracy assessments confirm this: AlphaMissense‑Optimized is benign, and the SGM‑Consensus (majority‑vote) is benign; Foldetta results are unavailable. Overall, the consensus of the majority of tools, including the high‑accuracy methods, points to a benign effect. This prediction is consistent with the absence of a ClinVar pathogenic classification, so there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | 6-33420283-T-C | -3.798 | Likely Benign | 0.111 | Likely Benign | Likely Benign | 0.210 | Likely Benign | -0.23 | Neutral | 0.267 | Benign | 0.029 | Benign | 4.07 | Benign | 0.00 | Affected | 4.32 | 1 | 0.2474 | 0.4925 | -1 | 1 | -0.8 | 10.04 | ||||||||||||||||||||||||||||||||||||
| c.19T>G | S7A 2D AIThe SynGAP1 missense variant S7A is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only SIFT predicts a pathogenic outcome. The high‑accuracy consensus, SGM‑Consensus, is derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, all of which are benign, so the consensus is benign. AlphaMissense‑Optimized also predicts benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Based on the available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.590140 | Disordered | 0.548467 | Binding | 0.386 | 0.922 | 0.750 | -3.613 | Likely Benign | 0.072 | Likely Benign | Likely Benign | 0.137 | Likely Benign | -0.10 | Neutral | 0.002 | Benign | 0.001 | Benign | 4.18 | Benign | 0.00 | Affected | 0.5485 | 0.4355 | Weaken | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||||||||||
| c.2000T>A | I667N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I667N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic impact. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized indicates pathogenicity; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports “Likely Pathogenic”; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenicity. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.083597 | Uncertain | 0.927 | 0.379 | 0.000 | -15.730 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 2.89 | Destabilizing | 0.2 | 2.84 | Destabilizing | 2.87 | Destabilizing | 2.56 | Destabilizing | 0.572 | Likely Pathogenic | -6.73 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 2.95 | Benign | 0.00 | Affected | 0.0841 | 0.0470 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.2000T>C | I667T 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant I667T is listed in ClinVar with an uncertain significance and is not reported in gnomAD. Functional prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all predict pathogenicity, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized scores the variant as pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts pathogenicity. No prediction or stability result is missing or inconclusive. Overall, the consensus of the available tools points to a pathogenic effect, which contrasts with the ClinVar designation of uncertain significance. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.083597 | Uncertain | 0.927 | 0.379 | 0.000 | Uncertain | 1 | -11.917 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 3.29 | Destabilizing | 0.1 | 2.44 | Destabilizing | 2.87 | Destabilizing | 2.23 | Destabilizing | 0.676 | Likely Pathogenic | -4.81 | Deleterious | 1.000 | Probably Damaging | 0.985 | Probably Damaging | 2.98 | Benign | 0.00 | Affected | 0.0967 | 0.0608 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||
| c.2000T>G | I667S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I667S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include only FATHMM, whereas all other evaluated algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify the variant as pathogenic. High‑accuracy methods further support a deleterious impact: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because the variant is not yet catalogued in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.083597 | Uncertain | 0.927 | 0.379 | 0.000 | -14.328 | Likely Pathogenic | 0.993 | Likely Pathogenic | Likely Pathogenic | 3.69 | Destabilizing | 0.1 | 3.97 | Destabilizing | 3.83 | Destabilizing | 2.48 | Destabilizing | 0.690 | Likely Pathogenic | -5.90 | Deleterious | 1.000 | Probably Damaging | 0.995 | Probably Damaging | 2.96 | Benign | 0.00 | Affected | 0.2462 | 0.0858 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.2001C>G | I667M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I667M is not reported in ClinVar (ClinVar ID None) and has no entry in gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Predictions from FoldX, Rosetta, Foldetta, and premPS are uncertain and are treated as unavailable. High‑accuracy methods give AlphaMissense‑Optimized a benign prediction, SGM‑Consensus a pathogenic prediction (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta an uncertain result. Overall, the majority of evidence—including the high‑accuracy consensus—points to a pathogenic impact. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.083597 | Uncertain | 0.927 | 0.379 | 0.000 | -10.679 | Likely Pathogenic | 0.579 | Likely Pathogenic | Likely Benign | 1.29 | Ambiguous | 0.3 | 1.68 | Ambiguous | 1.49 | Ambiguous | 0.92 | Ambiguous | 0.360 | Likely Benign | -2.90 | Deleterious | 1.000 | Probably Damaging | 0.993 | Probably Damaging | 2.98 | Benign | 0.00 | Affected | 0.0746 | 0.2612 | 2 | 1 | -2.6 | 18.03 | |||||||||||||||||||||||||||||
| c.2002T>A | S668T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S668T is not reported in ClinVar (ClinVar ID: None) and has no entries in gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, premPS, FATHMM, AlphaMissense‑Optimized, and polyPhen2_HumVar. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen2_HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates a likely pathogenic outcome; Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result. No evidence from FoldX or Rosetta alone is conclusive. Overall, the balance of predictions leans toward pathogenicity, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.247041 | Structured | 0.084935 | Uncertain | 0.922 | 0.370 | 0.000 | -11.860 | Likely Pathogenic | 0.703 | Likely Pathogenic | Likely Benign | 1.73 | Ambiguous | 0.7 | 0.89 | Ambiguous | 1.31 | Ambiguous | 0.27 | Likely Benign | 0.238 | Likely Benign | -2.99 | Deleterious | 0.844 | Possibly Damaging | 0.198 | Benign | 3.24 | Benign | 0.02 | Affected | 0.1212 | 0.5918 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.2002T>C | S668P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S668P has no ClinVar entry and is not reported in gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the only benign predictor, while the remaining 13 tools (SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify it as pathogenic or likely pathogenic. The premPS score is uncertain and therefore not used as evidence. High‑accuracy methods give consistent results: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports likely pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.247041 | Structured | 0.084935 | Uncertain | 0.922 | 0.370 | 0.000 | -13.452 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 6.40 | Destabilizing | 1.1 | 10.90 | Destabilizing | 8.65 | Destabilizing | 0.61 | Ambiguous | 0.612 | Likely Pathogenic | -4.82 | Deleterious | 0.998 | Probably Damaging | 0.790 | Possibly Damaging | 3.16 | Benign | 0.02 | Affected | 0.1979 | 0.5137 | 1 | -1 | -0.8 | 10.04 | |||||||||||||||||||||||||||||
| c.2002T>G | S668A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 S668A missense variant is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, Rosetta, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and Foldetta. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Two tools—AlphaMissense‑Default and FoldX—return uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Taking all evidence together, the majority of predictions (seven benign versus four pathogenic) and the two high‑accuracy benign calls suggest that the variant is most likely benign. This conclusion does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.247041 | Structured | 0.084935 | Uncertain | 0.922 | 0.370 | 0.000 | -12.011 | Likely Pathogenic | 0.506 | Ambiguous | Likely Benign | 0.73 | Ambiguous | 0.3 | -0.44 | Likely Benign | 0.15 | Likely Benign | 0.48 | Likely Benign | 0.399 | Likely Benign | -2.99 | Deleterious | 0.887 | Possibly Damaging | 0.738 | Possibly Damaging | 3.28 | Benign | 0.19 | Tolerated | 0.4866 | 0.3967 | 1 | 1 | 2.6 | -16.00 | ||||||||||||||||||||||||||||||
| c.2003C>A | S668Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S668Y is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include premPS and FATHMM, whereas the remaining 12 tools (REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict it to be pathogenic or likely pathogenic. High‑accuracy methods give consistent results: AlphaMissense‑Optimized scores it as pathogenic; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts pathogenicity. No predictions are missing or inconclusive. Based on the overwhelming majority of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.247041 | Structured | 0.084935 | Uncertain | 0.922 | 0.370 | 0.000 | -14.833 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 18.34 | Destabilizing | 6.2 | 12.69 | Destabilizing | 15.52 | Destabilizing | 0.06 | Likely Benign | 0.641 | Likely Pathogenic | -5.95 | Deleterious | 0.999 | Probably Damaging | 0.935 | Probably Damaging | 3.18 | Benign | 0.00 | Affected | 0.0647 | 0.5693 | -3 | -2 | -0.5 | 76.10 | |||||||||||||||||||||||||||||
| c.2003C>G | S668C 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant S668C is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split opinion: benign predictions come from premPS, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions are returned by REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus. Uncertain results are reported by FoldX, Rosetta, and Foldetta. High‑accuracy assessments indicate that AlphaMissense‑Optimized predicts a benign effect, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) supports a pathogenic outcome; Foldetta remains inconclusive. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion does not contradict the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.247041 | Structured | 0.084935 | Uncertain | 0.922 | 0.370 | 0.000 | -12.815 | Likely Pathogenic | 0.758 | Likely Pathogenic | Likely Benign | 1.31 | Ambiguous | 0.6 | 1.36 | Ambiguous | 1.34 | Ambiguous | 0.18 | Likely Benign | 0.503 | Likely Pathogenic | -4.99 | Deleterious | 0.999 | Probably Damaging | 0.944 | Probably Damaging | 3.27 | Benign | 0.02 | Affected | 0.0962 | 0.5528 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.2005A>C | N669H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N669H has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) are uncertain and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion is not contradicted by any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.142424 | Structured | 0.086615 | Uncertain | 0.872 | 0.380 | 0.000 | -10.364 | Likely Pathogenic | 0.421 | Ambiguous | Likely Benign | 1.26 | Ambiguous | 0.2 | 1.69 | Ambiguous | 1.48 | Ambiguous | 0.80 | Ambiguous | 0.432 | Likely Benign | -4.49 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.35 | Benign | 0.01 | Affected | 0.1732 | 0.4839 | 2 | 1 | 0.3 | 23.04 | ||||||||||||||||||||||||||||||
| c.2005A>G | N669D 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N669D has no ClinVar entry and is not reported in gnomAD. Functional prediction tools show mixed results: benign predictions come from REVEL, Rosetta, Foldetta, SIFT, FATHMM, and AlphaMissense‑Optimized, whereas pathogenic predictions are reported by premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, leans toward pathogenicity (3/4 votes). High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized predicts benign, SGM‑Consensus (majority vote) predicts pathogenic, and Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, predicts benign. Overall, the majority of high‑confidence tools (AlphaMissense‑Optimized, Foldetta, and the SGM‑Consensus majority) suggest a pathogenic effect, but the presence of several benign predictions introduces uncertainty. Based on the current computational evidence, the variant is most likely pathogenic, which does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.086615 | Uncertain | 0.872 | 0.380 | 0.000 | -10.384 | Likely Pathogenic | 0.674 | Likely Pathogenic | Likely Benign | 0.53 | Ambiguous | 0.2 | 0.00 | Likely Benign | 0.27 | Likely Benign | 1.00 | Destabilizing | 0.336 | Likely Benign | -4.45 | Deleterious | 0.999 | Probably Damaging | 0.990 | Probably Damaging | 3.50 | Benign | 0.07 | Tolerated | 0.2182 | 0.2827 | 2 | 1 | 0.0 | 0.98 | |||||||||||||||||||||||||||||
| c.2005A>T | N669Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N669Y is not reported in ClinVar and has no entries in gnomAD. Prediction tools that agree on a benign effect include FoldX, FATHMM, and premPS. Those that predict a pathogenic effect comprise SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. With eight pathogenic predictions versus three benign, the overall evidence points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.086615 | Uncertain | 0.872 | 0.380 | 0.000 | -13.548 | Likely Pathogenic | 0.802 | Likely Pathogenic | Ambiguous | 0.45 | Likely Benign | 0.4 | 1.29 | Ambiguous | 0.87 | Ambiguous | 0.21 | Likely Benign | 0.546 | Likely Pathogenic | -7.01 | Deleterious | 1.000 | Probably Damaging | 0.997 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.0666 | 0.4151 | -2 | -2 | 2.2 | 49.07 | |||||||||||||||||||||||||||||
| c.2006A>C | N669T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N669T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. FoldX, Rosetta, and Foldetta give uncertain results. High‑accuracy methods give a split verdict: AlphaMissense‑Optimized classifies the variant as benign, whereas the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) predicts it to be pathogenic; Foldetta remains inconclusive. Overall, the majority of tools favor a pathogenic interpretation, and this assessment does not contradict the ClinVar status, which currently has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.086615 | Uncertain | 0.872 | 0.380 | 0.000 | -9.875 | Likely Pathogenic | 0.565 | Likely Pathogenic | Likely Benign | 0.74 | Ambiguous | 0.1 | 0.68 | Ambiguous | 0.71 | Ambiguous | 0.49 | Likely Benign | 0.292 | Likely Benign | -5.25 | Deleterious | 0.991 | Probably Damaging | 0.962 | Probably Damaging | 3.50 | Benign | 0.17 | Tolerated | 0.1504 | 0.4803 | 0 | 0 | 2.8 | -13.00 | |||||||||||||||||||||||||||||
| c.2006A>G | N669S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N669S is not reported in ClinVar (ClinVar ID None) but is present in gnomAD (ID 6‑33441265‑A‑G). Functional prediction tools that agree on a benign effect include REVEL, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 benign vs 2 pathogenic), and Foldetta (combining FoldX‑MD and Rosetta) is also inconclusive. No folding‑stability metrics (FoldX, Rosetta, Foldetta) provide definitive evidence. Overall, the majority of predictions lean toward a benign impact, and this is consistent with the absence of a ClinVar pathogenic classification. Thus, the variant is most likely benign, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.142424 | Structured | 0.086615 | Uncertain | 0.872 | 0.380 | 0.000 | 6-33441265-A-G | 3 | 1.86e-6 | -8.369 | Likely Pathogenic | 0.187 | Likely Benign | Likely Benign | 0.55 | Ambiguous | 0.1 | 1.88 | Ambiguous | 1.22 | Ambiguous | 0.35 | Likely Benign | 0.210 | Likely Benign | -4.02 | Deleterious | 0.999 | Probably Damaging | 0.960 | Probably Damaging | 3.52 | Benign | 0.14 | Tolerated | 3.39 | 27 | 0.3521 | 0.4480 | 1 | 1 | 2.7 | -27.03 | |||||||||||||||||||||||||
| c.2006A>T | N669I 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N669I is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect include premPS and FATHMM, whereas the remaining ten tools—SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the majority‑vote SGM‑Consensus—predict a pathogenic outcome. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain (treated as unavailable), SGM‑Consensus as likely pathogenic, and Foldetta as uncertain (also treated as unavailable). The overall consensus of the available predictions leans strongly toward pathogenicity, and this conclusion does not conflict with the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.086615 | Uncertain | 0.872 | 0.380 | 0.000 | -13.324 | Likely Pathogenic | 0.862 | Likely Pathogenic | Ambiguous | 0.84 | Ambiguous | 0.0 | 1.09 | Ambiguous | 0.97 | Ambiguous | 0.31 | Likely Benign | 0.517 | Likely Pathogenic | -8.18 | Deleterious | 0.999 | Probably Damaging | 0.996 | Probably Damaging | 3.34 | Benign | 0.00 | Affected | 0.0749 | 0.4697 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.2007T>A | N669K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N669K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign predictions come from REVEL, FoldX, and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, SGM‑Consensus confirms a Likely Pathogenic status, and Foldetta, which integrates FoldX‑MD and Rosetta outputs, yields an uncertain result. Overall, the majority of evidence points toward a pathogenic impact, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.086615 | Uncertain | 0.872 | 0.380 | 0.000 | -10.797 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 0.39 | Likely Benign | 0.3 | 1.50 | Ambiguous | 0.95 | Ambiguous | 0.94 | Ambiguous | 0.243 | Likely Benign | -5.35 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 3.41 | Benign | 0.03 | Affected | 0.2647 | 0.3312 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.2007T>G | N669K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N669K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, FoldX, and FATHMM, whereas pathogenic calls are made by PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, labels the variant as Likely Pathogenic. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized predicts pathogenicity, SGM‑Consensus concurs, and the Foldetta stability analysis is inconclusive and therefore not used as evidence. No other tools provide definitive support for benignity. Consequently, the preponderance of evidence points to a pathogenic impact. This conclusion is not contradicted by ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.142424 | Structured | 0.086615 | Uncertain | 0.872 | 0.380 | 0.000 | -10.797 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 0.39 | Likely Benign | 0.3 | 1.50 | Ambiguous | 0.95 | Ambiguous | 0.94 | Ambiguous | 0.243 | Likely Benign | -5.35 | Deleterious | 0.999 | Probably Damaging | 0.989 | Probably Damaging | 3.41 | Benign | 0.03 | Affected | 0.2647 | 0.3312 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.2008C>A | L670M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L670M occurs in the GAP domain. ClinVar contains no entry for this variant, but it is present in gnomAD (ID 6‑33441267‑C‑A). Prediction tools that classify the variant as benign include REVEL, FoldX, premPS, PROVEAN, SIFT, AlphaMissense‑Default, AlphaMissense‑Optimized, FATHMM, Foldetta, and the SGM‑Consensus score (Likely Benign). Tools that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b; Rosetta gives an uncertain result. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points to a benign effect, and this is consistent with the absence of a ClinVar pathogenic classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.161087 | Structured | 0.090855 | Uncertain | 0.812 | 0.385 | 0.000 | 6-33441267-C-A | 2 | 1.24e-6 | -9.438 | Likely Pathogenic | 0.125 | Likely Benign | Likely Benign | -0.11 | Likely Benign | 0.0 | 0.70 | Ambiguous | 0.30 | Likely Benign | 0.06 | Likely Benign | 0.038 | Likely Benign | -0.16 | Neutral | 0.970 | Probably Damaging | 0.777 | Possibly Damaging | 3.40 | Benign | 0.25 | Tolerated | 3.39 | 27 | 0.0826 | 0.3849 | 2 | 4 | -1.9 | 18.03 | ||||||||||||||||||||||||
| c.2008C>G | L670V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense change L670V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score all indicate benign. Only ESM1b predicts pathogenicity, while FoldX, Rosetta, and Foldetta provide uncertain or inconclusive stability results. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is benign; and Foldetta remains uncertain. Taken together, the overwhelming majority of evidence supports a benign interpretation, and this conclusion does not conflict with the absence of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.161087 | Structured | 0.090855 | Uncertain | 0.812 | 0.385 | 0.000 | -9.829 | Likely Pathogenic | 0.113 | Likely Benign | Likely Benign | 1.45 | Ambiguous | 0.1 | 1.05 | Ambiguous | 1.25 | Ambiguous | 0.15 | Likely Benign | 0.025 | Likely Benign | -0.60 | Neutral | 0.127 | Benign | 0.023 | Benign | 3.67 | Benign | 0.74 | Tolerated | 0.1558 | 0.4108 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2009T>A | L670Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L670Q is not reported in ClinVar and has no entry in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. Predictions that are inconclusive (FoldX, Rosetta, Foldetta, premPS) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus also as Likely Benign, while Foldetta remains uncertain. Overall, the majority of available evidence points to a benign impact. This conclusion is not contradicted by ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.161087 | Structured | 0.090855 | Uncertain | 0.812 | 0.385 | 0.000 | -8.217 | Likely Pathogenic | 0.306 | Likely Benign | Likely Benign | 1.02 | Ambiguous | 0.1 | 1.21 | Ambiguous | 1.12 | Ambiguous | 0.96 | Ambiguous | 0.152 | Likely Benign | -1.12 | Neutral | 1.000 | Probably Damaging | 0.988 | Probably Damaging | 3.40 | Benign | 0.36 | Tolerated | 0.1088 | 0.1303 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||
| c.2009T>C | L670P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L670P is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, premPS, PROVEAN, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) all indicate benign. In contrast, polyPhen‑2 (HumDiv and HumVar) predict a pathogenic impact. Stability‑based methods are inconclusive: FoldX, Rosetta, and the combined Foldetta output are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Overall, the majority of evidence supports a benign classification, and this is consistent with the absence of a ClinVar assertion; there is no contradiction with ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.161087 | Structured | 0.090855 | Uncertain | 0.812 | 0.385 | 0.000 | -4.916 | Likely Benign | 0.237 | Likely Benign | Likely Benign | 0.56 | Ambiguous | 0.3 | 1.83 | Ambiguous | 1.20 | Ambiguous | -0.25 | Likely Benign | 0.211 | Likely Benign | 2.42 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.41 | Benign | 0.26 | Tolerated | 0.3590 | 0.1474 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2009T>G | L670R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L670R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Functional prediction tools that agree on a benign effect include REVEL, PROVEAN, SIFT, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic impact are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. The remaining tools—FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Default—return uncertain or inconclusive results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as benign, and Foldetta as uncertain. Overall, the majority of reliable predictions indicate a benign effect. There is no ClinVar annotation to contradict this assessment, so the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.161087 | Structured | 0.090855 | Uncertain | 0.812 | 0.385 | 0.000 | -9.801 | Likely Pathogenic | 0.552 | Ambiguous | Likely Benign | 0.64 | Ambiguous | 0.1 | 1.42 | Ambiguous | 1.03 | Ambiguous | 0.67 | Ambiguous | 0.193 | Likely Benign | -1.74 | Neutral | 0.993 | Probably Damaging | 0.755 | Possibly Damaging | 3.43 | Benign | 0.41 | Tolerated | 0.1327 | 0.1145 | -3 | -2 | -8.3 | 43.03 | ||||||||||||||||||||||||||||||
| c.200T>A | L67Q 2D AIThe SynGAP1 missense variant L67Q is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Tools that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign and the SGM‑Consensus as Likely Benign; Foldetta results are not available. Overall, the majority of evidence points to a benign impact. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.473668 | Uncertain | 0.428 | 0.761 | 0.125 | -3.948 | Likely Benign | 0.687 | Likely Pathogenic | Likely Benign | 0.115 | Likely Benign | -0.66 | Neutral | 0.943 | Possibly Damaging | 0.766 | Possibly Damaging | 4.10 | Benign | 0.00 | Affected | 0.0861 | 0.0719 | -2 | -2 | -7.3 | 14.97 | |||||||||||||||||||||||||||||||||||||||
| c.200T>C | L67P 2D AIThe SynGAP1 missense variant L67P is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a “Likely Benign” classification. High‑accuracy assessments show AlphaMissense‑Optimized as “Uncertain” and the SGM‑Consensus as “Likely Benign”; Foldetta results are unavailable. Overall, the majority of evidence points toward a benign impact. The variant’s predicted benign status does not contradict any ClinVar annotation, as none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.473668 | Uncertain | 0.428 | 0.761 | 0.125 | -2.852 | Likely Benign | 0.945 | Likely Pathogenic | Ambiguous | 0.150 | Likely Benign | -0.80 | Neutral | 0.943 | Possibly Damaging | 0.766 | Possibly Damaging | 4.07 | Benign | 0.00 | Affected | 0.3052 | 0.1382 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.200T>G | L67R 2D AIThe SynGAP1 missense variant L67R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, and FATHMM, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, classifies the variant as Likely Benign. High‑accuracy assessments show AlphaMissense‑Optimized as Uncertain, SGM‑Consensus as Likely Benign, and Foldetta results are unavailable. Overall, the balance of evidence leans toward a benign interpretation, with no conflict with ClinVar status because no ClinVar assertion exists. Thus, the variant is most likely benign based on current predictive tools. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.458154 | Structured | 0.473668 | Uncertain | 0.428 | 0.761 | 0.125 | -3.430 | Likely Benign | 0.814 | Likely Pathogenic | Ambiguous | 0.115 | Likely Benign | -0.84 | Neutral | 0.943 | Possibly Damaging | 0.766 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 0.1159 | 0.0719 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2011G>A | D671N 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant D671N is reported in gnomAD (6‑33441270‑G‑A) but has no ClinVar entry. Functional prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is labeled Likely Pathogenic. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, while the SGM Consensus remains pathogenic. Overall, the predictions are split, with a slight bias toward benign outcomes from the majority of tools, but the consensus pathogenic signal from SGM and several high‑confidence predictors suggests uncertainty. The variant is most likely benign based on the preponderance of benign predictions, and this does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | 6-33441270-G-A | -10.347 | Likely Pathogenic | 0.685 | Likely Pathogenic | Likely Benign | 0.18 | Likely Benign | 0.1 | 0.39 | Likely Benign | 0.29 | Likely Benign | 0.19 | Likely Benign | 0.184 | Likely Benign | -3.19 | Deleterious | 0.887 | Possibly Damaging | 0.592 | Possibly Damaging | 3.36 | Benign | 0.02 | Affected | 3.39 | 27 | 0.1298 | 0.6463 | 1 | 2 | 0.0 | -0.98 | ||||||||||||||||||||||||||
| c.2011G>C | D671H 2D 3DClick to see structure in 3D Viewer AISynGAP1 D671H is not reported in ClinVar and is absent from gnomAD. Benign predictions are provided by REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). High‑accuracy tools give mixed results: AlphaMissense‑Optimized is uncertain, SGM Consensus predicts pathogenic, and Foldetta predicts benign. Overall, the balance of evidence leans toward pathogenicity, and this does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | -11.501 | Likely Pathogenic | 0.940 | Likely Pathogenic | Ambiguous | 0.49 | Likely Benign | 0.0 | 0.29 | Likely Benign | 0.39 | Likely Benign | 0.09 | Likely Benign | 0.300 | Likely Benign | -4.35 | Deleterious | 0.999 | Probably Damaging | 0.939 | Probably Damaging | 3.30 | Benign | 0.01 | Affected | 0.1627 | 0.6509 | 1 | -1 | 0.3 | 22.05 | |||||||||||||||||||||||||||||
| c.2011G>T | D671Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D671Y missense variant is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split consensus: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy methods give a mixed signal: AlphaMissense‑Optimized and the SGM‑Consensus both indicate pathogenicity, while Foldetta predicts a benign effect on protein stability. Because the majority of standard predictors lean toward pathogenicity and the two high‑confidence tools also favor a deleterious outcome, the variant is most likely pathogenic. This assessment does not contradict ClinVar, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | -13.107 | Likely Pathogenic | 0.964 | Likely Pathogenic | Likely Pathogenic | 0.31 | Likely Benign | 0.1 | 0.05 | Likely Benign | 0.18 | Likely Benign | 0.11 | Likely Benign | 0.385 | Likely Benign | -6.05 | Deleterious | 1.000 | Probably Damaging | 0.961 | Probably Damaging | 3.29 | Benign | 0.00 | Affected | 0.0564 | 0.6376 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.2012A>C | D671A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D671A missense variant is not reported in ClinVar and has no entries in gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, premPS, and FATHMM. Those that predict a pathogenic effect comprise PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (derived from the majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the balance of evidence favors a pathogenic interpretation, with seven tools supporting pathogenicity versus four supporting benignity. This conclusion does not conflict with ClinVar, which contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | -11.709 | Likely Pathogenic | 0.911 | Likely Pathogenic | Ambiguous | 0.25 | Likely Benign | 0.1 | 0.79 | Ambiguous | 0.52 | Ambiguous | 0.10 | Likely Benign | 0.245 | Likely Benign | -5.08 | Deleterious | 0.980 | Probably Damaging | 0.804 | Possibly Damaging | 3.34 | Benign | 0.03 | Affected | 0.4056 | 0.5895 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.2012A>G | D671G 2D AIThe SynGAP1 D671G missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, and FATHMM, whereas a majority of tools predict a pathogenic impact: SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as Likely Pathogenic (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as uncertain. No evidence from these tools contradicts the pathogenic prediction. Overall, the balance of evidence favors a pathogenic classification for D671G, and this conclusion is consistent with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | -10.346 | Likely Pathogenic | 0.857 | Likely Pathogenic | Ambiguous | 0.50 | Ambiguous | 0.5 | 1.53 | Ambiguous | 1.02 | Ambiguous | 0.00 | Likely Benign | 0.279 | Likely Benign | -4.78 | Deleterious | 0.995 | Probably Damaging | 0.946 | Probably Damaging | 3.45 | Benign | 0.01 | Affected | 0.4032 | 0.5823 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.2012A>T | D671V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D671V missense variant is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM. Tools that predict a pathogenic effect include PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. The high‑accuracy consensus methods give a pathogenic signal: AlphaMissense‑Optimized is pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is also pathogenic. Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, is uncertain. Overall, the majority of predictions (seven pathogenic vs. three benign) support a pathogenic classification. There is no ClinVar entry to contradict this assessment, so the variant is most likely pathogenic based on the available computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | -12.376 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 0.63 | Ambiguous | 0.1 | 0.51 | Ambiguous | 0.57 | Ambiguous | 0.14 | Likely Benign | 0.379 | Likely Benign | -6.08 | Deleterious | 0.975 | Probably Damaging | 0.885 | Possibly Damaging | 3.31 | Benign | 0.01 | Affected | 0.0820 | 0.6651 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.2013C>A | D671E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D671E is not reported in ClinVar and is absent from gnomAD. Consensus predictions from multiple in‑silico tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and FATHMM all classify the substitution as benign. The only inconclusive result is AlphaMissense‑Default, which is listed as uncertain; all other high‑accuracy predictors support a benign outcome. In particular, AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports benign. No pathogenic predictions are present. Therefore, based on the available computational evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar annotation (none exists). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | -3.657 | Likely Benign | 0.371 | Ambiguous | Likely Benign | -0.17 | Likely Benign | 0.1 | 0.22 | Likely Benign | 0.03 | Likely Benign | 0.03 | Likely Benign | 0.066 | Likely Benign | -0.83 | Neutral | 0.013 | Benign | 0.009 | Benign | 3.47 | Benign | 0.53 | Tolerated | 0.1520 | 0.6388 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.2013C>G | D671E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D671E is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome. The high‑accuracy assessments are consistent: AlphaMissense‑Optimized indicates Benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also reports Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Benign. With all available evidence pointing to a benign effect and no ClinVar record to contradict this, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.194234 | Structured | 0.096749 | Uncertain | 0.677 | 0.370 | 0.000 | -3.657 | Likely Benign | 0.371 | Ambiguous | Likely Benign | -0.17 | Likely Benign | 0.1 | 0.22 | Likely Benign | 0.03 | Likely Benign | 0.03 | Likely Benign | 0.066 | Likely Benign | -0.83 | Neutral | 0.013 | Benign | 0.009 | Benign | 3.47 | Benign | 0.53 | Tolerated | 0.1520 | 0.6388 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.2014A>C | T672P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T672P missense variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect comprise FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Uncertain results come from premPS and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as pathogenic. No predictions are missing or inconclusive. Overall, the majority of evidence points to a pathogenic impact for T672P. This conclusion is not contradicted by ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.116183 | Structured | 0.102069 | Uncertain | 0.586 | 0.362 | 0.000 | -11.022 | Likely Pathogenic | 0.346 | Ambiguous | Likely Benign | 2.22 | Destabilizing | 0.2 | 5.54 | Destabilizing | 3.88 | Destabilizing | 0.55 | Ambiguous | 0.087 | Likely Benign | -4.20 | Deleterious | 0.968 | Probably Damaging | 0.824 | Possibly Damaging | 3.40 | Benign | 0.01 | Affected | 0.1988 | 0.5532 | 0 | -1 | -0.9 | -3.99 | ||||||||||||||||||||||||||||||
| c.2014A>T | T672S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T672S is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess pathogenicity uniformly indicate a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS (uncertain), PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as benign or likely benign. No tool predicts pathogenicity. High‑accuracy methods corroborate this: AlphaMissense‑Optimized reports benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) yields “Likely Benign”; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Consequently, the variant is most likely benign, and this assessment does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.116183 | Structured | 0.102069 | Uncertain | 0.586 | 0.362 | 0.000 | -4.932 | Likely Benign | 0.097 | Likely Benign | Likely Benign | -0.13 | Likely Benign | 0.1 | 0.29 | Likely Benign | 0.08 | Likely Benign | 0.61 | Ambiguous | 0.044 | Likely Benign | -2.32 | Neutral | 0.006 | Benign | 0.010 | Benign | 3.43 | Benign | 0.09 | Tolerated | 0.3005 | 0.4282 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.2015C>G | T672R 2D  3DClick to see structure in 3D Viewer AISynGAP1 missense variant T672R is not reported in ClinVar and is absent from gnomAD. Prediction tools that indicate a benign effect include REVEL, FoldX, Foldetta, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). Uncertain results from Rosetta and premPS are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, while the SGM Consensus indicates a likely pathogenic outcome. Overall, the majority of individual predictors lean toward pathogenicity, but the high‑accuracy tools provide conflicting benign signals. Therefore, the variant is most likely pathogenic based on the collective predictions, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.116183 | Structured | 0.102069 | Uncertain | 0.586 | 0.362 | 0.000 | -12.454 | Likely Pathogenic | 0.626 | Likely Pathogenic | Likely Benign | -0.48 | Likely Benign | 0.4 | 1.19 | Ambiguous | 0.36 | Likely Benign | 0.60 | Ambiguous | 0.084 | Likely Benign | -4.47 | Deleterious | 0.886 | Possibly Damaging | 0.377 | Benign | 3.43 | Benign | 0.02 | Affected | 0.0996 | 0.2919 | -1 | -1 | -3.8 | 55.08 | |||||||||||||||||||||||||||||
| c.2017C>A | L673I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L673I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. The remaining tools (FoldX, Rosetta, Foldetta, ESM1b) return uncertain or inconclusive results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as likely benign, and Foldetta as uncertain. Taken together, the majority of reliable predictors indicate a benign effect, and there is no conflict with ClinVar status (which has no entry). Therefore, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.104692 | Uncertain | 0.545 | 0.369 | 0.000 | -7.823 | In-Between | 0.099 | Likely Benign | Likely Benign | 1.51 | Ambiguous | 0.2 | 1.89 | Ambiguous | 1.70 | Ambiguous | -0.02 | Likely Benign | 0.036 | Likely Benign | -0.14 | Neutral | 0.535 | Possibly Damaging | 0.112 | Benign | 3.37 | Benign | 0.47 | Tolerated | 0.0910 | 0.3689 | 2 | 2 | 0.7 | 0.00 | |||||||||||||||||||||||||||||
| c.2017C>G | L673V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L673V is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include SGM‑Consensus (Likely Benign), REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic outcome are FoldX, Rosetta, Foldetta, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus as Likely Benign, and Foldetta as pathogenic, yielding one pathogenic versus two benign predictions. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.060549 | Structured | 0.104692 | Uncertain | 0.545 | 0.369 | 0.000 | -8.132 | Likely Pathogenic | 0.099 | Likely Benign | Likely Benign | 2.18 | Destabilizing | 0.3 | 2.10 | Destabilizing | 2.14 | Destabilizing | 0.10 | Likely Benign | 0.017 | Likely Benign | -0.58 | Neutral | 0.016 | Benign | 0.003 | Benign | 3.36 | Benign | 0.28 | Tolerated | 0.1448 | 0.3777 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2018T>A | L673Q 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L673Q is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Tools that agree on a pathogenic effect include Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. FoldX and Foldetta, as well as AlphaMissense‑Default, are inconclusive and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized predicting benign, while the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts pathogenic. Foldetta remains uncertain. Overall, the majority of available predictions (seven pathogenic vs. three benign) indicate that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar claim exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.060549 | Structured | 0.104692 | Uncertain | 0.545 | 0.369 | 0.000 | -11.963 | Likely Pathogenic | 0.445 | Ambiguous | Likely Benign | 1.56 | Ambiguous | 0.2 | 2.32 | Destabilizing | 1.94 | Ambiguous | 1.02 | Destabilizing | 0.190 | Likely Benign | -3.40 | Deleterious | 1.000 | Probably Damaging | 0.968 | Probably Damaging | 3.32 | Benign | 0.03 | Affected | 0.1045 | 0.1172 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||
| c.2018T>C | L673P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L673P is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. All other evaluated tools—SGM‑Consensus, FoldX, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—predict a pathogenic impact. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized indicates benign, but the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts Pathogenic. Taken together, the overwhelming majority of predictions, including the high‑accuracy tools, classify the variant as pathogenic. This conclusion is not contradicted by ClinVar, which contains no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.104692 | Uncertain | 0.545 | 0.369 | 0.000 | -12.160 | Likely Pathogenic | 0.622 | Likely Pathogenic | Likely Benign | 2.41 | Destabilizing | 0.3 | 1.63 | Ambiguous | 2.02 | Destabilizing | 1.17 | Destabilizing | 0.207 | Likely Benign | -4.10 | Deleterious | 1.000 | Probably Damaging | 0.978 | Probably Damaging | 3.37 | Benign | 0.02 | Affected | 0.3552 | 0.1474 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||
| c.2018T>G | L673R 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant L673R is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, polyPhen‑2 HumVar, FATHMM, and AlphaMissense‑Optimized; pathogenic calls come from Rosetta, premPS, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, and the SGM Consensus (majority vote). FoldX and Foldetta are inconclusive. High‑accuracy assessments give a benign result from AlphaMissense‑Optimized, a likely pathogenic verdict from the SGM Consensus, and an uncertain outcome from Foldetta. Overall, the majority of evidence points toward pathogenicity, and this conclusion does not conflict with the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.060549 | Structured | 0.104692 | Uncertain | 0.545 | 0.369 | 0.000 | -14.474 | Likely Pathogenic | 0.714 | Likely Pathogenic | Likely Benign | 1.10 | Ambiguous | 0.2 | 2.04 | Destabilizing | 1.57 | Ambiguous | 1.23 | Destabilizing | 0.178 | Likely Benign | -3.81 | Deleterious | 0.991 | Probably Damaging | 0.433 | Benign | 3.36 | Benign | 0.03 | Affected | 0.1348 | 0.1015 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||
| c.2020A>C | T674P 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 T674P missense change is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized; pathogenic predictions come from Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. The high‑accuracy AlphaMissense‑Optimized predicts benign, while Foldetta predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive because it yields a 2‑to‑2 split. No other high‑accuracy tool provides a definitive call. Consequently, the evidence is evenly split between benign and pathogenic, leaving the variant’s clinical significance uncertain. This uncertainty does not conflict with the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | -8.661 | Likely Pathogenic | 0.109 | Likely Benign | Likely Benign | 0.91 | Ambiguous | 0.6 | 4.12 | Destabilizing | 2.52 | Destabilizing | 0.12 | Likely Benign | 0.143 | Likely Benign | -2.69 | Deleterious | 0.995 | Probably Damaging | 0.891 | Possibly Damaging | 3.50 | Benign | 0.16 | Tolerated | 0.2039 | 0.6103 | 0 | -1 | -0.9 | -3.99 | ||||||||||||||||||||||||||||||
| c.2020A>G | T674A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T674A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools uniformly favor a benign effect: AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, SIFT, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, REVEL, FoldX, premPS, Foldetta, and the SGM‑Consensus score all indicate benign or likely benign. No tool predicts pathogenicity; the only inconclusive result comes from Rosetta, which is listed as uncertain. High‑accuracy methods corroborate the benign assessment: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Based on the aggregate predictions, the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | -5.915 | Likely Benign | 0.099 | Likely Benign | Likely Benign | -0.13 | Likely Benign | 0.0 | 0.89 | Ambiguous | 0.38 | Likely Benign | 0.29 | Likely Benign | 0.052 | Likely Benign | -1.48 | Neutral | 0.398 | Benign | 0.045 | Benign | 3.45 | Benign | 0.28 | Tolerated | 0.4042 | 0.4845 | 1 | 0 | 2.5 | -30.03 | |||||||||||||||||||||||||||||
| c.2020A>T | T674S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T674S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools uniformly indicate a benign effect: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, Rosetta, and AlphaMissense‑Default all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. With all available evidence pointing to a benign outcome and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | -5.320 | Likely Benign | 0.082 | Likely Benign | Likely Benign | -0.23 | Likely Benign | 0.0 | 0.44 | Likely Benign | 0.11 | Likely Benign | -0.14 | Likely Benign | 0.084 | Likely Benign | 0.47 | Neutral | 0.107 | Benign | 0.033 | Benign | 3.47 | Benign | 1.00 | Tolerated | 0.3329 | 0.4938 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.2021C>A | T674N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T674N is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, SIFT, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (Likely Benign). Only the two polyPhen‑2 implementations (HumDiv and HumVar) predict a pathogenic outcome; ESM1b is uncertain and does not influence the consensus. High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized reports Benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts Benign. No contradictory evidence is present. **Based on the aggregate predictions, the variant is most likely benign, and this conclusion does not conflict with the ClinVar status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | -7.839 | In-Between | 0.135 | Likely Benign | Likely Benign | -0.21 | Likely Benign | 0.0 | 0.15 | Likely Benign | -0.03 | Likely Benign | 0.19 | Likely Benign | 0.119 | Likely Benign | -0.71 | Neutral | 0.958 | Probably Damaging | 0.671 | Possibly Damaging | 3.44 | Benign | 0.17 | Tolerated | 0.1263 | 0.4979 | 0 | 0 | -2.8 | 13.00 | |||||||||||||||||||||||||||||
| c.2021C>G | T674S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T674S is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Functional prediction tools uniformly indicate a benign effect: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, Rosetta, and AlphaMissense‑Default all classify the change as benign. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields “Likely Benign”; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also reports a benign effect. With all available evidence pointing to a benign outcome and no conflicting ClinVar annotation, the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | -5.320 | Likely Benign | 0.082 | Likely Benign | Likely Benign | -0.23 | Likely Benign | 0.0 | 0.44 | Likely Benign | 0.11 | Likely Benign | -0.14 | Likely Benign | 0.088 | Likely Benign | 0.47 | Neutral | 0.107 | Benign | 0.033 | Benign | 3.47 | Benign | 1.00 | Tolerated | 0.3329 | 0.4938 | 1 | 1 | -0.1 | -14.03 | |||||||||||||||||||||||||||||
| c.2021C>T | T674I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant T674I is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (ID 6‑33441280‑C‑T). Functional prediction tools cluster into two groups: benign predictions come from REVEL, FoldX, Rosetta, Foldetta, premPS, SIFT, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from PROVEAN, polyPhen‑2 HumDiv, and ESM1b (polyPhen‑2 HumVar is benign). AlphaMissense‑Default is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic. Overall, the majority of evidence points to a benign effect, and this conclusion does not contradict the ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.109297 | Uncertain | 0.521 | 0.349 | 0.000 | 6-33441280-C-T | 2 | 1.24e-6 | -8.951 | Likely Pathogenic | 0.522 | Ambiguous | Likely Benign | -0.05 | Likely Benign | 0.1 | 0.28 | Likely Benign | 0.12 | Likely Benign | -0.04 | Likely Benign | 0.289 | Likely Benign | -3.18 | Deleterious | 0.981 | Probably Damaging | 0.444 | Benign | 3.46 | Benign | 0.11 | Tolerated | 3.40 | 24 | 0.0898 | 0.6945 | -1 | 0 | 5.2 | 12.05 | |||||||||||||||||||||||||
| c.2023A>C | N675H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N675H is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), and SIFT. Predictions that are inconclusive are Foldetta and premPS. High‑accuracy methods give a benign result from AlphaMissense‑Optimized, a benign consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN), and an uncertain result from Foldetta. Taken together, the majority of evidence points to a benign impact, and this does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.129801 | Structured | 0.111024 | Uncertain | 0.513 | 0.333 | 0.000 | -5.593 | Likely Benign | 0.254 | Likely Benign | Likely Benign | 3.06 | Destabilizing | 1.1 | -0.16 | Likely Benign | 1.45 | Ambiguous | 0.56 | Ambiguous | 0.186 | Likely Benign | -2.62 | Deleterious | 0.999 | Probably Damaging | 0.929 | Probably Damaging | 3.39 | Benign | 0.04 | Affected | 0.1478 | 0.7478 | 2 | 1 | 0.3 | 23.04 | |||||||||||||||||||||||||||||
| c.2023A>G | N675D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N675D is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, Rosetta, SIFT, FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and ESM1b. High‑accuracy assessments give AlphaMissense‑Optimized a benign call, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) a pathogenic call, and Foldetta an uncertain result. With an equal split of general predictions but a pathogenic majority in the high‑accuracy consensus, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.111024 | Uncertain | 0.513 | 0.333 | 0.000 | -12.829 | Likely Pathogenic | 0.497 | Ambiguous | Likely Benign | 1.41 | Ambiguous | 0.4 | -0.26 | Likely Benign | 0.58 | Ambiguous | 1.05 | Destabilizing | 0.246 | Likely Benign | -3.87 | Deleterious | 0.997 | Probably Damaging | 0.865 | Possibly Damaging | 3.53 | Benign | 0.17 | Tolerated | 0.1914 | 0.4901 | 2 | 1 | 0.0 | 0.98 | ||||||||||||||||||||||||||||||
| c.2023A>T | N675Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N675Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. Tools with uncertain or inconclusive results are Rosetta, Foldetta, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of available predictions (six pathogenic vs. four benign) indicate that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.111024 | Uncertain | 0.513 | 0.333 | 0.000 | -9.981 | Likely Pathogenic | 0.553 | Ambiguous | Likely Benign | 3.02 | Destabilizing | 1.3 | -0.81 | Ambiguous | 1.11 | Ambiguous | 0.19 | Likely Benign | 0.439 | Likely Benign | -5.86 | Deleterious | 1.000 | Probably Damaging | 0.955 | Probably Damaging | 3.38 | Benign | 0.01 | Affected | 0.0603 | 0.6417 | -2 | -2 | 2.2 | 49.07 | ||||||||||||||||||||||||||||||
| c.2024A>C | N675T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N675T is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and Foldetta. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. Uncertain results come from Rosetta and premPS. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. No evidence contradicts the ClinVar status. Overall, the balance of evidence, especially from the high‑accuracy tools, indicates that the variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.111024 | Uncertain | 0.513 | 0.333 | 0.000 | -10.636 | Likely Pathogenic | 0.208 | Likely Benign | Likely Benign | 0.36 | Likely Benign | 0.3 | -0.67 | Ambiguous | -0.16 | Likely Benign | 0.56 | Ambiguous | 0.258 | Likely Benign | -4.63 | Deleterious | 0.991 | Probably Damaging | 0.710 | Possibly Damaging | 3.49 | Benign | 0.05 | Affected | 0.1283 | 0.7910 | 0 | 0 | 2.8 | -13.00 | ||||||||||||||||||||||||||||||
| c.2024A>G | N675S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N675S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, FATHMM, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict a pathogenic outcome are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Two tools (premPS and ESM1b) return uncertain results. High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) yields a benign consensus; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. Taken together, the preponderance of evidence indicates that the variant is most likely benign, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.129801 | Structured | 0.111024 | Uncertain | 0.513 | 0.333 | 0.000 | -7.871 | In-Between | 0.081 | Likely Benign | Likely Benign | 0.37 | Likely Benign | 0.0 | -0.43 | Likely Benign | -0.03 | Likely Benign | 0.61 | Ambiguous | 0.157 | Likely Benign | -3.78 | Deleterious | 0.993 | Probably Damaging | 0.606 | Possibly Damaging | 3.50 | Benign | 0.11 | Tolerated | 0.3462 | 0.7587 | 1 | 1 | 2.7 | -27.03 | ||||||||||||||||||||||||||||||
| c.2024A>T | N675I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N675I is not reported in ClinVar (ClinVar ID None) and has no entries in gnomAD (gnomAD ID None). Prediction tools cluster into two groups: benign predictions come from REVEL, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized; pathogenic predictions come from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. FoldX and Rosetta give uncertain results and are treated as unavailable. High‑accuracy methods give mixed outcomes: AlphaMissense‑Optimized predicts benign, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) predicts likely pathogenic, and Foldetta predicts benign. Overall, the majority of tools (7/12) indicate pathogenicity, while 5/12 indicate benign. Thus, the variant is most likely pathogenic based on the current computational evidence, and this assessment does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.111024 | Uncertain | 0.513 | 0.333 | 0.000 | -13.254 | Likely Pathogenic | 0.574 | Likely Pathogenic | Likely Benign | 1.00 | Ambiguous | 0.1 | -0.97 | Ambiguous | 0.02 | Likely Benign | 0.41 | Likely Benign | 0.338 | Likely Benign | -7.37 | Deleterious | 0.999 | Probably Damaging | 0.955 | Probably Damaging | 3.37 | Benign | 0.00 | Affected | 0.0620 | 0.7188 | -2 | -3 | 8.0 | -0.94 | |||||||||||||||||||||||||||||
| c.2025C>A | N675K 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant N675K is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show a split: benign calls come from REVEL, Rosetta, and FATHMM, whereas pathogenic calls are made by FoldX, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. Uncertain results are reported for Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments give a mixed picture: AlphaMissense‑Optimized is inconclusive, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta remains uncertain. Overall, the majority of evidence points toward a pathogenic effect, and this conclusion does not conflict with ClinVar, which currently contains no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.111024 | Uncertain | 0.513 | 0.333 | 0.000 | -12.851 | Likely Pathogenic | 0.898 | Likely Pathogenic | Ambiguous | 2.88 | Destabilizing | 1.2 | 0.06 | Likely Benign | 1.47 | Ambiguous | 0.74 | Ambiguous | 0.177 | Likely Benign | -4.75 | Deleterious | 0.993 | Probably Damaging | 0.688 | Possibly Damaging | 3.44 | Benign | 0.02 | Affected | 0.2424 | 0.6040 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.2025C>G | N675K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant N675K is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM, whereas a majority (FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact. Tools with inconclusive results are Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the balance of evidence favors a pathogenic classification for N675K, and this conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.129801 | Structured | 0.111024 | Uncertain | 0.513 | 0.333 | 0.000 | -12.851 | Likely Pathogenic | 0.898 | Likely Pathogenic | Ambiguous | 2.88 | Destabilizing | 1.2 | 0.06 | Likely Benign | 1.47 | Ambiguous | 0.74 | Ambiguous | 0.177 | Likely Benign | -4.75 | Deleterious | 0.993 | Probably Damaging | 0.688 | Possibly Damaging | 3.44 | Benign | 0.02 | Affected | 0.2424 | 0.6040 | 1 | 0 | -0.4 | 14.07 | |||||||||||||||||||||||||||||
| c.2026A>C | S676R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, FATHMM, and polyPhen‑2 HumVar, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as inconclusive, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or folding stability result is missing; all available data are reported. Overall, the balance of evidence (six benign versus four pathogenic predictions, with the high‑accuracy Foldetta supporting benign) indicates that the variant is most likely benign. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -10.665 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 0.22 | Likely Benign | 0.3 | 0.74 | Ambiguous | 0.48 | Likely Benign | 0.45 | Likely Benign | 0.136 | Likely Benign | -2.34 | Neutral | 0.891 | Possibly Damaging | 0.278 | Benign | 3.40 | Benign | 0.02 | Affected | 0.0962 | 0.3454 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||
| c.2026A>G | S676G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and SIFT. Two tools (ESM1b and premPS) returned uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also favors benign, and Foldetta predicts a benign stability change. No prediction or folding result is missing or inconclusive. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict ClinVar status, as the variant is not yet classified in that database. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -7.249 | In-Between | 0.117 | Likely Benign | Likely Benign | 0.38 | Likely Benign | 0.1 | 0.25 | Likely Benign | 0.32 | Likely Benign | 0.67 | Ambiguous | 0.132 | Likely Benign | -2.74 | Deleterious | 0.855 | Possibly Damaging | 0.100 | Benign | 3.39 | Benign | 0.02 | Affected | 0.2772 | 0.4342 | 1 | 0 | 0.4 | -30.03 | ||||||||||||||||||||||||||||||
| c.2026A>T | S676C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676C is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen2_HumVar, and the SGM‑Consensus (majority vote). Tools that predict a pathogenic effect are polyPhen2_HumDiv, SIFT, and ESM1b. Rosetta and Foldetta give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM‑Consensus as likely benign, and Foldetta as uncertain. Based on the overall consensus of the available predictions, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -9.230 | Likely Pathogenic | 0.132 | Likely Benign | Likely Benign | 0.47 | Likely Benign | 0.1 | 0.77 | Ambiguous | 0.62 | Ambiguous | 0.15 | Likely Benign | 0.164 | Likely Benign | -2.45 | Neutral | 0.959 | Probably Damaging | 0.431 | Benign | 3.35 | Benign | 0.01 | Affected | 0.1113 | 0.6352 | 0 | -1 | 3.3 | 16.06 | |||||||||||||||||||||||||||||
| c.2027G>A | S676N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676N is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote). Only SIFT predicts a pathogenic outcome. Predictions that are inconclusive (FoldX, premPS, ESM1b) are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus, derived from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates benign. Foldetta, which integrates FoldX‑MD and Rosetta outputs, is unavailable because one component (FoldX) is uncertain. Overall, the majority of evidence points to a benign effect, and this is consistent with the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -7.486 | In-Between | 0.282 | Likely Benign | Likely Benign | 0.60 | Ambiguous | 0.3 | -0.21 | Likely Benign | 0.20 | Likely Benign | 0.55 | Ambiguous | 0.099 | Likely Benign | -1.63 | Neutral | 0.438 | Benign | 0.036 | Benign | 3.42 | Benign | 0.05 | Affected | 0.1454 | 0.4599 | 1 | 1 | -2.7 | 27.03 | |||||||||||||||||||||||||||||
| c.2027G>C | S676T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676T is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and Rosetta. No tool predicts a pathogenic outcome; the only inconclusive results are from FoldX (Uncertain) and Foldetta (Uncertain). High‑accuracy assessments further support a benign interpretation: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta remains Uncertain. Overall, the consensus of available predictions indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar status because the variant is not yet catalogued there. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -5.621 | Likely Benign | 0.092 | Likely Benign | Likely Benign | 0.79 | Ambiguous | 0.1 | 0.34 | Likely Benign | 0.57 | Ambiguous | -0.36 | Likely Benign | 0.098 | Likely Benign | 0.91 | Neutral | 0.050 | Benign | 0.017 | Benign | 3.52 | Benign | 1.00 | Tolerated | 0.1575 | 0.6286 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.2027G>T | S676I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676I is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, premPS, PROVEAN, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, and ESM1b. FoldX, Rosetta, Foldetta, and AlphaMissense‑Default are inconclusive. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, while Foldetta remains uncertain. Overall, the balance of evidence (five benign versus three pathogenic predictions) indicates that the variant is most likely benign, and this conclusion does not contradict any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -10.165 | Likely Pathogenic | 0.383 | Ambiguous | Likely Benign | 1.25 | Ambiguous | 0.3 | 1.12 | Ambiguous | 1.19 | Ambiguous | 0.14 | Likely Benign | 0.170 | Likely Benign | -2.46 | Neutral | 0.801 | Possibly Damaging | 0.063 | Benign | 3.38 | Benign | 0.02 | Affected | 0.0879 | 0.5720 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||
| c.2028C>A | S676R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, FATHMM, and polyPhen‑2 HumVar, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as inconclusive, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or folding stability result is missing; all available data are included. Overall, the balance of evidence (six benign versus four pathogenic predictions, with the high‑accuracy Foldetta supporting benign) indicates that the variant is most likely benign. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -10.665 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 0.22 | Likely Benign | 0.3 | 0.74 | Ambiguous | 0.48 | Likely Benign | 0.45 | Likely Benign | 0.157 | Likely Benign | -2.34 | Neutral | 0.891 | Possibly Damaging | 0.278 | Benign | 3.40 | Benign | 0.02 | Affected | 0.0962 | 0.3454 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||
| c.2028C>G | S676R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S676R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, PROVEAN, FATHMM, and polyPhen‑2 HumVar, while those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as inconclusive, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. No prediction or folding stability result is missing; all available data are included. Overall, the balance of evidence (six benign versus four pathogenic predictions, with the high‑accuracy Foldetta supporting benign) indicates that the variant is most likely benign. This conclusion is not contradicted by ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.209395 | Structured | 0.113632 | Uncertain | 0.551 | 0.338 | 0.125 | -10.665 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 0.22 | Likely Benign | 0.3 | 0.74 | Ambiguous | 0.48 | Likely Benign | 0.45 | Likely Benign | 0.156 | Likely Benign | -2.34 | Neutral | 0.891 | Possibly Damaging | 0.278 | Benign | 3.40 | Benign | 0.02 | Affected | 0.0962 | 0.3454 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||
| c.2029A>C | S677R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and FATHMM. Those that predict a pathogenic effect are FoldX, ESM1b, and AlphaMissense‑Default. Uncertain results come from Rosetta, Foldetta, and premPS. High‑accuracy assessments show AlphaMissense‑Optimized predicts a benign outcome, while the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta is also inconclusive. Overall, the majority of tools and the high‑accuracy benign prediction suggest the variant is most likely benign, and this assessment does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | -9.388 | Likely Pathogenic | 0.714 | Likely Pathogenic | Likely Benign | 2.59 | Destabilizing | 0.1 | 0.73 | Ambiguous | 1.66 | Ambiguous | 0.71 | Ambiguous | 0.104 | Likely Benign | -2.49 | Neutral | 0.385 | Benign | 0.037 | Benign | 3.31 | Benign | 0.12 | Tolerated | 0.1120 | 0.4195 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||
| c.2029A>G | S677G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677G is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the substitution as benign. No tool predicts pathogenicity. Predictions that are inconclusive—FoldX, Rosetta, Foldetta, and premPS—are treated as unavailable. High‑accuracy assessments reinforce the benign interpretation: AlphaMissense‑Optimized is benign; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates benign; Foldetta, which integrates FoldX‑MD and Rosetta outputs, remains uncertain. Overall, the evidence supports a benign classification for S677G, and this conclusion does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | -5.126 | Likely Benign | 0.060 | Likely Benign | Likely Benign | 0.98 | Ambiguous | 0.1 | 1.20 | Ambiguous | 1.09 | Ambiguous | 0.64 | Ambiguous | 0.126 | Likely Benign | -1.38 | Neutral | 0.002 | Benign | 0.001 | Benign | 3.30 | Benign | 0.26 | Tolerated | 0.2885 | 0.5325 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||
| c.2029A>T | S677C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677C is reported in ClinVar as Benign (ClinVar ID 2825814.0) and is not present in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, Foldetta, premPS, PROVEAN, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, polyPhen‑2 HumVar, and the SGM‑Consensus (Likely Benign). In contrast, polyPhen‑2 HumDiv, SIFT, and ESM1b predict a pathogenic impact. High‑accuracy predictors all support a benign outcome: AlphaMissense‑Optimized is benign, the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No prediction or folding‑stability result is missing or inconclusive. Based on the preponderance of evidence, the variant is most likely benign, and this assessment aligns with its ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | Benign | 1 | -8.496 | Likely Pathogenic | 0.076 | Likely Benign | Likely Benign | -0.51 | Ambiguous | 0.3 | -0.30 | Likely Benign | -0.41 | Likely Benign | 0.15 | Likely Benign | 0.153 | Likely Benign | -2.41 | Neutral | 0.932 | Possibly Damaging | 0.222 | Benign | 3.25 | Benign | 0.04 | Affected | 3.41 | 23 | 0.1375 | 0.6697 | -1 | 0 | 3.3 | 16.06 | |||||||||||||||||||||||||
| c.202C>A | L68M 2D AIThe SynGAP1 missense variant L68M is not reported in ClinVar and is absent from gnomAD. Consensus from multiple in‑silico predictors shows a split: benign calls come from REVEL, PROVEAN, ESM1b, FATHMM, and AlphaMissense‑Optimized, while pathogenic calls arise from polyPhen‑2 (HumDiv and HumVar) and SIFT. The AlphaMissense‑Default tool remains uncertain. High‑accuracy assessments reinforce the benign trend: AlphaMissense‑Optimized predicts benign, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) reports Likely Benign, and no Foldetta stability data are available. Overall, the majority of evidence points to a benign effect. This conclusion is consistent with the lack of ClinVar annotation and gnomAD presence, so there is no contradiction with existing clinical databases. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.470567 | Uncertain | 0.405 | 0.768 | 0.250 | -5.580 | Likely Benign | 0.442 | Ambiguous | Likely Benign | 0.094 | Likely Benign | -0.13 | Neutral | 0.824 | Possibly Damaging | 0.665 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 0.0565 | 0.2762 | 4 | 2 | -1.9 | 18.03 | |||||||||||||||||||||||||||||||||||||||
| c.202C>G | L68V 2D AIThe SynGAP1 missense variant L68V has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumVar, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are polyPhen‑2 HumDiv and SIFT, while AlphaMissense‑Default remains uncertain. The high‑accuracy consensus from SGM (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as Likely Benign, and AlphaMissense‑Optimized independently predicts Benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of evidence points to a benign impact, and this assessment does not contradict any ClinVar status because no ClinVar classification exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.470567 | Uncertain | 0.405 | 0.768 | 0.250 | -4.079 | Likely Benign | 0.470 | Ambiguous | Likely Benign | 0.028 | Likely Benign | -0.43 | Neutral | 0.458 | Possibly Damaging | 0.364 | Benign | 4.13 | Benign | 0.00 | Affected | 0.1077 | 0.2817 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||||||||||||
| c.2030G>A | S677N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677N is catalogued in gnomAD (6‑33441289‑G‑A) but has no ClinVar entry. Functional prediction tools uniformly indicate a benign effect: REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign, while premPS and ESM1b are uncertain. No tool predicts pathogenicity. High‑accuracy assessments corroborate this: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, is benign. Thus, all available evidence supports a benign classification, and there is no conflict with ClinVar status (which is absent). The variant is most likely benign. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | 6-33441289-G-A | 1 | 6.20e-7 | -7.955 | In-Between | 0.111 | Likely Benign | Likely Benign | 0.17 | Likely Benign | 0.5 | -0.15 | Likely Benign | 0.01 | Likely Benign | 0.89 | Ambiguous | 0.079 | Likely Benign | -2.05 | Neutral | 0.038 | Benign | 0.007 | Benign | 3.28 | Benign | 0.15 | Tolerated | 3.41 | 23 | 0.1763 | 0.5688 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||
| c.2030G>C | S677T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677T is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree that the substitution is benign: SIFT, PolyPhen‑2 (HumDiv and HumVar), REVEL, PROVEAN, premPS, FoldX, AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, and FATHMM all classify it as benign. No tool predicts pathogenicity. The only inconclusive results come from Rosetta (Uncertain) and Foldetta (Uncertain), which are treated as unavailable evidence. High‑accuracy assessments reinforce the benign prediction: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign, and Foldetta remains Uncertain. Overall, the consensus of available predictions indicates that S677T is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | -4.760 | Likely Benign | 0.063 | Likely Benign | Likely Benign | -0.17 | Likely Benign | 0.3 | -0.85 | Ambiguous | -0.51 | Ambiguous | 0.15 | Likely Benign | 0.070 | Likely Benign | -0.97 | Neutral | 0.008 | Benign | 0.007 | Benign | 3.32 | Benign | 0.30 | Tolerated | 0.1775 | 0.6985 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.2030G>T | S677I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign. Pathogenicity is suggested only by PROVEAN and SIFT, while Foldetta and ESM1b give uncertain results. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is benign, and Foldetta remains uncertain. Overall, the majority of evidence points to a benign impact, and this conclusion does not contradict the lack of ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | -7.942 | In-Between | 0.156 | Likely Benign | Likely Benign | -0.09 | Likely Benign | 0.0 | -2.25 | Stabilizing | -1.17 | Ambiguous | -0.01 | Likely Benign | 0.097 | Likely Benign | -2.81 | Deleterious | 0.002 | Benign | 0.002 | Benign | 3.34 | Benign | 0.05 | Affected | 0.1040 | 0.6463 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||
| c.2031T>A | S677R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict a pathogenic effect are FoldX, ESM1b, and AlphaMissense‑Default. Tools with uncertain or inconclusive results are Rosetta, Foldetta, and premPS. High‑accuracy methods give mixed signals: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta is uncertain. Overall, the majority of consensus tools (six benign vs. three pathogenic) lean toward a benign interpretation. This prediction does not contradict any ClinVar status, as none is available. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | -9.388 | Likely Pathogenic | 0.714 | Likely Pathogenic | Likely Benign | 2.59 | Destabilizing | 0.1 | 0.73 | Ambiguous | 1.66 | Ambiguous | 0.71 | Ambiguous | 0.150 | Likely Benign | -2.49 | Neutral | 0.385 | Benign | 0.037 | Benign | 3.31 | Benign | 0.12 | Tolerated | 0.1120 | 0.4195 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||
| c.2031T>G | S677R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S677R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and FATHMM. Those that predict a pathogenic effect are FoldX, ESM1b, and AlphaMissense‑Default. Tools with uncertain or inconclusive results are Rosetta, Foldetta, and premPS. High‑accuracy methods give mixed signals: AlphaMissense‑Optimized predicts benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑to‑2 split, and Foldetta is uncertain. Overall, the majority of consensus tools (six benign vs. three pathogenic) lean toward a benign interpretation. This prediction does not contradict any ClinVar status, as none is available. Thus, the variant is most likely benign based on current computational evidence. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.194234 | Structured | 0.115685 | Uncertain | 0.555 | 0.338 | 0.125 | -9.388 | Likely Pathogenic | 0.714 | Likely Pathogenic | Likely Benign | 2.59 | Destabilizing | 0.1 | 0.73 | Ambiguous | 1.66 | Ambiguous | 0.71 | Ambiguous | 0.150 | Likely Benign | -2.49 | Neutral | 0.385 | Benign | 0.037 | Benign | 3.31 | Benign | 0.12 | Tolerated | 0.1120 | 0.4195 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||
| c.2032A>C | S678R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta predicts a benign effect. Overall, the majority of tools (8 benign vs. 4 pathogenic) support a benign classification. Thus, the variant is most likely benign, and this assessment does not contradict any ClinVar status because no ClinVar entry exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | -9.708 | Likely Pathogenic | 0.878 | Likely Pathogenic | Ambiguous | -0.37 | Likely Benign | 0.2 | 0.48 | Likely Benign | 0.06 | Likely Benign | 0.32 | Likely Benign | 0.106 | Likely Benign | -2.07 | Neutral | 0.454 | Possibly Damaging | 0.057 | Benign | 3.44 | Benign | 0.02 | Affected | 0.0950 | 0.3519 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||
| c.2032A>G | S678G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678G is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include AlphaMissense‑Default, AlphaMissense‑Optimized, ESM1b, FATHMM, PROVEAN, PolyPhen‑2 HumDiv, PolyPhen‑2 HumVar, SIFT, REVEL, FoldX, premPS, and Foldetta. No tool predicts a pathogenic outcome; the only inconclusive result is from Rosetta, which is listed as “Uncertain.” High‑accuracy assessments further support a benign classification: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | -4.379 | Likely Benign | 0.069 | Likely Benign | Likely Benign | 0.24 | Likely Benign | 0.2 | 0.55 | Ambiguous | 0.40 | Likely Benign | -0.24 | Likely Benign | 0.082 | Likely Benign | 1.90 | Neutral | 0.000 | Benign | 0.001 | Benign | 3.43 | Benign | 1.00 | Tolerated | 0.2807 | 0.4961 | 1 | 0 | 0.4 | -30.03 | |||||||||||||||||||||||||||||
| c.2032A>T | S678C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678C is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that classify the variant as benign include REVEL, FoldX, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Those that predict pathogenicity are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, and SIFT. Tools with uncertain results are Rosetta and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) resolves to benign (2 benign vs. 1 pathogenic, 1 uncertain), and Foldetta also predicts benign. No prediction or folding stability result is missing or inconclusive. Overall, the preponderance of evidence indicates the variant is most likely benign, and this conclusion does not contradict any ClinVar status because the variant is not yet classified in ClinVar. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | -7.879 | In-Between | 0.095 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.2 | 0.55 | Ambiguous | 0.38 | Likely Benign | 0.35 | Likely Benign | 0.094 | Likely Benign | -3.31 | Deleterious | 0.947 | Possibly Damaging | 0.527 | Possibly Damaging | 3.37 | Benign | 0.01 | Affected | 0.1080 | 0.5875 | 0 | -1 | 3.3 | 16.06 | ||||||||||||||||||||||||||||||
| c.2033G>A | S678N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678N is not reported in ClinVar (ClinVar status: not listed) but is present in gnomAD (gnomAD ID 6‑33441292‑G‑A). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. No tool predicts a pathogenic outcome. High‑accuracy assessments are consistent: AlphaMissense‑Optimized predicts benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts benign. Overall, the variant is most likely benign, and this conclusion does not contradict ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | 6-33441292-G-A | 1 | 6.20e-7 | -3.355 | Likely Benign | 0.139 | Likely Benign | Likely Benign | -0.10 | Likely Benign | 0.1 | -0.47 | Likely Benign | -0.29 | Likely Benign | 0.38 | Likely Benign | 0.067 | Likely Benign | -0.63 | Neutral | 0.001 | Benign | 0.002 | Benign | 3.43 | Benign | 0.14 | Tolerated | 3.43 | 19 | 0.1435 | 0.4863 | 1 | 1 | -2.7 | 27.03 | ||||||||||||||||||||||||
| c.2033G>C | S678T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678T is not reported in ClinVar and is absent from gnomAD. All available in‑silico predictors classify the change as benign: REVEL, FoldX, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, FATHMM, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN). No tool predicts pathogenicity. High‑accuracy assessments likewise support a benign effect: AlphaMissense‑Optimized is benign, the SGM Consensus is “Likely Benign,” and Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign stability. Based on the unanimous computational evidence, the variant is most likely benign, and this conclusion is consistent with the absence of any ClinVar pathogenic annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | -5.389 | Likely Benign | 0.083 | Likely Benign | Likely Benign | 0.32 | Likely Benign | 0.1 | 0.41 | Likely Benign | 0.37 | Likely Benign | 0.14 | Likely Benign | 0.053 | Likely Benign | -1.82 | Neutral | 0.255 | Benign | 0.053 | Benign | 3.41 | Benign | 0.11 | Tolerated | 0.1494 | 0.6351 | 1 | 1 | 0.1 | 14.03 | |||||||||||||||||||||||||||||
| c.2033G>T | S678I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678I is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 (HumDiv and HumVar), FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, SIFT, and ESM1b. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta as benign, and the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive due to a 2‑vs‑2 split. Overall, the majority of evidence (10 benign vs. 3 pathogenic) supports a benign classification. This consensus does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | -9.735 | Likely Pathogenic | 0.253 | Likely Benign | Likely Benign | 0.36 | Likely Benign | 0.3 | -0.01 | Likely Benign | 0.18 | Likely Benign | -0.11 | Likely Benign | 0.119 | Likely Benign | -3.99 | Deleterious | 0.294 | Benign | 0.057 | Benign | 3.45 | Benign | 0.01 | Affected | 0.0941 | 0.5954 | -1 | -2 | 5.3 | 26.08 | ||||||||||||||||||||||||||||||
| c.2034C>A | S678R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678R is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, and FATHMM. Tools that predict a pathogenic effect are SIFT, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is classified as uncertain. High‑accuracy assessments show AlphaMissense‑Optimized uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive (2 pathogenic vs. 2 benign), and Foldetta predicts a benign effect. Overall, the majority of evidence (8 benign vs. 4 pathogenic) supports a benign classification. This conclusion does not contradict ClinVar status, as no ClinVar assertion exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | -9.708 | Likely Pathogenic | 0.878 | Likely Pathogenic | Ambiguous | -0.37 | Likely Benign | 0.2 | 0.48 | Likely Benign | 0.06 | Likely Benign | 0.32 | Likely Benign | 0.158 | Likely Benign | -2.07 | Neutral | 0.454 | Possibly Damaging | 0.057 | Benign | 3.44 | Benign | 0.02 | Affected | 0.0950 | 0.3519 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||
| c.2034C>G | S678R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant S678R is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are polyPhen‑2 HumDiv, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is classified as uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is inconclusive, and Foldetta predicts a benign stability change. Overall, the majority of evidence points to a benign impact. This conclusion does not contradict the ClinVar status, which currently has no classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.301917 | Structured | 0.123585 | Uncertain | 0.660 | 0.321 | 0.000 | -9.708 | Likely Pathogenic | 0.878 | Likely Pathogenic | Ambiguous | -0.37 | Likely Benign | 0.2 | 0.48 | Likely Benign | 0.06 | Likely Benign | 0.32 | Likely Benign | 0.157 | Likely Benign | -2.07 | Neutral | 0.454 | Possibly Damaging | 0.057 | Benign | 3.44 | Benign | 0.02 | Affected | 0.0950 | 0.3519 | 0 | -1 | -3.7 | 69.11 | ||||||||||||||||||||||||||||||
| c.2035T>A | F679I 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F679I is not reported in ClinVar and is absent from gnomAD. Functional prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM. In contrast, a majority of predictors—PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—consistently classify the substitution as pathogenic, and the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also reports it as Likely Pathogenic. High‑accuracy assessments are inconclusive: AlphaMissense‑Optimized is uncertain, Foldetta (combining FoldX‑MD and Rosetta outputs) is uncertain, and the SGM Consensus remains pathogenic. Because the uncertain results are treated as unavailable, the clear majority of predictions support pathogenicity. Thus, the variant is most likely pathogenic, and this assessment does not contradict ClinVar status, which currently has no entry for F679I. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.129316 | Uncertain | 0.700 | 0.320 | 0.000 | -12.620 | Likely Pathogenic | 0.935 | Likely Pathogenic | Ambiguous | 1.62 | Ambiguous | 0.4 | -0.27 | Likely Benign | 0.68 | Ambiguous | 0.88 | Ambiguous | 0.498 | Likely Benign | -5.91 | Deleterious | 0.993 | Probably Damaging | 0.977 | Probably Damaging | 3.59 | Benign | 0.01 | Affected | 0.1725 | 0.2322 | 1 | 0 | 1.7 | -34.02 | |||||||||||||||||||||||||||||
| c.2035T>C | F679L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F679L is not reported in ClinVar (ClinVar status: not listed) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include Rosetta, Foldetta, and FATHMM, while the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Two tools (FoldX and premPS) give uncertain results. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the preponderance of evidence points to a pathogenic effect for F679L, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.129316 | Uncertain | 0.700 | 0.320 | 0.000 | -11.395 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.07 | Ambiguous | 0.0 | -0.36 | Likely Benign | 0.36 | Likely Benign | 0.82 | Ambiguous | 0.506 | Likely Pathogenic | -5.91 | Deleterious | 0.982 | Probably Damaging | 0.952 | Probably Damaging | 3.54 | Benign | 0.03 | Affected | 0.1918 | 0.3493 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.2035T>G | F679V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F679V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, and FATHMM, whereas pathogenic predictions arise from PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments further support a deleterious effect: the SGM Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) reports the variant as Likely Pathogenic, while AlphaMissense‑Optimized and Foldetta provide inconclusive results and are treated as unavailable. Overall, the balance of evidence favors a pathogenic classification, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.129316 | Uncertain | 0.700 | 0.320 | 0.000 | -11.868 | Likely Pathogenic | 0.900 | Likely Pathogenic | Ambiguous | 1.92 | Ambiguous | 0.5 | 0.29 | Likely Benign | 1.11 | Ambiguous | 0.89 | Ambiguous | 0.497 | Likely Benign | -6.86 | Deleterious | 0.993 | Probably Damaging | 0.968 | Probably Damaging | 3.50 | Benign | 0.01 | Affected | 0.1830 | 0.2349 | -1 | -1 | 1.4 | -48.04 | |||||||||||||||||||||||||||||
| c.2036T>A | F679Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F679Y is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, SIFT, ESM1b, FATHMM, and AlphaMissense‑Optimized. Those that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, and polyPhen‑2 HumVar. Two tools give uncertain results: premPS and AlphaMissense‑Default. High‑accuracy assessments further support a benign outcome: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) resolves to benign; and Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, also predicts benign. Overall, the majority of evidence indicates that F679Y is most likely benign, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.222385 | Structured | 0.129316 | Uncertain | 0.700 | 0.320 | 0.000 | -5.842 | Likely Benign | 0.462 | Ambiguous | Likely Benign | 0.48 | Likely Benign | 0.2 | 0.13 | Likely Benign | 0.31 | Likely Benign | 0.71 | Ambiguous | 0.315 | Likely Benign | -2.71 | Deleterious | 0.993 | Probably Damaging | 0.952 | Probably Damaging | 3.47 | Benign | 0.14 | Tolerated | 0.1306 | 0.1954 | 7 | 3 | -4.1 | 16.00 | ||||||||||||||||||||||||||||||
| c.2036T>C | F679S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F679S is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: benign predictions are limited to FATHMM, while the remaining 12 tools (SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) all classify the variant as pathogenic. FoldX reports an uncertain outcome and is therefore not counted as evidence. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also predicts pathogenicity. Based on the preponderance of pathogenic predictions and the absence of any benign consensus, the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar status (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.129316 | Uncertain | 0.700 | 0.320 | 0.000 | -12.159 | Likely Pathogenic | 0.963 | Likely Pathogenic | Likely Pathogenic | 1.86 | Ambiguous | 0.4 | 2.20 | Destabilizing | 2.03 | Destabilizing | 1.28 | Destabilizing | 0.575 | Likely Pathogenic | -7.86 | Deleterious | 0.998 | Probably Damaging | 0.986 | Probably Damaging | 3.50 | Benign | 0.04 | Affected | 0.4276 | 0.0200 | -3 | -2 | -3.6 | -60.10 | |||||||||||||||||||||||||||||
| c.2036T>G | F679C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F679C has no ClinVar entry and is not reported in gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, whereas SGM‑Consensus, REVEL, Rosetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the variant as pathogenic. FoldX and Foldetta are uncertain and are treated as unavailable evidence. High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta remains inconclusive. Overall, the preponderance of evidence points to a pathogenic impact for F679C. This prediction is not contradicted by ClinVar status, which currently lacks any classification for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.129316 | Uncertain | 0.700 | 0.320 | 0.000 | -10.269 | Likely Pathogenic | 0.958 | Likely Pathogenic | Likely Pathogenic | 1.65 | Ambiguous | 0.3 | 2.02 | Destabilizing | 1.84 | Ambiguous | 1.17 | Destabilizing | 0.532 | Likely Pathogenic | -7.86 | Deleterious | 0.999 | Probably Damaging | 0.993 | Probably Damaging | 3.40 | Benign | 0.00 | Affected | 0.2344 | 0.0949 | -4 | -2 | -0.3 | -44.04 | |||||||||||||||||||||||||||||
| c.2037T>A | F679L 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant F679L is not reported in ClinVar and is absent from gnomAD. Functional prediction tools cluster into two groups: benign predictions come from REVEL, Rosetta, FATHMM, and Foldetta, whereas pathogenic predictions are made by SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. Uncertain results from FoldX and premPS are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, SGM‑Consensus as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) as benign. The majority of evidence points toward a pathogenic effect, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.129316 | Uncertain | 0.700 | 0.320 | 0.000 | -11.395 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.07 | Ambiguous | 0.0 | -0.36 | Likely Benign | 0.36 | Likely Benign | 0.82 | Ambiguous | 0.287 | Likely Benign | -5.91 | Deleterious | 0.982 | Probably Damaging | 0.952 | Probably Damaging | 3.54 | Benign | 0.03 | Affected | 0.1918 | 0.3493 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.2037T>G | F679L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant F679L is not reported in ClinVar (ClinVar status: none) and is absent from gnomAD (gnomAD ID: none). Prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM, whereas a majority of tools (PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact; FoldX and premPS are inconclusive. High‑accuracy methods give a mixed picture: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also predicts pathogenic, while Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, the balance of evidence favors a pathogenic classification, and this assessment does not contradict any ClinVar annotation because no ClinVar claim exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.222385 | Structured | 0.129316 | Uncertain | 0.700 | 0.320 | 0.000 | -11.395 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.07 | Ambiguous | 0.0 | -0.36 | Likely Benign | 0.36 | Likely Benign | 0.82 | Ambiguous | 0.287 | Likely Benign | -5.91 | Deleterious | 0.982 | Probably Damaging | 0.952 | Probably Damaging | 3.54 | Benign | 0.03 | Affected | 0.1918 | 0.3493 | 2 | 0 | 1.0 | -34.02 | |||||||||||||||||||||||||||||
| c.2038G>A | E680K 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E680K missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, polyPhen‑2 HumVar, and FATHMM. Those that predict a pathogenic effect are SIFT, PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as Benign. With a majority of individual tools and the SGM‑Consensus indicating pathogenicity, the variant is most likely pathogenic. This assessment does not contradict ClinVar status, as no ClinVar classification exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.209395 | Structured | 0.136843 | Uncertain | 0.636 | 0.320 | 0.000 | -12.728 | Likely Pathogenic | 0.901 | Likely Pathogenic | Ambiguous | -0.10 | Likely Benign | 0.4 | -0.15 | Likely Benign | -0.13 | Likely Benign | 0.33 | Likely Benign | 0.417 | Likely Benign | -3.54 | Deleterious | 0.959 | Probably Damaging | 0.411 | Benign | 3.49 | Benign | 0.02 | Affected | 0.3048 | 0.7553 | 0 | 1 | -0.4 | -0.94 | |||||||||||||||||||||||||||||
| c.2038G>C | E680Q 2D AIThe SynGAP1 missense variant E680Q is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, FoldX, Rosetta, premPS, SIFT, FATHMM, AlphaMissense‑Optimized, and Foldetta. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of evidence points toward a benign impact for E680Q. This conclusion is not contradicted by ClinVar, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.209395 | Structured | 0.136843 | Uncertain | 0.636 | 0.320 | 0.000 | -10.502 | Likely Pathogenic | 0.742 | Likely Pathogenic | Likely Benign | -0.01 | Likely Benign | 0.7 | -0.01 | Likely Benign | -0.01 | Likely Benign | -0.10 | Likely Benign | 0.239 | Likely Benign | -2.58 | Deleterious | 0.981 | Probably Damaging | 0.483 | Possibly Damaging | 3.47 | Benign | 0.15 | Tolerated | 0.1751 | 0.7241 | 2 | 2 | 0.0 | -0.98 | |||||||||||||||||||||||||||||
| c.2039A>C | E680A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E680A missense variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, premPS, SIFT, and FATHMM. Those that predict a pathogenic impact are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. Two tools give uncertain results: Rosetta and AlphaMissense‑Optimized. High‑accuracy assessments show that the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) classifies the variant as likely pathogenic, whereas Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, predicts a benign effect. AlphaMissense‑Optimized remains inconclusive. Overall, the majority of predictions lean toward pathogenicity, but the conflicting high‑accuracy results leave the classification uncertain. The variant is most likely pathogenic based on the prevailing evidence, and this assessment does not contradict the ClinVar status, which currently has no entry for this change. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.209395 | Structured | 0.136843 | Uncertain | 0.636 | 0.320 | 0.000 | -11.338 | Likely Pathogenic | 0.819 | Likely Pathogenic | Ambiguous | 0.34 | Likely Benign | 0.2 | 0.59 | Ambiguous | 0.47 | Likely Benign | 0.30 | Likely Benign | 0.444 | Likely Benign | -5.22 | Deleterious | 0.935 | Possibly Damaging | 0.490 | Possibly Damaging | 3.47 | Benign | 0.09 | Tolerated | 0.3931 | 0.6989 | 0 | -1 | 5.3 | -58.04 | |||||||||||||||||||||||||||||
| c.2039A>G | E680G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant E680G is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, Rosetta, Foldetta, premPS, FATHMM, and AlphaMissense‑Optimized. Those that predict pathogenicity are SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized as benign, SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as benign. Overall, the majority of tools lean toward a pathogenic effect, with high‑accuracy methods split but tipping toward pathogenicity. The variant’s status in ClinVar is unknown, so there is no contradiction between the predictions and existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.209395 | Structured | 0.136843 | Uncertain | 0.636 | 0.320 | 0.000 | -11.396 | Likely Pathogenic | 0.743 | Likely Pathogenic | Likely Benign | 0.67 | Ambiguous | 0.2 | 0.11 | Likely Benign | 0.39 | Likely Benign | 0.44 | Likely Benign | 0.411 | Likely Benign | -5.48 | Deleterious | 0.998 | Probably Damaging | 0.739 | Possibly Damaging | 3.47 | Benign | 0.01 | Affected | 0.2708 | 0.6228 | 0 | -2 | 3.1 | -72.06 | |||||||||||||||||||||||||||||
| c.2039A>T | E680V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 E680V missense change is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, FATHMM, premPS, and Foldetta. Tools that predict a pathogenic effect are SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. AlphaMissense‑Optimized is uncertain, and Rosetta is inconclusive. High‑accuracy methods give mixed results: AlphaMissense‑Optimized is uncertain; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely pathogenic; Foldetta (combining FoldX‑MD and Rosetta outputs) predicts benign. Overall, more tools (seven) predict pathogenicity than benign (five), and the high‑accuracy consensus leans toward pathogenic. Thus, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status because none is available. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.209395 | Structured | 0.136843 | Uncertain | 0.636 | 0.320 | 0.000 | -12.051 | Likely Pathogenic | 0.936 | Likely Pathogenic | Ambiguous | 0.46 | Likely Benign | 0.3 | -1.08 | Ambiguous | -0.31 | Likely Benign | 0.18 | Likely Benign | 0.454 | Likely Benign | -6.21 | Deleterious | 0.988 | Probably Damaging | 0.606 | Possibly Damaging | 3.47 | Benign | 0.01 | Affected | 0.1091 | 0.7518 | -2 | -2 | 7.7 | -29.98 | |||||||||||||||||||||||||||||
| c.203T>A | L68Q 2D AIThe SynGAP1 missense variant L68Q is listed in gnomAD (6‑33425811‑T‑A) but has no ClinVar entry. Functional prediction tools cluster into two groups: benign predictions from REVEL, PROVEAN, ESM1b, and FATHMM; pathogenic predictions from polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and AlphaMissense‑Default. The SGM‑Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports a likely benign outcome, reflecting the majority of benign calls. High‑accuracy assessments are limited: AlphaMissense‑Optimized is uncertain, and Foldetta results are not available. Based on the available evidence, the variant is most likely benign, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.470567 | Uncertain | 0.405 | 0.768 | 0.250 | 6-33425811-T-A | 5 | 3.10e-6 | -3.436 | Likely Benign | 0.826 | Likely Pathogenic | Ambiguous | 0.119 | Likely Benign | -0.14 | Neutral | 0.943 | Possibly Damaging | 0.766 | Possibly Damaging | 4.15 | Benign | 0.00 | Affected | 4.32 | 1 | 0.0864 | 0.0919 | -2 | -2 | -7.3 | 14.97 | ||||||||||||||||||||||||||||||||||
| c.203T>C | L68P 2D AIThe SynGAP1 missense variant L68P is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (Likely Benign). Tools that predict pathogenicity are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, while the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) indicates likely benign. Foldetta, a protein‑folding stability method combining FoldX‑MD and Rosetta outputs, has no available result for this variant. Overall, the majority of predictions lean toward pathogenic, but the consensus from SGM and the high‑accuracy AlphaMissense‑Optimized are contradictory, leaving the variant’s clinical significance uncertain. No conflict exists with ClinVar status, which has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.470567 | Uncertain | 0.405 | 0.768 | 0.250 | -2.122 | Likely Benign | 0.956 | Likely Pathogenic | Likely Pathogenic | 0.143 | Likely Benign | -0.57 | Neutral | 0.943 | Possibly Damaging | 0.766 | Possibly Damaging | 4.09 | Benign | 0.00 | Affected | 0.2827 | 0.1382 | -3 | -3 | -5.4 | -16.04 | |||||||||||||||||||||||||||||||||||||||
| c.203T>G | L68R 2D AIThe SynGAP1 missense variant L68R is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD (no gnomAD ID). Prediction tools that agree on a benign effect include REVEL, PROVEAN, ESM1b, FATHMM, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), which collectively suggest a likely benign impact. In contrast, polyPhen‑2 (HumDiv and HumVar), SIFT, and AlphaMissense‑Default predict a pathogenic effect. The high‑accuracy AlphaMissense‑Optimized result is uncertain, and Foldetta stability analysis is unavailable. Overall, the balance of evidence leans toward a benign interpretation, and this assessment does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | 0.534167 | Disordered | 0.470567 | Uncertain | 0.405 | 0.768 | 0.250 | -3.427 | Likely Benign | 0.865 | Likely Pathogenic | Ambiguous | 0.147 | Likely Benign | -0.61 | Neutral | 0.943 | Possibly Damaging | 0.766 | Possibly Damaging | 4.13 | Benign | 0.00 | Affected | 0.1178 | 0.0519 | -3 | -2 | -8.3 | 43.03 | |||||||||||||||||||||||||||||||||||||||
| c.2040G>C | E680D 2D AIThe SynGAP1 missense variant E680D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a benign effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify it as benign. Only polyPhen‑2 HumDiv predicts a pathogenic outcome. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely benign, and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No prediction or folding stability result is missing or inconclusive. Based on the collective evidence, the variant is most likely benign, and this conclusion does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.209395 | Structured | 0.136843 | Uncertain | 0.636 | 0.320 | 0.000 | -5.019 | Likely Benign | 0.289 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.8 | 0.27 | Likely Benign | 0.24 | Likely Benign | -0.10 | Likely Benign | 0.121 | Likely Benign | -0.64 | Neutral | 0.906 | Possibly Damaging | 0.295 | Benign | 3.66 | Benign | 0.32 | Tolerated | 0.2199 | 0.5208 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.2040G>T | E680D 2D AIThe SynGAP1 missense variant E680D is not reported in ClinVar (no ClinVar ID) and is absent from gnomAD. Functional prediction tools largely agree on a benign effect: REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumVar, SIFT, ESM1b, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict benign or likely benign. Only polyPhen‑2 HumDiv flags the variant as pathogenic, representing the sole discordant prediction. High‑accuracy assessments reinforce the benign consensus: AlphaMissense‑Optimized is benign; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Benign; and Foldetta (combining FoldX‑MD and Rosetta outputs) is benign. No prediction or folding‑stability result is missing or inconclusive. **Based on the aggregate predictions, the variant is most likely benign, and this conclusion is not contradicted by any ClinVar status.** Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Benign | GAP | 0.209395 | Structured | 0.136843 | Uncertain | 0.636 | 0.320 | 0.000 | -5.019 | Likely Benign | 0.289 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.8 | 0.27 | Likely Benign | 0.24 | Likely Benign | -0.10 | Likely Benign | 0.121 | Likely Benign | -0.64 | Neutral | 0.906 | Possibly Damaging | 0.295 | Benign | 3.66 | Benign | 0.32 | Tolerated | 0.2199 | 0.5208 | 3 | 2 | 0.0 | -14.03 | |||||||||||||||||||||||||||||
| c.2041G>A | G681S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G681S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, premPS, SIFT, and FATHMM, while those that predict a pathogenic outcome are SGM‑Consensus, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, and AlphaMissense‑Default. High‑accuracy assessments show AlphaMissense‑Optimized classifying the variant as benign, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) labeling it likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) yielding an uncertain result. Overall, the majority of tools lean toward pathogenicity, but the presence of a benign prediction from AlphaMissense‑Optimized and an uncertain Foldetta score leaves the assessment inconclusive. No ClinVar entry exists, so there is no contradiction with clinical database status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | -9.913 | Likely Pathogenic | 0.716 | Likely Pathogenic | Likely Benign | 2.11 | Destabilizing | 1.3 | -0.23 | Likely Benign | 0.94 | Ambiguous | 0.41 | Likely Benign | 0.483 | Likely Benign | -5.99 | Deleterious | 0.997 | Probably Damaging | 0.780 | Possibly Damaging | 3.45 | Benign | 0.08 | Tolerated | 0.2591 | 0.4584 | 1 | 0 | -0.4 | 30.03 | |||||||||||||||||||||||||||||
| c.2041G>C | G681R 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G681R is listed in gnomAD (6-33441300-G-C) but has no ClinVar entry. In silico predictors largely converge on a deleterious effect: benign calls are limited to FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—report pathogenicity. premPS is inconclusive. High‑accuracy assessments reinforce this trend: AlphaMissense‑Optimized predicts pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) indicates pathogenic folding instability. No prediction tool suggests a benign outcome, and the variant’s presence in gnomAD does not alter the consensus. Thus, the variant is most likely pathogenic, with no conflict with ClinVar status because no ClinVar claim exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | 6-33441300-G-C | 1 | 6.20e-7 | -12.170 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 2.25 | Destabilizing | 1.7 | 5.46 | Destabilizing | 3.86 | Destabilizing | 0.99 | Ambiguous | 0.556 | Likely Pathogenic | -7.98 | Deleterious | 0.999 | Probably Damaging | 0.928 | Probably Damaging | 3.42 | Benign | 0.00 | Affected | 3.43 | 14 | 0.1022 | 0.3879 | -2 | -3 | -4.1 | 99.14 | ||||||||||||||||||||||||
| c.2041G>T | G681C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G681C is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all classify it as pathogenic, while only FATHMM predicts a benign outcome. Uncertain calls come from FoldX, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments reinforce the pathogenic signal: the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is pathogenic, Foldetta (combining FoldX‑MD and Rosetta) is pathogenic, and AlphaMissense‑Optimized remains inconclusive. Overall, the evidence strongly favors a pathogenic interpretation, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | -12.374 | Likely Pathogenic | 0.941 | Likely Pathogenic | Ambiguous | 1.89 | Ambiguous | 1.3 | 2.63 | Destabilizing | 2.26 | Destabilizing | 0.66 | Ambiguous | 0.554 | Likely Pathogenic | -8.98 | Deleterious | 1.000 | Probably Damaging | 0.959 | Probably Damaging | 3.33 | Benign | 0.00 | Affected | 0.1194 | 0.3886 | -3 | -3 | 2.9 | 46.09 | |||||||||||||||||||||||||||||
| c.2042G>A | G681D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G681D is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: benign predictions come from REVEL and FATHMM, whereas the remaining 11 tools (FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) and the SGM‑Consensus score all predict pathogenicity. The high‑accuracy methods reinforce this view: AlphaMissense‑Optimized returns a pathogenic classification; the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta stability outputs, also predicts a pathogenic effect. premPS is inconclusive and is treated as unavailable. Taken together, the overwhelming majority of evidence points to a pathogenic impact for G681D. This conclusion is consistent with the absence of a ClinVar entry, so there is no contradiction with existing clinical annotations. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | -12.451 | Likely Pathogenic | 0.966 | Likely Pathogenic | Likely Pathogenic | 2.62 | Destabilizing | 1.5 | 4.54 | Destabilizing | 3.58 | Destabilizing | 0.96 | Ambiguous | 0.471 | Likely Benign | -6.98 | Deleterious | 0.999 | Probably Damaging | 0.840 | Possibly Damaging | 3.45 | Benign | 0.00 | Affected | 0.1697 | 0.1695 | 1 | -1 | -3.1 | 58.04 | |||||||||||||||||||||||||||||
| c.2042G>C | G681A 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant G681A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools show mixed results: benign calls come from REVEL, polyPhen‑2 HumVar, SIFT, and FATHMM, while pathogenic calls are made by PROVEAN, polyPhen‑2 HumDiv, ESM1b, and AlphaMissense‑Default. Predictions labeled uncertain include FoldX, Rosetta, Foldetta, premPS, and AlphaMissense‑Optimized. High‑accuracy assessments further clarify the picture: AlphaMissense‑Optimized remains uncertain; the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as likely pathogenic; Foldetta, which integrates FoldX‑MD and Rosetta outputs, is also uncertain. Overall, the preponderance of evidence points to a pathogenic effect for G681A. This conclusion is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | -11.958 | Likely Pathogenic | 0.836 | Likely Pathogenic | Ambiguous | 1.77 | Ambiguous | 1.0 | 0.89 | Ambiguous | 1.33 | Ambiguous | 0.68 | Ambiguous | 0.354 | Likely Benign | -5.99 | Deleterious | 0.968 | Probably Damaging | 0.427 | Benign | 3.41 | Benign | 0.07 | Tolerated | 0.3879 | 0.4401 | 1 | 0 | 2.2 | 14.03 | |||||||||||||||||||||||||||||
| c.2042G>T | G681V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant G681V is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect are limited to FATHMM, whereas the majority of algorithms—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default—classify the change as pathogenic. High‑accuracy methods give the following results: AlphaMissense‑Optimized is uncertain; SGM‑Consensus predicts a likely pathogenic effect; Foldetta, which integrates FoldX‑MD and Rosetta stability calculations, reports a pathogenic outcome. No other high‑confidence predictions are available. Taken together, the consensus of pathogenic predictions outweighs the single benign call, indicating that G681V is most likely pathogenic. This assessment is not contradicted by ClinVar, which contains no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.301917 | Structured | 0.140647 | Uncertain | 0.694 | 0.320 | 0.000 | -14.043 | Likely Pathogenic | 0.953 | Likely Pathogenic | Ambiguous | 3.21 | Destabilizing | 2.0 | 6.12 | Destabilizing | 4.67 | Destabilizing | 0.64 | Ambiguous | 0.572 | Likely Pathogenic | -8.98 | Deleterious | 0.999 | Probably Damaging | 0.928 | Probably Damaging | 3.33 | Benign | 0.01 | Affected | 0.1350 | 0.3840 | -1 | -3 | 4.6 | 42.08 | |||||||||||||||||||||||||||||
| c.2044T>A | Y682N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 Y682N variant is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include only FATHMM, while the majority of tools (SGM‑Consensus, REVEL, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict a pathogenic impact; FoldX and AlphaMissense‑Optimized are uncertain. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM‑Consensus as likely pathogenic (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN), and Foldetta as pathogenic. Overall, the evidence strongly favors a pathogenic effect, and this conclusion does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.141467 | Uncertain | 0.758 | 0.328 | 0.000 | -11.734 | Likely Pathogenic | 0.859 | Likely Pathogenic | Ambiguous | 1.86 | Ambiguous | 0.1 | 2.22 | Destabilizing | 2.04 | Destabilizing | 1.54 | Destabilizing | 0.564 | Likely Pathogenic | -8.61 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.34 | Benign | 0.02 | Affected | 0.2354 | 0.0928 | -2 | -2 | -2.2 | -49.07 | |||||||||||||||||||||||||||||
| c.2044T>C | Y682H 2D 3DClick to see structure in 3D Viewer AISynGAP1 missense variant Y682H is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify the variant as benign include REVEL and FATHMM, whereas the majority of other in silico predictors (premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default) predict it to be pathogenic. The high‑accuracy consensus method SGM Consensus, which aggregates AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, reports the variant as Likely Pathogenic. AlphaMissense‑Optimized and the protein‑folding stability predictor Foldetta both return uncertain results, and FoldX and Rosetta individually are inconclusive. Overall, the preponderance of pathogenic predictions outweighs the benign ones, indicating that Y682H is most likely pathogenic. This assessment does not contradict ClinVar status, as the variant has no ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.141467 | Uncertain | 0.758 | 0.328 | 0.000 | -9.255 | Likely Pathogenic | 0.902 | Likely Pathogenic | Ambiguous | 1.78 | Ambiguous | 0.0 | 0.56 | Ambiguous | 1.17 | Ambiguous | 1.23 | Destabilizing | 0.399 | Likely Benign | -4.58 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.32 | Benign | 0.03 | Affected | 0.2405 | 0.0868 | 0 | 2 | -1.9 | -26.03 | |||||||||||||||||||||||||||||
| c.2044T>G | Y682D 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y682D is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: FATHMM is the sole benign predictor, whereas SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all classify the change as pathogenic. The high‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports a pathogenic effect. With no ClinVar assertion to oppose these findings, the variant is most likely pathogenic. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.141467 | Uncertain | 0.758 | 0.328 | 0.000 | -15.094 | Likely Pathogenic | 0.957 | Likely Pathogenic | Likely Pathogenic | 2.81 | Destabilizing | 0.4 | 3.06 | Destabilizing | 2.94 | Destabilizing | 0.96 | Ambiguous | 0.639 | Likely Pathogenic | -9.60 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.32 | Benign | 0.01 | Affected | 0.4191 | 0.0760 | -4 | -3 | -2.2 | -48.09 | |||||||||||||||||||||||||||||
| c.2045A>C | Y682S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y682S has no ClinVar entry and is not reported in gnomAD. Prediction tools that agree on a benign effect are SIFT and FATHMM, whereas the majority of tools predict a pathogenic impact: REVEL, FoldX, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, ESM1b, AlphaMissense‑Default, and the SGM‑Consensus score (Likely Pathogenic). High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. Overall, the preponderance of evidence points to a pathogenic effect for Y682S, and this conclusion does not contradict the ClinVar status, which is currently unreported. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.141467 | Uncertain | 0.758 | 0.328 | 0.000 | -11.058 | Likely Pathogenic | 0.894 | Likely Pathogenic | Ambiguous | 2.12 | Destabilizing | 0.1 | 1.12 | Ambiguous | 1.62 | Ambiguous | 0.88 | Ambiguous | 0.552 | Likely Pathogenic | -8.64 | Deleterious | 1.000 | Probably Damaging | 0.999 | Probably Damaging | 3.42 | Benign | 0.12 | Tolerated | 0.4487 | 0.2343 | -3 | -2 | 0.5 | -76.10 | |||||||||||||||||||||||||||||
| c.2045A>G | Y682C 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y682C is not reported in ClinVar and is absent from gnomAD. Prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all predict pathogenicity, while only FATHMM predicts a benign outcome. The remaining tools (FoldX, Rosetta, Foldetta, AlphaMissense‑Optimized) are uncertain. High‑accuracy assessments reinforce this trend: the SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is pathogenic; AlphaMissense‑Optimized is uncertain; and Foldetta is uncertain. Overall, the preponderance of evidence points to a pathogenic impact for Y682C. This conclusion is consistent with the lack of ClinVar annotation, so there is no contradiction with existing clinical data. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.206376 | Structured | 0.141467 | Uncertain | 0.758 | 0.328 | 0.000 | -10.023 | Likely Pathogenic | 0.793 | Likely Pathogenic | Ambiguous | 1.79 | Ambiguous | 0.1 | 1.51 | Ambiguous | 1.65 | Ambiguous | 1.11 | Destabilizing | 0.559 | Likely Pathogenic | -8.71 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.33 | Benign | 0.01 | Affected | 0.2949 | 0.2399 | 0 | -2 | 3.8 | -60.04 | |||||||||||||||||||||||||||||
| c.2045A>T | Y682F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant Y682F is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect include REVEL, FoldX, Rosetta, Foldetta, premPS, FATHMM, AlphaMissense‑Default, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, and ESM1b. The SGM Consensus, which is a majority vote among AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, is inconclusive (two benign vs. two pathogenic votes) and is treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign, Foldetta (combining FoldX‑MD and Rosetta outputs) as benign, and the SGM Consensus remains unavailable. Overall, the balance of evidence—both from general predictors and from the high‑accuracy tools—leans toward a benign classification. This conclusion does not contradict any ClinVar annotation, as none exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.206376 | Structured | 0.141467 | Uncertain | 0.758 | 0.328 | 0.000 | -9.740 | Likely Pathogenic | 0.225 | Likely Benign | Likely Benign | 0.21 | Likely Benign | 0.1 | -0.09 | Likely Benign | 0.06 | Likely Benign | 0.42 | Likely Benign | 0.278 | Likely Benign | -3.72 | Deleterious | 0.997 | Probably Damaging | 0.947 | Probably Damaging | 3.40 | Benign | 0.04 | Affected | 0.2452 | 0.3662 | 7 | 3 | 4.1 | -16.00 | ||||||||||||||||||||||||||||||
| c.2047A>C | I683L 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I683L is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, PROVEAN, polyPhen‑2 (HumDiv and HumVar), FATHMM, and AlphaMissense‑Optimized. Tools that predict a pathogenic effect are SIFT and ESM1b. Remaining methods (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results and are treated as unavailable. High‑accuracy assessments show AlphaMissense‑Optimized as benign; the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) also yields a benign prediction, while Foldetta remains uncertain. Overall, the balance of evidence favors a benign impact for I683L, and this conclusion does not contradict any ClinVar annotation (none is available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.200174 | Structured | 0.143268 | Uncertain | 0.848 | 0.314 | 0.000 | -9.988 | Likely Pathogenic | 0.488 | Ambiguous | Likely Benign | 0.63 | Ambiguous | 0.1 | 0.76 | Ambiguous | 0.70 | Ambiguous | 0.69 | Ambiguous | 0.286 | Likely Benign | -2.00 | Neutral | 0.011 | Benign | 0.056 | Benign | 3.43 | Benign | 0.04 | Affected | 0.0957 | 0.3185 | 2 | 2 | -0.7 | 0.00 | ||||||||||||||||||||||||||||||
| c.2047A>T | I683F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I683F is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, Rosetta, and FATHMM, whereas a majority of tools (SGM‑Consensus, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default) predict a pathogenic impact. High‑accuracy assessments show the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) as likely pathogenic, while AlphaMissense‑Optimized and Foldetta are inconclusive and thus treated as unavailable evidence. Overall, the balance of evidence favors a pathogenic classification for I683F, and this assessment does not contradict the current ClinVar status, which has no entry for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.143268 | Uncertain | 0.848 | 0.314 | 0.000 | -12.781 | Likely Pathogenic | 0.828 | Likely Pathogenic | Ambiguous | 1.38 | Ambiguous | 0.1 | -0.23 | Likely Benign | 0.58 | Ambiguous | 0.59 | Ambiguous | 0.481 | Likely Benign | -3.99 | Deleterious | 0.971 | Probably Damaging | 0.499 | Possibly Damaging | 3.27 | Benign | 0.01 | Affected | 0.0770 | 0.2307 | 1 | 0 | -1.7 | 34.02 | |||||||||||||||||||||||||||||
| c.2048T>A | I683N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I683N is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default all predict pathogenicity, whereas only FATHMM predicts a benign outcome. High‑accuracy assessments reinforce the pathogenic signal: AlphaMissense‑Optimized returns a pathogenic score, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is labeled Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta stability outputs) also indicates pathogenicity. No predictions are missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation (none available). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.143268 | Uncertain | 0.848 | 0.314 | 0.000 | -12.120 | Likely Pathogenic | 0.974 | Likely Pathogenic | Likely Pathogenic | 2.18 | Destabilizing | 0.1 | 2.08 | Destabilizing | 2.13 | Destabilizing | 1.46 | Destabilizing | 0.546 | Likely Pathogenic | -6.87 | Deleterious | 1.000 | Probably Damaging | 0.992 | Probably Damaging | 3.26 | Benign | 0.01 | Affected | 0.0978 | 0.1133 | -2 | -3 | -8.0 | 0.94 | |||||||||||||||||||||||||||||
| c.2048T>C | I683T 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I683T has no ClinVar record and is not reported in gnomAD. Prediction tools that agree on a benign effect include SIFT, FATHMM, and AlphaMissense‑Optimized, whereas a majority of tools predict pathogenicity: SGM‑Consensus, REVEL, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), ESM1b, and AlphaMissense‑Default. High‑accuracy assessments further support this pattern: AlphaMissense‑Optimized classifies the variant as benign, while the SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates it is likely pathogenic; the Foldetta stability analysis is inconclusive and therefore unavailable. Taken together, the preponderance of evidence points to a pathogenic effect for I683T. This conclusion does not contradict ClinVar status, which currently contains no classification for the variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.143268 | Uncertain | 0.848 | 0.314 | 0.000 | -9.891 | Likely Pathogenic | 0.775 | Likely Pathogenic | Likely Benign | 1.67 | Ambiguous | 0.1 | 1.35 | Ambiguous | 1.51 | Ambiguous | 1.25 | Destabilizing | 0.548 | Likely Pathogenic | -4.77 | Deleterious | 0.999 | Probably Damaging | 0.981 | Probably Damaging | 3.29 | Benign | 0.08 | Tolerated | 0.1090 | 0.0880 | 0 | -1 | -5.2 | -12.05 | |||||||||||||||||||||||||||||
| c.2048T>G | I683S 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant I683S is not reported in ClinVar (ClinVar ID None) and is absent from gnomAD (gnomAD ID None). Prediction tools that agree on a benign effect are limited to FATHMM, whereas the majority of tools predict a pathogenic impact: SGM‑Consensus, REVEL, FoldX, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. High‑accuracy methods give a consistent pathogenic signal: AlphaMissense‑Optimized is uncertain, SGM‑Consensus (a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is Pathogenic. No prediction or folding stability result is missing or inconclusive; all available evidence points to a deleterious effect. Based on the collective predictions, the variant is most likely pathogenic, and this assessment does not contradict the ClinVar status, which currently has no classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.200174 | Structured | 0.143268 | Uncertain | 0.848 | 0.314 | 0.000 | -11.443 | Likely Pathogenic | 0.950 | Likely Pathogenic | Ambiguous | 2.53 | Destabilizing | 0.2 | 1.94 | Ambiguous | 2.24 | Destabilizing | 1.35 | Destabilizing | 0.552 | Likely Pathogenic | -5.88 | Deleterious | 1.000 | Probably Damaging | 0.989 | Probably Damaging | 3.29 | Benign | 0.05 | Affected | 0.1936 | 0.1320 | -1 | -2 | -5.3 | -26.08 | |||||||||||||||||||||||||||||
| c.2049C>G | I683M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 I683M variant is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FATHMM, and AlphaMissense‑Optimized. Those that agree on a pathogenic effect are PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, and ESM1b. The remaining tools (FoldX, Rosetta, Foldetta, premPS, AlphaMissense‑Default) give uncertain or inconclusive results and are treated as unavailable evidence. High‑accuracy assessments show AlphaMissense‑Optimized as benign, the SGM Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) as pathogenic, and Foldetta as uncertain. Overall, the majority of definitive predictions (five pathogenic vs. three benign) point to a pathogenic impact. Thus, the variant is most likely pathogenic, and this conclusion does not contradict the ClinVar status, which currently has no entry for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.200174 | Structured | 0.143268 | Uncertain | 0.848 | 0.314 | 0.000 | -9.010 | Likely Pathogenic | 0.424 | Ambiguous | Likely Benign | 0.69 | Ambiguous | 0.1 | 0.68 | Ambiguous | 0.69 | Ambiguous | 0.74 | Ambiguous | 0.296 | Likely Benign | -2.88 | Deleterious | 0.999 | Probably Damaging | 0.986 | Probably Damaging | 3.30 | Benign | 0.01 | Affected | 0.0933 | 0.2662 | 2 | 1 | -2.6 | 18.03 | ||||||||||||||||||||||||||||||
| c.2050G>A | D684N 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684N is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that indicate a benign effect include REVEL, premPS, and FATHMM, whereas the majority of tools predict a pathogenic outcome: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus (majority vote from AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN). High‑accuracy assessments reinforce this view: AlphaMissense‑Optimized classifies the variant as pathogenic, the SGM‑Consensus also reports it as likely pathogenic, and the Foldetta stability analysis is inconclusive. Protein‑stability predictors FoldX and Rosetta likewise return uncertain results. Overall, the preponderance of evidence points to a pathogenic effect, which contradicts the current ClinVar designation of uncertainty. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | Uncertain | 1 | -13.155 | Likely Pathogenic | 0.985 | Likely Pathogenic | Likely Pathogenic | 1.47 | Ambiguous | 0.8 | 1.76 | Ambiguous | 1.62 | Ambiguous | 0.37 | Likely Benign | 0.382 | Likely Benign | -4.99 | Deleterious | 0.999 | Probably Damaging | 0.746 | Possibly Damaging | 3.39 | Benign | 0.01 | Affected | 0.1157 | 0.6373 | 2 | 1 | 0.0 | -0.98 | |||||||||||||||||||||||||||
| c.2050G>C | D684H 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684H is listed in ClinVar with an “Uncertain” status and is not reported in gnomAD. Prediction tools that agree on a benign effect include only FATHMM; all other evaluated algorithms (REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) predict a pathogenic impact, and the SGM‑Consensus score is “Likely Pathogenic.” High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No predictions are inconclusive or missing. Based on the overwhelming majority of pathogenic predictions, the variant is most likely pathogenic, which does not contradict the current ClinVar “Uncertain” classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | Uncertain | 1 | -14.194 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.36 | Destabilizing | 1.0 | 2.95 | Destabilizing | 3.16 | Destabilizing | 0.55 | Ambiguous | 0.613 | Likely Pathogenic | -6.98 | Deleterious | 1.000 | Probably Damaging | 0.972 | Probably Damaging | 3.36 | Benign | 0.00 | Affected | 3.42 | 17 | 0.1344 | 0.6618 | -1 | 1 | 0.3 | 22.05 | |||||||||||||||||||||||||
| c.2050G>T | D684Y 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684Y is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that assess the variant’s effect largely agree on a deleterious outcome: SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized all predict pathogenicity. Only premPS and FATHMM predict a benign effect. High‑accuracy methods reinforce the pathogenic signal: AlphaMissense‑Optimized is pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic; and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. No prediction or folding stability result is missing or inconclusive. Based on the overwhelming consensus of pathogenic predictions, the variant is most likely pathogenic, and this assessment does not contradict the absence of ClinVar reporting. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | -15.224 | Likely Pathogenic | 0.994 | Likely Pathogenic | Likely Pathogenic | 3.65 | Destabilizing | 1.5 | 2.12 | Destabilizing | 2.89 | Destabilizing | -0.06 | Likely Benign | 0.600 | Likely Pathogenic | -8.98 | Deleterious | 1.000 | Probably Damaging | 0.963 | Probably Damaging | 3.44 | Benign | 0.00 | Affected | 0.0575 | 0.6564 | -4 | -3 | 2.2 | 48.09 | |||||||||||||||||||||||||||||
| c.2051A>C | D684A 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684A is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely converge on a deleterious effect: pathogenic calls are made by REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while only premPS and FATHMM predict a benign outcome. High‑accuracy assessments reinforce the pathogenic interpretation: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No evidence is missing or inconclusive. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | -14.873 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.34 | Destabilizing | 1.1 | 2.85 | Destabilizing | 3.10 | Destabilizing | 0.28 | Likely Benign | 0.547 | Likely Pathogenic | -7.98 | Deleterious | 0.994 | Probably Damaging | 0.758 | Possibly Damaging | 3.42 | Benign | 0.01 | Affected | 0.3477 | 0.5423 | 0 | -2 | 5.3 | -44.01 | |||||||||||||||||||||||||||||
| c.2051A>G | D684G 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684G is not reported in ClinVar and is absent from gnomAD. Prediction tools that assess the variant’s effect largely agree on a deleterious outcome. Benign predictions come from premPS and FATHMM, whereas the remaining 12 tools—including REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—classify it as pathogenic. High‑accuracy methods reinforce this view: AlphaMissense‑Optimized predicts pathogenicity, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) also reports pathogenic. No prediction is inconclusive or missing. Consequently, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar annotation because none exists. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | -14.238 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.34 | Destabilizing | 1.0 | 4.07 | Destabilizing | 3.71 | Destabilizing | -0.30 | Likely Benign | 0.561 | Likely Pathogenic | -6.98 | Deleterious | 0.999 | Probably Damaging | 0.935 | Probably Damaging | 3.37 | Benign | 0.01 | Affected | 0.3686 | 0.5403 | 1 | -1 | 3.1 | -58.04 | |||||||||||||||||||||||||||||
| c.2051A>T | D684V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684V is not reported in ClinVar and is absent from gnomAD. Functional prediction tools largely agree on a deleterious effect: pathogenic calls come from REVEL, FoldX, Rosetta, Foldetta, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized, while only premPS and FATHMM predict a benign outcome. High‑accuracy assessments reinforce the pathogenic interpretation: AlphaMissense‑Optimized is pathogenic, the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. No evidence suggests a benign effect, and the lack of ClinVar annotation means there is no conflicting clinical classification. Therefore, the variant is most likely pathogenic, with no contradiction to ClinVar status. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | -16.128 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 3.86 | Destabilizing | 1.1 | 2.06 | Destabilizing | 2.96 | Destabilizing | 0.07 | Likely Benign | 0.601 | Likely Pathogenic | -8.98 | Deleterious | 0.901 | Possibly Damaging | 0.480 | Possibly Damaging | 3.44 | Benign | 0.00 | Affected | 0.0775 | 0.6209 | -2 | -3 | 7.7 | -15.96 | |||||||||||||||||||||||||||||
| c.2052C>A | D684E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 D684E missense variant has no ClinVar entry and is not reported in gnomAD. Functional prediction tools that agree on a benign effect include REVEL, polyPhen‑2 HumVar, and FATHMM. In contrast, a majority of predictors (SGM‑Consensus, FoldX, Foldetta, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized) indicate a pathogenic impact; predictions from Rosetta and premPS are inconclusive and are treated as unavailable. High‑accuracy assessments further support pathogenicity: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is likely pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) is pathogenic. Based on the consensus of these tools, the variant is most likely pathogenic, and this assessment does not contradict any ClinVar status (none reported). Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | -9.506 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.88 | Destabilizing | 0.9 | 1.48 | Ambiguous | 2.18 | Destabilizing | 0.66 | Ambiguous | 0.362 | Likely Benign | -3.99 | Deleterious | 0.910 | Possibly Damaging | 0.210 | Benign | 3.37 | Benign | 0.01 | Affected | 0.1316 | 0.6187 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.2052C>G | D684E 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant D684E is not reported in ClinVar and is absent from gnomAD. Prediction tools that classify it as benign include REVEL, polyPhen‑2 HumVar, and FATHMM, whereas the majority of algorithms predict a deleterious effect: FoldX, Foldetta, PROVEAN, polyPhen‑2 HumDiv, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus score (Likely Pathogenic). Two methods (Rosetta and premPS) returned uncertain results. High‑accuracy assessments further support a damaging impact: AlphaMissense‑Optimized is pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is Likely Pathogenic, and Foldetta (combining FoldX‑MD and Rosetta) is pathogenic. Overall, the computational evidence overwhelmingly indicates that D684E is pathogenic, and this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.254060 | Structured | 0.153798 | Uncertain | 0.870 | 0.282 | 0.000 | -9.506 | Likely Pathogenic | 0.996 | Likely Pathogenic | Likely Pathogenic | 2.88 | Destabilizing | 0.9 | 1.48 | Ambiguous | 2.18 | Destabilizing | 0.66 | Ambiguous | 0.362 | Likely Benign | -3.99 | Deleterious | 0.910 | Possibly Damaging | 0.210 | Benign | 3.37 | Benign | 0.01 | Affected | 0.1316 | 0.6187 | 3 | 2 | 0.0 | 14.03 | |||||||||||||||||||||||||||||
| c.2053T>A | L685M 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L685M is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include REVEL, FoldX, PROVEAN, and FATHMM, whereas those that predict a pathogenic effect are polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, and AlphaMissense‑Default. Uncertain predictions come from premPS, Foldetta, Rosetta, and AlphaMissense‑Optimized. High‑accuracy methods give inconclusive results: AlphaMissense‑Optimized is uncertain; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, PROVEAN) is a 2‑to‑2 tie and therefore uncertain; Foldetta also reports an uncertain stability change. Consequently, the overall computational evidence is mixed, with a slight tilt toward pathogenicity. Thus, the variant is most likely pathogenic based on predictions, and this assessment does not contradict the absence of a ClinVar entry. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | GAP | 0.175930 | Structured | 0.162061 | Uncertain | 0.913 | 0.280 | 0.000 | -10.790 | Likely Pathogenic | 0.902 | Likely Pathogenic | Ambiguous | 0.35 | Likely Benign | 0.1 | 1.01 | Ambiguous | 0.68 | Ambiguous | 0.83 | Ambiguous | 0.281 | Likely Benign | -2.00 | Neutral | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.29 | Benign | 0.01 | Affected | 0.0814 | 0.2758 | 4 | 2 | -1.9 | 18.03 | ||||||||||||||||||||||||||||||
| c.2053T>G | L685V 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L685V is not reported in ClinVar (ClinVar ID = None) and is absent from gnomAD (gnomAD ID = None). Prediction tools that agree on a benign effect include REVEL and FATHMM, whereas a majority of tools predict a pathogenic impact: PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, and AlphaMissense‑Default. The SGM Consensus, derived from a majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN, also indicates a likely pathogenic effect. High‑accuracy assessments show AlphaMissense‑Optimized as uncertain, SGM Consensus as pathogenic, and Foldetta (combining FoldX‑MD and Rosetta outputs) as uncertain. No other folding‑stability methods provide definitive evidence. Overall, the preponderance of pathogenic predictions, including the SGM Consensus, suggests that the variant is most likely pathogenic; this conclusion is not contradicted by any ClinVar annotation. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.162061 | Uncertain | 0.913 | 0.280 | 0.000 | -11.418 | Likely Pathogenic | 0.935 | Likely Pathogenic | Ambiguous | 1.87 | Ambiguous | 0.0 | 1.15 | Ambiguous | 1.51 | Ambiguous | 0.97 | Ambiguous | 0.214 | Likely Benign | -2.99 | Deleterious | 0.993 | Probably Damaging | 0.694 | Possibly Damaging | 3.33 | Benign | 0.02 | Affected | 0.1414 | 0.3010 | 2 | 1 | 0.4 | -14.03 | |||||||||||||||||||||||||||||
| c.2054T>C | L685S 2D  3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L685S is not reported in ClinVar (ClinVar ID: None) and is absent from gnomAD (gnomAD ID: None). Prediction tools that agree on a benign effect include only FATHMM, whereas the remaining tools—SGM‑Consensus, REVEL, FoldX, Rosetta, Foldetta, premPS, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized—consistently predict a pathogenic or likely pathogenic impact. High‑accuracy assessments further support this: AlphaMissense‑Optimized predicts pathogenic; the SGM Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) indicates likely pathogenic; and Foldetta, which integrates FoldX‑MD and Rosetta outputs, also predicts pathogenic. Based on the collective evidence, the variant is most likely pathogenic, and this conclusion does not contradict any existing ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.162061 | Uncertain | 0.913 | 0.280 | 0.000 | -12.303 | Likely Pathogenic | 0.999 | Likely Pathogenic | Likely Pathogenic | 3.08 | Destabilizing | 0.2 | 2.95 | Destabilizing | 3.02 | Destabilizing | 1.24 | Destabilizing | 0.520 | Likely Pathogenic | -5.99 | Deleterious | 1.000 | Probably Damaging | 0.996 | Probably Damaging | 3.27 | Benign | 0.01 | Affected | 0.2844 | 0.0505 | -3 | -2 | -4.6 | -26.08 | |||||||||||||||||||||||||||||
| c.2054T>G | L685W 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L685W is not reported in ClinVar and is absent from gnomAD. Prediction tools that agree on a benign effect include only FATHMM, while the majority of tools (REVEL, premPS, PROVEAN, polyPhen‑2 HumDiv, polyPhen‑2 HumVar, SIFT, ESM1b, AlphaMissense‑Default, AlphaMissense‑Optimized, and the SGM‑Consensus) predict a pathogenic impact. FoldX, Rosetta, and Foldetta provide uncertain results and are not considered evidence for either side. High‑accuracy assessments show AlphaMissense‑Optimized as pathogenic, the SGM‑Consensus as likely pathogenic, and Foldetta as uncertain. Taken together, the preponderance of evidence indicates that the variant is most likely pathogenic, and this conclusion does not contradict any ClinVar annotation because no ClinVar entry exists for this variant. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.162061 | Uncertain | 0.913 | 0.280 | 0.000 | -15.885 | Likely Pathogenic | 0.998 | Likely Pathogenic | Likely Pathogenic | 1.53 | Ambiguous | 0.2 | 1.14 | Ambiguous | 1.34 | Ambiguous | 1.14 | Destabilizing | 0.509 | Likely Pathogenic | -5.99 | Deleterious | 1.000 | Probably Damaging | 0.984 | Probably Damaging | 3.23 | Benign | 0.00 | Affected | 0.0700 | 0.2328 | -2 | -2 | -4.7 | 73.05 | |||||||||||||||||||||||||||||
| c.2055G>C | L685F 2D 3DClick to see structure in 3D Viewer AIThe SynGAP1 missense variant L685F is not reported in ClinVar and has no gnomAD entry. Functional prediction tools cluster into two groups: benign predictions come from REVEL, premPS, and FATHMM, whereas pathogenic predictions arise from SGM‑Consensus, PROVEAN, polyPhen‑2 (HumDiv and HumVar), SIFT, ESM1b, AlphaMissense‑Default, and AlphaMissense‑Optimized. High‑accuracy assessments further support a deleterious effect: AlphaMissense‑Optimized classifies the change as pathogenic, the SGM‑Consensus (majority vote of AlphaMissense‑Default, ESM1b, FATHMM, and PROVEAN) also indicates likely pathogenic, while Foldetta’s stability analysis is inconclusive. FoldX and Rosetta predictions are uncertain and are treated as unavailable. Overall, the preponderance of evidence points to a pathogenic impact for the variant, and this conclusion does not conflict with the absence of a ClinVar classification. Disclaimer: This summary was generated using AI and should be interpreted alongside expert review. | Likely Pathogenic | GAP | 0.175930 | Structured | 0.162061 | Uncertain | 0.913 | 0.280 | 0.000 | -12.304 | Likely Pathogenic | 0.992 | Likely Pathogenic | Likely Pathogenic | 1.62 | Ambiguous | 0.2 | 1.80 | Ambiguous | 1.71 | Ambiguous | 0.50 | Likely Benign | 0.300 | Likely Benign | -3.99 | Deleterious | 0.999 | Probably Damaging | 0.895 | Possibly Damaging | 3.33 | Benign | 0.01 | Affected | 0.0724 | 0.2367 | 2 | 0 | -1.0 | 34.02 | |||||||||||||||||||||||||||||
Found 8751 rows. Show 800 rows per page. Page 3/11 « Previous | Next »